Embed Size (px)
Citation preview

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 1/7
2246 Macro mo lecu les 1985 18, 2246-2252
(15) Rehage, G.; Goldbach, G. Ber. Bunsenges.Phys. Chem. 1966,70, 1144.
(16) Goldbach, G.; Rehage,G. J. Polym. Sci., Part C 1967, 16,2289.(17) Kovacs, A. J. J . Polym. Sci. 1958, 30, 131.(18) Hozumi, S.; Wakabayashi, T.; Sugihara, S. Polym. J. 1970, 1
632.(19) Uchidoi, M.; Adachi, K.; Ishida, Y. Polym. J. 1978, 10, 161.(20) Adachi, K.; Kotaka, T. Polym. J. 1982, 1 4 959.(21) Kogowski, G. J.; Filisko, F. E. Macromolecules, in press.(22) Kogowski, G. J.; Robertson, R. E.; Filisko, F. E., to be pub-
lished.(23) Oels, H. J.; Rehage, G. Macromolecules 1977, 10 1036.(24) Rehage, G. Ber. Bunsenges. Phys. Chem.1970, 74, 796.(25) Fillers, R. W.; Tschoegl, N. W. Trans. SOC . heol. 1977,21, 51.(26) Moonan,W. K.; Tschoegl, N. W. Macromolecules 1983,16,55.(27) Cogswell, F. N.; McGowan, J. C. Br. Polym. J . 1972, 4 183.(28) Plazek, D. J. J . Phys . Chem. 1965, 69, 3480.(29) Plazek, D. J.; O Rourke, V. M. J . Polym. Sci., Part A - 2 1971,
9, 209.(30) Plazek, D. J. Polym J . 1980, 12, 43.
Theoretical Study of th e Influence of the Molecular Weight onthe M aximum T ensile Strengthof Polymer Fibers
Yves Termonia,* Paul Meakin, and Paul SmithCentral Research and Developm ent Depar tm ent, Exper imenta l Stat ion , E. I . du Pont deNemo urs and Com pany, Inc. , Wilmingto n , Delaware 19898. Received March 9, 1985
ABSTRACT: A stochastic Monte Carlo approach, based on the kinetic theory of fracture, has been usedto study the axial maximum tensile strength of polymer fibers. The approach is entirely microscopic andthe inhomogeneous distribution of the external stress among atomic bonds near the chain ends is explicitlytaken into account. Both primary and secondary bonds are assumed to break during fracture of the polymerfiber. The approach has been applied to perfectly oriented and ordered polyethylene fibers, for whichapproximate values of the model's parameters were obtained from experiment. Stress-strain curves havebeen calculated for several fibers of various (monodisperse) molecular weights and predictions for the tensilestrength have been compared to experimental values.
1. IntroductionValues of the maximum tensile modulus have been
measured or calculated for many polymers' and haveproven to be very useful in set ting goals for polymer ma-ter ia ls re~earch.~-~ he maximum tensile strength ofpolymers is a less satisfying issue. Some authors , withwidely different results, have addressed the theoreticalultimate breaking stress of single chain^.^-^Such treat-ments, however, are only of value for hypothetical speci-mens comprising infinitely long chain molecules and arenot necessarily relevant for finite molecular weights. Ex-perimental tensile strength data, obtained for drawn fibershave been extrapolated to infinitely small fiber diameter8and this procedure has led sometimes to agreement withsome of the above-mentioned values for the breaking stressof a single chain. Some att empts have been made to de-scribe the development of tensile strength with draw ra-tio9J0, and purely empirical extrapolations of th e tensilestrength to the theoretical modulus have also been re-ported. J2 Despite various theoretical and some empiricalefforts, no relation for relatively simple issues such as theeffect of the molecular weight on the maximum tensile
strength has appeared in the polymer literature, other thanFlory's expre~sion'~ or th e tensile strength of cross-linkedisotropic rubbers. All in all, the maximum tensile strengthof polymeric materials of finite molecular weight and therelation to the ultimate breaking stress of infinitely longchains remains an obscure, ill-understood topic.
A theoretical study of the tensile strength of polymerfibers is an extremely complex problem. Many structuralparameters, such as amorphous defects, trapped entan-glements, chain ends, misalignment, etc., need to be con-sidered. In fact, a detailed knowledge of the fiber structureis required on a molecular level. But , even in the presenceof such information, a theoretical treatmen t of the tensileproperties remains an enormous task. For that reason, allprevious theories have made many simplifying assumptions
0024-9297/85/2218-2246 01.50/0
with regard to the structure and the failure mechanism.For example, classical continuum mechanics, e.g., Grif-fith-type approaches,14 fail to take into account the inho-mogeneous nature of the structures of interest. Kinetictheories of fracture commonly focus on only one mode offailure, Le., merely primary bond breakage,15J6 or pureslippage, J8 while both mechanisms are known to operateduring fracture of polymer fibers.lg Other kinetic theories20ignore the important contributions of the intermolecularinteractions altogether.
Due to the complexity of a general theoretical treatmentof the strength of polymer fibers, the present study isrestricted to the case of ideal fibers made of a perfectlyordered array of fully extended macromolecules, with nodefects other than chain ends resulting from finite mo-lecular weight. As such, the study deals with the maximumaxial tensile strength, which is of obvious interest, muchlike the knowledge of the theoretical modulus has provento be. The present approach, which is based on the kinetictheory of fracture, is entirely microscopic and the inho-mogeneous distribution of the external stress among at-omic bonds near the chain ends is explicitly taken into
account. Th e bonds are viewed as coupled oscillators ina stat e of constant thermal vibration. The basic eventsare controlled by thermally activated bond breakage andthe accumulation of those events leads to crack formationand, ultimately, to the breakdown of the fiber. The bondbreakages are simulated by a Monte Carlo process on athree-dimensional array of nodes, representing the ele-mentary repetition unit of a polymer. These nodes arejoined in the and z directions by weak secondary bonds,e.g., hydrogen or van der Waals forces, whereas in the ydirection, stronger bonds account for the primary (C-C)forces. Our model thus resembles that of Dobrodumov andEl'yashevitch2l except for the fact th at, in the latter, i t wasassumed tha t only primary bonds can break. Here, incontrast, we allow fiber fracture to proceed by the breaking
985 American Chemical Society

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 2/7
Macromolecules, Vol. 18 No. 11 1985
Figure I. Two-dimensional representation of the array of nodeaand of bonds. K , and K zare the elastic constants or the primaryand for the secondary bonds, respectively.
of both primary and secondary bonds.This approach has been applied to perfectly oriented
and ordered polyethylene for which approximate valuesof the model's parameters can be obtained from experi-ment. With the help of ow zero-parameter model, we havecalculated the stress-strain curves for several fibers ofvarious (monodisperse) molecular weights, and predictionsfor the tensile streng th have been compared to extrapo-lated experimental values.
2. Stochastic ModelIn the kinetic theory of fracture, a bond in th e absence
of external mechanical forces breaks whenever it becomesexcited beyond a certain level, U , he activation energy ofthe bond. For molecular chains in th e process of beingstrained, there is overriding experimental evidence frominfrared studiesz2 hat the activation energy barrier islinearly decreased by the local stress. Thus, n the presenceof stress, the rate of breakage of a bond i is given by
(1)
where r is the thermal vibration frequency, T i s the (ab-solute) temperature, p s an activation volume whose lineardimension is of the order of the bond length, and ui s thelocal stress
2)
Here, Ki and ti are the elastic constant and the local strain,
respectively. Note that , strictly speaking, the bilinear formf l u should be a scalar. However, thi s is of litt le concernto the simulation, for which real numbers are substitutedfor the products pu .
The above kinetic model has been simulated on athree-dimensional xy.4 array of up to 6 X 6 X loo0 nodesrepresenting the elementary repetition units of the polymerchains. (For very long chains, each node is made to cor-respond to more than one repetition unit; see section 4.)The number of chains per cross-sectional area thusamounts up to 36. The nodes are joined in t he r and zdirections by secondary bonds, e.g., hydrogen or van derWaals forces, having an elastic constant K,. Only near-est-neighbor interactions are considerded. In the y di-rection, stronger forces with elastic constant K , account
u i = T exp[(-Ui + u ; ) / k r ]
I. = K.* .L
Maximum Tensile Strength of Polymer Fibers 2247
for th e primary (C-C) bonds. A two-dimensional repre-sentation of the array of nodes is given in Figure 1. Inthe absence of stress, the nodes are in contact with eachother along they axis so that the length of a node actuallyrepresents the length of an unstrained C-C bond. Weassume these nodes to be incompressible. For simplicity,we also assume that the displacements of the networknodes along the coordinate axes are mutually independ-entz l and we focus on the displacement along th e y axis,along which the fiber is strained. Thus, the strain values6 (see eq 2) are for elongations along the y axis and the yrepresent either an axial tensile strain (for primary bonds)or a shear strain (for secondary bonds). These strain valuesare in percent of the length of a node along the y axis.
The simulation is star ted from an unstrained array inwhich all the bonds are unbroken, except for few brokenC-C bonds along th e y axis, which account for the finitemolecular weight of the polymer chains. Th e unbrokenbonds are visited at random by a Monte Carlo lottery thatbreaks a bond according to a probability
pi = Ui/UIn. . (3)
where u is the rate of breakage of the most strained bondin th e array. After each visit of a bond, the time t isincremented by l / [u -n t ) ] , where n ( t ) s the tot numberof intact bonds a t time t. After a small time interval 6 thas elapsed, the bond-breaking process is stopped. Ofcourse, 6t must be chosen much smaller than t he shortesttime for a single bond breaking; i.e., 6 t << l / u - This hasled us o take 6t = 0.01 s which was found to be sufficientlysmall to ensure that our results are 6t independent. Thesample is then strained along the y axis by a small constantamount and the network is relaxed to its minimum-energyconfiguration, using the method described in section 3.This leads to displacements of the nodes along they axis.After that step, secondary bonds are allowed to re-formbetween nodes coming in perfect register to each other, towithin a small error 6y. The alternation of breaking andrelaxation steps for the secondary bonds should be
equivalent to the use of a sinusoidal potential for the shearforces. Primary covalent bonds, however, are assumed notto re-f~rm.~' he system is then relaxed again and theaxial stress at one end of the fiber is calculated. TheMonte Carlo process of bond breaking is then restartedfor another time interval 6 t . And so on and so forth untilthe sample breaks. This procedure, performed a t a con-stant strain rate, yields a stress-strain curve, the maximumof which is referred to as th e maximum tensile strengthof the fiber for a given molecular weight under specifiedtest conditions .
Results of a simulation for a two-dimensional r-y arrayare illustrated in Figure 2 for a low (case a) and for a high(case b) elongation, respectively. Cylindrical boundaryconditions along the x axis were imposed. Note the de-
velopment of a large fracture focus in Figure Zb, whichappears as a result of an acceleration of its growth due tothe high concentration of stresses at it s boundary.
3. Relaxation MethodThe relaxation of t he system t o it s minimum-energy
configuration is realized in a sequence of steps. A t eachstep, a node i is visited an d i ts y ordinate is adjusted soas to to bring to zero the net residual stress r )
ri = .E + K , t j X ri. 4)bonds j nodes i
acting on that node. The first term on the right-hand sideof eq 4 is due to the elastic stress acting on the bonds joriginating from node i, with j = 1 or j = 0 depending

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 3/7
22 Macromolecules, Vol.18 No. 11, 1985
on whether bond j is intac t or broken. Also, K , = K1 andK , = K z for a primary and for a secondary bond, respec-tively. l is the local strain measured by the difference inthey coordinates of the two ends of the bond. Th at lattervalue is in percent of the length of a node. Th e secondterm on the right-hand side of eq 4 accounts for the ex-terna l forces acting on node i when in contact with any ofits two neighbors i along the y axis. Indeed, we recall ourassumption that the nodes are incompressible, i.e., thatthe length of a C-C bond cannot be decreased below its
equilibrium value in the absence of stress. Of course,= 1 or tIt= 0, depending on whether contact with node iis made or not.
It is important to note th at bringing to zero the residualstress rr for a node often results in an automatic increaseof ( the absolute value of the residuals for the neighboringnodes. Therefore, the liquidation of all the residuals inthe array is a very computer time-consuming process. Forthat reason, the liquidation of the residuals has beenperformed by using two well-known computational de-v i c e ~ , ~ ~hich considerably speed u p the convergence ofthe calculations. The first device, known as overrelaxation,consists of n ot just reducing to zero the residual rr for anode but, instead, deliberately going further to give to rLa sign th at is opposite th at of the residuals for th e neigh-
boring nodes. Th e second, and more import ant, device isknown as block relaxation and consists of adjusting theordinates of more than one node a t a time. Relaxation ofthe system using the above two devices was considered tobe completed when the largest residual stress for a nodefell below a few percent of the average stress for a bond.
4. Results: Application to PolyethyleneTh e stochastic model described in sections 2 and 3 has
been applied to a hypothetical structure of perfectly or-iented and ordered polyethylene, for which approximatevalues of the parameters in eq 1 and 2 are available. Forsecondary bonds the shear modulus K 2 = 3 GPaz6 and wetook U = 0.65 kcal/mol with = (2.5 A)3; for primarybonds the axial modulus K1 = 300 GPa,z6 U = 25 kcal/
= (1.54 A 3. The atomic vibrationfrequency was chosen equal to lo1 s-l a t T = 300 K.Thu s, there are no free parameters in the model. Ad-mittedly, t he lit erature reveals a relatively wide scatter inth e values for these parameters . Our value for U of theprimary bonds of polyethylene is low compared to disso-ciation energies of -80 kcal/mol commonly attributed tothe C-C bond. However, th e choice U = 25 kcal/mol ismotivated by the fact th at the lat ter value, reported in ref22, has been obtained from a thermal fluctuation approachto fracture, like the pre~ent.~' ote also that activationvolumes are rather difficult to measure experimentally andthe selection U = 25 kcal/mol is further consistent withtaking the lowest value, = (1.54 for the activationvolume of the C-C bond. Th at uncertainty in the ex-perimental data has led us to investigate the sensitivityof the calculated tensile strength to the values chosen forthe parameters. It was found that the maximum stress isnot appreciably affected by a n increase or a decrease ofT by a factor of 10. Neither does it change significantlywhen taking U = 80 kcal/mol together with = (2.75 AI3for primary bonds. A choice U = 80 kcal/mol with =(1.54 A)3leads to notably higher values of the strength onlyat M > 2 x 105 (see Discussion). Finally, i t should be notedtha t, for reasons of computat ional time and memory, animportant struct ural assumption was made in the appli-cation of our model to polyethylene. Namely, in the model,the coordination number in the x-z plane is 4, whereas thecentral chain in the crystalline orthorhombic polyethylene
and we chose
148
a b
Figure 2. Resul ts of the s imula t ionfor a two-dimensional arrayof 20 X 100 nodes: case a, 150 strain; case b, 300% s t ra in . Theprobabilities for breaking primary an d secondary bonds in thesehighly s t re tched samples were chosen equal to exp(c2)for s im-plicity. C ylindrical boun dary condition s along th e axis wereimposed.
8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 4/7
Macromolecules, Vol. 18, N o . 11, 1985
2 2 04
6 o3
Maximum Tensile Strength of Polymer Fibers 2249
4 105r I I I I I \
I
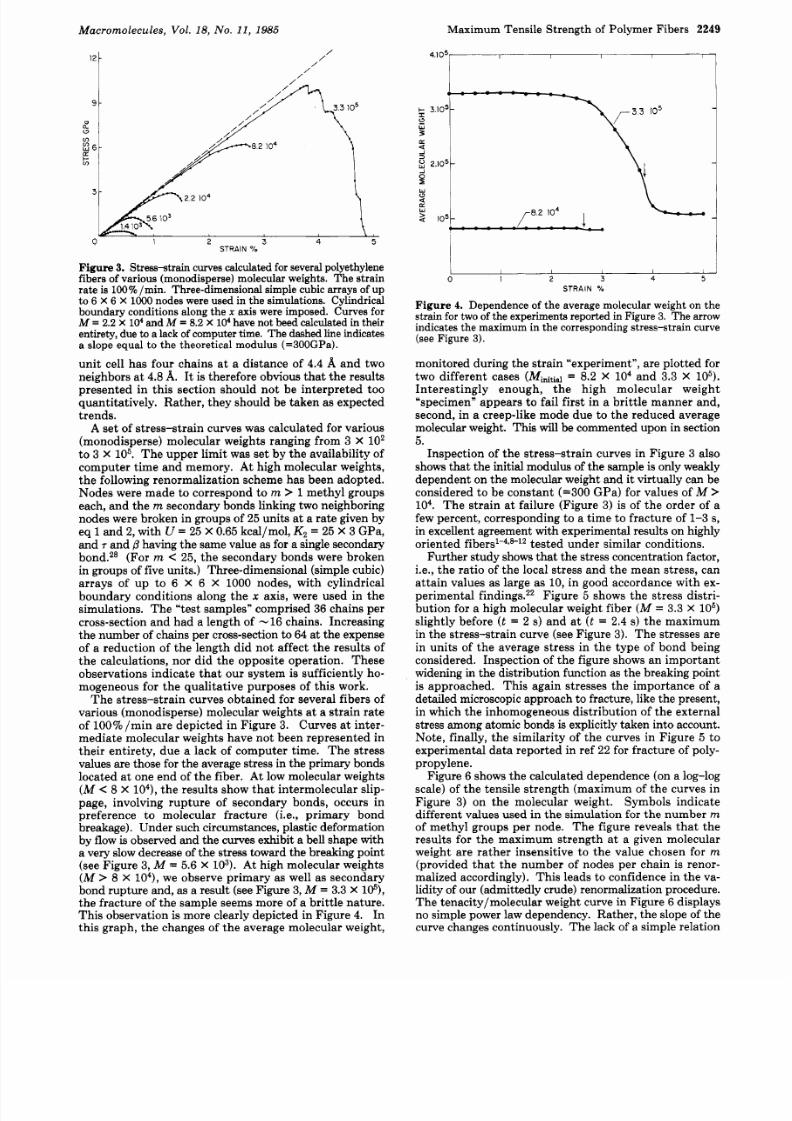
Figure 3. Streas-strain curves calculated for several polyethylenefibers of various (monodisperse) molecular weights. The strainrate is 100% min. Three-dimensional simple cubic arrays of upto 6 X 6 X lo00 nodes were used in the simulations. Cylindricalboundary conditions along the axis were imposed. Curves forM = 2.2 X lo4 and M = 8.2 X 104 have not beed calculated in theirentirety, due t a lack of computer time. The dashed line indicatesa slope equal t o the theoretical modulus (=300GPa).
unit cell has four chains at a distance of 4.4 A and twoneighbors at 4.8 A. It is therefore obvious that t he resultspresented in this section should not be interpreted tooquantitatively. Rather , they should be taken as expectedtrends.
A set of stress-strain curves was calculated for various(monodisperse) molecular weights ranging from 3 X l o2to 3 X lo5.The upper limit was set by the availability ofcomputer time and memory. At high molecular weights,the following renormalization scheme has been adopted.Nodes were made to correspond to m > 1 methyl groupseach, and the m secondary bonds linking two neighboringnodes were broken in groups of 25 units a t a rate given byeq 1 and 2, with U = 25 X 0.65 kcal/mol, K z = 25 X 3 GPa,and and having the same value as for a single secondary
bond.28 (For m < 25, the secondary bonds were brokenin groups of five units.) Three-dimensional (simple cubic)arrays of up to 6 X 6 X 1000 nodes, with cylindricalboundary conditions along the x axis, were used in thesimulations. The “test samples” comprised 36 chains percross-section and had a length of -16 chains. Increasingthe number of chains per cross-section to 64 at the expenseof a reduction of the length did not affect the results ofthe calculations, nor did the opposite operation. Theseobservations indicate that our system is sufficiently ho-mogeneous for the qualitative purposes of this work.
The stress-strain curves obtained for several fibers ofvarious (monodisperse) molecular weights at a strain rateof 100 min are depicted in Figure 3. Curves a t inter-mediate molecular weights have not been represented intheir entirety, due a lack of computer time. The stressvalues are those for the average stress in the primary bondslocated a t one end of the fiber. At low molecular weights(M C 8 X lo4), he results show th at intermolecular slip-page, involving rupture of secondary bonds, occurs inpreference to molecular fracture (i.e., primary bondbreakage). Under such circumstances, plastic deformationby flow is observed and the curves exhibit a bell shape witha very slow decrease of the stress toward the breaking point(see Figure 3, M = 5.6 X lo3). A t high molecular weights
M > 8 X lo4),we observe primary as well as secondarybond rupture and, as a result (see Figure 3, M = 3.3 X le),the fracture of the sample seems more of a brittle nature.Thi s observation is more clearly depicted in Figure 4. Inthis graph, the changes of the average molecular weight,
W3
2.105
0L
w
W
2 1 0 5 I2 o4
I I 1 1 1
0 2 3 4 5S T RAI N
Figure 4. Dependence of the average molecular weight on thestrain for two ofthe experiments reported in Figure 3. The arrowindicates the maximum in the corresponding stress-strain curve(see Figure 3).
monitored during the st rain “experiment”, are plotted fortwo different cases (Minitid 8.2 X l o 4 and 3.3 X lo5).Interestingly enough, the high molecular weight“specimen” appears to fail first in a brittle manner a nd,second, in a creep-like mode due to the reduced averagemolecular weight. This wil lbe commented upon in section5.
Inspection of the stress-strain curves in Figure 3 alsoshows that the initial modulus of the sample is only weaklydependent on the molecular weight and it virtually can beconsidered to be constant (=300 GPa) for values of M >lo4 . The strain at failure (Figure 3) is of the order of afew percent, corresponding to a time to fracture of 1-3 s,in excellent agreement with experimental results on highlyoriented fiber~’-~@l~ested under similar conditions.
Further study shows tha t the stress concentration factor,i.e., the ratio of the local stress and the mean stress, canatta in values as large as 10, in good accordance with ex-perimental findings.22 Figure 5 shows the stress distri-bution for a high molecular weight fiber M = 3.3 X lo5)slightly before t = 2 s) and at t = 2.4 s) the maximumin the stress-strain curve (see Figure 3). The stresses arein units of the average stress in th e type of bond beingconsidered. Inspection of the figure shows an importantwidening in the distribution function as the breaking pointis approached. This again stresses the importance of adetailed microscopic approach to fracture, like the present,in which the inhomogeneous distribution of the externalstress among atomic bonds is explicitly taken into account.Note, finally, the similarity of the curves in Figure 5 toexperimental data reported in ref 22 for fracture of poly-propylene.
Figure 6 shows th e calculated dependence (on a log-logscale) of the tensile strength (maximum of the curves inFigure 3) on the molecular weight. Symbols indicatedifferent values used in the simulation for the number mof methyl groups per node. The figure reveals th at theresults for the maximum strength at a given molecularweight are rather insensitive to the value chosen for m(provided tha t t he number of nodes per chain is renor-malized accordingly). This leads to confidence in th e va-lidity of our (admittedly crude) renormalization procedure.The tenacity/molecular weight curve in Figure 6 displaysno simple power law dependency. Rather, the slope of thecurve changes continuously. The lack of a simple relation

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 5/7
2250 Termonia et al.
I
Macromolecules, Vol. 18, No. 11, 1985
proceeds through slippage at low molecular weights (M <8 lo4); 4) chain frac ture occurs at M > 8 X lo4; 5) atM > 3 X 105, failure predominantly involves chain fracture,whereas for 8 X lo4 C M C 3 X lo5 ,both creep and bondrupture occur; (6) straining at 100 min results in failurewithin a few seconds at an elongation of a few percent; (7)the elongation a t break varies systematically with molec-ular weight and ranges from 1 to 5 for M = 6 lo3 o5 X lo5; 8) the local stress concentration factor can at tainvalues as high as 10; (9) there is no simple relation between
the tenacity and th e chain length due to changing modesof failure with molecular weight; (10) in the molecularweight range from 2 X lo4 o lo6, he tenacity varies withM approximately as u - iP see Figure 5); and (11)extrapolation of the tensile strength to infinite molecularweight yields a value of about 24 GPa (at 8% strain), whichlies within the range of previously reported calculations.“’At this point, i t is worth noting that the l atter value, ob-tained with U = 25 kcal/mol and 3 = (1.54 A 3for theprimary bonds is also recovered with U = 80 kcal/mol and/3 = (2.75 A 3. However, taking U = 80 kcal/mol and 3 =(1.54 A 3leads to an unrealistic&‘ ultimate tensile strengthof -138 GPa (at 46 stra in). We have also performedcalculations in which the activation energy barrier U (seeeq 1) is decreased by the elastic energy (force constanttimes square of displacement) stored in the bond. Takingfor the C-C bond a force constant equal to K,A/r with anaxial modulus K1 = 300 GPa, a chain cross-section A =18 A2,and a bond length r = 1.54 A leads to an ultimatetensile strength of around 42 GPa (at 14 strain).
Each and everyone of these theoretical results can bereadily substantiated with experimental data on highlyoriented polyethylene fibers. It is well-known that lowmolecular weight polyethylene fibers exhibit creep duringtensile loading.29 Also well established is that the modulusof drawn fibers of medium molecular weight dependsprimarily on the draw ratio and not sensitively on the chainlength.30 Chain fracture during tensile tests of mediumand high molecular weight polyethylene has been observedin electron spin resonance experiment^.^ A host of ex-perimental data indicates that short-term (1-10 s) tensilefracture of highly oriented polyethylene fibers occurs atelongations of a few percentz4 and that the strain a t breakincreases with increasing molecular eight.^^,^^ Remark-ably, the empirical power law ( u -M0.4) orrelating themolecular weight with the tenacity of moderately orientedfibers (Young’s modulus = 50 GPa), found in an earlierexperimental for 5 x IO4 < fi, < 3.5 x lo6, isrecovered in the present theoretical work, albeit for aperfectly ordered structure. These outstanding qualitativeagreements between the model a t hand and the relatedexperimental observations, in our view, greatly support itsusefulness.
With regard to quantitative predictions, i.e., the actualbreaking stress and the molecular weights where the modesof fracture change, caution should be exercised. First ofall, as was pointed out in the previous section, the valuesof the model’s parameters are not established with greataccuracy. Moreover, a number of assumptions were madein the model, many of them to save computer time andmemory. It is nonetheless tempting to compare someexperimental values against the “predicted” tenacities.First of all, one should bear in mind that the presenttheoretical work concerns the maximum tensile strength,that is, the tenacity of a perfectly ordered, flawlessstructure of monodisperse macromolecules. Experimentalresults are available only for highly oriented fibers ofheterodisperse molecular weight that have moduli lower
\I 1
loo
O I L - -L--L L U U l d L l l L L L A _J
102 lo3 io4 lo5 IO6
Figure 6. Dependence of the strength (maxima of the curves inFigure 3) on t he molecular weight for monodisperse polyethylenefibers. The symbols are for different values for the number mof methyl groups per node. V ) m = 5; 0 ) m = 2 5 ; 0) = 100;
X ) m = 200; H) m = 400; A ) m = 800; 0 ) = 1600; A ) m =3200. In order to speed up the calculations for the case m = 5 ,we took T = lo4.
is clearly due to the molecular weight dependence of themodulus at very low M <lo4) and, a t higher M , to thenature of the failure process, which gradually changes fromslippage to chain fracture.5. Discussion
The present kinetic model for fracture of perfectly or-dered and fully extended polyethylene chains has revealedthe following results: (1) at very low molecular weights,the initial axial modulus depends on the chain length; (2)a t M > lo4, he modulus is virtually constant; (3) failure
MOLECULAR WEIGHT

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 6/7
Macromolecules, Vol. 18 No. 11, 1985
tha n th e theoretical values. For example, highly drawnpolyethylene fibers nowadays are routinely p r o d ~ c e d ~ ~ ~ ~ ~ ’ ~with an axial Young’s modulus E of 100-150 GPa, u = 3-5GPa, and = 3-5 . Th e weight-average molecular weightof the polymers used is typically in t he range (1-5) X lo6.M s usually not reported but supposedly is of the orderof (1-5) X 105. More recently, data have been publishedgJOon fibers with the following room temperature properties:E = 200 GPa and u = 7GPa for M w 2 lo6.Assuming,to a fir st approximation, t hat the tenacity varies linearly
with the modulus, all of these aforementioned values wouldcorrespond at K1 = 300 GPa to u = 10 GPa for perfectlyordered and extended polyethylene having a weight-av-erage molecular weight in t he million range and a,, f theorder of lo5. Inspection of the graph in Figure 5 shows apredicted value of u = 9 GPa and u = 13 GPa for, re-spectively, M = 2.5 X lo5 and M = lo6 (monodisperse).Previous extrapolations” of the tenacity of oriented fibersto a modulus of 300 GPa resulted in a value u = 8.5 GPafor A?w = 1.5 X l o6 and it& = 2 X lo5. The admittedlyfortuitous, agreement between extrapolated tenacities ofexperimental fibers and the present calculations providesat l east confidence with regard t o the choice of t he se t ofvalues for th e model’s parameters.
A few comments should be devoted to the size of the“te st sample” in the present work. As was mentionedbefore, the sample typically comprised 6 X 6 X 16 chains.In comparison with th e dimensions of experimental fibers,which usually contain ( -2 X lo4) X (2 X lo4) X (2 X lo4)chains, our dimensions appear exceptionally small. Th eobvious question arises how changing the specimen sizeaffects the calculations. After all, it is well-known th atfiber length and cross-sectional area have a dramatic effecton its mechanical properties.% It was pointed out in t heprevious section that our system appeared to be homoge-neous; i.e., no size effects were observed. The very strongdependence of sample size on the propert ies of actual fibersoriginates in the fact th at fiber failure is commonly dom-inated by gross flaws.34 In the absence of such flaws, as
in the present model, indeed no effect is expected.Finally, the shape of the stress-strain curves after thepoint of maximum stress (see Figure 3) deserves somediscussion. These curves are typified by a rapid , but notsudden, decrease in stress with increasing strain. To ourknowledge, such behavior is not commonly observed intensile tes ts of high-modulus/ high-strength polyethylene.The latter display abrupt, catastrophic failure. Thepresent work indicates that failure of high molecularweight specimens is accompanied by substantial chainfracture, which reduces the chain length (see also Figure4) o levels where chain slippage occurs. The calculatedshape of th e stress-strain curves is thus a logical andplausible consequence of the various modes of failure. Onepossible reason for the absence of such “tails” on thestress-strain curves of actual fibers is th e difference instructure between the perfect model fibers and experi-mental filaments. The la tte r are known to contain somedefects, apar t from chain ends, the major one being trap -ped entanglements. Trapped entanglements provide lat-eral bonds in the fibers, which will reduce slippage.
In summary, we have presented a kinetic approach tothe fracture of a perfectly ordered and oriented polymerfiber. The model is microscopic in natu re but is notburdened with the need to provide a detailed structure atthe atomic level. Instead, th e fiber is represented by anarray of nodes joined by oscillating bonds, and bond-breaking events are treated as thermally activated pro-cesses. Our approach has been successfully applied to
Maximum Tensile Strength of Polymer Fibers 2251
oriented polyethylene filaments and both qualitative and(fortuitious) quantitative agreement between theoreticaland related experimental observations has been found.Th e main disadvantages of our model are that i t dependson paramet ers which may be difficult to measure or cal-culate for various polymers and that large amounts ofcomputer time ar e required. For thi s reason, we believethat the model will probably be of more help in relatingthe physical characteristics of polymer molecules (molec-ular weight distribution, orientat ion, topology, etc.) to their
ultimate mechanical properties than in relating chemicalstructure to them.
Acknowledgment. We thank Dr. A. D. English for acritical reading of the manuscript.
Registry No. Polyethylene (homopolymer) ,9002-88-4.
References and Notes1) H. H . K ausch, “Polymer Fra cture”, Springer-Verlag, Berlin,
1978.(2) See, e.g., “Ultra-High Modulus Polymers”, A. Ciferri andI. M.
Ward, Eds., Applied Science Publishers, Lon don,1979.(3) P. Smith and P. J. Lemstra, J. Mater. Sci., 15, 505 (1980).(4) W. Wu and W. B. Black, Polym. Eng. Sci., 19 1163 (1979).(5) K. E. Perepelkin,Angew. Makromol. Chem.,22, 181 (1972).(6) D. P. Boudreaux,J. Polym. Sci., Polym. Phys. Ed.,11, 1285
(1973).(7) B. Crist, M.A. Rafner, A. J. Brower, and J. R. Sabin,J . Appl .
Phys. , 50, 6047 (1979).(8) J. Smook. W. Hammersma. and A.J. Pennines. J . Mater. Sci..
19, 1359 i1984).(9) A. V. Savitakii. I. A. Gorshkova. G. N. Shm ikk. an d I. L. Fro-,
lova, Vysokomol. Soedin., Ser.B , 25, 352 (1983).
A. F. Ioffe, Polym. Bull., 12, 195 (1984).
19, 1007 (1981 ).
(10) A. V. Savitskii,I. A. Gorshkova, I. L. Frolova,G. N. Smikk, and
(11) P. Smith andP. J. Lemstra, J. Polym. Sci., Polym. Phys. Ed.,
(12) T. Ohta, Polym. Eng. Sci. ,23, 697 (1983).(13) P. J. Flory, J. Am. Chem. SOC. ,7, 2048 (1945).(14) A. A. Griffith, Philos. Trans. R. S OC . ondon,221,163 (1921).(15) S. N. Zhurkov andT. P. Sanhirova. Dokl. Akad. Nauk SSS R.
101, 237 (19 55).(16) F. Bueche. J. A m l . P hv s. .26. 1133 (1955): 28. 784 (1957): 29.. “ I I I
1231 (1958).(17) A. Tobolsky and H. Eyring, J. Chem. Phys. , 11, 125 (1943).
(18) J. R. Schaefgen, T. I. Bair, J. W. Ballou, S. L. Kwolek, P. W.Morgan, M. Panar, andJ. Zimmerman, in ref 2, p 199.(19) Reference 1, Chapter 3.(20) H. H. Kau sch and C. C. Hsiao,J. App l. Phys.,39,4915 (1968).(21) A. V. Dobrodumov and A. M. El’yashevitch,Sou. Phys.-Solid
S ta t e Engl. Transl.), 15, 1259 (1973).(22) S. N. Zhurkov, V. I. Vettegren, V. E. Korsukov, andI. I. No-
vak, “Proceedings of the 2nd International Conference onFracture”, Brighton, Chapma n and H all Ltd. London,1969, p545. S. N. Zhurkov an d V. E. Korsukov, J . Polym. Sci., Polym.Phys. Ed. , 12, 385 (1974); Sou. Phys.-Solid State Engl.Transl.), 15, 1379 (1974).
(23) We chose Sy = 0.1 , which, for a typical node made of100methyl groups (see section4) , corresponds t o a sh ear strain ofaround 10% for a single van der Waals bond. T ha t latter valuecompares favorably with the5 thermal am plitude of the vander Waals bond in polyethylene (see H. H. Kausch andJ.Becht, in “Deformation a nd Fract ure of High Polymers”, H.
H. Kausch et al., Eds., Plenum Press, New York,1973, 317).As far as we could determine, a decrease of6y by a factor of10 does not significantly affectour results for the maximumstrength. We have also performed calculations in which thesecondary bonds are re-formed a t a rate given by eq1, with u= 0. These two alternative procedures for the reformation ofth e secondary bonds lead, within experimen tal error,to similarresults for the maximum strength.
(24) E. H. Andrews andP. E. Reed, Adu. Polym. Sci.,27, 1 (1978).(25) D. N. de G. Allen, Relaxation Methods,Mc Graw Hill,N. Y
(1954).(26) I. M. Ward, “Mechanical Properties of Solid Polymers”,2nd
ed., Wiley, New York, 1983, p 270.(27) Suc h a low value for the activation energy isalso confirmed by
therm al degrada tion experimen ts, which are seen to proceedin two stages: one characterized by a low activation energy(25-30 kcal/mol) and the other by a higher value(50-60kcal/ mo l). See V.R. Regel, A. V. Amelin, 0. F. Pozdnyakov,

8/10/2019 Termonia 1985 - MM - Influence MW on Max Tensile Strength
http://slidepdf.com/reader/full/termonia-1985-mm-influence-mw-on-max-tensile-strength 7/7
2252 Macromolecules 1985, 18, 2252-2258
T. P. Sanfirova, and A. F. Ioffe, Preprint of the International M. Pierce, and J. L. Wesley, Polym. Commun., 24 130 (1983).Symposium on Macromolecules, Helsinki, Finland, Jul 2-7, (30) G. Capaccio, A. G. Gibson, and I. M. Ward, ref 2, p 12.1972, Vol. 5, 163. (31) See ref 1, Chapter 7.
(28) In that new representation, he stochastic process still visits (32) W. G. Perkins, N. J. Capiati, and R. S. Porter, Polym. Sci.primary bonds with the same probability as secondary bonds, Eng. 16 200 (1976).since one must not overlook the fact that each node itself (33) P. Smith, P. J . Lemstra, and J. P. L. Pijpers, J. Polym. Sci.,actually represents m primary bonds aligned in series along the Polym. Phys. Ed. 0, 2229 (1982).y axis. (34) A. Kelly, Strong Solids , 2nd ed., Clarendon Press, Oxford,
(29) D. L. M. Cansfield, I. M. Ward, D. W. Woods, A. Buckley, J. 1973, Chapter 2.
Adsorption of Polyacrylamide on Solid Surfaces. Kinetics of theEstablishment of Adsorption Equilibrium
E. Pefferkorn, A. Carroy, and R. Varoqui*
Centre de Recherche5 sur les Macromol6cules (CNRS), 67083 Strasbourg Cedex, France.Received March 13. 1985
ABSTRACT The kinetics of the establishment of adsorption equilibria of polyacrylamide onto chemicallymodified glass beads was studied by a radioactive tracer method using 3H-labeled polymers. The adsorptionrates were determined as a function of temperature and surface affinity by varying the surface density ofthe interacting sites. The experimental conditions were chosen to minimize diffusion effects. Quantitative
analysis shows that adsorption is governed by the structure of the surface layer; in particular, the adsorptionrate is proportional to the number of free interacting surface sites. The role of the diffuse polymer layer asa barrier rate-limiting factor is also discussed.
IntroductionIn previous work we measured the rate of exchange of
adsorbed 3H-labeled polymers with nonlabeled polymersin the bulk and analyzed the adsorption rates in the regionof adsorption equi1ibrium.l In this paper, we discuss thefactors involved in the establishment of adsorption equi-librium.
A study of the rate of establishment of adsorptionequilibrium of polymers at solid/liquid interfaces is offundamental importance in many areas (polymer pro-cessing, flocculation of colloidal particles, adhesion, coating,etc.) From a conceptual poin t of view, th e adsorption ofpolymers implies rate-controlling factors that are not en-countered with ordinary molecules. In particular, becauseof the ir chainlike structure, t he adsorbed surface films ofpolymers are structura lly similar to the surface of a gel,and the rate of adsorption depends not only on themechanism of adherence to the surface by multiple pointsof physical adsorption bu t also on the r ate of penetrationof a gellike layer. Adsorption rates of polymers have beenstudied in th e past.2 However, the most interesting fea-tures were often blurred by trivial ones (for example, masstransfer by diffusion of polymers to the surface, whichdepends heavily on the hydrodynamic conditions, presents
no interest on its own). Furthermore, th e experimentalconditions were frequently such that equilibrium wasrapidly established in a few minutes or even a few sec-onds3p4 and , owing to experimental difficulties, the kineticparamete rs could not be assessed with certainty.
In this study of the adsorption of polyacrylamide inaqueous solution on the surface of modified silica particles,the experimental conditions were chosen to ensure a cer-tain number of meaningful points: (i) the natu re anddensity of the surface sites interacting with the polymerwere characterized quant itatively, and one objective wasto correlate the adsorption kinetics with the active sitedensity; (ii) experimental conditions were chosen to min-imize bulk diffusion effects (very often, adsorption wascarried out in a semiinfinite and unstirred medium by
0024-9297/85/2218-2252 01.50/0
Table ICharacteristics of Glass Beads
9i compspec surface [AlOH]/
sample code mean diam, pm area, cm*.g-' [SiOH]AS0 34 780 0AS1 34 780 5AS2 34 780 1 2
transferring adsorbent plates into a vessel containing the
polymer solution); (iii) a very sensitive determination ofadsorption was obtained via 3H labeling.
Experimental Section1. Polymer. Radioactive polyacrylamide was prepared by
radical polymerization in the presence of traces of acrolein. Afterfractionation, the resulting material was reacted in aqueous so-lution with tritiated potassium borohydride to yield the followingchemical structure (details are given in ref 1):
-( C p - H )m,- CH2 H m2
ICHOH
Ic=o
I3HHz
3m , / q = 10
The characteristics of the sample used in this study are as follows:M, = 1.2 X lo6;polydispersity (GPC) = 1.25;specific radioactivity= 1.12 x lo8counts.min-'.g-'.
2. Adsorbents. Nonporous glass beads (Verre e t Industrie)of 34-pm average diameter were used as the adsorbent. Thesurface was first made hydrophilic by soaking for 24 h in hothydrochloric acid medium
and was then treated with aluminum chloride in chloroform inorder to replace some silanols (SiOH) by aluminum chloride
985 American Chemical Society





























