Embed Size (px)
Citation preview

Targeting the active B-catenin pathway totreat cancer cells
Hadas Dvory-Sobol,1,2 Eyal Sagiv,1,2
Diana Kazanov,1 Avri Ben-Ze’ev,3 andNadir Arber1,2
1Integrated Cancer Prevention Center, Tel Aviv SouraskyMedical Center; 2Sackler School of Medicine, Tel-Aviv University,Tel-Aviv, Israel; 3Department of Molecular Cell Biology,Weizmann Institute of Science, Rehovot, Israel
AbstractThe adenomatous polyposis coli or b-catenin genes arefrequently mutated in colorectal cancer cells, resulting inoncogenic activation of B-catenin signaling. We tried toestablish in vitro and in vivo models for selectivelykilling human cancer cells with an activated B-catenin/T-cell factor (Tcf) pathway. We used a recombinantadenovirus that carries a lethal gene [p53-up-regulatedmodulator of apoptosis (PUMA)] under the control of aB-catenin/Tcf–responsive promoter (AdTOP-PUMA) toselectively target human colorectal cancer cells(SW480, HCT116, DLD-1, and LS174T), hepatocellularcarcinoma (HepG2), and gastric cancer cells (AGS) inwhich the B-catenin/Tcf pathway is activated, andcompared its efficiency in killing cancer cells in whichthis pathway is inactive or only weakly active. AdFOP-PUMA, carrying a mutant Tcf-binding site, was used ascontrol virus. The combined effect of AdTOP-PUMA withseveral chemotherapeutic agents (5-florouracil, doxorubi-cin, and paclitaxel) was also evaluated. The effect ofAdTOP-PUMA on colorectal cancer cells was alsoexamined in nude mice: SW480 cells were infected withthe AdTOP-PUMA and AdFOP-PUMA, and then inoculat-ed s.c. into nude mice. The TOP-PUMA adenovirusinhibited cell growth in a dose-dependent fashion,depending on the signaling activity of B-catenin. Thegrowth of cells displaying high levels of active B-catenin/Tcf signaling was inhibited after infection with AdTOP-
PUMA, whereas that of cells with low levels of B-cateninsignaling was not. Growth inhibition was associated withinduction of apoptosis. Chemotherapy synergisticallyenhanced the effect of AdTOP-PUMA. A combination ofthe adenovirus system with standard therapy mayimprove the efficacy and reduce the toxicity of therapyin humans. [Mol Cancer Ther 2006;5(11):2861–71]
Introductionh-Catenin is a multifunctional protein serving as a majorstructural component of cell-to-cell adherens junctions. Inaddition, it also acts as an important signaling molecule inthe Wnt pathway that plays a key role in embryogenesisand tumorigenesis (1–3).In the absence of Wnt signaling, the cytoplasmic level of
h-catenin is kept low through interaction with a proteincomplex [containing GSK3h-glycogen synthase kinase 3h,axin and adenomatous polyposis coli (APC)] that canphosphorylate h-catenin and target it to ubiquitin-mediatedproteasomal degradation (4). Activation of Wnt signalingleads to inactivation of GSK3h, resulting in cytoplasmicaccumulation of h-catenin (5). The increase in h-cateninlevel is followed by its translocation into the nucleus,where in complex with members of the T-cell factor (Tcf)/lymphocyte enhancer–binding factor family of transcrip-tion factors it activates the expression of target genes (6).The APC tumor suppressor is mutated in f80% of the
familial adenomatous polyposis syndrome and sporadiccolorectal cancer patients (7). Loss of APC is believed to beone of the earliest initiating events in multistage colorectalcarcinogenesis (8). Mutant APC loses its ability to direct h-catenin to degradation, resulting in nuclear accumulationand inappropriate activation of h-catenin–mediated trans-activation. Mutations in h-catenin in the GSK-3h phosphor-ylation sites have been identified in 50% of colorectalcancer cases that retain wild-type APC (9–11). c-MYC (12)and cyclin D1 (13, 14), which positively regulate cellproliferation, are target genes of h-catenin/Tcf with directimplications in tumorigenesis (15–19). Activating h-cateninmutations have also been identified in a variety of othertumors, including melanomas (20), hepatocellular carcino-mas (21), skin (22), breast (23), and prostate cancers (24),whereas the h-catenin–Tcf pathway is not activated inmost normal tissues. Therefore, a therapeutic strategy thattargets this pathway could be applied to patients withprimary or metastatic colorectal cancer.p53-up-regulated modulator of apoptosis (PUMA) is a
potent mediator of the p53 apoptotic response (25, 26).It belongs to the group of BH3-only proteins that havebeen shown to function by dimerization with other BH3domain–containing proteins, including Bcl-2 and Bcl-XL,that results in the release of cytochrome c from
Received 3/6/06; revised 8/19/06; accepted 9/11/06.
Grant support: Israel Cancer Association (N. Arber), Israel ScienceFoundation (A. Ben-Ze’ev), and the German-Israel Foundation for ScientificResearch and Development (A. Ben-Ze’ev).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
Note: This work was part of the requirements of Hadas Dvory-Sobol forher Ph.D. degree at the Sackler School of Medicine at Tel Aviv University.
Requests for reprints: Nadir Arber, Director-Integrated Cancer PreventionCenter, Tel-Aviv Medical Center, 6 Weizmann Street, Tel-Aviv 64239,Israel. Phone: 972-3-6974968; Fax: 972-3-6950339.E-mail: [email protected] or [email protected]
Copyright C 2006 American Association for Cancer Research.
doi:10.1158/1535-7163.MCT-06-0122
2861
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

mitochondria and induction of apoptosis by activation ofcaspase-3 and caspase-9 (27).A substantial limitation of conventional cancer chemo-
therapy and radiotherapy is the toxicity of these agents tonormal tissue. The toxicity of currently available genedelivery systems to the normal cell population results fromtheir toxicity to the normal cell population.Here,we proposea novel gene therapy approach that selectively expresses alethal gene by targeting the active h-catenin–Tcf pathway inhuman colorectal, gastric, and hepatic cancer cells. More-over, we show that combining this strategy with standardchemotherapy results in a synergistic growth inhibition ofcolorectal cancer cells that overcomes cancer cell resistanceto therapy. This approach may pave the way to a noveltreatment of primary and metastatic colorectal cancer.
Materials andMethodsCell CultureHuman colorectal cancer (SW480, DLD-1, HT-29,
HCT116, LS174T), gastric (AGS), hepatic (HepG2,SK-Hep-1), pancreatic (Colo357, Panc-1), and embryonickidney (293) cell lines were obtained from the AmericanType Culture Collection (Manassas, VA). They werecultured in DMEM (Sigma, Rehovot, Israel) containing5% to 10% fetal bovine serum (Biological Industries, BeitHaemek, Israel), 1% penicillin, and 1% streptomycin, at37jC, in an atmosphere of 95% oxygen and 5% CO2
(complete medium).
Construction of Plasmids and AdenoviralVectorsTwo sets of h-catenin/Tcf–responsive promoters were
generated: one contains wild-type Tcf/lymphocyte enhanc-er–binding factor binding sites fused with cFos (TOP-cFos-Luc-TOPFLASH) and the other, the SV40 (TOP-SV40-Luc)minimal promoter upstream to a luciferase (Luc) reportergene. The corresponding control plasmids were constructedfor each promoter by replacing the TOP oligomers withmutant Tcf-binding oligomers (FOP), e.g., FOP-cFos-Luc(FOPFLASH) and FOP-SV40-Luc (see the TOP and FOPsequences in Fig. 1A). To construct the TOP/FOP-cFos-Lucplasmids, an XbaI fragment containing the TOP-cFos, andthe FOP-cFos from TOPFLASH and FOPFLASH plasmids(generous gifts from Hans Clevers, Utrecht University,Utrecht, the Netherlands), was cloned into the NheIsite upstream to the Luc gene in the pGL3-basic plasmid(Promega, Rehovot, Israel). To construct the TOP/FOP-SV40-Luc plasmids, the BglII-NheI TOP and FOP fragmentswere cloned into the NheI and BglII sites upstream of theSV40 minimal promoter in the pGL3-promoter plasmid(Promega).The AdEasy system (28) was used to generate the
AdTOP-PUMA and AdFOP-PUMA (AdTOP/FOP-PUMA)adenoviruses. The TOP and FOP sequences were obtainedfrom the TOP-cFos-Luc and FOP-cFos-Luc plasmids andcloned into the shuttle vector-pAd-Track. The pAdTrackalso contains a green fluorescent protein (GFP) gene underthe control of the cytomegalovirus (CMV) promoter(Fig. 1B). The blunted HA-PUMA fragment (containing
the human PUMA cDNA fused to a double hemagglutinin-epitope tag) from pCEP4-PUMA (a generous gift from BertVogelstein, Johns Hopkins Oncology Center, Baltimore,MD) was cloned downstream to the TOP/FOP elementsin the pAd-Track vector. The resultant plasmids weredesignated pAdTrack-TOP/FOP-PUMA. These shuttlevectors were linearized with PmeI and cotransformed withE1-deleted adenoviral backbone AdEasy-1 into the compe-tent bacterial strain BJ5183, which enables efficient recom-bination. A panel of Ad-TOP-PUMA and Ad-FOP-PUMArecombinant adenoviruses were generated.
Adenovirus Production andTiteringTo produce viruses, 4 Ag PacI-linearized adenoviral
DNA was transfected into 50% to 70% confluent 293 cellsin 10-cm dishes using LipofectAMINE and Plus Reagents(Invitrogen Life Technologies, Carlsbad, CA). Between 5and 7 days posttransfection, colonies expressing GFP wereobserved under a fluorescent microscope, the cells wereharvested and lysed in PBS by four cycles of freeze/thaw/vortex (Fig. 1C). The supernatant was collected and half ofit was used to reinfect 50% to 70% confluent 293 cells.Viruses were collected 2 to 3 days postinfection when acytopathic effect became evident. Further amplificationand concentration of the virus stocks was achievedthrough several rounds of infection. To titer the viruses,50% to 70% confluent 293 cells in 96-well dishes wereinfected with serial dilutions of the virus stocks. GFP-positive colonies were counted 5 days postinfections. Thecontrol Ad-CMV-GFP adenovirus containing the GFP geneunder the control of a full-length CMV promoter was a
Figure 1. Construction of adenoviruses. A, h-catenin/Tcf activatablepromoters containing TOP or FOP sequences. B, schematic representa-tion of different adenovirus constructs. C, the Pac I-digested recombi-nant adenoviral vector pAdTOP-PUMA was transfected into 293 cellsand GFP expression was visualized by fluorescence microscopy at theindicated times. Comet-like adenovirus–producing foci became apparentafter 5 to 7 d.
Targeting activated b-Catenin in Human Cancer Cells2862
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

kind gift of Hila Giladi (Hadassah School of Medicine,Jerusalem, Israel) and was amplified in 293 cells.Human cDNA of caspase-8, Bak , and Bax were a kind gift
from Atan Gross (Weizmann Institute of Science, Rehovot,Israel). The expression constructs of PUMA and PUMA-DBH3, a mutant PUMA without activity, were generousgifts from Bert Vogelstein. PKGIh that encodes a mutantPKG sequence with an NH2-terminal truncation (29), was agift from I. Bernard Weinstein (Columbia University, NewYork, NY). This deletion renders PKG independent ofcyclic guanosine 3¶,5¶-monophosphate, and it is constitu-tively active.
LuciferaseAssaysTransfections were done using LipofectAMINE and Plus
Reagents (Invitrogen) according to the instructions fromthe manufacturer. A total of 5 � 105 cells were seeded insix-well plates. The next day, 50% confluent dishes werecotransfected with 1 Ag vectors plus 0.1 Ag pRL-TK(Promega). Luc assay was done 24 hours posttransfection.Briefly, cells were washed once with PBS and lysed in400 AL lysis buffer for 15 minutes at room temperature.The lysates were centrifuged at 14,000 rpm for 5 minutes,and 20 AL of each lysate were used to measure Lucreporter gene expression. Luc activity was normalized toRenilla Luc activity from a parallel cotransfection of pRL-TK (Dual Luc system, Promega). All experiments weredone in triplicate at least thrice and gave similar results.
The Potency of PUMA in Cell KillingSW480 cells were transfected with constructs encoding
for PUMA or a mutant PUMA-DBH3, without activity.Cells were harvested 24 hours after transfection, and anequal number of cells was diluted in duplicates (1:10 and1:25) into 10-cm dishes and grown under hygromycin Bselection for 3 weeks, after which the cells were fixed andstained with 0.2% Coomassie blue, 50% methanol, 10%acetic acid, and 40% H2O.
Cell Viability AssaysBetween 2 � 104 and 5 � 104 cells in 100 AL complete
medium were plated in 96-well dishes. The next day, sixwells were infected with each adenovirus at a differentmultiplicity of infection (0.1–50 MOI). Cell viability wasassessed bymethylene blue staining after 48 hours. The cellswere washed once with PBS and fixed in 150 ALformaldehyde (4%) for 2 hours at room temperature,washed with 0.1 mol/L sodium borate (pH 8.5), and stainedwith 0.5% methylene blue for 10 minutes, then washed withtap water and 150 AL of 0.1 mol/L HCl to dilute the cell-bound dye. Absorbance was measured at 590 nm. Cellviability is expressed as percentage absorbance relative tomock-infected cells. The average of at least two independentexperiments with six replicates was recorded.
ChemicalsPaclitaxel, doxorubicin, and 5-florouracil were obtained
from Sigma. Cells were infected with AdTOP-PUMA,AdFOP-PUMA, or Ad-CMV-GFP (5 MOI); after 5 hourswere treated with 0.05 Amol/L paclitaxel, 1 Amol/Ldoxorubicin, or 0.05 Amol/L 5-florouracil; and were thencultured for 48 hours.
Western Blot AnalysisInfected cells were harvested and protein concentrations
were determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). An equal amount of protein fromeach lysate was analyzed by SDS-PAGE and the proteinswere transferred to hybond-C extra nitrocellulose mem-branes (Amersham Life Science, Buckinghamshire, UnitedKingdom). Membranes were blocked with buffer contain-ing 5% low-fat milk and 0.05% Tween 20 in PBS for 1 hour,incubated with primary antibodies for 1 hour withperoxidase-conjugated secondary antibodies, and devel-oped with a Supersignal West Pico chemiluminescentsubstrate (Pierce, Rockford, IL). Antibodies against hemag-glutinin, actin, h-catenin, and caspase-3 were purchasedfrom Santa Cruz Biotechnology (Santa Cruz, CA).
Apoptosis AnalysisFlow Cytometry. Cells were plated at 5 � 106/10-cm
dish 24 hours before infection, and were infected withrecombinant adenoviral vectors at 5 MOI. Twenty-fourhours later, both adherent and floating cells wereharvested, washed with PBS, and fixed in 80% ethanolfor 1 hour and stained with propidium iodide for anal-ysis of DNA content. The number of subdiploid cells,representing apoptotic cells, was quantified by FACScanusing the CellQuest software (Becton Dickinson Immuno-cytometry Systems, San Jose, CA). Necrotic cells wereexcluded by staining with trypan blue. The average of atleast three independent experiments with two replicateswas recorded.ssDNA. For the ssDNA assay, 104 cells were seeded in
96-well microplates, and after 24 hours infected withrecombinant adenoviral vectors at 5 MOI. The followingday, the ssDNA Apoptosis ELISA kit was used (ChemiconInternational, Inc., Temecula, CA). Based on the selective,formamide-induced denaturation of DNA, this methodidentifies apoptotic cells (30), by staining ssDNA using amixture of anti-ssDNA monoclonal antibody and peroxi-dase-conjugated anti-mouse IgM. The average of at leasttwo independent experiments with two replicates wasrecorded.
Fluorogenic assay for caspase-3 activity.Cells were plated at 1 � 106 per well onto a six-well
plate 24 hours before infection, and were infected withrecombinant adenoviral vectors at 5 MOI. Cells wereharvested with a rubber policeman; washed; resuspendedin 50 mmol/L Tris-HCl buffer (pH 7.4), 1 mmol/L EDTA,and 10 mmol/L EGTA; and lysed by three successivefreeze-thaw cycles on dry ice. Cell lysates were centrifugedat 20,000 � g for 5 minutes, and the supernatants werestored at �70jC. The protein concentration of each samplewas determined using the Bradford Bio-Rad protein assay.For caspase-3 activity, a total of 50 Ag protein wasincubated with 50 mmol/L ac-DEVD-AMC (from BIOMOLResearch Laboratories, Plymouth Meeting, PA) at 37jC, for30 minutes in the dark. The release of 7-amino-4-methyl-coumarine was monitored by a spectrofluorometer usingan excitation wavelength of 360 nm and an emissionwavelength of 460 nm.
Molecular Cancer Therapeutics 2863
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

Tumorigenic AssaysCD1 nude mice housed in sterile cages were handled
under aseptic conditions. The animals were maintained infacilities approved by the Israeli Association for Accredi-tation of Laboratory Animal Care and in accordance withcurrent regulations and the standard of care of the IsraeliMinistry of Health. SW480 colorectal cancer cells wereinfected with 5 MOI of adenoviral vectors in serum-freemedium. Five hours after infection, the medium waschanged to 10% fetal bovine serum medium and theinfected cells were incubated at 37jC overnight. Twenty-four hours after adding the virus, the cells were trypsinizedand inoculated s.c. on the backs of nude mice (3 � 106
per animal). The number of mice with tumors (incidence)and their volume (tumor burden) were determined afterdifferent times.
Statistical AnalysisStatistical analysis was done by using InStat software
version 3.01 (GraphPad Software, Inc., San Diego, CA). Inthe tissue culture experiments, the comparison betweentwo samples was done using Student’s t test and betweenmore than two samples using one-way ordinary parametricANOVA followed by Tukey-Kramer multiple comparisontest. For all statistical tests, preliminary evaluation of thehomoscadacity and normality of the compared sampleswas done using Bartlett and Kolmogorov-Smirnov tests,respectively.
ResultsB-Catenin/Tcf ^Mediated Luc Activity in Different
Colorectal Cancer Cellsh-Catenin/Tcf–dependent activity was determined in
human cell lines displaying different levels of h-catenin(Fig. 2A). These cell lines harbored mutant APC proteins,except for HCT116 that have a deletion at residue S45 ofthe h-catenin protein (11). To evaluate which h-catenin/Tcfreporter construct is more readily detectable in these cells,the h-catenin/Tcf–responsive promoters fused to SV40and cFos minimal promoters were used using the luci-ferase (Luc) assay. h-Catenin–activated promoters contain-ing four copies of the Tcf-binding site (TOP) fused to eitherthe cFos or the minimal SV40 promoter were analyzed.The TOP-cFos (TOPFLASH) construct exhibited higheractivity than TOP-SV40 and was therefore used in thefollowing experiments. The relative activity of TOPFLASHis shown in Table 1. Luc activity, determined as foldinduction of TOPFLASH, was 4.1- to 25.3-fold higher thanthat of the control FOPFLASH in colorectal cancer cells(SW480, DLD-1, HCT116, and LS174T), whereas HT-29cells did not show significant transcriptional activation ofthis reporter construct (Table 1).
PUMA Induces Cell Death in SW480 CellsTo identify the most potent proapoptotic gene in
colorectal cancer cells, we tested several full-length cDNAs,including those encoding for Bak, caspase-8, PUMA,
Figure 2. PUMA suppresses the growth of colon cancer cells. A, comparison of h-catenin levels in different cell types. Cell extracts were prepared fromcolorectal cancer lines (SW480, HCT116, DLD-1, and HT-29). Twenty micrograms of protein from each sample were subjected to Western blot analysisusing anti-h-catenin and anti-h-actin antibodies. B, 293 cells were transiently transfected with human cDNA of caspase-8, Bid, Bak, Bax, PUMA, PKGIh,and pcDNA3. Forty-eight hours after transfection, the percentage of apoptotic (sub-G1) cells was determined by FACS analysis. *, P < 0.05, significantlydifferent from control. **, P < 0.01, significantly different from control. C, SW480 cell lines were transfected with constructs encoding PUMA andPUMA-DBH3. Cells were harvested 24 h after transfection, and equal cell numbers were diluted in duplicates in 10-cm dishes and grown under selection inhygromycin B for 3 wks, then fixed and stained with Coomassie blue. Colony formation in representative dishes (1:10 and 1:25 dilution) of transfectedSW480 cells is shown. D, the number of colonies formed with cells infected with PUMA and PUMA-DBH3 is shown. *, P < 0.05, significantly differentfrom PUMA-DBH3.
Targeting activated b-Catenin in Human Cancer Cells2864
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

PKGIh, Bax, and Bid that we transfected into 293 cells.Forty-eight hours after transfection, the number of sub-diploid DNA-containing cells, representing apoptotic cells,was quantified by FACScan (Fig. 2B). PUMA and Baxinduced the highest apoptotic activity. We have chosenPUMA because it was more effective in SW480 cells, whichwe used in the following studies (Fig. 2C).To determine the effect of PUMA expression on colon
cancer cell growth, an expression vector containing PUMAunder the control of the CMV promoter and a hygromycin
B –resistant gene (pCEP4-PUMA) was transfected intoSW480 cells. PUMA-DBH3 encoding for a nonfunctionalPUMA (without its BH3 domain) was used as control. Theresults shown in Fig. 2C and D show a drastic reduction incolony formation by cells after transfection with PUMAcompared with the mutant PUMA vector.Next, we used an adenoviral vector selected for gene
delivery with PUMA placed downstream to the cFosminimal promoter. The promoter contained either thewild-type (AdTOP-PUMA) or the mutant (AdFOP-PUMA)
Figure 3. AdTOP-PUMA suppresses the survival of colon cancer cells. A, SW480, DLD-1, HCT116, and HT-29 cells were infected with AdTOP-PUMA,AdFOP-PUMA, and Ad-CMV-GFP adenoviruses in 96-well culture plates. Cell viability expressed as percentage absorbance relative to mock-infected cellswas measured by methylene blue staining 48 h after adenoviral infection. Average of at least two independent experiments with six replicates. Statisticaldifference was observed between AdTOP-PUMA and the control groups (AdFOP-PUMA and Ad-CMV-GFP; ***P < 0.001; **P < 0.01). B, the number ofviable cells is proportional to the intensity of methylene blue staining shown here for SW480 cells 48 h after adenoviral infection. C, SW480 cells48 h following infection with either AdFOP-PUMA (left ) or AdTOP-PUMA (right ). AdTOP-PUMA and AdFOP-PUMA also contain a GFP gene under thecontrol of a CMV promoter. GFP expression was visualized by fluorescence microscopy. D, HT-29 and DLD-1 cells were infected (at 5 MOI) with eitherAdFOP-PUMA (left ) or AdTOP-PUMA (right ) and GFP expression was visualized by fluorescence microscopy after 48 h.
Table 1. B-catenin/Tcf–regulated transcription activity
Cell line Fold Luc activity (TOP/FOP) Cell line Fold Luc activity (TOP/FOP)
SW480 25.3 HepG2 63.5HCT116 4.1 SK-Hep-1 1.6DLD-1 4.8 AGS 5.5HT-29 1.5 Colo357 1.1LS174T 12.9 Panc-1 0.9
NOTE: Luc activities were assayed after 24 hours and plotted as fold activation of TOP-cFos-Luc relative to that of FOP-cFos-Luc.
Molecular Cancer Therapeutics 2865
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

Tcf/lymphocyte enhancer–binding factor binding sites.The Ad-CMV-GFP vector was used as control for viraltoxicity. The ability of AdTOP-PUMA and AdFOP-PUMAadenoviral vectors to kill cells with different levels ofh-catenin signaling was evaluated by cell viability assays48 hours after infection with adenoviruses at varyingdoses (Fig. 3A). Cells displaying elevated h-catenin tran-sactivation (Table 1), such as SW480, HCT116, and DLD-1,were killed efficiently by infection with AdTOP-PUMA,in a dose-dependent manner. Although the infectionefficiency of the adenoviral vectors in the human colorectalcancer cell line HT-29 was high (shown by the number ofGFP positive cells in Fig. 3D), neither AdTOP-PUMA norAdFOP-PUMA caused cell death in this cell line (Fig. 3A),most probably because the level of h-catenin/Tcf trans-activation in these cells is low (Table 1). The numberof viable cells after infection with AdTOP-PUMA wasproportional to the methylene blue color intensity as shownfor SW480 cells infected with adenoviral constructs(Fig. 3B). Representative pictures of SW480, DLD-1, andHT-29 cells, 48 hours after infection with either AdTOP-PUMA or AdFOP-PUMA, are shown in Fig. 3C.Next, the number of cells in the sub-G1 population was
determined by fluorescence-activated cell sorting (FACS)analysis in the different colorectal cancer cell lines infectedwith the various adenovirus constructs. By this method,SW480 and HCT116 cells displayed only a very lownumber of cells in the sub-G1 population (representing
apoptotic cells) 24 hours after treatment with AdFOP-PUMA or Ad-CMV-GFP (Fig. 4A). Apoptosis induced byAdTOP-PUMA was detected in SW480, DLD-1, andHCT116 cells 48 hours after infection, by the ssDNA assay,but not in HT-29 cells (Fig. 4B). The number of apoptoticcells was proportional to the increase in methylene bluecolor intensity as shown for SW480 cells infected withadenoviral constructs and stained with this dye after48 hours (Fig. 4C). Taken together, the results of thesedifferent approaches suggest that AdTOP-PUMA is capa-ble of inducing the death of cells that have elevated h-catenin/Tcf transcriptional activity.
Activated B-Catenin/Tcf Signaling Induces Apoptosisby Up-regulating PUMAExpressionIn SW480, HCT116, and DLD-1 cells, high levels of
PUMA protein were detected after infection with AdTOP-PUMA, but not after AdFOP-PUMA or Ad-CMV-GFPinfection (Fig. 5A). HT29 colorectal cancer cells that expresslower levels of h-catenin and low levels of h-catenin–Tcftransactivation did not express the PUMA protein afterinfection with AdTOP-PUMA (Fig. 5A). The infection ofSW480 cells with AdTOP-PUMA induced PUMA expres-sion as early as 12 hours after infection, as shown inFig. 5B. Induction of apoptosis by AdTOP-PUMA wasapparently mediated by the activation of caspases. Westernblot analysis of SW480 cells after infection with AdTOP-PUMA, but not with AdFOP-PUMA or Ad-CMV-PUMA,detected a cleaved form of caspases-3 (p17; Fig. 5C). Also,
Figure 4. Cell killing by AdTOP-PUMA adenovirus. A, SW480 and HCT116 cells were infected with AdTOP-PUMA, AdFOP-PUMA, or Ad-CMV-GFPadenovirus constructs at 5 MOI. Percentage apoptotic (sub-G1) cells was determined by FACS analysis 24 h after treatment. Significantly different from thecontrol groups (no virus, AdFOP-PUMA, Ad-CMV-GFP; *P < 0.05; **P < 0.01). B, induction of apoptosis determined by the apoptosis ELISA kitdescribed in Materials and Methods in cultures grown in 96-well plates and infected with adenoviruses for 48 h. *, significantly different from the controlgroups (no virus, P < 0.01; AdFOP-PUMA, P < 0.05; Ad-CMV-GFP, P < 0.01). #, significantly different from the control groups (no virus, P < 0.01;AdFOP-PUMA, P < 0.05; Ad-CMV-GFP, P < 0.05). &, significantly different from the control groups (no virus, P < 0.001; AdFOP-PUMA, P < 0.001;Ad-CMV-GFP, P < 0.001). C, color intensity is proportional the number of apoptotic SW480 cells 48 h after adenoviral infection.
Targeting activated b-Catenin in Human Cancer Cells2866
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

caspase-3 activation occurred 48 hours following infectionwith AdTOP-PUMA, but not after infection with controlviruses (AdFOP-PUMA or Ad-CMV-PUMA; Table 2).
AdTOP-PUMA Inhibits Tumor Growth In vivo
Next, we evaluated whether AdTOP-PUMA infection caninduce a cell killing effect in s.c. tumors established in nudemice with colorectal cancer cells. SW480 cells were infectedwith adenoviruses in vitro , harvested after 24 hours, andthen inoculated s.c. into nude mice. As shown in Table 3,mice that were injected with SW480 cells that were infectedwith AdTOP-PUMA virus failed to develop tumors. In con-trast, AdFOP-PUMA and Ad-CMV-GFP did not suppressthe growth of the implanted SW480 cells.
B-Catenin Activates Expression of PUMA fromWild-Type Tcf/Lymphocyte Enhancer ^Binding Factor ^Responsive Constructs in Other Tumor CellsThe effect of AdTOP-PUMA was determined in other
tumor cell lines with hyperactive h-catenin/Tcf signaling.Luc activity was analyzed in AGS (gastric cancer), HepG2(hepatocellular carcinoma), and LS174T (a colorectalcancer) cell lines. The results (Table 1) show that thesecell lines displayed significant levels of h-catenin/Tcfactivity. In contrast, pancreatic cells (Colo357 and Panc-1)and SK-Hep-1 (hepatocellular carcinoma) showed only
basal levels of h-catenin/Tcf activity. Next, we examinedthe effect of Ad-PUMA constructs on these cells. As shownin Fig. 6A, AGS, HepG2, and LS174T were efficiently killedby AdTOP-PUMA infection, whereas Colo357 and Panc-1cells were resistant, despite the high adenovirus infectionefficiency in these cells (Fig. 6A and B).
AdTOP-PUMA and Chemotherapy SynergisticallyInduce Tumor Cell DeathWe investigated whether the combination of AdTOP-
PUMA adenovirus and chemotherapeutic agents more
Figure 5. Up-regulation of PUMA after infection of cells with AdTOP-PUMA adenovirus. A, SW480, HCT116, DLD-1, and HT-29 cells were infectedwith AdTOP-PUMA, AdFOP-PUMA, or Ad-CMV-GFP (Ad-GFP) adenoviruses. Cells were harvested after 48 h and the lysates, normalized for proteinconcentration, were analyzed by Western blotting using antihemagglutinin (for PUMA detection) and anti-h-actin antibodies. Cells were treated with PBS(left lane in each blot ). B, immunoblot analysis of PUMA protein in SW480 cells 12, 24, 36, and 48 h postinfection with AdTOP-PUMA (5 MOI). C,Western blot analysis of caspase-3 activation in SW480 cells 24 h after infection. The antibody detected both the procaspase-3 (32 kDa) and the cleavedfragment of caspase-3 (17 kDa).
Table 2. Caspase-3 is activated by the AdTOP-PUMA construct
Construct Caspase activity
AdTOP-PUMA 38,070 F 193AdFOP-PUMA 9,342 F 1744AdCMV-GFP 14,515 F 409No treatment 11,363 F 317
NOTE: SW480 cells (1 � 106 per well) were seeded onto a six-well plate.Twenty-four hours later, 5 MOI of the various viruses were added to themedium for 48 hours. Caspase-3 activity was measured using thefluorometric assay described in Materials and Methods. The numbersrepresent the fluorometric signal F SD (P < 0.01).
Molecular Cancer Therapeutics 2867
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

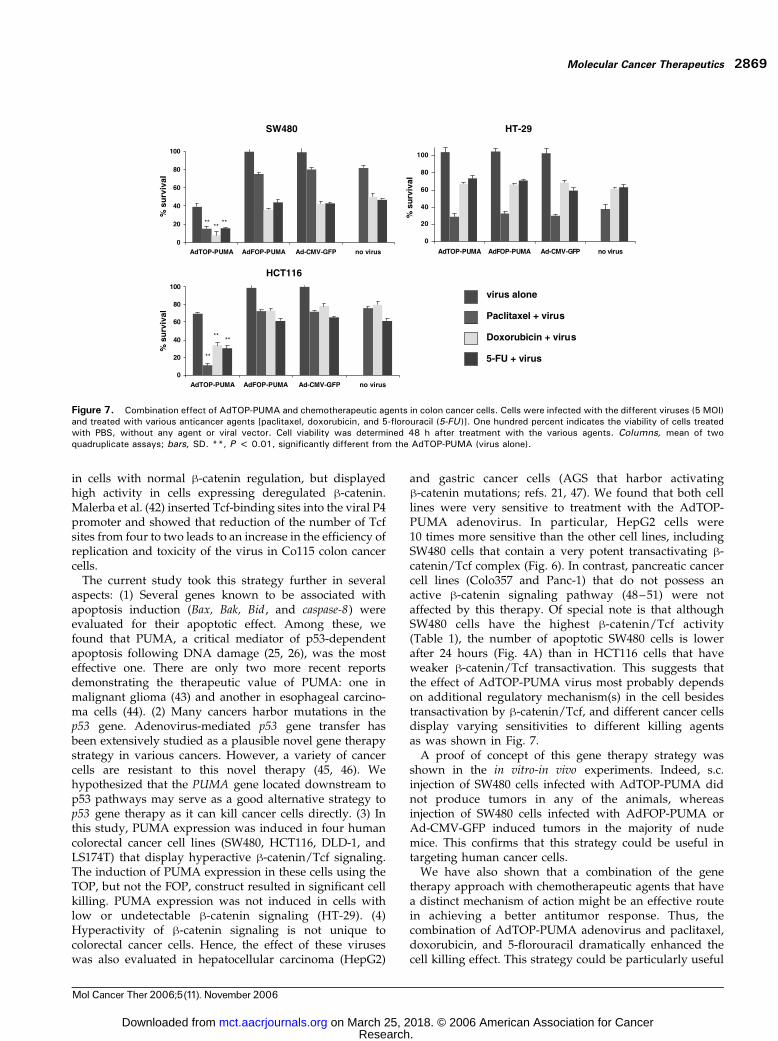
efficiently induce apoptosis in colorectal cancer cells thaneach treatment alone. HCT116, SW480, and HT29 cellswere infected with either AdTOP-PUMA, AdFOP-PUMA,or Ad-CMV-GFP adenoviruses (at 5 MOI) and cultured for48 hours in the presence or absence of paclitaxel (0.05Amol/L), doxorubicin (1 Amol/L), or 5-florouracil (0.05Amol/L). AdTOP-PUMA adenovirus dramatically en-hanced the killing effect by all three chemotherapeuticagents in both SW480 and HCT116 cells, but not in HT29cells (Fig. 7). The efficacy of the chemotherapeutic agentswas not augmented by exposure of the different cells toeither Ad-CMV-GFP or AdFOP-PUMA adenovirus con-structs (Fig. 7).
DiscussionAlthough gene therapy–based clinical trials showedsignificant success (31–33) in a variety of human tumors,thus far they failed to show a significant therapeutic effectin colorectal cancer. By contrast, in cell lines and inexperimental animals, remarkable results, including com-plete regression of the tumor, have been shown by thisapproach (34, 35). The lack of efficacy of this approach inhumans might result from the poor targeting selectivity ofthe vectors that could lead to low levels of expressionof the transferred gene in tumor cells and to high toxicityof the gene product in normal cells.
Adenoviral systems are easy to produce and they havea highly effective nuclear entry mechanism and displayvery low pathogenicity in humans (36). The adenoviralvectors can transduce cells in vivo and they do not integrateinto the host cell genome (36). Mulvihill et al. (37)conducted a phase I clinical trial of adenoviral administra-tion using ONYX-015 that consists of an adenovirusexpressing a deletion mutant of E1B-55 that selectivelyreplicates in and lyses tumor cells displaying a mutant p53.The virus was injected through hepatic arterial cathetersto patients with colorectal cancer liver metastases, and waswell tolerated at doses up to 1011 plaque-forming units. Aphase I clinical trial using the Escherichia coli cytosinedeaminase for the treatment of metastatic colon cancer iscurrently being conducted (38).In the present study, we show that targeting h-catenin/
Tcf responsive transcription can selectively and efficientlykill tumor cells displaying high levels of h-catenin/Tcftransactivation, with minimal toxicity to cells displayinglow levels of h-catenin–Tcf signaling. Similar strategieswere used and recently reported by others (39, 40). In Chenand McCormick’s work (39), an adenoviral vector, AdWt-Fd, containing the thymidine kinase promoter carrying theproapoptotic gene Fadd , selectively killed colorectal cancercells in vitro . Kwong et al. (40) used an in vitro-in vivoanimal model similar to the one used in the present study.They showed selective killing of DLD-1 colorectal cancercells in an ex vivo animal model, by the adenoviral vectorAdTOP-CMV-TK that contains a herpes simplex virusthymidine kinase gene under the control of a h-catenin/Tcf–responsive promoter linked to a minimal CMV promoter.Lipinski et al. (41) optimized the activity and specificityprofile of a synthetic catenin-dependent promoter byvarying its basal promoter, the number of Tcf-binding sites,and the distance between them and the basal promoter. Theoptimal promoter showed virtually undetectable expression
Table 3. AdTOP-PUMA suppresses the growth of tumors formedby SW480 cells displaying hyperactive B-catenin
No. mice Adenovirus Mice with tumor
8 AdTOP-PUMA 0* (0%)8 AdFOP-PUMA 3 (37.5%)7 Ad-CMV-PUMA 4 (57.1%)
*Number of mice with s.c. tumors after 8 weeks.
Figure 6. The effect of AdTOP-PUMA in different tumor cell lines. A, AGS (gastric cancer), HepG2 (hepatocellular carcinoma), LS174T (colon cancer),and Colo357 and Panc-1 (pancreatic cancer) cells were infected with AdTOP-PUMA, AdFOP-PUMA, and Ad-CMV-GFP viruses. Cell viability was measured48 h after adenoviral infection by methylene blue staining. Cell viability is expressed as percentage absorbance relative to mock-infected cells. Average ofat least two independent experiments with six replicates. Statistical difference was observed between AdTOP-PUMA and the control groups (AdFOP-PUMA and Ad-CMV-GFP) at 5, 10, and 25 MOI (***P < 0.001, **P < 0.01, *P < 0.05). B, Colo357 and Panc-1 cells were infected (at 5 MOI) witheither AdFOP-PUMA (left ) or AdTOP-PUMA (right ), and GFP expression was visualized by fluorescence microscopy after 48 h.
Targeting activated b-Catenin in Human Cancer Cells2868
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from
in cells with normal h-catenin regulation, but displayedhigh activity in cells expressing deregulated h-catenin.Malerba et al. (42) inserted Tcf-binding sites into the viral P4promoter and showed that reduction of the number of Tcfsites from four to two leads to an increase in the efficiency ofreplication and toxicity of the virus in Co115 colon cancercells.The current study took this strategy further in several
aspects: (1) Several genes known to be associated withapoptosis induction (Bax, Bak, Bid , and caspase-8) wereevaluated for their apoptotic effect. Among these, wefound that PUMA, a critical mediator of p53-dependentapoptosis following DNA damage (25, 26), was the mosteffective one. There are only two more recent reportsdemonstrating the therapeutic value of PUMA: one inmalignant glioma (43) and another in esophageal carcino-ma cells (44). (2) Many cancers harbor mutations in thep53 gene. Adenovirus-mediated p53 gene transfer hasbeen extensively studied as a plausible novel gene therapystrategy in various cancers. However, a variety of cancercells are resistant to this novel therapy (45, 46). Wehypothesized that the PUMA gene located downstream top53 pathways may serve as a good alternative strategy top53 gene therapy as it can kill cancer cells directly. (3) Inthis study, PUMA expression was induced in four humancolorectal cancer cell lines (SW480, HCT116, DLD-1, andLS174T) that display hyperactive h-catenin/Tcf signaling.The induction of PUMA expression in these cells using theTOP, but not the FOP, construct resulted in significant cellkilling. PUMA expression was not induced in cells withlow or undetectable h-catenin signaling (HT-29). (4)Hyperactivity of h-catenin signaling is not unique tocolorectal cancer cells. Hence, the effect of these viruseswas also evaluated in hepatocellular carcinoma (HepG2)
and gastric cancer cells (AGS that harbor activatingh-catenin mutations; refs. 21, 47). We found that both celllines were very sensitive to treatment with the AdTOP-PUMA adenovirus. In particular, HepG2 cells were10 times more sensitive than the other cell lines, includingSW480 cells that contain a very potent transactivating h-catenin/Tcf complex (Fig. 6). In contrast, pancreatic cancercell lines (Colo357 and Panc-1) that do not possess anactive h-catenin signaling pathway (48–51) were notaffected by this therapy. Of special note is that althoughSW480 cells have the highest h-catenin/Tcf activity(Table 1), the number of apoptotic SW480 cells is lowerafter 24 hours (Fig. 4A) than in HCT116 cells that haveweaker h-catenin/Tcf transactivation. This suggests thatthe effect of AdTOP-PUMA virus most probably dependson additional regulatory mechanism(s) in the cell besidestransactivation by h-catenin/Tcf, and different cancer cellsdisplay varying sensitivities to different killing agentsas was shown in Fig. 7.A proof of concept of this gene therapy strategy was
shown in the in vitro-in vivo experiments. Indeed, s.c.injection of SW480 cells infected with AdTOP-PUMA didnot produce tumors in any of the animals, whereasinjection of SW480 cells infected with AdFOP-PUMA orAd-CMV-GFP induced tumors in the majority of nudemice. This confirms that this strategy could be useful intargeting human cancer cells.We have also shown that a combination of the gene
therapy approach with chemotherapeutic agents that havea distinct mechanism of action might be an effective routein achieving a better antitumor response. Thus, thecombination of AdTOP-PUMA adenovirus and paclitaxel,doxorubicin, and 5-florouracil dramatically enhanced thecell killing effect. This strategy could be particularly useful
Figure 7. Combination effect of AdTOP-PUMA and chemotherapeutic agents in colon cancer cells. Cells were infected with the different viruses (5 MOI)and treated with various anticancer agents [paclitaxel, doxorubicin, and 5-florouracil (5-FU)]. One hundred percent indicates the viability of cells treatedwith PBS, without any agent or viral vector. Cell viability was determined 48 h after treatment with the various agents. Columns, mean of twoquadruplicate assays; bars, SD. **, P < 0.01, significantly different from the AdTOP-PUMA (virus alone).
Molecular Cancer Therapeutics 2869
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

in the treatment of chemotherapy-resistant colorectalcancer (52) because it might minimize the toxicity of thisregimen. Adenoviruses practically infect all types of cellsin the body and, therefore, PUMA will probably beactivated only in cells with hyperactive h-catenin signaling.Hence, in the clinical setting, combination therapy usingadenovirus and chemotherapeutic agents would be suit-able not only for the metastatic disease, but also at stages IIand III of the disease, aiming to eradicate microscopicresidual tumor cells.Recently, we showed that a Ras-responsive element
selectively kills tumor cells without affecting the growthand survival of normal cells (53). Introduction of cell deathgenes (bax, caspase-8 , and mutant PKG) under the control ofa promoter containing wild-type Ets and activator protein-1binding sites resulted in preferential killing of cells dis-playing a mutant, but not a normal, Ras pathway.In conclusion, this strategy involving an active h-catenin/
Tcf pathway is not a cell- or organ-specific approach, butcould be applied to a much wider range of cases becausehyperactive h-catenin/Tcf signaling is found in a significantpercentage of almost all types of cancer (2, 54). The resultsof the current study may pave the way to new combinedapproaches with chemotherapy in the setting of all stagesof colorectal cancer with activated h-catenin signaling.
Acknowledgments
We thank Moshe Oren (Weizmann Institute of Science, Israel) for helpfuladvice and thorough discussions, Hila Giladi (Hebrew University, Jerusa-lem, Israel) for assistance with adenovirus production, and Naham Kariv(Tel Aviv University, Israel) for assistance with the animal studies.
References
1. Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: proteininteractions and their implications for cadherin function. J Cell Biochem1996;61:514–23.
2. Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-cateninadhesion system in signaling and cancer. J Clin Invest 2002;109:987–91.
3. Wodarz A, Nusse R. Mechanisms of Wnt signaling in development.Annu Rev Cell Dev Biol 1998;14:59–88.
4. Polakis P. Wnt signaling and cancer. Genes Dev 2002;14:1837–51.
5. Eastman Q, Grosschedl R. Regulation of LEF-1/Tcf transcription factorsby Wnt and other signals. Curr Opin Cell Biol 1999;11:233–40.
6. Behrens J, von Kries JP, Kuhl M, et al. Functional interaction ofh-catenin with the transcription factor LEF-1. Nature 1996;382:638–42.
7. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis.Cell 1990;61:759–67.
8. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer.Cell 1996;87:159–70.
9. Morin PJ, Sparks AB, Korinek V, et al. Activation of h-catenin-Tcfsignaling in colon cancer by mutations in h-catenin or APC. Science 1997;275:1787–90.
10. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis ofthe APC/h-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130–4.
11. Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. h-Cateninmutations in cell lines established from human colorectal cancers. ProcNatl Acad Sci U S A 1997;94:10330–4.
12. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target ofthe APC pathway. Science 1998;281:1509–12.
13. Tetsu O, McCormick F. h-Catenin regulates expression of cyclin D1 incolon carcinoma cells. Nature 1999;398:422–6.
14. Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is atarget of the h-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 1999;96:5522–7.
15. Sikora K, Chan S, Evan G, et al. c-Myc oncogene expression incolorectal cancer. Cancer 1987;59:1289–95.
16. Erisman MD, Scott JK, Watt RA, Astrin SM. The c-myc protein isconstitutively expressed at elevated levels in colorectal carcinoma celllines. Oncogene 1998;2:367–78.
17. Finley GG, Schulz NT, Hill SA, Geiser JR, Pipas JM, Meisler AI.Expression of the myc gene family in different stages of human colorectalcancer. Oncogene 1989;4:963–71.
18. Imaseki H, Hayashi H, Taira M, et al. Expression of c-myc oncogene incolorectal polyps as a biological marker for monitoring malignant potential.Cancer 1989;64:704–9.
19. Arber N, Hibshoosh H, Moss SF, et al. Increased expression of cyclinD1 is an early event in multistage colorectal carcinogenesis. Gastroenter-ology 1996;110:669–74.
20. Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P.Stabilization of h-catenin by genetic defects in melanoma cell lines.Science 1997;275:1790–2.
21. de La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations ofthe h-catenin gene are frequent in mouse and human hepatocellularcarcinomas. Proc Natl Acad Sci U S A 1998;95:8847–51.
22. Chan EF, Gat U, McNiff JM, Fuchs E. A common human skin tumouris caused by activating mutations in h-catenin. Nat Genet 1999;21:410–3.
23. Lin SY, Xia W, Wang JC, et al. h-Catenin, a novel prognostic markerfor breast cancer: its roles in cyclin D1 expression and cancer progression.Proc Natl Acad Sci U S A 2000;97:4262–6.
24. Voeller HJ, Truica CI, Gelmann EP. h-Catenin mutations in humanprostate cancer. Cancer Res 1998;58:2520–3.
25. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMAinduces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001;7:673–82.
26. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is inducedby p53. Mol Cell 2001;7:683–94.
27. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12.
28. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B.A simplified system for generating recombinant adenoviruses. Proc NatlAcad Sci U S A 1998;95:2509–14.
29. Soh JW, Mao Y, Liu L, Thompson WJ, Pamukcu R, Weinstein IB.Protein kinase G activates the JNK1 pathway via phosphorylation ofMEKK1. J Biol Chem 2001;276:16406–10.
30. Frankfurt OS, Krishan A. Identification of apoptotic cells byformamide-induced DNA denaturation in condensed chromatin. J Histo-chem Cytochem 2001;49:369–78.
31. Nemunaitis J, Swisher SG, Timmons T, et al. Adenovirus-mediatedp53 gene transfer in sequence with cisplatin to tumors of patients withnon-small-cell lung cancer. J Clin Oncol 2000;18:609–22.
32. Buller RE, Runnebaum IB, Karlan BY, et al. A phase I/II trial of rAd/p53(SCH 58500) gene replacement in recurrent ovarian cancer. Cancer GeneTher 2002;9:553–66.
33. Swisher SG, Roth JA, Komaki R, et al. Induction of p53-regulatedgenes and tumor regression in lung cancer patients after intratumoraldelivery of adenoviral p53 (INGN 201) and radiation therapy. Clin CancerRes 2003;9:93–101.
34. Mullen JT, Donahue JM, Chandrasekhar S, et al. Oncolysis by viralreplication and inhibition of angiogenesis by a replication-conditionalherpes simplex virus that expresses mouse endostatin. Cancer 2004;101:869–77.
35. Qin XQ, Tao N, Dergay A, et al. Interferon-h gene therapy inhibitstumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci U S A 1998;95:14411–6.
36. Volpers C, Kochanek S. Adenoviral vectors for gene transfer andtherapy. J Gene Med 2004;6 Suppl 1:S164–71.
37. Mulvihill S, Warren R, Venook A, et al. Safety and feasibility ofinjection with an E1B-55 kDa gene-deleted, replication-selective adenovi-rus (ONYX-015) into primary carcinomas of the pancreas: a phase I trial.Gene Ther 2001;8:308–15.
Targeting activated b-Catenin in Human Cancer Cells2870
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

38. Crystal RG, Hirschowitz E, Lieberman M, et al. Phase I study ofdirect administration of a replication deficient adenovirus vector contain-ing the E. coli cytosine deaminase gene to metastatic colon carcinoma ofthe liver in association with the oral administration of the pro-drug5-fluorocytosine. Hum Gene Ther 1997;8:985–1001.
39. Chen RH, McCormick F. Selective targeting to the hyperactiveh-catenin/T-cell factor pathway in colon cancer cells. Cancer Res 2001;61:4445–9.
40. Kwong KY, Zou Y, Day CP, Hung MC. The suppression of coloncancer cell growth in nude mice by targeting h-catenin/Tcf pathway.Oncogene 2002;21:8340–6.
41. Lipinski KS, Djeha HA, Gawn J, et al. Optimization of a synthetich-catenin-dependent promoter for tumor-specific cancer gene therapy.Mol Ther 2004;10:150–61.
42. Malerba M, Nikolova D, Cornelis J, Iggo R. Targeting of autonomousparvoviruses to colon cancer by insertion of Tcf sites in the P4 promoter.Cancer Gene Ther 2005;13:273–80.
43. Ito H, Kanzawa T, Miyoshi T, et al. Therapeutic efficacy of PUMA formalignant glioma cells regardless of p53 status. Hum Gene Ther 2005;16:685–98.
44. Wang H, Qian H, Yu J, et al. Administration of PUMA adenovirusincreases the sensitivity of esophageal cancer cells to anticancer drugs.Cancer Biol Ther 2006;5:380–5.
45. Harris MP, Sutjipto S, Wills KN, et al. Adenovirus-mediated p53 genetransfer inhibits growth of human tumor cells expressing mutant p53protein. Cancer Gene Ther 1996;3:121–30.
46. Kagawa S, Gu J, Swisher SG. Antitumor effect of adenovirus-mediated Bax gene transfer on p53-sensitive and p53-resistant cancerlines. Cancer Res 2000;60:1157–61.
47. Ebert MP, Yu J, Hoffmann J, et al. Loss of h-catenin expression inmetastatic gastric cancer. J Clin Oncol 2003;21:1708–14.
48. Lowy AM, Fenoglio-Preiser, Kim OJ, et al. Dysregulation of h-cateninexpression correlates with tumor differentiation in pancreatic ductadenocarcinoma. Ann Surg Oncol 2003;10:284–90.
49. Gerdes B, Ramaswamy A, Simon B, et al. Analysis of h-catenin genemutations in pancreatic tumors. Digestion 1999;60:544–8.
50. Ikenoue T, Ijichi H, Kato N, et al. Analysis of the h-catenin/T cell factorsignaling pathway in 36 gastrointestinal and liver cancer cells. JpnJ Cancer Res 2002;93:1213–20.
51. Caca K, Kolligs FT, Ji X, et al. Beta- and g-catenin mutations, but notE-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factortranscriptional deregulation in gastric and pancreatic cancer. Cell GrowthDiffer 1999;10:369–76.
52. de Angelis PM, Fjell B, Kravik KL, Haug T, Tunheim SH, Reichelt W.Molecular characterizations of derivatives of HCT116 colorectal cancercells that are resistant to the chemotherapeutic agent 5-fluorouracil. IntJ Oncol 2004;24:1279–88.
53. Dvory-Sobol H, Kazanov D, Arber N. Gene targeting approach toselectively kill colon cancer cells, with hyperactive K-Ras pathway.Biomed Pharmacother 2005;59 Suppl 2:S370–4.
54. Polakis P. Wnt signaling and cancer. Genes Dev 2000;14:1837–51.
Molecular Cancer Therapeutics 2871
Mol Cancer Ther 2006;5(11). November 2006
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from

2006;5:2861-2871. Mol Cancer Ther Hadas Dvory-Sobol, Eyal Sagiv, Diana Kazanov, et al.
-catenin pathway to treat cancer cellsβTargeting the active
Updated version
http://mct.aacrjournals.org/content/5/11/2861
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/5/11/2861.full#ref-list-1
This article cites 54 articles, 20 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/5/11/2861.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/5/11/2861To request permission to re-use all or part of this article, use this link
Research. on March 25, 2018. © 2006 American Association for Cancermct.aacrjournals.org Downloaded from





























