Embed Size (px)
Citation preview

OLIGONUCLEOTIDES 15:36–50 (2005)© Mary Ann Liebert, Inc.
Targeting and Regulation of the HER-2/neu OncogenePromoter with Bis-Peptide Nucleic Acids
AMY J. ZIEMBA, ZHANNA V. ZHILINA, YULIA KROTOVA-KHAN, LENKA STANKOVA, and SCOT W. EBBINGHAUS
ABSTRACT
Antigene oligonucleotides have the potential to regulate gene expression through site-specific DNAbinding. However, in vivo applications have been hindered by inefficient cellular uptake, degrada-tion, and strand displacement. Peptide nucleic acids (PNAs) address several of these problems, asthey are resistant to degradation and bind DNA with high affinity. We designed two cationic pyrim-idine bis-PNAs (cpy-PNAs) to target the polypurine tract of the HER-2/neu promoter and comparedthem to an unmodified phosphodiester triplex-forming oligonucleotide (TFO1) and a TFO-nitrogenmustard conjugate (TFO2). PNA1 contains a �2 charge and bound two adjacent 9-bp target se-quences with high affinity and specificity, but only at low pH. PNA2 contains a �5 charge and boundone 11-bp target with high affinity up to pH 7.4, but with lower specificity. The PNA:DNA:PNAtriplex formed by these cpy-bis-PNAs presented a stable barrier to DNA polymerase extension. Thecpy-bis-PNAs and the TFO-alkylator conjugate prevented HER-2/neu transcription in a reportergene assay (TFO2 � PNA1 � PNA2 �� TFO1). Both PNAs and TFOs were effective at binding thetarget sequence in naked genomic DNA, but only the TFO-alkylator (TFO2) and the more cationicPNA (PNA2) were detected at the endogenous HER-2/neu promoter in permeabilized cells. Thiswork demonstrates the potential for preventing HER-2/neu gene expression with cpy-bis-PNAs in tu-mor cells.
INTRODUCTION
OLIGONUCLEOTIDES (OLIGOS) have the potential totarget DNA site specifically and regulate gene ex-
pression, prevent DNA replication, and induce site-spe-cific mutations (Guntaka et al., 2003). These propertiescould ultimately lead to new treatments for a wide vari-ety of genetic and infectious diseases, including cancerand HIV infection (Cho-Chung, 2002; Laskin and San-dler, 2004; Orr, 2001; Rudin et al., 2004). Oligos thatare designed to target DNA in order to interfere withgene expression are known as antigene oligos. Triplex-forming oligos (TFOs) are antigene oligos that bind topolypurine-polypyrimidine DNA tracts and form basetriplets by Hoogsteen bonding to double-stranded DNA
(dsDNA). Triplex-forming oligonucleotides have beendemonstrated to recognize and bind target sequenceswithin the structured chromatin of permeabilized cells(Giovannangeli 1997) or isolated nuclei (Besch et al.,2002). Further evidence of the ability of TFOs to bind totarget sequences within living cells comes from the abil-ity of TFOs to mediate mutagenesis at the target site inyeast cells (Barre et al., 2000), cultured mammalian cells(Vasquez et al., 1999, 2001; Wang et al., 1996), and mice(Vasquez et al., 2000). TFO binding to the target se-quence in intact mammalian cells was also demonstratedby site-specific, TFO-directed strand cleavage (Sedel-nikova et al., 2002). Although mutagenesis or cleavageevents are infrequent, they have unambiguously demon-strated that a TFO can bind to a target sequence in living
Arizona Cancer Center, Tucson, AZ 85724.
36

cells. TFOs have also been shown to prevent transcrip-tion initiation or elongation from plasmid DNA trans-fected into cells (Faria et al., 2001; Kim et al., 1998;Ziemba et al., 2003), but few studies have evaluated theregulation of endogenous gene expression by TFOs. Onepromising study demonstrated biologic activity in cellsby a TFO and also indicated specific target interactions(Majumdar et al., 2003). Other recent reports demon-strated antigene effects of TFOs at endogenous targets incell culture (Besch et al., 2002; Carbone et al., 2003,2004) but did not confirm site-specific binding in viablecells. To date, antigene activity by a TFO has not beendemonstrated to be caused by direct interactions at the in-tended target sequence in living cells.
Peptide nucleic acids (PNAs) are DNA mimics inwhich the sugar-phosphate backbone of DNA is replacedby an uncharged aminoethylglycine backbone. PNAswere first reported in 1991 (Nielsen et al., 1991) and havesince been shown to interact with single-stranded, dou-ble-stranded, hairpin, and quadruplex DNA, as well asRNA (Armitage, 2003). PNAs have great potential as anantigene therapy for targeting of chromosomal DNA(Kaihatsu et al., 2004b). Compared with DNA and manyDNA analogs, PNAs have a higher affinity for comple-mentary DNA and RNA sequences, bind less to proteins,and are less susceptible to degradation by nucleases orproteases (Demidov et al., 1994; Uhlmann, 1998). TheDNA:DNA:DNA triple helix is recognized by DNA re-pair proteins (Vasquez et al., 2002) and displaced by he-licases (Maine and Kodadek, 1994), but PNAs are lesssusceptible to unwinding by helicases (Bastide et al.,1999; Tackett et al., 2002) and block DNA polymeraseelongation when bound to duplex DNA (Smolina et al.,2003). Pyrimidine-rich PNAs can bind to a polypur-ine DNA sequence by strand invasion and form aPNA:DNA:PNA triple helix, differentiating PNAs fromDNA-binding oligos described to date. In this complex,one PNA molecule binds the purine-rich DNA strand byHoogsteen bonds while a second PNA molecule binds byWatson-Crick base pairing, displacing the noncomple-mentary pyrimidine DNA strand (Egholm et al., 1993;Nielsen, 2001; Nielsen et al., 1991). A theoretical analy-sis indicated that the Hoogsteen base pairing occurs first,followed by strand invasion and Watson-Crick base pair-ing, resulting in formation of a P-loop (Kuhn et al.,1999). Local distortion of the double helix appears to be the rate-limiting step for PNA invasion (Bentin andNielsen, 1996; Demidov et al., 1995; Larsen and Nielsen,1996). The rate of PNA binding and strand invasion maybe improved by linking the two single pyrimidine-richPNA molecules by a flexible linker (bis-PNAs) and theinclusion of cationic lysine residues (cationic pyrimidinePNAs, cpy-PNAs) (Griffith et al., 1995). Finally, the for-mation of a pyrimidine motif triple helix requires proto-nation of cytosines and is facilitated at neutral pH by J-
TARGETING THE HER-2/NEU PROMOTER 37
base (pseudoisocytosine) substitutions (Egholm et al.,1995).
In cell-free models and in tissue culture, PNAs canboth inhibit and activate transcription, prevent DNAreplication, and induce mutations in chromosomal DNA(Diviacco et al., 2001; Faruqi et al., 1998; Larsen andNielsen, 1996; Mollegaard et al., 1994; Nielsen et al.,1994; Wang et al., 1999, 2001; Zhao et al., 2003). Onedrawback is that PNAs are more difficult to deliver to cultured cells than DNA oligos because their neutrallycharged backbone (Chinnery et al., 1999) is not amenableto PNA inclusion in most of the commonly used DNAtransfection reagents. Recently, several groups haveshown that PNAs conjugated to either lipophilic moietiesor to cell-penetrating peptides (CPPs) can effectively de-liver PNAs to cultured cells (Kaihatsu et al., 2004a; Kop-pelhus and Nielsen, 2003). The HER-2/neu oncogene isoverexpressed in numerous types of human cancer, andits overexpression often correlates with poor clinical out-come (Cirisano and Karlan, 1996; Jardines et al., 1993).The apparent involvement of HER-2/neu in tumor pro-gression and metastasis makes it an attractive target forcancer therapeutics. The HER-2/neu promoter contains apolypurine tract within a region of transcriptional impor-tance, overlapping a transcription initiator sequence andjust upstream of the TATAA box, both of which are ca-pable of independently controlling transcription initiation(Mizuguchi et al., 1994). TFOs targeting this polypurinetract can disrupt transcription factor binding and preventtranscription (Ebbinghaus et al., 1993; Noonberg et al.,1994; Porumb et al., 1996; Ziemba et al., 2003). How-ever, we have observed previously that a triple helixformed within this sequence does not persist in cells un-less third strand binding is stabilized by covalent adductswith the target sequence using a TFO-alkylator conjugate(Ziemba et al., 2003). In the present work, we designedbis-PNAs predicted to bind to the HER-2/neu promoterwith sufficient stability to mediate antigene effects incells without the need for covalent binding to the targetsequence. We demonstrate that two different bis-PNAmolecules can target the HER-2/neu promoter and inhibitHER-2 transcription comparable to a TFO-alkylator con-jugate in a reporter plasmid. PNA binding was detectedat the endogenous HER-2/neu promoter in permeabilizedbreast cancer cells with an amplified HER-2/neu gene.
MATERIALS AND METHODS
Oligonucleotide synthesis
PNAs were purchased from Applied Biosystems (Fos-ter City, CA) (PNA1) and Biosynthesis, Inc. (Lewisville,TX) (PNA2) and the sequences are PNA1, NH2-TC-CTCCTCC-OOO-Lys-CCTCCTCCT; PNA2, NH2-Lys-

Lys-CCTCCTCCTCC-OOO-CCTCCTCCTC-Lys-Lys.The PNA arms were separated by three linker molecules(O, 2-amino-ethoxy-2-ethoxyacetic acid). Lysine (lys)residues were incorporated where indicated. The PNAswere aliquoted in siliconized tubes, stored at 4°C for lessthan 2 months, and preheated to 50°C for 10 minutesprior to use. DNA oligos were purchased from QiagenOperon (Chatsworth, CA) and gel purified before use.TFO sequences are TFO1, 3�-AGGAGAAGGAGGAG-GCGGAGGAGGAGGG; TFO2, 3�-PAM-GAGAAG-GAGGAGGCGGAGGAGGAGG-PAM. TFO2 was syn-thesized with a C6-amino group at both the 5�-end and3�-end, conjugated to phenylacetic acid mustard (PAM),and purified by reverse-phase high-performance liquidchromatography (HPLC) as previously described(Ziemba et al., 2003). Guanine residues in TFO2 weresubstituted with 8-aza-7-deazaguanine (PPG) to preventself-association of the TFO (Ebbinghaus et al., 1999;Lampe et al., 1997).
Electrophoretic mobility shift assays (EMSAs)
Synthetic duplexes were formed with complementaryoligos that were end-labeled on either the pyrimidine orthe purine strand, annealed, and gel purified. The du-plexes represented the native HER-2/neu promoter se-quence from �214 to �249 (Fig. 1), the HER-2/neu promoter sequence with 1-bp mismatches in the PNAbinding sites (at �223 and �234), or a purine-rich con-trol containing a PNA binding site with a 3-bp mismatch.In addition, an asymmetric HER-2/neu duplex (51-merpyrimidine strand and 65-mer purine strand) was end-la-beled on both strands to simultaneously demonstratePNA binding to the purine strand and dissociation of thepyrimidine strand. Labeled duplexes (0.01 �M) were in-cubated with increasing concentrations of PNA (0.1nM–1 �M). PNA binding reactions were performed in 10mM NaPO4 buffer, pH 6.5–7.4, at 37°C for 4 hours. Thecomplexes were separated on a 12% native polyacry-lamide gel and visualized by autoradiography. Becausebis-PNA-induced invasion complexes form with highstability, the dissociation rate is low and often unmeasur-able (Bentin et al., 2003). Therefore, true thermodynamicequilibrium constants are not practical. In these experi-ments, the PNA binding affinity is described by the PNAconcentration leading to conversion of 50% of the dou-ble-stranded target to single-stranded pyrimidine strandplus PNA:DNA complexes (C50).
DNase I and S1 digestions
For DNase I footprinting on the purine strand, a 250-bpPstI/XmaI fragment of the HER-2/neu promoter was ex-cised from pGL3/HNP410. The pGL3/HNP410 constructcontains the HER-2/neu promoter from �410 to �1 rela-tive to the translation start site driving luciferase expres-
ZIEMBA ET AL.38
sion (pGL3 basic) (Promega, Madison, WI). The restric-tion fragment was end-labeled on the top strand down-stream of the target sequence by Klenow fill-in, and 0.1�M labeled probe was incubated with 0.01–1.0 �MPNA, representing a 0.1–10� molar ratio of PNA, at pH6.5, or with 1 �M (10� molar ratio) of TFO1 in 1�TBM (90 mM Tris, pH 7.4, 90 mM borate, 10 mMMgCl2). The reactions (0.01 �M final) were incubatedwith 2 �G dIdC in balance buffer (75 mM KCl, 20 mMTris, pH 7.4) for 2 minutes at ambient temperature. Anequal volume (10 �l) of DNase dilution buffer (10 mMMgCl2, 10 mM CaCl2) containing 0.1 U DNase I (Invit-rogen, San Diego, CA) was added for 30 seconds at ambient temperature. Reactions were immediately termi-nated in formamide containing 20 mM EDTA and sepa-rated on a denaturing 8% sequencing gel. A Maxam andGilbert G-reaction was performed to identify the pro-tected guanine residues (Sambrook et al., 2001). Compe-tition experiments were performed by incubating 0.1 �MPNA2 (1�) with the labeled HER-2/neu promoter re-striction fragment and either wild-type or purine-richcontrol duplexes as cold competitors. The cold competi-tor duplexes were added at 10-fold molar excess to theradiolabeled HER-2/neu promoter, either just before in-cubation with PNA2 or for 4 hours after the PNA was al-ready bound to the radiolabeled HER-2/neu promoter.
For S1 probing of the pyrimidine strand, a 280-bpEcoRI/SmaI fragment of the pGL3/HNP410 plasmid wasend-labeled on the bottom strand upstream of the targetsequence by Klenow fill-in. The fragment was incubatedwith the indicated concentration of PNA1 or TFO1 at37°C for 4 hours or overnight. The reaction was incu-bated with 10 U S1 endonuclease in 0.5� S1 endonucle-ase buffer (Fisher Scientific, Pittsburgh, PA) at ambienttemperature for 20 minutes. The reaction conditions wereoptimized so that 50% of the full-length fragment wascleaved by S1 endonuclease. Reactions were terminatedin formamide containing 50 mM EDTA and separated ona denaturing 8% sequencing gel. A Maxam and GilbertC-reaction was performed to identify the location of theS1 sensitive sites (Sambrook et al., 2001).
DNA polymerase arrest assay
DNA polymerase arrest assays are a variation of a de-scribed technique (Han et al., 1999; Weitzmann et al.,1996). The 86-mer template strand for primer extensioncontains the polypurine tract of the HER-2/neu pro-moter from �249 to �212 flanked by a primer binding (TCCAACTATGTATAC) and tail sequence (AGCG-GCACGCAATTGCTATAGTGAGTCGTATTA) (Fig.4A). End-labeled primer (0.67 �M) and template strand(0.33 �M) were annealed in a 30-�l final volume. Thisprimer-template heteroduplex was used for PNA bindingand primer extension. For TFO binding, end-labeled

TARGETING THE HER-2/NEU PROMOTER 39
primer and a 36-mer complement (0.67 �M) to the poly-purine tract of the HER-2/neu promoter were both an-nealed to the 86-mer template. Annealed heteroduplexeswere gel purified to remove excess primer or pyrimidinecomplement.
PNAs were mixed with 0.01 �M template-primer mix-ture in a 10-�l final volume (NaPO4, pH 6.5 or 7.4) andincubated for 4 hours at 37°C before adding 1 mM MgCl2
for DNA polymerase extension. TFOs were mixed with0.01 �M promoter primer-template mixture in a final vol-ume of 10 �l 1�TBM and incubated for 4 hours at 37°C.DNA polymerase extension was performed by adding0.15 mM dNTPs and 5 U Taq DNA polymerase (Fermen-tas, Hanover, MD) for 20 minutes at 37°C. Taq was se-lected for these assays so that future studies could be per-formed to evaluate PNA stability at higher temperatures,but only 37°C was used here. The reactions were stoppedby adding formamide loading buffer with 10 mM EDTAand 10 mM NaOH, and the extension products were re-solved on a denaturing 8% sequencing gel. To identify thesites of polymerase arrest, a chain termination sequencingreaction was performed with the same primer-templatemixture using ThermoSequenase (USB, Cleveland, OH)according to the manufacturer’s instructions.
Transient transfections
HeLa cells were purchased from the American TypeCulture Collection (ATCC, Rockville, MD) and culturedas recommended. Site-directed mutagenesis was per-formed (Quickchange, Stratagene, La Jolla, CA) to gen-erate mutant promoter plasmids using the normal plas-mid pGL3/HNP410, and 6-bp mutations were introducedby engineering an SphI restriction site at target 1 from�237 to �232 (�237 plasmid) or at target 2 from �225to �220 (�225 plasmid). Oligos (1–10 �M) were incu-bated with plasmids (2 �g) at 37°C for 4 hours in either10 mM NaPO4 buffer at pH 6.5 (for PNAs) or 1�TBM atpH 7.4 (for TFOs) and then mixed with lipofectamine(Invitrogen) and the internal control pRLSV40 renilla lu-ciferase vector (Promega) for transfection into HeLacells. Dual luciferase Assays (Promega) were performedat 24–72 hours, HER-2/neu-driven luciferase activitywas normalized for transfection efficiency, and the dataare presented relative to luciferase activity from un-treated pGL3/HNP410 as previously described (Ziembaet al., 2003).
Southern blot protection assay
Genomic DNA (gDNA) from SK-BR-3 cells (15 �g)was heated to 50°C for 20 minutes and was incubatedwith PNAs in 10 mM NaPO4 buffer (pH 6.5 or 7.4) for up to 24 hours. The TFO reaction was performed in1�TBM without prior heating of gDNA. The gDNA wasthen digested with 1 U BbvI/�g DNA for 2 hours at
37°C, separated on an agarose gel, and transferred to anylon membrane. High stringency Southern hybridiza-tion was performed using a 300-bp internally radiola-beled PstI/ApaI fragment of the HER-2/neu promoter(Sambrook et al., 2001).
Digitonin permeabilization
Digitonin permeabilization was a slight modificationof previously published methods (Duverger et al., 1995;Giovannangeli et al., 1997). Briefly, SK-BR-3 cells weretrypsinized and washed in phosphate-buffered saline(PBS) prior to treatment. Two million cells were incu-bated with 40 �g/ml digitonin (Sigma, St. Louis, MO) inpermeabilization buffer (Giovannangeli et al., 1997) atambient temperature for 5 minutes, washed in digitonin-free buffer, and tested for permeabilization efficiency bytrypan blue staining. Permeabilized cells were then incu-bated with 1 �M TFO2 in triplex-forming buffer (10 mMTris, pH 7.4, 50 mM NaCl, 10 mM MgCl2) or with 10�M PNA in NaPO4 buffer (10 mM NaPO4, pH 6.5–7.0,50 mM NaCl) at 37°C for 4 hours with gentle rocking.Prior to genomic DNA isolation, cells were washed threetimes with 1� PBS, and gDNA was isolated, digestedwith BbvI, and subjected to Southern hybridization as de-scribed.
RESULTS
PNA design and target sequences
The target sequence for PNA binding (Fig. 1) is a poly-purine-polypyrimidine tract in the HER-2/neu promoter(�218–�245 relative to the translation start site) thatoverlaps an initiator-like sequence and lies between theTATA and CCAAT boxes (Mizuguchi et al., 1994). Inour recent work targeting this polypurine tract withTFOs, we demonstrated that an unmodified TFO (TFO1)rapidly dissociated from the target sequence in a plasmidthat was transfected into tumor cells. To prevent HER-2/neu transcription, it was necessary to modify the TFOat both ends with a DNA alkylating agent, and triplexformation with this TFO-alkylator conjugate (TFO2) pre-vented recombinant helicase activity and persisted incells for up to 72 hours (Ziemba et al., 2003). For thisreason, we designed cpy-bis-PNAs (PNA1, PNA2) to de-termine if the strand invasion complex was sufficientlystable to alleviate the need for covalent modification ofthe target sequence.
PNA1 contains two linked 9-mer with the sequence(TCC)3-(CCT)3 designed to target (AGG)3 sequences bythe formation of a PNA:DNA:PNA triple helix. The tar-get sequence contains two adjacent runs of AGG repeats(target 1 and target 2), so that two PNAs could bind simultaneously to the target sequence. PNA2 contains

ZIEMBA ET AL.40
linked 10-mer and 11-mer arms designed to bind to target2 in order to increase binding specificity (Fig. 1B). Ap-pending multiple lysine residues to cpy-bis-PNAs accel-erates invasion complex formation (Egholm et al., 1995;Griffith et al., 1995), and we compared binding affinityand specificity of PNA1 with a single lysine residue inthe linker to PNA2 with four lysine residues, two on eachterminus. PNA binding and the level of HER-2/neu pro-moter suppression in plasmid DNA in cells was comparedwith purine motif TFOs that were either unmodified(TFO1) or modified to contain 8-aza-7-deazaguanine sub-stitutions and C6-PAM at the 5� and 3� termini (TFO2).Mismatched target sequences were designed to containeither a 1-bp alteration in the center of each target or apolypurine tract from an unrelated sequence containing a3-bp mismatch (Fig. 1C).
PNA binding affinity and specificity
Pyrimidine-rich PNAs invade duplex DNA in a pH-de-pendent manner (Demidov et al., 1995; Griffith et al.,1995). Therefore, EMSAs were performed at differentpHs using an end-labeled duplex target (Fig. 2). Figure2A illustrates the duplex target end-labeled on the pyrim-idine strand and incubated with PNAs at pH 6.5, and theduplex is end-labeled on the purine-rich strand and incu-bated with PNAs at pH 7.4 in Figure 2B. For comparisonof relative binding by PNAs under different conditions,the C50, defined as the concentration of PNA that pro-
duces 50% strand invasion under a given set of experi-mental conditions, is presented (Fig. 2D) (Bentin et al.,2003). PNA1 demonstrated pH-dependent strand inva-sion, with a high affinity for the target duplex at slightlyacid or neutral pH (C50 � 5 nM at pH 6.5 and 30 nM atpH 7.0) and a sharp decrease in binding at slightly alka-line pH (C50 � 1 �M at pH 7.4). In contrast, PNA2 dem-onstrated a high affinity for the target sequence over thispH range (C50 � 2 nM at pH 6.5 and 40 nM at pH 7.4).Higher PNA concentrations caused the formation of sev-eral low mobility complexes with the purine strand. Mul-tiple low mobility complexes are formed and explainedby bis-PNA orientation, number of interacting mole-cules, direction of linker wrapping, and degree of disso-ciation of the labeled duplexes (Hansen et al., 2001).PNA binding was rapid, and strand invasion with theshort 36-mer duplex at pH 6.5 appeared to be complete at1 hour (data not shown).
Binding studies with the single mismatched target du-plex demonstrate that PNA1 has approximately 1000-fold higher specificity for the native duplex target thanfor the single mismatch. In contrast, PNA2 was less spe-cific, with only 5–10-fold higher affinity for the nativeduplex than for the single base pair mismatch. Bindingstudies with an unrelated polypurine-polypyrimidine se-quence containing three mismatches demonstrated nobinding by PNA1 at any pH. In contrast, PNA2 boundthis unrelated sequence with moderate affinity at low pH(C50 � 0.1 �M at pH 6.5) but not at pH 7.4 (C50 � 1
FIG. 1. PNA binding targets. (A) Schematic of PNA1 interactions with the HER-2/neu promoter duplex targets. The individualbis-PNA targets are shown in bold. The sequence illustrated from �249 to �214 represent the oligos used in mobility shift assays.The TATAA (�201 to �205) and CCAAT (�249 to �253) boxes are shown relative to the translation start site (double arrow).The major transcription start site (single arrow) is located at �120. (B) Schematic of PNA2 invasion complex with the HER-2/neupromoter. PNA2 was designed to target an 11-bp stretch of target 2. (C) Single base pair mutations (transition G to A) were incor-porated in the center of each PNA target sequence to generate a 1-bp mismatch duplex. The target was assured to contain no intro-duced PNA target sites. A random purine-rich duplex was also generated containing a 3-bp mismatch.
A
B
C

TARGETING THE HER-2/NEU PROMOTER 41
�M). Together, these data demonstrate that increasingthe length and number of positive charges in a cpy-bis-PNA increases the binding affinity and at least partiallyovercomes the need for low pH for pyrimidine-richPNAs to bind DNA. The improved binding, however, isat the cost of a fairly dramatic loss of target specificity.EMSAs were also performed with a longer, asymmetricduplex containing a 5� overhang on the purine strand andwith both strands labeled to demonstrate that these PNAsbind to the purine strand and displace the pyrimidinestrand (Fig. 2C) in a concentration-dependent manner.
DNase I footprinting was performed to determine theprecise PNA binding sites for PNA1 and PNA2 (Fig.3A). A 280-bp promoter fragment was end-labeled on thepurine-rich strand and incubated with PNA or TFO.Complete protection of both target 1 and target 2 was de-tected within 30 minutes using a 10-fold molar excess ofPNA1, and no nonspecific interactions were detected upto 24 hours. The HER-2/neu promoter target sequencewas also incubated with a concentration gradient result-ing in a 0.1–10� molar ratio of both PNAs (0.01–1 �M).Complete protection of both targets was detected at a 1:1molar ratio for both PNA1 and PNA2, further demosntrat-ing that PNA2 can bind with relatively high affinity tothe single mismatch target sequence. The DNase I pro-tection included both PNA targets, the 3 interveningbases, and partial protection of 4–6 bases on each side ofthe targets (FIg. 3D). The PNA2 footprint appears to ex-tend a few nucleotides farther upstream of target 1, likelydue to interaction with another naturally occurring 1-bpmismatch target between �244 and �234. Competition
experiments were performed with PNA2 to determine ifan excess of specific targets or nonspecific DNA inter-feres with PNA binding (FIg. 3B). Once bound to theHER-2/neu promoter, neither an excess of the specificcompetitor (the native duplex target, WT) nor an excessof the nonspecific competitor (purine-rich control, MM)could displace PNA2 from its binding sites. In contrast, a10-fold excess of specific competitor prevented PNA2from binding to the HER-2/neu promoter when addedimmediately prior to incubation with the radiolabeledHER-2/neu promoter fragment, whereas a 10-fold excessof the nonspecific competitor did not prevent binding tothe labeled promoter fragment. These data suggest thatPNA2 binding to the specific target sequence in theHER-2/neu promoter occurs more rapidly than binding tothe nonspecific competitor DNA.
S1 endonuclease was used to demonstrate strand dis-placement after PNA binding by digesting the exposedssDNA loop (Demidov et al., 1993). PNA1 caused site-specific pyrimidine strand cleavage of the HER-2/neupromoter in both PNA target sites (Fig. 3C). The regionof cleavage extended a few base pairs upstream of thetarget sequences at high PNA concentrations (Fig. 3C),but no other sites in the 280-bp promoter fragment weresensitive to S1 cleavage. No significant difference in thecleavage pattern was identified between 4-hour andovernight reaction times at any PNA concentration, fur-ther indicating a rapid rate of invasion complex forma-tion. TFO1 did not displace the pyrimidine strand, as expected. Taken together, these data show the site speci-ficity of strand invasion for the target sequence and dem-
FIG. 2. EMSAs of bis-PNA binding to normal vs. mutated HER-2/neu duplex targets. Reactions were incubated in NaPO4 andisolated on native polyacrylamide gels containing 1� Tris-acetate buffer, pH 6.5 (A) vs. pH 7.4 (B). The radiolabeled duplexeswere incubated with increasing 10-fold concentrations of PNA1 or PNA2 from 0.1 nM to 100 nM (A) or 1 nM to 1 �M (B). Anasymmetric radiolabeled duplex (C) was also incubated with 0.1 nM–10 �M PNA1. Single-stranded (ss) and duplex (ds) DNAwere included as mobility markers. (D) The C50 values (nM) are demonstrated for PNA1 vs. PNA2 on the native and mismatchedHER-2/neu duplexes.
A
C D
B

ZIEMBA ET AL.42
onstrate that the mechanism of PNA binding to duplexDNA involves the formation of a D-loop by the pyrimi-dine strand.
Inhibition of DNA polymerase extension
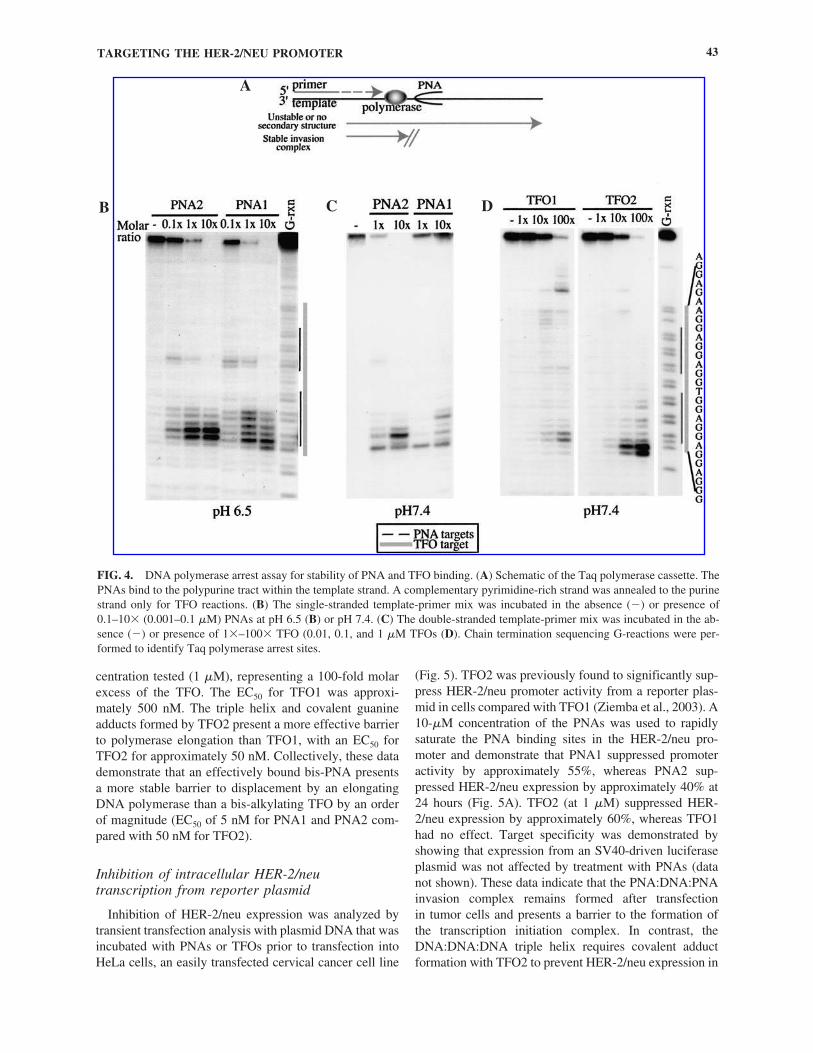
The ability of the bis-PNAs to inhibit the extension ofa DNA polymerase was evaluated using a Taq poly-merase arrest assay. Figure 4B demonstrates concentra-tion-dependent arrest of labeled primer extension by Taq DNA polymerase due to the binding of both PNAs to a single-stranded template and formation of aPNA:DNA:PNA triple helix. At 0.01 �M PNA, the tem-plate is in excess, and polymerase extension was partiallyblocked at both PNA binding sites. PNA1 and PNA2demonstrated similar arrest patterns at pH 6.5 (FIg. 4B),whereas at pH 7.4 (Fig. 4C), only PNA2 blocked poly-merase extension. PNA1 appears to have multiple poly-merase stop sites at the downstream site due to the pres-ence of two overlapping binding sequences within target2. PNA2 has only a single target sequence within the
same region. When the PNA is in molar excess (1 �M)relative to the template, only the first arrest site is ob-served, with no full-length primer extension product.These results demonstrate that the PNA:DNA:PNA triplehelix is very resistant to displacement by DNA poly-merase and that PNA2 can form this structure at physio-logic pH, whereas PNA1 cannot. We estimate the oligoconcentration leading to 50% polymerase arrest (EC50)relative to full-length primer extension product for bothPNAs is approximately 5 nM at pH 6.5 and �1 �M forPNA1 and �100 nM for PNA2 at pH 7.4.
Figure 4D depicts the ability of an unmodified or bis-alkylating TFO to arrest Taq polymerase elongation. Forthese experiments, a double-stranded template was re-quired for triple helix formation, and a 36-mer pyrimi-dine-rich complement was annealed to the template toform a double-stranded region of the template for TFObinding. The complementary strand did not present a bar-rier to Taq polymerase elongation (untreated controllanes, 0, Fig. 4C). The triple helix formed by TFO1 par-tially blocked polymerase elongation at the highest con-
FIG. 3. Binding specificity and displaced-loop probing. (A) DNase I footprint analysis was performed in the absence of oligo(Unt) or with TFO1 or PNA. A Maxam-Gilbert G-reaction was performed to identify the protected guanine bases. PNA1 was in-cubated at a 10-fold excess (1 �M) and incubated with the 250-bp radiolabeled target for 30 minutes, 4 hours, or overnight. Con-centration gradients of PNA2 and PNA1 were also performed, ranging from 0.1� to 10� (0.1 �M–10 �M). (B) DNase I footprintanalysis with competing target DNA. Equimolar ratios of PNA2 and the radiolabeled target were incubated with a 10-fold excessof the native (WT) or 3-bp mismatch (MM) duplexes (from EMSA analysis). The competitor duplex was added either after a 4-hour incubation of the PNA and radiolabeled fragment or just before their incubation. (C) S1 endonuclease was used to probe forsingle-stranded regions in the pyrimidine-rich strand of a radiolabeled fragment. Invasion complexes were formed using10�–0.01� (10 �M–0.01 �M) PNA1. (D) Illustration of the PNA binding site on the purine strand and the displacement loop onthe pyrimidine strand of the HER-2/neu promoter. S1 cleavage sites are illustrated with an x. PNA binding sites are from �229 to�219 and �239 to �231. A third hypersensitive region (*) is detected at high concentrations of PNA1.
A B C
D
TARGETING THE HER-2/NEU PROMOTER 43
centration tested (1 �M), representing a 100-fold molarexcess of the TFO. The EC50 for TFO1 was approxi-mately 500 nM. The triple helix and covalent guanineadducts formed by TFO2 present a more effective barrierto polymerase elongation than TFO1, with an EC50 forTFO2 for approximately 50 nM. Collectively, these datademonstrate that an effectively bound bis-PNA presentsa more stable barrier to displacement by an elongatingDNA polymerase than a bis-alkylating TFO by an orderof magnitude (EC50 of 5 nM for PNA1 and PNA2 com-pared with 50 nM for TFO2).
Inhibition of intracellular HER-2/neutranscription from reporter plasmid
Inhibition of HER-2/neu expression was analyzed bytransient transfection analysis with plasmid DNA that wasincubated with PNAs or TFOs prior to transfection intoHeLa cells, an easily transfected cervical cancer cell line
(Fig. 5). TFO2 was previously found to significantly sup-press HER-2/neu promoter activity from a reporter plas-mid in cells compared with TFO1 (Ziemba et al., 2003). A10-�M concentration of the PNAs was used to rapidlysaturate the PNA binding sites in the HER-2/neu pro-moter and demonstrate that PNA1 suppressed promoteractivity by approximately 55%, whereas PNA2 sup-pressed HER-2/neu expression by approximately 40% at24 hours (Fig. 5A). TFO2 (at 1 �M) suppressed HER-2/neu expression by approximately 60%, whereas TFO1had no effect. Target specificity was demonstrated byshowing that expression from an SV40-driven luciferaseplasmid was not affected by treatment with PNAs (datanot shown). These data indicate that the PNA:DNA:PNA invasion complex remains formed after transfection in tumor cells and presents a barrier to the formation ofthe transcription initiation complex. In contrast, theDNA:DNA:DNA triple helix requires covalent adductformation with TFO2 to prevent HER-2/neu expression in
FIG. 4. DNA polymerase arrest assay for stability of PNA and TFO binding. (A) Schematic of the Taq polymerase cassette. ThePNAs bind to the polypurine tract within the template strand. A complementary pyrimidine-rich strand was annealed to the purinestrand only for TFO reactions. (B) The single-stranded template-primer mix was incubated in the absence (�) or presence of0.1–10� (0.001–0.1 �M) PNAs at pH 6.5 (B) or pH 7.4. (C) The double-stranded template-primer mix was incubated in the ab-sence (�) or presence of 1�–100� TFO (0.01, 0.1, and 1 �M TFOs (D). Chain termination sequencing G-reactions were per-formed to identify Taq polymerase arrest sites.
A
B C D

ZIEMBA ET AL.44
cells, likely because of intracellular dissociation of TFO1after transfection (Ziemba et al., 2003). To evaluate thespecificity of PNA binding and the relative importance ofthe two potential PNA binding sites, PNA invasion com-plexes were formed in HER-2/neu luciferase constructswith mutations in either target 1, the upstream binding site(�237 to �232, designated �237 HER-2), or in target 2,the downstream binding site (�225 to �220, designated�225 HER-2). PNA1 and PNA2 both suppressed HER-2/neu expression by approximately 40% when bound onlyto target 2 in the �237 HER-2 construct. PNA1 bindingonly to target 1 had a small effect on HER-2/neu expres-sion from the �225 HER-2 construct (�10%), whereasPNA2 had a greater effect on HER-2/neu transcription(�30%), even though its intended binding site was dis-rupted in this construct. These data suggest that PNA2 can
bind to target 1 with a single mismatch, consistent withthe relatively high affinity of PNA2 for a single mismatchtarget sequence (Fig. 2). These data also show that target-ing the downstream portion of the polypurine tract is morerobust than targeting the upstream region and suggest thattarget 2 is more important to HER-2/neu transcription. Itis interesting to note that a well-characterized ets bindingsite (EBS) is 7 bp downstream from target 2 (Scott et al.,2000), and previous studies of TFOs targeting the poly-purine tract demonstrated that a TFO could prevent PU.1binding to the adjacent EBS in competition binding stud-ies (Noonberg et al., 1994).
The stability of PNA and TFO-alkylator binding incells was evaluated by time course analysis of HER-2/neu luciferase activity (Fig. 5B). For this analysis, theplasmid was incubated with 1 �M PNA or TFO, repre-senting a 15-fold molar excess of oligo/plasmid. PNA2 atthis concentration suppressed HER-2/neu transcriptionby only 30%–35%, but this level of suppression was re-tained up to 72 hours. PNA1 and TFO2 suppressed HER-2/neu expression by 55%–60%, but the magnitude ofsuppression of HER-2/neu expression by both PNA1 andTFO2 decreased over time to a similar degree, so that at72 hours, HER-2/neu expression was only reduced by20%–30%, favoring TFO2. To test for the possibility ofplasmid degradation as a result of the presence of thePNA clamp, we performed restriction digestion andSouthern analysis on PNA1-treated vs. untreated plasmidextracted from cells at 24–72 hours after transfection. Wedid not observe degradation of the plasmid due to thepresence of the PNA (data not shown), nor have we pre-viously observed degradation of a transiently transfectedplasmid due to the presence of a TFO-alkylator (Faria etal., 2001; Kim, et al., 1998; Ziemba et al., 2003). Collec-tively, these data demonstrate that a PNA:DNA:PNAcomplex can remain stably associated in cells and pre-vent transcription initiation from a reporter plasmid. Anincrease in the number of positive charges in a cpy-bis-PNA appears to decrease the specificity of PNA bindingbut increase the stability of the invasion complex in thismodel system to a degree that is comparable if not supe-rior to a TFO bound with dual covalent adducts.
PNA binding to HER-2/neu promoter in gDNA
Oligonucleotide binding to the HER-2/neu target canbe detected by preventing restriction endonuclease cleav-age of an ideally located BbvI site within the PNA targetsequence (Fig. 6). Digestion of the HER-2/neu promoterwith BbvI results in two fragments of 108 bp and 178 bp,whereas TFO or PNA binding prevents cleavage at theinternal BbvI site, resulting in a single 286-bp fragment(Fig. 6A). Restriction fragments are detected by Southernblot analysis, and the relative intensities of the 286-bpfragment compared to the 178-bp and 108-bp fragments
A
B
FIG. 5. Transient transfection analysis. (A) Transient trans-fection analysis in HeLa cells was performed to determine theeffects of the PNAs on HER-2/neu promoter activity in HeLacells. Plasmid DNA (0.05 �M) was incubated with an excess ofPNA (10 �M) or TFO (1 �M) to ensure maximal suppressionlevels (TFO amount previously identified as maximal necessaryconcentration). Transfection analysis was used to determinePNA effect on promoter activity in plasmid DNA containingmutations disrupting PNA target 1 (�237 HER-2) vs. target 2(�225 HER-2). Untreated native or mutant plasmid was delin-eated as 100% promoter activity, and experiments were per-formed in triplicate. (B) Time course analysis was performed24–72 hours posttransfection using 1 �M oligonucleotides andthe native HER-2 plasmid.

TARGETING THE HER-2/NEU PROMOTER 45
can be used to determine the amount of oligo bindingsemiquantitatively. Southern blot protection assays wereperformed to directly demonstrate PNA binding to theHER-2/neu promoter in naked gDNA and in tumor cellspermeabilized with digitonin (Fig. 6B,C). gDNA washeated to 50°C for 20 minutes to promote duplex openingprior to incubation with PNAs at 37°C. gDNA was incu-bated with PNAs for 4 hours at pH 7.4, and efficientbinding to the HER-2/neu promoter was observed withPNA2 at a high concentration (10 �M) but was not ob-served with PNA1 at any concentration. With prolongedincubation and lower pH, PNA binding to the HER-2/neupromoter in gDNA was demonstrated at 1 �M for bothPNA1 and PNA2. Under these conditions, the HER-
2/neu promoter is completely bound by PNA2 at 24hours and approximately 50% bound by PNA1 at 24hours. To determine if TFO or PNA binding was de-tectable in the endogenous HER-2/neu promoter in tumorcells, we performed this assay in digitonin-permeabilizedSK-BR-3 cells in which the HER-2/neu gene is amplifiedapproximately 8-fold (Fig. 6C). Permeabilized cells wereincubated with TFO2 (1 �M) for 4 hours in a buffer con-taining 10 mM MgCl2, and binding to the HER-2/neupromoter was observed at a level of approximately 50%.Permeabilized SK-BR-3 cells were suspended with 10�M PNA1 or PNA2 in a buffer adjusted to pH 6.5 or 7.0for 4 hours. Binding to the endogenous HER-2/neu pro-moter by PNA2, but not PNA1, was detectable (approxi-
FIG. 6. Southern blot protection assays. (A) Schematic of the HER-2/neu promoter and BbvI restriction sites. BbvI recognizesthe sequence shown in italics and cleaves between the PNA target sites (arrows). In the absence of PNA binding, two restrictionfragments of 108 bp and 178 bp are detected. In the presence of bound oligo, the internal BbvI site is protected from cleavage, andone 286-bp pair fragment is detected. (B) Naked gDNA from SK-BR-3 cells was preheated to 50°C for 20 minutes prior to PNAincubation. Reactions contained the indicated concentrations of PNA at either pH 6.5 or 7.4. (C) Digitonin-permeabilized SK-BR-3 cells were incubated with 10 �M PNA or 1 �M TFO for 4 hours. gDNA was isolated and subjected to Southern blot analysis.
A
B
C

mately 5% at pH 6.5, trace at pH 7.0) in permeabilizedcells.
In summary, these studies demonstrate site-specifictargeting of the HER-2/neu promoter with cpy-bis-PNAs.PNA1 can bind to two adjacent polypurine repeat se-quences in the HER-2/neu promotor with high affinityand specificity, leading to significant suppression ofHER-2/new transcription, but this PNA lacks a sufficientnumber of positive charges to bind to gDNA efficientlyand binds only at a low pH. PNA2 can also target theHER-2/neu promoter with high affinity and because of agreater number of lysines, can bind efficiently to gDNAas well as short synthetic sequences at physiologic pH,but at the expense of a loss of specificity. The improve-ment in binding affinity allows for PNA2 to bind to its in-tended target sequence in the HER-2/neu promoter inpermeabilized cells.
DISCUSSION
In spite of obstacles to the development of effective ther-apeutic nucleic acid drugs, the ability to control specificgene expression using antigene oligos remains of substan-tial interest. In fact, a great deal of current research focuseson understanding the molecular mechanisms of nucleicacid interactions and developing new high-affinity oligonu-cleotides and oligonucleotide mimics (Demidov and Frank-Kamenetskii, 2004). The current study directly comparestwo antigene oligo strategies, a DNA-based TFO conju-gated to covalent DNA-modifying agents and bis-PNAsdesigned to form a stable strand invasion triple helixwithout conjugation to additional agents, all targeting thesame polypurine tract in the HER-2/neu promoter. Thesedata demonstrate the ability of bis-PNAs to strand invadeat this polypurine tract and prevent HER-2/neu transcrip-tion in tumor cells when the strand invasion complex ispreformed in a reporter plasmid. Interestingly, these datademonstrate the relative importance of targeting the dis-tal portion of the polypurine tract, suggesting that strandinvasion and alteration of the double helix at this locationmay be more likely to disrupt transcription factor bindingto an adjacent EBS. PNA1, designed to bind to two adja-cent polypurine repeats, is able to prevent HER-2/neutranscription from plasmid DNA to a degree similar tothat of a TFO-alkylator conjugate, whereas PNA2, de-signed to bind to only one of these sites, is slightly lesseffective. The resistance of the PNA:DNA:PNA triplehelix to displacement by DNA helicases has been de-scribed previously (Bastide et al., 1999; Tackett et al.,2002). Our studies expand on these observations anddemonstrate that the PNA:DNA:PNA triple helix is morestable to displacement by an elongating DNA poly-merase than a DNA:DNA:DNA triple helix with guanineadducts at both ends. The persistence of promoter sup-
ZIEMBA ET AL.46
pression provides an indirect measurement of the stabil-ity of the invasion complex within cells and demonstratethat the PNA invasion complex is at least as stable incells as a TFO stabilized by the formation of dual gua-nine adducts and may be moreso with more cationicPNAs.
The pyrimidine bis-PNAs used in these studies havethe drawback of pH dependence in the formation of theinvasion complex because of the need for cytosine proto-nation for the Hoogsteen bonds involved in forming theinvasion triple helix, a problem that may be overcome byincorporating a modified cytosine base, such as the J-base, in place of cytosine (Demidov and Frank-Kamenetskii, 2001). Only recently has the synthesis of PNAmolecules containing the J-base substitution becomecommercially available. In the present studies, the addi-tion of three more positive charges dramatically in-creased target binding at neutral pH, but at the expense oflower target specificity. The optimal number of lysinesfor a given sequence will balance high affinity againstspecificity. Increasing the number of positive charges ina cpy-bis-PNA molecule also accelerates strand invasionby improving the electrostatic interaction of the other-wise neutrally charged PNA with the phosphodiesterbackbone of DNA (Griffith et al., 1995; Kuhn et al.,1998). This concept was demonstrated in our studieswhere a PNA with four lysines bound to its genomic tar-get more effectively than a PNA with one lysine even atlow pH. The binding of both PNAs to the HER-2/neu tar-get sequence in gDNA was slower than the binding ofTFO, likely due to the presence of an increase in poten-tial binding sites for this PNA, a large increase in nontar-get DNA, and the need for strand opening (Abibi et al.,2004). Some groups are currently designing PNA modifi-cations that should improve binding to duplex DNA tar-gets of both homopurine and mixed sequence motifs(Pokorski et al., 2004; Smolina and Demidov, 2003).
In vivo applications of antigene oligos of any composi-tion remain hindered by inefficient uptake in cells. Inspite of demonstrating the uptake of fluorescently labeledPNA conjugated to the well-characterized Antennapediacell-penetrating peptide, we have been unable to demon-strate any antigene effects on endogenous HER-2/neu ex-pression thus far. In these studies, we have directly dem-onstrated that a cpy-bis-PNA is capable of binding to theHER-2/neu promoter in permeabilized tumor cells, sug-gesting that with effective delivery and optimal PNA de-sign, antigene effects might be observed. In our studies, aTFO bound to the endogenous target sequence muchmore efficiently, suggesting that this region of DNA isprobably accessible to antigene oligo binding. A caution-ary note has been raised in the interpretation of datademonstrating intracellular binding by a TFO when liga-tion-mediated PCR (LM-PCR) is used (Becker and Ma-her, 1999). Contamination of the DNA preparations with

the TFO was found to inhibit ligation of the unidirec-tional linker in an apparently site-specific manner.Oligonucleotide adherence to the cell surface is a well-rec-ognized source of artifact with fluorescently labeledoligonucleotides and can be minimized by careful wash-ing. In our studies, we washed cell pellets twice prior toDNA extractions. In experiments in which TFO2 wasadded to the permeabilized cells in a mock treatment justprior to pelleting and washing, we observed no evidenceof TFO binding (data not shown). We believe that this as-say is a powerful tool for detecting site-specific bindingby TFOs and PNAs in cells, and this method is readilyapplicable to correlate reports of antigene activity withDNA binding by the oligonucleotide.
It is possible that the nuclear membrane is an addi-tional barrier to PNA uptake. Whereas DNA oligos andanalogs microinjected into the cytoplasm rapidly distrib-ute to the nucleus (Leonetti et al., 1991) and, in fact, un-dergo active transport into the nucleus through nuclearpores (Lorenz et al., 2000), the subcellular trafficking ofPNA oligomers is poorly characterized, and nuclear up-take may be less efficient (Gray et al., 1997). Some stud-ies have demonstrated that a nuclear localization signalimproves the uptake and antisense or antigene activity ofPNAs (Cutrona et al., 2000; Gait, 2003; Rapozzi et al.,2002). Nuclear localization signals are generally com-posed of cationic amino acids, and these molecules mayalso promote strand invasion. More likely, PNA bindingis rate limited by transient opening of the duplex for ef-fective strand invasion, whereas TFO binding is not.Prior studies have demonstrated that bis-PNA bindingcan be accelerated more than 10-fold by a DNA-bindingagent that probably does so by inducing distortions of thedouble helix to promote PNA invasion (Mollegaard etal., 2000).
In summary, these data demonstrate that bis-PNAshave some advantages over TFOs as antigen oligos in theformation of stable invasion complexes but have somedrawbacks in the efficiency and specificity of complexformation under physiologic conditions. In order to over-come these drawbacks, the length and charge of a cpy-bis-PNA must be optimized to balance specificity againstefficient DNA binding. We infer from the marked effectof pH on strand invasion by PNA1, a cpy-bis-PNA withonly a single lysine, that Hoogsteen binding precedesand, in fact, drives strand invasion. Thus, modification ofthe Hoogsteen strand of the bis-PNA with the J-base im-proves triplex formation and may speed the formation ofthe strand invasion complex. However, once formed,even without the J-base, the invasion complex is highlystable even under conditions not favorable to the initialformation of the complex (as shown by the persistence ofinhibition of HER-2/neu transcription by the bis-PNA incells). We postulate that further improvements in triplexformation, for example, by conjugation of the Hoogsteen
TARGETING THE HER-2/NEU PROMOTER 47
strand of the bis-PNA to a covalent DNA-modifyingagent, such as a DNA alkylator, will improve the effi-ciency of strand invasion. We believe that the currentdata provide a foundation for such improvements anddemonstrate the potential of an optimally designed bis-PNA to suppress HER-2/neu overexpression.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH(CA85306) and the Flinn Foundation (1580).
REFERENCES
ABIBI, A., PROTOZANOVA, E., DEMIDOV, V.V., andFRANK-KAMENETSKII, M.D. (2004). Specific versusnonspecific binding of cationic PNAs to duplex DNA. Bio-phys. J. 86, 3070–3078.
ARMITAGE, B.A. (2003). The impact of nucleic acid sec-ondary structure on PNA hybridization. Drug Discov. Today8, 222–228.
BARRE, F.X., AIT-SI-ALI, S., GIOVANNANGELI, C.,LUIS, R., ROBIN, P., PRITCHARD, L.L., HÉLÈNE, C.,and HAREL-BELLAN, A. (2000). Unambiguous demon-stration of triple helix-directed gene modification. Proc. Natl.Acad. Sci. USA 97, 3084–3088.
BASTIDE, L., BOEHMER, P.E., VILLANI, G., and LEBLEU,B. (1999). Inhibition of a DNA-helicase by peptide nucleicacids. Nucleic Acids Res. 27, 551–554.
BECKER, N.A., and MAHER, L.J., 3rd. (1999). LMPCR for de-tection of oligonucleotide-directed triple helix formation: Acautionary note. Antisense Nucleic Acid Drug Dev. 9,313–316.
BENTIN, T., LARSEN, H.J., and NIELSEN, P.E. (2003).Combined triplex/duplex invasion of double-stranded DNAby “tail-clamp” peptide nucleic acid. Biochemistry 42,13987–13995.
BENTIN, T., and NIELSEN, P.E. (1996). Enhanced peptidenucleic acid binding to supercoiled DNA: Possible implica-tions for DNA “breathing” dynamics. Biochemistry 35,8863–8869.
BESCH, R., GIOVANNANGELI, C., KAMMERBAUER, C.,and DEGITZ, K. (2002). Specific inhibition of ICAM-1 ex-pression mediated by gene targeting with triplex-formingoligonucleotides. J. Biol. Chem. 277, 32473–32479.
CARBONE, G.M., McGUFFIE, E.M., COLLIER, A., andCATAPANO, C.V. (2003). Selective inhibition of transcrip-tion of the Ets2 gene in prostate cancer cells by a triplex-forming oligonucleotide. Nucleic Acids Res. 31, 833–843.
CARBONE, G.M., McGUFFIE, E., NAPOLI, S., FLANAGAN,C.E., DEMBECH, C., NEGRI, U., ARCAMONE, F., CAPO-BIANCO, M.L., and CATAPANO, C.V. (2004). DNA bind-ing and antigene activity of a daunomycin-conjugatedtriplex-forming oligonucleotide targeting the P2 promoter ofthe human c-myc gene. Nucleic Acids Res. 32, 2396–2410.
CHINNERY, P.F., TAYLOR, R.W., DIEKERT, K., LILL, R.,TURNBULL, D.M., and LIGHTOWLERS, R.N. (1999).

Peptide nucleic acid delivery to human mitochondria. GeneTher. 6, 1919–1928.
CHO-CHUNG, Y.S. (2002). Antisense DNAs as targeted ther-apeutics for cancer: No longer a dream. Curr. Opin. Invest.Drugs 3, 934–939.
CIRISANO, F.D., and KARLAN, B.Y. (1996). The role of theHER-2/neu oncogene in gynecologic cancers. J. Soc. Gy-necol. Invest. 3, 99–105.
CUTRONA, G., CARPANETO, E.M., ULIVI, M., RON-CELLA, S., LANDT, O., FERRARINI, M., and BOFFA,L.C. (2000). Effects in live cells of a c-myc anti-gene PNAlinked to a nuclear localization signal. Nat. Biotechnol. 18,300–303.
DEMIDOV, V.V., and FRANK-KAMENETSKII, M.D.(2001). Sequence-specific targeting of duplex DNA by pep-tide nucleic acids via triplex strand invasion. Methods 23,108–122.
DEMIDOV, V., and FRANK-KAMENETSKII, M.D. (2004).Two sides of the coin: Affinity and specificity of nucleic acidinteractions. Trends Biochem. Sci. 29, 62–71.
DEMIDOV, V.V., FRANK-KAMENETSKII, M.D., EGHOLM,M., BUCHARDT, O., and NIELSEN, P.E. (1993). Sequenceselective double-strand DNA cleavage by peptide nucleicacid (PNA) targeting using nuclease S1. Nucleic Acids Res.21, 2103–2107.
DEMIDOV, V.V., POTAMAN, V.N., FRANK-KAMENET-SKII, M.D., EGHOLM, M., BUCHARD, O., SONNICH-SEN, S.H., and NIELSEN, P.E. (1994). Stability of peptidenucleic acids in human serum and cellular extracts. Biochem.Pharmacol. 48, 1310–1313.
DEMIDOV, V.V., YAVNILOVICH, M.V., BELOTSERKOV-SKII, B.P., FRANK-KAMENETSKII, M.D., and NIELSEN,P.E. (1995). Kinetics and mechanism of polyamide (“pep-tide”) nucleic acid binding to duplex DNA. Proc. Natl. Acad.Sci. USA 92, 2637–2641.
DIVIACCO, S., RAPOZZI, V., XODO, L., HÉLÈNE, C.,QUADRIFOGLIO, F., and GIOVANNANGELI, C. (2001).Site-directed inhibition of DNA replication by triple helixformation. FASEB J. 15, 2660–2668.
DUVERGER, E., PELLERIN-MENDES, C., MAYER, R.,ROCHE, A.C., and MONSIGNY, M. (1995). Nuclear importof glycoconjugates is distinct from the classical NLS path-way. J. Cell Sci. 108, 1325–1332.
EBBINGHAUS, S.W., FORTINBERRY, H., and GAMPER,H.B., Jr. (1999). Inhibition of transcription elongation in the HER-2/neu coding sequence by triplex-directed cova-lent modification of the template strand. Biochemistry 38,619–628.
EBBINGHAUS, S.W., GEE, J.E., RODU, B., MAYFIELD,C.A., SANDERS, G., and MILLER, D.M. (1993). Triplexformation inhibits HER-2/neu transcription in vitro. J. Clin.Invest. 92, 2433–2439.
EGHOLM, M., BUCHARDT, O., CHRISTENSEN, L.,BEHRENS, C., FREIER, S.M., DRIVER, D.A., BERG, R.H.,KIM, S.K., NORDEN, B., and NIELSEN, P.E. (1993). PNAhybridizes to complementary oligonucleotides obeying theWatson-Crick hydrogen-bonding rules. Nature 365, 566–568.
EGHOLM, M., CHRISTENSEN, L., DUEHOLM, K.L.,BUCHARDT, O., COULL, J., and NIELSEN, P.E. (1995).Efficient pH-independent sequence-specific DNA binding by
ZIEMBA ET AL.48
pseudoisocytosine-containing bis-PNA. Nucleic Acids Res.23, 217–222.
FARIA, M., WOOD, C.D., WHITE, M.R., HÉLÈNE, C., andGIOVANNANGELI, C. (2001). Transcription inhibition in-duced by modified triple helix-forming oligonucleotides: Aquantitative assay for evaluation in cells. J. Mol. Biol. 306,15–24.
FARUQI, A.F., EGHOLM, M., and GLAZER, P.M. (1998).Peptide nucleic acid-targeted mutagenesis of a chromosomalgene in mouse cells. Proc. Natl. Acad. Sci. USA 95, 1398–1403.
GAIT, M.J. (2003). Peptide-mediated cellular delivery of anti-sense oligonucleotides and their analogues. Cell. Mol. LifeSci. 60, 844–853.
GIOVANNANGELI, C., DIVIACCO, S., LABROUSSE,V.,GRYAZNOV, S., CHARNEAU, P., and HÉLÈNE, C.(1997). Accessibility of nuclear DNA to triplex-formingoligonucleotides: The integrated HIV-1 provirus as a target.Proc. Natl. Acad. Sci. USA 94, 79–84.
GRAY, G.D., BASU, S., and WICKSTROM, E. (1997). Trans-formed and immortalized cellular uptake of oligodeoxynucleo-side phosphorothioates, 3�-alkylamino oligodeoxynucleo-tides, 2�-O-methyl oligoribonucleotides, oligodeoxynucleosidemethylphosphonates, and peptide nucleic acids. Biochem.Pharmacol. 53, 1465–1476.
GRIFFITH, M.C., RISEN, L.M., GREIG, M.J., LESNIK, E.A.,SPRANKLE, K.G., GRIFFERY, R.H., KIELY, J.S., andFREIER, S.M. (1995). Single and bis-peptide nucleic acidsas triplexing agents: Binding and stoichiometry. J. Am.Chem. Soc. 117, 831–832.
GUNTAKA, R.V., VARMA, B.R., and WEBER, K.T. (2003).Triplex-forming oligonucleotides as modulators of gene ex-pression. Int. J. Biochem. Cell Biol. 35, 22–31.
HAN, H., HURLEY, L.H., and SALAZAR, M. (1999). A DNApolymerase stop assay for G-quadruplex-interactive com-pounds. Nucleic Acids Res. 27, 537–542.
HANSEN, G.I., BENTIN, T., LARSEN, H.J., and NIELSEN,P.E. (2001). Structural isomers of bis-PNA bound to a targetin duplex DNA. J. Mol. Biol. 307, 67–74.
JARDINES, L., WEISS, M., FOWBLE, B., and GREENE, M.(1993). neu(c-erbB-2/HER2) and the epidermal growth fac-tor receptor (EGFR) in breast cancer. Pathobiology 61,268–282.
KAIHATSU, K., HUFFMAN, K.E., and COREY, D.R.(2004a). Intracellular uptake and inhibition of gene expres-sion by PNAs and PNA-peptide conjugates. Biochemistry43, 14340–14347.
KAIHATSU, K., JANOWSKI, B.A., and COREY, D.R.(2004b). Recognition of chromosomal DNA by PNAs.Chem. Biol. 11, 749–758.
KIM, H.G., REDDOCH, J.F., MAYFIELD, C., EBBING-HAUS, S., VIGNESWARAN, N., THOMAS, S., JONES,D.E., Jr., and MILLER, D.M. (1998). Inhibition of transcrip-tion of the human c-myc proto-oncogene by intermoleculartriplex. Biochemistry 37, 2299–2304.
KOPPELHUS, U., and NIELSEN, P.E. (2003). Cellular deliv-ery of peptide nucleic acid (PNA). Adv. Drug Deliv. Rev. 55,267–280.
KUHN, H., DEMIDOV, V.V., FRANK-KAMENETSKII,M.D., and NIELSEN, P.E. (1998). Kinetic sequence discrim-

ination of cationic bis-PNAs upon targeting of double-stranded DNA. Nucleic Acids Res. 26, 582–587.
KUHN, H., DEMIDOV, V.V., NIELSEN, P.E., and FRANK-KAMENETSKII, M.D. (1999). An experimental study ofmechanism and specificity of peptide nucleic acid (PNA)binding to duplex DNA. J. Mol. Biol. 5, 1337–1345.
LAMPE, J.N., KUTYAVIN, I.V., RHINEHART, R., REED,M.W., MEYER, R.B., and GAMPER, H.B., Jr. (1997). Fac-tors influencing the extent and selectivity of alkylationwithin triplexes by reactive G/A motif oligonucleotides. Nu-cleic Acids Res. 25, 4123–4131.
LARSEN, H.J., and NIELSEN, P.E. (1996). Transcription-me-diated binding of peptide nucleic acid (PNA) to double-stranded DNA: Sequence-specific suicide transcription. Nu-cleic Acids Res. 24, 458–463.
LASKIN, J.J., and SANDLER, A.B. (2004). Epidermal growthfactor receptor: A promising target in solid tumours. CancerTreat Rev. 30, 1–17.
LEONETTI, J.P., MECHTI, N., DEGOLS, G., GAGNOR, C.,and LEBLEU, B. (1991). Intracellular distribution of mi-croinjected antisense oligonucleotides. Proc. Natl. Acad. Sci.USA 88, 2702–2706.
LORENZ, P., MISTELI, T., BAKER, B.F., BENNETT, C.F.,and SPECTOR, D.L. (2000). Nucleocytoplasmic shuttling: A novel in vivo property of antisense phosphorothioate oligo-deoxynucleotides. Nucleic Acids Res. 28, 582–592.
MAINE, I.P., and KODADEK, T. (1994). Efficient unwindingof triplex DNA by a DNA helicase. Biochem. Biophys. Res.Commun. 204, 1119–1124.
MAJUMDAR, A., PURI, N., McCOLLUM, N., RICHARDS,S., CUENOUD, B., MILLER, P., and SEIDMAN, M.M.(2003). Gene targeting by triple helix-forming oligonu-cleotides. Ann. NY Acad. Sci. 1002, 141–153.
MIZUGUCHI, G., KANEI-ISHII, C., SAWAZAKI, T.,HORIKOSHI, M., ROEDER, R.G., YAMAMOTO, T., andISHII, S. (1994). Independent control of transcription initia-tions from two sites by an initiator-like element and TATAbox in the human c-erbB-2 promoter. FEBS Lett. 348,80–88.
MOLLEGAARD, N.E., BAILLY, C., WARING, M.J., andNIELSEN, P.E. (2000). Quinoxaline antibiotics enhancepeptide nucleic acid binding to double-stranded DNA. Bio-chemistry 39, 9502–9507.
MOLLEGAARD, N.E., BUCHARDT, O., EGHOLM, M., andNIELSEN, P.E. (1994). Peptide nucleic acid. DNA stranddisplacement loops as artificial transcription promoters.Proc. Natl. Acad. Sci. USA 91, 3892–3895.
NIELSEN, P.E. (2001). Targeting double-stranded DNA withpeptide nucleic acid (PNA). Curr. Med. Chem. 8, 545–550.
NIELSEN, P.E., EGHOLM, M., BERG, R.H., and BUCHARDT,O. (1991). Sequence-selective recognition of DNA by stranddisplacement with a thymine-substituted polyamide. Science254, 1497–1500.
NIELSEN, P.E., EGHOLM, M., and BUCHARDT, O. (1994).Sequence-specific transcription arrest by peptide nucleic acidbound to the DNA template strand. Gene 149, 139–145.
NOONBERG, S.B., SCOTT, G.K., HUNT, C.A., HOGAN,M.E., and BENZ, C.C. (1994). Inhibition of transcriptionfactor binding to the HER2 promoter by triplex-formingoligodeoxyribonucleotides. Gene 149, 123–126.
TARGETING THE HER-2/NEU PROMOTER 49
ORR, R.M. (2001). Technology evaluation: Fomivirsen, IsisPharmaceuticals Inc/CIBA vision. Curr. Opin. Mol. Ther. 3,288–294.
POKORSKI, J.K., WITSCHI, M.A., PURNELL, B.L., and AP-PELLA, D.H. (2004). (S,S)-Trans-cyclopentane-constrainedpeptide nucleic acids. A general backbone modification thatimproves binding affinity and sequence specificity. J. Am.Chem. Soc. 126, 15067–15073.
PORUMB, H., GOUSSET, H., LETELLIER, R., SALLE, V.,BRIANE, D., VASSY, J., AMOR-GUERET, M., ISRAEL,L., and TAILLANDIER, E. (1996). Temporary ex vivo inhi-bition of the expression of the human oncogene HER2(NEU) by a triple helix-forming oligonucleotide. CancerRes. 56, 515–522.
RAPOZZI, V., BURM, B.E., COGOI, S., VAN DER MAREL,G.A., VAN BOOM, J.H., QUADRIFOGLIO, F., andXODO, L.E. (2002). Antiproliferative effect in chronicmyeloid leukaemia cells by antisense peptide nucleic acids.Nucleic Acids Res. 30, 3712–3721.
RUDIN, C.M., KOZLOFF, M., HOFFMAN, P.C., EDEL-MAN, M.J., KARNAUSKAS, R., TOMEK, R., SZETO, L.,and VOKES, E.E. (2004). Phase I study of G3139, a bcl-2antisense oligonucleotide, combined with carboplatin andetoposide in patients with small-cell lung cancer. J. Clin. On-col. 22, 1110–1117.
SAMBROOK, J.A.R., and RUSSELL, D.W. (2001). MolecularCloning, A Laboratory Manual. Cold Spring Harbor, NY:Cold Spring Harbor Laboratory Press.
SCOTT, G.K., CHANG, C.H., ERNY, K.M,. XU, F., FRED-ERICKS, W.J., RAUSCHER, F.J., 3rd, THOR, A.D., andBENZ, C.C. (2000). Ets regulation of the erbB2 promoter.Oncogene 19, 6490–6502.
SEDELNIKOVA, O.A., KARAMYCHEV, V.N., PANYUTIN,I.G., and NEUMANN, R.D. (2002). Sequence-specific genecleavage in intact mammalian cells by 125I-labeled triplex-forming oligonucleotides conjugated with nuclear localiza-tion signal peptide. Antisense Nucleic Acid Drug Dev. 12,43–49.
SMOLINA, I.V., and DEMIDOV, V.V. (2003). Sequence-uni-versal recognition of duplex DNA by oligonucleotides viapseudocomplementarity and helix invasion. Chem. Biol. 10,591–595.
SMOLINA, I.V., DEMIDOV, V.V., and FRANK-KAMENET-SKII, M.D. (2003). Pausing of DNA polymerases on duplexDNA templates due to ligand binding in vitro. J. Mol. Biol.326, 1113–1125.
TACKETT, A.J., COREY, D.R., and RANEY, K.D. (2002).Non-Watson-Crick interactions between PNA and DNA in-hibit the ATPase activity of bacteriophage T4 Dda helicase.Nucleic Acids Res. 30, 950–957.
UHLMANN, E. (1998). Peptide nucleic acids (PNA) and PNA-DNA chimeras: From high binding affinity towards biologi-cal function. Biol. Chem. 379, 1045–1052.
VASQUEZ, K.M., CHRISTENSEN, J., LI, L., FINCH, R.A.,and GLAZER, P.M. (2002). Human XPA and RPA DNA re-pair proteins participate in specific recognition of triplex-in-duced helical distortions. Proc. Natl. Acad. Sci. USA 99,5848–5853.
VASQUEZ, K.M., DAGLE, J.M., WEEKS, D.L., andGLAZER, P.M. (2001). Chromosome targeting at short poly-

ZIEMBA ET AL.50
purine sites by cationic triplex-forming oligonucleotides. J.Biol. Chem. 276, 38536–38541.
VASQUEZ, K.M., NARAYANAN, L., and GLAZER, P.M.(2000). Specific mutations induced by triplex-formingoligonucleotides in mice. Science 290, 530–533.
VASQUEZ, K.M., WANG, G., HAVRE, P.A., and GLAZER,P.M. (1999). Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic AcidsRes. 27, 1176–1181.
WANG, G., JING, K., BALCZON, R., and XU, X. (2001).Defining the peptide nucleic acids (PNA) length requirementfor PNA binding-induced transcription and gene expression.J. Mol. Biol. 313, 933–940.
WANG, G., SEIDMAN, M.M., and GLAZER, P.M. (1996).Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science 271,802–805.
WANG, G., XU, X., PACE, B., DEAN, D.A., GLAZER, P.M.,CHAN, P., GOODMAN, S.R., and SHOKOLENKO, I.(1999). Peptide nucleic acid (PNA) binding-mediated induc-tion of human gamma-globin gene expression. Nucleic AcidsRes. 27, 2806–2813.
WEITZMANN, M.N., WOODFORD, K.J., and USDIN, K.(1996). The development and use of a DNA polymerase
arrest assay for the evaluation of parameters affecting intrastrand tetraplex formation. J. Biol. Chem. 271, 20958–20964.
ZHAO, X., KAIHATSU, K., and COREY, D.R. (2003). Inhibi-tion of transcription by bisPNA-peptide conjugates. Nucleo-sides Nucleotides Nucleic Acids 22, 535–546.
ZIEMBA, A.J., REED, M.W., RANEY, K.D., BYRD, A.B.,and EBBINGHAUS, S.W. (2003). A bis-alkylating triplex-forming oligonucleotide inhibits intracellular reporter geneexpression and prevents triplex unwinding due to helicaseactivity. Biochemistry 42, 5013–5024.
Address reprint requests to:Dr. Scot W. EbbinghausArizona Cancer Center
1515 North Campbell AvenueTucson, AZ 85724
E-mail: [email protected]
Received December 20, 2004; accepted in revisedform January 24, 2005.

This article has been cited by:
1. Victor Meidan, Judith Glezer, Sharona Salomon, Yechezkel Sidi, Yechezkel Barenholz, Jack Cohen, Gila Lilling. 2006. SpecificLipoplex-Mediated Antisense Against Bcl-2 in Breast Cancer Cells: A Comparison between Different Formulations. Journal ofLiposome Research 16:1, 27-43. [CrossRef]




![Presentazione standard di PowerPointmedia.aiom.it/userfiles/files/doc/AIOM-Servizi/slide/...paronychia, dermatitis, stomatitis,..) 63% (vs 41%) Oncogene Addicted [ARCHER 1050] Oncogene](https://img.dokumen.tips/doc/110x75/5e8e913ff7852e421e584c5b/presentazione-standard-di-paronychia-dermatitis-stomatitis-63-vs-41.jpg)







![Chromosomal Rearrangements and Oncogene Amplification ... · [CANCER RESEARCH 61, 1214–1219, February 1, 2001] Chromosomal Rearrangements and Oncogene Amplification Precede Aneuploidization](https://img.dokumen.tips/doc/110x75/6044c5937a1f9344c165f56e/chromosomal-rearrangements-and-oncogene-amplification-cancer-research-61-1214a1219.jpg)
















