Embed Size (px)
Citation preview

of June 14, 2018.This information is current as
ModelImmunity in an Autochthonous CancerLigands Activates Spontaneous Antitumor Targeted Liposomal Delivery of TLR9
O'Donoghue and Ruth GanssChristopher R. Parish, Günter J. Hämmerling, Helen Juliana Hamzah, Joseph G. Altin, Thomas Herringson,
http://www.jimmunol.org/content/183/2/1091doi: 10.4049/jimmunol.0900736June 2009;
2009; 183:1091-1098; Prepublished online 26J Immunol
MaterialSupplementary
6.DC1http://www.jimmunol.org/content/suppl/2009/06/26/jimmunol.090073
Referenceshttp://www.jimmunol.org/content/183/2/1091.full#ref-list-1
, 17 of which you can access for free at: cites 36 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2009 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Targeted Liposomal Delivery of TLR9 Ligands ActivatesSpontaneous Antitumor Immunity in an AutochthonousCancer Model1
Juliana Hamzah,* Joseph G. Altin,† Thomas Herringson,† Christopher R. Parish,‡
Gunter J. Hammerling,§ Helen O’Donoghue,* and Ruth Ganss2*
Accessibility of tumors for highly effective local treatment represents a major challenge for anticancer therapy. Immunostimu-latory oligodeoxynucleotides (ODN) with CpG motifs are ligands of TLR9, which prime spontaneous antitumor immunity, but areless effective when applied systemically. We therefore developed a liposome-based agent for selective delivery of CpG-ODN intothe tumor environment. A peptide that specifically targets angiogenic endothelial cells in a transgenic tumor model for islet cellcarcinogenesis was engrafted into CpG-ODN containing liposomes. Intravenous injection of these liposomes resulted in specificaccumulation around tumor vessels, increased uptake by tumor-resident macrophages, and retention over time. In contrast,nontargeted liposomes did not localize to the tumor vasculature. Consequently, only vascular targeting of CpG-ODN liposomesprovoked a marked inflammatory response at vessel walls with enhanced CD8� and CD4� T cell infiltration and, importantly,activation of spontaneous, tumor-specific cytotoxicity. In a therapeutic setting, 40% of tumor-bearing, transgenic mice survivedbeyond week 45 after systemic administration of vascular-directed CpG-ODN liposomes. In contrast, control mice survived up to30 wk. Therapeutic efficacy was further improved by increasing the frequency of tumor-specific effector cells through adoptivetransfers. NK cells and CD8� T cells were major effectors which induced tumor cell death and acted in conjunction with anti-vascular effects. Thus, tumor homing with CpG-ODN-loaded liposomes is as potent as direct injection of free CpG-ODN and hasthe potential to overcome some major limitations of conventional CpG-ODN monotherapy. The Journal of Immunology, 2009,183: 1091–1098.
O ligonucleotides containing unmethylated CpG dinucle-otides (CpG oligodeoxynucelotides (CpG-ODN)3) re-sembling bacterial DNA have long been recognized as
immune-stimulating agents. Cells are able to detect unmethylatedCpGs via TLR9, an endosomal receptor expressed on a limitednumber of immune cells (1). In mice, monocytes are directly ac-tivated by CpG motifs to secrete the Th1-like cytokine IL-12 andtype I IFNs, whereas NK cells respond with increased lytic activity
and IFN-� secretion. In addition, TLR9 stimulation drives den-dritic cell and B cell maturation and thus enhances their Ag pre-sentation capability. CpG-ODN are prime candidates for antican-cer vaccination because they are, as single agents, able to modulateinnate and adaptive immunity.
Naturally occurring antitumor immunity is often weak andineffective at rejecting solid tumors. In preclinical mouse mod-els, adjuvant CpG-ODN therapy primes antitumor cytotoxicityand IFN-� secretion by lymphocytes which is highly effective inpreventing tumor formation in a prophylactic setting (2). CpG-ODN is also used as a potent single agent (3–7). In these mod-els, CpG-ODN is directly injected into s.c. transplantation tu-mors and is sufficient to reject small tumors. Surprisingly, localCpG-ODN monotherapy is potent enough to activate spontane-ous anticancer immunity mediated by innate and adaptive im-mune cells, in particular NK and CD8� T cells (3, 5). However,highest efficacy can only be achieved with direct intratumoralinjection into s.c. tumors, whereas injection into the tumor-freeflank is less effective (5, 6). Similarly, in a mouse model ofmammary adenocarcinoma, direct intramammary CpG-ODNapplication markedly impairs tumor growth (8), whereas sys-temic application only prevents tumor formation but has noimpact on established tumors (9). Moreover, it has been dem-onstrated that systemic application of CpG-ODN in mice sup-presses T cell activity through IDO induction in spleen (10).This raises the question of how effective systemic CpG-ODNmonotherapy would be in dealing with tumors which are notaccessible for intratumoral injections. This is especially rele-vant since several TLR9 agonists are now being assessed inclinical trials ranging from vaccine adjuvants and combinationtherapies to local or systemic monotherapies (11).
*Western Australian Institute for Medical Research, University of Western AustraliaCentre for Medical Research, Perth, Australia; †Biochemistry and Molecular Biology,School of Biology and ‡Division of Immunology and Genetics, John Curtin School ofMedical Research, ANU College of Medicine, Biology and Environment, AustralianNational University, Canberra; and §Department of Molecular Immunology, GermanCancer Research Center, Heidelberg, Germany
Received for publication March 6, 2009. Accepted for publication May 14, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by grants from the Medical Research Foundation, RoyalPerth Hospital, the National Health and Medical Research Council (Project Grants572578 to R.G. and 316949 to J.G.A.), the Cancer Council Western Australia (toJ.H.), and start-up funds from the Western Australian Institute for Medical Researchand the University of Western Australia (to R.G.).2 Address correspondence and reprint requests to Dr. Ruth Ganss, Western AustralianInstitute for Medical Research, Rear, 50 Murray Street, Medical Research FoundationBuilding, Level 5, Perth, WA 6000, Australia. E-mail address: [email protected] Abbreviations used in this paper: ODN, oligodeoxynucleotide; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOTAP, 1,2-dioleoyl-3-trimethylammonium propane;NTA3-DTDA, 3(nitrotriacetic acid)ditetradecylamine; DSPE-mPEG, 1,2-disteroyl-sn-glycerophosphoethanolamine-N-[methoxy(polyethyleglycol)-2000]; DODAP, 1,2-dio-leoyl-3-dimethylammonium propane; OG-488-DHPE, Oregon Green-488 1,2-dihexade-canoyl-sn-glycero-3-phosphoethanolamine gene promoter; Tag, SV40 Large T antigen;RIP, rat insulin.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0900736
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

In the RIP1-Tag5-transgenic mouse model of pancreatic islettumorigenesis, developing insulinomas are deeply embedded in theexocrine pancreas and thus out of reach for intratumoral injections.We found that systemic application of CpG-ODN as a single agenthad no impact on established tumors and hence survival of trans-genic mice (12). Nevertheless, we observed that systemically ap-plied CpG-ODN had indirect effects on the tumor environmentthrough local uptake by macrophages. We now show that thera-peutic success can be enhanced if CpG-ODN was enriched andretained in the tumor microenvironment. We used a vascular tar-geting peptide (RGR peptide) which specifically homes to tumorblood vessels (13) and has been successfully used for intratumoraltherapy (14). To combine tumor-specific targeting with delivery ofTLR9 ligands, CpG-ODN was packaged into RGR peptide-coatedstealth liposomes. These liposomes were formulated for efficientincorporation of CpG-ODN and contained a sterically stabilizinglipid to enhance circulation time in vivo; the engrafted RGR pep-tide mediated specific binding to tumor endothelium and lipid ves-icle retention in the tumor environment. We further demonstrate agreatly improved therapeutic outcome of systemically applied, tu-mor-targeted CpG-ODN liposomes through stimulation of sponta-neous antitumor immunity and antivascular effects.
Materials and MethodsMice
RIP1-Tag5 transgenic mice (provided by D. Hanahan, University of Cal-ifornia San Francisco, CA) were used on a C3HeBFe background unlessstated otherwise (15). For adoptive transfer experiments, mice transgenicfor a TCR that recognizes Tag presented by the MHC class I moleculeH-2Kk (referred to as TagTCR8 (16); provided by T. Geiger, St. JudeChildren’s Research Hospital, Memphis, TN and R. Flavell, Yale Univer-sity, New Haven, CT) or the MHC class II molecule I-A (TagTCR1 (17);provided by I. Forster, Heinrich-Heine University, Dusseldorf, Germany)bred on a C3HeBFe background were used. All mice were kept underspecific pathogen-free conditions at the University of Western Australiaand all experimental protocols were approved by the Animal Ethics Com-mittee of the University of Western Australia.
Reagents
The phospholipids 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)as well as 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) and cho-lesterol were obtained from Sigma-Aldrich. The chelator lipid 3(nitrilotri-acetic acid)ditetradecylamine (NTA3-DTDA) was produced in the Re-search School of Chemistry (Australian National University) as describedpreviously (18, 19). 1,2-Disteroyl-sn-glycero-3-phophoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-mPEG2000) and 1,2-dioleoyl-3-dimethylammonium propane (DODAP) were obtained from Avanti PolarLipids. The tracer lipid Oregon Green-488 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (OG-488-DHPE) was from Invitrogen. NiSO4 wasused in all additions of Ni2� to lipid preparations. The two peptides forengraftment onto liposomes were produced with their amino acid sequencebeing: RGR, (His)13-GGGGGQKLISEEDLGGGGGCRGRRST and L2,GHHPHGHHPH. The L2 peptide, a sequence of 10 aa found in the plasmaprotein histidine-rich glycoprotein, was used as control since it isknown to bind to Ni-NTA3-DTDA with high avidity and can blocknonspecific interactions of Ni-NTA-DTDA-liposomes with cells. Thephosphorothioate-stabilized oligonucleotide used for incorporation intopeptide-engrafted liposomes was TCCATGACGTTCCTGATGCT, re-ferred to as CpG-ODN 1668.
Liposome synthesis and characterization
Preparation of liposomes. Peptide-targeted NTA3-DTDA-liposomes con-taining encapsulated CpG-ODN were produced by a method similar to thatused for producing targeted liposomes containing small interfering RNA(T. Herringson and J. G. Altin, manuscript submitted for publication).Briefly, stock solutions of lipids in ethanol (stored at �80oC) were mixedto give DODAP:DOTAP:DOPE:cholesterol:DSPE-mPEG2000 in the ratioof 45:2:13:30:10 mol%. For some experiments. the fluorochrome-labeledtracer lipid OG-488-DHPE (used at 1 mol%) also was included in the lipidmixture to permit liposome tracking. Lipids were dried under a stream ofnitrogen gas and liposomes were produced by suspending the lipids in
distilled water by sonication as previously described (18). Stock suspen-sions of NTA3-DTDA containing a 3-fold molar excess of NiSO4 in PBSwere also prepared by sonication. Liposome suspensions were stored for upto 1 wk at 4oC; storage for a longer time was at �20°C. Liposomes werethawed and briefly resonicated before use in experiments.Encapsulation of CpG. The ionizable lipid DODAP, a lipid component ofthe liposomes, was used to drive CpG-ODN encapsulation. Liposomes (to-tal lipid concentration, 4 mM) were prepared as above and first acidified bythe addition of glycine buffer (final pH �4.5). After 5 min, CpG-ODN (5�M) was added to give a CpG-ODN:lipid ratio of 1:10 (w/w). After mixingand incubating for 15 min, the CpG-ODN liposome mixture was sonicatedfor 20–30 s and then incubated for a further 30 min at room temperature.Agarose gel electrophoresis indicated that the efficiency of CpG-ODN en-capsulation under these conditions was reproducibly at �50% (T. Her-ringson and J. G. Altin, manuscript submitted for publication).
Incorporation of NTA3-DTDA and engraftment of peptides
To incorporate the chelator lipid NTA3-DTDA into CpG-ODN liposomesproduced as above, the liposomes were first neutralized by adding sodiumphosphate buffer and 10� PBS to give 1� PBS salts in the final suspen-sion. Ni-NTA3-DTDA (1 mol% total lipid) was then added and the sus-pension was mixed and incubated for 30 min at 37oC to allow incorpora-tion of the NTA3-DTDA. The NTA3-DTDA-containing liposomes wereengrafted with either L2 (control) or RGR (targeting peptide) by incubatingwith the indicated peptide for 30 min at room temperature with occasionalmixing. The amount of RGR-targeting peptide used was optimized to givethe greatest increase in cell fluorescence in binding assays with NIH-3T3cells (data not shown). As judged from their ability to target fluorochrome-conjugated CpG-ODN to cells, engrafted liposomes were stable for at least4 wk when stored at 4oC. For use in experiments liposomes were usedwithin 4 wk of their preparation.
ELISA
Primary murine splenocytes (5 � 105 cells) were incubated with variousdilutions of CpG-ODN and liposome-engrafted CpG-ODN in RPMI 1640culture medium for 18 h in a 37°C tissue culture incubator. Supernatantswere harvested and IL-12 expression was analyzed in a sandwich ELISA(R&D Systems). Streptavidin-HRP (Vector Laboratories) and substrate forperoxidase-conjugated secondary Ab (FAST OPD tablets; Sigma-Aldrich)were used for detection in an ELISA plate reader (Victor 1420 MultilabelCounter; PerkinElmer).
Treatment of RIP1-Tag5 mice
For histology, 27-wk-old transgenic RIP1-Tag5 mice were treated over 2wk with biweekly i.v. injections of 100 �l of liposomes, peptide-liposome-CpG-ODN vesicles, or CpG-ODN in PBS (5 �g total). Mice were sacri-ficed on day 14 and tumors were harvested. For survival analyses, 22- to23-wk-old RIP1-Tag5 mice were treated over a period of 10 wk with bi-weekly i.v. injections of 50 �l of liposome/CpG-ODN mixtures as de-scribed above and survival monitored up to week 45. In addition, adoptivetransfers with combined 2.5 � 106 in vitro-activated TagTCR lymph nodecells and 2.5 � 106 TagTCR8-derived splenocytes were performed every2 wk in some treatment groups. In vitro proliferation and adoptive transfersare described elsewhere (14). Briefly, TagTCR8 splenocytes or TagTCR1lymph node cells were activated in vitro for 3 days with 10 U of rIL-2/mland 25 mM Tag peptide 362–568 (SEFLIEKRI for TagTCR8 cells) or 25nM Tag peptide 362–384 (TNRFNDLLDRMDIMFGSTGSADI forTagTCR1 cells) before injection. For Ab depletion studies, anti-L3T4 (anti-CD4�, rat anti-mouse GK1.5), anti-Lyt.2 (anti-CD8�, rat anti-mouse 53-6.7), and TM-�1 (anti-IL2R-�, rat IgG2b, selectively abrogates cytolyticactivity of NK cells in various mouse strains, including C3H (20)) Abswere produced in a miniPERM bioreactor and column-purified. GK1.5 andanti-Lyt.2 Abs and irrelevant rat IgG control (0.5 mg) were injected i.p. for3 consecutive days before RGR-CpG-liposome treatment, followed byweekly injections of Abs for 8 wk. One mg of TM-�1 Abs was injected i.p.before RGR-CpG-liposome treatment, followed by 0.5 mg of Ab every 2wk for a period of 8 wk. Depletion efficacy was monitored by FACSanalysis.
Histology
For localization of peptide-tagged liposomal vesicles, 100 �l of OregonGreen-488-labeled liposomes were injected into tumor-bearing mice andmice were sacrificed at 2-, 24-, and 48-h time points. Tumors were em-bedded in Tissue-Tek OCT compound (Sakura Finetek) and 7-�m sectionswere analyzed using fluorescent microscopy. For fluorescence detection ofblood vessels and tumor-resident macrophages, the following Abs were
1092 INTRATUMORAL DELIVERY OF CpG LIPOSOMES
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
used: anti-CD31 (Mec 13.3, 10 �g/ml; BD Pharmingen), anti-ICAM-1(YNI/1.7.4, 10 �g/ml; American Type Culture Collections), anti-CD68(FA/11, 10 �g/ml, Abcam), followed by cyanin 3-conjugated IgG (F(ab�)2
goat anti-rat (3 �g/ml, Jackson ImmunoResearch Laboratories). Immuno-histochemistry was performed as described (15) with the following Abs at10 �g/ml (BD Pharmingen): anti-CD4 (GK1.5), anti-CD8 (Ly-2), anti-CD11b (M1/70), and anti-CD45 (30-F11). Primary Abs were detected withanti-rat or anti-donkey biotinylated secondary Abs (Vector Laboratories)followed by an ABC elite kit (Vector Laboratories). TUNEL assay wasperformed on frozen sections using an in situ cell death detection kit-fluorescein (Roche). Quantification was performed on tumor sections usinga �20 objective lens. Five independent areas per tumor were selected,digitally photographed, and positive signals counted using automated soft-ware (Image-Pro Plus 5.0; Media Cybernetics).
In vivo killing assay
The in vivo CTL assay was performed as described (12). For this assay, theF1 generation of RIP1-Tag5/C3H mice and C57BL/6 mice (referred to asRIP1-Tag5/F1) were used. Briefly, 1 � 107 splenocytes/ml were loadedwith H2-Kb-restricted Tag peptide IV (404–411, VVYDFLKL) or leftwithout peptide. Targets were labeled with CSFE (Molecular Probes andInvitrogen) in a final concentration of 0.75 �M (high, with peptide IV) or0.075 �M (low, without peptide IV). One � 107 cells of each populationwere injected i.v. into recipient mice. CTL activity was assessed 18 h afterthe adoptive transfer using FACS analysis.
Flow cytometry
Spleen cell suspensions were prepared from mice injected with OregonGreen-488-labeled liposomes after 2, 24, and 48 h. One � 106 spleen cellswere analyzed on a FACScan (BD Biosciences).
Statistical analysis
Cumulative survival was calculated by the Kaplan-Meier method and an-alyzed by the log-rank test. Student’s t test was used for all other statisticalevaluation. Statistical values are presented as mean � SEM. A p value of0.05 was considered statistically significant.
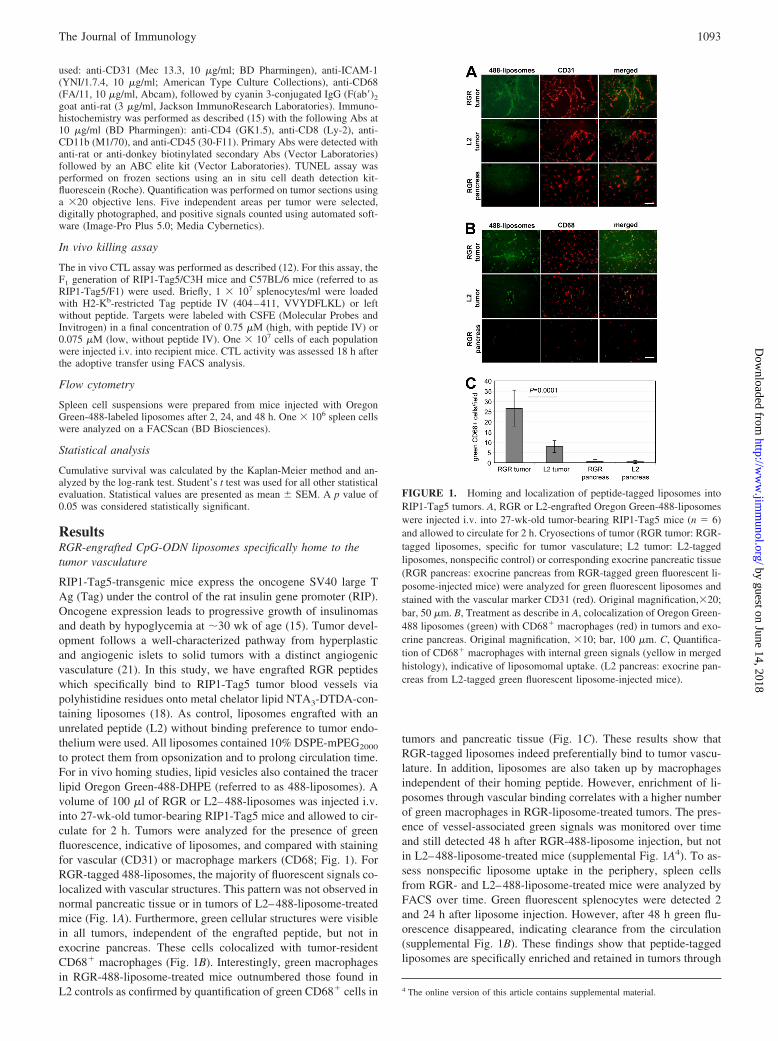
ResultsRGR-engrafted CpG-ODN liposomes specifically home to thetumor vasculature
RIP1-Tag5-transgenic mice express the oncogene SV40 large TAg (Tag) under the control of the rat insulin gene promoter (RIP).Oncogene expression leads to progressive growth of insulinomasand death by hypoglycemia at �30 wk of age (15). Tumor devel-opment follows a well-characterized pathway from hyperplasticand angiogenic islets to solid tumors with a distinct angiogenicvasculature (21). In this study, we have engrafted RGR peptideswhich specifically bind to RIP1-Tag5 tumor blood vessels viapolyhistidine residues onto metal chelator lipid NTA3-DTDA-con-taining liposomes (18). As control, liposomes engrafted with anunrelated peptide (L2) without binding preference to tumor endo-thelium were used. All liposomes contained 10% DSPE-mPEG2000
to protect them from opsonization and to prolong circulation time.For in vivo homing studies, lipid vesicles also contained the tracerlipid Oregon Green-488-DHPE (referred to as 488-liposomes). Avolume of 100 �l of RGR or L2–488-liposomes was injected i.v.into 27-wk-old tumor-bearing RIP1-Tag5 mice and allowed to cir-culate for 2 h. Tumors were analyzed for the presence of greenfluorescence, indicative of liposomes, and compared with stainingfor vascular (CD31) or macrophage markers (CD68; Fig. 1). ForRGR-tagged 488-liposomes, the majority of fluorescent signals co-localized with vascular structures. This pattern was not observed innormal pancreatic tissue or in tumors of L2–488-liposome-treatedmice (Fig. 1A). Furthermore, green cellular structures were visiblein all tumors, independent of the engrafted peptide, but not inexocrine pancreas. These cells colocalized with tumor-residentCD68� macrophages (Fig. 1B). Interestingly, green macrophagesin RGR-488-liposome-treated mice outnumbered those found inL2 controls as confirmed by quantification of green CD68� cells in
tumors and pancreatic tissue (Fig. 1C). These results show thatRGR-tagged liposomes indeed preferentially bind to tumor vascu-lature. In addition, liposomes are also taken up by macrophagesindependent of their homing peptide. However, enrichment of li-posomes through vascular binding correlates with a higher numberof green macrophages in RGR-liposome-treated tumors. The pres-ence of vessel-associated green signals was monitored over timeand still detected 48 h after RGR-488-liposome injection, but notin L2–488-liposome-treated mice (supplemental Fig. 1A4). To as-sess nonspecific liposome uptake in the periphery, spleen cellsfrom RGR- and L2–488-liposome-treated mice were analyzed byFACS over time. Green fluorescent splenocytes were detected 2and 24 h after liposome injection. However, after 48 h green flu-orescence disappeared, indicating clearance from the circulation(supplemental Fig. 1B). These findings show that peptide-taggedliposomes are specifically enriched and retained in tumors through
4 The online version of this article contains supplemental material.
FIGURE 1. Homing and localization of peptide-tagged liposomes intoRIP1-Tag5 tumors. A, RGR or L2-engrafted Oregon Green-488-liposomeswere injected i.v. into 27-wk-old tumor-bearing RIP1-Tag5 mice (n � 6)and allowed to circulate for 2 h. Cryosections of tumor (RGR tumor: RGR-tagged liposomes, specific for tumor vasculature; L2 tumor: L2-taggedliposomes, nonspecific control) or corresponding exocrine pancreatic tissue(RGR pancreas: exocrine pancreas from RGR-tagged green fluorescent li-posome-injected mice) were analyzed for green fluorescent liposomes andstained with the vascular marker CD31 (red). Original magnification,�20;bar, 50 �m. B, Treatment as describe in A, colocalization of Oregon Green-488 liposomes (green) with CD68� macrophages (red) in tumors and exo-crine pancreas. Original magnification, �10; bar, 100 �m. C, Quantifica-tion of CD68� macrophages with internal green signals (yellow in mergedhistology), indicative of liposomomal uptake. (L2 pancreas: exocrine pan-creas from L2-tagged green fluorescent liposome-injected mice).
1093The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
vascular targeting, but some are also taken up by the reticuloen-dothelial system via phagocytosis.
Intratumoral CpG-ODN elicits vessel wall inflammation inRIP1-Tag5 tumors
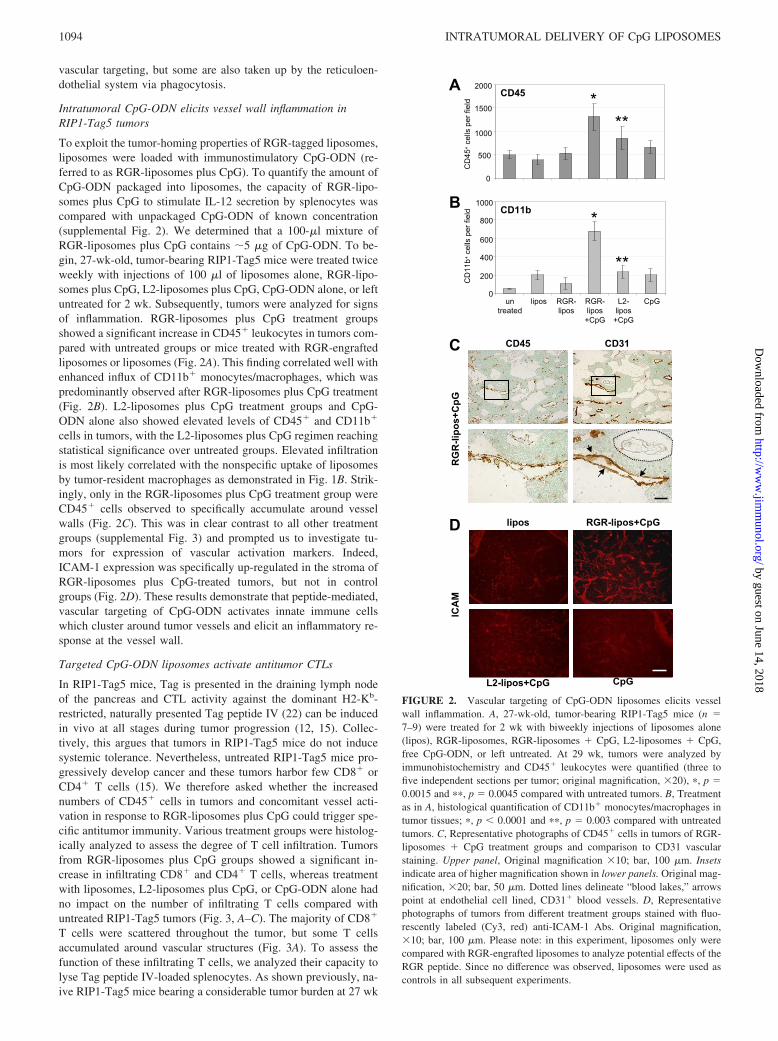
To exploit the tumor-homing properties of RGR-tagged liposomes,liposomes were loaded with immunostimulatory CpG-ODN (re-ferred to as RGR-liposomes plus CpG). To quantify the amount ofCpG-ODN packaged into liposomes, the capacity of RGR-lipo-somes plus CpG to stimulate IL-12 secretion by splenocytes wascompared with unpackaged CpG-ODN of known concentration(supplemental Fig. 2). We determined that a 100-�l mixture ofRGR-liposomes plus CpG contains �5 �g of CpG-ODN. To be-gin, 27-wk-old, tumor-bearing RIP1-Tag5 mice were treated twiceweekly with injections of 100 �l of liposomes alone, RGR-lipo-somes plus CpG, L2-liposomes plus CpG, CpG-ODN alone, or leftuntreated for 2 wk. Subsequently, tumors were analyzed for signsof inflammation. RGR-liposomes plus CpG treatment groupsshowed a significant increase in CD45� leukocytes in tumors com-pared with untreated groups or mice treated with RGR-engraftedliposomes or liposomes (Fig. 2A). This finding correlated well withenhanced influx of CD11b� monocytes/macrophages, which waspredominantly observed after RGR-liposomes plus CpG treatment(Fig. 2B). L2-liposomes plus CpG treatment groups and CpG-ODN alone also showed elevated levels of CD45� and CD11b�
cells in tumors, with the L2-liposomes plus CpG regimen reachingstatistical significance over untreated groups. Elevated infiltrationis most likely correlated with the nonspecific uptake of liposomesby tumor-resident macrophages as demonstrated in Fig. 1B. Strik-ingly, only in the RGR-liposomes plus CpG treatment group wereCD45� cells observed to specifically accumulate around vesselwalls (Fig. 2C). This was in clear contrast to all other treatmentgroups (supplemental Fig. 3) and prompted us to investigate tu-mors for expression of vascular activation markers. Indeed,ICAM-1 expression was specifically up-regulated in the stroma ofRGR-liposomes plus CpG-treated tumors, but not in controlgroups (Fig. 2D). These results demonstrate that peptide-mediated,vascular targeting of CpG-ODN activates innate immune cellswhich cluster around tumor vessels and elicit an inflammatory re-sponse at the vessel wall.
Targeted CpG-ODN liposomes activate antitumor CTLs
In RIP1-Tag5 mice, Tag is presented in the draining lymph nodeof the pancreas and CTL activity against the dominant H2-Kb-restricted, naturally presented Tag peptide IV (22) can be inducedin vivo at all stages during tumor progression (12, 15). Collec-tively, this argues that tumors in RIP1-Tag5 mice do not inducesystemic tolerance. Nevertheless, untreated RIP1-Tag5 mice pro-gressively develop cancer and these tumors harbor few CD8� orCD4� T cells (15). We therefore asked whether the increasednumbers of CD45� cells in tumors and concomitant vessel acti-vation in response to RGR-liposomes plus CpG could trigger spe-cific antitumor immunity. Various treatment groups were histolog-ically analyzed to assess the degree of T cell infiltration. Tumorsfrom RGR-liposomes plus CpG groups showed a significant in-crease in infiltrating CD8� and CD4� T cells, whereas treatmentwith liposomes, L2-liposomes plus CpG, or CpG-ODN alone hadno impact on the number of infiltrating T cells compared withuntreated RIP1-Tag5 tumors (Fig. 3, A–C). The majority of CD8�
T cells were scattered throughout the tumor, but some T cellsaccumulated around vascular structures (Fig. 3A). To assess thefunction of these infiltrating T cells, we analyzed their capacity tolyse Tag peptide IV-loaded splenocytes. As shown previously, na-ive RIP1-Tag5 mice bearing a considerable tumor burden at 27 wk
FIGURE 2. Vascular targeting of CpG-ODN liposomes elicits vesselwall inflammation. A, 27-wk-old, tumor-bearing RIP1-Tag5 mice (n �7–9) were treated for 2 wk with biweekly injections of liposomes alone(lipos), RGR-liposomes, RGR-liposomes � CpG, L2-liposomes � CpG,free CpG-ODN, or left untreated. At 29 wk, tumors were analyzed byimmunohistochemistry and CD45� leukocytes were quantified (three tofive independent sections per tumor; original magnification, �20), �, p �0.0015 and ��, p � 0.0045 compared with untreated tumors. B, Treatmentas in A, histological quantification of CD11b� monocytes/macrophages intumor tissues; �, p � 0.0001 and ��, p � 0.003 compared with untreatedtumors. C, Representative photographs of CD45� cells in tumors of RGR-liposomes � CpG treatment groups and comparison to CD31 vascularstaining. Upper panel, Original magnification �10; bar, 100 �m. Insetsindicate area of higher magnification shown in lower panels. Original mag-nification, �20; bar, 50 �m. Dotted lines delineate “blood lakes,” arrowspoint at endothelial cell lined, CD31� blood vessels. D, Representativephotographs of tumors from different treatment groups stained with fluo-rescently labeled (Cy3, red) anti-ICAM-1 Abs. Original magnification,�10; bar, 100 �m. Please note: in this experiment, liposomes only werecompared with RGR-engrafted liposomes to analyze potential effects of theRGR peptide. Since no difference was observed, liposomes were used ascontrols in all subsequent experiments.
1094 INTRATUMORAL DELIVERY OF CpG LIPOSOMES
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

of age were unable to mount a tumor-specific immune response.Similarly, treatment with liposomes or CpG-ODN alone was in-sufficient to prime Tag-specific cell lysis (Fig. 3D and Ref. 12).L2-liposomes plus CpG treatment groups showed a consistent, butvery low in vivo killing. Strikingly, however, treatment with RGR-liposomes plus CpG elicited a Tag-specific CTL activity (Fig. 3D)correlating with the increased CD8� and CD4� immune cells inthe tumors (Fig. 3A). This result is remarkable, since systemicCpG-ODN alone activates an innate immune response, in partic-ular NK cells, but fails to elicit tumor-specific CTL activity (12).Therefore, CpG-ODN-containing liposomes when targeted intosolid tumors have superior effects by attracting innate immunecells, activating tumor vessels, and, in addition, eliciting tumor-specific adaptive immunity.
Specific tumor targeting of CpG-ODN is therapeutically effective
Having monitored the effects of a 2-wk treatment of RGR-lipo-somes plus CpG in transgenic mice with a late-stage tumor burden,we next assessed whether these short-term effects would translate
into meaningful survival benefits. Untreated RIP1-Tag5 mice suc-cumb to insulinomas at 30 � 2 wk. Adoptive transfers of activatedCD4� and CD8� anti-Tag T cells fail to prolong the survival ofthese mice since activated effectors are unable to penetrate intosolid tumors (12, 15). To test whether liposomes alone or in com-bination with adoptive transfers of CD4� and CD8� anti-Tag Tcells had any effect on tumor growth, tumor-bearing 22- to 23-wk-old RIP1-Tag5 mice were treated over 10 wk. Not surprisingly,these treatments were ineffective with no survival benefits (Fig.4A). Similarly, repeated, systemic injection of CpG-ODN alonehad no impact on survival (Fig. 4B), whereas L2-liposomes plusCpG demonstrated a statistically significant survival advantage(mean survival 32 � 2 wk, p � 0.012 compared with untreatedmice) most likely due to limited intratumoral uptake of CpG-ODNpackaged in liposomes (Fig. 1). Remarkably, however, in RGR-liposomes plus CpG treatment groups, all mice survived beyondweek 35 and 30% were still alive when the experiment was ter-minated at week 45 (mean survival 39 � 4 wk).
After RGR-liposomes plus CpG treatment, tumor infiltration byCD4� and CD8� T cells was significant but moderate (Fig. 3A).Thus, we postulated that therapeutic success could be improved byincreasing effector cell frequency. To test this, RIP1-Tag5 micewere treated with CpG-ODN alone, L2-liposomes plus CpG, andRGR-liposomes plus CpG in combination with adoptive transfersof preactivated anti-Tag CD4� and CD8� T cells. In all threetreatment groups, therapeutic efficacy was considerably enhancedwhen compared with single treatments without immunotherapy(Fig. 4C), demonstrating that effector cells are indeed limiting. The
FIGURE 3. Activation of spontaneous antitumor immunity after vascu-lar targeting of CpG-ODN liposomes (lipos) into RIP1-Tag5 tumors. A,Tumors were harvested from untreated RIP1-Tag5 mice (n � 7–9) or afterbiweekly treatment with liposomes, RGR-liposomes � CpG, L2-liposomes �CpG, and free CpG-ODN and stained with anti-CD8 Abs. Representativephotographs are shown for liposomal-only and RGR-liposome � CpGtreatment groups. Upper panel, Original magnification, �4; bar, 250 �m.Insets indicate area of higher magnification shown in lower panels, originalmagnification 20x, bar length, 50 �m. Arrows point at a vascular structure.B, Quantification of infiltrating CD8� T cells. �, p � 0.005 and ��, p �0.015 compared with untreated. C, Quantification of infiltrating CD4� Tcells. �, p � 0.0005 and ��, p � 0.002 compared with untreated. D, Un-treated 29-wk-old RIP1-Tag5 (C3H � C57BL/6)F1 mice (n � 3–5) or afterbiweekly treatment were assessed for in vivo CTL activity against theTag-specific peptide IV. Right panel, Combined in vivo kill data for spleencells and a pool of tumor-draining pancreatic lymph nodes (panc LN) of alltreatment groups. Left panel, Representative histograms and percent spe-cific kill of CFSEhigh splenocytes for three treatment groups.
C
B
A
FIGURE 4. RIP1-Tag5 mice treated with tumor-targeted CpG-ODNcontaining liposomes survive long term. A, Kaplan-Meier survival analysisof untreated RIP1-Tag5 mice and mice injected biweekly for 10 wk withliposomes (lipos) or a combination of liposomes and adoptive transfers of invitro-activated Tag-specific CD4� and CD8� T cells, n � 8. B, Correspondingsurvival analysis of RIP1-Tag5 mice treated with L2-liposomes � CpG, RGR-liposomes � CpG, or free CpG-ODN, n � 8–11. p � 0.0001 for RGR-liposomes � CpG compared with untreated; p � 0.012 for L2-liposomes �CpG compared with untreated. C, Treatment groups as in B, but combinedwith adoptive transfers. p � 0.0001 for RGR-liposomes � CpG � ad T, p �0.0037 for RGR-liposomes � CpG � ad T, and p � 0.004 for CpG � ad T,all compared with untreated RIP1-Tag5 mice.
1095The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

best outcome was achieved when RGR-liposomes plus CpG treat-ment was combined with adoptive transfers which resulted in 70%long-term survivors. Importantly, a comparable dose of systemicCpG-ODN alone was ineffective and had limited efficacy in com-bination with immunotherapy (Fig. 4C).
CD8� T cells and NK cells are major effectors for long-termsurvival
Having established considerable therapeutic efficacy of intratu-moral RGR-liposomes plus CpG, we attempted to identify theeffector cell population responsible for long-term survival. Totest potential effectors of the adaptive immune system, CD4�
and CD8� T cells were depleted during RGR-liposomes plusCpG treatment of tumor-bearing RIP1-Tag5 mice. A 10-wktreatment regimen was started at the age of 23 wk and survivalwas monitored up to week 45. Without depletion or with controlIgG Ab injection, 30 – 40% of RGR-liposomes plus CpG-treatedRIP1-Tag5 mice survived beyond week 45 (Figs. 4B and 5).Depletion of CD4� T cells had no impact on the survival rate(Fig. 5A), whereas depletion of CD8� T cells significantly re-duced survival benefits, with death occurring at 38 � 3 wk (Fig.5B, p � 0.0078 compared with IgG-treated controls). Althoughthere were no long-term survivors, this result is still signifi-cantly better compared with untreated RIP1-Tag5 mice, imply-ing that CD8� T cells are not the only important effectors. SinceCpG-ODN are known to be potent activators of the innate im-mune system, including NK cells (5, 12), we also depleted NKcells in RGR-liposomes plus CpG-treated RIP1-Tag5 mice. NK
cell depletion reduced survival to the same extent as CD8� Tcell depletion, with survival to 37 � 3 wk, which was still betterthan that of control RIP1-Tag5 mice (Fig. 5B; p � 0.0047).Since both CD8� and NK effector cells impact on survival, weassessed possible synergistic effects in RGR-liposomes plusCpG treatment groups with combined CD8� and NK cell de-pletion. Long-term survival was significantly impaired and micesuccumbed to tumors at the age of 34 � 4 wk (Fig. 5C; p �0.0006 compared with IgG controls). This result demonstratesthat CD8� and NK cells are indeed major players for intratu-moral CpG-ODN liposomal therapy. However, combined de-pletion was not sufficient to reduce survival to that of untreatedexperimental groups ( p � 0.024, CD8�/NK depletion com-pared with untreated controls). This implies that tumor-targetedCpG-ODN liposomes exert even more complex effects, possiblywithin the tumor environment, which impact on tumor growthand overall survival.
FIGURE 5. NK cells and CD8� T cells are major effectors in long-termsurviving transgenic mice. A, Kaplan-Meier survival analysis of untreatedRIP1-Tag5 mice (n � 8) and mice treated with biweekly RGR-liposomes(lipos) � CpG injections under depletion of CD4� T cells (n � 5). B,RGR-liposomes � CpG treatment combined with depletion of CD8� Tcells or NK cells or injection of an irrelevant IgG as control, n � 8. p �0.0078, CD8� T cell depletion compared with IgG controls and p �0.0047, NK depletion vs IgG controls. C, RGR-liposomes � CpG treat-ment in combination with double depletion of CD8� T cells and NK cells,IgG controls, and untreated RIP1-Tag5 mice (as shown in A), n � 8. p �0.0006, NK/CD8� cell depletion vs IgG controls and p � 0.024, NK/CD8�
cell depletion vs untreated mice.FIGURE 6. Antivascular effects and tumor cell apoptosis after targetedCpG-ODN liposomal treatment. A, Quantification of vessel density in tu-mors of untreated 29-wk-old RIP1-Tag5 mice (n � 7–9) or after biweeklytreatment with liposomes (lipos), RGR-liposomes � CpG, L2-lipo-somes � CpG, or free CpG-ODN. Vessels were stained with anti-CD31Abs. �, p � 0.0015 compared with untreated tumors. B, Quantification ofapoptotic events in corresponding tumor samples as analyzed by TUNELassay on frozen tissue sections. �, p � 0.0001 and ��, p � 0.01 comparedwith untreated tumors. C, Representative photographs of tumors from li-posomal and RGR-liposomes � CpG treatment groups. Left panel,TUNEL histology; Original magnification, �10; bar, 100 �m. Right panel,Costaining of apoptotic cells (TUNEL, green) and CD31� blood vessels(red). Original magnification, �100; bar, 10 �m; arrows indicate TUNELsignals in close proximity to vascular structures.
1096 INTRATUMORAL DELIVERY OF CpG LIPOSOMES
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Antivascular effects of intratumoral CpG-ODN liposomes
Untreated RIP1-Tag5 tumors display vessels that are heteroge-neous in caliber and lack architectural hierarchy. In addition, in-tratumoral “blood lakes” are described, which are nonendothe-lium-lined cavities (23, 24). A striking feature of RGR-liposomesplus CpG-treated RIP1-Tag5 tumors was the presence of predom-inantly large vessels on a background of increased numbers ofblood lakes (Fig. 2C). This observation prompted us to quantifyCD31-positive vascular structures in different treatment groups.This indeed confirmed a significant reduction in blood vessel den-sity specifically with RGR-liposomes plus CpG treatment (Fig.6A). We further assessed apoptotic events in 29-wk-old RIP1-Tag5tumors after a 2-wk treatment period and found a marked increasein TUNEL� cells throughout tumor tissues when mice were re-peatedly injected with RGR-liposomes plus CpG (Fig. 6B). In theRGR-liposomes plus CpG group but not control tumors, someof these signals were in close vicinity to CD31-positive vessels(Fig. 6C). This demonstrates that intratumorally enriched CpG-ODN activates vessels but also affects endothelial cell survival.Although the sequence of events is difficult to determine, weshow here that innate immune cell and vessel activationstrongly correlate with influx of adaptive immune cells, antitu-mor CTL activity, and increased numbers of apoptotic cells.Vessel death may occur simultaneously or as a consequence ofvessel activation. These antivascular effects are specific to tu-mor-targeted CpG-ODN and contribute to CD8�- and NK cell-mediated tumor immunity, resulting in delayed tumor growthand long-term survival benefits.
DiscussionWe and others have previously shown that CpG-ODN mono-therapy is sufficient to eradicate s.c. growing tumors when directlyinjected into the tumor mass. However, systemic application ofCpG-ODN is only partially effective for murine transplantationtumors and therapeutically inefficient in an autochthonous tumormodel (5, 12). In this study, we show that the therapeutic efficacyof CpG-ODN is substantially enhanced by targeted liposomal de-livery into the tumor environment.
Liposomes are particularly useful as carrier of drugs, proteins,or nucleic acids, including CpG-ODN (25–27). More advanceddevelopments combine the drug delivery capacity of liposomeswith direct targeting of tumor cells or stroma via mAbs or peptides(28, 29). We have recently described a new methodology whichuses metal chelator lipids to engraft polyhistidine-tagged proteinsto the surface of liposomes (18, 30). This approach was used hereto anchor the tumor vessel-specific RGR peptide with a chain ofHis residues onto liposomes. Targeting of angiogenic vessels is anattractive approach to specifically deliver reagents into tumors, es-pecially if tumor-specific targeting molecules have not been iden-tified. Several investigators have described in vivo tumor-homingcapacities of liposomes tagged with tumor endothelium-specificpeptides or Abs such as RGD and NGR peptides and VCAM-1-specific Abs (31–33). Similar to these studies, we also observedthat vascular-directed lipid vesicles localize to different intratu-moral compartments compared with nonspecific control liposomes(31, 33). Lipid particles coated with cyclic RGD as targeting li-gand for �v�3 integrin, for instance, specifically bind to the tumorvessel wall, whereas control liposomes extravasate into tumor pa-renchyma (31). VCAM-1-specific targeting of angiogenic vesselsresults in selective binding to tumor vasculature as opposed tononspecific accumulation in tumor tissue by controls (33). In con-trast to these studies, however, we observed that specific vasculartargeting also enhanced liposome uptake by tumor-resident mac-
rophages resulting in an overall increase of phagocytosis in thetumor environment.
We further used tumor vascular targeting to investigate the ther-apeutic efficacy of an immune stimulatory agent, CpG-ODN, in anovel and so far unexplored context. Free CpG-ODN has beenextensively analyzed as adjuvant and direct stimulator of antitumorimmunity. More recently, CpG-ODNs were encapsulated in vari-ous lipid formulations with superior immunostimulatory activity.CpG-ODN-containing liposomes when administered i.v. or s.c.trigger higher plasma cytokine levels, NK cell activation, and amore vigorous adaptive immune response compared with freeCpG-ODN (25–27). This is most likely due to enhanced uptake ofliposomes by APCs, including macrophages. Interestingly, lipo-some-CpG-ODN-Ag complexes are sufficient to cross-primeCD8� T cells in vivo, independent of CD4� T cell help (26). Asa vaccine adjuvant, immunization with liposomal CpG-ODN incombination with tumor Ag such as a melanoma-specific peptidedelayed the growth of established B16 melanomas (26). Althoughthe enhanced antitumor effects of liposomal CpG-ODN is encour-aging, its use as vaccine adjuvant requires coadministration of tu-mor-associated Ags. In contrast, direct intratumoral injections offree CpG-ODN does not require prior identification of tumor Ags,but this administration route is limited to anatomically accessibletumors. Our approach enables high concentration targeting intoinaccessible tumor environments.
One of the major findings we report is that tumor-targeted CpG-ODN-containing liposomes were sufficient to prime a tumor Ag-specific cytotoxic T cell response. This is in stark contrast to i.v.injection of free CpG-ODN in the same mouse model which ef-fectively primes innate immunity but fails to elicit tumor-specificadaptive immunity (12). Interestingly, our data mimic the resultsof intratumoral injections of free CpG into s.c. growing tumorswhich were rejected through a concerted action of NK and CD8�
effectors (5). However, through vascular-directed CpG-ODN lipo-somes, we have now developed an improved strategy where treat-ment of autochthonous tumors is efficient through the circulation,with important clinical implications for many deeply embeddedprimary cancers and metastases.
A striking feature of RGR-liposomes plus CpG treatment wasthe inflammatory response of the vessel wall. This in turn corre-lated with significantly increased leukocyte entry into tumor tissuecompared with controls. Furthermore, adoptively transferred anti-tumor T cells reach the tumor side under RGR-liposomes plusCpG treatment but not without manipulation of the tumor envi-ronment as shown previously (12, 15, 24, 34). Once in the tumor,these effector cells encounter a substantial number of tissue-resi-dent, CpG-ODN� macrophages; activation of their endosomalTLR9 receptors most likely amplifies immune activation further.Long-term survival was mainly mediated by CD8� and NK cellsand presumably direct tumor cell lysis. Moreover, enrichment ofCpG-ODN liposomes on the vessel wall reduced vascular densityover time and thus indirect antivascular effects also impacted ontumor growth. In summary, vascular-directed CpG-ODN lipo-somes activate innate and adaptive immunity and alter the tumorenvironment, which results in greatly improved therapeutic effi-cacy compared with free CpG-ODN injections.
The ability to combine intratumoral targeting with immunemodulation using liposome technology has enormous potential forclinical evaluation. We envision that other combinatorial ap-proaches could result in further survival benefits. For instance,peptide-tagged and nontagged liposomes could be loaded withCpG-ODN and tumor Ag for coadministration. In this setting,
1097The Journal of Immunology
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

CpG-ODN would act locally and as a vaccine adjuvant. Synergis-tic action of local CpG-ODN with radiotherapy or depletion ofregulatory T cells has also been reported (35, 36).
AcknowledgmentsWe thank D. Hanahan, I. Forster, T. Geiger, and R. Flavell for transgenicmice and Yew Choong Wai for contributions to some experiments. Wethank Lipotek Pty Ltd for providing the NTA3-DTDA for researchpurposes.
DisclosuresJ.G.A. and C.R.P. declare a commercial interest in Lipotek Pty Ltd.
References1. Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto,
K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptorrecognizes bacterial DNA. Nature 408: 740–745.
2. Jahrsdorfer, B., and G. J. Weiner. 2003. CpG oligodeoxynucleotides for immunestimulation in cancer immunotherapy. Curr. Opin. Invest. Drugs 4: 686–690.
3. Carpentier, A. F., L. Chen, F. Maltonti, and J. Y. Delattre. 1999. Oligode-oxynucleotides containing CpG motifs can induce rejection of a neuroblastoma inmice. Cancer Res. 59: 5429–5432.
4. Carpentier, A. F., J. Xie, K. Mokhtari, and J. Y. Delattre. 2000. Successful treat-ment of intracranial gliomas in rat by oligodeoxynucleotides containing CpGmotifs. Clin. Cancer Res. 6: 2469–2473.
5. Kawarada, Y., R. Ganss, N. Garbi, T. Sacher, B. Arnold, and G. J. Hammerling.2001. NK� and CD8� T cell-mediated eradication of established tumors byperitumoral injection of CpG-containing oligodeoxynucleotides. J. Immunol.167: 5247–5253.
6. Heckelsmiller, K., K. Rall, S. Beck, A. Schlamp, J. Seiderer, B. Jahrsdorfer,A. Krug, S. Rothenfusser, S. Endres, and G. Hartmann. 2002. Peritumoral CpGDNA elicits a coordinated response of CD8 T cells and innate effectors to cureestablished tumors in a murine colon carcinoma model. J. Immunol. 169:3892–3899.
7. Baines, J., and E. Celis. 2003. Immune-mediated tumor regression induced byCpG-containing oligodeoxynucleotides. Clin. Cancer Res. 9: 2693–2700.
8. Mastini, C., P. D. Becker, M. Iezzi, C. Curcio, P. Musiani, G. Forni, F. Cavallo,and C. A. Guzman. 2008. Intramammary application of non-methylated-CpGoligodeoxynucleotides (CpG) inhibits both local and systemic mammary carci-nogenesis in female BALB/c Her-2/neu transgenic mice. Curr. Cancer DrugTargets 8: 230–242.
9. Sfondrini, L., D. Besusso, C. Rumio, M. Rodolfo, S. Menard, and A. Balsari.2002. Prevention of spontaneous mammary adenocarcinoma in HER-2/neu trans-genic mice by foreign DNA. FASEB J. 16: 1749–1754.
10. Wingender, G., N. Garbi, B. Schumak, F. Jungerkes, E. Endl, D. von Bubnoff,J. Steitz, J. Striegler, G. Moldenhauer, T. Tuting, et al. 2006. Systemic applicationof CpG-rich DNA suppresses adaptive T cell immunity via induction of IDO.Eur. J. Immunol. 36: 12–20.
11. Krieg, A. M. 2008. Toll-like receptor 9 (TLR9) agonists in the treatment ofcancer. Oncogene 27: 161–167.
12. Garbi, N., B. Arnold, S. Gordon, G. J. Hammerling, and R. Ganss. 2004. CpGmotifs as proinflammatory factors render autochthonous tumors permissive forinfiltration and destruction. J. Immunol. 172: 5861–5869.
13. Joyce, J. A., P. Laakkonen, M. Bernasconi, G. Bergers, E. Ruoslahti, andD. Hanahan. 2003. Stage-specific vascular markers revealed by phage display ina mouse model of pancreatic islet tumorigenesis. Cancer Cell 4: 393–403.
14. Hamzah, J., D. Nelson, G. Moldenhauer, B. Arnold, G. J. Hammerling, andR. Ganss. 2008. Vascular targeting of anti-CD40 antibodies and IL-2 into au-tochthonous tumors enhances immunotherapy in mice. J. Clin. Invest. 118:1691–1699.
15. Ganss, R., and D. Hanahan. 1998. Tumor microenvironment can restrict the ef-fectiveness of activated antitumor lymphocytes. Cancer Res. 58: 4673–4681.
16. Geiger, T., L. R. Gooding, and R. A. Flavell. 1992. T-cell responsiveness to anoncogenic peripheral protein and spontaneous autoimmunity in transgenic mice.Proc. Natl. Acad. Sci. USA 89: 2985–2989.
17. Forster, I., R. Hirose, J. M. Arbeit, B. E. Clausen, and D. Hanahan. 1995. Limitedcapacity for tolerization of CD4� T cells specific for a pancreatic � cell neo-antigen. Immunity 2: 573–585.
18. van Broekhoven, C. L., and J. G. Altin. 2005. The novel chelator lipid 3(nitrilo-triacetic acid)-ditetradecylamine (NTA3-DTDA) promotes stable binding of His-tagged proteins to liposomal membranes: potent anti-tumor responses induced bysimultaneously targeting antigen, cytokine and costimulatory signals to T cells.Biochim. Biophys. Acta 1716: 104–116.
19. Altin, J., M. Banwell, P. Coghlan, C. Easton, M. Nairn, and D. Offermann. 2006.Synthesis of NTA3-DTDA-A chelator-lipid that promotes stable binding of His-tagged proteins to membranes. Aust. J. Chem. 59: 302–306.
20. Tanaka, T., F. Kitamura, Y. Nagasaka, K. Kuida, H. Suwa, and M. Miyasaka.1993. Selective long-term elimination of natural killer cells in vivo by an anti-interleukin 2 receptor � chain monoclonal antibody in mice. J. Exp. Med. 178:1103–1107.
21. Hanahan, D. 1985. Heritable formation of pancreatic �-cell tumours in transgenicmice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315:115–122.
22. Schell, T. D., and S. S. Tevethia. 2001. Control of advanced choroid plexustumors in SV40 T antigen transgenic mice following priming of donor CD8� Tlymphocytes by the endogenous tumor antigen. J. Immunol. 167: 6947–6956.
23. Ryschich, E., J. Schmidt, G. J. Hammerling, E. Klar, and R. Ganss. 2002. Trans-formation of the microvascular system during multistage tumorigenesis. Int. J.Cancer 97: 719–725.
24. Hamzah, J., M. Jugold, F. Kiessling, P. Rigby, M. Manzur, H. H. Marti, T. Rabie,S. Kaden, H. J. Grone, G. J. Hammerling, et al. 2008. Vascular normalization inRgs5-deficient tumours promotes immune destruction. Nature 453: 410–414.
25. Mui, B., S. G. Raney, S. C. Semple, and M. J. Hope. 2001. Immune stimulationby a CpG-containing oligodeoxynucleotide is enhanced when encapsulated anddelivered in lipid particles. J. Pharmacol. Exp. Ther. 298: 1185–1192.
26. Zaks, K., M. Jordan, A. Guth, K. Sellins, R. Kedl, A. Izzo, C. Bosio, and S. Dow.2006. Efficient immunization and cross-priming by vaccine adjuvants containingTLR3 or TLR9 agonists complexed to cationic liposomes. J. Immunol. 176:7335–7345.
27. de Jong, S., G. Chikh, L. Sekirov, S. Raney, S. Semple, S. Klimuk, N. Yuan,M. Hope, P. Cullis, and Y. Tam. 2007. Encapsulation in liposomal nanoparticlesenhances the immunostimulatory, adjuvant and anti-tumor activity of subcutane-ously administered CpG ODN. Cancer Immunol. Immunother. 56: 1251–1264.
28. Park, J. W., C. C. Benz, and F. J. Martin. 2004. Future directions of liposome-and immunoliposome-based cancer therapeutics. Semin. Oncol. 31: 196–205.
29. Altin, J. G., and C. R. Parish. 2006. Liposomal vaccines: targeting the delivery ofantigen. Methods 40: 39–52.
30. Altin, J. G., F. A. White, and C. J. Easton. 2001. Synthesis of the chelator lipidnitrilotriacetic acid ditetradecylamine (NTA-DTDA) and its use with the IAsysbiosensor to study receptor-ligand interactions on model membranes. Biochim.Biophys. Acta 1513: 131–148.
31. Mulder, W. J., G. J. Strijkers, J. W. Habets, E. J. Bleeker, D. W. van der Schaft,G. Storm, G. A. Koning, A. W. Griffioen, and K. Nicolay. 2005. MR molecularimaging and fluorescence microscopy for identification of activated tumor endo-thelium using a bimodal lipidic nanoparticle. FASEB J. 19: 2008–2010.
32. Pastorino, F., C. Brignole, D. Di Paolo, B. Nico, A. Pezzolo, D. Marimpietri,G. Pagnan, F. Piccardi, M. Cilli, R. Longhi, et al. 2006. Targeting liposomalchemotherapy via both tumor cell-specific and tumor vasculature-specific ligandspotentiates therapeutic efficacy. Cancer Res. 66: 10073–10082.
33. Gosk, S., T. Moos, C. Gottstein, and G. Bendas. 2008. VCAM-1 directed im-munoliposomes selectively target tumor vasculature in vivo. Biochim. Biophys.Acta 1778: 854–863.
34. Ganss, R., E. Ryschich, E. Klar, B. Arnold, and G. J. Hammerling. 2002. Com-bination of T-cell therapy and trigger of inflammation induces remodeling of thevasculature and tumor eradication. Cancer Res. 62: 1462–1470.
35. Meng, Y., A. F. Carpentier, L. Chen, G. Boisserie, J. M. Simon, J. J. Mazeron,and J. Y. Delattre. 2005. Successful combination of local CpG-ODN and radio-therapy in malignant glioma. Int. J. Cancer 116: 992–997.
36. Sharma, S., A. L. Dominguez, S. Z. Manrique, F. Cavallo, S. Sakaguchi, andJ. Lustgarten. 2008. Systemic targeting of CpG-ODN to the tumor microenvi-ronment with anti-neu-CpG hybrid molecule and T regulatory cell depletion in-duces memory responses in BALB-neuT tolerant mice. Cancer Res. 68:7530–7540.
1098 INTRATUMORAL DELIVERY OF CpG LIPOSOMES
by guest on June 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from





























