Embed Size (px)
Citation preview

SYNTHETIC GENERATION OF AN AZIDE-BEARNING N-MUSTARD COFACTOR MIMIC OF S-ADENOSYL-L-METHIONINE
BY
VAN MAI
A Thesis Submitted to the Graduate Faculty of
WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES
in Partial Fulfillment of the Requirements
for the Degree of
MASTER OF SCIENCE
Chemistry
May 2011
Winston-Salem, North Carolina
Approved By:
Lindsay R. Comstock, Ph.D., Advisor
Rebecca W. Alexander, Ph.D.
S. Bruce King, Ph.D.

ii
This work is dedicated to my parents-
for their love and sacrifice.

iii
ACKNOWLEDGEMENTS
First and foremost, I would like to thank my parents for their love and support
throughout my graduate studies. I wish to thank my father for giving everything he had
to fight liver cancer for more than a year and for supporting me throughout his impossible
ordeal. During that difficult time, I realized that nothing in the world would ever be as
tough as what my father was forced to endure and I have learned to appreciate life and its
many ups and downs much more thanks to him. I am very fortunate to have two
wonderful sisters who are always there for me. Also, I am very grateful to my Uncle Da
and Aunt Khang who have given me the opportunity to live and study in the United
States, without whom I do not know where I would be.
I would like to sincerely thank Dr. Comstock for her support and advice, which
she has provided to me since the first day I joined Wake Forest University. I wish to
thank her from the bottom of my heart for her confidence in me, for being so patient
while guiding and helping me every step of the way, and, above all, for being there for
me during my father’s illness. I am proud to be her first graduate student, and I wish her
nothing but the best in all her future work.
I would also like to thank Dr. King and Dr. Alexander for serving as members on
my thesis committee. I thank Dr. Wright for all his patient help and advice regarding the
use of NMR and LC/MS as well as data analysis. To Dr. Tomlinson and Dr.
Guddneppanavar, I am grateful for their helpful discussion and advice.
Finally, I wish to thank my very supportive boyfriend and all my friends for being
there for me, and making my time at Wake Forest University a wonderful experience.

iv
TABLE OF CONTENTS
ABBREVIATIONS ………………………………………………………………….….vi
TABLE OF FIGURES …………………………………………………………………ix
TABLE OF SCHEMES ………………………………………………………………...x
ABSTRACT ……………………………………………………………………………xi
CHAPTER 1. INTRODUCTION ………………………………………………………..1
1.1 Biological Methylation ………………………………………………………1
1.1.1 Protein Methylation ………………………………………………3
1.1.2 DNA Methylation ………………………………………………...5
1.2 Detection of Biological Methylation ……………………………………….7
1.3 Development of SAM-Based Cofactor Mimics …………………………….9
1.3.1 Development of Aziridine Adenosine ……………………………9
1.3.2 Development of Azide-Functionalized Aziridine Adenosines …...10
1.3.3 Development of Nitrogen-Mustard Analogs ……………………12
1.4 Conclusions ………………………………………………………………....14
CHAPTER 2. DESIGN AND SYNTHESIS OF AN AZIDE-BEARING N-MUSTARD COFACTOR MIMIC OF SAM ……………………………………….15
2.1 Introduction ………………………………………………………………...15
2.2 Synthetic Analysis of the Azide-Bearing N-Mustard Adenosine 2.1……16
2.3 Synthetic Approach ………………………………………………………...18
2.3.1 Initial Synthetic Approach ………………………………………..18
2.3.2 An Alternate Approach Using TES Protecting Group ……………23
2.3.3 An Improved Method for Installation of Azide Functionality ……27

v
2.3.4 A More Efficient Pathway to Obtain 8-Azido-5′-Phthalimide Adenosine 2.32 …………………………………………………..29
2.3.5 NMR, MS, and HPLC Analysis of 2.1 …………………………...33
2.4 Biochemical Analysis of 2.1 Using a Restriction/Protection Assay …….....37
2.5 Conclusions ………………………………………………………………...39
2.6 Synthetic Procedures And Compound Characterizations …………………..41
REFERENCES …………………………………………………………………………58
APPENDIX …………………………………………………………………………….61
CIRRICULUM VITAE ………………………………………………………………...80

vi
ABBREVIATIONS
Commonly used abbreviations are listed below.
ACN acetonitrile
AcOH acetic acid
Boc tert-butoxycarbonyl
C Celsius
CDCl3 chloroform-d
CH2Cl2 dichloromethane
DCC N,N'-Dicyclohexylcarbodiimide
DIAD diisopropyl azodicarboxylate
DIBAL-H diisobutylaluminum hydride
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
DNMT’s DNA methyltransferases
Et2O diethyl ether
EtOH ethanol
EtOAc ethyl acetate
g gram
h hour
H2O water
HCl hydrochloric acid
HPLC high performance liquid chromatography
J coupling constant

vii
LC/MS liquid chromatography/mass spectrometry
µ micro
M molar
MeOH methanol
mg milligram
min minute
mmol millimoles
MS mass spectrometry
MTase methyltransferase
NaCNBH3 sodium cyanoborohydride
NaOAc sodium acetate
NaHCO3 sodium hydrogen carbonate
Na2SO4 sodium sulfate
NH4Cl ammonium chloride
NMR nuclear magnetic resonance
PCR polymerase chain reaction
Pet Ether petroleum ether
PPh3 triphenylphosphine
rt room temperature
SAM S-adenosyl-L-methionine
TBS tert-butyl dimethylsilyl
TEA triethylamine
TES triethylsilyl

viii
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography

ix
TABLE OF FIGURES Figure 1.1. Methylated Residues of DNA ………………………………………………………...6
Figure 1.2. 8-Azido Aziridine Compounds …………………………………………………..…11
Figure 1.3. N-Mustard Analog ………………………………………………………………….....13
Figure 1.4. Proposed Structure of Azide-Bearing N-Mustard Cofactor Mimic ………....14
Figure 2.1. 8-Azido-5′-N-Mustard Hydrochloride Salt …………………………………..…...15
Figure 2.2. The t-Butyl-Containing Byproduct of Global Deprotection at Room Temperature ………………….........................................................................................32
Figure 2.3. Hydrolyzed Compounds Formed during MS …………………………….............33
Figure 2.4. Formation of Aziridinium (2.42) and Methyl Ether (2.43) during MS …......34
Figure 2.5. Analytical HPLC Chromatograms of 2.1 before (I) and after Isolation (II) ………….…………………………………………………………….…...36
Figure 2.6. M.TaqI Restriction/Protection Assay with Increasing Cofactor ………….....39

x
TABLE OF SCHEMES
Scheme 1.1. The Synthesis of S-Adenosyl-L-Methionine (SAM) ……………….…………...1
Scheme 1.2. SAM-Dependent Methylation …..……………………………….……………….....2
Scheme 1.3. Hydrolysis of S-Adenosyl-L-Homocysteine Followed by Methylation of Homocysteine ...........................................................................................3 Scheme 1.4. Sequence-Specific Modification of DNA by M.TaqI with SAM and Cofactor 1.5 .................................................................................................10 Scheme 1.5. Formation of Aziridinium at Physiological pH ………..……………………....13
Scheme 2.1. Cofactor 2.1 as a Probe for Post-Alkylation Modification …...……………...16
Scheme 2.2. Synthetic Analysis of the Azide-Bearing N-Mustard Cofactor 2.1 .................................................................................................................17 Scheme 2.3. Unsuccessful Bromination .…….…….……………………………………………..18
Scheme 2.4. Synthesis of Protected Aldehyde …….…………………………………...............19
Scheme 2.5. Proposed Synthetic Pathway to Azide-Containing N-Mustard Cofactor 2.1 ………………………………………………………………………….21 Scheme 2.6. Successful Iodination and TES Deprotection ………..…………………………24
Scheme 2.7. Alternate Synthetic Pathway with TES Protection ……..……………………..25
Scheme 2.8. Unexpected Mono-TES and Di-TES Deprotection during Azidation ..…...26
Scheme 2.9. NaCNBH3 Can Reduce Azide to the Primary Amine ………….…………….28
Scheme 2.10. Revised Synthetic Pathway Incorporating Azide Earlier …………………29
Scheme 2.11. Improved Synthesis of 8-Azido 5′-Phthalimide Adenosine 2.32 ………..30

xi
ABSTRACT
The synthesis of an azide-bearing N-mustard cofactor, 8-azido-5′-
(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine ammonium hydrochloride (2.1),
has been accomplished with a small amount of impurity in several steps from
commercially available 2′,3′-isopropylidene adenosine (2.2). This cofactor was designed
to efficiently mimic S-adenosyl-L-methionine (SAM) by incorporating an N-mustard and
an amino acid moiety surrounding an adenosine core. In addition, an azide functionality
was introduced for post-alkylation modification. The crucial factors that led to this
success were (1) the choice of the proper alcohol protecting group and (2) the installation
of the azide functionality at the C8 position of the adenine base prior to the incorporation
of N-mustard and amino acid moieties. Cofactor 2.1 was found to be effectively
transferred onto DNA by M.TaqI, providing an azide-bearing modified DNA that will
allow for subsequent chemoselective ligation chemistry that is hypothesized to facilitate
the detection of biological methylation sites.

CHAPTER 1. INTRODUCTION
1.1 Biological Methylation
In a number of biological processes, methylation is considered an essential post-
translational modification for normal cellular function and development, as well as for
the regulation of gene expression and DNA replication. It was in the 1940s that Du
Vigneaud and colleagues thought they had found that a significant fraction of the methyl
groups in cell metabolites derived from methionine (1.1) – one of the two essential amino
acids containing sulfur. Yet about ten years later, Cantoni and colleagues discovered that
the actual methyl donor is the ATP-activated form of methionine, S-adenosyl-L-
methionine (SAM), 1.2,1 with the chemical structure shown in Scheme 1.1.
Scheme 1.1. The Synthesis of S-Adenosyl-L-Methionine (SAM).
The process of how methionine is metabolized to generate SAM involves the
participation of ATP and an enzyme called methionine adenosyltransferase; this enzyme
is responsible for the transfer of the adenosyl portion of ATP onto methionine. Binding
to a charged sulfur, the SAM methyl group becomes highly reactive, thus making SAM
an important biological methylating agent. As a product of this catalysis, SAM (also
1

called activated methionine) serves as a precursor for numerous methylation transfer
reactions in all living organisms.2
Biological methylation is the replacement of a hydrogen atom with a methyl
group and these reactions are catalyzed by enzymes called methyltransferases.
Particularly, two primary types of methylation that are known to play an important role in
diseases include DNA methylation and protein methylation. As a general mechanism, the
activated methyl group from SAM is transferred to nucleophilic atoms such as nitrogen,
oxygen, carbon, or sulfur within DNA, RNA, proteins, lipids, polysaccharides, or small
molecules, as shown in Scheme 1.2.2
Scheme 1.2. SAM-Dependent Methylation.
Some initial studies have demonstrated that O-, N-, and S- methylations occur via
a straightforward SN2 nucleophilic attack by the lone electron pairs of oxygen, nitrogen,
or sulfur from substrates on the methyl group of SAM. Upon methylation, the
methylated substrate is released to yield S-adenosyl-L-homocysteine (SAH), 1.3, which is
subsequently hydrolyzed to form adenosine and homocysteine (1.4) by S-
adenosylhomocysteine hydrolase (Scheme 1.3). The homocysteine can then be
2

methylated to re-form methionine (1.1) via a reaction with 5N methyltetrahydrofolate
(5N-methyl-THF).2
H2O Adenosine
1.4
HSO
O
NH3
1.1
H3CS
O
O
NH3
5N-methyl-THF THF
SAH
Scheme 1.3. Hydrolysis of S-Adenosyl-L-Homocysteine Followed by
Methylation of Homocysteine.
1.1.1 Protein Methylation
Carried out by several classes of highly substrate-specific protein
methyltransferases, protein methylation is the transfer of the methyl group from SAM to
either the carboxyl groups or the nitrogen groups of amino acids. Only N- or O-
methylation has been found in proteins. For example, protein methylation is
predominantly found on lysine and arginine residues, but has also been found on histidine,
proline, and carboxyl groups.3 Studies have shown that modifications to these amino
acids in histones and other proteins affect the regulation of transcription. Particularly
with histones, modifications were found to occur on only lysine and arginine residues and
appear to play an important role in the regulation of chromatin structure and gene
transcription.4
In eukaryotic cells, a highly condensed structure of chromatin is formed by a
repeating, complex unit called a nucleosome, where an octamer of histones is wrapped by
complimentary strands of DNA. The core histones consist of a pair each of H3, H4, H2A,
3

and H2B proteins; each of these proteins is rich in positively charged amino acids, such
as lysine and arginine. This contributes to the compaction of chromatin due to the
interaction of histone proteins with the negatively charged DNA backbone.5 In nature,
histone tails protrude out of the nucleosome itself; therefore, they are subjected to
methylation as well as other covalent modifications that are necessary for the regulation
of gene activity, including acetylation.3
When being interfered with by methylation, even though the charge of histone
residues should not be affected, the affinity of histone for DNA may be affected by an
increase in size and hydrophobicity of the histone by the addition of methyl groups. This
modification may not only affect the function of histones, but may also impact the
establishment of heterochromatin and euchromatin, which are the tightly and lightly
packed regions of chromatin, respectively. Modification in chromatin structure may
affect the function of proteins involved in the control of gene expression, and thus may
either activate or repress gene expression.3
While less is known about the mechanism of arginine methylation, it has been
found that arginine methylation can promote or inhibit specific intermolecular
interactions that play an important role in signal transduction pathways that regulate
transcription.3 Lysine methylation is reported to inhibit the binding of proteins to histone
tails and block additional posttranslational modifications of the histone tails.
Alternatively, lysine methylation has been shown to recruit other proteins as well as
chromatin modifying enzymes.3 Ultimately, lysine and arginine methylation were both
found to be able to either facilitate or inhibit transcription depending on the specific site
on the histone tails, the position, and the methylation level of lysine or arginine.6,7
4

Recent studies have shown that methylation of Lys-4 and Arg-17 of histone H3, and Arg-
3 of histone H4, have been associated with transcription activation, whereas methylation
of Lys-9 of histone H3 has contributed to gene silencing.3 In addition to many
methylation events, other post-translational modifications of histone tails, such as
acetylation and phosphorylation, also contribute to the alteration of chromatin structure
that may lead to the activation or repression of transcription. This complex system of
histone modification is known as the histone code.3
1.1.2 DNA Methylation
Playing a crucial role in controlling gene expression, a particular level of DNA
methylation is essential for normal development of cells.8 It is the addition of methyl
groups from SAM to either cytosine or adenine residues at specific sites on DNA,
catalyzed by a group of sequence-specific enzymes called DNA methyltransferases
(DNMT’s). DNA methyltransferases are categorized into two classes based on the atom,
either C or N, which is modified: N-DNA methyltransferase and C-DNA
methyltransferase. In nature, N-DNA methyltransferase catalyzes the methylation of the
amino group on either N6 of adenine to form N6-methyladenine (N6mA) or N4 of
cytosine to give N4-methylcytosine (N4mC). Alternatively, C-DNA methyltransferase
catalyzes the methylation of C5 of cytosine to make C5-methylcytosine (5mC), as shown
in Figure 1.1. It was discovered that only 5mC was found in eukaryotes, whereas
methylation of both cytosine and adenine bases occur in prokaryotes.9 In general, after
DNA replication, a daughter strand of DNA inherits the methylation pattern of a parental
DNA; this may affect the genes controlling cell division if aberrant methylation occurs.
5

N
NN
N
N
N
N
NH2
O
N
N
N
O
H
R
H3C
R
N6mA N4mC5mC
HH3CH3C
R
Figure 1.1. Methylated Residues of DNA. R = Ribose Sugar.
DNA methylation usually occurs within CpG islands, CG dinucleotide-rich
regions of a DNA sequence, which are found upstream of the promoter region of the gene.
When high levels of methylated cytosine are observed in these islands, repression of
transcription occurs and shuts off gene expression through a cascade event. When these
CpG islands are lacking methylated cytosine residues, correlation with DNA
transcriptional activation has been indicated.10
There are two classes of abnormal DNA methylation: hypermethylation and
hypomethylation. The mechanism by which methylated CpG islands repress
transcription involves the binding of methyl-CpG binding proteins, which bind
selectively to stretches of methylated DNA, and thus sterically reduce the access of the
transcriptional machinery required to transcribe a DNA sequence to mRNA. These
methyl-CpG binding proteins, in turn, recruit transcriptional repressors such as histone
deacetylases (HDACs) and other chromatin remodeling proteins. This series of events
results in a condensed DNA structure, as well as a compact state of chromatin, that makes
DNA less accessible for transcription factors. Different from hypermethylation,
hypomethylation events usually occur in CpG islands where cytosines are normally
6

heavily methylated. Even though the biological effects of hypomethylation are not
understood as well compared to hypermethylation, a net loss of 5mC content may result
in an increase in transcription and an overexpression of associated genes, and thus an
increase in genomic instability.11
Both hypermethylation and hypomethylation may lead to the onset of certain
diseases, including cancer. Cells can become tumorous when hypermethylation develops
at the CpG islands, rendering certain tumor suppressor genes inactive.12 With regard to
hypomethylation, it has been documented that a 35-60 % reduction in 5mC was observed
in most tumors, including colorectal cancer cells.13 Studies have also demonstrated that
the degree of hypomethylation correlates with disease progression. For example, the
5mC content was found to be normal at a benign stage of human prostate cancer, but was
significantly lower once it had reached the metastatic state.14 Currently, the causes of
DNA hypermethylation and hypomethylation and the precise mechanisms of how they
occur are being explored.
1.2 Detection of Biological Methylation
Even though methylation plays an important role in the regulation of gene
expression, the methyl group itself has limited utility for practical applications. Methyl
groups are small and nearly devoid of functionality. Therefore, identifying methyl
groups in complex biological systems has been a challenge. For example, labeling
methyl groups with tritium and carbon-14 introduces additional problems related to
safety, stability, and disposal of radioactive isotopes.15 Consequently, many improved
methodologies have been designed to efficiently ascertain the status of a methylation
7

event. Today, some newly developed techniques are being used in combination with
established methods for detection of DNA and protein methylation.
The pattern of DNA methylation can be determined using several methods, each
with its own advantages and disadvantages. One traditional chemical conversion method
relies on base modification by treating the DNA with bisulfite (HSO3¯), hydrazine (N2H4)
or permanganate (MnO4¯) to specifically detect the position of 5mC.16 Bisulfite
deaminates cytosine on single-stranded DNA to generate uracil, whereas permanganate
reacts with 5mC and thymine. Hydrazine reacts with cytosine and thymine on either
single-stranded or double-stranded DNA. The modified DNA can be amplified via
quantitative and qualitative PCR techniques followed by DNA sequence analysis to
analyze methylation status.17 For screening large sections of a DNA sequence,
methylation-sensitive restriction endonucleases (MSREs) were used to identify
methylated bases at specific sites of DNA, in addition to the use of bisulfite.16
Compared to DNA methylation, detection of protein methylation has proven to be
more of a challenge. One reliable method is to incorporate radioactive [3H]-methyl
groups followed by enzymatic digestion with proteases. Then, high-performance liquid
chromatography (HPLC) and mass spectrometry (MS) are used for separation of peptide
fragments and mass analysis.18 Alternatively, antibodies can also be utilized to detect
protein methylation at specific residues. Today, antibodies tailored for specific proteins
are commercially available. However, antibodies are not available for most proteins, and
generating a new antibody for an unidentified protein is expensive and time consuming.
8

1.3 Development of SAM-Based Cofactor Mimics
In an attempt to develop an alternate method to identify sites of biological
methylation that does not rely on traditional chemical modification or radioactive
isotopes, the development of a substance that is capable of modifying either nucleic acids
or proteins in a MTase-dependent manner may serve as an alternate means to identify
sites of biological methylation. That is, a small molecule which is designed to mimic
SAM, but yield a larger, more easily-detected product, is predicted to be useful as a probe
of methylation of various substrates. More interestingly, the incorporation of a unique
functionality and/or affinity tag or fluorophore onto these small molecules is anticipated
to hold promising results.
1.3.1 Development of Aziridine Adenosine
In 1998, E. Weinhold and co-workers utilized the natural ability of DNA
methyltransferases to recognize and modify DNA in a sequence-specific fashion to
design an aziridine-based cofactor mimic as an alternative to SAM. Specifically, 5′-
aziridinio-5′-deoxy adenosine (1.5) was synthesized as a mimic of SAM.20 Ultimately,
this cofactor has been shown to be suitable for sequence-specific modification of DNA
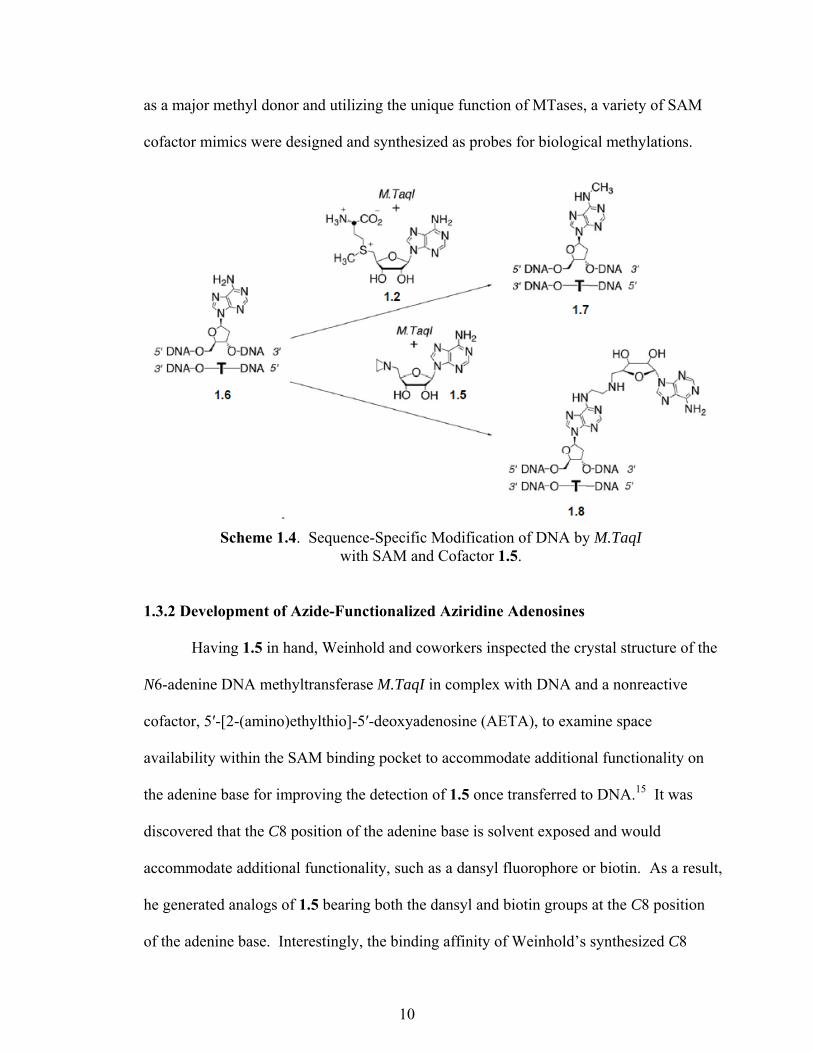
when catalyzed by a DNA methyltransferase. As shown in Scheme 1.4, M.TaqI from
Thermus aquaticus, recognizes the specific sequence of DNA as 5′-TCGA-3′ and
catalyzes the transfer of the activated methyl group from SAM onto the amino group of
adenine 1.6 to provide methylated adenine 1.7. Interestingly, in comparison, nucleophilic
attack on the aziridine ring of 1.5 leads to ring opening and subsequent coupling of the
whole cofactor to the target adenine to provide 1.8. By employing the versatility of SAM
9
as a major methyl donor and utilizing the unique function of MTases, a variety of SAM
cofactor mimics were designed and synthesized as probes for biological methylations.
Scheme 1.4. Sequence-Specific Modification of DNA by M.TaqI with SAM and Cofactor 1.5.
1.3.2 Development of Azide-Functionalized Aziridine Adenosines
Having 1.5 in hand, Weinhold and coworkers inspected the crystal structure of the
N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a nonreactive
cofactor, 5′-[2-(amino)ethylthio]-5′-deoxyadenosine (AETA), to examine space
availability within the SAM binding pocket to accommodate additional functionality on
the adenine base for improving the detection of 1.5 once transferred to DNA.15 It was
discovered that the C8 position of the adenine base is solvent exposed and would
accommodate additional functionality, such as a dansyl fluorophore or biotin. As a result,
he generated analogs of 1.5 bearing both the dansyl and biotin groups at the C8 position
of the adenine base. Interestingly, the binding affinity of Weinhold’s synthesized C8
10

biotinylated aziridine cofactor to M.TaqI has been shown to be only four-fold lower than
the reported affinity of SAM.21 This result is evidence in support of previous work by Dr.
Lindsay R. Comstock to incorporate azide functionalities at the C8 position of the
adenine base for post-synthetic modification.
Dr. Lindsay Comstock has successfully synthesized two aziridine-based cofactor
mimics: 8-azido-5′-aziridinio-5′-deoxy adenosine (1.9) and 8-(4′′-azido-butylamino)-5′-
aziridinio-5′-deoxy adenosine (1.10), as shown in Figure 1.2.22,23 Not only does the
azide functionality have the advantage of being rarely seen in naturally occurring
compounds, but it also exhibits sufficient stability at room temperature and does not react
significantly with biological functional groups. Due to its small size, its incorporation at
the C8 position of the adenine base can be easily accommodated within the active site of
the MTase. After obtaining 1.9 and 1.10, Dr. Lindsay Comstock evaluated their utility in
a MTase-dependent DNA fashion by performing both a DPAGE assay using 32P-labelled
oligonucleotides and a restriction/protection assay with plasmid DNA.23,24 In comparison
to the original aziridine-based cofactor 1.5, results from both assays demonstrated that
N
NN
N
NH2
O
OHHO
N
N
NN
N
NH2
O
OHHO
NN3 HN
N3
1.9 1.10
Figure 1.2. 8-Azido Aziridine Compounds.
11

1.9 and 1.10 are more effective cofactors compared to 1.5. Additionally, Dr. Lindsay R.
Comstock established that DNA modified with 1.9 or 1.10 was capable of undergoing
chemoselective ligations, such as the Staudinger ligation and “Click” chemistry, to
generate a uniquely ligated DNA product bearing an affinity tag that would facilitate the
detection of sites of DNA methylation.24,25
1.3.3 Development of Nitrogen-Mustard Analogs
In 1997, the crystal structure of a binary complex of the DNA MTase M.TaqI with
SAM was obtained by Weinhold and co-workers, which provided evidence of how the
amino acid moiety of SAM contributes to the cofactor-enzyme binding interaction. 26
The methionine amino acid moiety of SAM is held in position by three types of
interactions with MTase.26 It was shown that the carboxylate group is hydrogen bonded
to Thr 23, whereas the ammonium group forms a salt-bridge to Glu 45, Ala 47, and Cys
48. Also, the alkyl chain has some hydrophobic interaction with Pro 52, Pro 106, and Pro
107. Even though the amino acid side chain does not directly engage in the formation of
a covalent bond between cofactor and DNA, it was proven to play a crucial role in
MTase-dependent alkylation. This was concluded from the work of Rajski and Weller,
who generated and evaluated an N-mustard analog of SAM (Figure 1.3): 5′-
(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine ammonium hydrochloride (1.11).27
Different from the previously generated aziridine and azide cofactor mimics, 1.11
is a hydrochloride salt with an N-mustard and an amino acid moiety on the 5′ position of
the ribose sugar. Since 1.11 possessed a high structure homology to SAM, its amino acid
side chain is expected to have similar types of interactions as those exhibited in the
12

NI
CO2HH3N
N
NN
N
NH2
O
OHHO
H
1.11
Figure 1.3. N-Mustard Analog.
binary complex of SAM and M.TaqI. As a result, this particular compound has been
shown to have a strong binding affinity to MTase and has been efficiently transferred to
DNA by various MTases, compared to previously reported aziridine-based cofactors.27
Therefore, to provide a compatible cofactor-enzyme interaction with the SAM-M.TaqI
complex, the presence of the amino acid moiety is essential.
Another crucial part of the structure of 1.11 is the N-mustard moiety. Under
physiological conditions, the N-mustard is deprotonated and rapidly forms a highly
reactive aziridinium ion, as shown in Scheme 1.5, upon elimination of the iodide by an
intramolecular nucleophilic substitution. The aziridinium is known to be a highly
reactive alkylating agent and can serve to alkylate biomolecules, such as DNA, protein,
and small molecules, through a MTase-dependent mechanism.28
Scheme 1.5. Formation of Aziridinium at Physiological pH.
13

1.4 Conclusions
In this work, further advancement of compounds 1.9 and 1.11 was made by
generating a “second generation analog” of SAM containing a ligatable handle in a way
that would not only provide an improved, efficient coupling reaction catalyzed by
methyltransferase, but also further allow chemical modification for easy detection in
biological complexes. As shown in Figure 1.4, this new SAM-based cofactor mimic was
engineered to bring together the advantage of having an azide functional group for
carrying out subsequent chemoselective ligation reactions on the alkylated product, in
addition to incorporating the amino acid moiety for improved cofactor-MTase
interactions.
This cofactor is expected to yield minimal disruption in the cofactor-MTase
interactions and undergo a rapid coupling reaction with the substrate under biological
conditions. It is anticipated that future biological studies using this new cofactor will
provide essential information necessary to develop novel methods to identify sites of
methylation. At the same time, it is also anticipated to aid in the development of a better
understanding of biological methylation and support future studies of how erroneous
methylation can lead to different stages of diseases.
Figure 1.4. Proposed Structure of Azide-Bearing N-Mustard Cofactor Mimic.
NI
CO2HH3N
N
NN
N
NH2
O
OHHO
N3
H
14

CHAPTER 2. DESIGN AND SYNTHESIS OF AN AZIDE-BEARING N-MUSTARD COFACTOR MIMIC OF SAM
2.1 Introduction
Figure 2.1. 8-Azido-5′-N-Mustard Hydrochloride Salt.
The azide-bearing N-mustard analog 2.1 was designed to serve as a new cofactor
bearing a high structural similarity to SAM. Together, the moieties incorporated into 2.1
are anticipated to form an efficient cofactor for methyltransferase-driven alkylation of
biomolecules, such as small molecules, DNA, or proteins. On the 5′-C of the ribose
sugar, two functionalities were necessary. The first was an amino acid moiety to improve
the MTase-cofactor interaction. Second, the N-mustard moiety was needed as a means to
form an aziridinium ion under physiological conditions, allowing for the coupling of the
cofactor and substrate. Additionally, the incorporation of an azide functionality on the
adenine base provides the advantage of further ligation chemistry, which can be
performed after coupling 2.1 to substrate (as illustrated in Scheme 2.1) to generate a
modified substrate bearing an affinity tag or a fluorophore.
15

2.2 Synthetic Analysis of the Azide-Bearing N-Mustard Adenosine 2.1
Based on the synthetic steps that were used in generating 1.9 and 1.11, a synthetic
analysis to synthesize cofactor mimic 2.1 was constructed (see Scheme 2.2).22,27 The
fundamental plan that underlies the synthesis of 2.1 was to start with commercially
available 2′,3′-isopropylidene adenosine (2.2) and then incorporate necessary
functionalities to mimic SAM. Upon evaluating the final desired structure 2.1, several
transformations were necessary to incorporate the amino acid functionality at the 5′
position of the ribose sugar and to install the azide on the C8 position of the adenine base.
Based on literature precedence, azide installation would need to occur prior to
derivatization of the ribose sugar. Ultimately, it was envisioned that ethanolamine 2.5
would serve as a valuable intermediate to carry out the requisite reductive amination with
aldehyde 2.8 to produce 2.9. This step to incorporate the amino acid functionality was
seen as a potentially difficult step in the synthesis, as the use of sodium
cyanoborohydride was foreseen to reduce the azide functional group to a primary amine.
Thus, an alternate pathway to introduce the azide after incorporating the amino acid
moiety was part of the strategy in developing the first synthetic approach towards 2.1.
Once the core structure of the cofactor mimic was generated (2.9), subsequent iodination
and global deprotection was anticipated to yield the desired product.
Scheme 2.1. Cofactor 2.1 as a Probe for Post-Alkylation Modification.
16

Scheme 2.2. Synthetic Analysis of the Azide-Bearing N-Mustard Cofactor 2.1.
An additional consideration that was taken into account in the synthetic analysis
depicted in Scheme 2.2 was the choice of protecting groups on the ribose sugar.
Choosing an optimal protecting group was very important due to its potential impact on
the solubility of pathway intermediates, such as 2.4, 2.5, and 2.9. Since these protecting
groups needed to be present throughout the entire synthetic pathway, their removal in the
final step, along with the Boc and t-butyl ester, needed to be facile. The tert-
butyldimethylsilyl (TBS) was deemed to be a good starting point due to its recent use in
synthesizing SAM cofactor mimic 1.9.
17

2.3 Synthetic Approach
2.3.1 Initial Synthetic Approach
Two similar synthetic schemes were initially proposed to synthesize azide
containing N-mustard cofactor 2.1. Both schemes were identical, except that in one,
bromination was carried out in the first few steps of the reaction sequence, while in the
other, performed by Charles E. Hendrick, the bromination step was carried out much later
in the scheme. Hendrick successfully synthesized a fully N-protected amine, 5′-(N-Boc-
diaminobutyric acid O-tBu ester)-N-ethanolamine-2′,3′-OTBS adenosine, as shown in
Scheme 2.3.29 However, his attempt to brominate the C8 position of the adenine base
was unsuccessful due to decomposition of material, as well as the formation of a side
product, as indicated by both TLC and 1H NMR analysis. The alternate pathway, where
bromination was carried out in an earlier step, proceeded without difficulty. Based on
these results, bromination of the adenine base was determined to be optimal if performed
prior to derivatizing the ribose sugar with the amino acid functionality.
NHO
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
NHO
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
BrBr2, NaOAc
Dioxane
Scheme 2.3. Unsuccessful Bromination.
Prior to proceeding with the synthesis of cofactor 2.1, preparation of the aldehyde
2.8, a key component for the reductive amination step, needed to be carried out.
Following literature precedence, thioester 2.7 was prepared from a commercially
18

available α-tert-butyl (S)-N-(tert-butoxycarbonyl) aspartate 2.6 by treatment with
ethanethiol, DCC, and DMAP in an overall yield of 94 %, as shown in Scheme 2.4.30 In
the following step, synthesis of aldehyde 2.8 was first attempted using Bergmeier’s
method via a slow addition of triethylsilane to thioester 2.7 in CH2Cl2 containing 10 %
palladium on carbon at 0ºC.30 This procedure gave about 90 % yield of product.
However, the reaction was unable to be re-produced after the first few attempts, as either
unreacted thioester 2.7 or contaminated aldehyde was obtained.
Scheme 2.4. Synthesis of Protected Aldehyde.
While no conclusion was drawn as to why contaminants were present in the
reactions performed in CH2Cl2, an alternate means to reduce the thioester to the aldehyde
was investigated. Ultimately, it was determined that acetone was a better choice of
solvent in this case. Subsequent reduction to aldehyde 2.8 was successfully carried out
using triethylsilane and a catalytic amount of 10 % palladium on carbon in acetone at 0°C,
as done by Fukuyama.31 This highly efficient reduction method provided product in
98 % yield without further purification. Meanwhile, it was observed that the reaction in
acetone was sensitive to environmental conditions, as the reaction only worked during the
last two summer seasons.
19

While preparing the key aldehyde, the synthetic pathway depicted in Scheme 2.5
was followed to generate N-mustard cofactor mimic 2.1. Beginning with commercially
available 2′,3′-isopropylidene adenosine, 2.2, the phthalimide group was introduced at the
5′-position of the ribose sugar using Mitsunobu chemistry.22,32 Subsequent
isopropylidene cleavage using aqueous TFA was carried out and followed by reprotection
of the resulting diol as TBS ethers to generate 2.10.22 As a result of these first three
reactions, phthalimide 2.10 was obtained in 62 % overall yield without the need of any
purification. Bromination was then carried out at the C8 of the adenine base under mildly
acidic conditions, providing 2.11 in 91 % yield.22 Upon treatment of the phthalimide
2.11 with ethylenediamine, unmasking of the primary amine was carried out in 70 %
yield to provide 2.12.22 The resulting amine was utilized in a SN2 reaction with
methylbromoacetate to generate 2.13.23,33 Following reduction of the methylester in the
presence of DIBALH to amino alcohol 2.14,34 reductive amination with aldehyde 2.8, in
the presence of sodium cyanoborohydride and acetic acid, provided the fully N-protected
amino alcohol 2.15.27,33
After obtaining the desired reductive amination product, the azide functionality
was introduced to generate 2.16 using NaN3 at 85ºC via bromine displacement.35 Due to
similar polarities between 2.15 and 2.16, it was difficult to monitor reaction progression,
as the Rf values for both materials were similar during TLC analysis. Therefore, it was
necessary to optimize the reaction conditions to push the reaction to completion and
deplete all residual starting material that could not be separated from the desired product.
Thus, it was determined that the reaction needed to be stirred overnight above 80ºC.
20

DIBALH
Br2, NaOAc
N
NN
N
NH2
O
OTBSTBSO
N
O
O
N
NN
N
NH2
O
OTBSTBSO
H2NBr
N
NN
N
NH2
O
OTBSTBSO
HNHOO
MeOBr
Br
NI
CO2HH3N
N
NN
N
NH2
O
OHHO
NHO
CBocHN
N
NN
N
NH2
O
OTBSTBSO
PPh3, Imidazole, I2 HCl / Dioxane
O
O
CBocHNO
O
O
H
AcOH, NaCNBH3Br
NHO
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
N3
N3
NaN3
H2N NH2
N
NN
N
NH2
O
OO
1. Phthalimide, PPh3, DIAD, THF
2. TFA/H2O/THF3. TBSCl, Imidazole, DMF
N
NN
N
NH2
O
OTBSTBSO
N
O
O
Br
N
NN
N
NH2
O
OTBSTBSO
HNMeO Br
O
THF, 0oC
HO
DMSO, 85oC
TEA, THF
EtOH, 70oCDioxane
CH2Cl2, OoC
NI
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
N3
2.2
2.10
2.11 2.12
2.13 2.14
2.15 2.16
2.12.17
2.8
H
Scheme 2.5. Proposed Synthetic Pathway to Azide-Containing N-Mustard Cofactor 2.1.
21

Additionally, it was necessary to keep the temperature below 95ºC, as the product was
found to decompose at high temperatures.
The final two steps of the synthetic pathway required iodination of alcohol 2.16,
followed by a global deprotection. Based on literature precedence,27 both of these steps
were expected to proceed smoothly in providing the desired azide-containing N-mustard
cofactor 2.1. Although iodination of the amino alcohol 2.16 proceeded easily using a
mixture of iodine, PPh3, and imidazole to successfully obtain 2.17, its deprotection turned
out to be very challenging. It was anticipated upon subjection to HCl-dioxane, that all
protecting groups would be efficiently removed and a liquid-liquid extraction with
CH2Cl2 would suffice as a purification step due to the polar nature of 2.1. Unfortunately,
upon careful analysis of the resulting product mixture, it was determined that incomplete
deprotection of the TBS protecting groups on the ribose sugar resulted. Based on TLC
analysis, the majority of the adenosine material remained in the organic layer during the
extraction step. This layer was found, after NMR analysis, to contain adenosine material
where the Boc and t-butyl protecting groups were removed, but the TBS groups remained.
Consequently, only a small fraction of the material was extracted into the aqueous layer,
which contained some adenosine material and TBS byproducts, and the yield of the final
product (2.1) was low.
In an attempt to determine whether complete deprotection of the TBS protecting
groups could be achieved, several methodologies were explored. The first approach used
two strong acids (concentrated HCl in acetone and TFA in CH2Cl2) as a means to carry
out the global deprotection.36,37 Unfortunately, these methods failed to remove the TBS
groups and most of the unprotected adenosine remained in the organic layer following
22

extraction. A second approach subjected 2.17 to Dowex-H+(a weak acid) in acetonitrile
with gentle stirring for 3 days.38 Again, this method resulted in an incomplete removal of
the TBS groups, as observed by NMR analysis. Lastly, a two-step method was attempted
to efficiently deprotect the silyl groups using TBAF, followed by the deprotection of Boc
and t-butyl upon treatment with HCl in dioxane.22,27 When iodinated product 2.17 was
subjected to TBAF, silyl groups were cleaved off as expected. However, residual TBAF
salt remained in the organic layer even after a liquid-liquid extraction. Column
chromatography of this mixture reduced the amount of TBAF salt, but a significant
amount of this salt was still present in the organic layer, as evidenced by NMR analysis.
The remainder of this salt would present a problem in the following extraction, after the
Boc and t-butyl deprotection, due to its presence in the aqueous layer along with the
desired product. Because of time constraints, further investigation of this problem was
relegated to a later date if it became necessary. Even though TBAF is commonly used
for TBS deprotection in general, it was found that TBAF was not a suitable reagent in
this case. Thus, exploration of an alternate protecting group to replace the TBS group
was necessary at this stage to continue designing a promising pathway to 2.1.
2.3.2 An Alternate Approach Using TES Protecting Group
With the unsuccessful removal of the TBS protecting groups, an alternate
synthetic strategy to obtain compound 2.1 was needed. Both literature precedence27 and
preparation of 2.19 in our laboratory by Dr. Lindsay Comstock has demonstrated that the
triethylsilyl (TES) protecting group can be easily removed using HCl in dioxane, as
shown in Scheme 2.6. Upon carrying out this cleavage, very promising results were
23

obtained by 1H NMR, showing the desired product in high yield without any trace of the
TES protecting group. With the TES group known to be less stable than the TBS
protecting group, an alternate strategy incorporating the TES protecting group was
developed as a means to synthesize the desired azide-containing N-mustard cofactor
mimic 2.1.
Scheme 2.6. Successful Iodination and TES Deprotection.
Using the basis of the synthetic route described in Scheme 2.5, a new synthetic
pathway incorporating the TES protecting group, rather than TBS was developed. As
shown in Scheme 2.7, the synthetic procedures were similar to those described
previously, with the exception of the TES protecting group. Preparation of intermediates
2.20 through 2.25 were facile and proceeded as carried out in the previous synthetic
pathway using the TBS protecting groups (Scheme 2.5). It should be noted that the
installation of the bromine on phthalimide 2.20 required a reaction time of 6 h, whereas
the TBS-containing phthalimide only needed 3 h.
Following the successful reductive amination to yield 2.25, the next step in the
synthesis was to install the azide at the C8 position of the adenine base via a nucleophilic
substitution with sodium azide to generate 2.26. TLC analysis of the reaction revealed
several spots, suggesting the formation of side-products along with 2.26. MS and
24

1H NMR analysis of the reaction material after column chromatography surprisingly
revealed only a small amount of the desired product 2.26 which co-eluted with the mono-
Scheme 2.7. Alternate Synthetic Pathway with TES Protection.
25

TES protected product(s) (2.27a and/or 2.27b), as shown in Scheme 2.8. Additionally,
the corresponding diol 2.28 was also isolated. This reaction was performed multiple
times in order to confirm that the deprotection of the TES protecting groups was a real
phenomenon. With the majority of the generated 2.26 being converted to the mono-TES
protected product(s), as indicated by TLC analysis, this reaction was determined as an
inefficient means to generate 2.26.
Scheme 2.8. Unexpected Mono-TES and Di-TES Deprotection during Azidation.
In an attempt to determine the cause of TES deprotection during the installation of
the azide, it was first hypothesized that the resulting bromide generated from the
displacement reaction may have caused the cleavage of TES groups, this cleavage did not
occur during the bromination of phthalimide 2.20, as shown in Scheme 2.7. To rule out
that high temperature does not affect the stability of the TES ethers, 2.25, in DMF, was
26

subjected to increasing temperature starting at 40ºC with an incremental increase of 20°C
every 2 h. The reaction was stopped after stirring at 87ºC for an hour. Predictably, 1H
NMR showed the presence of both TES ethers on compound 2.25. At this stage, the
cause of this cleavage is still unclear, but it clearly impeded the formation of the desired
product 2.26.
2.3.3 An Improved Method for Installation of Azide Functionality
Based on the difficulties in installation of the azide on the adenine base, an
alternative approach was envisioned to incorporate the azide earlier in the synthetic
pathway prior to derivatizing the ribose sugar at the 5′-position. While this is a feasible
option based on literature precedence,22 one major concern with such a synthetic route
would be whether or not the use of NaCNBH3 would reduce the azide to a primary amine
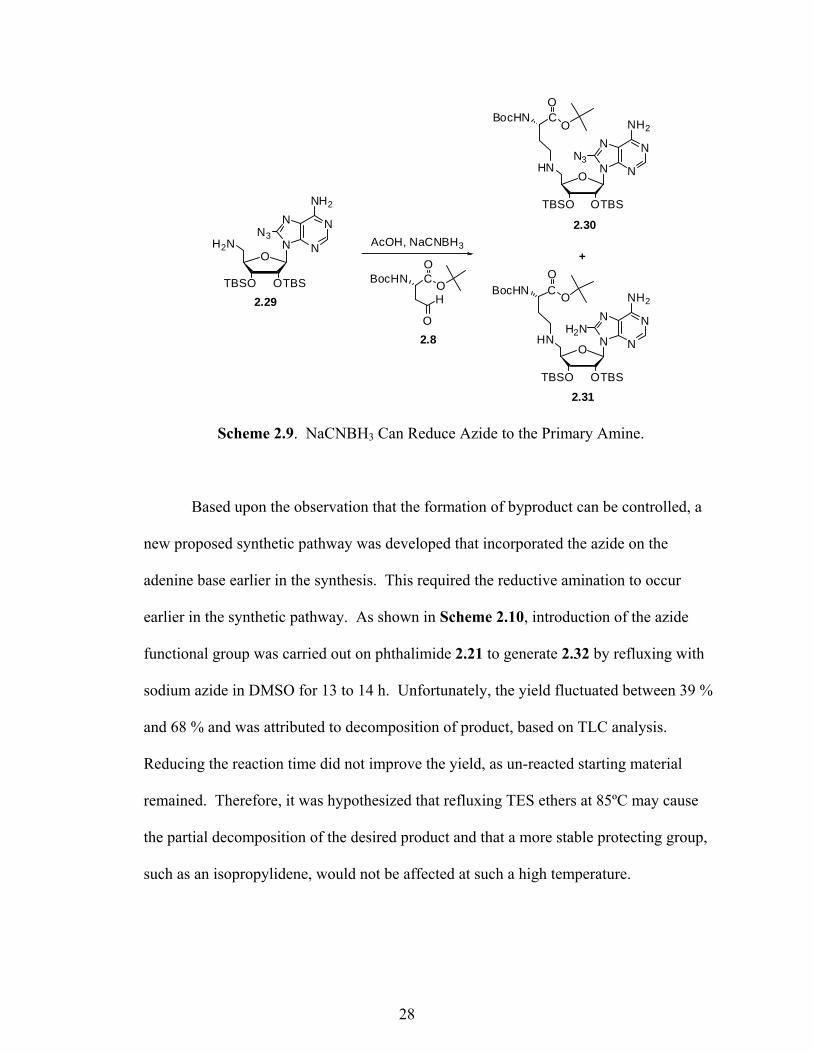
during the reductive amination. To explore this possibility, an available 8-azido-5′-
amino-2′,3′-OTBS adenosine (2.29)22 synthesized by Dr. Lindsay Comstock was used as
a model compound. As shown in Scheme 2.9, reductive amination with aldehyde 2.8 and
NaCNBH3 consequently produced both the desired azide product 2.30 and reduced
product 2.31, in 32 % and 31 % yields, respectively. At this point, optimization of the
reaction conditions to yield more of the desired product and less of the primary amine
side-product was essential. By reducing the number of equivalents of NaCNBH3 from
1.5 to 1.1 equivalents, a 55 % yield of azide 2.30 was achieved.
27
N
NN
N
NH2
O
OTBSTBSO
H2NN3
HN
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
CBocHNO
O
O
H
AcOH, NaCNBH3
N3
HN
CBocHN
N
NN
N
NH2
O
OTBSTBSO
O
O
H2N
+
2.29
2.30
2.31
2.8
Scheme 2.9. NaCNBH3 Can Reduce Azide to the Primary Amine.
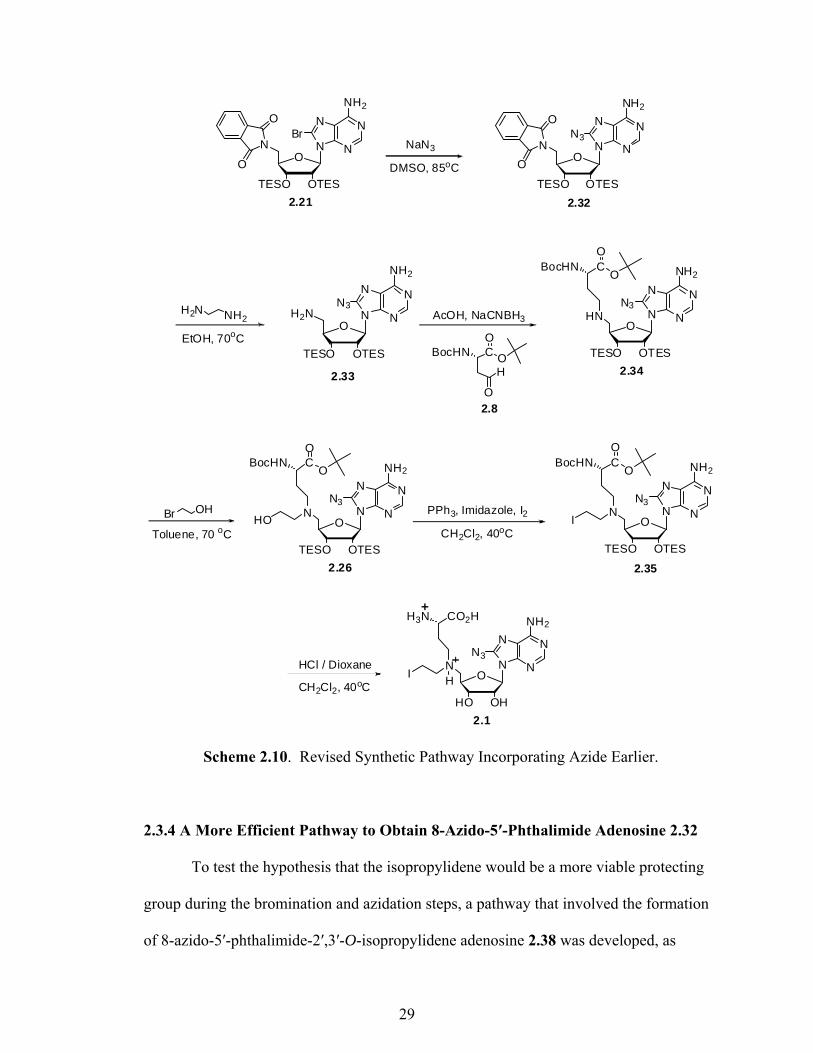
Based upon the observation that the formation of byproduct can be controlled, a
new proposed synthetic pathway was developed that incorporated the azide on the
adenine base earlier in the synthesis. This required the reductive amination to occur
earlier in the synthetic pathway. As shown in Scheme 2.10, introduction of the azide
functional group was carried out on phthalimide 2.21 to generate 2.32 by refluxing with
sodium azide in DMSO for 13 to 14 h. Unfortunately, the yield fluctuated between 39 %
and 68 % and was attributed to decomposition of product, based on TLC analysis.
Reducing the reaction time did not improve the yield, as un-reacted starting material
remained. Therefore, it was hypothesized that refluxing TES ethers at 85ºC may cause
the partial decomposition of the desired product and that a more stable protecting group,
such as an isopropylidene, would not be affected at such a high temperature.
28
H2N NH2
CBocHNO
O
O
H
AcOH, NaCNBH3 HN
CBocHN
N
NN
N
NH2
O
OTESTESO
O
N3
Br OH
Toluene, 70 oC
NI
CO2HH3N
N
NN
N
NH2
O
OHHO
PPh3, Imidazole, I2
HCl / Dioxane
NHO
CBocHN
N
NN
N
NH2
O
OTESTESO
O
N3
N3
EtOH, 70oC
O
O
CH2Cl2, 40oC
NI
CBocHN
N
NN
N
NH2
O
OTESTESO
O
N3
O
2.33 2.34
2.26 2.35
2.1
N
NN
N
NH2
O
OTESTESO
H2NN3
H
2.8
CH2Cl2, 40oC
N
NN
N
NH2
O
OTESTESO
N
O
O
Br
2.21 2.32
N
NN
N
NH2
O
OTESTESO
N
O
O
N3NaN3
DMSO, 85oC
Scheme 2.10. Revised Synthetic Pathway Incorporating Azide Earlier.
2.3.4 A More Efficient Pathway to Obtain 8-Azido-5′-Phthalimide Adenosine 2.32
To test the hypothesis that the isopropylidene would be a more viable protecting
group during the bromination and azidation steps, a pathway that involved the formation
of 8-azido-5′-phthalimide-2′,3′-O-isopropylidene adenosine 2.38 was developed, as
29

shown in Scheme 2.11. Generation of this intermediate was carried out easily using the
chemistry previously employed to install the bromide and azide. Additionally, 2.38
underwent facile isopropylidene deprotection and subsequent re-protection as the TES
ethers, to produce 2.32. This pathway provided 2.32 in a yield of 86 %, which is much
higher than when the TES protecting groups were present.
Scheme 2.11. Improved Synthesis of 8-Azido-5′-Phthalimide Adenosine 2.32.
After obtaining 2.32 in a sufficient yield, continuation of the synthetic pathway
(see Scheme 2.10) upon reduction of phthalimide 2.32 to primary amine 2.33 was carried
out in the presence of ethylenediamine. Introduction of the amino acid moiety via
reductive amination was then conducted via treatment with aldehyde 2.8 and NaCNBH3.
30

As a result, 2.34 was obtained in 51 % yield after the elimination of the primary amine
byproduct using column chromatography. Upon successful addition of the amino acid
moiety to the structure, 5′-alkylation of 2.34 was performed using a haloethanol.22
Reflux of compound 2.34 with 10 equivalents of iodoethanol at 70ºC for 2 days gave a
low yield of desired product 2.26. In addition to unreacted starting material, the
formation of a secondary alkylated product, which resulted from the addition of a second
ethyl alcohol to the nitrogen on the 5´-position of the ribose sugar, was also observed by
TLC. Purification of 2.26 by column chromatography proved to be difficult, as co-
migration of the product, starting material, and the secondary alkylated product occurred
in the column. Column optimization by pre-treating the silica with TEA gave 2.26 in
20 % yield.
With the difficulty in purifying 2.26, as well as the low yield of product, it was
concluded that iodoethanol perhaps was not the best haloethanol to utilize for the
requisite alkylation. Upon turning to bromoethanol as an alternate reagent, product yields
were more promising and reaction optimization was carried out by varying the equivalent
amounts of alkylating reagent. Overall, it was determined that the optimal yield of 2.26
could be obtained with 13 equivalents of bromoethanol at 70ºC in the presence of N,N-
diisopropylethylamine. Upon stirring the reaction for 1 day while monitoring by TLC, it
was determined that most of the starting material was converted to product, with an
estimated ratio of product to secondary alkylated product that was approximately 1:1. It
was also observed that as the reaction stirred for longer periods of time, a larger
percentage of secondary alkylated product tended to form; therefore, a reaction time of
one day was found to be key to allow all of the starting material to react, while forming
31

the least amount of secondary alkylated product. Ultimately, 2.26 was obtained in a
higher yield of 55 % after purification by column chromatography.
Having successfully synthesized 2.26, iodination followed by global deprotection
was attempted. Although literature indicated that the iodination and deprotection steps
should occur at 0°C,27 several difficulties were encountered under these conditions.
Iodination consistently resulted in unreacted starting material and was independent of
reaction time, even at room temperature. Interestingly, it was found that a gentle reflux
(40°C) for 30 min facilitated iodination in a near quantitative yield, as determined by MS.
The global deprotection of the successfully iodinated product was not quantitative, as
side products were present. The identities of the t-butyl containing byproduct 2.39, in
addition to desired product 2.1, were confirmed by both NMR and MS analysis (see
Figure 2.2).
Figure 2.2. The t-Butyl-Containing Byproduct of Global Deprotection at Room Temperature.
Additionally, analysis of this mixture of products by LC/MS showed only a small
trace of 2.1 and mostly two hydrolyzed compounds: hydrolyzed form of the t-butyl-
containing product (2.40) and hydrolyzed form of the desired product (2.41), as shown in
Figure 2.3. Based on these results, material obtained from the reaction was found to be
32

unstable and easily underwent hydrolysis in an acidic solution of 0.1 % formic acid in
water. The presence of hydrolyzed byproduct 2.40 confirmed the formation of 2.39 as a
side product of an incomplete global deprotection, prior to LC/MS analysis. Therefore,
further optimization of the deprotection reaction conditions was necessary to eliminate
the formation of 2.39. Ultimately, full deprotection of the iodinated product was
achieved under a gentle reflux (at 40ºC) in CH2Cl2 for 3 h.
Figure 2.3. Hydrolyzed Compounds Formed during MS.
2.3.5 NMR, MS, and HPLC Analysis of 2.1
After successfully optimizing the reaction conditions for both the iodination and
deprotection, final product 2.1 was analyzed using a combination of 1H-NMR, MS, and
HPLC. Analysis of the 1H-NMR spectrum showed that 2.1 was 82 % pure, based on
peak integration. In addition to 2.1, a side product was indicated by a smaller set of
peaks. Further analysis of the product following MS indicated the desired mass of 2.1,
along with several other masses which were hypothesized to correspond to the observed
side products and product fragments. Careful analysis indicated the presence of
aziridinium 2.42 (Figure 2.4), as well as the hydrolyzed product 2.41 (see Figure 2.3).
33

Figure 2.4. Formation of Aziridinium (2.42) and Methyl Ether (2.43) during MS.
Due to the highly reactive nature of 2.1, a small amount of both the aziridinium
and hydrolyzed product was expected, as 2.1 can be easily converted to aziridinium 2.42
under physiological pH. Additionally, it was also observed that when the MS sample was
prepared in methanol, the mass of methyl ether 2.43 (Figure 2.4) was present. However,
when analyzed in an aqueous 0.1 % TFA matrix, this mass was absent from the spectrum.
As a confirmation that this product was the result of MS analysis, examination of the 1H-
NMR spectrum of 2.1 in deuterated methanol did not indicate that 2.43 was present.
Based on this evidence, it was concluded that the conditions during MS analysis initiated
the formation of methyl ether 2.43. Of all the mass peaks present in the spectrum, the
only mass that was not accounted for was at 471.3. Although the identity of this product
was not known, it was hypothesized that the byproduct observed by 1H-NMR may bear
that mass. As a means to improve the purity of 2.1, crystallizations were attempted using
the vial-in-a-vial diffusion method with solvent combinations of MeOH and EtOAc at
0°C and -20°C, but this method was unsuccessful. Many factors such as the choice of
solvents, solvent viscosity, temperature, and others can affect the crystallization process.
Various tests on these conditions were impractical to perform due to a limited amount of
2.1 and time.
34

At the same time, RP-HPLC was used to analyze and quantify 2.1. Experiments
were performed using a reverse-phase C18 analytical column. Initially, analytical
chromatographic conditions for the separation of 2.1 were investigated empirically by
employing the use of different sized analytical columns, in combination with various
gradient elution systems, in an attempt to find an optimum separation method.
Experiments were carried out using a mobile phase of 0.1 % formic acid/acetonitrile and
indicated that 2.1 quickly hydrolyzed over the course of 40 min and was completely
degraded after 1.5 h. Ultimately, it was found that switching to a 0.1 % TFA/acetonitrile
system significantly slowed the hydrolysis process, such that most of the material was
degraded after 24 h. Based on these results, it was determined that the material must be
kept cold and in the solid state for long-term storage. Additionally, acidic solutions of
2.1 should be made fresh right before their use in a biochemical assay.
The development of an analytical-scale HPLC separation method required the
optimization of several parameters. During the first several attempts, analysis of 2.1
using a short analytical column (C18, 5µm, 4.6 x 100 mm) resulted in a poor separation
of 2.1 from other components. Variations in the gradient system were explored, but no
significant effect on the separation of 2.1 was observed. Upon switching to a longer
column (C18, 5µm, 4.6 x 250 mm), the resulting chromatograms indicated the presence
of multiple peaks with baseline resolution, but were not spaced far enough for practical
fraction collection. Upon decreasing the amount of ACN used in the gradient elution, a
reasonable analytical separation method was devised, as shown on chromatogram I of
Figure 2.5. This method achieved a chromatographic profile of the sample that exhibited
the best separation of the material. Each of the major peaks (A, B, C, D, E, and F) was
35

collected, immediately frozen to halt hydrolysis, and then lyophilized for identification
by MS.
Figure 2.5. Analytical HPLC Chromatograms of 2.1 before (I) and after Isolation (II). Eluent: 0.1 % TFA in H2O /ACN gradient; Flow: 1.0 mL/min; UV Detector @ 254 nm.
Careful analysis of the mass spectrum for each of the collected peaks led to the
conclusion that peak D was the unknown side product carrying a mass of 471.3 and E
was the desired product 2.1. Other peaks, including A, B, C, and F, were determined to
36

be contaminants from the MilliQ water, such as phthalates (plasticizers), which have been
reported to be commonly found in mobile phase solvents.39 This finding was consistent
with results from both NMR and MS analysis of the starting material prior to HPLC
separation, as well as the MS analysis of a MilliQ water sample that mimicked HPLC
conditions. Based on peak areas of E and D, the ratio of 2.1 to unknown side product
was calculated to be 58:42 (58 % 2.1). This was somewhat lower than the 82 % purity of
2.1 calculated based upon NMR integration, but was anticipated due to natural hydrolysis
of 2.1 under HPLC conditions.
Ultimately, this separation method has allowed for the successful isolation of 2.1
from the analytical column. As shown in chromatogram II of Figure 2.5, the collected
product was nearly pure and provides evidence that, in the near future, this separation
methodology can be optimized and employed for a semi-preparative HPLC isolation. On
a larger scale, it is anticipated that isolation of sufficient amounts of 2.1 for use in future
biological assays will be possible.
2.4 Biochemical Analysis of 2.1 Using a Restriction/Protection Assay
To analyze the ability of 2.1 to function as a cofactor mimic of SAM, a
restriction/protection assay was performed to determine whether 2.1 is capable of
undergoing M.TaqI-dependent alkylation with circular pUC19 plasmid DNA bearing a
5′-TCGA-3′ recognition sequence. If so, the effectiveness of 2.1 can be evaluated via a
comparison to novel cofactor 1.11.
The general idea of this assay is to observe how the activity of the restriction
enzyme R.TaqI is affected as the concentration of cofactor 2.1 is varied in the presence of
37

the methyltransferase M.TaqI. If 2.1 is an effective cofactor for M.TaqI, it will be
transferred onto the DNA, interfering with the activity of R.TaqI. As a result, restriction
at R.TaqI recognition sites will decrease, resulting in a smaller concentration of DNA
fragments and the retention of full-length DNA.
Analysis of the experiment following agarose gel electrophoresis, as shown in
Figure 2.6, indicated that 2.1 was as effective as 1.11 in undergoing M.TaqI-dependent
DNA alkylation. In the absence of M.TaqI, no protection of the DNA was observed, as
evidenced by the presence of three smaller DNA fragments (lane 3 and 7). When the
concentration of 1.11 and 2.1 were increased from 10 to 100 µM, an increase in DNA
protection was observed. The bands of the smaller restriction fragments were less intense
and the band corresponding to fully-linearized DNA was more pronounced (lanes 4-6 and
8-10) compared to the control lanes 2, 3, and 7. With comparable amounts of full-length
pUC19 in both lanes 6 and 10 (as evident by an increased protection from R.TaqI), it was
concluded that the efficiency of 2.1 was comparable to 1.11. This result was expected, as
the only structural difference between 2.1 and 1.11 is the addition of the azide
functionality and the presence of this azide should not significantly affect the binding
interaction of 2.1 and M.TaqI.
38

Figure 2.6. M.TaqI Restriction/Protection Assay with Increasing Cofactor. Reaction mixtures were prepared by addition of appropriate stock solutions to a total volume of 20 μL containing R.EcoRI-linearized pUC19 (in duplex), M.TaqI, 1.11, and 2.1 in buffer at the appropriate concentration. Gel content 1. 100 bp. ladder; 2. Linearized pUC19; 3. DNA, 100 μM 1.11, R.TaqI; 4. DNA, 10 μM 1.11, M.TaqI, R.TaqI; 5. DNA, 50 μM 1.11, M.TaqI, R.TaqI; 6. DNA, 100 μM 1.11, M.TaqI, R.TaqI; 7. DNA, 100 μM 2.1, R.TaqI; 8. DNA, 10 μM 2.1, M.TaqI, R.TaqI; 9. DNA, 50 μM 2.1, M.TaqI, R.TaqI; 10. DNA, 100 μM 2.1, M.TaqI, R.TaqI.
2.5 Conclusions
In conclusion, the 8-azido-N-mustard cofactor 2.1 was successfully synthesized
based on the structural advantages exhibited in the two reported cofactors, 1.9 and 1.11.
Along with the N-mustard that functions to make 2.1 an alkylating agent, the amino acid
functionality was incorporated on the ribose sugar to enhance the binding of the cofactor
39

to the MTase, resulting in an efficient MTase-dependent alkylation with DNA. Having
an azide functionality on modified DNA allows ligation chemistry to be applied in the
study of detection sites of methylation.
In the synthetic work of generating 2.1, the use of protecting groups and several
key transformations were investigated and found to be vital in obtaining the 2.1. After
the deprotection of isopropylidene, triethylsilyl (TES) protecting group was found to
function best. TES not only provides good solubility and stability to most of the
intermediates, but also underwent a global deprotection reaction together with Boc and t-
butyl protecting groups without much difficulty. Isopropylidene, a more stable protecting
group than TES, was also found to be of great use in the first few steps to generate 2.32
in a relatively high yield. In performing modifications on the C8 of the adenine base,
bromination followed by azidation was determined to be necessary prior to the
derivatization of the ribose sugar with N-mustard and amino acid functionalities.
Interestingly, the last two steps in generating 2.1, iodination and deprotection, were found
to be time and temperature dependent, as both reactions need to be heated to 40°C for a
specific period of time.
Even though synthesized 2.1 was not pure, it is hypothesized that once the
identity of this byproduct can be determined, it may be possible for a synthetic strategy to
be devised in order to hinder its formation and, therefore, increase the reaction yield of
2.1. Due to time constraints, however, the identity of this compound is still under
investigation. Meanwhile, based on the achieved analytical separation of 2.1, the
development of a method for preparative purification by HPLC will be underway.
40

Additionally, the results from the restriction/protection assay revealed that 2.1
was enzymatically transferred by M.TaqI to plasmid DNA. Similarly to the previously
reported N-mustard cofactor 1.11, 2.1 also functions as an efficient cofactor for DNA
MTase M.TaqI. In the future, additional experiments will be designed and performed on
smaller substrates, such as oligonucleotides, to confirm that DNA protection by 2.1 is a
MTase-dependent event. These experiments will also serve to investigate if any other
non-enzymatic modification is occurring, beside the MTase-dependent alkylation. Since
2.1 features an azide functionality, DNA modified by 2.1 is hypothesized to be capable of
undergoing subsequent ligation chemistry. Therefore, it is anticipated that 2.1 will be
useful as a biochemical tool which will aid in the future detection of methylation sites.
Ultimately, it is anticipated that this new cofactor will have utility in future work to
elucidate the mechanism of methylation in a complex, biological system. More
information gathered from the biological applications of 2.1 would help to gain a better
understanding of the relationship between diseases and aberrant DNA and protein
methylation.
2.6 Synthetic Procedures and Compound Characterizations
General:
All reagents were purchased from commercial sources and used without
additional purification. All reactions were carried out under an inert atmosphere of argon
unless otherwise indicated. Anhydrous solvents were obtained from a Meyer solvent
system, except for DMSO and toluene, which were purchased. Column flash
chromatography was performed on silica gel obtained from Sorbtech (60 Ǻ, 230-400
41

mesh). Analytical TLC was conducted on silica gel plates (Sorbent Technologies) with
detection by ninhydrin or uber and/or UV light. 1H-NMR and 13C-NMR spectra were
recorded on a Bruker 300 MHz spectrometer using solvent as the internal reference.
Chemical shifts are reported in ppm, in δ units. LC/MS data was obtained from Agilent
1100 series LC/MSD. HRMS-ESI spectra were obtained from COSMIC Lab (Old
Dominion University, Norfolk, VA) on a Bruker 12 Tesla APEX –Qe FTICR-MS with an
Apollo II ion source.
Analytical HPLC was performed using a Waters 1525 binary pump with 717 plus
autosampler, a 2489 UV/VIS detector set to detect absorbance at 254 nm, and Empower
Pro software. An AlltimaTM C18 column (100 Å, 5 μm, 250 x 4.6 mm, Grace Discovery)
was used. All mobile phases were filtered through a 0.22 µm Durapore membrane filter
prior to use. Compound separation utilized a gradient system comprising of 0.1 % TFA
in water (solvent A) and HPLC grade ACN (solvent B) using a flow rate of 1.0 mL/min.
The gradient was run isocratically with 5 % B for 2 min followed by a linear gradient of
5-10 % B over a 16-min period. The gradient was then increased to 100 % B over the
next 1 min and ran isocratically for an additional 6 min. Under these conditions, azide-
bearing N-mustard adenosine (2.1) eluted at 17 min.
Restriction/Protection Assay:
Commercially-available pUC19 (New England Biolabs) was linearized with
R.EcoRI (New England Biolabs), followed by heat-inactivation at 65ºC for 15 min prior
to further plasmid use (carried out by Dr. Lindsay Comstock) according to
manufacturer’s protocol (final concentration of 0.2 μg/μL or 114 nM). Following a
42

reported procedure,23,24 assay reaction mixtures were prepared by the addition of
appropriate stock solutions to a total volume of 20 μL in M.TaqI buffer (supplied by New
England Biolabs). The final DNA concentration was 28.5 nM; the final concentration of
M.TaqI (New England Biolabs) was 200 nM and cofactor concentration ranged from 10
M to 100 M. All reactions were heated at 65ºC for 4 h followed by cooling to rt.
Methyltransferase-dependent DNA alkylation was analyzed by the addition of R.TaqI
(2U in an additional 10 μL M.TaqI buffer), followed by incubation at 65°C for 1 h. Upon
cooling to rt, Proteinase K (New England Biolabs) (0.02U in 5 μL H2O) was added to
each reaction and incubated at 37ºC for 1h. Agarose gel loading dye was added to each
reaction and the extent of alkylation was visualized by electrophoresis on a 2 % agarose
gel ran at 130V for 1 h.
Compound Characterization:
5′-Phthalimide-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine (2.10)
Following previously reported procedures,22,32 phthalimide (1.974 g, 13.420 mmol) and
PPh3 (3.417 g, 13.029 mmol) were added to commercially available 2′,3′-isopropylidene
adenosine 2.2 (4.004 g, 13.029 mmol) in dry 45 mL THF. After stirring at rt for 10 min,
DIAD (2.565 mL, 13.029 mmol) was added to the mixture and stirred an additional 2.5 h.
The white precipitate was filtered off and washed with cold Et2O to yield the product that
was taken forward. 5′-Phthalimide-5′-deoxy-2′,3′-isopropylidene adenosine (4.010 g,
9.189 mmol) was dissolved in 90 mL 3:1:1 TFA/H2O/THF and stirred for 2 h. The
solvent was evaporated in vacuo and co-evaporated with EtOH (x3). The resulting
material was dissolved in 20 mL dry DMF, followed by the addition of imidazole (3.128
43

g, 45.945 mmol) and TBSCl (3.047 g, 20.216 mmol). The reaction was stirred overnight,
followed by aqueous workup (NH4Cl (x2), EtOAc, brine), dried over Na2SO4, and
evaporated in vacuo. Column chromatography (4:4:2:1 Hexanes/EtOAc/CH2Cl2/MeOH)
yielded product (5.02 g, 62 %). The product was confirmed by spectroscopic comparison
to previously reported 1H NMR data.
8-Bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine (2.11)
Compound was synthesized using a previously reported procedure:22 To 5′-phthalimide-
5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine 2.10 (2.007 g, 3.212 mmol) in
48.2 mL 7:4 dioxane/0.5 M NaOAc (pH 5.2) was added Br2 (331 µL, 6.425 mmol). The
reaction was stirred for 3 h, followed by aqueous workup (Na2S2O3, CH2Cl2, brine), dried
over Na2SO4, and evaporated in vacuo. Column chromatography (1:1 Hexanes/EtOAc to
8:8:2:1 Hexanes/EtOAc/CH2Cl2/MeOH) yielded product (2.053 g, 91 %). The
compound was identified by spectroscopic confirmation to previously reported 1H NMR
data.
8-Bromo-5′-amine-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine (2.12)
Following a general procedure,22 ethylenediamine (668 µL, 9.984 mmol) was added to 8-
bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine 2.11
(1.4053 g, 1.997 mmol) in 65.5 mL EtOH. The reaction was heated at 70ºC and stirred
for 5 h. The solvent was evaporated in vacuo and chromatographed on silica pre-treated
with 2 % TEA (4:2:1 EtOAc/CH2Cl2/MeOH) to yield product (0.8071 g, 70 %). 1H
NMR (CDCl3) δ 8.29 (s, 1H), 5.99 (d, J = 6.4 Hz, 1H), 5.57 (bs, 2H), 5.26 (dd, J = 6.6,
44

4.7 Hz, 1H), 4.41 (dd, J = 4.6, 2.4 Hz, 1 H), 4.09 (m, 1H), 3.04 (m, 2H), 1.65 (bs, 2H),
0.96 (s, 9H), 0.80 (s, 9H), 0.14 (s, 3H), 0.14 (s, 3H), -0.08 (s, 3H), -0.43 (s, 3H).
8-Bromo-5′-amino-acetic acid methyl ester-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl)
adenosine (2.13)
Compound was prepared using a general procedure:23,33 To 8-bromo-5′-amine-5′-deoxy-
2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine 2.12 (0.8255 g, 1.439 mmol) in 7.93 mL
dry THF was added TEA (240 µL, 1.727 mmol). Methylbromoacetate (164 µL, 1.727
mmol) in 4 mL dry THF was then added drop wise to the solution. The reaction mixture
was stirred overnight. The resulting precipitate was filtered off and the organic was
evaporated in vacuo. Column chromatography (3:1 EtOAc/ CH2Cl2 to 15:5:1
EtOAc/CH2Cl2/MeOH) yielded product (0.7697 g, 83 %). 1H NMR (CDCl3) δ 8.28 (s,
1H), 5.98 (d, J = 6.7 Hz, 1H), 5.48 (bs, 2H), 5.30 (dd, J = 6.7, 4.7 Hz, 1H), 4.38 (dd, J =
4.7, 2.2 Hz, 1H), 4.18 (m, 1H), 3.70 (s, 3H), 3.46 (d, J = 2.2 Hz, 2H), 2.99 (dd, J = 12.3,
3.7 Hz, 1H), 2.92 (dd, J = 12.3, 6.3 Hz, 1H), 1.58 (bs, 1H), 0.95 (s, 9H), 0.79 (s, 9H),
0.13 (s, 3H), 0.13 (s, 3H), -0.10 (s, 3H), -0.45 (s, 3H).
8-Bromo-5′-ethanolamine -5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine
(2.14)
Compound was synthesized in adaptation of a procedure reported in the literature:34 To 8-
bromo-5′-amino-acetic acid methyl ester-5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl)
adenosine 2.13 (0.7697 g, 1.192 mmol) in 25.6 mL dry THF at 0°C was slowly added 1M
DIBAL-H (5.96 mmol). The reaction was kept cold for a few min, then warmed to rt,
45

and stirred for an additional 5 h. Saturated potassium sodium tartrate tetrahydrate (25.6
mL) was added to the reaction and stirred vigorously overnight. The organic was washed
(sat. Rochelle’s salt, H2O, EtOAc, brine), dried over Na2SO4, and evaporated in vacuo to
obtain product (0.7139 g, 97 %). 1H NMR (CDCl3) δ 8.27 (s, 1H), 6.00 (d, J = 7.0 Hz,
1H), 5.59 (bs, 2H), 5.29 (dd, J = 6.9, 4.7 Hz, 1H), 4.35 (dd, J = 4.6, 1.9 Hz, 1H), 4.21 (m,
1H), 3.67 (t, J = 5.2 Hz, 2H), 2.95 (m, 2H), 2.83 (ddt, J = 42, 12.1, 5.2 Hz, 2H), 1.90 (bs,
1H), 0.96 (s, 9H), 0.78 (s, 9H), 0.14 (s, 3H), 0.13 (s, 3H), -0.11 (s, 3H), -0.44 (s, 3H).
α-tert-Butyl β-S-ethyl(S)-N-(tert-butoxycarbonyl) thioaspartate (2.7)
Compound was prepared using a previously reported procedure:30 To a solution of α-
tert-butyl (S)-N-(tert-butoxycarbonyl) aspartate 2.6 (2.166 g, 7.485 mmol) in 14.8 mL
dry CH2Cl2 was added DCC (1.853 g, 8.981 mmol), ethanethiol (1.56 mL, 22.45 mmol),
and DMAP (0.0914 g, 0.7485 mmol). The reaction mixture was stirred at rt for 3 h,
filtered, evaporated in vacuo, and column chromatographed (10% EtOAc in Hexanes) to
give product as a clear oil (2.342 g, 94 %). The product was confirmed by spectroscopic
comparison to previously reported 1H NMR data.
tert-Butyl (S)-2-[N-(tert-butoxycarbonyl)amino]-4-oxobutanoate (2.8)
In adaptation of a previously reported procedure,31 a solution of α-tert-butyl β-S-
ethyl(S)-N-(tert-butoxycarbonyl) thioaspartate 2.7 (0.5809 g, 1.742 mmol) in 19 mL
acetone at 0ºC was added 10 % palladium on carbon (74.2 mg, 0.0697 mmol).
Triethylsilane (835 µL, 5.226 mmol) was slowly added to the solution. After the reaction
mixture had been stirred for 45 min at 0ºC, it was filtered through a short-pad of Celite.
46

The filtrate was evaporated in vacuo to afford the product as a colorless oil (0.467 g,
98 %). Upon drying under a vacuum for several days, this oil turned into a solid. The
product was confirmed by spectroscopic comparison to previously reported 1H NMR
data.30
8-Bromo-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-ethanolamine-5′-deoxy-
2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine (2.15)
Following a general procedure:27,33 To a solution of 8-bromo-5′-ethanolamine -5′-deoxy-
2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine 2.14 (0.3995 g, 0.6467 mmol) and tert-
butyl (S)-2-[N-(tert-butoxycarbonyl)amino]-4-oxobutanoate 2.8 (0.1607 g, 0.5879 mmol)
in 2.82 mL dry MeOH was added NaCNBH3 (0.0554 g, 0.8819 mmol) and AcOH (33.7
µL, 0.5879 mmol). The reaction mixture was stirred overnight at rt. After diluting the
reaction with EtOAc and NaHCO3, the organic layer was washed with NaHCO3, dried
over Na2SO4, and evaporated in vacuo. Column chromatography (10:4:2:1 Pet
Ether/EtOAc/CH2Cl2/MeOH) yielded product (0.3529 g, 72 %). 1H NMR (CDCl3) δ
8.27 (s, 1H), 5.94 (d, J = 5.2 Hz, 1H), 5.67 (bs, 2H), 5.43 (bd, J = 7.8 Hz, 1H), 5.31 (t, J =
4.9 Hz, 1H), 4.39 (t, J = 4.1 Hz, 1H), 4.18 (m, 2H), 3.52 (m, 2H), 2.95 (m, 2H), 2.63 (m,
4H), 1.94 (m, 1H), 1.70 (m, 1H), 1.42 (s, 9H), 1.42 (s, 9H), 0.96 (s, 9H), 0.81 (s, 9H),
0.16 (s, 3H), 0.14 (s, 3H), -0.06 (s, 3H), -0.32 (s, 3H).
47

8-Azido-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-ethanolamine-5′-deoxy-
2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine (2.16)
Following a reported preparation,35 NaN3 (0.0558 g, 0.8580 mmol) was added to a
solution of 8-bromo-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-ethanolamine-
5′-deoxy-2′,3′-bis-(O-tert-butyldimethylsilyl) adenosine 2.15 (0.1877 g, 0.2145 mmol) in
1.82 mL DMSO. The reaction mixture was heated to 85°C and stirred overnight,
followed by an aqueous workup (NaHCO3, CH2Cl2, brine). Material was dried over
Na2SO4, evaporated in vacuo, and column chromatographed (8:4:2:1 Pet Ether/
EtOAc/CH2Cl2/MeOH) to yield product (0.0865 g, 48 %). 1H NMR (CDCl3) δ 8.21 (s,
1H), 5.72 (d, J = 5.0 Hz, 1H), 5.57 (bs, 2H), 5.48 (bd, J = 7.9 Hz, 1H), 5.08 (t, J = 4.4 Hz,
1H), 4.38 (t, J = 3.6 Hz, 1H), 4.13 (m, 2H), 3.51 (m, 2H), 2.90 (m, 2H), 2.62 (m, 4H),
1.92 (m, 1H), 1.67 (m, 1H), 1.41 (s, 18H), 0.94 (s, 9H), 0.81 (s, 9H), 0.13 (s, 3H), 0.12 (s,
3H), -0.06 (s, 3H), -0.28 (s, 3H).
5′-Phthalimide-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.20)
Following a reported procedure:22,32,40 To the commercially available 2′,3′-isopropylidene
adenosine 2.2 (1.005 g, 3.257 mmol) in dry 11 mL THF was added phthalimide (0.4953 g,
3.366 mmol) and PPh3 (0.8522 g, 3.250 mmol). After stirring at rt for 10 min, DIAD
(640 µL, 3.250 mmol) was added to the mixture and stirred an additional 2.5 h. The
white precipitate was filtered off and washed with cold Et2O to yield product that was
taken forward. 5′-Phthalimide-5′-deoxy-2′,3′-isopropylidene adenosine (1.175 g, 2.693
mmol) was dissolved in 27 mL 3:1:1 TFA/H2O/THF and stirred for 2 h. The solvent was
evaporated in vacuo and co-evaporated with EtOH (x3). The resulting material was
48

dissolved in 5.93 mL dry DMF, followed by the addition of imidazole (0.9167 g, 13.47
mmol) and TESCl (994 µL, 5.925 mmol). The reaction was stirred overnight, followed
by aqueous workup (NH4Cl (x2), EtOAc, brine), dried over Na2SO4, and evaporated in
vacuo. Column chromatography (4:4:2:1 Hexanes/EtOAc/CH2Cl2/MeOH) yielded
product (1.148 g, 68 %). 1H NMR (CDCl3) δ 8.12 (s, 1H), 8.00 (s, 1H), 7.86 (m, 2H),
7.73 (m, 2H), 5.86 (d, J = 6.4 Hz, 1H), 5.84 (bs, 2H), 5.28 (dd, J = 6.3, 4.1 Hz, 1H), 4.32
(m, 2H), 4.24 (dd, J = 13.7, 7.2 Hz, 1H), 3.93 (dd, J = 13.7, 5.4 Hz, 1H), 0.93 (t, J = 7.8
Hz, 9H), 0.80 (t, J = 7.9 Hz, 9H), 0.61 (q, J = 8.0 Hz, 6H), 0.38 (qq, J = 15, 7.6 Hz, 6H);
13C NMR(CDCl3) δ 167.0, 154.8, 151.7, 148.7, 139.8, 133.3, 131.2, 122.7, 120.0, 88.7,
82.8, 73.9, 73.2, 40.3, 7.5, 7.2, 5.6, 5.2.
8-Bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.21) Compound was synthesized using a previously reported procedure:22 To 5′-phthalimide-
5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine 2.20 (0.9923 g, 1.588 mmol) in 23.8 mL 7:4
dioxane/0.5 M NaOAc (pH 5.2) was added Br2 (164 µL, 3.176 mmol). The reaction was
stirred for 6 h, followed by aqueous workup (Na2S2O3, CH2Cl2, brine), dried over Na2SO4,
and evaporated in vacuo. Column chromatography (8:1 EtOAc/CH2Cl2) yielded product
(0.6953 g, 62 %). 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.82 (m, 2H), 7.72 (m, 2H), 5.96 (d, J
= 6.8 Hz, 1H), 5.93 (bs, 2H), 5.74 (dd, J = 6.7, 4.5 Hz, 1H), 4.51 ( dd, J = 4.3, 1.2 Hz,
1H), 4.31 (m, 2H), 3.91 (dd, J = 13.0, 3.1 Hz, 1H), 0.90 (t, J = 7.9 Hz, 9H), 0.79 (t, J =
7.9 Hz, 9H), 0.59 (q, J = 8.0 Hz, 6H), 0.33 (qq, J = 15, 7.6 Hz, 6H); 13C NMR(CDCl3) δ
167.0, 153.5, 151.5, 149.8,133.2, 131.2, 128.0, 122.6, 119.8, 89.9, 83.8, 73.9, 71.2, 39.7,
7.5, 7.2, 5.7, 5.2.
49

8-Bromo-5′-amine-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.22)
Following a general procedure:22 To 8-bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-
triethylsilyl) adenosine 2.21 (0.509 g, 0.7232 mmol) in 23.5 mL EtOH was added
ethylenediamine (242 µL, 3.616 mmol). The reaction was heated to 70°C and stirred for
5 h. The solvent was evaporated in vacuo and chromatographed on silica pre-treated with
2 % TEA (4:2:1 EtOAc/CH2Cl2/MeOH) to yield product (0.3059 g, 74 %). 1H NMR
(CDCl3) δ 8.25 (s, 1H), 6.11 (bs, 2H), 5.97 (d, J = 6.5 Hz, 1H), 5.37 (dd, J = 6.4, 4.5 Hz,
1H), 4.39 (dd, J = 4.1, 2.8 Hz, 1H), 4.05 (m, 1H), 3.00 (d, J = 5.0 Hz, 2H), 1.66 (bs, 2H),
1.01 (t, J = 7.9 Hz, 9H), 0.78 (t, J = 7.9 Hz, 9H), 0.67 (q, J = 7.9 Hz, 6H), 0.34 (qq, J = 15,
7.6 Hz, 6H).
8-Bromo-5′-amino-acetic acid methyl ester-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine
(2.23)
Compound was prepared using a general procedure:23,33 To 8-bromo-5′-amine-5′-deoxy-
2′,3′-bis-(O-triethylsilyl) adenosine 2.22 (0.3059 g, 0.5332 mmol) in 2.94 mL dry THF
was added TEA (89.1 µL, 0.6399 mmol). Methylbromoacetate (61 µL, 0.6399 mmol) in
1.48 mL dry THF was then added drop wise to the solution. The reaction mixture was
stirred overnight. The resulting precipitate was then filtered off and the organic was
evaporated in vacuo. Column chromatography (3:1 EtOAc/ CH2Cl2 to 15:5:1
EtOAc/CH2Cl2/MeOH) yielded product (0.2678 g, 78 %). 1H NMR (CDCl3) δ 8.26 (s,
1H), 5.97 (d, J = 6.7 Hz, 1H), 5.93 (bs, 2H), 5.41 (dd, J = 6.6, 4.7 Hz, 1H), 4.38 (dd, J =
4.6, 2.2 Hz, 1H), 4.16 ( m, 1H), 3.68 (s, 3H), 3.43 (d, J = 2.3 Hz, 2H), 2.98 (dd, J = 12.4,
50

3.5 Hz, 1H), 2.90 (dd, J = 12.4, 6.5 Hz, 1H), 2.56 (bs, 1H), 1.01 (t, J = 7.9 Hz, 9H), 0.77
(t, J = 7.9 Hz, 9H), 0.67 (q, J = 7.7 Hz, 6H), 0.32 (qq, J = 15, 7.6 Hz, 6H).
8-Bromo-5′-ethanolamine -5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.24)
Compound was synthesized in adaptation of a literature procedure:34 To 8-bromo-5′-
amino-acetic acid methyl ester-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine 2.23 (0.2678
g, 0.4147 mmol) in 7.41 mL dry THF at 0°C was slowly added 1M DIBAL-H (2.074
mmol). The reaction was kept cold for a few minutes, then warmed to rt, and stirred for
an additional 5 h. Saturated potassium sodium tartrate tetrahydrate (8.91 mL) was added
to the reaction and stirred vigorously overnight. The organic was washed (sat. Rochelle’s
salt, H2O, EtOAc, brine), dried over Na2SO4, and evaporated in vacuo to obtain product
(0.2383 g, 93 %). 1H NMR (CDCl3) δ 8.23 (s, 1H), 6.26 (bs, 2H), 5.98 (d, J = 6.9 Hz,
1H), 5.37 (dd, J = 6.9, 4.6 Hz, 1H), 4.35 (dd, J = 4.6, 1.8 Hz, 1H), 4.22 (m, 1H), 3.67 (t, J
= 5.2 Hz, 2H), 3.45 (bs, 1H), 3.00 (dd, J = 12, 6.1 Hz, 1H), 2.94 (dd, J = 13, 3.5 Hz, 1H),
2.82 (ddt, J = 37, 12.4, 5.3 Hz, 2H), 1.00 (t, J = 7.9 Hz, 9H), 0.76 (t, J = 7.9 Hz, 9H), 0.66
(q, J = 7.8 Hz, 6H), 0.30 (qq, J = 15, 7.6 Hz, 6H).
8-Bromo-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-ethanolamine-5′-deoxy-
2′,3′-bis-(O-triethylsilyl) adenosine (2.25)
Following a general procedure,27,33 NaCNBH3 (0.01425 g, 0.2267 mmol) and AcOH (8.7
µL, 0.1511 mmol) were added to a solution of 8-bromo-5′-ethanolamine -5′-deoxy-2′,3′-
bis-(O-triethylsilyl) adenosine 2.24 (0.1027 g, 0.1663 mmol) and tert-butyl (S)-2-[N-
(tert-butoxycarbonyl)amino]-4-oxobutanoate 2.8 (0.0413 g, 0.1511 mmol) in 724 µL dry
51

MeOH. The reaction mixture was stirred overnight at rt. After diluting the reaction with
EtOAc and NaHCO3, the organic layer was washed with NaHCO3, dried over Na2SO4,
and evaporated in vacuo. Column chromatography (10:4:2:1 Pet
Ether/EtOAc/CH2Cl2/MeOH) yielded product (0.0899 g, 62 %). 1H NMR (CDCl3) δ
8.25 (s, 1H), 5.97 (bs, 2H), 5.93 (d, J = 5.3 Hz, 1H), 5.44 (bs, 1H), 5.40 (dd, J = 9.9, 5.0
Hz, 1H), 4.37 (t, J = 4.1 Hz, 1H), 4.16 (m, 2H), 3.50 (m, 2H), 2.90 (m, 2H), 2.62 (m, 4H),
1.93 (m, 1H), 1.68 (m, 1H), 1.42 (s, 9H), 1.41 (s, 9H), 1.01 (t, J = 7.9 Hz, 9H), 0.80 (t, J =
7.9 Hz, 9H), 0.67 (q, J = 7.8 Hz, 6H), 0.38 (qq, J = 15, 7.6 Hz, 6H).
8-Azido-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.32)
Following previously reported procedures,22,32 8-azido-5′-phthalimide-5′-deoxy-2′,3′-bis-
(O-isopropylidene) adenosine 2.38 (2.6318 g, 5.5124 mmol) was dissolved in 55.3 mL
3:1:1 TFA/H2O/THF and stirred for 2.5 h. The solvent was evaporated in vacuo and co-
evaporated with EtOH (x3). The resulting material was dissolved in 12.1 mL dry DMF,
followed by the addition of imidazole (1.876 g, 27.562 mmol) and TESCl (2.035 mL,
12.128 mmol). The reaction was stirred overnight, followed by aqueous workup (NH4Cl
(x2), EtOAc, brine), dried over Na2SO4, and evaporated in vacuo. Column
chromatography (8:4:2:0.5 Hexanes/EtOAc/CH2Cl2/MeOH to 8:4:2:1 Hexanes/EtOAc/
CH2Cl2/MeOH) yielded product (3.139 g, 86 %). 1H NMR (CDCl3) δ 7.99 (s, 1H), 7.83
(m, 2H), 7.72 (m, 2H), 5.79 (d, J = 6.9 Hz, 1H), 5.54 (dd, J = 6.9, 4.5 Hz, 1H), 5.47 (bs,
2H), 4.47 (dd, J = 4.5, 1.3 Hz, 1H), 4.25 (m, 2H), 3.91(dd, J = 17, 7.6 Hz, 1H), 0.92 (t, J
= 7.9 Hz, 9H), 0.81 (t, J = 7.9 Hz, 9H), 0.60 (q, J = 7.7 Hz, 6H), 0.37 (qq, J = 15, 7.6 Hz,
6H). 13C NMR (CDCl3) δ 167.0, 152.6, 150.7, 149.4, 145.3, 133.3, 131.2, 122.6, 117.5,
52

87.0, 83.5, 73.9, 71.3, 39.9, 7.5, 7.4, 5.7, 5.3. HRMS-ESI: calcd for C30H43N9O5Si2 (M +
Na+) 688.2818, obsd 688.2805.
8-Azido-5′-amine-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine (2.33)
Following a general procedure:22 To 8-azido-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-
triethylsilyl) adenosine 2.32 (0.9172 g, 1.377 mmol) in 45 mL EtOH was added
ethylenediamine (460 µL, 6.887 mmol). The reaction was heated to 70°C and stirred for
5 h. The solvent was evaporated in vacuo and chromatographed on silica pre-treated with
2% TEA (4:2:1 EtOAc/CH2Cl2/MeOH) to yield product (0.5655 g, 77 %). 1H NMR
(CDCl3) δ 8.23 (s, 1H), 5.77 (d, J = 6.3 Hz, 1H), 5.55 (bs, 2H), 5.24 (dd, J = 6.3, 4.8 Hz,
1H), 4.37 (dd, J = 4.7, 2.8 Hz, 1H), 4.02 (m, 1H), 3.02 (dd, J = 13, 4.0 Hz, 1H), 2.97 (dd,
J = 13, 5.8 Hz, 1H), 1.72 (bs, 2H), 1.02 (t, J = 7.9 Hz, 9H), 0.81 (t, J = 7.9 Hz, 9H), 0.69
(q, J = 7.7 Hz, 6H), 0.38 (qq, J = 15, 7.6 Hz, 6H). 13C NMR (CDCl3) δ 152.9, 150.8,
149.3, 145.1, 117.6, 87.5, 86.9, 73.3, 72.7, 44.2, 7.6, 7.3, 5.8, 5.3. HRMS-ESI: calcd for
C22H41N9O3Si2 (M + Na+) 558.2763, obsd 558.2763.
8-Azido-5′-N-Boc-diaminobutyric acid-O-tert-butyl ester-5′-deoxy-2′,3′-bis-(O-
triethylsilyl) adenosine (2.34)
In an adaptation of a reported procedure,27,33 NaCNBH3 (0.0663 g, 1.056 mmol) and
AcOH (55 µL, 0.9595 mmol) were added to a solution of 8-azido-5′-amine-5′-deoxy-
2′,3′-bis-(O-triethylsilyl) adenosine 2.33 (0.5655 g, 1.056 mmol) and tert-butyl (S)-2-[N-
(tert-butoxycarbonyl)amino]-4-oxobutanoate 2.8 (0.2623 g, 0.9595 mmol) in 5.6 mL dry
MeOH. The reaction mixture was stirred for 2 h. After diluting the reaction with EtOAc
53

and NaHCO3, the organic layer was washed with NaHCO3, dried over Na2SO4, and
evaporated in vacuo. Column chromatography (12:4:2:0.6 Pet
Ether/EtOAc/CH2Cl2/MeOH) yielded product (0.4311 g, 51 %). 1H NMR (CDCl3) δ
8.21 (s, 1H), 5.76 (d, J = 6.7 Hz, 1H), 5.56 (d, J = 6.5 Hz, 2H), 5.43 (d, J = 8.0 Hz, 1H),
5.25 (dd, J = 6.5, 4.8 Hz, 1H), 4.35 (dd, J = 4.6, 2.2 Hz, 1H), 4.26 (m, 1H), 4.12 (m, 1H),
2.87 (d, J = 4.9 Hz, 2H), 2.68 (m, 2H), 2.16 (bs, 1H), 1.96 (m, 1H), 1.76 (m, 1H), 1.45 (s,
9H), 1.44(s, 9H), 1.01 (t, J = 7.9 Hz, 9H), 0.80 (t, J = 7.9 Hz, 9H), 0.68 (q, J = 7.8 Hz,
6H), 0.35 (qq, J = 15, 7.6 Hz, 6H). 13C NMR (CDCl3) δ 170.8, 154.4, 152.8, 150.7, 149.3,
145.2, 117.7, 87.4, 85.2, 81.5, 79.3, 74.0, 72.5, 53.0, 51.8, 46.3, 33.4, 28.8, 28.5, 7.6, 7.2,
5.8, 5.3. HRMS-ESI: calcd for C35H64N10O7Si2 (M + Na+) 815.4390, obsd 815.4390.
5′-Phthalimide-5′-deoxy-2′,3′-bis-(O-isopropylidene) adenosine (2.36) Following a reported procedure,32 phthalimide (2.322 g, 15.787 mmol) and PPh3 (4.020 g,
15.327 mmol) were added to a solution of commercially available 2′,3′-isopropylidene
adenosine 2.2 ( 4.710 g, 15.327 mmol) in dry 53 mL THF. After stirring at rt for 15 min,
DIAD (3.02 mL, 15.327 mmol) was added to the mixture and stirred an additional 3 h.
Upon cooling with ice, the white precipitate was filtered off and washed with cold Et2O
to yield product (5.123 g, 77 %). The product was confirmed by spectroscopic
comparison to previously reported 1H NMR data.
8-Bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-isopropylidene) adenosine (2.37) Compound was synthesized using a previously reported procedure:22 To 5′-phthalimide-
5′-deoxy-2′,3′-bis-(O-isopropylidene) adenosine 2.36 (5.123 g, 11.74 mmol) in 176 mL
7:4 dioxane/0.5 M NaOAc (pH 5.2) was added Br2 (1.21 mL, 23.48 mmol). The reaction
54

was stirred for 3.5 h, followed by an aqueous workup (Na2S2O3, CH2Cl2, brine), dried
over Na2SO4, and evaporated in vacuo. Column chromatography (32:4:1 EtOAc/
CH2Cl2/MeOH) yielded product (5.879 g, 97 %). 1H NMR (CDCl3) δ 8.04 (s, 1H), 7.76
(m, 2H), 7.68 (m, 2H), 6.16 (d, J = 1.3 Hz, 1H), 5.97 (bs, 2H), 5.74 (dd, J = 6.2, 1.3 Hz,
1H), 5.34 (dd, J = 6.2, 3.2 Hz, 1H), 4.54 (m, 1H), 3.99 (dd, J = 14.1, 5.7 Hz, 1H), 3.87
(dd, J = 14.1, 6.8 Hz, 1H), 1.57 (s, 3H), 1.38 (s, 3H). 13C NMR (CDCl3) δ 167.9, 154.4,
152.7, 150.2, 133.8, 131.8, 127.5, 123.2, 120.0, 114.0, 91.1, 85.7, 83.5, 82.6, 39.3, 27.0,
25.3. HRMS-ESI: calcd for C21H19BrN6O5 (M + Na+) 537.0493, obsd 537.0485.
8-Azido-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-isopropylidene) adenosine (2.38)
Following a reported preparation,35 NaN3 (1.602 g, 24.619 mmol) was added to a solution
of 8-bromo-5′-phthalimide-5′-deoxy-2′,3′-bis-(O-isopropylidene) adenosine 2.37 (3.172 g,
6.155 mmol) in 51.9 mL DMSO. The reaction mixture was heated to 85°C and stirred
for 10.5 h, followed by an aqueous workup (NaHCO3, CH2Cl2, brine). Material was
dried over Na2SO4, evaporated in vacuo, and column chromatographed (16:2:1
EtOAc/CH2Cl2/MeOH) to yield product (2.632 g, 90 %). 1H NMR (CDCl3) δ 8.01 (s,
1H), 7.76 (m, 2H), 7.68 (m, 2H), 5.98 (d, J = 1.5 Hz, 1H), 5.64 (bs, 2H), 5.60 (dd, J = 6.3,
1.5 Hz, 1H), 5.25 (dd, J = 6.3, 3.3 Hz, 1H), 4.50 (m, 1H), 3.99 (dd, J = 14.1, 5.7 Hz, 1H),
3.89 (dd, J = 14.1, 6.7 Hz, 1H), 1.55 (s, 3H), 1.36 (s, 3H). 13C NMR (CDCl3) δ 167.9,
153.6, 151.7, 149.6, 145.1, 133.8, 131.8, 123.1, 117.7, 114.1, 88.4, 85.2, 83.4, 82.5, 39.4,
27.0, 25.4. HRMS-ESI: calcd for C21H19N9O5 (M + Na+) 500.1401, obsd 500.1395.
55

8-Azido-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-ethanolamine-5′-deoxy-
2′,3′-bis-(O-triethylsilyl) adenosine (2.26)
Following a reported preparation,22 N,N-diisopropylethylamine (1.086 mL, 6.2351 mmol)
and 2-bromoethanol (443 µL, 6.2351 mmol) were added to a solution of 8-azido-5′-N-
Boc-diaminobutyric acid-O-tert-butyl ester-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine
2.34 (0.3804 g, 0.4796 mmol) in 2.514 mL dry toluene. The reaction was heated to 70oC
and stirred for 27 h, followed by an aqueous workup (NaHCO3, EtOAc, brine). Material
was dried over Na2SO4, evaporated in vacuo, and column chromatographed on silica pre-
treated with 1.5 % TEA (1:1.5 Hexanes/ EtOAc to 1:2 Hexanes/ EtOAc, each solvent
system was pre-treated with 0.5 % TEA) to yield product (0.2294 g, 57 %).
Note: Recently, another solvent system was found to be more efficient: 1:0.5 Pet/
EtOAc to 1:2 Pet/ EtOAc. Silica and solvent were not pre-treated with TEA. 1H NMR
(CDCl3) δ 8.20 (s,1H), 5.73 (d, J = 5.3 Hz, 1H), 5.69 (bs, 2H), 5.43 (d, J = 8.2 Hz, 1H),
5.19 (t, J = 4.9 Hz, 1H), 4.34 (t, J = 4.1 Hz, 1H), 4.12 (m, 2H), 3.50 (m, 2H), 2.88 (m,
2H), 2.62 (m, 4H), 1.91 (m, 1H), 1.68 (m, 1H), 1.42 (s, 9H), 1.42 (s, 9H), 1.00 (t, J = 7.9
Hz, 9H), 0.81 (t, J = 7.9 Hz, 9H), 0.66 (q, J = 7.8 Hz, 6H), 0.39 (qq, J = 15, 7.5 Hz, 6H).
13C NMR (CDCl3) δ 171.9, 155.4, 153.6, 151.6, 150.0, 145.9, 118.1, 87.9, 83.0, 81.7,
79.5, 74.4, 72.4, 59.1, 56.5, 56.1, 52.8, 50.5, 29.6, 28.2, 27.9, 6.8, 6.5, 4.9, 4.5. HRMS-
ESI: calcd for C37H68N10O8Si2 (M + H+) 837.4833, obsd 837.4810.
56

8-Azido-5′-(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine ammonium
hydrochloride (2.1)
In an adaptation of a reported procedure,27 I2 (0.06527 g, 0.2572 mmol) was added to
triphenylphosphine (0.06528 g, 0.2489 mmol) and imidazole (0.01695 g, 0.2489 mmol)
in 653 µL CH2Cl2 at 0°C. 8-Azido-5′-(N-Boc-diaminobutyric acid-O-tert-butyl ester)-N-
ethanolamine-5′-deoxy-2′,3′-bis-(O-triethylsilyl) adenosine 2.26 (0.1371 g, 0.1638 mmol)
in CH2Cl2 (653 µL) was then added. The reaction mixture was heated to 40°C and stirred
for 30 min. The reaction was diluted with ice-chilled CH2Cl2 and H2O, and the organic
layer was washed with H2O (3x). After evaporating in vacuo, HCl-dioxane (4N, 942 µL)
was added to the material in CH2Cl2 (1.884 mL) and the mixture was heated to 40°C and
stirred for 3 h. Ice-chilled H2O was added, and the aqueous layer was extracted with
CH2Cl2 several times prior to lyophilization to afford a light yellow solid. This was
dissolved in MeOH, and EtOAc was added dropwise to precipitate 2.1 as a light yellow
solid (0.0719 g, 78 %). 1H NMR (CD3OD) δ 8.49 (s, 1H), 5.99 (d, J = 3.7 Hz, 1H), 4.85
(m, 1H), 4.53 (m, 2H), 4.19 (m, 1H), 3.97–3.67 (m, 4H), 3.65–3.46 (m, 4H), 2.49 (m, 1H),
2.40 (m, 1H). 13C NMR (CD3OD) δ 170.4, 150.2, 149.9, 149.7, 144.9, 119.1, 91.0, 80.2,
73.7, 73.5, 57.6, 56.0, 51.4, 49.1, 26.0, -8.39. HRMS-ESI: calcd for C16H24IN10O5+ (M+)
563.0970, obsd 563.0969.
57

REFERENCES
(1) Kresge, N.; Tabor, H.; Simoni, R. D.; Hill, R. L. J Biol Chem 2005, 280, e35. (2) Lu, S. C. Int J Biochem Cell Biol 2000, 32, 391-5. (3) Lee, D. Y.; Teyssier, C.; Strahl, B. D.; Stallcup, M. R. Endocr Rev 2005, 26,
147-70. (4) Zhang, Y.; Reinberg, D. Genes & Development 2001, 15, 2343-2360. (5) Stallcup, M. R. Oncogene 2001, 20, 3014-3020. (6) Sims, R. J., 3rd; Nishioka, K.; Reinberg, D. Trends Genet 2003, 19, 629-39. (7) Wysocka, J.; Allis, C. D.; Coonrod, S. Front Biosci 2006, 11, 344-55. (8) Prokhortchouk, E.; Defossez, P. A. Biochim Biophys Acta 2008, 1783, 2167-73. (9) Klose, R. J.; Bird, A. P. Trends Biochem Sci 2006, 31, 89-97. (10) Jurkowska, R. Z.; Jeltsch, A. Methods Mol Biol, 649, 149-61. (11) Robertson, K. D. Oncogene 2001, 20, 3139-55. (12) Esteller, M. Nat Rev Genet 2007, 8, 286-98. (13) Shen, R.; Tao, L.; Xu, Y.; Chang, S.; Van Brocklyn, J.; Gao, J. X. Int J Clin Exp
Pathol 2009, 2, 21-33. (14) Bedford, M. T.; van Helden, P. D. Cancer Res 1987, 47, 5274-6. (15) Pljevaljcic, G.; Schmidt, F.; Weinhold, E. Chembiochem 2004, 5, 265-269. (16) Rein, T.; DePamphilis, M. L.; Zorbas, H. Nucleic Acids Res 1998, 26, 2255-64. (17) Sulewska, A.; Niklinska, W.; Kozlowski, M.; Minarowski, L.; Naumnik, W.;
Niklinski, J.; Dabrowska, K.; Chyczewski, L. Folia Histochem Cytobiol 2007, 45, 315-24.
(18) Snijders, A. P.; Hung, M. L.; Wilson, S. A.; Dickman, M. J. J Am Soc Mass
Spectrom, 21, 88-96. (19) Liang, Z.; Wong, R. P.; Li, L. H.; Jiang, H.; Xiao, H.; Li, G. Proteome Sci 2008,
6, 2.
58

(20) Pignot, M.; Siethoff, C.; Linscheid, M.; Weinhold, E. Angewandte Chemie-International Edition 1998, 37, 2888-2891.
(21) Weinhold, E.; Pljevaljcic, G.; Schmidt, F.; Scheidig, A. J.; Lurz, R. Chembiochem
2007, 8, 1516-1519. (22) Comstock, L. R.; Rajski, S. R. J Org Chem 2004, 69, 1425-8. (23) Comstock, L. R.; Rajski, S. R. Nucleic Acids Res 2005, 33, 1644-52. (24) Comstock, L. R. Ph.D. Dissertation, University of Wisconsin-Madison, 2005. (25) Comstock, L. R.; Rajski, S. R. J Am Chem Soc 2005, 127, 14136-7. (26) Schluckebier, G.; Kozak, M.; Bleimling, N.; Weinhold, E.; Saenger, W. J Mol
Biol 1997, 265, 56-67. (27) Weller, R. L.; Rajski, S. R. Chembiochem 2006, 7, 243-5. (28) Osborne, T.; Roska, R. L. W.; Rajski, S. R.; Thompson, P. R. Journal of the
American Chemical Society 2008, 130, 4574. (29) Hendrick, E. C. Unpublished results, 2009. (30) Bergmeier, S. C.; Cobas, A. A.; Rapoport, H. J. Org. Chem. 1993, 58, 2369. (31) Fukuyama, T.; Lin, S.-C.; Li, L. J. Am. Chem. Soc. 1990, 112, 7050-7051. (32) Kolb, M.; Danzin, C.; Barth, J.; Claverie, N. J Med Chem 1982, 25, 550-6. (33) Rajski, S. R.; Comstock, L. R.; Weller, R. L. In U.S. Patent 20070161007, 2007. (34) Gano, K. W.; Monbouquette, H. G.; Myles, D. C. Tetrahedron Letters 2001, 42,
2249-2251. (35) Holmes, R. E.; Robins, R. K. J. Am. Chem. Soc. 1965, 87, 1772. (36) Eggert, J. P. W.; Luning, U.; Nather, C. European Journal of Organic Chemistry
2005, 1107-1112. (37) Yamazaki, S.; Iwata, Y.; Fukushima, Y. Organic & Biomolecular Chemistry 2009,
7, 655-659. (38) Comstock, L. R.; Denu, J. M. Org Biomol Chem 2007, 5, 3087-91.
59

(39) Keith, L. H.; Jones-Lepp, T. L.; Needham, L. L. Analysis of environmental endocrine disruptors; American Chemical Society: Washington, DC, 2000.
(40) Petersen, S. G.; Rajski, S. R. Journal of Organic Chemistry 2005, 70, 5833-5839.
60

APPENDIX
NUCLEAR MAGNETIC RESONANCE SPECTRA
61
o ~ ,
o o
o
o LO
o to
o
o ex:)
o E ' 0..
0>0..
62

--N Z~
I~t-Z _ C/)
zyZ3l~ M ~ 0 .-: m 0 N
C/) m
O~~ I-
0 al
::2:
-
-
~
~
=
.-=l
-j
__ :ii
-~
.-...............
..;
--------------- " - ~
=
1
-
1
~
r r
0
r- ~ t-t-t-
t-0
1- 6 t-
t-
t-
e-o
I-~
e-e-e-e-
o I- N t-
e-e-f-
0 r- . C"'l
f-
f-
f-
f-0 r- ..,,:
f-
r- :; t-r r t-
o r-- <ci t-
f-
f-
t-o
I- r-: t-
e-e-f-
r-f-
e-e-
o <Xl
~
~ t-
t-o~ 'E (J)a.
t- a.
63

o ...... ,
o o
o
o N
o <0
o 00
o E . a. cna.
64

o .,.... ,
o o
o
o N
o LO
o <0
0 00
0 en
~ .,.... -~ E a. a.
65

N Z" I-Hz Z Cf)
- ID
I-ZYZ~O X <') 0
Z 0 o Cf)
' ID
o=~\-/~ I-I 0 t.l I 0
ID
CD "": N
---------------
'-------------
------------
~ ,
o N
0 '<t
0 LI'i
0 to
o ex:>
o E (j)~
66
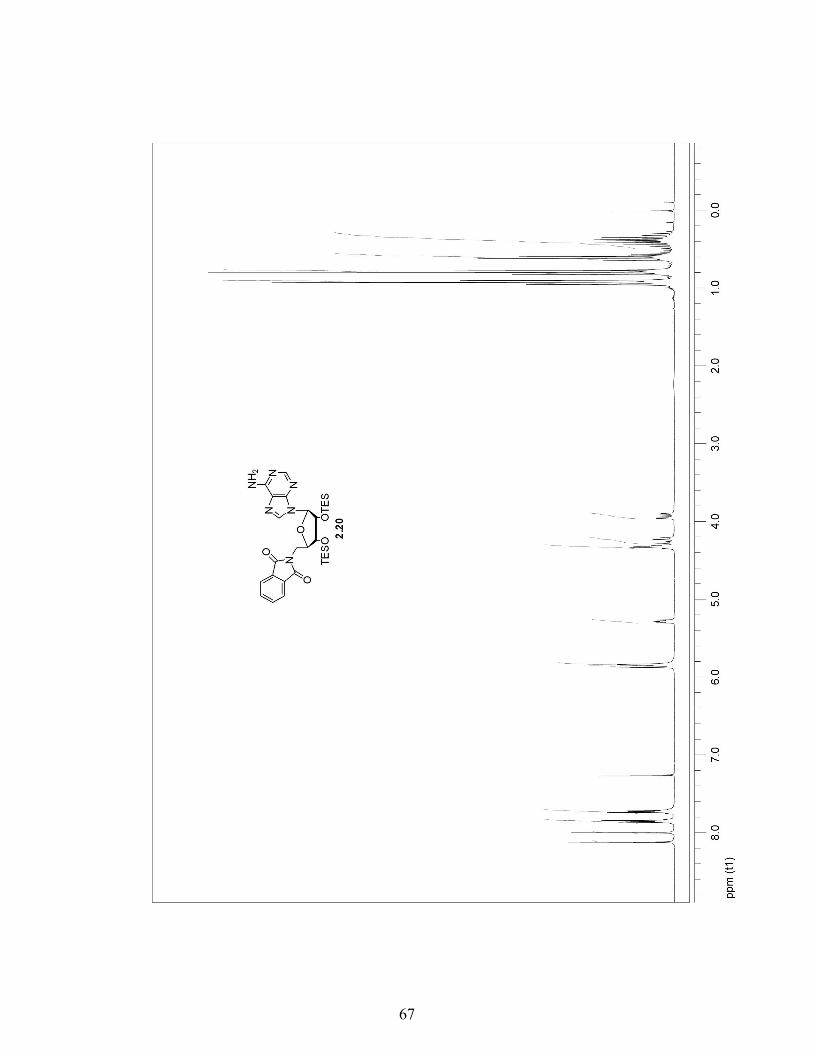
67

~
--------
N z I~ z z
C/) w ZyZ,Db ....
-------------m 0 N
0 N
~ ~ -----------------------: I 0 I-
---------------- -
~
~
~
---'
---<
~
-
== d
r-0
r- ""': , r-r-
r-r-r- 0
0 r-
r-
r-r-r-
r-r-r-r-r-
r-r-r-r-r-
r-r-r-r-r-
r-r-r-r-
r-r-r-r-r-
r-r-r-r-r-
r-r-r-r-r-
r-r-r-r-
r-->-
o
o N
o C')
o o.ri
o cD
o CO
o E ' Q.
(J)Q.
68
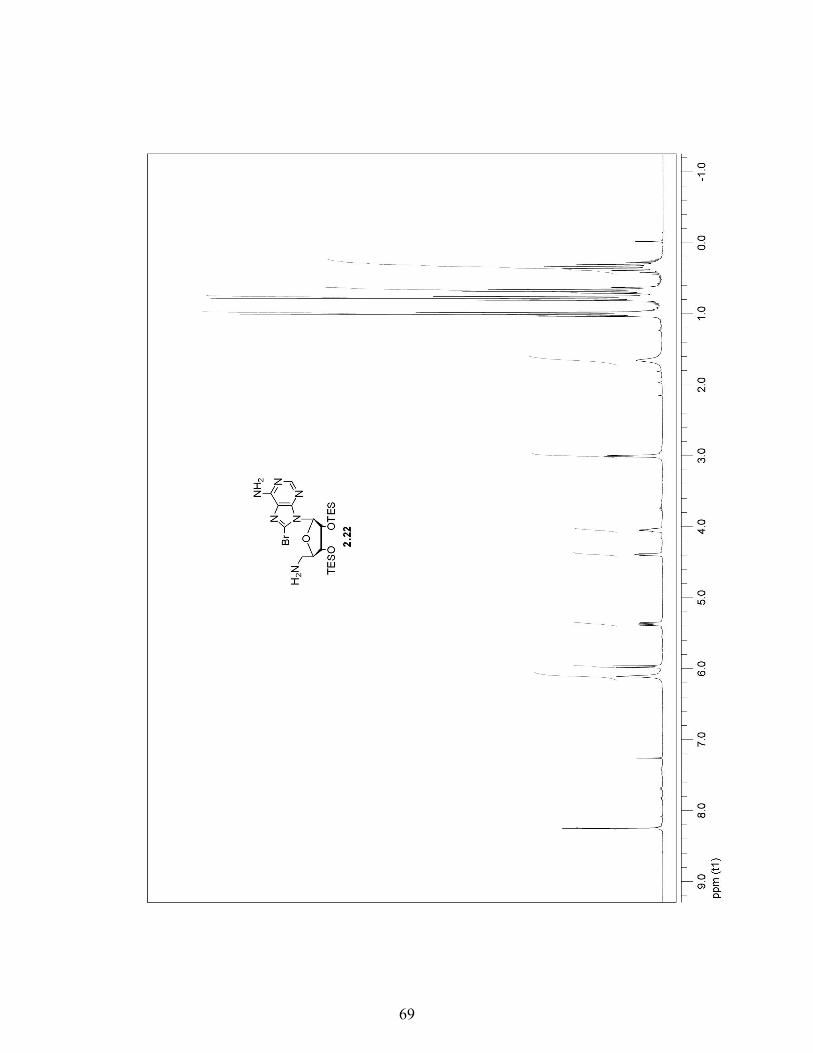
o ...... ,
o o
o
o N
o (")
o ui
o <0
0 <Xl
0 (J)
...... -E 0.. 0..
69

-----
-
-
N Z
~~ C/)
zyz3l~ M as 0 N
0 N
C/) ==<z w o I I-
0 -OJ
:2:
----- --=; ")
-'
~ 1 ~
1
] ---------------
----- 1 -
-
-
f-
f-
f-
f-
o c:i
o
f- ~ N
f----- 0 (")
f- ~ ....
f- O L!')
f- 0 <0
0 r--:
f- O co
~
~
f-o~ 0> E
0.. 0..
70

----
'" Z
I~ Z Z C/) w ZyZ31b '<t
OJ a <'! a N
C/) !:z w I I-
a I
~
~ )
~
-=<
-----~~~ -1 ]
------------------------------
--------1 ---------
--- ~ ~
-
f-
f----f-
f----
f-
f-
f----
f-
f-
o ...... ,
o o
o
o N
f- o l!)
1--0 (!)
f- o r-
1--0 00
f-
f- o 0)
~ ...... -E a. a.
71

---------------- -----
o ..-,
o o
o
o N
o <D
0 ex)
0 (J)
~
..--~
E 0.. 0..
72
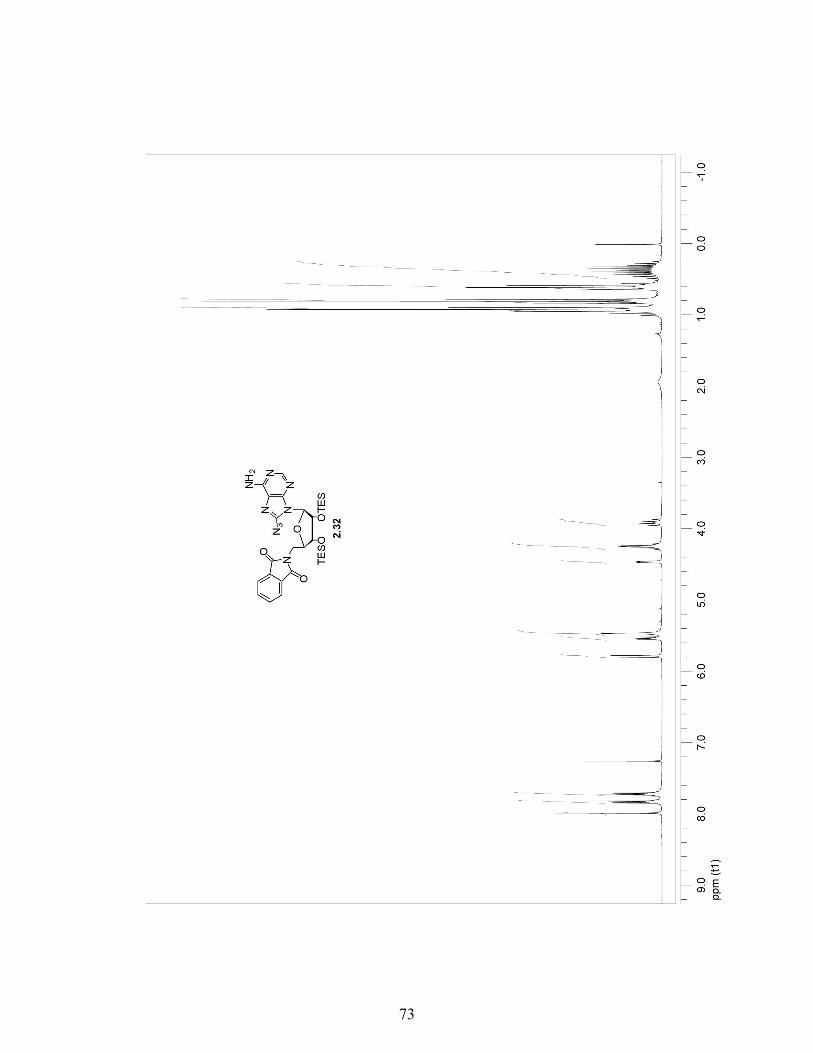
---------
C'J Z
I~ z z (J) w Z']r y 0 N Z 0 C'?
N
2t§ -Z I-
-;/ 0
~ I
~
-------- ~
-<;
------------- J ~
-=1 '-----------
--------------= ~ ~
r-r-r-r-r-
r-r-r-r-r-r-
r-
r-r-r-r-
r-r-r-r-r-f-
r-r-r-r-f-
f-
r-r-r-f-
f-
f-
r-r-f-
f-
f-
f-
r-r-f-
f-
f-
r-
r-r-f-
f-
r-f-
o 0
o o
o
o N
o (")
o L!)
o <D
0 00
0 (J)
.,.... -E a. a.
73

o ...... ,
o o
o
o N
o LO
o cD
0 00
0 (J')
~ ...... ~
E a. a.
74
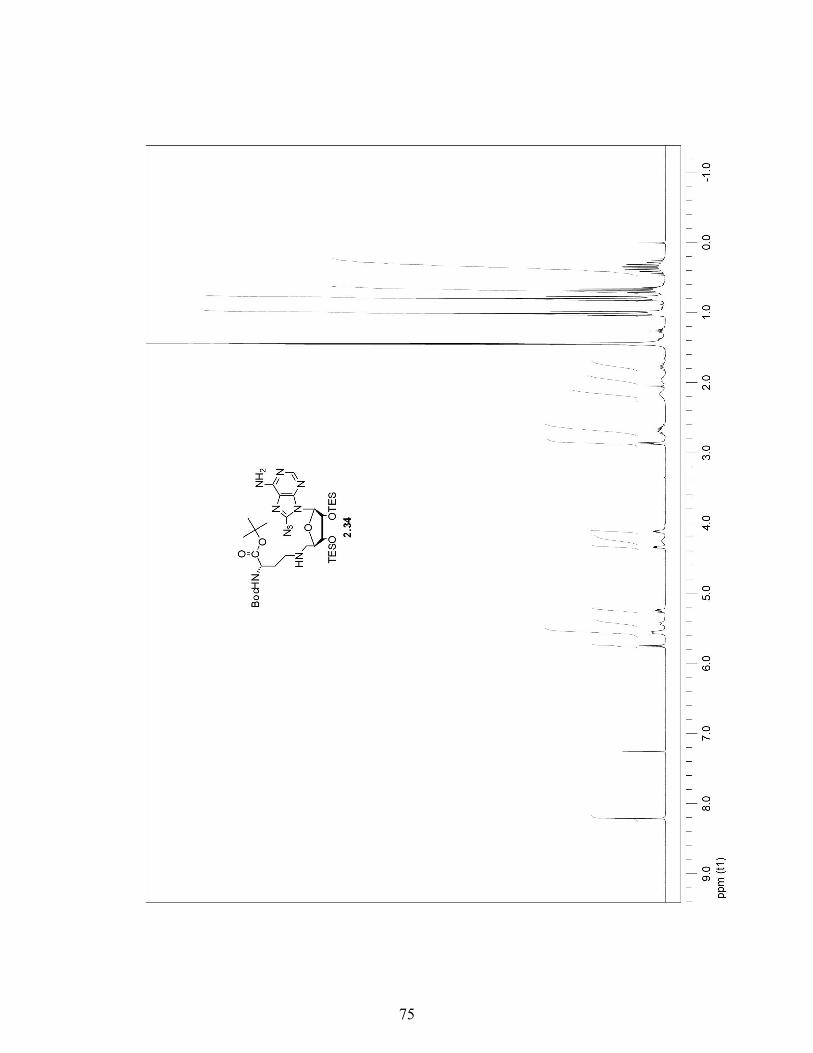
-------
N Z
2~ - U) w
ZyZ~b ;! ~ M O . Z 0"'" P U)
O'()~~ ~ .:i
Z I o 0
Ol
-
~ ------- J
J
~ ~ ~
~ -~
~ ~
~
---------------~ ---------------
--------------~ ---- , ~
C-
f-~ ~ ,
C-
C-
C-
C-
f- 0 0
r-r-r-
0 f- ,...:
f-
f-
f-
f-0 f- . C\I
f-
f-
f-
f-0 f- . <'1
f-
f-
C-
c-0 r- . "<t
c-C-
C-
C-o
r- Lti C-
C-
c-r-r-r-
r-r-I
r-r-I
I
r--r-r-I
o <0
~ r-r-r-
oE. m E
0.. 0..
L
75

E 0.. 0..
76

N Z \\ ~Hz
----------------------Z~]l:x CO M N
--------------2t V" 0
~ I
------------ -
-
~ \
d ~
-
-
~
J 1
~ J
= J ~
0 ' ei
0 r- ......
0 ' N
r- ;j
0 ' .,f
0 ' Lei
0 r- cci
,
-
r-
o co
...... -E 0.. 0..
77

N Z~
I~z Z Cf)
-Zy~ Zy 0 ~ \/ <") 0 N /\ Z 0
o Cf) I w
O=ULrZ I-
~. ~ 15 I In
o CO
E 0.. 0..
78

o ci
o
o N
o o.ri
o to
o cO
o~ . ...... (J)~
E 0.. 0..
79

CURRICULUM VITAE
VAN MAI
BORN: October 28, 1984, Dalat, Vietnam UNDERGRADUATE STUDY: Wingate University
Wingate, North Carolina B.S., Chemistry, May 2008
GRADUATE STUDY: Wake Forest University Winston-Salem, North Carolina M.S. candidate, May 2011
SCHOLARTIC AND PROFESSIONAL EXPERIENCE:
Teaching Assistant, Wake Forest University, 2009-2011. Intern, National Gypsum Company, Summer 2008. HONORS AND AWARDS:
Summa Cum Laude, Wingate University, 2008. Travel award to the 59th Southeastern Regional Meeting of the American Chemical Society, Greenville, SC, October 2007. North Carolina Rotary Scholarship, 2007. President’s List Recognition, 2006-2007. Inaugurated into Apha Chi Honor Scholarship Society, 2006-2007.
80





























