Embed Size (px)
Citation preview

Indi an Journal of Chemistry Vol. 40B, April 200 I, pp. 278-283
Synthesis of hormothamnione and 6-desmethoxyhormothamnione
Niveta Jainl, Geetu Gambhir2 & H G Krishnamurty*2
'Div ision of Environmentl Sciences, NRL Builing, IARI , New Delhi 11001 2
Dcpart ment of Chemistry, University of Delhi , Delhi li D 007
Received 24 Novell/bel' 1999; accepted (revised) 23 Ocrober 2000
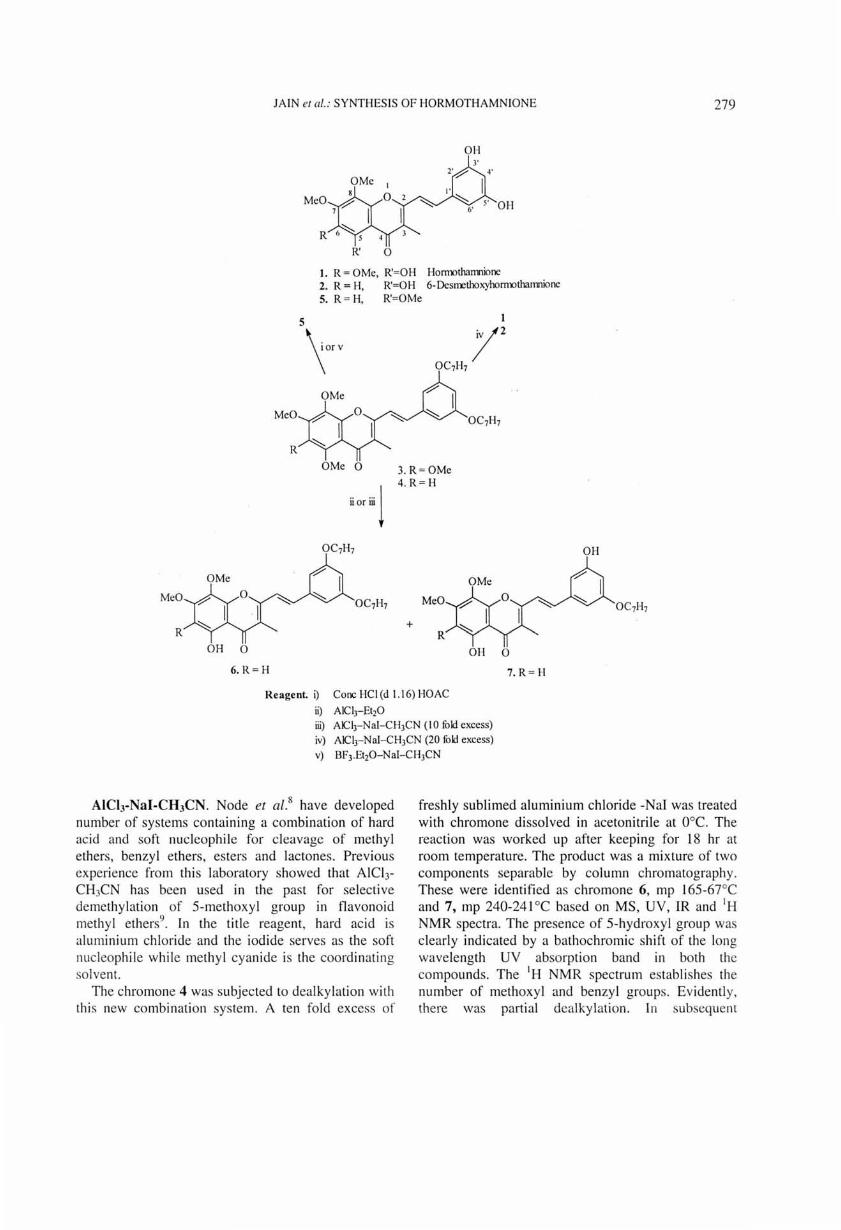
The antineoplast ic styrylchrolllones horlllo thalllnione 1 and 6-deslllethoxyhorlllothalllnione 2 are readi ly prepared by selective and si multaneous delllethylation and debenzylation of chrolllones 3 and 4 using excess AICI.1-Nal in CH)CN.
Hormothamnione 1 and 6-desmethoxyhormothamnione 2, the first natura lly occurring styrylchromones were isolated from the marine cryptophyte Chrysophaeul1/ tay/oril.2 . Hormothamni one 1 is except ionally cytotoxic to P388 lymphocytic leukemia and HL-60 promyelocy tic leukem ia ce ll lines ill vitro and appears to be a selecti ve inhi bitor of RNA sy nthesis I . 6-Des methoxyhormothamnione 2 showed cytotoxicity to 9 KB cell lines. The pharmaco logical act ivities and potential medicinal uses of styry lchromones have not yet been reported in detail.
The isolation and purification of the styrylchromones 1 and 2 is beset with great difficulties in view of very low concentrations in the rare algae. The potent biological activity has stimulated great interest in practical laboratory synthesis of such compounds for extensive studies related to their biological activities. Three syntheses of hormothamnione have been described3.5. The key intermediate is the styrylchromone 3. The crucial step in the synthesis of 1 and 2 is selective de methylation at C-5 and debenzylation at C-3' and C-5' positions of the chromone 3. Previous workers have utilised HClAcOH and boron trichloride as dealkylating agent for this purpose3.5. BCh is very expensive and requires careful handling. The HCl-AcOH agent in our experiments gave variable results. So it is of interest to explore alternative reagents for the purpose. We investigated the use of anhydrous aluminium chloride, the traditional dealkylating agent in a suitable solvent and also reexamined the use of HOAc-HCI reagent.
Results and Discussion 6-Desmethoxyhormothamnione 2. Dealkylation
with cone. HCI. The styrylchromone 4 required for the synthesis of 6-desmethoxyhormothamnione was prepared using Baker-Venketaraman transformation
(B VT)6 and oxidati ve cyclisation of cinnamylidene . h 7 propJOp en one .
The reaction between the chromone 4 and HOAc-HCI (d 1.16, E. Merck) (4:1, v/v) on a steambath for 2 hr gave a mixture of severa l products (TLC) with partial deco mposi tion. A change in HOAc - HCI ratio (2 : I , v/v) under the same conditions gave a ye ll ow solid , mp 2 10-14°C. The UV spectrum of the product resembled the paren t chro mone. The IR spectrum displayed a strong absorption at 1620, 1595 cm- I and other bands indicated the conjugated carbonyl of the chromone. The IH NMR spectrum showed a singlet at 8 2.1 for 3-methyl group, a si nglet at 8 3.9 for one methoxyl, a singlet at 8 4 .0 for two methoxy i groups and other aromatic signals but free from O-benzyl groups. These observations are consistent with the structure shown in formula S. The product gave a diacetate whose MS and IH NMR fully supported the above structural assignment.
Dealkylation with anhydrous aluminium chloride
AIClrEther. A mixture of anhydrous aluminium chloride (9-10 mole equivalent) and the chromone (4, 1 mole equivalent) was stirred in dry ether for about 18 hr at room temperature. Standard work-up gave a mix ture of atleast three phenolic ,.:;ompounds. This mixture gave an intense colour wi th ferric chloride indicating the presence of hydrogen bonded 5-hydroxyl group. Separation of the components of the mixture was not undertaken but modification of the reaction was attempted. The use of twenty fold excess aluminium chloride and reaction time of 24 hr gave compound 6, mp 165-67°C and compound 7 mp 240-41 °C. These were separated by preparative TLC. The structures of these products are discussed under dealkylation with AlCI3-NaI-CH3CN.
JAIN el al.: SYNTHESIS OF HORMOTHAMNIONE 279
OH
OMe
MeO 6'
R' 0
1. R = OMe, R'=OH Hom¥lthamnione 2. R = H, R'=OH 6-Desrrethoxyhom¥lthamnione 5. R = H, R'=OMe
MeO OC7H7
R
0 3. R= OMe
;;" ili I 4. R=H
OH
OMe OMe
OC7H7 MeO OC7H7
MeO
+ R R
o OH 0
6. R=H 7. R=H
Reagent. i) Cone HCI (d 1.16) HOAC
u) AIClrEt20 iii) AlClrNaI-CH)CN (10 fold excess) iv) AlClrNaI-CH)CN (20 fold excess) v) BF) .Et20-NaI-CH)CN
AICh-NaI-CH3CN. Node et al.8 have developed number of systems containing a combination of hard acid and soft nucleophile for cleavage of methyl ethers, benzyl ethers, esters and lactones. Previous experience from this laboratory showed that AICI)CH)CN has been used in the past for selective demethylation of 5-methoxyl group in flavonoid methyl ethers9
. In the title reagent, hard acid is aluminium chloride and the iodide serves as the soft nucleophile while methyl cyanide is the coordinating solvent.
The chromone 4 was subjected to dealkylation with this new combination system. A ten fold excess of
freshly sublimed aluminium chloride -NaI was treated with chromone dissolved in acetonitrile at O°c. The reaction was worked up after keeping for 18 hr at room temperature. The product was a mixture of two components separable by column chromatography. These were identified as chromone 6, mp 165-67°C and 7, mp 240-241 °C based on MS, UV, IR and IH NMR spectra. The presence of 5-hydroxyl group was clearly indicated by a bathochromic shift of the long wavelength UV absorption band in both the compounds. The I H NMR spectrum establishes the number of methoxyl and benzyl groups. Evidently, there was partial dcalkylation. In subsequent

280 INDIAN J CHEM., SEC B, APRIL 2001
experiments it was found that reaction time could be shortened and the chromone 2 became the chief product on employing 20 fold excess of AICI) - Nal in 55% yield.
The properties of the synthetic 2 were identical with the properties reported for natural 6-desmethoxyhormothamni one including the triacetate.
Dealkylation using BF3.EtzO-NaI-CH3CN. A combination of BF).Et20 -Il-Bu4NI was reported to be useful for selective cleavage of ethers 10. Later, the system BF3.Et20-Nal-CH3CN has been developed for selecti ve cleavage of benzy l ethers. The reacti vity order is benzyl ether »> aliphatic methyl ether» phenol methyl ether. Thi s combinat ion of hard acid (BF3) and soft base (Nal) has now been studied with the ex pectati on that 5-methoxy l group may also be vu lnerable towards this reagent.
In a typi cal procedure, one mmole of the chromone was d issolved in anhydrous acetonitrile and then treated with anh ydrous sodiu m iodide (5-6 mmole equi va lent). The mi xture was cooled to O°C and freshly dist illed boron trifl uoride etherate (5-6 mmole equi va lent) was added (O°C) . The reac tion mi xture was kept <It room temperature fo r 18-20 hr. Usual work-up gave a dark brown solid which required ex tensive chromatographi c puri fication. The product was found to be identi ca l with 5,7,8-trimethoxy-3',5'dihydroxystyrylchromone 5 , previously obtained by using HCl-HOAc.
Subsequent experiments indicated that this system does not cause demethylation of the 5-methoxyl in fl avonoids such as 5,7,4'-trimethoxyflavone. Evidently, the system BF3.Et20-Nal-CH3CN appears to be good only for O-debenzylation . Moreover, there are other experimental difficulties associated with this reagent. A re-examination of the scope of the system for dealkylation in a wide variety of phenolic natural products is in order.
Hormothamnione 1. The styrylchromone 3 was prepared by condensation of 2,3-dimethyl-5,6,7,8-tetramethoxychromone) with 3,5-dibenzyloxybenzaldehyde. The synthesis of hormothamnione was accomplished by dealkylation of chromone 3 using 30 fold excess of AlCI3-Nal in methyl cyanide. The properties of the resulting product are consistent (IH NMR, JR, UV and UV shift with AlCl)) with the styrylchromone 1.
Conclusion Selective and simultaneous demethylation (at C-5)
and debenzylation of the chromones 3 and 4 are
possible using excess aluminium chloride-sodium iodide in acetonitrile. The system AlCh-NaI-CH)CN is a good alternative to BCI). The AlCb-ether and AcOH-HCl reagents offer a way to prepare certain new derivatives such as 5, 6 and 7.
Experimental Section General. Melting points were taken in open
capillaries in sulphuric acid bath and in Thomas Hoover Unimelt capillary melting point apparatus and are uncorrected. IR spectra were recorded in KB r on a Shimadzu IR-435 and Perkin-Elmer FT-171O spectrophotometer; I H NMR spectra on a PerkinElmer R-32 [90 MHz, Jeol PMX 60 SI (60 MH z)l spectrophotometer in CDCb using T MS as internal re ference; and mass spectra on a Hewlett Packard 5939 A mass spectrometer. Column chromatography was carri ed out over silica gel (60- 120 mesh).
6-Desmcthoxyhormothamnione
Baker-Venketaraman Transformation 2-(3,5-Dibenzyloxycinnamyloxy)-3,4,6-trirnethoxy
propiophenone. A mi xture of 3,5-dibenzyloxycinnamoyl chloride (1.5 g, 3 .96 x 10-3 moles), pyridine ( IOmL) and 2-hydroxy-3,4,6-trimethoxypropiophenone (0.9g, 3.75 x 10-) moles) was heated at 60-70°C for 3 hr. The mixture was then poured into ice-water with stirring and acidified with di!. HC!. The product was collected by filtration , washed with 5% aq. alkali and water. The ester was obtained as white solid. It was crystallised from ethanol , mp lIS-16°C yield 1.5 g (65 %); IR:1730, 1635 cm· l
; IH NMR:81.I(t, 3H, CH3), 2.8 (q , 2H, CH2), 3.8 (s, 3H , OCH)), 3.85 (s, 3H, OCH)), 3.95 (s, 3H , OCH3) , 5.1 (s, 4H, 2xCH2Ph), 6.6 (d, IH, }=16Hz Ha), 6.5-7.0 (m, unresolved, 4H, Ar-H), 7.1 (s, IOH, 2xCH2Ph), 7.8 (d, IH, }=16Hz, Hf3).
Diketone. The above ester (1.5 g , 2.6 x 10-3 moles) was dissolved in dry pyridine (10 mL) and powdered anhydrous KOH (6 g) was added to the above solution. The reaction mixture was stirred vigorously at 60-65°C till a yellow paste was formed. The reaction mixture was diluted with ice-cold water and acidified with di!. HC!. The orange yellow solid thus obtained was collected by filtrati on, yield 0.84 g (56%). The crude diketone was used for next step without further purification .
Chromone 4. A solution of diketone (I g) and ptoluenesulphonic acid (2 g) in dry benzene (15 mL) was refluxed for 4 hr. The reaction mixture was

JAIN et al.: SYNTHESIS OF HORMOTHAMNIONE 281
cooled and diluted with cold water. The organic layer was separated, washed with aq. alkali (5%), water, dried and evaporated. The resulting chromone was crystallised from methanol as yellow solid, mp 185-8rC, yield 0.63 g (65%); IR: 1640, 1590 cm'l; IH NMR: 8 2.0 (s, 3H, CH3), 3.8 (s, 3H, OCH3), 3.85 (s, 6H, 2xOCH3), 5.1 (s, 4H, 2xCH2Ph), 6.1-7.6 (m, 6H, H-6, 2',4',6' and CH=CH), 7.1 (s, lOH, CH2Ph-H) MS: mlz 564 00, M+, C3sH3207), 563 (45), 549 (2), 536 (3 1), 275 (7), 162 (8), 91 (l00).
By oxidative cyclisation of cinnamylidene propiophenone
3,5-Dibenzyloxycinnamaldehyde (i) 3,5-Dibenzyloxycinnamic acid . A mixture of
3,5-dibenzyloxybenzaldehydell (3g, 9.4x 10'3 moles) , malonic acid (Jg, 9.4x lO,3 moles) and ~-alanine (0.3 g) in dry pyridine was heated on a water-bath for 4 hr. The reaction mixture was diluted with ice-cold water and acidified (dil. HCI). Crystallisation from ethanol gave white solid, mp 144-45 DC. yield 2.9 g (85 %); IR: 1680, 1630, 1590 cnfl; IH NMR: 8 4.9 (s, 4H , 2xCH2Ph), 6.33 (d, IH, 1=16Hz, Ha), 6.4 (s, IH, H-4), 6.8 (s, 2H, H-2 and 6) , 7.2 (s, lOH, 2xPh-H), 7.3 (d, IH, 1=16 Hz, H~), 12.3 (s, IH, -COOH); MS: mlz 360 (15, M+, C23H200 4), 269 (24),178 (40), 91 (80).
(ii) Methyl-3,5-dibenzyloxycinnamate. To 3,5-dibenzyloxycinnamic acid (3g, 8xlO,3 moles) in absolute methanol (25 mL) was added conc. H2S04 (1 mL) and gently refluxed for 6 hr. Standard work-up gave the ester as white solid, crystallised from petroleum, mp 72-73 DC, yield 2.5 g (80%); IR: 1680, 1600 cm· 1
; IH NMR: 83.8 (s, 3H, -COOCH3), 4.9 (s, 4H, 2xCH2Ph), 6.3 (s, IH, H-4), 6.4 (d, IH, 1=16 Hz, Ha), 6.8 (s, 2H, H-2 and 6), 7.3 (s, lOH, 2xPh-H), 7.5 (d, IH, 1=16 Hz, H~).
(iii) 3,5-Dibenzyloxycinnamyl alcohol. The methyl cinnamate was reduced with LAH in THF. The cinnamyl alcohol obtained was crystallised from ethanol as white solid, mp 82-83 DC, yield 1.3 g (56%); IR: 1600 cm-I; IH NMR: 8 4.0 (d, 2H, -CH20H), 4.9 (s, 4H, 2xCH2Ph), 6.3 (s, IH, H-4) , 6.5 (d, IH, 1=16 Hz, Ha), 6.9 (s, 2H, H-2 and 6), 7.2 (s, 10H, 2xPh-H), 7.4 (d, IH, 1=16 Hz, H~) .
(iv) 3,5-Dibenzyloxycinnamaldehyde. Pyridinium chlorochromate (1.5 g, 6.5x l0-3 moles) was added to a solution of cinnamyl alcohol (1.2 g, 3.4xlO,3 moles) in dry dichloromethane (30 mL) and gently refluxed for 6 hr. The reaction mixture was poured into
saturated brine solution and then repeatedly extracted with dichloromethane. The dichloromethane solution was filtered through a small bed of Si02 gel. The cinnamaldehyde was obtained as a light yellow oil, yield 0.9 g (75%); IR: 1680, 1600 cm-I; 'H NMR: 8 4.8 (s, 4H, 2xCH2Ph), 6.5 (s, IH, H-4) , 6.6 (dd, IH , 1=16 and 6 Hz, Ha), 6.8 (s, 2H, H-2 and 6), 7.2 (s, lOH, 2xPh-H), 7.7 (d, IH, 1=16 Hz, H~) , 9.8 (d, IH, 1=6 Hz, -CHO) (Found: C, 79.81; H,:6.25; C23H200 3 requires C,80.23; H, 5.81 %) .
2-Hydroxy-3,4,6-trimethoxy-l-(3' ,5' -dibenzyloxycinnamylidenepropiophenone). A mixture of 2-hydroxy-3,4,6-trimethoxypropiophenone (0.15 g, 6.3xl0-4 moles) and 3,5-dibenzyloxycinnamaldehyde (0.206 g, 6.0x I0-4 moles) in ethanol (15 mL) was refluxed with anhydrous Ba(OHh (0.3 g) for 8 hr. The excess alcohol was removed under vacuum, diluted with ice-water followed by acidification. It was extracted with EtOAc, dried and evaporated . The product was purified by column chromatography using pet. ether-ethyl acetate gradient. Golden yellow oil was obtained, yield 0.25 g (70%); AC20-H2S04: Red; IR: 1630 cm· l; IHNMR: 82.08 (d, 3H, 1=IHz, CH3), 3.82, 3.85 and 3.88 (s, each, 9H, 3xOCH3), 5.05 (s, 4H, 2 x CH2Ph); 6.0 (S, 1 H, H-5) , 6.7(m, 3H, C=CH-CH=CH-), 7.2 (s, lOH, 2xCH2-Ph), 7.4(m, 3H, Ar-H),9.6(s, 1 H, OH); MS: mlz 566 (M+' C3s H340 7).
Cyclisation of cinnamylidene propiophenone. A mixture of cinnamylidene propiophenone (0.1 g, 1.8 x 10-4 moles) in dimethyl sulphoxide (4 mL) and iodine (10 mg) was heated at l20DC for 2 hr. It was then cooled and treated with aqueous sodium thiosulphate (10%, lOmL). The chromone was collected by filtration, washed with water and crystallized from ethanol,mp 185-87DC, yield 0.07 g(68%). The chromone was identical in all respects with the chromone 4 prepared by BVT.
Dealkylation (i) HCI-AcOH. The chromone 4 (0.5 g, 9 x 10.4
moles) was heated with conc.HCI (2 mL, d 1.16, E.Merck) and glacial AcOH (4 mL) at 100DC for 2 hr. Usual work-up gave chromone 5 which was purified by column chromatography (Si02, 60-120 mesh) using EtOAc-benzene (3: 17,v/v) as an eluant. Compound 5 was obtained as pale yellow solid, mp 21O-14°C, yield 0.15 g (44%), IH NMR:82.1 (s, 3H, CH3) 3.9(s, 3H, OMe), 4.0(s, 6H, 2 x OMe), 6.5(s, 1 H, H-6), 6.3-6.6(m, 3H, Ar-H), 6.9(d, 1 H, 1 = 16 Hz, Ha), 7.4, (d, IH, 1= 16 Hz, H~); MS: mlz 384(M+ ,

282 INDIAN J CHEM., SEC B, APRIL 2001
20 %, C2IH2007), 383 (100),369 (81), 356 (27), 350 (22), 211 (7) , 195 (27), 167 (85).
Acetate of 5. The above chromone 5 (O.lg, 2.4xlO'4 moles) was acetylated with pyridine (2 mL) and acetic anhydride (0,5 mL) at room temperature overnight. The acetate was crystallised from ethanol, mp 189-90°C, yield 0,09 g; IH NMR: 2.1 (s, 3H, CH3), 2.3 (s, 6H, 2 x COCH3), 3.9 (s, 3H, OCH3), 4.1 (s, 6H, 2xOCH3), 6.4 (s, IH, H-6), 7.2 (d, IH, 1=16 Hz, Hf3), 7.4 (m, 3H, H-2',6',4'), 7.7 (d, IH, 1=16 Hz, Hf3 ); MS: mlz 468(27, M+, C25H~409), 467 (100),452 (70), 438 (19), 410 (23), 368 (11), 211 (8), 195 (20), 167 (43).
(ii) AICh-Ether. The chromone 4 (O.lg, 1.7 x 10'4 moles), ether (25 mL) and anhydrous AICI3 (0.29 g, 2.5x 10'3 moles) were stirred at room temperature for 24 hr. Ether was distilled off and the brownish aluminium chloride complex decomposed with icecold HCI and decomposition completed by warming on a water-bath at 45°C.The dark yellow product obtained was filtered, washed with dil. HCI, water and dried. The solid was chromatographed over silica gel with benzene-ethyl acetate gradient yielding compound 6, mp 165-67°C and compound 7, mp 240-41°C.
(iii) AICh-Nal-chromone-CH3CN. To a stirred solution of 4 (0.1 g, 1.7x lO-4 moles) in dry acetonitrile was added aluminium chloride (0.2 g, 1.7xlO,3 moles) and sodium iodide (0.25 g, 1.7xlO,3 moles) and sodium iodide (0.25 g, 1.7xlO'3) at O°c. The stirring was continued for 18 hr while the temperature was gradually raised to room temperature. The reaction mixture was carefully added into water and extracted with ethyl acetate. The organic phase was washed with 15% aqueous sodium thiosulphate and water. Removal of the solvent gave a yellow solid. The solid was chromatographed over silica gel with benzeneethyl acetate gradient and two main compounds 6 and 7 were isolated.
3-Methyl-5-hydroxy-7, 8-dimethoxy-3', 5'-dibenzyloxy-2-styrylchromone 6'. mp 165-67°C, cone. H2S04: orange color; UV: Amax (logE): 265 (3.1), 350 (4.24); +AICI3: 365; IR : 1635, 1600, 1580 cm'l and other bands; MS: mlz (%) 550(4, M+, C34H300 7), 549 (10),534 (6),459 (43),181(7), 91 (100).
3-Methyl-5,5' -dihydroxy-7, 8-dimethoxy-3' -benzyloxystyrylchromone 7. mp 240-41 °C; conc. H2S04 :
orange color; FeCI3: dark green brown color; UV: Amax (logE): 260 (3.01), 350 (4.3); +AICI3: 368; IR: 1630, 1600 cm' l; MS: mlz (%) 460 (18, M+,
C27H2407), 459 (46), 445 (15),444 (44), 368 (5), 181 (5), 91 (100).
AICh-Nal-chromone-CH3CN. 7 ~8-Dimethoxy-5, 3' ,5' -trihydroxy-2-styrylchromone (6-desmethyoxyhormothamnone 2). The chromone 4 (0.1 g, 1.7 x 10-4 moles), aluminium chloride (0.4 g, 3.4 x 10'3 moles) and sodium iodide (0.5 g, 3.4 x 10'3 moles) in dry acetonitrile were stirred at room temperature for 12 hr. Usual work-up gave a yellow sol id. The solid was chromatgraphed over silica gel using benzeneethyl acetate gradient. Fractions elu ted were pooled and evaporated yielding chromatographically pure substances. Compound 7, mp 240-41 °C (0.015 g) and compound 2, mp 255-60°C, yield 0.04g (55%); UV:Amax (log E): 265(3.01), 350 (4.3); +AICI3: 368; IR: 1630 cm'l; MS: mlz (%) 370 (18 , M+,C20HI807) , 356 (64), 342 (20), 230 (42), 138 (18).
Acetate. The above chromone 2 was acetylated with pyridine and acetic anhydride at room temperature overnight. The product was chromatographed (silica gel) using petroleum-ethyl acetate (4: 1; v/v), mp 182-84°C (lit? mp 184-86°C) ; UV:Amax (logE): 326 (4.75); IR: 1770, 1620 cm'l and other bands; IH NMR: 8 2.1 (s, 3H, CH3), 2.32 (s, 6H, 2 x OCOCH3), 2.4 (s, 3H, OCOCH3), 3.9 and 3.95 (s, each 3H, 2 x OCH3), 6.6 (s, IH, H-6), 6.9 (2, 1 H, H-4'), 7.0 (d, IH, 1=16 Hz, Ha), 7.2 (d, 2H, H-2",6'), 7.6 (d, IH, 1=16 Hz, Hf3).
BF3.Et20-NaI-CH3CN. To a stirred solution of the chromone 4 (0.1 g, 1.7 x 10'4 moles) and anhydrous sodium iodide (0.102g, 6.8 x 10'4 moles) in dry acetonitrile at O°C was added slowly a solution of freshly distilled borontrifluoride et erate (0.1 mL, 8.5 x 10'4 moles) in acetonitrile during 10-15 min. The mixture was stirred at O°C and then at room temperature for 20 hr. The reaction mixture was poured into ice-cold water (20 mL), treated with aqueous sodium thiosulphate (15%, 0.5 mL) and extracted. The combined organic extracts on evaporation yielded a yellow solid product. Column chromatography (silica gel) using benzene-ethyl acetate gradient gave a fraction eluted with C6H6-
EtOAc (3: 1) which was rechromatographed to get a pale yellow compound 5, mp 2 I 2- 14 °C; H2S04:
orange colour; FeCb: slight intensification of yellow colour.
Acetate. The above chromone was acetylated lI sing pyridine-acetic anhydride by stirring overnight at room temperature. The reaction mixture on usual work-up gave acetate of 5; mp 189-90°C.

JAIN et 01.: SYNTHESIS OF HORMOTHAMNIONE 283
Hormothamnione. Dealkylation (excess AICh NaI in CH3CN). The chromone 33 (0.1 g, 1.7 X 10.4
moles), aluminium chloride (0.65 g, 5.1 x 10.3 mole) and sodium iodide (0.102 g, 6.8 x 10'4 moles) in dry acetonitrile were stirred at room temperature for 24 hr. Usual work-up gave a yellow solid. The solid was chromatographed over silica gel usi ng benzeneethyl acetate gradient. The major yellow band was eluted by C6H6-EtOAc; mp 250°C (lit. I mp 270°C, natural hormothamnion), yield 0.04 g (55%). FeCI3: dark green brown colour; IR: 3400, 1635 cm'l; UV: Amax (log E): 295 (4.05),353(4.29); +AICI3: 370; MS: m/z 400 (M+ C2IH200g).
Acetate. The chromone was acetylated with pyridine and acetic anhydride at room temperature overnight. The acetate was purified by column chromatography (silica gel) using petroleum-ethyl acetate (4:1, v/v), mp 199-201 oC (lit. ' mp 198-200°C for the acetate from natural hormothamnione); IR: 1780, 1635 cm'l ; 'H NMR:8 2. 17 (s, 3H, CH3) , 2.35 (s, 6H, 2 x OCOCH3), 2.5 (s, 3H, COCH3), 3.89, 4.0,4,2 (s, 3H each, OCH3), 6.9 (s, IH, H-4), 7.0 (d, IH, J=16 Hz, Ha), 7.2 (s, 2H, H-2',6'), 7.6 (d, IH, J=16 Hz, H~); MS: mlz 526 (M+, C27H26011).
Acknowledgement We thank CSIR, New Delhi for providing Research
Fellowship to two of us (NJ and GG).
References I Gerwick W H, Albert Lopez, Van Duyne G D, Clardy J, Ortiz
W & Baez A, Tetrahedron Lett, 27(18), 1986, 1979.
2 Gerwick W H, J Nat Prod, 52(2), 1989, 252.
3 Ricardo Alonso & Arnold Brossi , Tetrahedron Lett, 29, 1988, 735.
4 Ayyangar N R, Khan R A & Deshpande V H, Tetrahedron Lett, 29, 1988, 2347 .
5 McGarry L W & Detty M R, J Org Gem, 55, 1990,4349.
6 (a) Baker W, J Chem Soc, 1933,1381.
(b) Mahal H S & Venketaraman K, J Chelll Soc, 1934, 1767.
7 Doshi A K, Soni P A & Ghiya B J, Indian J Chell/, 25 B. 1986,759.
8 Node M, Ohta K, Kajimoto T, Nishide K, Fujita E & Fuji K,Che11l Pharlll Bull, 31( 11), 1983, 4178.
9 Krishnamurty H G & Seshadri T R, J scient, ind Res, 198. 1962,115.
10 Krishnamurty H G. Synthesis and ring isolll eric change ill flavallones and conversion of anthoxanthins illlo anthocyallidills .Ph.D Thesis University of Delhi,1962 , pp.19.
II Mandai A K,Soni N R & Ratnam K R, Synthesis, 1985, 274. 12 Millen J, Riley T N, Waters I W & Hamrick M E, J Med
Chem, 28, 1985, 12.