Embed Size (px)
Citation preview

0020-1685/03/3907- $25.00 © 2003
MAIK “Nauka
/Interperiodica”0687
Inorganic Materials, Vol. 39, No. 7, 2003, pp. 687–693. Translated from Neorganicheskie Materialy, Vol. 39, No. 7, 2003, pp. 812–819.Original Russian Text Copyright © 2003 by Molchanov, Maksimov, Maksimovskaya, Goidin, Buyanov.
INTRODUCTION
Heteropoly acids (HPAs) with the general formula
H
a
Z
b
M
c
O
n
(Z = heteroatom; M = Mo, W, V in theirhigher oxidation states) are effective catalysts [1–6]. Atpresent, HPAs are used in a number of commercial pro-cesses: hydration of olefins, tetrahydrofuran polymer-ization, alkylation of phenols, oxidation of metacrolein,and others [2, 5]. In recent years, there has been intenseinterest in HPA salts as acidic catalysts [3–6].
Although the high effectiveness of HPA catalystshas been documented in a large number of studies, theircommercial application is limited by the lack of simple,environmentally safe processes for the production ofHPAs and by their high production cost, which is asso-ciated, in large measure, with the complexity of theexisting preparation procedures. The latter include thepreparation of salts and subsequent acid extraction withethers and also long-term boiling of an oxide suspen-sion. These processes are accompanied by the forma-tion of large amounts of acid waste and noxious gasesand are fire-hazardous and power-consuming. More-over, they involve a large number of stages and ensureonly a limited yield of HPAs (60–80%) [7, 8].
An attractive alternative is the direct synthesis ofHPAs via the reaction of molybdenum, tungsten, andvanadium oxides with compounds of heteroatoms andwater. Such reactions do take place during long-term(tens of hours) boiling in the case of some phosphomo-lybdic and phosphomolybdovanadic HPAs, but theiryields are rather low [7–9].
The rate of reactions with the participation of oxidescan be raised via mechanochemical activation (MCA),one of the most effective means of accelerating chemi-cal processes [10]. The purpose of this work was todevelop a procedure for the preparation of HPAs (pri-marily, those with the Keggin structure,
H
a
ZM
12
O
40
)with the use of MCA and to study the processesinvolved.
EXPERIMENTAL
MCA of individual oxides or oxide mixtures wasconducted in an AGO-2 planetary centrifugal mill atrotation rates of 10–17 rps, using 150-cm
3
stainless-steel grinding vessels and steel balls 5 mm in diameter.The ball load was 0.2 kg, and the sample weight wasvaried in the range 0.005–0.1 kg. All of the startingchemicals were of analytical grade.
HPA solutions were prepared by dissolving acti-vated oxides or oxide mixtures in aqueous phosphoricacid or water at a fixed temperature. If prolonged heat-ing was needed, we used a reflux condenser. To isolatea solid HPA, the solvent was evaporated to dryness. Theformation of HPAs was followed using NMR and IRspectroscopic techniques.
51
V and
31
P NMR spectra were measured on a BrukerMSL-400 spectrometer at 105.2 and 161.98 MHz,respectively, using VOCl
3
and H
3
PO
4
as standards forchemical-shift calibration.
Synthesis of Heteropoly Acids and Their Salts Using Mechanochemical Activation
V. V. Molchanov, G. M. Maksimov, R. I. Maksimovskaya, V. V. Goidin, and R. A. Buyanov
Boreskov Institute of Catalysis, Siberian Division, Russian Academy of Sciences, pr. Akademika Lavrent’eva 5, Novosibirsk, 630090 Russia
e-mail: [email protected]
Received November 12, 2002; in final form, February 5, 2003
Abstract
—A method is proposed for the synthesis of heteropoly acids from oxides of molybdenum, tungsten,and vanadium via mechanochemical activation. The fundamental principles of this approach to the synthesis ofheteropoly acids containing different ligands and heteroatoms are formulated. The new V
2
O
5
· n
MoO
3
com-pounds synthesized in this work are found to be highly reactive with phosphoric acid, which is due to the unsat-urated coordination of the vanadium cations and the low structural perfection of these compounds. The appli-cation area of the proposed method is established. It appears to be most effective in the synthesis of phospho-molybdovanadic and phosphomolybdic heteropoly acids. In some instances, heteropoly acids can be preparedby solid-state reactions, which makes it possible to use V
2
O
5
· n
MoO
3
compounds with
n
≤
6 as startingreagents. The method has a number of important advantages: the process is waste-free, requires a shorter syn-thesis time and involves a smaller number of steps as compared to the existing processes, affords an increasedyield of heteropoly acids, and involves no explosion- or fire-hazardous steps.

688
INORGANIC MATERIALS
Vol. 39
No. 7
2003
MOLCHANOV
et al
.
IR spectra were taken with a Specord-75IR spectro-photometer, using the KBr disk method.
RESULTS AND DISCUSSION
Our experiments demonstrate that MCA is an effec-tive means of enhancing the reactivity of tungsten,molybdenum, and vanadium oxides. For example, theactivation of individual molybdenum and vanadiumoxides notably enhanced their reactivity with hydrogenperoxide solutions: the oxides could easily be dissolvedto give peroxy acids. MCA also raised the reactivity ofmolybdenum and tungsten oxides with H
3
PO
4
solu-tions. As a result, we could obtain solutions of phos-phoric HPAs.
Note that, in our experiments,
H
3
PW
12
O
40
has forthe first time been prepared by the direct reaction oftungsten oxide with an H
3
PO
4
solution. Similar synthe-ses were described in [9, 11], but, in those studies,phosphotungstic HPAs were obtained with the use of afreshly precipitated tungstic acid rather than tungstenoxide.
MCA has an even stronger effect on the reactivity(solubility) of mixtures of molybdenum and vanadiumoxides, which react with H
3
PO
4
solutions more rapidlythan do activated molybdenum oxides. The reactivity ofthe mixtures increases with vanadium oxide content(Table 1).
Understanding the mechanisms responsible for theincrease in reactivity upon MCA or upon an increase invanadium content is of key importance in developingthe procedure for HPA synthesis. This led us to focus onthe processes involved in MCA.
MCA of mixtures of molybdenum and vanadiumoxides leads to the formation of phases of variable com-position with the general formula
V
2
O
5
·
n
MoO
3
, where
n
= 2–22. These phases are close in structure to orthor-hombic MoO
3
and consist of alternating layers typicalof the structures of molybdenum and vanadium oxides[12]. The formation of these phases seems to be respon-sible for the enhanced reactivity of the activated mix-
tures, which can be understood in terms of the structureof vanadium oxide. According to Enjalbert and Galy[13], its structure is made up of polyhedral bilayersformed by chains of square pyramids. Every two pyra-mids share an edge, and the apices of neighboring pairspoint in opposite directions. Upon the formation ofbonds between the layers, the apical oxygens approachthe bases of the pyramids in the adjacent layer to formoctahedra. Breaking of interlayer bonds leads to theformation of fragments consisting of chains of squarepyramids. The unsaturated coordination seems to facil-itate the incorporation of PO
4
groups, which leads tothe formation of Keggin anions. One of the oxygens ofeach PO
4
group falls in the nearest neighbor environ-ment of a vanadium atom, and the coordination of thelatter becomes octahedral. In the structure of molybde-num oxide, the octahedra remains intact upon thebreaking of interlayer bonds, and the incorporation ofPO
4
groups requires more complex structural rear-rangements.
In what follows, this picture will be corroborated byIR spectroscopy data. At the same time, the formationof HPAs via dissolution is also possible. It is wellknown that MCA increases both the dissolution rateand the solubility of substances [10]. Indeed, weobserve dissolution of small amounts of activatedmolybdenum and vanadium oxides upon heating of theaqueous suspension. This mechanism, however, is inca-pable of accounting for some of our results.
The new molybdenum–vanadium oxide compoundsare metastable, as evidenced by the reduction in theirreactivity during long-term (more than six months)storage and their dissociation into the constituentoxides upon heating. We believe that the high reactivityof the new compounds, as well as their metastability,results from a high defect density. Their low structuralperfection is a consequence of both the MCA process,which produces large amounts of structural defects, andtheir unusual structure, made up of alternating octahe-dral (MoO
3
) and square-pyramidal (V
2
O
5
) layers.
The reactivity of tungsten–vanadium oxide com-pounds with H
3
PO
4
also increases upon MCA, but theHPA yield is rather low here. Preliminary results sug-gest that, in this system, MCA also gives rise to chem-ical interaction between oxides with the formation ofnew compounds. The same is evidenced by x-ray dif-fraction (XRD) data for a sample of composition1.5V
2
O
5
· 9WO
3
: the XRD pattern of this sample showsextremely weak diffraction peaks from V
2
O
5
, similar towhat was found in the V
2
O
5
–MoO
3
system [12].
In the
51
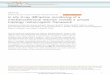
V MAS NMR spectra of the compounds inquestion, the peaks characteristic of V
2
O
5
are also veryweak (Fig. 1). Since molybdenum and tungsten oxidesdiffer in crystal structure (tungsten oxide has a frame-work, rather than layered, structure of the ReO
3
type),the amount of groups capable of transforming into theKeggin anion is here much lower in comparison with
Table 1.
Effect of the activated-mixture composition onthe time needed for its dissolution in aqueous H
3
PO
4
withthe formation of a 10% HPA solution (MCA time, 1800 s;rotation rate, 11 rps; ball load/sample weight ratio, 40;(Mo + V) : P = 12)
Composition
T
, K
τ
, s
V
2
O
5
· 22MoO
3
353 420
V
2
O
5
· 10MoO
3
353 240
1.5V
2
O
5
· 9MoO
3
353 10
V
2
O
5
· 4MoO
3
298 15
V
2
O
5
· 2MoO
3
298 10

INORGANIC MATERIALS
Vol. 39
No. 7
2003
SYNTHESIS OF HETEROPOLY ACIDS AND THEIR SALTS 689
the molybdenum–vanadium compounds, which seemsto be responsible for the low HPA yield even after high-energy activation.
Taking advantage of MCA, we developed a proce-dure for the preparation of HPAs which is particularlyeffective in the case of phosphomolybdovanadic andphosphomolybdic HPAs. This procedure involves onlytwo or three steps: MCA of individual oxides or oxidemixtures, reaction of activated oxides with an aqueoussolution containing an appropriate amount of H
3
PO
4
,and (occasionally) isolation of solid acids by boilingdown the solution. In this way, we synthesized a numberof HPAs with the general formulas
H
3 +
m
PM
12 –
m
V
m
O
40
and
H
3
PMo
12 –
m
W
m
O
40
, where M = Mo or W and
m
=0–4, and also
H
6
P
2
Mo
18
O
62
.
To optimize the conditions of HPA synthesis, weexamined the effects of the MCA duration, rotation rateof the grinding vessel, ball load/sample ratio, dissolu-tion temperature, and H
3
PO
4
concentration on the reac-tion rate and conversion.
The MCA time was found to have a significanteffect on the reactivity of oxides (Tables 2, 3). At MCAtimes shorter than 600 s, the desired HPA yield couldnot be attained.
Accelerating the rotation rate of the grinding vesselhas a significant effect on the reactivity of WO
3
andWO
3
+ V
2
O
5
mixtures and reduces the activation timenecessary for the desired HPA yield (Table 4). Oxidesactivated at rotation rates below 17 rps do not react withH
3
PO
4
solutions. Note that, even after activation, tung-sten oxide and its mixtures with vanadium and molyb-denum oxides have low reactivities, and the HPA yieldin terms of tungsten is as low as 10–12% even afterlong-term boiling.
At fast rotation rates, the three oxides lose anamount of oxygen. As a result, subsequent reactionwith an H
3
PO
4
solution yields reduced HPAs, which arereadily oxidized, e.g., by hydrogen peroxide.
The production rate of a planetary mill depends onthe amount of oxides being activated, while the activa-tion efficiency increases with decreasing sample/ballload ratio. For this reason, it is important to optimizethis ratio so as to ensure high activation efficiency.
The optimal sample/ball load ratio was found to be1 : 5 or smaller. At larger ratios, the reaction betweenoxides and aqueous H
3
PO
4
took much longer times(Table 5).
We also optimized the parameters of the reactionbetween oxides and H
3PO4 solutions. With stoichio-metric amounts of oxides and H3PO4, we obtainedH3 + mPMo12 – mVmO40 acids, independent of the reac-tion temperature, which influenced the dissolution rateonly. The solution concentration could be as high as60 wt %. Although, in earlier studies [8], only HPAswith m = 0–3 were identified, we also observed com-plete dissolution of oxide mixtures with m = 4–6.
According to 51V NMR data, the resulting solutionscontained mixtures of HPAs with m = 1–4, and the
excess vanadium was present in the form of V .Tungstovanadic HPAs were only formed upon long-term boiling of an oxide suspension in an H3PO4 solu-tion, but the yield was rather low even when H3PO4 waspresent in large excess of the HPA stoichiometry.According to 31P NMR data, the reaction between acti-vated mixtures and H3PO4 yields mixtures of isomers ofphosphomolybdovanadic and phosphotungstovanadicHPAs with the Keggin structure, which have the samecomposition as in other syntheses [14].
The composition of the dissolution products of acti-vated molybdenum oxide in aqueous H3PO4 dependsstrongly on process conditions: temperature, MoO3content of the suspension, and Mo : P ratio. AtMo : P ≤ 12, complete dissolution can be only achievedat MoO3 concentrations in the suspension no higherthan 10 wt %. At Mo : P = 12, an MoO3 content of10 wt %, and a solution temperature of 100°C, the mainreaction product (up to 75 wt %) is H3PMo12O40. Boil-ing down the solution raises the concentration of the
O2+
2000150010005000–500
2
1
Chemical shift, ppm
Fig. 1. 51V MAS NMR spectra of (1) V2O5 and(2) 1.5V2O5 · 9WO3 after MCA.
Table 2. Effect of the MCA duration on the time needed for ox-ide dissolution with the formation of a 10% H7PMo8V4O40 so-lution (rotation rate, 11 rps; ball load/sample weight ratio, 40)
τMCA, s T, K τ, s
600 373 7200
1200 373 900
1800 373 6
2400 373 6
1800 323 10
1800 298 15

690
INORGANIC MATERIALS Vol. 39 No. 7 2003
MOLCHANOV et al.
acid, and it can be isolated with an acceptable yield. Atlower MoO3 concentrations, or temperatures, or Mo : Pratios, the solutions contain a mixture of up to five dif-ferent phosphomolybdic HPAs, of which onlyH3PMo12O40 and H6P2Mo18O62 could be identified withcertainty (31P NMR chemical shifts of –3.7 and−2.9 ppm, respectively). Such solutions are in a non-equilibrium state. Aging for about one month ensurescomplete equilibration, and unreacted H3PO4 disap-pears. In the range 9 < Mo : P < 12, the solution agedfor a month contains only the above two acids. AtMo : P = 9, after aging for a month the solution con-tains H6P2Mo18O62 of 94–98% purity [15].
By boiling down the solution, this HPA can be iso-lated in solid form. At Mo : P < 9, the solutions aged for
about one year also contain only two acids:H6P2Mo18O62 and, surprisingly enough, H6P2Mo5O23
(31P NMR chemical shift of +1.4 ppm). The relativeamounts of these acids are determined by the Mo : Pratio. For example, at Mo : P = 6, the H6P2Mo18O62 :H6P2Mo5O23 molar ratio is ≈1.2. However, the latteracid could not be recovered from the solution becauseof its instability.
The drawback to all of the known synthesis meth-ods, including the one with the use of MCA, is thatsolid HPAs must be recovered from solution. In view ofthis, we attempted solid-state synthesis via mecha-nochemical interaction between oxides or between thenew compounds identified here and compounds con-taining a heteroatom.
MCA of molybdenum oxide with an H3PO4 solutionled to a high degree of MoO3 reduction, and no HPAwas formed. In the mechanochemical reaction betweenpreactivated molybdenum oxide and phosphoric acid,the degree of MoO3 reduction was substantially lower,but no HPA was formed either. Room-temperature stor-age of samples for one month and exposure of the acti-vated mixture to water vapor also did not lead to the for-mation of HPAs.
Attempts to synthesize H3 + mPMo12 – mVmO40 HPAswere more successful. According to IR spectroscopydata, the MCA of mixtures of molybdenum and vana-dium oxides and phosphoric acid or a mixture ofV2O5 · nMoO3 compounds (n = 10 and 22) and phos-phoric acid leads to the formation of structures close insymmetry to the Keggin anion (Fig. 2). Such mixturesare however difficult to dissolve in water at room tem-perature. For this reason, we could not identify HPAswith certainty. The IR data confirm the above assump-tion that V2O5 · nMoO3 compounds contain structuralelements similar to some fragments of the Kegginanion. Heating such mixtures in water, we obtain solu-tions of the corresponding HPAs. The rate of this pro-cess is comparable to the rate of V2O5 · nMoO3 dissolu-tion in aqueous H3PO4.
H3 + mPMo12 – mVmO40 HPAs with m = 3 and 4 couldbe prepared by solid-state reactions between oxides andH3PO4. This is confirmed by IR spectra of activatedmixtures (Fig. 2), which show absorptions characteris-tic of the Keggin anion [16]. The resultant samplesreadily dissolve in water at room temperature. Accord-ing to IR results, HPAs are formed after just 60 s of acti-vation. 31P MAS NMR data confirm the formation ofHPAs via solid-state reactions (Fig. 3).
Salts of HPAs can be prepared in a similar manner.For example, MCA of a mixture of V2O5 · 4MoO3 andNa3PO4 leads to the formation of Na3H4PMo8V4O40(Fig. 3). Note that water plays an important role in thisreaction. HPAs and their salts can be prepared via MCAonly if the mixture being activated contains an amountof water sufficient for the formation of a crystalhydrate. In anhydrous mixtures, no HPAs are formed.
Table 3. Effect of the MCA duration on the time needed forMoO3 dissolution in aqueous H3PO4 (Mo : P = 11) with theformation of a 10% solution of phosphomolybdic HPAs (ro-tation rate, 17 rps; ball load/sample weight ratio, 40)
τMCA, s T, K τ, s
60 373 600
180 373 240
300 373 60
600 373 10
600 298 600
Table 4. Effect of the rotation rate of the grinding vessel onthe time needed for MoO3 dissolution in aqueous H3PO4 withthe formation of a 10% H3PMo12O40 solution (solution tem-perature, 373 K; ball load/sample weight ratio, 40)
Rotation rate, rps τMCA, s
11 1800
14 1500
17 1020
Table 5. Effect of the ball load/MoO3 weight ratio on thetime needed for MoO3 dissolution with the formation ofa 10% solution of phosphomolybdic HPAs (Mo : P = 11) (ro-tation rate, 17 rps; MCA duration, 1200 s; solution tempera-ture, 373 K)
Ball load/MoO3 weight ratio τ, s
40 10
20 10
10 15
5 60
2 600

INORGANIC MATERIALS Vol. 39 No. 7 2003
SYNTHESIS OF HETEROPOLY ACIDS AND THEIR SALTS 691
The most likely reason is that vigorous mechanicalmixing of a wet mixture creates a state which is close innature to an incongruent melt or to a state called apseudoliquid HPA [4]. This enhances the reactivity ofthe substances involved. That neither phosphomolyb-dovanadic HPAs nor their salts can be prepared viaMCA of anhydrous mixtures is also associated with thefact that these compounds are only stable in the form ofcrystal hydrates [8].
The formation of HPAs and their salts via solid-statereactions can hardly be accounted for by hydrolyticprocesses because the amount of water in the mixtureswas sufficient for the formation of crystal hydrates butinsufficient for dissolution: the amount of oxides wasabout ten times that of water. Hydrolytic processes alsocannot account for the marked difference in reactivitybetween V2O5 · 10MoO3 and V2O5 · 4MoO3, since thesecompounds are close in solubility in water, while therates of HPA formation from these compounds differ byorders of magnitude.
In addition to phosphoric HPAs, we attempted toprepare acids containing Si as a heteroatom. Mixturesof SiO2 and oxide precursors for ligands were subjectedto MCA and then heated in water with stirring. Theresultant solutions were boiled down. The formation ofHPAs was checked by IR spectroscopy and by deter-mining the amounts of dissolved oxides.
In the mechanochemical synthesis of H4SiMo12O40from stoichiometric mixtures of SiO2 and MoO3, theyield of the HPA is very low. Increasing the percentageof SiO2 increases the HPA yield (Table 6). The likelyreason for this behavior is that, at a higher SiO2 content,the probability of a SiO2 particle coming in contact withmolybdenum oxide is also higher.
A similar effect of the starting-mixture compositionon the HPA yield was observed in the synthesis of sili-cotungstic HPAs (Table 7), but the yields of
H4SiW12O40 and H6SiW10V2O40 were not very high.Note that, in our experiments, silicotungstic HPAs, aswell as phosphotungstic HPAs, have for the first timebeen synthesized by the direct reaction between oxides.
In addition to the reactions above, we have shownfor the first time that it is possible in principle to syn-thesize molybdic HPAs containing other heteroatoms,in particular Al and Fe. Such acids were formed onlywhen aluminum and iron hydroxides were used as start-ing reagents. With oxides, no HPAs were obtained. Inthe instance of aluminum, the synthetic procedure issimilar to that for the silicomolybdic HPA. To obtain aferromolybdic HPA, the activation of the starting mix-ture must be followed by treatment with a hydrogenperoxide solution during heating. Neither of these acidscan be isolated in pure form because both are stableonly in solution. For this reason, they were isolated andidentified in the form of tetrabutylammonium salts.According to IR absorption data (Fig. 4), we obtained atetrabutylammonium salt of a new HPA with the com-position H5FeMo12O40. The IR spectrum showed a setof absorptions characteristic of anions with the Keggin
11001000800600500300
12
400 700 900 1200
Wavenumber, cm–1
3
45
Fig. 2. IR spectra of V2O5 · nMoO3 + H3PO4 mixtures withn = (1) 22, (2) 10, and (3–5) 6 after MCA for (1, 2) 5, (3) 1,(4) 2, and (5) 3 min.
30200–10–20–30
1
2
10
Chemical shift, ppm
3
Fig. 3. 31P MAS NMR spectra of (2) V2O5 · 4MoO3 +H3PO4 and (3) V2O5 · 4MoO3 + Na3PO4 · 10H2O mixturesafter MCA; (1) Na3H4PMo8V4O40 standard.
Table 6. Effect of the starting-mixture composition on theyield of silicomolybdic HPA (MCA duration, 1800 s; rota-tion rate of grinding vessels, 10 rps; boiling of the suspensionin water for 1800 s)
Weight percent Weight percent of MoO3 converted to HPASiO2 MoO3
1.0 99.0 10.2
7.7 92.3 36.7
17.2 82.8 60.3
33.3 66.7 78.5

692
INORGANIC MATERIALS Vol. 39 No. 7 2003
MOLCHANOV et al.
structure [16, 17]. In the case of Al, we obtained a mix-ture of salts of the HPAs H5AlMo12O40 and H9AlMo6O24,which can be distinguished in IR spectra [17].
We also attempted to synthesize HPAs with otherheteroatoms through dissolution or by solid-state reac-tions. The MCA of mixtures of molybdenum oxide andoxides of Sb, Ce, Ti, Ga, Se, and B (or boric acid), fol-lowed by heating in water, did not lead to the formationof HPAs. The MCA of a mixture of molybdenum oxideand iodic acid led to the formation of a mixture whichreadily dissolved in water at room temperature to forma weakly acidic solution. The acids formed were iso-lated in the form of a tetrabutylammonium salt.According to IR spectroscopy data, we obtained a mix-ture of isopolymolybdates rather than an iodomolyb-dic HPA.
Analysis of the successful and unsuccessful prepa-rations with the use of MCA indicates that HPAs can besynthesized if the starting compounds have layeredcrystal structures or if such structures are produced byMCA, as occurs in tungsten oxide owing to the forma-
tion of crystallographic shear planes. Otherwise, eitherno reaction takes place or other compounds are formed.
MCA was also used to synthesize salts of HPAs byreacting the acids with metal carbonates. The MCA ofmixtures of H3PW12O40 or H3PMo12O40 and Cs2CO3 orNH4HCO3 led to the formation of two or three substi-tuted insoluble salts of HPAs. In a similar manner, weobtained Cs1.5Ce0.5PW12O40. A more complex reactiontook place during the MCA of mixtures of phospho-tungstic HPA and bismuth and cerium carbonates.According to IR spectroscopy data, the activated mix-tures contained no HPA. The spectra showed absorp-tion bands characteristic of WO3, attesting to dissocia-tion of the heteropolyanion. Moistening and subse-quent drying of the mixtures led to the formation of theparent HPA and also Ce(III,IV)-containing HPAs. Acti-vation of mixtures of phosphotungstic HPA and SnO2led to irreversible decomposition of the acid. The MCAof H3PW12O40 produced no structural changes. Theonly result was a slight increase in specific surface.
CONCLUSIONS
The fundamental principles of the application ofMCA in the synthesis of HPAs containing differentligands and heteroatoms were formulated. The newV2O5 · nMoO3 compounds synthesized in this workwere found to be highly reactive with phosphoric acid,which is due to the unsaturated coordination of thevanadium cations and the low structural perfection ofthese compounds.
We suppose that the formation of phosphomolybdo-vanadic HPAs may follow two different mechanisms. Akey feature of one of them is the incorporation of PO4groups into the surrounding of the coordination-unsat-urated vanadium as a result of the breaking of bondsbetween vanadium oxide layers. The other mechanismis oxide dissolution followed by hydrolytic formationof Keggin anions.
The application area of the approach in question isestablished. It appears to be most effective in the syn-thesis of phosphomolybdovanadic and phosphomolyb-dic HPAs. For successful application of MCA, the start-ing compounds must have layered crystal structures or,at least, such structures must be produced by MCA. Insome instances, HPAs can be prepared by solid-statereactions, which makes it possible to use V2O5 · nMoO3compounds with n ≤ 6 as starting reagents.
The proposed approach to the preparation of HPAsand related catalysts [15, 18–21] has a number ofimportant advantages: the process is waste-free,requires a shorter synthesis time and involves a smallernumber of steps as compared to the existing methods,affords an increased yield of HPAs and reduced powerconsumption, and involves no explosion- or fire-haz-ardous steps.
Table 7. Effect of the starting-mixture composition on theyield of silicotungstic HPAs (MCA duration, 1800 s; rotationrate of grinding vessels, 10 rps; boiling of the suspension inwater for 1800 s)
Weight percent Weight percent of oxides converted to HPASiO2 WO3
8.1 91.9 7.7
17.0 83.0 9.2
29.0 71.0 13.4
SiO2 V2O5 WO3
8.1 6.7 85.2 9.1
17.0 6.0 77.0 19.6
29.0 5.2 65.8 20.4
12001000800600400
1
2
Wavenumber, cm–1
Fig. 4. IR spectra of tetrabutylammonium salts of (1) ferro-and (2) aluminomolybdic HPAs.

INORGANIC MATERIALS Vol. 39 No. 7 2003
SYNTHESIS OF HETEROPOLY ACIDS AND THEIR SALTS 693
ACKNOWLEDGMENTSThis work was supported by the Russian Foundation
for Basic Research (Support to Leading ScientificSchools Program), grant no. 00-15-97440).
REFERENCES1. Ono, Y., Heteropolyacid Catalysis—a Unique Blend of
Acid–Base and Redox Properties, Perspectives in Catal-ysis: Chemistry for the 21st Century, Thomas, J.M. andZamaraev, K.I., Eds., Oxford: Blackwell Scientific,1992, pp. 431–463.
2. Kozhevnikov, I.V., Catalysts for Fine Chemical Synthe-sis: 2. Catalysis by Polyoxometalates, Chichester: Wiley,2002.
3. Moffat, J.B., Metal–Oxygen Clusters: The Surface andCatalytic Properties of Heteropoly Oxometalates, NewYork: Plenum, 2001.
4. Okuhara, T., Mizuno, N., and Misono, M., CatalyticChemistry of Heteropoly Compounds, Adv. Catal., 1996,vol. 41, pp. 113–252.
5. Misono, M., Unique Acid Catalysis of HeteropolyCompounds (Heteropolyoxometalates) in the SolidState, Chem. Commun. (Cambridge), 2001, no. 13,pp. 1141–1152.
6. Okuhara, T., New Catalytic Function of HeteropolyCompounds as Solid Acids, Catal. Today, 2002, vol. 73,no. 1/2, pp. 167–176.
7. Handbuch der präparativen anorganischen Chemie indrei Bänden, von Brauer, G., Ed., Stuttgart: FerdinandEnke, 1981, 3rd ed. Translated under the title Rukovod-stvo po neorganicheskomu sintezu, Moscow: Mir, 1986,vol. 6.
8. Maksimov, G.M., Advances in the Synthesis of Polyox-ometalates and Investigation of Heteropoly Acids, Usp.Khim., 1995, vol. 64, no. 5, pp. 480–496.
9. Nikitina, E.N., Geteropolisoedineniya (HeteropolyCompounds), Moscow: Goskhimizdat, 1962.
10. Avvakumov, E.G., Mekhanicheskie metody aktivatsiikhimicheskikh protsessov (Mechanical Activation ofChemical Processes), Novosibirsk: Nauka, 1986.
11. Gu, Y., Active White Powdery Tungstic Acid—Its Prep-aration, Nature, and Wide Variety of Reaction Products,Proc. I Int. Conf. on Metal and Material Science: Tung-sten, Titanium, Rare Earths, and Antimony (Beijing,1988), Oxford, 1989, vol. 1, pp. 213–216.
12. Molchanov, V.V., Plyasova, L.M., Goidin, V.V., et al.,New Compounds in the V2O5–MoO3 System, Neorg.Mater., 1995, vol. 31, no. 9, pp. 1225–1229 [Inorg.Mater. (Engl. Transl.), vol. 31, no. 9, pp. 1121–1124].
13. Enjalbert, R. and Galy, J., Refinement of the Structure ofV2O5, Acta Crystallogr., Sect. C: Cryst. Struct. Com-mun., 1986, vol. 42, no. 11, pp. 1467–1469.
14. Odyakov, V.F., Zhizhina, E.G., Maksimovskaya, R.I.,and Matveev, K.I., Novel Methods for Synthesis ofPhosphomolybdovanadic Heteropoly Acids, Kinet.Katal., 1995, vol. 36, no. 5, pp. 795–800.
15. Maksimov, G.M., Maksimovskaya, R.I., Kozhevni-kov, I.V., et al., RF Patent 2019513, Byull. Izobret.,1994, no. 17, p. 81.
16. Rocchiccioli-Deltcheff, C. and Thouvenot, R., Com-
plexes métalliques des hétéropolyanions α-XM11
(X = Si, P; M = Mo, W): étude des modifications struc-turales du ligand au moyen de la spectrométrie de vibra-tion, J. Chem. Res., 1977, pp. 549–571.
17. Kazanskii, L.P. and Golubev, A.M., Vibrational Spectraof Heteropolyanions of Different Structure Types, inKhimiya soedinenii Mo(VI) i W(VI) (Chemistry ofMo(VI) and W(VI) Compounds), Novosibirsk, 1979,pp. 66–84.
18. Maksimov, G.M., Molchanov, V.V., and Goidin, V.V.,Novel Technologies of Heteropoly Acids and RelatedCatalysts, Khim. Prom–st. (Moscow), 1998, no. 10,pp. 599–601.
19. Molchanov, V.V., Maksimov, G.M., Goidin, V.V., et al.,RF Patent 2076070, Byull. Izobret., 1997, no. 9, p. 151.
20. Molchanov, V.V., Maksimov, G.M., Goidin, V.V., et al.,RF Patent 2076071, Byull. Izobret., 1997, no. 9, p. 151.
21. Maksimov, G.M., Timofeeva, M.N., Molchanov, V.V.,et al., RF Patent 2080923, Byull. Izobret., 1997, no. 16,p. 78.
O39n–