Embed Size (px)
Citation preview

Bioorganic & Medicinal Chemistry Letters 24 (2014) 4948–4953
Contents lists available at ScienceDirect
Bioorganic & Medicinal Chemistry Letters
journal homepage: www.elsevier .com/ locate/bmcl
Synthesis and preliminary investigations into novel1,2,3-triazole-derived androgen receptor antagonists inspiredby bicalutamide
http://dx.doi.org/10.1016/j.bmcl.2014.09.0360960-894X/� 2014 Elsevier Ltd. All rights reserved.
⇑ Corresponding author. Tel.: +61 (3) 52272767; fax: +61 (3) 52271045.E-mail address: [email protected] (L.C. Henderson).
Jarrad M. Altimari a,d, Birunthi Niranjan b, Gail P. Risbridger b, Stephanie S. Schweiker c, Anna E. Lohning c,Luke C. Henderson a,d,⇑a Strategic Research Center for Chemistry and Biotechnology, Deakin University, Pigdons Road, Waurn Ponds Campus, Geelong 3216, Victoria, Australiab Department of Anatomy and Developmental Biology, Faculty of Medicine, Nursing & Health Sciences, Monash University, Victoria 3800, Australiac Faculty of Health Sciences and Medicine, Bond University, Gold Coast 4229, Queensland, Australiad Institute for Frontier Materials, Deakin University, Pigdons Road, Waurn Ponds Campus, Geelong 3216, Victoria, Australia
a r t i c l e i n f o
Article history:Received 5 August 2014Revised 9 September 2014Accepted 11 September 2014Available online 19 September 2014
Keywords:TriazoleClick chemistryAndrogen receptorMolecular modelingBicalutamideProstate cancer
a b s t r a c t
A versatile and high yielding synthesis of novel androgen receptor (AR) antagonists is presented. Usingthis methodology, six 1,4-substituted-1,2,3-triazole derived bicalutamide mimics were synthesised infive steps and in isolated overall yields from 41% to 85%. Evaluation of these compounds for their anti-proliferative properties against androgen dependent (LNCaP) and independent (PC-3) cells showed prom-ising IC50 values of 34–45 lM and 29–151 lM, respectively. The data suggest that the latter compoundsmay be an excellent starting point for the development of prostate cancer therapeutics for both androgendependent and independent forms of this disease. Docking of these compounds (each enantiomer) in sil-ico into the T877A mutated androgen receptor, as possessed by LNCaP cells, was also undertaken.
� 2014 Elsevier Ltd. All rights reserved.
Cancer of the prostate is the most commonly diagnosed cancerin men worldwide and is the fourth most common cancer diag-nosed overall.1 Treatment of prostate cancer is currently carriedout via several different methods including castration, chemother-apy, radiation therapy and androgen blockade.2,3 The developmentof androgen receptor antagonists falls under androgen blockadetherapy and compounds which display this activity are commonlyclassed as steroidal or non-steroidal. Commonly used non-steroidalAR antagonists, flutamide 1a and bicalutamide 2 have seen greatsuccess in the clinic, note that flutamide 1a is a pro-drug whichis metabolised in vivo to hydroxyflutamide 1b (Fig. 1—left). Theextremely electron deficient 3,4-substituted aryl ring is a featurewhich is present in many AR antagonists, and often consists of atrifluoromethyl group at the 3-position while a nitrile or nitrogroup is present at the 4-position, though others are known.4–11
Accessing aryl withdrawn N-phenyl amides via peptide couplingcan be challenging, providing moderate yields due to the non-nucleophilic anilinic nitrogen.12–16 In our own experience,11 these
compounds have proven troublesome for common amide forma-tion protocols, giving low-to-moderate yields (typically 20–45%).
As such we believe that replacement of the amide moiety of 2with a 1,2,3-triazole (accessed via very high yielding ‘click chemis-try’)17,18 may have synthetic and biological value (Fig. 1—right).Replacement of the amide serves dual purposes in this study; (i)increasing the synthetic yield and (ii) probing the suitability of the1,2,3-triazole as an isosteric replacement of the N-phenyl amide.The incorporation of a 1,2,3-triazole as an amide isostere is a widelyadopted strategy within medicinal chemistry and has been exten-sively used in peptidomimetics.19–29 Another benefit of thisapproach is minimising the peptidic nature of these compounds,which can result in resistance to in vivo metabolism. One of manymetabolic pathways for flutamide 1a and hydroxyflutamide 1band several other SARMs (Selective Androgen Receptor Modulators)is via cleavage of the anilinicAamide bond and the consistent pres-ence of electron deficient amides within SARM scaffolds has furtherjustified our investigation into this novel class of scaffolds.30–32 Inaddition, the relative ease to install azides and alkynes into smallmolecules makes this an attractive avenue of investigation.
We recently reported the development of 1,2,3-triazole derivedflutamide derivatives which showed moderate antagonistic behavior

O O
S
R
1. SO2Cl2
2. RSH, NEt3S
R
1. LaCl3.2LiCl
2 . MgBr
4 5, R= F, 94% (over 2 steps) 7, R = F (99%)8, R = H (79%)6, R= H, 92% (over 2 steps)
S
R
9, R = F (98%)10, R = H (73%)
Oxone ®HO HO OO
Scheme 1. Synthesis of tertiary alcohols 9 and 10.
Table 1Synthesis of triazoles 12a–f
HO
12
NN N
R3
R2
N3R3
R2
+S
HO
9 R1 = F10 R1 = H
CuSO4, Ascorbic acid
MW, 30 minutes, 100 oC
11
R1
O O S
R1
O O
Entry Alcohol R1 R2 R3 Product Yield (%)
1 9 F CF3 CN 12a 942 9 F CF3 NO2 12b 923 9 F H COOEt 12c 674 9 F H COOH 12d Trace5 10 H CF3 CN 12e 786 10 H CF3 NO2 12f 78
NH
O2N
F3C
O
R
1a, R = H1b, R = OH
NH
NC
F3C
O
S
HO
2
S
HO3
NN N
R2
R3
N3
R2
R3 HO
S
R1
O
O CuAAc+
F
OO R1
O O
Figure 1. Currently used androgen receptor antagonists (left) and the target compounds of this study (right).
J. M. Altimari et al. / Bioorg. Med. Chem. Lett. 24 (2014) 4948–4953 4949
towards the androgen receptor, and our purpose of this study wasto extend this concept to more advanced molecular scaffolds toincrease biological efficacy.33 Herein we present a high yieldingand rapid synthesis of bicalutamide 2 inspired androgen receptorantagonists incorporating a 1,2,3-triazole replacement for the N-phenyl amide. The in silico docking of these compounds into thehuman androgen receptor (hAR) has been carried out, and theirdetermined IC50 values for androgen responsive (LNCaP) andandrogen independent (PC-3) cell lines is also presented.
Synthesis of target triazole AR antagonists: When designing thesynthetic route to these compounds, we considered the ability tovary the structural and electronic features at both ends of the tar-get scaffold (e.g., R1–3 of 3, Fig. 1) an important feature as thiswould allow rapid access to a large volume of chemical space forSAR studies. Our synthesis began with the in situ generation of2-chloroacetone from acetone 4, followed by treatment with 4-flurothiophenol or thiophenol to give either thioethers 5 or 6 ingood yield (Scheme 1), thus demonstrating the simple change inthiol (e.g., alkyl, aryl, substituted aryl) can easily generate a diversenature of analogues. Treatment of ketone 5 with ethynyl Grignardgave no conversion to the corresponding 1,2-addition adduct 7.Taking ketone 5, further attempts at promoting 1,2-addition wereundertaken though elevated temperatures and extended reactiontimes, again, giving no trace of 7. Repeating this reaction in thepresence of anhydrous CeCl3, a common strategy used to promote
1,2-addition via Lewis acid activation, gave a 17% conversion toalcohol 7.34–36 Continuing on the use of additives, stirring 5 withLaCl3�2LiCl in THF for 15 min prior to the addition of ethynyl Grig-nard gave the desired tertiary alcohol 7, in quantitative yield and>95% crude purity.
The reason for this dramatic change in yield is unknown, thoughit has been suggested that the presence of LaCl2�2LiCl attenuatesthe basicity of Grignard reagents, effectively promoting 1,2-addi-tion.37 This methodology was also successful for the 1,2-additionof ketone 6 to give alcohol 8, in good yield. Rapid oxidation ofthioethers 7 and 8 to the corresponding sulfones 9 and 10 wasachieved in excellent yield by treatment with Oxone�. With 9and 10 in hand, our attention turned to the formation of thetriazole moiety using a ‘click’ reaction with azides which werecommercially available or on-hand in our laboratory from otherstudies.38
Formation of triazoles 12a and 12b (Table 1, entries 1 and 2)proceeded in excellent yield while formation of 12c was still ingood yield it was markedly lower (Table 1, entry 3), though stillin high enough yield for evaluation purposes. Attempts at using4-azidocarboxylic acid in this protocol led to no success only givingtraces of the desired compound, therefore we accessed 12d viaester hydrolysis of 12c in good yield (71%). Triazoles 12e and 12fwere obtained in good yield using the same CuCAAC protocol whenemploying alcohol 10 as the alkyne reaction partner.

R NO
O
N
NN
R
HH
HVs R
O
O
N
NN
R
HH
H
H
H
H
H
Figure 2. Geometric and electronic similarities between the NO2 functionality anda carboxylate interacting with a guanidium moiety.
Table 2IC50 values for 12a–f against LNCaP and PC-3 cell lines with bicalutamide 2 forcomparison
Entry Compound IC50 (lM)
LNCaP PC-3
1 12a 45 >3002 12b 36 >3003 12c >300 >3004 12d >300 >3005 12e 151 1146 12f 34 297 2 30 58
4950 J. M. Altimari et al. / Bioorg. Med. Chem. Lett. 24 (2014) 4948–4953
Our desire to install a carboxylic acid moiety in 12d was fromprevious work showing that the NO2 moiety of hydroxyflutamide1b interacts with the guanidine unit of ARG75212 and the carboxylicacid possesses a similar trigonal planar structure. Guanidine–carboxylate complementarity (Fig. 2) has been used to encourageintramolecular interaction in the solid state, self-assembly, as a chi-ral selective transport agent and has featured among other applica-tions.39–45 Therefore we were interested to see if this same principlecould be used to encourage small molecule–receptor interaction inthis case, and with a robust synthesis and these compounds in-handour focus shifted to the in vitro evaluation of these compounds.
Biological evaluation in LNCaP and PC-3 cells: Androgen receptordependent (LNCaP) and androgen independent cell lines (PC-3)were selected to evaluate 12a–f for their capability to inhibitandrogen mediated growth in vitro.46 Compounds 12a and 12bdisplayed moderate amounts of inhibitory activity in the LNCaPcell line with IC50 values of 45 and 36 lM, respectively (Table 2,entries 1 and 2) and displayed no activity against the PC-3 cell line.This level of inhibition was comparable to that of bicalutamide 2which was also assessed in this same assay (Table 2, entry 7) givingand IC50 of 30 lM, though in this instance, 2 did possess antiprolif-erative properties against the PC-3 cell line, albeit slightly weakerat 58 lM.
We were disappointed in the lack of activities of 12c and 12d(Table 2, entries 3 and 4) against either cell line, as 12d had excel-lent predicted binding to the androgen receptor (see ESI). It isimportant to remember that a high in silico predicted pKD doesnot necessarily correlate to receptor antagonism, as other pro-cesses must be taken into account, for example, binding not initi-ating conformational changes to affect antagonism or insufficientability of these compounds to pass through the cell wall to accessthe androgen receptor.
Interestingly, vastly different biological properties wereobserved for compounds 12e and 12f, for example, 12e (Table 2,entry 5) displayed a 3-fold decrease in activity (vs 12a its aryl fluo-rine analogue) for LNCaP cells, though unlike 12a some (albeitweak) activity was seen towards the PC-3 cell line (Table 2, entry5). Conversely, 12f (Table 2, entry 6) showed marginally increasedactivity (vs 12b) towards LNCaP cells (cf. 34 vs 36 lM). Addition-ally, 12f was only marginally less active than bicalutamide 2against LNCaP cells, though 12f was more active towards PC-3 thanany other compound in this study, including bicalutamide 2(Table 2, entry 7). This is an interesting outcome as the only struc-
tural variation between 12a/b and 12e/f is the removal of a fluorinesubstituent on the sulfonyl aryl ring, which seemingly has ‘turnedon’ the anti-proliferative properties towards PC-3 cells. We are cur-rently undertaking further analogue development by derivatisa-tion of this scaffold at the 4-position on the aryl sulfone groupand results of that study will be reported in due course.
With biological data in hand for these compounds we proceededto dock these compounds into the human androgen receptor ligandbinding domain (hARLBD) possessing a single point mutation ofT877A. This mutation corresponds to the AR expressed by LNCaPcells, thus allowing for correlation between the in vitro activityand computational models.
Docking of triazoles 12a–f into the human androgen receptorligand binding domain: As all compounds in this study were evalu-ated as racemic mixtures we considered it pertinent to dock bothenantiomers of each compound into the human Androgen ReceptorLigand Binding Domain (hARLBD), as at this time this point we donot know if the compounds 12a–f have a eutomeric (more potentenantiomer) or distomeric (less potent enantiomer) isomer or ifboth enantiomers interact with the hAR equally. Note thatalthough bicalutamide 2 is administered as a racemic mixture, itis well known that only the R-bicalutamide is responsible for theantagonistic effect (eutomer), the S-enantiomer undergoing stere-oselective metabolism (distomer).47 The hARLBD of 2OZ7.pdb 48
was selected as, structurally, it correlates with the T877A mutanthAR thus corresponding to the AR possessed by the LNCaP cells.This mutation produces a more promiscuous binding site able toaccommodate a broader range of ligands, and removal of the thre-onine 877 residue removes a key hydrogen bonding moiety. Theligand set comprised of hydroxyflutamide 1b, bicalutamide 2 (R-isomer only) and compounds 12a–f (R and S isomers) as well ascyproterone acetate (CPY) 13 co-crystallised with the protein in2OZ7.pdb.
In the interest of brevity we have restricted the discussion ofdocking studies to the novel compounds which showed someanti-proliferative nature (12a, b, e, and f). For the remainingdocked images, including hydroxyflutamide 1b and bicalutamide2, refer to the ESI. Docking was performed using Surflex-dockwithin SYBYL-X version 2.049 and was validated by superimposingthe docked CPY with its original conformation.50 As a comparisonto the data presented below, the predicted pKD for R-bicalutamide2 was 5.5.
Docking 12a (R) into the hARBLD shows four hydrogen bonds,two at both ends of the binding domain (Fig. 3—left). ASN705 inter-acts with both the oxygen of the phenyl sulfonyl group and N3 ofthe triazole while the 4-phenylnitrile group, relative to the triazolemoiety, forms two hydrogen bonds with the guanidine unit ofARG752. The corresponding S-enantiomer, 12 (S) (Fig. 3—right),still interacts with ASN705, though due to the inverted stereo-chemistry, the triazole no longer participates in any interactionswith surrounding residues. The lack of hydrogen bond from N3on the triazole is compensated for by the trifluoromethyl groupwhich now interacts with GLN711. Again, this orientation is pre-sumably a result of stereochemical inversion, note that the nitrilegroup still interacts with ARG752.
Interestingly, replacement of the 4-cyano group of 12a withthat of a nitro moiety (12b) results in a completely different bind-ing mode (Fig. 4). In these instances, both 12b (R) and (S) fold ontothemselves to participate in a pi-stacking interaction with the N-phenyl triazole moiety. The oxygen of the nitro group moiety of12b (R) (Fig. 4—right) interacts with GLN711 and maximising thehydrophobic interactions through this folded results in a good pre-dicted pKD of 5.8. Interestingly, 12b (S) conformation has the N-phenyl triazole rotated so that the trifluoromethyl group, in addi-tion to the nitro functionality, interacts with GLN711, at the
12a (R) 12a (S)
pKD 5.7 pKD 6.0
Figure 3. Docked images of 12a (R and S enantiomers) in the hARLBD with predicted pKD.
12b (R) 12b (S)
pKD 5.8 pKD 5.3
Figure 4. Docked images 12b in the hARLBD with predicted pKD.
12e (R) 12e (S)
pKD 6.1 pKD 6.1
Figure 5. Docked images of 12e in the hARLBD with predicted pKD.
J. M. Altimari et al. / Bioorg. Med. Chem. Lett. 24 (2014) 4948–4953 4951
expense of optimizing hydrophobic interactions, leading to a good(though reduced) pKD of 5.3.
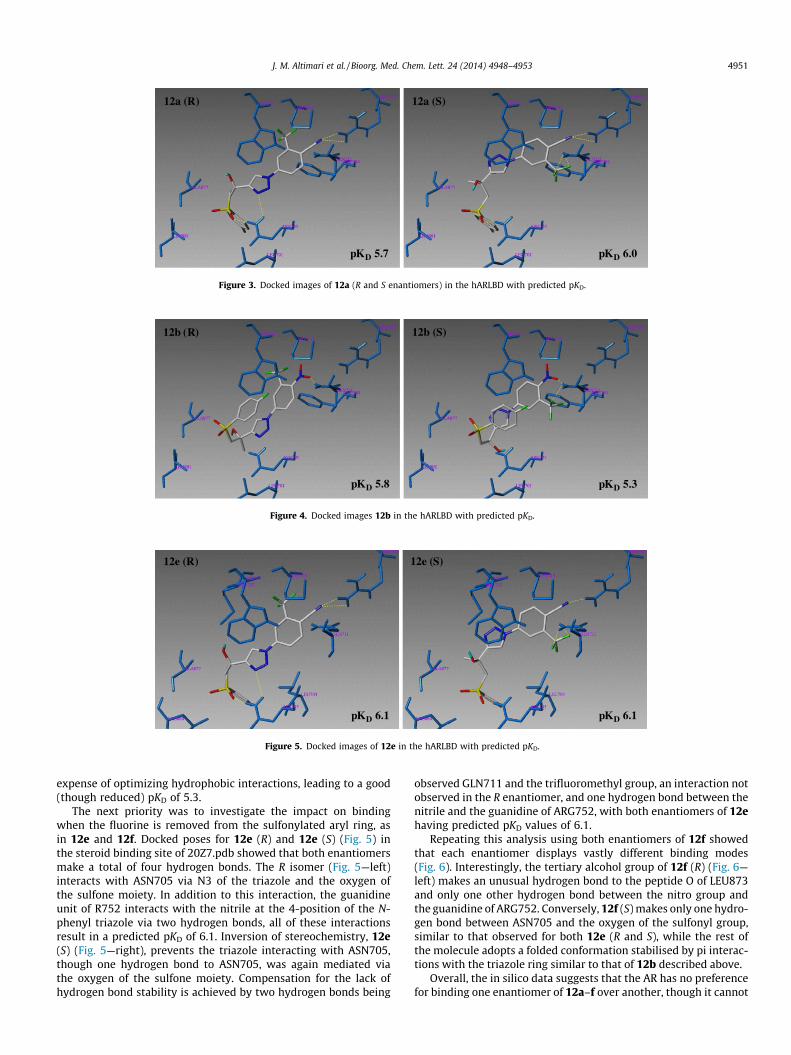
The next priority was to investigate the impact on bindingwhen the fluorine is removed from the sulfonylated aryl ring, asin 12e and 12f. Docked poses for 12e (R) and 12e (S) (Fig. 5) inthe steroid binding site of 20Z7.pdb showed that both enantiomersmake a total of four hydrogen bonds. The R isomer (Fig. 5—left)interacts with ASN705 via N3 of the triazole and the oxygen ofthe sulfone moiety. In addition to this interaction, the guanidineunit of R752 interacts with the nitrile at the 4-position of the N-phenyl triazole via two hydrogen bonds, all of these interactionsresult in a predicted pKD of 6.1. Inversion of stereochemistry, 12e(S) (Fig. 5—right), prevents the triazole interacting with ASN705,though one hydrogen bond to ASN705, was again mediated viathe oxygen of the sulfone moiety. Compensation for the lack ofhydrogen bond stability is achieved by two hydrogen bonds being
observed GLN711 and the trifluoromethyl group, an interaction notobserved in the R enantiomer, and one hydrogen bond between thenitrile and the guanidine of ARG752, with both enantiomers of 12ehaving predicted pKD values of 6.1.
Repeating this analysis using both enantiomers of 12f showedthat each enantiomer displays vastly different binding modes(Fig. 6). Interestingly, the tertiary alcohol group of 12f (R) (Fig. 6—left) makes an unusual hydrogen bond to the peptide O of LEU873and only one other hydrogen bond between the nitro group andthe guanidine of ARG752. Conversely, 12f (S) makes only one hydro-gen bond between ASN705 and the oxygen of the sulfonyl group,similar to that observed for both 12e (R and S), while the rest ofthe molecule adopts a folded conformation stabilised by pi interac-tions with the triazole ring similar to that of 12b described above.
Overall, the in silico data suggests that the AR has no preferencefor binding one enantiomer of 12a–f over another, though it cannot

12f (R) 12f (S)
pKD 4.3 pKD 5.2
Figure 6. Docked images of 12f in the hARLBD with predicted pKD.
4952 J. M. Altimari et al. / Bioorg. Med. Chem. Lett. 24 (2014) 4948–4953
be assumed that each enantiomer is responsible for the antagonis-tic binding of the AR in vitro. We are currently developing strategiesto facilitate enantiomeric resolution of these compounds tounequivocally determine this property, and results will be reportedin due course.
The variation in biological data obtained from these com-pounds highlights the importance of being able to rapidly con-struct and vary the peripheral structure of this scaffold as ithas already led to intriguing results. These triazoles possessedsimilar growth inhibitory properties, compared to that of bicalu-tamide 2 under the same assay conditions. We were pleased withthis outcome as the presented synthesis is amenable to a myriadof derivatisation at both termini of the target scaffold. For exam-ple, substitution of the starting ketone 4, thiophenol or phenylazide will provide further access to a large area of chemical spacefor optimisation of the anti-proliferative properties towards hor-mone responsive prostate cancer.
In conclusion, we have presented a novel approach to access-ing lead compounds for novel prostate cancer therapeutics. Thisapproach uses a high yielding synthetic pathway, providingpotential access to a huge number of novel compounds. In thiswork we present the design, synthesis and in silico docking ofthese compounds, 12a–f, which incorporate a 1,2,3-triazole asan amide isostere and possess promising anti-proliferative prop-erties against LNCaP cells (IC50 34–45 lM) and PC-3 cells (IC50
29–151 lM). Docking studies in the hARLBD suggests that eachof the enantiomers for 12a, b, e, and f are likely to bind intothe hARBLD as no substantially different binding affinities werenoted between stereoisomers. Enantiomeric resolution of thesecompounds is currently being attempted and receptor bindingstudies of the chirally enriched compounds are currently beingsought after and will be presented in the future. The ability for12e and 12f to inhibit the growth of PC-3 cells may be due tothe removal of fluorine on the sulfonylated phenyl group, a veryminor structural change for the introduction of biological activitytowards PC-3 cells. We are currently synthesizing analogous scaf-folds of those presented here which are suitable for rapid ana-logue generation to probe this property. The results of thatstudy will be reported in due course.
Acknowledgments
The authors gratefully acknowledge the CASS Foundation, Stra-tegic Research Center for Chemistry and Biotechnology, the Facultyof Science Engineering and Built Environment, and the School ofLife and Environmental Sciences for the funding to carry out thiswork. We thank Metabolomics Australia for the use of their HRMSservice. We would also like to thank the Australian Government foran APA scholarship to J.A.
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.bmcl.2014.09.036.
References and notes
1. Siegel, R.; Ma, J.; Zhao, Z.; Jemal, A. CA-Cancer J. Clin. 2014, 64, 9.2. Allan, G. F.; Sui, Z. Nucl. Recept. Signaling 2003, 1, 1.3. Haendler, B.; Cleve, A. Mol. Cell. Endocrinol. 2012, 352, 72.4. Hwang, D. J.; Yang, J.; Xu, H.; Rakov, I. M.; Mohler, M. L.; Dalton, J. T.; Miller, D.
D. Bioorg. Med. Chem. 2006, 14, 6525.5. Yoshino, H.; Sato, H.; Tachibana, K.; Shiraishi, T.; Nakamura, M.; Ohta, M.;
Ishikura, N.; Nagamuta, M.; Onuma, E.; Nakagawa, T.; Arai, S.; Ahn, K.-H.; Jung,K.-Y.; Kawata, H. Bioorg. Med. Chem. 2010, 18, 3159.
6. Yamamoto, S.; Tomita, N.; Suzuki, Y.; Suzaki, T.; Kaku, T.; Hara, T.; Yamaoka,M.; Kanzaki, N.; Hasuoka, A.; Baba, A.; Ito, M. Bioorg. Med. Chem. 2012, 20, 2338.
7. Li, H.; Ren, X.; Leblanc, E.; Frewin, K.; Rennie, P. S.; Cherkasov, A. J. Chem. Inf.Model. 2013, 53, 123.
8. Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J.ACS Med. Chem. Lett. 2013, 4, 180.
9. Yamamoto, S.; Tomita, N.; Suzuki, Y.; Suzaki, T.; Kaku, T.; Hara, T.; Yamaoka,M.; Kanzaki, N.; Hasuoka, A.; Baba, A.; Ito, M. Bioorg. Med. Chem. 2012, 20, 2338.
10. Zhang, X.; Li, X.; Allan, G. F.; Sbriscia, T.; Linton, O.; Lundeen, S. G.; Sui, Z. J. Med.Chem. 2007, 50, 3857.
11. Yoshino, H.; Sato, H.; Shiraishi, T.; Tachibana, K.; Emura, T.; Honma, A.;Ishikura, N.; Tsunenari, T.; Watanabe, M.; Nishimoto, A.; Nakamura, R.;Nakagawa, T.; Ohta, M.; Takata, N.; Furumoto, K.; Kimura, K.; Kawata, H.Bioorg. Med. Chem. 2010, 18, 8150.
12. Henderson, L. C.; Altimari, J. M.; Dyson, G.; Servinis, L.; Niranjan, B.; Risbridger,G. P. Bioorg. Chem. 2012, 40, 1.
13. Shi, Q.; Wada, K.; Ohkoshi, E.; Lin, L.; Huang, R.; Morris-Natschke, S. L.; Goto,M.; Lee, K.-H. Bioorg. Med. Chem. 2012, 20, 4020.
14. Schragl, K. M.; Forsdahl, G.; Gmeiner, G.; Enev, V. S.; Gaertner, P. TetrahedronLett. 2013, 54, 2239.
15. Jacobson, O.; Bechor, Y.; Icar, A.; Novak, N.; Birman, A.; Marom, H.; Fadeeva, L.;Golan, E.; Leibovitch, I.; Gutman, M.; Even-Sapir, E.; Chisin, R.; Gozinb, M.;Mishani, E. Bioorg. Med. Chem. 2005, 13, 6195.
16. Lee, I.-Y.; Gruber, T. D.; Samuels, A.; Yun, M.; Nama, B.; Kang, M.; Crowley,K.; Winterroth, B.; Boshoff, H. I.; Barry, C. E., III Bioorg. Med. Chem. 2013, 21,114.
17. Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K.B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210.
18. Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem. Int. Ed.2002, 41, 2596.
19. Pedersen, D. S.; Abell, A. Eur. J. Org. Chem. 2011, 2399.20. Horne, W. S.; Olsen, C. A.; Beierle, J. M.; Montero, A.; Ghadiri, M. R. Angew.
Chem. 2009, 121, 4812.21. Kuijpers, B. H. M.; Groothuys, S.; Soede, A. C.; Laverman, P.; Boerman, O. C.; van
Delft, F. L.; Rutjes, F. P. J. T. Bioconjugate Chem. 2007, 18, 1847.22. Davis, M. R.; Singh, E. K.; Wahyudi, H.; Alexander, L. D.; Kunick, J. B.; Nazarov, L.
A.; Fairweather, K. A.; Giltrap, A. M.; Jolliffe, K. A.; McAlpine, S. R. Tetrahedron2012, 68, 1029.
23. Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057.24. Jochim, A. L.; Miller, S. E.; Angelo, N. G.; Arora, P. S. Bioorg. Med. Chem. Lett.
2009, 19, 6023.25. Horne, W. S.; Yadav, M. K.; Stout, C. D.; Ghadiri, M. R. J. Am. Chem. Soc. 2004,
126, 15366.26. Bock, V. D.; Speijer, D.; Hiemstra, H.; van Maarseveen, J. H. Org. Biomol. Chem.
2007, 5, 971.27. Hou, J.; Liu, X.; Shen, J.; Zhao, G.; Wang, P. G. Expert Opin. Drug Discov. 2012, 7,
489.

J. M. Altimari et al. / Bioorg. Med. Chem. Lett. 24 (2014) 4948–4953 4953
28. Proteau-Gagné, A.; Rochon, K.; Roy, M.; Albert, P.-J.; Guérin, B.; Gendron, L.;Dory, Y. L. Bioorg. Med. Chem. Lett. 2013, 23, 5267.
29. Rathwell, K.; Sperry, J.; Brimble, M. A. Tetrahedron 2010, 66, 4002.30. Kim, J.; Wang, R.; Veverka, K. A.; Dalton, J. T. Xenobiotica 2013, 43, 993.31. Gao, W.; Wu, Z.; Bohl, C. E.; Yang, J.; Miller, D. D.; Dalton, J. T. Drug Metab.
Dispos. 2006, 34, 243.32. Fukami, T.; Yokoi, T. Drug Metab. Pharmacokinet. 2012, 27, 466.33. Altimari, J. M.; Niranjan, B.; Risbridger, G. P.; Schweiker, S. S.; Lohning, A. E.;
Henderson, L. C. Bioorg. Med. Chem. 2014, 22, 2692.34. Bartoli, G.; Marcantoni, E.; Marcolini, M.; Sambri, L. Chem. Rev. 2010, 110,
6104.35. Henderson, L. C.; Loughlin, W. A.; Jenkins, I. D.; Healy, P. C.; Campetelli, M. R. J.
Org. Chem. 2006, 71, 2384.36. Loughlin, W. A.; Jenkins, I. D.; Henderson, L. C.; Campetelli, M. R.; Healy, P. C. J.
Org. Chem. 2008, 73, 3435.37. Krasovskiy, A.; Kopp, F.; Knochel, P. Angew. Chem. Int. Ed. 2006, 45, 497.38. See ESI.39. Rether, C.; Schmuck, C. Eur. J. Org. Chem. 2011, 1459.40. Nanubolu, J. B.; Sridhar, B.; Ravikumar, K.; Sawant, K. D.; Naik, T. A.; Patkar, L.
N.; Cherukuvadac, S.; Sreedhar, B. CrystEngComm 2013, 15, 4448.
41. Fameau, A.-L.; Houinsou-Houssou, B.; Ventureira, J. L.; Navailles, L.; Nallet, F.;Novales, B.; Douliez, J.-P. Langmuir 2011, 27, 4505.
42. Yadav, V. N.; Görbitz, C. H. CrystEngComm 2013, 15, 7321.43. Beck, C. L.; Berg, S. A.; Winter, A. H. Org. Biomol. Chem. 2013, 11, 5827.44. Rether, C.; Sicking, W.; Boese, R.; Schmuck, C. Beilstein J. Org. Chem. 2010, 6.
http://dx.doi.org/10.3762/bjoc.6.3.45. Breccia, P.; Van Gool, M.; Pèrez-Fernández, R.; Martin-Santamaria, S.; Gago, F.;
Prados, P.; de Mendoza, J. J. Am. Chem. Soc. 2003, 125, 8270.46. All IC50 values for novel compounds were carried out and determined under
the same protocol and within the same laboratories (GenScriptUSA) tominimise potential variation. Testing was carried out 10 dilutions (dilutionfactor 2) starting at 300 lM.
47. Mukherjee, A.; Kirkovsky, L.; Yao, X. T.; Yates, R. C.; Miller, D. D.; Dalton, J. T.Xenobiotica 1996, 26, 117.
48. Bohl, C. E.; Wu, Z.; Miller, D. D.; Bell, C. E.; Dalton, J. T. J. Biol. Chem. 2007, 282,13648.
49. SYBYL-X 2.0, Tripos International, 1699 South Hanley Rd., St. Louis, Missouri63144, USA.
50. Note: An RMSD of less than 2 Å is widely accepted to indicate a successfuldocking process, this work an RMSD of 0.949 Å was obtained.








![Bicalutamide 2013.06.17 양혜란. Bicalutamide 화학명 N-[4-Cyano-3-(trifluoromethyl)phenyl]-3-(4-fluorophenyl) sulfonyl-2-hydroxy-2-methylpropanamide 분류 항암제 약리](https://img.dokumen.tips/doc/110x75/5a4d1b187f8b9ab059992821/bicalutamide-20130617-bicalutamide-n-4-cyano-3-trifluoromethylphenyl-3-4-fluorophenyl.jpg)




















