Embed Size (px)
Citation preview

SYNTHESES, REACTIVITY, AND PHYSICAL PROPERTIES OF SPIRO-
TRICYCLIC PORPHODIMETHENES AND PORPHYRINS WITH EXOCYCLIC RINGS
By
IVANA BOŽIDAREVIĆ
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2004

Copyright 2004
by
Ivana Božidarević

In everlasting memory of my Father, Dragan, and my Grandparents Zlata, Boško, Vuka
and Spasa.
With all my love to my families Ćirić and Božidarević

ACKNOWLEDGMENTS
For becoming the chemist I am I have to thank my teachers, mentors and
colleagues; for becoming the person I am, I have to thank my family and friends.
The one who got me involved in chemistry when I was thirteen was my seventh-
and eight-grade chem. Teacher – Vera Kujačić. Milka Dokić successfully took over
when she became my high school chemistry teacher. My first lab TA in college, Dr.
Tibor Sabo, became my BS Thesis mentor 4 years later, and I learned a lot from him. I
learned much more when I started taking graduate courses at UF, and for that I have to
thank to Dr. Richardson, Dr. Talham, Dr. Abboud and Dr. Scott who taught these classes.
Even though I joined the Scott group relatively early in the Fall 1999, I kept the
desk in the X-ray lab that was assigned tome in the summer, and I was ‘hiding ’ there
until I started doing research in May 2000. During that time, Dr. Khalil Abboud was the
person I could always count on if I needed help, advice, or if I just wanted to talk. He
played a great part in my relatively quick adjustment to the new country, people, and
customs.
The biggest thanks, of course, go to my advisor Prof. Michael Scott for his
guidance, help, understanding and infinite patience. Working with him has been a great,
rewarding experience I learned a lot from. During the work on my Ph.D. thesis, there
were a few people, other than Mike who had ideas, explanations, questions and
instruments that helped my research. For that, I would like to thank Prof. Lisa McElwee-
iv

White, Prof. Dan Talham, Prof Kirk Schanze, Prof. Mark Meisel and my dear friend Dr.
Ksenija Haskins-Glušac.
The person who set up basis for my dissertation and taught me a lot about
porphyrin and porphodimethene syntheses was Dr, Michael Harmjanz, who will make a
great professor at University of New Orleans starting this fall. My experience in the
Scott group would not be what it is, if there weren’t for the past and the present group
members, so I need to thank Dr. Andrew Cottone for helping me set up and start working
in the lab and Dr. Matt Peters for answering my questions about how things work for
solid two years. Dan and Jen were here when I came, and left shortly after, Cooper and
Eric were around for a couple of years, Dolores, Javier, Pieter and Hanna came and left,
but all of these people made the work experience more enjoyable one for me. For
making our labs a better place to be these days I have to thank Nela, Ranjan, Ozge, Eric,
Erik, Priya, Claudia, Flo, Isaac, Candace and Hubert. The last person on this list is
someone who deserves more acknowledgements than I can provide right now, so I’ll just
say that I cannot imagine getting through the past five years without a lab mate like that.
Special thanks for loving me unconditionally, letting me become who I am, and
making my childhood a happy one go to my parents, Andjelka and Dragan, my
grandparents Boško, Zlata, Vuka and Spasa, and my little brother Dejan who taught me
how to fight for what I want. During past seven years, another family became very
important in my life, and I would like to thank my in laws Nada and Vlada for their
kindness, love and support. Lastly, I have to thank my husband, Nebojša, for his love,
patience and support that helped me overcome the obstacles, and always managed to put
a smile on my face.
v

These five years in Gainesville brought me probably more friends than I can
account for right now, and I will try mentioning them all, but I hope I will be forgiven if I
forget someone. There is no way I could tell how much all of them mean to me and why,
so I will just list their names in the order of appearance and thank them all for being here
when I needed them: Tamara, Isa, Ana I., Janina, Iwona, Luk, Josef, Celeste, Corey,
Ljubisa, Ksenija, Aleksa J., Ilka, Elon, Ana M., Andy, Balsa, Milan, Aleksa O., Vesna,
Jamshid, Feruza…
vi

TABLE OF CONTENTS page ACKNOWLEDGMENTS ................................................................................................. iv
LIST OF TABLES...............................................................................................................x
LIST OF FIGURES ........................................................................................................... xi
ABSTRACT..................................................................................................................... xiii
CHAPTER 1 INTRODUCTION TO PORPHODIMETHENES ......................................................1
Tetrapyrrolic Macrocycles............................................................................................1 Porphodimethene Syntheses .........................................................................................2 Solid-state and Solution Structures...............................................................................7 Electronic Properties...................................................................................................10 Electrochemistry .........................................................................................................13 Reactivity....................................................................................................................13
2 PORPHODIMETHENE SYNTHESES.....................................................................16
Introduction.................................................................................................................16 Results and Discussion ...............................................................................................19 Conclusions.................................................................................................................25 Experimental...............................................................................................................25
General Procedures..............................................................................................25 Chromatography ..................................................................................................26 Syntheses of 2-4 and 2-5 .....................................................................................26 Synthesis of 2-6 ...................................................................................................27 Syntheses of 2-7 and 2-8 .....................................................................................28 X-ray Crystallography .........................................................................................30
3 METALLATION AND RING-OPENING REACTIONS........................................31
Introduction.................................................................................................................31 Results and Discussion ...............................................................................................32
vii

Metallation of Porphodimethenes........................................................................32 Structure of Metalloporphodimethenes. ..............................................................34
Palladium anthracenone porphodimethene ..................................................34 Palladium pyrenone porphodimethene.........................................................35 Palladium and platinum phenanthrenone porphodimethenes.......................37 Copper phenanthrenone porphodimethenes .................................................39 Nickel phenanthrenone porphodimethene....................................................41 Summary of the Structural Data...................................................................41
Reactivity of Porphodimethenes..........................................................................42 Conclusions.................................................................................................................48 Experimental...............................................................................................................49
General Procedures..............................................................................................49 Chromatography ..................................................................................................49 Synthesis of 3-2 ...................................................................................................49 Synthesis of 3-4 ...................................................................................................50 Synthesis of 3-5 ...................................................................................................51 Synthesis of 3-8 ...................................................................................................52 Synthesis of 3-9 ...................................................................................................52 Synthesis of 3-11 .................................................................................................53 Synthesis of 3-12 .................................................................................................53 Synthesis of 3-13 .................................................................................................54 Synthesis of 3-14 and 3-15 ..................................................................................54 X-ray Crystallography .........................................................................................55
4 PHOTOPHYSISCAL PROPERTIES OF PORPHODIMETHENES .......................59
Introduction.................................................................................................................59 Fluorescence Spectroscopy .................................................................................64 Phosphorescence Emission..................................................................................65 Transient Absorption ...........................................................................................66
Conclusions.................................................................................................................68 Experimental...............................................................................................................69
5 SYNTHESES OF PORPHYRINS WITH EXOCYCLIC RING SYSTEMS............71
Introduction.................................................................................................................71 Results and Discussion ...............................................................................................71
Cyclooctanone Porphyrins...................................................................................73 Cyclohexannone Porphyrins................................................................................78
Conclusions.................................................................................................................81 Experimental...............................................................................................................82
General Procedures..............................................................................................82 Chromatography ..................................................................................................82 Synthesis of cis-5-5 and trans-5-5.......................................................................82 Synthesis of 5-8 ...................................................................................................83
viii

Synthesis of 5-9 ...................................................................................................84 Synthesis of 5-10 .................................................................................................84 Synthesis of cis-5-11 and trans-5-11...................................................................85 X-ray Crystallography .........................................................................................86
6 PHOTOPHYSICAL PROPERTIES OF PORPHYRINS WITH EXOCYCLIC RING SYSTEMS ..................................................................................................................89
Introduction.................................................................................................................89 Cyclooctanone Porphyrins...................................................................................93 Cyclohexanone Porphyrins..................................................................................97 Azulenone Porphyrins .........................................................................................98
Conclusions...............................................................................................................101 Experimental.............................................................................................................102
7 SUMMARY.............................................................................................................104
LIST OF REFERENCES.................................................................................................106
BIOGRAPHICAL SKETCH ...........................................................................................111
ix

LIST OF TABLES
Table page 2-1. Selected bond lengths and angles for 2-5, 2-6 and 2-8 ...........................................23
2-2. Crystallographic data...............................................................................................29
3-1. Selected parameters from the solid-state structures of metalloporphodimethenes. ..................................................................43
3-2. Crystallographic data for compounds 3-2, 3-4, 3-7 and 3-8 ...................................56
3-3. Crystallographic data for compounds 3-9, 3-11, 3-12 and 3-15 .............................57
4-1. Selected UV-Vis absorption data for the free-base and metalloporphodimethenes. The presence of metals in macrocyclic ring induces the absorption maximum to shift towards longer wavelengths. ............................................................................61
4-2. The values of fluorescence emission maxima and quantum yields for selected free-base porphodimethenes. The fluorescence is very weak.........................................65
5-1. Selected bond lengths for trans-5-5 ........................................................................77
5-2. Crystallographic data for trans-5-5 and 5-10..........................................................87
6-1. Summary of photophysical data..............................................................................94
x

LIST OF FIGURES
Figure page 1-1 Depiction of four examples of tetrapyrrolic macrocycles.. ........................................1
1-2 Illustration of redox relationships between tetrapyrrolic macrocycles.. ....................2
1-3 Schematic representation of a porphyrin spectrum . ................................................11
1-4 UV-vis spectra of a porphyrin (---) and a porphodimethene (—).. ..........................11
1-5 Nickel porphodomethene and porphyrin MO diagrams...........................................12
2-1 Diagram of the solid-state structure of 2-5...............................................................20
2-2 Diagram of the solid-state structure of 2-6...............................................................22
2-3 Diagram of the solid-state structure of 2-8...............................................................22
2-4 Diagrams of the porphodimethene cores of 2-5, 2-8 and 2-6...................................24
2-5 The highly symmetric nature of the 1H NMR spectrum of 2-5 illustrates the fast flexing of the molecule in solution at room temperature. ........................................24
3-1 Diagram of the solid-state structure of 3-2...............................................................34
3-2 Diagram of the solid-state structure of 3-4...............................................................36
3-3 Diagram of the solid-state structure of 3-7...............................................................37
3-4 Diagram of the solid-state structure of 3-8...............................................................38
3-5 Diagram of the solid-state structure of 3-9...............................................................40
3-6 Diagram of the solid-state structure of 3-11.............................................................40
3-7 Diagram of the solid-state structure of 3-12.............................................................42
3-8 Diagram of the solid-state structure of 3-14.............................................................46
4-1: Illustration of the UV-Vis absorption spectra of free base porphodimethenes.. ........62
xi

4-2 Illustration of the absorption spectra of metalloporphodimethenes.. .......................62
4-3 Diagram of solid-state structure of 4-1b. .................................................................64
4-4 Depiction of phosphorescence emission for 4-1 and 4-2. ........................................67
4-5 Depiction of transient absorption of 4-1 and 4-2.. ...................................................68
5-1 Illustration of the reaction progress for synthesis of 5-4..........................................74
5-2 Diagrams (side view on the bottom) of the solid-state structure of trans-5-5. .......76
5-3 Diagram of porphyrins with exocyclic rings synthesized in Callot’s lab. ...............78
5-4 Diagram of the solid-state structure of 5-10.............................................................81
6-1 Diagram of the porphyrins with exocyclic rings used for the photophysical measurements reported herein..................................................................................90
6-2 The cycloheptanone porphyrins exhibit red-shifts in the absorption spectra...........91
6-3 Depiction of the phosphorescence emission of cis-6-1 and trans-6-1 .....................92
6-4 Illustration of transient absorption of cis-6-1 and trans-6-1. ...................................94
6-5 Diagram of the absorption spectrum of the mixture of cis-6-2 and trans-6-2 highlights the coincidence of their Soret bands at 438 nm. .....................................95
6-6 Diagram of the phosphorescence emission of the mxture of cis-6-2 and trans-6-2. The emission is quenched by saturation with air. ...................................96
6-7 Depiction of transient absorption of cyclooctanone porphyrins.. ............................97
6-8 Diagram of the UV-Vis spectra of trans-6-3 and cis-6-3.. ......................................98
6-9 Illustration of the room temperature phosphorescence emission of trans-6-3.........99
6-10 Transient absorption of cis-6-3 and trans-6-3..........................................................99
6-11 Diagram of the electronic absorption of cis-6-4 and trans-6-4..............................100
xii

Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
SYNTHESES, REACTIVITY, AND PHYSICAL PROPERTIES OF SPIRO-TRICYCLIC PORPHODIMETHENES AND PORPHYRINS WITH EXOCYCLIC
RINGS
By
Ivana Božidarević
August 2004
Chair: Michael J. Scott Major Department: Chemistry
The MacDonald [2+2] condensation under Lindsey reaction conditions was
successfully employed towards the syntheses of spiro-tricyclic meso-aryl substituted
porphodimethenes from different 5-aryldipyrromethanes and aromatic vicinal diketones.
Depending on the diketone used, porphodimethenes capable of or resistant to ring
opening at the spiro-lock were prepared. The reactivity of porphodimethenes susceptible
to ring opening was studied. The porphodimethenes were metallated using palladium,
platinum, copper, nickel and zinc salts. The metal complexes were characterized and
their solid-state structures compared and analyzed.
Spiro-tricyclic porphodimethenes were used to synthesize unprecedented palladium
porphyrins with exocyclic eight-membered rings, while the synthesis of related
porphyrins with six-membered rings was accomplished through somewhat modified
literature procedures. Palladium porphyrins with six, seven and eight membered rings
were used for photophysical studies. These molecules have interesting electronic
xiii

properties, resulting in red-shifted absorption maxima in UV-Vis spectra. The results of
photophysical measurements performed on both the porphodimethenes and the
porphyrins are presented, and the dependence of the porphyrin photophysical properties
on the exocyclic ring size is discussed. The measurements include steady state emission
at room temperature and low temperature, transient absorption and singlet oxygen
quantum yield.
xiv

CHAPTER 1 INTRODUCTION TO PORPHODIMETHENES
Tetrapyrrolic Macrocycles
Tetrapyrrolic macrocycles (Fig 1-1) play a number of critical biological roles and
their importance has inspired an intensive research effort concerning artificial systems
that model the natural counterparts. One of the most abundant tetrapyrrols found in
nature, porphyrins, are cross-conjugated, planar ligands that are ubiquitous in living
systems, facilitating electron transfer and photosynthesis.1 In these macrocycles, the
pyrrolic carbon atoms are defined as either α or β, where the α carbons make up a part of
macrocycle core. The remaining carbon atoms in the core form the bridges between the
pyrrolic groups and these are referred to as meso carbons (Fig 1-1).
N HN
NH N
NH N
N NH
N N
N NH H
H HN HN
NH N
Porphyrin Chlorin Porphodimethene Porphyrinogen
α
βmesoβ
Figure 1-1. Depiction of four examples of tetrapyrrolic macrocycles. Pyrrolic carbons are
defined as α or β while the bridging carbons are called meso in all the tetrapyrroles.
The porphyrins are cross-conjugated, aromatic molecules containing an 18-
anulene system. Porphodimethenes differ from porphyrins in as much as they have
saturated carbons at two non-adjacent meso positions. The two sp3 carbon atoms cause
the macrocycle to adopt a “roof-like” folded structure, breaking the aromaticity and
1

2
disrupting the electronic communication between the two dipyrromethene halves. The
two halves of the porphodimethene macrocycle are still conjugated and the interplanar
angle between them is called a roof-angle.
Porphodimethene Syntheses
Although porphodimethenes were long recognized to be intermediates in the
oxidation pathway from porphyrinogens to porphyrins (Fig 1-2), a synthetic scheme for
their production was only first reported in 1974 by Buchler and Puppe.2 Buchler
reasoned that alkylation of meso carbons in the aromatic porphyrin could produce
porphodimethenes, and indeed, the reductive alkylation of zinc octaethyl porphyrin 1-1
resulted in formation of zinc porphodimethene 1-2 (Scheme 1-1).
N N
NH HN
NH N
N NH
NH HN
NH HN
N HN
NH N
Porphyrin
Porphodimethene
Porphyrinogen
N HN
NH HN
N HN
NH HN
Phlorin Porphomethene
2H+ 2e-
2H+
2e-2H+
2e-
-2H+
-2e--2H+
-2e--2H+
-2e-
Figure 1-2. Illustration of redox relationships between tetrapyrrolic macrocycles.
Porphyrins represent the most oxidized form of tetrapyrroles.
The presence of methyl groups at positions 5 and 15 (saturated meso-carbons)
prevented rapid oxidation of the macrocycle, allowing for the isolation of the first air-
stable porphodimethenes.

3
N N
N N1. 2e-
2. R-XN N
N N
R H
R H
(R = CH3, X = Br, I)
Zn Zn
1-1 1-2 Scheme 1-1. Depiction of the first stable synthesis of a porphodimethene. Buchler and
coworkers applied reductive alkylation of a zinc porphyrin to obtain the porphodimethene.
Over the years, Buchler et al. expanded the scope of this reaction to various
metalloporphodimethenes bearing different alkyl substituents on the saturated meso
carbon atoms.3-7 A significant library of X-ray structural data was collected and
electrochemical properties of these compounds were studied, but owing to the difficulties
associated with their isolation and separation, the reactivity studies of the alkyl
metalloporphodimethenes were never reported.
An alternative porphyrin route to porphodimethenes was discovered by Fontecave
et al. in 1984.8 During the catalytic reduction of allyl bromide by sodium ascorbate or
sodium dithionate, the catalyst, tetraphenylporphyrinato iron(III)chloride (Fe(TPP)Cl),
underwent a slow transformation to a porphodimethene species. Based on this
observation, a larger scale reaction of Fe(TPP)Cl with allyl bromide and sodium
dithionate (Scheme 1-2) was undertaken, and after demetallation with TFA, a stable free
base porphodimethene was obtained in 80% yield, as a complex mixture of anti and syn
axial and equatorial isomers.
Even though this study gave some insight into the iron porphyrin catalyzed
reduction mechanism, the synthetic method for porphodimethene preparation was not
explored further, due to the difficulties presented by the formation of product mixtures.

4
N N
N N
Ph
Ph
Ph
Ph Fe
Cl
N HN
NH N
Ph
Ph
Ph
Ph
RR
1) CH2CHCH2Br
2) TFA
R=CH2CHCH2
1-3 Scheme 1-2. Representation of reductive alkylation of FeTPPCl. Allylbromide catalyzes
this porphodimethene forming reaction.
In 1999 and 2000, several reports of new procedures for porphodimethene
synthesis appeared in the literature. The Floriani lab employed reductive dealkylation of
tin porphyrinogen to obtain hexaalkyl tin porphodimethene (Scheme 1-4).9 Using a
MacDonald’s [2+2] type condensation between a dipyrromethene and acenaphthenone,
our group reported the first synthesis of spiro-tricyclic porphodimethenes (Scheme 1-3).10
Almost concurrently with our report, another method for the high yield of
porphodimethenes was published. The Sessler group condensed dipyrromethane with an
excess of acetone to form a mixture of pyrrolic macrocycles from which
porphodimethene could be isolated in high yield (Scheme 1-4).11
R
NH NH
O
O
NH NN HN
R
R
O
O NH NN HN
R
R
O O1. TFA
2
2. DDQ
2
+
Anti Syn1-51-4
Scheme 1-3. Illustration of condensation of mesityl dipyrromethane and
acethnaphthenequinone to form syn and anti porphodimethenes. The isomers are easily separated by column chromatography.

5
N N
N N SnCl4(THF)2
R=alkylSn
R R
R R
R
R R
R
THF
THFN N
N N
Sn
R R
R R
R R
Cl
Cl
Ar
NH HN NH N
N HN
Ar
Ar
+ 1. acid2. DDQ
O
40 fold excess
2
1-7
1-6
Scheme 1-4. Depiction of syntheses of alkyl-substituted porphodimethenes. Alkyl
substituents prevent oxidation at meso carbons
The porphodimethenes illustrated in Scheme 1-4 contain alkyl groups at saturated meso
carbons; hence, they are ill suited for further functionalization. Our group was interested
in studying porphodimethenes with aromatic substituents at sp3 carbons, and the synthetic
pathway outlined in Scheme 1-3 will be further elaborated in the following chapters.
Recent developments in porphodimethene syntheses include the reaction of meso or
β- substituted porphyrins with alkyl lithium and iodo alkyl reagents,12 as well as the
condensation of pyrroles or dipyrromethanes with bulky aldehydes (Scheme 1-5).13, 14
The first method, consisting of two consecutive alkylation steps was developed by Senge
and coworkers,12 and it allows for facile isolation of asymmetrically substituted
porphodimethenes (Scheme 1-5).

6
N N
N N
Ph
Ph
Ni
Et Et
Et
Et
EtEt
Et
Et
N N
N N
Ph
Ph
Ni
Et Et
Et
Et
EtEt
Et
Et1. n-BuLi2. n-C6H13I n-C6H13n-Bu
HH
1-8 Scheme 1-5. Illustration of alkylation of nickel porphyrin. The use of alkyllithium and
alkyliodide reagents enables the synthesis of asymmetrically substituted porphodimethenes
The same group employed a condensation reaction between pivaldehyde and
pyrrole to prepare t-butyl substituted compounds.12 The Kim group made an interesting
choice of bulky reagent using ferrocene aldehyde for the condensation reaction with
dipyrromethane (Scheme 1-6).14 The resulting porphodimethene was stable to light and
oxidants in the absence of acid, but it readily formed a porphyrin upon irradiation in
acidic solution under anaerobic conditions.
NH NH2 Fe
HO
2+
N HN
NH NFcFc
HH
H+
1-10
NH
+ PhCHO + t-BuCHO
N HN
NH Nt-But-Bu
HH
1-9
Ph
Ph
H+
Scheme 1-6.Representation of condensation of pyrrole and dipyrromethane with bulky aldehydes. Oxidation of porphodimethenes to porphyrins is prevented by steric hindrance at the meso positions.

7
Solid-state and Solution Structures
From a detailed examination of the available crystallographic data for
porphodimethenes, a variety of important structural parameters can be identified. In
1974, Buchler’s group reported the first porphodimethene crystal structure of nickel
dimethyl-octaethyl porphodimethene,15 and over the next several years, the group
reported structures of related porphodimethenes with different metals (Scheme 1-7).2, 3, 7,
16, 17 All of these metalloporphodimethenes adopt a roof-like folded structure and the
roof angles between the two dipyrromethane halves of the molecule range between 128º
and 146º.
N N
N N
R H
R H
M
1-11: M = Ni, R = Me1-12: M = TiO, R = Me1-13: M = FeCl, R = Me1-14: M = OsCO, R = Me1-15: M = MnN, R = Me
Scheme 1-7. Illustration of metalloporphodimethenes with determined solid-state
structures. All of the dimethyl-octaethylporphodimethenenes adopt a roof-like folded structure in the solid state.
The metal center adopts a square planar arrangement with the four pyrrolic nitrogens in
1-11, while the geometry about the metals in 1-12 through 1-15 is square pyramidal.
Metal-nitrogen bond lengths vary from 1.902(5) Å in nickel porphodimethene to 2.113(3)
Å in the titanium oxo species. In all of the structures, methyl substituents on sp3 meso
carbons are in syn-diaxial conformation, which is crucial for the porphodimethene
stability under oxidative conditions. Interestingly, only one free-base porphodimethene
has been reported previous to 1999, and it had isopropyl substituents on the meso
carbons.18 The Buchler group initially thought the compound was a mixture of syn-

8
diaxial (aa), syn-diequatorial (ee) and anti (ae) isomers (Scheme 1-8),19 but out of these
three isomers, the syn-diaxial(aa) was proven to be the most stable, since the presence of
the alkyl groups at equatorial position on meso carbons increases the steric hindrance at
the periphery of a porphodimethene. Careful column chromatography of the reaction
mixture on alumina allowed for separation of the minor isomer fraction (the primary
product of the reaction was, as expected, the aa isomer).18 In the solid-state this molecule
possesses a slightly different geometry with two meso-substituents oriented trans to each
other and locked in an intermediate conformation between axial and equatorial. With the
roof angle of 180º, this planar stereoisomer was named diagonal (dd).
RH
R
HH
RH
H
R
RR
R
H
H
H
R
ae ee aa dd Scheme 1-8. Diagram of possible porphodimethene stereoisomers. In most cases aa is the
single isomer isolated from the porphodimethene reaction
More recent examples of porphodimethene solid-states structures have included
macrocycles with six meso substituents coordinating different transition metals (Scheme
1-9)20 and metalloporphodimethenes with long alkyl substituents on sp3 carbons (Scheme
1-10).12
N N
N NEtEt
EtEt
Et
Et
M
N N
N NEtEt
EtEt
Et
Et
M
N N
N NEtEt
EtEt
Et
Et
M
L L
L
1-16: M = Fe1-17: M = Co1-18: M = Ni
1-19: M = Co, L = Py1-20: M = Mn, L = THF
1-21: M = Mn, L = THF1-22: M = Mn, L = Py1-23: M = Fe, L = THF1-24: M = Mn, L = Py
Scheme 1-9. Depiction of several structurally characterized metalloporphodimethenes.

9
1-25: R = n-Bu1-26: R = H
N N
N N
n-Bu H
n-Bu H
MR n-Bu
Scheme 1-10. Illustration of two meso-substituted octaethyl porphodimethenes
In the recently characterized examples of porphodimethenes the bond lengths and
roof angles are consistent with the values described by Buchler except for roof angles in
1-23 and 1-24 - 180º, 1-20- 168.8º, 1-16- 149.3º, and 1-18-116.9º. In the case of Ni(II),
the smaller ionic radius of the metal causes the macrocycle to adopt a ruffled, saddle
shaped structure. Solid-state structural parameters of spiro-tricyclic porphodimethenes
are in agreement with those previously listed, and they will be further discussed in
chapters 2 and 3.
Owing to the lack of aromaticity within the tetrapyrrolic ring, several resonances in
the 1H NMR spectra of porphodimethenes are significantly shifted in comparison to the
fully aromatic porphyrin counterparts. In β-substituted porphodimethenes, the signals
arising from the meso hydrogens can be found in the region around 6.5 ppm for protons
on sp2 carbons and between 3.5 and 5.5 ppm for protons on sp3 carbons.5, 18 In addition,
the resonances for the β-pyrrolic protons in meso-substituted porphodimethenes always
occur as doublets between 5.5 and 7.2 ppm with coupling constants of approximately 4.5
Hz, and their position is influenced by the nature of meso substituents.21 When the
metals are inserted into the macrocycle, the separation between the pyrrolic doublets
increases in comparison to the free- bases.21 These changes can be attributed to both the
electronic modification of the dipyrromethane halves and the altered configuration of the

10
molecule induced by metal binding. In comparison to meso-substituted porphyrins, the
resonances for these β-hydrogens are significantly shifted upfield. The ring current in
porphyrins also induces a large shift of the signals for the pyrrolic hydrogens to between
–4 and –2 ppm, but since they lack this influence, the analogues NH resonances in
porphodimethenes are shifted far downfield to between 12 and 14 ppm.
Oftentimes, porphodimethenes exhibit 1H NMR spectra that look far more
symmetrical than would be expected upon inspection of their solid-state structures,21,22
and the ability of the macrocycle to flex along the axis defined by the two saturated meso
carbons may contribute to this phenomenon. In most instances, the roof-like fold of the
porphodimethene is not detectable on the NMR time-scale and the 1H NMR spectra
rather resemble the more symmetrical structure. Nickel porphodimethene 1-18 and the
free-base analog, on the other hand, feature three distinct sets of signals for the six ethyl
substituents at room temperature, but at 310 K the three signals collapse into two
resonances, further supporting the flexing of the porphodimethenes in solution.23
Electronic Properties
From a detailed comparison of porphodimethene properties to the closely related,
well-studied porphyrins, a clearer, more complete picture of the features for the former
compounds can be assembled. The electronic spectra of porphyrins exhibit two
characteristic absorptions: a strong band around 400 nm (Soret or B band) and weak
absorption bands between 550 and 650 nm (Q-bands).24 Figure 1-3 illustrates a
molecular orbital diagram and a typical absorption spectrum of a porphyrin, exemplified
here by octaethylporphyrinatozinc(II).

11
N N
N NZn
Q
B
S2
S1
S0
eg y (LUMO) eg x (LUMO)
a1u (HOMO) a2u (HOMO - 1)
a1ua2u
eg
400 500 600 nm
Et
Et
Et
Et
Et
EtEt
Et
a b c d Figure 1-3. Schematic representation of a porphyrin spectrum:
a)Octaethylporphyrinatozinc(II); b)Molecular orbitals; c)States; d)Absorption spectrum (adapted from Anderson, H.L. Chem. Commun. 1999, 2323-2330).
These spectral features arise from π−π* transitions that mix together by
configurational interaction and the constructive interference of the two results in a strong
Soret or B band, while the destructive interference gives rise to weaker Q-bands.25 As
can be seen in Fig 1-4, a typical porphodimethene spectrum has a broad absorption at
about 440 nm instead of a sharp Soret feature. The porphodimethenes also lack the long
wavelength Q-bands characteristic of porphyrins.
350 450 550 650
Wavelength (nm) Figure 1-4. UV-vis spectra of a porphyrin (---) and a porphodimethene (—). Typical
porphodimethene absorption is red-shifted in comparison to a porphyrin and has a smaller extinction coefficient.

12
N
N
N
N
HH
H
HNi
H
H
N
N
N
NH HNi
H
H
b2a1a2
a1b1b2b1a2
a1
eg
b1g
a1gega1ua2u
a1gdx2-y2
dxy
dxzdyz
dz2
pp
pp
Figure 1-5. Nickel porphodomethene and porphyrin MO diagrams. (adapted from Re, N.;
Bonomo, L.; Da Silva, C.; Solari, E.; Scopelliti, R.; Floriani, C., Chemistry-a European Journal 2001, 7, (12), 2536-2546)
Based on the MO diagrams exemplified in Fig 1-5, the absorption spectra of nickel
porphyrin and porphodimethene differ primarily for two reasons:
• The energies of the metal orbitals (especially dxy) in the porphodimethene complex are higher
• The degenerate porphyrin eg (dxz, dyz) orbitals split into two inequivalent b1 and b2 orbitals in the porphodimethene.
The significant increase in the energy of dxy orbital can be attributed to the smaller
M-N core size in virtually all metalloporphodimethenes when compared to corresponding
porphyrins, while the loss of degeneracy of the porphyrin eg(π*) orbital is caused by
lowering the macrocycle symmetry. Since the main absorptions in both porphyrin and
porphodimethene spectra originate from π−π* transitions, the aforementioned changes in
the relative energies of these orbitals lead to the red shift of the main porphodimethene
absorption with respect to the porphyrin Soret band.20 The presence of the two saturated
carbon atoms in tetrapyrrolic macrocycle breaks the aromaticity and disrupts the

13
electronic communication between the two halves of the porphodimethene molecule, and
therefore the UV-Vis spectra of these compounds resemble the sum of the two
dipyrromethene absorptions rather than the porphyrin absorption spectra.26
Electrochemistry
Previous to 1999, the electrochemical properties of porphodimethenes were
relatively unexplored with the exception of several molecules synthesized in Buchler’s
group and the ferrocenyl porphodimethene 1-10. In Ni(OEPMe2) 1-11, two reversible
oxidations and one reduction were found with respective half-wave potentials of 1.01 V,
0.64 V, and -1.52 V (vs. SCE). Surprisingly, even under oxidative potentials, the
porphodimethene did not dehydrogenate to give a porphyrin.26
Unlike square planar metals, axially bound iron and cobalt porphodimethenes
exhibit only a single oxidation and reduction in the presence of excess axial ligand, and
the potentials for these events are highly dependent on the nature of both the central
metal and the axial ligand.27 When the metal ion is incorporated into the meso-
substituent as is the case in the ferrocenyl porphodimethene 1-10 shown in Scheme 1-6
the potential for the metal-based oxidation for the porphyrin is reduced to values close to
zero (0.02 and 0.23 V, vs. SCE), due to effective stabilization of the monocation through
an extended π-system.14
Reactivity
The identity of the central atom as well as the presence and type of the axial
ligands on the metal influence the reactivity of the alkyl substituted
metalloporphodimethenes. Starting from the tin hexaethylporphodimethene 1-6,
magnesium and dilithium derivatives can be obtained by transmetallation.
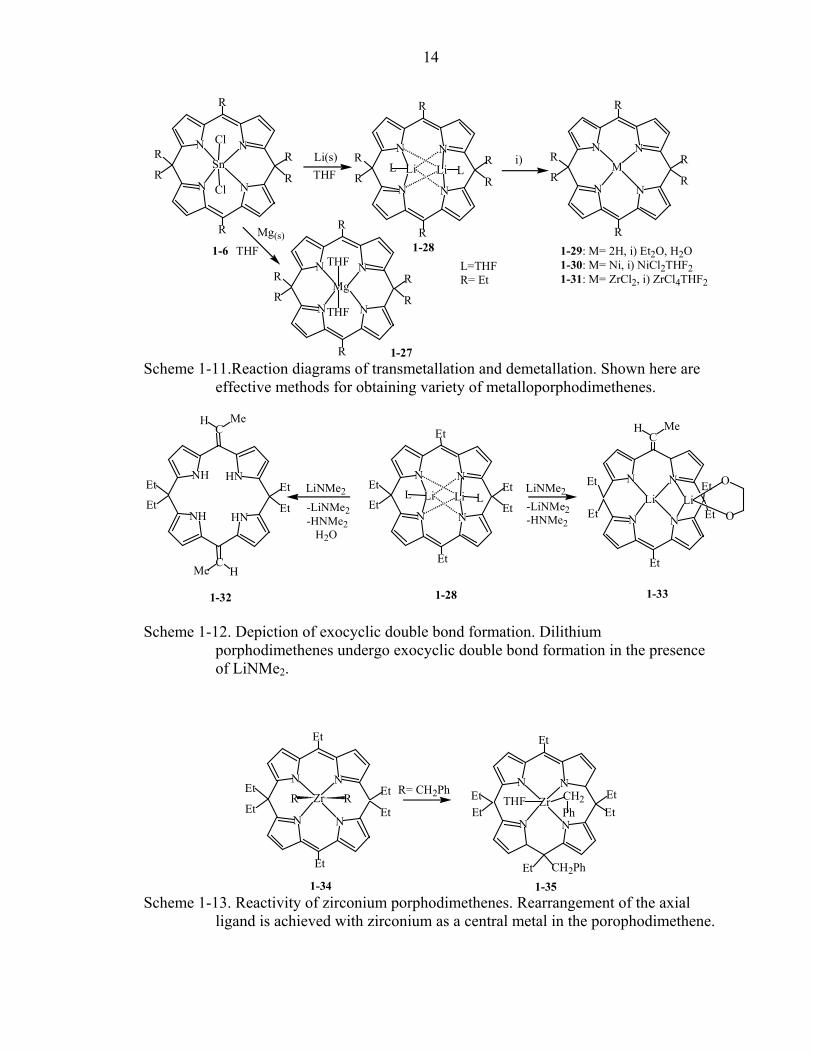
14
N N
N NRR
RR
R
R
Sn
Cl
Cl N N
N NRR
RR
R
R
N N
N NRR
RR
R
R
MLi Li LLLi(s) i)
1-29: M= 2H, i) Et2O, H2O1-30: M= Ni, i) NiCl2THF21-31: M= ZrCl2, i) ZrCl4THF2
N N
N NRR
RR
R
R
Mg
THF
THF
Mg(s)
L=THFR= Et
1-6
1-27
1-28
THF
THF
Scheme 1-11.Reaction diagrams of transmetallation and demetallation. Shown here are
effective methods for obtaining variety of metalloporphodimethenes.
N N
N NEtEt
EtEt
Et
Et
Li Li LL LiNMe2-LiNMe2-HNMe2 N N
N N EtEt
EtEt
C
Et
Li LiNH HN
NH HNEtEt
EtEt
O
O
MeH
LiNMe2
-LiNMe2-HNMe2 H2O
CMeH
CMe H
1-32 1-28 1-33
Scheme 1-12. Depiction of exocyclic double bond formation. Dilithium
porphodimethenes undergo exocyclic double bond formation in the presence of LiNMe2.
N N
N NEtEt
EtEt
Et
Et
Zr RR
N N
N NEt
Et
Et
Et
Zr CH2Ph Et
Et THF
CH2Ph
R= CH2Ph
1-351-34 Scheme 1-13. Reactivity of zirconium porphodimethenes. Rearrangement of the axial
ligand is achieved with zirconium as a central metal in the porophodimethene.

15
Many different metalloporphodimethenes are accessible upon further
transmetallation of the dilithium species. (Scheme 1-11).23, 28 If the addition-elimination
reaction sequence is performed on the dilithium porphodimethene 1-28, it gives rise to
tetrapyrrolic species containing one or two exocyclic double bonds on the meso carbon
atoms (Scheme 1-12).29
Furthermore, porphomethenes and porphyrinogens can be generated from
zirconium and nickel porphodimethenes. Zirconium porphodimethene 1-34 undergoes
reductive alkylation at one of the sp2 meso carbons to form porphomethene species 1-35
(Scheme 1-13),28 while the nickel porphodimethene 1-36 acts as an electrophile in
reactions with different nucleophiles to form the porphyrinogens as illustrated in Scheme
1-14.
N N
N NEtEt
EtEt
Et
Et
Ni
N N
N NEtEt
EtEt
Et
Et
Nii)
R
R
i) LiCH2CN, R=CH2CNi) BuLi, R= Bui) LiHBEt3, R= H
Li(THF)2
(THF)2Li1-36 1-37
Scheme 1-14. Illustration of porphyrinogen formation. These reactions result in porphyrinogens with substitution patterns slightly different than the ones used to make hexaethyl nickelporphodimethene.
Unlike their alkyl substituted counterparts, spiro-tricyclic porphodimethenes are
easily metallated in one step using zinc, nickel, copper, palladium or platinum salts, and
they offer the unique ability to open the spiro-locks affording functionalized
porphyrins.10, 21 The conditions and the products of the ring-opening reactions will be
discussed in detail in chapters 3 and 5.

CHAPTER 2 PORPHODIMETHENE SYNTHESES
Introduction
The condensation of 5-aryl dipyrromethanes with aromatic aldehydes followed by
oxidation with DDQ was developed in 1984 by Lindsey30 as an efficient method for
syntheses of trans-meso substituted porphyrins (Scheme 2-1). The first step of this
reaction generates a porphyrinogen species, which is then easily oxidized to a porphyrin.
NH HN
NH HNAr
Ar
R
R
H
H
H
H
3 DDQ3 DDQH2
NH N
N HNAr Ar
R
R
2 Ar-CHOAcid
rt
NH NH
HR
2
+
Scheme 2-1. Representation of Lindsey condensation reaction.
NHNH
OO
O
HN O
OMe
COOMe
COOMe+ 2
Scheme 2-2. Illustration of reactivity of vicinal diketones
If the porphyrinogen oxidation can somehow be prevented at the two meso
positions, the reaction should produce a porphodimethene instead of a porphyrin. With
this issue in mind and inspired by the observation that aromatic vicinal diketones react in
a manner similar to aldehydes in condensation with pyrroles31 (Scheme 2-2), a synthetic
method for making meso-aryl substituted porphodimethenes was developed in our lab by
Dr. Michael Harmjanz.32 The condensation of acenaphthenequinone with 5-
16

17
aryldipyrromethanes followed by oxidation with DDQ resulted in the formation of
porphodimethenes. These macrocycles could be further transformed to give porphyrins
bearing two 8-functionalized naphthalene spacers upon reaction with base or sodium
boron hydride. The porphyrins have shown unusual electrochemical properties33 and
have been found to be excellent building blocks for heterometallic, one-dimensional
arrays.10 Initially, meso-aryl substituted porphodimethenes were prepared using 5-
aryldipyrromethanes and acenaphthenequinone containing carbonyl groups on a five-
membered ring (Scheme 2-3).
Several synthetic pathways, including reductive alkylation of porphyrins2 and
oxidative dealkylation of porphirinogens,23 can be used to generate meso-alkyl
substituted porphodimethenes, but these methods have been restricted to
porphodimethenes containing aliphatic substituents on the sp3 carbons.
HNNH
O
O 2.DDQ1.TFA
O
O O
NH NN HN
R
R
2
O
NH NN HN
R
R2R
+
R =
Br COOMe
ClCl
OMeOMeMeO
F F
t-But-Bu
Scheme 2-3. Depiction of the first spiro-tricyclic porphodimethenes.
More recently, Harmjanz employed aceanthrenequinone and phenanthrenequinone
in condensation reactions with 5-mesityldipyrromethane to obtain novel meso-aryl

18
porphodimethenes,22 and the work presented here with pyrrene-4,5-dione complements
his efforts to expand the scope of this reaction (Scheme 2-3).
O
O2.DDQ1.TFA
O
OOO
NH NN HN
R
RNH NN HN
R
R
Anti (4%)Syn (1%)
HNNH 2.DDQ1.TFA O
O
Anti (14%)
NH NN HN
R
R
R=Mesityl
O
O
2.DDQ1.TFA
OO O
Syn (4%)
NH NN HN
R
R2
Anti (18%)
ONH NN HN
R
R
2-1
2-2
2-4 2-5
2-3
2
2
O
O2
Scheme 2-4. Diagram of condensation reactions of different vicinal diketones.
We have demonstrated the use of different polycyclic vicinal diketones, with
carbonyl groups on both five- and six-membered rings, for preparing novel spiro-tricyclic
meso-aryl substituted porphodimethenes. The porphodimethene products were
characterized (including solid-state structure and fluorescence measurements), and tested
for ring opening at the spiro-lock. Since 2-3 was chosen as a subject of further reactivity
studies, analogues of this porphodimethene bearing aromatic substituents different then
mesityl were also synthesized (Scheme 2-5), including 2-8 as the only isolated syn isomer
of a phenanthrenone porphodimethene.

19
HNNH
2.DDQ1.TFA
O
O
NH NN HN
R
R2
O
O2+
HNNH
ClCl2.DDQ1.TFA
O
O
NH NN HN
R
R2
O
O2+
t-But-BuR =
R =
t-But-Bu
ClCl
O
NH NN HN
R
R
O
2-6
2-7 2-8
Scheme 2-5. Illustration of novel phenanthrenone porphodimethenes.
Results and Discussion
As demonstrated by Harmjanz et al.,22 porphodimethenes 2-1, 2-2 and 2-3 (see
Scheme 2-4) can be synthesized by a [2+2] MacDonald type condensation reaction of
aceanthrenequinone and phenanthrenequinone with 5-mesityldipyrromethane in the
presence of TFA as a catalyst and DDQ as an oxidant. The work presented here adds
pyrenone-4,5-dione to the list of aromatic polycyclic vicinal diketones that can be used in
the synthesis of porphodimethenes. Unlike acenaphthenequinone, phenanthrenequinone
and pyrene-4,5-dione both contain carbonyl groups on six-membered rings, and these two
molecules were selected to examine the influence of six-membered rings of the vicinal
diketones on the chemistry of the porphodimethenes (Scheme 2-3).
Aceanthrenequinone22, acetnaphthenequinone21 and pyrene-4,5-dione react with 5-
mesityldipyrromethane to yield both the syn and the anti isomer. Surprisingly,
condensation of phenanthrenequinone with 5-mesityldipyrromethane or 5-(3,5-di-tert-
butyl-phenyl) dipyrromethane gives exclusively the anti isomer.

20
The solid-state structure of 2-5 is shown in Figure 2-1. The porphodimethene
skeleton shares meso-carbons 5 and 15 as spiro centers with the 4 position of pyrenone.
The polycyclic substituents are aligned trans and oriented anti to each other. The
presence of two saturated carbons (5 and 15) causes the molecule to fold and adopt a
roof-like structure with an inter-planar roof-angle of 138.0(1)°.
Figure 2-1. Diagram of the solid-state structure of 2-5 (40% probability; carbon atoms
depicted with arbitrary radii). Hydrogen atoms are omitted for clarity.
The two dipyrromethane halves deviate only slightly from planarity (0.04 Å and
0.10 Å), while the four pyrrole rings are completely planar (mean deviation from the
plane is less then 0.01 Å). Bond lengths between α and meso carbons range from
1.396(9) Å for the unsaturated meso carbon to 1.555(9) Å for the aliphatic carbons. The
meso substituents are nearly perpendicular to the porphodimethene core with interplanar
angles between pyrenone moieties and the macrocylic ring of 88.3(1)º and 89.6(1)º, while

21
the angles between mesityl groups and tetrapyrrols are 83.4(2)º and 78.9(2)º. Selected
bond lengths and angles are listed in Table 2-2.
The X-ray structure of 2-6 is illustrated in Figure 2-2. Even though the
phenanthrenone substituent is less rigid than the pyrenone, the polyaromatic backbone
still retains planarity and forms the angle of 83.9(0)º with the porphodimethene core. The
molecule is virtually flat, with the roof-angle stretched out to 180º, and the mean
deviation from the plane defined by 20 carbon and 4 nitrogen atoms of the core being
0.075 Å. Bond lengths between α and meso carbons range from 1.377(3) Å for the
unsaturated meso carbon to 1.516(2) Å for the aliphatic one. The angle between di-t-
butyl-phenyl substituents and the porphodimethene core is 70.5(0)º, showing that these
substituents are somewhat more rotated toward the plane of the macrocycle than the
mesityl moieties in 2-5. The higher degree of rotation is attributed to less steric
hindrance due to the presence of hydrogens in 2 and 6 positions, instead of methyl
groups, and this phenomenon has previously been noted in meso substituted porphyrins.25
The angles on saturated carbons within the porphodimethene ring are 118.4(1)°, and this
number tends to change dramatically upon insertion of the metal in the macrocycle (vide
infra). Selected bond lengths and angles are listed in Table 2-2.
As illustrated in Figure 2-3, compound 2-8 represents the only example of
phenathrenone substituted porphodimethene in which the two oxygens point in the same
direction, forming the syn isomer. In this species, the distance between the oxygens is
3.691 Å, which is comparable to the distances found in other syn spiro-tricyclic
porphodimethenes.

22
Figure 2-2. Diagram of the solid-state structure of 2-6. (40% probability; carbon atoms
depicted with arbitrary radii). Hydrogen atoms are omitted for clarity. Primed and non-primed atoms are related by center of inversion.
Figure 2-3. Diagram of the solid-state structure of 2-8. (40% probability; carbon atoms
depicted with arbitrary radii). Hydrogen atoms are omitted for clarity.

23
The roof angle in this structure is 137.4(1)º, with the dipyrromethene mean plane
deviations of 0.042 Å and 0.094 Å. The angles between the phenanthrenone substituents
and the porphodimethene core are 89.0(0)º and 82.4(1)º, proving that the orientation of
the substituents does not affect them significantly. The same angles in the related
compound 2-6 are 83.9(0)º. The angles between the substituents on the sp2 meso carbon
atoms and the macrocycle are 85.4(1)º and 85.9(1)º, closer to those in 2-5 then in 2-6.
These findings are in agreement with the previously discussed relationship between the
degree of rotation and the presence of the substituents in 2 and 6 positions of the phenyl
ring. If the meso substituents are disregarded, the tetrapyrrolic cores of the
porphodimethenes with six-membered rings on the spiro-locks have very similar
structural parameters for compounds 2-5 and 2-8, while the core of 2-6 appears to be
significantly flattened compared to the first two. This is discrepancy, caused by the
difference in crystal packing is illustrated in Figure 2-4 and quantified by selected
parameters in Table 2-1.
Table 2-1. Selected bond lengths and angles for 2-5, 2-6 and 2-8 2-5 2-6 2-8
N1-C1 1.409(8) 1.391(5) 1.317(2)
N1-C4 1.351(8) 1.337(5) 1.417(2)
C1-C20 1.416(9) 1.405(6) 1.377(3)
C4-C5 1.531(9) 1.512(5) 1.512(5)
O1-C33 1.194(5) 1.216(2)
C4-C5-C6 113.9(5) 111.9(3) 118.4(2)
C14-C15-C16 113.9(5) 115.1(3)

24
Figure 2-4. Diagrams of the porphodimethene cores of 2-5, 2-8 and 2-6 (40% probability; carbon atoms depicted with arbitrary radii). Primed and non-primed atoms are related by center of inversion
2-6
2-82-5
NH NN HN
R
R
O
O
2-5
R= a a
b
cc
a
b
c
Figure 2-5. The 1H NMR spectrum indicates highly symmetric nature of 2-5 illustrating the fast flexing of the molecule in solution at room temperature.

25
Based on an examination of the solid-state structure, the sets of aromatic
substituents in the anti-isomers (2-3, 2-5) should be asymmetrical due to the roof-like
fold in the molecule, but both compounds exhibit equivalent resonance in 1H NMR for
the meso-bound polyaromatic systems as well as the mesityl substituents (as illustrated in
Figure 2-5), consistent with the fast flexing of a porphodimethene macrocycle as
observed in 1H NMR described earlier for related acenaphthenone derivatives.10
Conclusions
The use of different vicinal aromatic diketones for the syntheses of the spiro-
tricyclic porphodimethenes has been demonstrated. The aromatic groups on the
porphodimethene sp2 meso carbons can be varied easily, by changing the 5-aryl
substituents on the dipyrromethane starting material. Other molecules used for
phenanthrenone porphodimethene synthesis, but not included in this thesis, are 4-t-
butylphenyl and 3,4,5-trimethoxyphenyl dipyrromethanes.
Even though the reaction conditions were the same for all the porphodimethenes
reported herein, different ratios of cis and trans isomers were obtained, depending on the
identity of the diketone and the aryl group of the dipyrromethane. The spiro-tricyclic
porphodimethenes were tested for metallation and the ring opening at sp3 meso carbons,
and the details of the reactivity studies are described in chapters 3 and 5.
Experimental
General Procedures.
NMR spectra were recorded on Varian Mercury or VXR 300 MHz
spectrometers. UV-Vis spectra were recorded with a Varian Cary 50 spectrophotometer.
High resolution mass spec analyses were performed by University of Florida Mass Spec
services using FAB or ESI as ionization method. The compounds 5-mesityl

26
dipyrromethane, pyrene-4,5-dione, 5-(2,6-dichlorophenyl)-dipyrromethane and 5-(3,5-di-
tert-butyl-phenyl) dipyrromethane were prepared following the literature procedures.34-36
All solvents were used as purchased, unless otherwise specified.
Chromatography
Absorption column chromatography was performed using neutral
alumina (Aldrich, Brockman I ~ 158 mesh, 58Ǻ) or chromatographic silica gel (Fisher,
200 – 425 mesh).
Syntheses of 2-4 and 2-5
A sample of 2.000 g (8.62 mmol) of pyrene-4,5-dione and 2.276 g (8.62 mmol) of
5-mesityldipyrromethane were dissolved in 930 ml of CH2Cl2. Trifluoroacetic acid (1.19
ml, 14.61 mmol) was added, and the reaction mixture was stirred for 50 minutes at room
temperature. A portion of 1.909 g (8.62 mmol) of DDQ was then added, and the mixture
was stirred for another 20 minutes. The volume was reduced by 90%, and the mixture
was loaded onto alumina column and eluted with CH2Cl2. The first orange fraction was
collected. The solvent was evaporated, and the residue redissolved in a minimal amount
of o-dichlorobenzene and placed on a silica column. Separation was achieved with
benzene as eluent. Compound 2-4 was isolated as the second orange fraction. Removal
of the solvent yielded 2-4 as an orange solid (0.041g, 1%). UV-Vis [o-dichlorobenzene,
λmax (log ε)] 442 nm (4.97). mp 240ºC (dec). 1H NMR (300 MHz, o-dichlorobenzene-d4):
δ = 13.71 (s, 2H), 8.01 (d, 2H J = 7.49 Hz), 7.71 (d, 2H J = 7.79 Hz) , 7.44 – 7.37 (m,
4H) 7.54 (d, 2H, J = 8.99 Hz), 7.43 (d, 2H J = 8.39 Hz), 7.38 (d , 2H J = 7.49 Hz), 7.26
(dd, 2H J1 = J2 = 7.49 Hz), 6.47 (s, 2H), 6.37 (s, 2H), 5.91 (bs, 4H), 5.39 (d, 4H J = 4.20
Hz), 1.93 (s, 6H), 1.65 (s, 6H), 1.46 (s, 6H). HRMS (FAB) calcd. for MH+ (C68H49O2N4):
953.3855. Found 953.3833.

27
The anti isomer 2-5 was collected as the first fraction from the silica column in the
reaction procedure described for 2-4. Yield: 0.178 g (4 %). UV-Vis [o-dichlorobenzene,
λmax (log ε)] 440 nm (4.94). mp 320ºC (dec). 1H NMR (300 MHz, o-dichlorobenzene-d4):
δ = 13.82 (s, 2H), 8.53 (d, 2H J = 7.49 Hz), 8.33 (d, 2H J =7.49 Hz), 7.76 (d, 2H J = 8.08
Hz), 7.66 (d, 2H J1 = J2 = 7.79 Hz), 7.55 (d, 2H, J = 7.79 Hz), 7.48 (d, 2H 8.99 Hz), 7.42
– 7.34 (m, 4H), 6.40 (s, 4H), 5.95 (d, 4H J = 3.90 Hz), 5.71 (d, 4H, J = 4.20 Hz), 1.91 (s,
6H), 1.54 (s, 12H). HRMS (FAB) calcd. for MH+ (C68H49O2N4): 953.3855. Found
953.3869. Single crystals were grown by slow evaporation of a saturated solution of 2-5
in o-dichlorobenzene.
Synthesis of 2-6
Compound 2-6 was prepared following the literature procedure for 2-3.
Phenanthrenequinone (1.142 g, 5.49 mmol) and 5-(3,5-di-tert-butyl-phenyl)
dipyrromethane (1.832 g, 5.49 mmol) were dissolved in 550 ml of methylene chloride. A
sample of 0.75 ml (9.21 mmol) of TFA was added dropwise. The solution was stirred at
room temperature for two hours prior to addition of 1.238 g (5.49 mol) of DDQ, and the
mixture was stirred for an additional hour. Volume was then reduced to 10%, and the
solution was filtered through an alumina column with methylene chloride. The solvent
was evaporated under vacuum to yield 0.473 g (16 %) of orange solid. Slow diffusion of
pentane in a saturated chloroform solution of 2-6 afforded small, single crystals. UV-Vis
[methylene chloride, λmax (log ε)] 438 nm (4.90). 1H NMR (300 MHz, CDCl3) δ =
13.41 (s, 2H), 8.39 (dd, 4H, J1 = J2 = 9.0 Hz ), 8.14 (d, 4H, J = 7.8 Hz), 7.76-7.68 (m,
4H), 7.57-7.54 (m,4H), 7.40 (s, 2H), 7.24 (s, 4H), 6.36 (d, 4H, J = 4.2 Hz), 5.85 (d, 4H, J

28
= 3.9 Hz), 1.26 (s, 36H). HRMS (FAB) calcd. for MH+ (C74H69O2N4): 1045.5420. Found
1045.5438.
Syntheses of 2-7 and 2-8
A 1.429 g (6.87 mmol) portion of phenanthrenequinone and 2.000 g (6.87 mmol)
of 5-(o-dichlorophenyl) dipyrromethane were dissolved in 400 ml of methylene chloride
and 0.78 ml (9.58 mmol) of TFA was added dropwise to the solution. The solution was
stirred for 90 minutes at the room temperature and DDQ (1.670 g, 7.35 mmol) was
added. The reaction mixture was stirred for an additional hour. Excess DDQ was filtered
off, and the solution volume was reduced to 10%, and filtered through neutral alumina.
The first, orange fraction was collected, and the solvent was removed. The solid residue
was washed with toluene and filtered. The filtrate was loaded onto silica column. The
first orange fraction that came of the column with toluene as an eluent was collected, and
the solvent was evaporated to yield orange-brown powder. Yield: 0.340 g (9 %). UV-
Vis [toluene, λmax (log ε)] 433 nm (5.04). 1H NMR (300 MHz, CDCl3) δ = 13.17 (s, 2H),
8.25 (dd, 2H J1 = 1.5 Hz, J2 = 7.8 Hz), 8.16-8.11 (m, 4H), 7.90 (dd, 2H, J1 = 7.5 Hz, J2 =
1.8 Hz), 7.74 (ddd 4H, J1 = 1.5 Hz, J2 = J3 = 7.5 Hz), 7.54 – 7.44 (m, 6H),7.35 –7.32 (m,
4H) 6.11 (d, 4H, J = 3.9 Hz), 5.72 (d, 4H, J = 4.2 Hz). HRMS (FAB) calcd. for
MH+(C58H33N4O2Cl4, monoisotopic peak) 957.1358. Found: 957.1358.
The compound 2-8 was collected as precipitate from the toluene washing and
filtration in the reaction procedure described for 2-7. Yield: 0.110 g (3 %). UV-Vis
[toluene, λmax (log ε)] 440 nm (4.89). 1H NMR (300 MHz, CDCl3) δ = 13.27 (s, 2H),
8.40 (dd, 2H J1 = 1.2 Hz, J2 = 7.8 Hz), 8.17 (dd, 4H, J1 = 10.8 Hz, J2 = 8.4 Hz),
7.71(ddd, 2H J1 = 1.5 Hz, J2 = J3 = 7.5 Hz ), 7.57 – 7.43 (m, 6H), 7.39 – 7.31 (m, 6H),

29
7.27 (s, 2H) 6.11 (bs, 4H), 5.61 (d, 4H, J = 3.9 Hz). HRMS (FAB) calcd. for
MH+(C58H33N4O2Cl4, monoisotopic peak) 957.1358. Found: 957.1131
Table 2-2. Crystallographic data
2-5·4C6H4Cl2 2-6·2CHCl3 2-8·CH2Cl2
Formula C92H64Cl8N4O2 C76H70O2N4Cl6 C59H34O2N4Cl6
Formula weight 1541.07 1284.53 1029.58
Crystal system Monoclinic Monoclinic Triclinic
Space group P21/n C2/c P1
Z 4 4 2
Temp, K 173(2) 173(2) 193(2)
Dcalc/ gcm-3 1.350 1.276 1.289
a, Å 20.934(6) 29.624(1) 13.009(1)
b, Å 16.421(4) 12.557(1) 13.350(1)
c, Å 22.497(8) 19.250(1) 17.885(1)
a, deg - - 73.142(1)
β, deg 101.358(15) 111.071(1) 69.193(1)
γ, deg - - 68.285(1)
V Å3 7582(4) 6682(2) 2652.2(2)
µ, mm-1 0.352 0.307 0.369
Uniq. data coll./obs. 11196/7181 5885/4458 12126/7652
R1 [I > 2σ(I)data]a 0.0966 0.0566 0.0856
wR2 [I > 2σ(I)data]b 0.2395 0.1331 0.2635 a R1 = Σ||Fo| - |Fc||/ Σ| Fo| bwR2 = { Σ[w (Fo
2 – Fc2)2/ Σ[w ( Fo
2)2}

30
X-ray Crystallography
Unit cell dimensions were obtained (Table 2-1) and intensity data collected by Prof.
Michael Scott on a Siemens CCD SMART diffractometer at low temperature, with
monochromatic Mo-Kα X-rays (λ = 0.71073 Å). The data collections nominally covered
over a hemisphere of reciprocal space, by a combination of three sets of exposures; each
set had a different φ angle for the crystal and each exposure covered 0.3° in ω. The
crystal to detector distance was 5.0 cm. The data sets were corrected empirically for
absorption using SADABS.37 The structures were solved using the Bruker SHELXTL
software package for the PC, by direct method option of SHELXS.
The space groups were determined from an examination of the systematic
absences in the data, and the successful solution and refinement of the structure
confirmed these assignments. All hydrogen atoms were assigned idealized locations and
were given a thermal parameter equivalent to 1.2 or 1.5 times the thermal parameter of
the carbon atom to which they were attached. For the methyl groups, where the location
of the hydrogen atoms was uncertain, the AFIX 137 card was used to allow the hydrogen
atoms to rotate to the maximum area of residual density, while fixing their geometry.
Relevant crystallographic data are listed in Table 2-1.

CHAPTER 3 METALLATION AND RING-OPENING REACTIONS
Introduction
The insertion of metals into porphyrin macrocycles has frequently been employed
in syntheses, and the reaction conditions to prepare these complexes range from mild for
high yielding syntheses of manganese and iron derivatives38 to very harsh for moderate
yielding synthesis of palladium and platinum porphyrins.39 The resulting
metalloporphyrins often have different properties and reactivities than the porphyrin
precursors.1 The number of reported free-base porphodimethenes is very limited18, 40, 41,
due to the fact that the most porphodimethenes synthesized thus far can be obtained from
reductive alkylation of metalloporphyrins19 or oxidative dealkylation of
metalloporphyrinogens.9 The macrocycles isolated from these reactions are inherently
metallated, and in order to change the metal center, transmetallation of the product9 or the
starting material18 must be employed. The 2+2 MacDonald’s type condensation of 5-aryl
dipyrromethanes and vicinal diketones provides a number of free-base porphodimethenes
that can be metallated in one step, and the presence of keto-groups on the spiro-locks
introduces a new mode of reactivity to this class of tetrapyrolles. We were interested in
comparing the properties and reactivities of metalloporphodimethenes and their free-base
precursors.
Other than our entry in the field of the porphodimethene syntheses, all
porphodimethenes prepared thus far feature alkyl substituents at the sp3 meso carbons,
making these ill-suited for porphyrin forming reactions. With an interest in producing
31

32
compounds that could be used as precursors to otherwise inaccessible porphyrins, we
prepared the spiro-tricyclic porphodimethenes introduced in Chapter 2. These synthons
were designed to be susceptible to ring opening and rearrangement reactions producing
porphyrins bearing pendant functional groups or porphyrin with fused exocyclic rings.
Results and Discussion
Metallation of Porphodimethenes
Free-base porphodimethenes depicted in Scheme 3-1 are easily metallated using
nickel, zinc, copper, palladium or platinum salts. With the two sp3 carbons incorporated
into the macrocyclic ring, porhodimethenes are inherently more flexible than the
porphyrins, and can easily accommodate metals of different sizes.6
The insertion of metals within the porphodimethene macrocycle is generally a high
yielding reaction which can be easily monitored by UV-Vis spectroscopy, since the
typical porphodimethene absorbtion shifts from 430-440 nm to longer wavelengths (470-
510 nm).21 In addition, many of the characteristic features in the 1HNMR spectra of the
porphodimethene change upon metallation. For instance, the separation between the two
doublets arising from the pyrrolic protons in the 1H NMR spectrum of 3-7 decreases in
comparison to the metal free porphodimethene (0.67 ppm versus 0.53 ppm). The
disappearance of the singlet for the NH protons in the free-base provides perhaps the
most diagnostic change on going from the free-base to metalloporphodimethene. Altered
electronic situation within the dipyrromethene halves as well as a modification of the
structural configuration of the macrocycle upon metallation, clearly evident in a
comparison of the solid-state structures of free-base and metalloporphodimethenes, are
likely responsible for the observed change in the spectroscopy

33
O
O
NH N
N HN
R
RO
O
N N
N N
R
R
M
3-6: R = Mesityl
3-11: M = Cu; i) Cu(OAc)2 in MeOH, CH2Cl2, rt3-12: M = Ni; i) Ni(OAc)2 in MeOH, CH2Cl2, rt
3-7: M = Pd; i) Pd(PhCN)2Cl2, CH2Cl2, rt
3-10: R = 3,5-(di-t-butyl)-phenyl
O
O
NH N
N HN
R
R O
O
N N
N N
R
R
M
3-3 3-4: M = Pd; i) Pd(PhCN)2Cl2, reflux in xylenes3-5: M = Zn; i) Zn(OAc)2in MeOH, reflux in CHCl3
i)
R = Mesityl
3-8: M = Pt; i) PtCl2, reflux in PhCN
O
O
O
O
3-1: R = Mesityl 3-2: M = Pd; i) Pd(PhCN)2Cl2, reflux in PhCN
NH N
N HN
R
R N N
N N
R
R
M
3-9: M = Cu; i) Cu(OAc)2 in MeOH, CH2Cl2, rt
3-13: M = Pd; i) Pd(PhCN)2Cl2, CH2Cl2, rt
i)
i)
Scheme 3-1. Illustration of porphodimethene metallation reactions.
As previously outlined in Chapter 1, the properties of porphyrin and
porphodimethene macrocycles are markedly different. The metallation of the macrocycle
with palladium is more facile for porphodimethenes in comparison to porphyrins, with
shorter reaction times, better yields and stoichiometric amounts of the metal, most likely

34
due to the increased flexibility of the porphodimethene macrocycle. Porphodimethenes
can accommodate metals of different sizes imposing less strain on the structure than the
corresponding porphyrins. Due to the presence of saturated carbon atoms in the
macrocyclic ring, the metal-nitrogen bonds in palladium porphodimethenes are somewhat
longer than in their porphyrin counterparts.20 Although platinum porphodimethene has
very similar structure to its palladium analog (vide infra), the insertion of platinum is
limited by the low solubility of Pt(II) salts in the common solvents and harsher conditions
(i.e. refluxing in benzonitrile for 7 days ) are required, resulting in reduced yield.
Structure of Metalloporphodimethenes.
Palladium anthracenone porphodimethene
The crystal structures of various metalloporphodimethenes best illustrate the
flexibility these macrocycles exhibit in accommodating different metal ions.
Figure 3-1. Diagram of the solid-state structure of 3-2 (40% probability; carbon atoms are
depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity.

35
The solid-state structure of 3-2 is shown in Figure 3-1. This molecule adopts a
roof-like folded structure, with the interplanar angle of 136.4 °. The bond angles on the
saturated carbons within the macrocycle are somewhat greater than for an ideal
tetrahedron, but they do not exceed 113.8(6)° due to the strain imposed by the presence
of five-membered rings at the spiro-locks and the rigidity of the anthracene backbone.
Palladium adopts a square-planar arrangement, with bond lengths ranging from 2.001(6)
Å to 2.007(6) Å, and bond angles between 88.7(2)° and 90.4(2)°. The 20 carbon atoms
and the four nitrogens within the tetrapyrrolic ring define the mean plane of the
porphodimethene core, and the average deviation of the 24 atoms from the plane is 0.339
Å, with a maximum deviation being 1.005(8) Å for the sp3 carbon in the
porphodimethene core. Palladium is situated 0.212(2) Å above the mean plane of the
porphodimethene core. The 1H NMR spectrum of this molecule is consistent with a fast
flexing of the molecule in solution, equilibrating between the two roof-like folded
structures. Structural parameters, such as roof angle, palladium –nitrogen bond lengths
and angles, and the displacement of palladium from the plane defined by four nitrogens
for the related palladium porphodimethene with naphthenone substituents at the spiro-
lock42 are coincident to the ones discussed here. The correlation of these parameters
suggests that the additional six-membered ring in the anthracenone substituent compared
to the naphthenone moiety does not have significant influence on the solid-state structure
of 3-2.
Palladium pyrenone porphodimethene
Palladium porphodimethene 3-4 has a six-membered ring with the rigid pyrenone
backbone at the spiro-lock. The pyrenone substituents are orthogonal to the plane of the
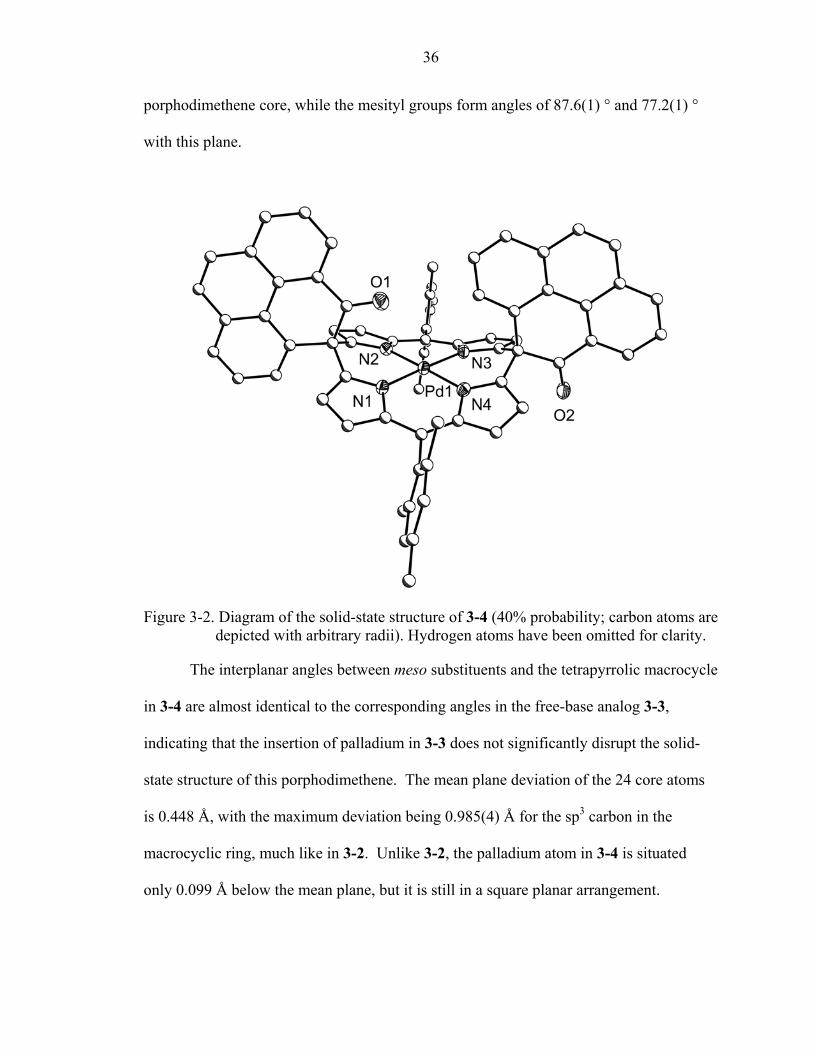
36
porphodimethene core, while the mesityl groups form angles of 87.6(1) ° and 77.2(1) °
with this plane.
Figure 3-2. Diagram of the solid-state structure of 3-4 (40% probability; carbon atoms are depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity.
The interplanar angles between meso substituents and the tetrapyrrolic macrocycle
in 3-4 are almost identical to the corresponding angles in the free-base analog 3-3,
indicating that the insertion of palladium in 3-3 does not significantly disrupt the solid-
state structure of this porphodimethene. The mean plane deviation of the 24 core atoms
is 0.448 Å, with the maximum deviation being 0.985(4) Å for the sp3 carbon in the
macrocyclic ring, much like in 3-2. Unlike 3-2, the palladium atom in 3-4 is situated
only 0.099 Å below the mean plane, but it is still in a square planar arrangement.

37
Palladium and platinum phenanthrenone porphodimethenes
The increased flexibility of phenanthrenone substituents at the spiro-locks
compared to the pyrenone or anthracenone allows the porphodimethene core in 3-7 to
flatten out in the presence of palladium forcing angles of nearly 118° on the saturated
carbons within the ring
Figure 3-3. Diagram of the solid-state structure of 3-7 (40% probability; carbon atoms are
depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity. Primed and non-primed atoms are related by crystalographically imposed center of inversion.
. As can be seen from the figure 3-3, palladium is in a square planar conformation,
with angles of 90.2(1) ° and 89.8(1) ° between the neighboring nitrogens. The palladium-
nitrogen bonds average 2.037(6) Å, while the average deviation from the mean plane of
the 24 core atoms is only 0.052 Å. The angles between porphodimethene core and the

38
substituents are 88.1(0) º for the phenanthrenone and 71.1(0) ° for the mesityl moieties.
The 1H NMR spectrum is in good agreement with the solid-state structure.
Figure 3-4. Diagram of the solid-state structure of 3-8 (40% probability; carbon atoms are
depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity. Primed and non-primed atoms are related by crystallographically imposed center of inversion.
The structure of the platinum porphodimethene 3-8 (Figure 3-3) is, as expected,
very similar to the palladium analog, 3-7. The palladium nitrogen bond lengths in 3-7 are
the same as the platinum nitrogen bond lengths in 3-8 within the estimated standard
deviation. The porphodimethene core is virtually flat with the average mean plane
deviation of only 0.038 Å for the 24 core atoms and the platinum is situated within the
mean plane. The bond angles on the sp3 meso carbons within the porphodimethene ring

39
once again extend to almost 118 °. The angles between the porphodimethene core and
the mesityl substituents in 3-8 are slightly greater than in 3-7 (75.8(1)° vs. 71.1(0) °).
Copper phenanthrenone porphodimethenes
Even though the smaller size of copper compared to palladium and platinum can be
expected to induce roof-like folding in the structure of 3-9, the core of this
porphodimethene is also flat with the average mean plane deviation even smaller than in
3-7 or 3-8 (0.038 Å). The copper adopts a square planar geometry and is situated 0.001
Å above the mean plane of the core. Metal-nitrogen bonds in the copper
porphodimethene are somewhat shorter (2.025 Å) than in platinum and palladium
analogs. The meso substituents form angles of 76.8 ° (mesityl) and 87.0 °
(phenanthrenones) with the porphodimethene ring.
The solid-state structure of 3-11 is illustrated in Figure 3-5. The smaller size of
copper does, in this case, induce a roof-like folding of the macrocycle resulting in an.
angle of 131.7(8)° between the two dipyrromethene halves of the molecule (Figure 3-6).
Metal ligand bonds in 3-11 are shorter (1.966(3) Å to 1.992(3) Å) than in previously
discussed palladium, platinum and copper species, causing the saddle shape of the
molecule. Crystal packing forces can account for different geometries of the two copper
porphodimethenes. Phenathrenone substituents in 3-11 are almost perpendicular to the
porphodimethene core, while the di-t-butyl substituents are somewhat tilted forming the
angles of 75.1(1)° and 56.7(1)° with the core
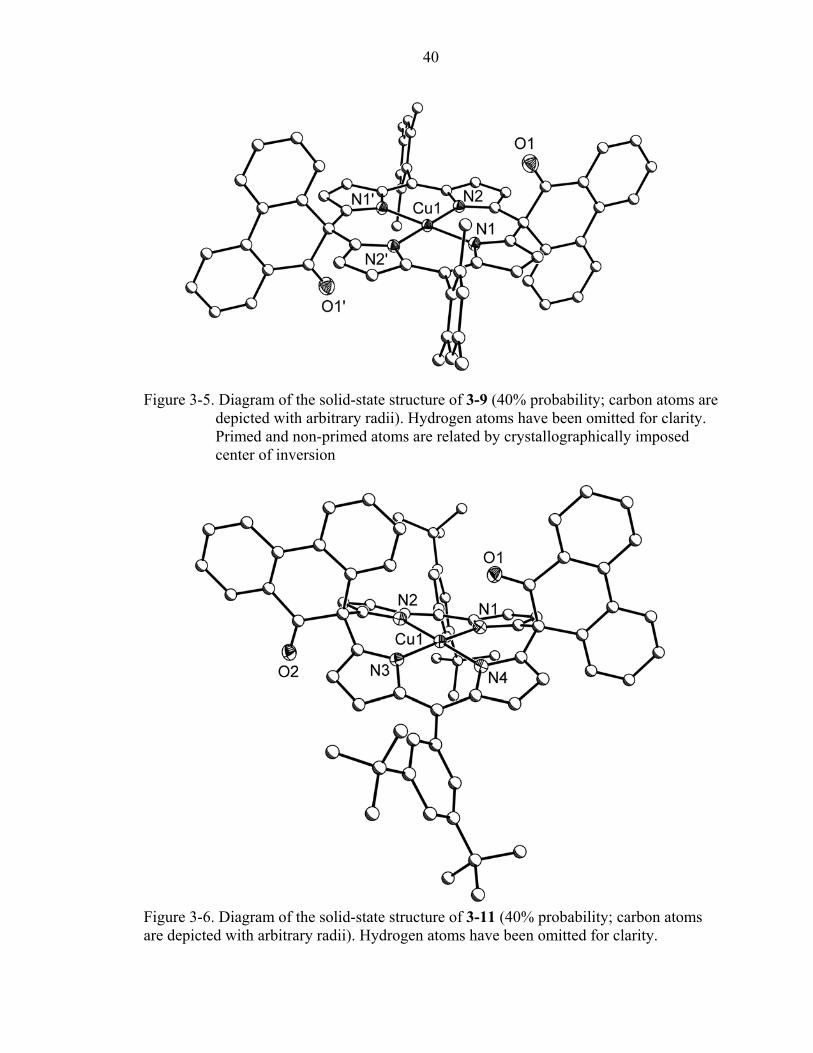
40
Figure 3-5. Diagram of the solid-state structure of 3-9 (40% probability; carbon atoms are depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity. Primed and non-primed atoms are related by crystallographically imposed center of inversion
Figure 3-6. Diagram of the solid-state structure of 3-11 (40% probability; carbon atoms are depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity.

41
Nickel phenanthrenone porphodimethene
The small size of nickel(II) causes a severe ruffling of the porphodimethene core
upon metallation (Figure 3-5). In the solid-state structure, nickel maintains the square-
planar configuration with metal-nitrogen bond lengths ranging from 1.884(5) to 1.910(5)
Å and angles of 89.2(2)° to 90.9(2), but it is displaced from the porphodimethene core
mean plane by 0.134 Å. The roof-angle in this structure is smaller than in other
porphodimethenes reported here (124.9(1)°). Normally, the presence of nickel in
porphodimethenes locks the roof-like conformation in the solution, and the 1H NMR
spectra of the compounds are more complex.20 Compound 3-12 exhibited a similar
tendency for broad spectral features in 1H NMR spectrum at room temperature, and sharp
resonances were only evident upon heating to 105°. While nickel porphodimethenes with
five membered rings on the spiro-locks do not flex at all at room temperature21, the six-
membered ring of the phenanthrenone in 3-12 is less restraining and at elevated
temperatures this molecule flexes fast enough to display a less complex 1H NMR
spectrum, indicative of a symmetrical molecule - similar to its palladium analog 3-13.
Summary of the Structural Data
Selected bond lengths and angles as well as the interplanar angles and mean plane
deviations listed in Table 3-1 illustrate some of the trends observed in the solid state
structures of the spiro-tricyclic metalloporphodimethenes. The decrease in the metal-
nitrogen bond lengths is accompanied by the decrease in the roof angle. The short nickel-
nitrogen bonds induce a ruffling of the macrocycle that results in a roof angle of only
125°. The metal-nitrogen bonds are the longest in virtually flat palladium
phenanthrenone porphodimethenes (2.037 Å), and they become shorter when the roof-
like folding of the porphodimethene is imposed by more rigid anthracenone and pyrenone

42
substituents. The length of the copper-nitrogen bonds also varies considerably depending
on the conformation of the macrocycle. In the flat copper porphodimethene 3-9 metal-
nitrogen bonds are 0.044 Å longer then in the roof-like folded molecule 3-11. The
central metal does not have much influence on the angles of the substituents at C10 and
C20. These angles seem to be more dependant on the nature of the substituents
themselves. The mesityl groups form angles of 72.0 ° to 87.6 ° with the
porphodimethene core in all the structures, while the angles of di-t-butyl-phenyls are
more flexible and range from 56.9 ° to 80.2 °.
Figure 3-7. Diagram of the solid-state structure of 3-12 (40% probability; carbon atoms
are depicted with arbitrary radii). Hydrogen atoms and the disordered t-butyl groups have been omitted for clarity.
Reactivity of Porphodimethenes
Apart from chelating metals, spiro-tricyclic porphodimethenes exhibit reactivity
towards ring-opening, resulting in functionalized porphyrins.21, 32 A related reaction was
described in 1986 by Chang and Kondylis31 for ring-opening of an acetnatphthenone

Table 3-1. Selected parameters from the solid-state structures of metalloporphodimethenes.
43
3-2 3-4 3-7 3-8 3-9 3-11 3-12
M-N (avg.) 2.005 (6) Å 2.004(4) Å 2.037(6) Å 2.037(3) 2.025(2) 1.981(3) Å 1.900(3) Å
N-M-N (avg.) 89.9(2) ° 90.0(2) ° 90.0(1) ° 90.0(1) ° 90.0(1 ) ° 90.0(1) ° 90.0(1) °
C4-C5-C6 113.3(8) ° 112.8(4) ° 117.7(1) ° 117.4(3) ° 117.4(3) ° 114.0(3) ° 110.3(3) º
C14-C15-C16 113.8(6) ° 113.2(4) ° - - - 111.3(3) ° 109.1(3) º
Roof angle 135.6(2) ° 136.8(1) ° 180.0 ° 180.0 ° 179.8 ° 131.7(8)° 124.9(1) º
Average mean plane deviation
0.339 Å 0.448 Å 0.052 Å 0.038 Å 0.012 Å 0.344 Å 0.536 Å
Maximum mean plane deviation
1.005(8) Å 0.985(4) Å 0.140 Å 0.121 Å 0.031 Å 0.881(4)Å 1.133(4) Å
M-mean plane 0.212(2) Å - 0.099 (1) Å 0.000 Å 0.000 Å 0.014 Å 0.134(1)Å 0.140(1)

44
dipyrromethane in refluxing 30% potassium hydroxide. Following this approach,
treatment of porphodimethenes with strong bases such as KOH in the presence of
dioxygen afforded porphyrins with two carboxylic functionalities.21 If sodium
borohydride is used instead of KOH, the ring-opening reaction yields porphyrin with two
benzyl alcohol groups (Scheme 3-2). In view of the ease at which functionalized
porphyrins can be isolated from spiro-ticyclic porphodimethenes, we undertook further
studies to establish the influence of the ring-size at the spiro-lock as well as the identity
of the groups on the meso positions on the reactivity of the macrocycle. The ring-
opening reactions were first tested on the naphthenone porphodimethenes with five-
membered rings on the spiro-locks,21 and the same conditions were successfully
employed to convert phenanthrenone porphodimethene with six-membered rings at the
spiro-locks to corresponding bis-functionalized porphyrins (Scheme 3-3).
Metalloporphodimethenes react in the same fashion in the presence of base or NaBH4 and
oxygen to form bisfunctionalized metalloporphyrins.21
Ring opening reactions of dispiro porphodimethenes appear to be driven by the
desire of the macrocycle to achieve fully aromatic 18-annulene ring system and we have
utilized that driving force to explore the reactivity of porphodimethenes with six-
membered ring on the spiro-lock. Both the pyrenone derivative 3-3 and the
phenanthrenone derivative 3-6 have a six-membered ring at the spiro-lock, but their
reactivities are considerably different.

45
HN
N
R
N
HN
R
1. NaOMe, THF, rt
2. O2
O
ONH
N
R
N
HN
R
1. NaBH4, THF, rt
2. O2, HCl
R = Mesityl
MeO
OMe
O
O
HO
OH
NH N
N HN
R
R
O
O NH N
N HN
R
R
Scheme 3-2. Diagram of the ring-opening of the acenaphthenone porphodimethene. Both
the dialcohol and the diester formation are almost quantitative.
HN
N
R
N
HN
R
1. NaOMe, THF, rt
2. O2
1. NaBH4, THF, rt
2. O2, HCl
R = Mesityl
CO2Me
MeO2C
HN
N
R
N
HN
R
CH2OH
HOH2C
O
O
NH N
N HN
R
R
O
O
NH N
N HN
R
R
3-14
Scheme 3-3. Diagram of ring-opening of the phenanthrenone porphodimethene.
Resulting α,β porphyrins interconvert to corresponding α,α isomers upon heating in toluene.

46
In addition to previously demonstrated ring-opening reaction of 3-6 to form
biphenyl- porphyrin dialcohol (the first example of biphenyl substituted porphyrin),22 we
have been able to obtain corresponding porphyrindiester derivatives shown in Scheme 3-
4. Much like the porphyrin dialcohol, the diester 3-14, which is the only isomer formed
in the ring-opening reaction, interconverts to α,α atropoisomer upon heating in toluene.
The interconversion reaches equilibrium at the isomer ratio 42: 58 (α,α : α,β).
The solid state structure of porphyrin 3-14 (Figure 3-8) shows that the biphenyl
moieties are twisted for 82.9(1) ° with respect to the porphyrin ring and the phenyl
groups within the biphenyls are rotated 51.6(1) ° relative to each other. This
conformation places the two carboxy carbons 3.104(3) Å above and below the porphyrin
plane, reducing unfavorable interactions of the methoxy groups with the electron rich
macrocycle.
Figure 3-8. Diagram of the solid-state structure of 3-14 (40% probability; carbon atoms
are depicted with arbitrary radii). Hydrogen atoms have been omitted for clarity. Primed and non-primed atoms are related by center of inversion

47
The rigidity of the pyrene backbone, on the other hand, caused the compound 3-3
to be stable under harsh conditions (KOH in refluxing THF, or H2SO4 in refluxing o-
dichlorobenzene) and resistant to ring opening and porphyrin formation in the presence of
NaBH4 or KMnO4. If the six-membered rings were to open, the pyrenones would be
converted into 10-functionalized phenanthrene groups, and the carboxylates (or hydoxy
groups) would be forced to point directly at the ring of the resulting porphyrin, as
illustrated in Scheme 3-4. These unfavorable interactions appear to preclude the ring
opening at the spiro-lock of compound 3-3.
Another type of ring-opening reaction specific to metal containing spiro-tricyclic
porphodimethenes is a radical initiated rearrangement of carbon-carbon bonds to yield
porphyrins with exocyclic keto-rings.
1. NaBH4, THF, rt2. O2, HCl
O
O
NH N
N HN
R
R
HO
NH N
R
N HN
OH
R
3-3 Scheme 3-4. Illustration of the ring-opening reaction of 3-3. The resulting porphyrin
would have two functional groups pointing towards the macrocyclic ring.
The reaction conditions for this transformation are mild for naphthenone
porphodimethenes and very harsh for phenanthrenone porphodimethenes (Scheme 3-5).
Syntheses of porphyrins with exocyclic rings will be discussed in detail in Chapter 5.
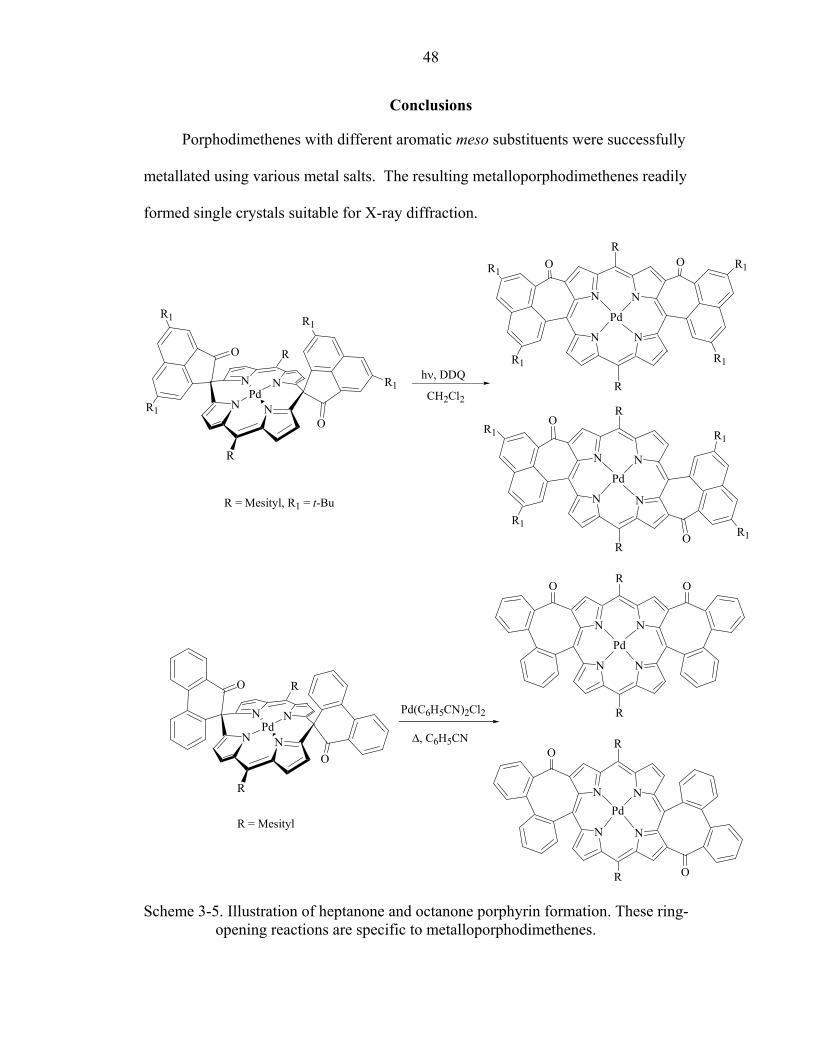
48
Conclusions
Porphodimethenes with different aromatic meso substituents were successfully
metallated using various metal salts. The resulting metalloporphodimethenes readily
formed single crystals suitable for X-ray diffraction.
N N
N N
R
R
O
Pd
O
∆, C6H5CN
Pd(C6H5CN)2Cl2
N
N
RO
N
N
O
R
N
N
RO
N
N
OR
Pd
Pd
R = Mesityl
O
O
R1
R1
R1
R1
N N
N N
R
R
Pd
N
N
N
N
Pd
OO
R
R
R1
R1
R1
R1
N
N
N
N
Pd
O
O
R
RR1
R1R1
R1
hν, DDQ
CH2Cl2
R = Mesityl, R1 = t-Bu
Scheme 3-5. Illustration of heptanone and octanone porphyrin formation. These ring-
opening reactions are specific to metalloporphodimethenes.

49
The dissimilarities in the solid-state structures of the metalloporphodimethenes
arise from the differences in the central metal atom as well as the nature of the
substituents at saturated meso carbons and the size of the ring at the spiro-lock.
The nature of the substituents at spiro-locks also determines the reactivity of the
porphodimethenes. The presence of phenanthrenones at saturated meso carbons is
suitable for ring-opening reactions resulting in bis functionalized porphyrins.
Porphodimethenes with pyrenone substituents at the spiro-locks, on the other hand, are
unreactive under the ring-opening reaction conditions. In view of their unique stability,
these macrocycles should be suitable for further functionalization of carbonyl groups
without altering the porphodimethene backbone.
Experimental
General Procedures.
NMR spectra were recorded on Varian Mercury or VXR 300 MHz
spectrometers. UV-Vis spectra were recorded with a Varian Cary 50 spectrophotometer.
High resolution mass spec analyses were performed by University of Florida Mass Spec
services using FAB or ESI as ionization method. All solvents were used as purchased,
unless otherwise specified.
Chromatography
Absorption column chromatography was performed using neutral alumina
(Aldrich, Brockman I ~ 158 mesh, 58 Ǻ) or chromatographic silica gel (Fisher, 200 – 425
mesh).
Synthesis of 3-2
A portion of 0.024g (0.02 mmol) of 3-1 was dissolved in 20 ml of xylenes and
0.015 g (0.04 mmol) of Pd(C6H5CN)2Cl2 in methanol was added. The mixture was

50
refluxed for 3 days, and the solvent was evaporated under vacuum. The solid residue was
redissolved in a minimal amount of methylene chloride and purified through a silica gel
column. The first bright orange fraction was collected, and the solvent was evaporated to
give 0.022g (83%) of dark orange solid. 1H NMR (300 MHz, CDCl3) : δ = 8 .70 (d, 2H,
J = 7.5 Hz), 8.63 (d, 2H, J = 6.9 Hz), 8.24 (J = 6.6 Hz), 7.98 – 7.79 (m, 10H), 6.80 (s,
4H), 6.27 (d, 4H, J = 4.5 Hz), 5.70 (d, 4H, J = 4.5 Hz), 2.26 (s, 6H), 2.06 (s, 12H). UV-
Vis [methylene chloride, λmax(logε) ] 491 nm (4.51). Anal.Calcd. for
C68H46N4O2Pd·2CH2Cl2: C, 68.62; H, 4.12; N, 4.58. Found: C, 66.88; H, 3.86; N, 4.48 MS
(MALDI-DIOS) calcd. for M+ (C68H46O2N4Pd): 1056. Found 1056. Experimental
isotope pattern matched the theoretical isotope pattern. Slow diffusion of pentane into a
saturated chloroform solution of 3-2 yielded single crystals suitable for diffraction
studies.
Synthesis of 3-4
A sample of 0.155 (0.16 mmol) g of 3-3 and 0.070 g (0.25 mmol)of
Pd(C6H5CN)Cl2 was dissolved in 50 ml of benzonitrile, and the mixture was refluxed for
90 minutes. The solvent was removed under vacuum, and the solid was washed with
methylene chloride – toluene (5-1) and filtered. The filtrate was preadsorbed on silica gel
and purified through a silica column with methylene chloride. The leading orange band
was collected, and the solvent was evaporated to give dark orange solid. Yield 87 %.
Slow evaporation of toluene solution of 3-4 afforded large single crystals. UV-Vis
[toluene, λmax (log ε)] 501nm (4.86). 1HNMR (300 MHz, CDCl3): 9.36 (d, 2H, J = 8.1
Hz), 8.96 (d, 2H, J = 6.6 Hz), 8.77 (s, 2H), 8.19 (d, 2H, J = 8.4 Hz), 8.08 (s, 1H), 8.05 (s,

51
1H), 7.90 (dd, 2H, J1 = 6.6 Hz J2 = 11.7 Hz), 7.84- 7.79 (m, 2H), 7.67-7.61 (m, 2H), 6.83
(s, 4H), 6.36 (d, 4H, J = 4.5 Hz), 6.03 (d, 4H, J= 4.2 Hz), 2.28 (s, 6H), 2.07 (s, 12H).
Synthesis of 3-5
A sample of 0.065g (0.07 mmol) of 3-3 was dissolved in 60 ml of chloroform, and
5 ml of a saturated methanolic solution of Zn(OAc)2 was added. The reaction mixture
was refluxed overnight, cooled to the room temperature, washed with water and dried
over Na2SO4. The solvent was evaporated to yield 0.065 g (94 %) of green solid. UV-Vis
[methylene chloride, λmax (log ε)] 474 nm (4.99). 1H NMR (300 MHz, o-C6D4Cl2): 8.59
(d, 2H, J = 7.5 Hz), 8.38 (d, 2H, J = 7.5 Hz ), 8.18 (d, 2H, J = 6.9 Hz), 7.93 – 7.71 (m, 10
H), 6.85 (s, 4H), 6.50 (d, 4H, J = 3.9 Hz), 6.02 (d, 4H, J = 4.2 Hz), 2.33 (s, 6H), 2.13 (s,
12H). HRMS (FAB) calcd. for MH+ (C68H47O2N4Zn) 1015.2990. Found: 1015.3021.
Synthesis of 3-7.
A sample of 0.010 g (0.03 mmol) of Pd(C6H5CN)2Cl2 was heated in 50 ml of
refluxing dry toluene until it completely dissolved. After cooling to 70 ºC, 0.025 g (0.03
mmol) of phenanthrenone porphodimethene 3-6 was added to the solution, and the
reaction mixture was heated to 90 ºC and kept at this temperature for 2.5 hours. The
solution was filtered through a plug of silica, and the solvent was evaporated under
vacuum yielding 0.027 g (97 %) of dark orange powder. The product was recrystallized
from chloroform/pentane. UV-Vis [methylene chloride, λmax (log ε)] 489 nm (5.03) .
Anal.Calcd. for C64H46N4O2Pd·2CHCl3: C, 63.51; H, 3.88; N, 4.49. Found: C,63.22; H,
3.43; N, 4.49. 1H NMR (300 MHz, CDCl3) δ = 8.28 (dd, 4H, J1 = J2 = 7.2 Hz), 8.14 (dd,
4H, J1 = 8.1 Hz, J2 = 11.1 Hz), 7.74 (dd, 2H, J1 = J2 = 7.2 Hz), 7.54 – 7.43 (m, 6H), 6.82
(s, 4H), 6.29 (d, 4H, J = 4.5 Hz), 5.74 (d, 4H, J = 4.5 Hz), 2.28 (s, 6H), 2.06 (s, 12H).

52
HRMS (ESI-FT-ICR) calcd. for M+ (C64H46O2N4Pd): 1008.2672. Found 1008.2665.
Slow diffusion of pentane in a concentrated acetonitrile solution of 3-7 produced large
single crystals.
Synthesis of 3-8
A sample of 0.050 g (0.05 mmol) of 3-6 was dissolved in 20 ml of benzonitrile
and 0.046 mg (0.16 mmol) of PtCl2 was added. The solution was heated to reflux, and
after 7 days of refluxing, the solvent was removed under vacuum. The solid residue was
redissolved in a minimal amount of methylene chloride. The solution was columned over
silica with methylene chloride : toluene 1: 1. The second, orange band was collected, and
the solvent was evaporated. The residue was recrystalized from methylene chloride -
hexanes. Slow diffusion of pentane into a saturated methylene chloride solution of 3-8
afforded crystals suitable for X-ray diffraction. Yield 0.010 g (16 %). UV-Vis [toluene,
λmax (log ε)] 496 nm (5.01). 1HNMR (300 MHz, CDCl3) δ = 8.26 (d, 2H, J = 7.50 Hz ),
8.18 – 8.11 (m, 6H), 7.75 (dd, 2H, J1 = J2 = 7.20 Hz), 6.83 (s, 4H), 6.36 (d, 2H, J = 4.50
Hz), 5.82 (d, 4H, J = 4.50 Hz), 2.29 (s, 6H), 2.07 (s, 12H). HRMS (FAB) calcd. for MH+
(C64H47O2N4Pt): 1098.3347. Found 1098.3352.
Synthesis of 3-9
A portion of 0.025 g (0.03 mmol) of 3-6 was dissolved in 20 ml of methylene
chloride and 10 ml of a saturated methanolic solution of Cu(OAc)2 was added. The
reaction mixture was stirred at room temperature for 40 minutes. The solution was then
filtered through a silica gel plug. Evaporation of the solvent resulted in the isolation of
0.027g (99 %) of red-orange powder. Slow diffusion of pentane into a saturated
cloroform solution of 3-9 produced large single crystals. UV-Vis [methylene chloride,

53
λmax (log ε)] 482 nm (5.15). HRMS (FAB) calcd. for MH+ (C64H47O2N4Cu): 966.2995.
Found 966.2989.
Synthesis of 3-11
A portion of 0.050 g (0.05 mmol) of 3-10 was dissolved in 40 ml of methylene
chloride and 10 ml of saturated methanolic solution of Cu(OAc)2 was added. The
reaction mixture was stirred at room temperature for 40 minutes. The solution was then
filtered through a plug of silica. Evaporation of the solvent resulted in the isolation of
0.052 g (98 %) of red-orange powder. UV-Vis [methylene chloride λmax (log ε)] 480 nm
(4.79). HRMS (FAB) calcd. for MH+ (C74H67O2N4Cu): 1106.4560. Found 1107.4130
(isotope distribution corresponds to combination of M+ and MH+). Slow diffusion of
pentanes into a chloroform saturated solution of 3-11 afforded crystals suitable for
diffraction studies.
Synthesis of 3-12
A sample of 0.050 g (0.05 mmol) of 3-10 was dissolved in 30 ml of methylene
chloride and 8 ml of saturated solution of Ni(OAc)2 in methanol was added. The reaction
mixture was refluxed overnight, washed with water, and dried over Na2SO4. The solvent
was evaporated to yield 0.035 g (66 %) of dark orange solid. Slow diffusion of ether into
a chloroform solution saturated with 3-12 produced X-ray quality crystals. UV-Vis
[toluene, λmax(log ε)] 487 nm (4.40), 433 nm (4.33). 1H NMR (300 MHz, toluene-d8,
105°C) δ = 9.74 (bs, 2H), 8.39 (d, 2H, J = 7.5 Hz ), 7.79 – 7.67 (m, 6H), 7.47- 7.45 (s,
2H), 7.29 – 7.20 (m, 8H),7.03 (s, 2H, under the solvent peak) 6.48 (d, 4H, J = 4.5 Hz),
5.90 (d, 4H, J = 4.5 Hz), 1.19 (s, 36H). Anal.Calcd. for C74H66N4O2Ni·2CHCl3: C, 68.08;

54
H, 5.11; N, 4.18. Found: C,68.22; H, 4.73; N, 4.28. HRMS (FAB) calcd. for MH+
(C74H67O2N4Ni): 1101.4617. Found 1101.4526.
Synthesis of 3-13
A sample of 0.025 g (0.02 mmol) of 3-10 was dissolved in 15 ml of methylene
chloride and a solution of Pd(PhCN)2Cl2 (0.010 g in 2 ml of methanol) was added. The
reaction mixture was stirred at room temperature until the starting material was consumed
(48 h). The solution was then concentrated and purified over silica column with
methylene chloride. The first orange band was collected, and the solvent was evaporated.
Yield: 0.021 g (76 %). 1H NMR (300 MHz, CDCl3) δ = 8.47 (d, 2H, J = 7.8 Hz), 8.33
(d, 2H, J = 7.6 Hz ), 8.18 – 8.11 (m, 4H), 7.74 (dd, 2H, J1 = J2 = 7.5 Hz), 7.57 (dd, 2H,
J1 = J2 = 7.5 Hz), 7.50 - 7.44 (m, 4H), 7.39 (s, 2H), 7.25 (s, 2H), 7.18 (s, 2H), 6.45 (d,
4H, J = 4.5 Hz), 5.81 (d, 4H, J = 4.5 Hz), 1.26 (s, 36H). UV-Vis [methylene chloride,
λmax (log ε)] 487 nm (5.30). MS (MALDI-DIOS) calcd. for M+ (C74H66O2N4Pd): 1148.
Found 1148. Experimental isotope pattern matched the theoretical isotope pattern.
Synthesis of 3-14 and 3-15
A sample of 0.025 g (0.63 mmol) of sodium was dissolved in 5 ml of methanol and
10 ml of THF. Following the dissolution of sodium, 0.027 g (0.03 mmol) of 3-6 was
added, and the reaction mixture was stirred under argon. After two hours of stirrting,
oxygen was bubbled through the solution, followed after 5 minutes, with 15 ml of water
and 35 ml of methylene chloride. The organic layer was collected, washed with water,
and dried over Na2SO4. The solvent was removed under vacuum, and the solid residue
was redissolved in a minimal amount of methylene chloride and purified through a plug
of silica. The first, green-purple band was collected and the solvent evaporated under
vacuum. Yield: 0.025 g (94%). UV-Vis [methylene chloride, λmax (log ε)] 422 nm

55
(5.53), 553 nm (3.64), 594 nm (3.61), 649 nm (3.27) . Anal.Calcd. for C66H54N4O4: C,
81.96; H, 5.63; N, 5.79. Found: C,81.87; H, 5.48; N, 5.31. 1H NMR (300 MHz, CDCl3) δ
= 9.04 (bs, 2H ), 8.70 (bs, 2H ), 8.50 (bs, 4H), 8.01 (d, 2H, J = 7.5 Hz), 7.78 (dd, 2H, J1 =
J2 = 7.5 Hz), 7.66 - 7.61 (m, 4H), 7.28 – 7.25 (m, 8H ), 6.61 – 6.51 (m, 4H), 3.67 (s, 6H),
2.61 (s, 6H), 1.73 (s, 12H), -2.86 (s, 2H). HRMS (FAB) calcd. for M+ (C66H55N4O4)
966.4145. Found: 966.4170
Upon heating the solution of 3-14 in toluene, interconversion to α,β isomer 3-15
was observed. Interconversion was complete after 2 hours of reflux and the ratio of the
two isomers was estimated by integration of the NMR resonances to be approximately
42: 58 α,α : α,β.
An analyticaly pure sample of 3-15 was separated from 3-14 on a silica column
using toluene : methylene chloride 1:1 as eluent. Compound 3-15 was collected as the
second fraction. UV-Vis [methylene chloride, λmax(log ε)] 423 nm (5.67), 518 nm (3.56)
553 nm (3.50), 650 nm (2.85). 1H NMR (300 MHz, CDCl3) δ = 9.00 (bs, 2H ), 8.68 (bs,
2H ), 8.49 (bs, 4H), 8.16 (d, 2H, J = 7.2 Hz), 7.81 (dd, 2H, J1 = J2 = 7.2 Hz), 7.72 - 7.65
(m, 4H), 7.28 – 7.25 (m, 4H ), 7.05 (bs, 4H), 6.39 (d, 2H, J = 7.2 Hz), 3.57 (s, 6H), 2.61
(s, 6H), 1.73 (s, 12H), -2.93 (s, 2H) . HRMS (FAB) calcd. for MH+ (C66H55N4O4)
967.4223. Found: 967.4224. Slow diffusion of pentane in the methylene chloride
solution of 3-15 produced small single crystals.
X-ray Crystallography
Unit cell dimensions were obtained (Tables 3-2 and 3-3) and intensity data
collected by Prof. Michael Scott on a Siemens CCD SMART diffractometer at low
temperature, with monochromatic Mo-Kα X-rays (λ = 0.71073 Å).

56
Table 3-2. Crystallographic data for compounds 3-2, 3-4, 3-7 and 3-8 3-2· CHCl3 3-4· 2C7H8 3-7 3-8·2CH2Cl2
Formula C69H47Cl3N4O2Pd C82H62N4O2Pd C64H46N4O2Pd C66H50Cl4N4O2Pt
Formula weight 1176.86 1241.76 1009.45 1267.99
Crystal system Triclinic Monoclinic Monoclinic Monoclinic
Space group P1 P21/c P21/n P2/m
Z 2 4 2 2
Temp, K 173(2) 173(2) 193(2) 193(2)
Dcalc gcm-3 1.116 1.358 1.474 1.523
a Å 13.361(2) 15.0316(9) 11.003(7) 13.806(6)
b Å 16.996(3) 15.0316(9) 13.186(8) 14.359(6)
c Å 17.448(3) 14.5815(9) 15.691(10) 15.511(7)
α, deg 69.749(4) - - -
β, deg 87.155(4) 91.8500(10) 92.229(15) 115.974(10)
γ, deg 70.886(3) - - -
V Å3 3503.2(10) 6074.0(7) 2275(2) 2764(2)
µ, mm-1 0.420 0.361 0.463 0.278
Uniq. data coll./obs. 9644/7387 10030/5762 15066/5278 6266/4678
R1[I > 2σ(I)data]a 0.1050 0.0453 0.0322 0.0283
wR2[I > 2σ(I)data]b 0.2894 0.1076 0.0893 0.0762 a R1 = Σ||Fo| - |Fc||/ Σ| Fo| bwR2 = { Σ[w (Fo
2 – Fc2)2/ Σ[w ( Fo
2)2}

57
Table 3-3. Crystallographic data for compounds 3-9, 3-11, 3-12 and 3-15 3-9·2CHCl3 3-11· CHCl3 3-12· ½C4H10O 3-15
Formula C66H48Cl6N4O2Cu C75H67Cl3CuN4O2 C75H71N4O2.5Ni C66H54N4O4
Formula weight 1205.32 1226.22 1127.11 967.19
Crystal system Tetragonal Monoclinic Monoclinic Monoclinic
Space group I41/a P21/c P21/n P21/c
Z 8 4 4 2
Temp, K 173(2) 193(2) 193(2) 193(2)
Dcalc gcm-3 1.455 1.302 1.013 1.252
a Å 30.220(2) 24.305(1) 15.134(1) 14.152(6)
b Å 30.220(2) 13.659(1) 17.449(1) 12.700(6)
c Å 12.046(1) 19.031(1) 26.541(2) 14.344(6)
β, deg 90.0 98.022(1) 97.660(2) 95.542(1)
V Å3 11001(1) 6256(1) 6946(1) 2566(2)
µ, mm-1 0.740 0.528 0.321 0.078
Uniq. data coll./obs. 33993/6312 14762/8270 16351/ 17653/6008
R1[I > 2σ(I)data]a 0.0618 0.0715 0.0863 0.0503
wR2[I > 2σ(I)data]b 0.1653 0.1739 0.2262 0.1374 a R1 = Σ||Fo| - |Fc||/ Σ| Fo| bwR2 = { Σ[w (Fo
2 – Fc2)2/ Σ[w ( Fo
2)2}

58
The data collections nominally covered over a hemisphere of reciprocal space, by a
combination of three sets of exposures; each set had a different φ angle for the crystal and
each exposure covered 0.3° in ω. The crystal to detector distance was 5.0 cm. The data
sets were corrected empirically for absorption using SADABS.37 The structure was
solved using the Bruker SHELXTL software package for the PC, by direct method option
of SHELXS. The space group was determined from an examination of the systematic
absences in the data, and the successful solution and refinement of the structure
confirmed these assignments. All hydrogen atoms were assigned idealized locations and
were given a thermal parameter equivalent to 1.2 or 1.5 times the thermal parameter of
the carbon atom to which it were attached. For the methyl groups, where the location of
the hydrogen atoms was uncertain, the AFIX 137 card was used to allow the hydrogen
atoms to rotate to the maximum area of residual density, while fixing their geometry.

CHAPTER 4 PHOTOPHYSISCAL PROPERTIES OF PORPHODIMETHENES
Introduction
Although porphodimethenes have long been recognized to be intermediates in the
oxidation pathway of porphyrinogens to porphyrins, the first synthetic scheme for their
production was only reported in the 1974.2 Despite the success of Buchler, investigations
of the physical properties of porphodimethenes have been hampered by the difficulties
associated with the synthesis and separation of these partially reduced porphyrins. Since
new, simple procedures for the isolation of multigram quantities of porphodimethenes,
are now available, we undertook a detailed examination of their physical and chemical
properties. To date, structural,23 theoretical,40 magnetic,41 and electrochemical27 studies
have been conducted on metalloporphodimethenes, and herein we present the first
detailed study of the photophysical properties, including steady-state emission
measurements, transient absorption, and the quantum yield of singlet oxygen generation
of this unique class of tetrapyrrolic macrocycles. While only two porphodimethenes (4-2
and 4-14) were chosen for detailed photophysical examination, electronic absorption
spectra of all the previously reported metalloporphodimethenes prepared in our lab and
the fluorescence emission of several free-bases (Scheme 4-1) were also measured and
will be discussed in this chapter.
59

60
O
O
N N
N N
R
R
M
4-11: M = Pd; R = Mesityl4-12: M = Zn; R = Mesityl
4-10: M = 2H; R = Mesityl
O
4-16: M = 2H; R = Mesityl
O
N N
N N
R
R
M
O
O
4-1: M = Pd; R = Mesityl; R1 = t-butyl4-1a: M = Pd; R = p-Tolyl; R1 = H
N N
N N
R
R
M
O O
N N
N N
R
R
M
4-13: M = 2H; R = Mesityl
O
O
N N
N N
R
R
M
4-4: M = Pt; R = Mesityl4-5: M = Cu; R = Mesityl
4-7: M = Pd; R = 3,5-di-t-butyl-phenyl4-8: M = Cu; R = 3,5-di-t-butyl-phenyl4-9: M = Ni; R = 3,5-di-t-butyl-phenyl
4-3: M = 2H; R = Mesityl4-2: M = Pd; R = Mesityl
4-6: M = 2H; R = 3,5-di-t-butyl-phenyl
O
O
4-15: M = Pd; R = Mesityl4-14: M = 2H; R = Mesityl
N N
N N
R
R
M
R1
R1 R1
R1
Scheme 4-1 Depiction of free-base and metalloporphodimethenes with measured
electronic absorption spectra.
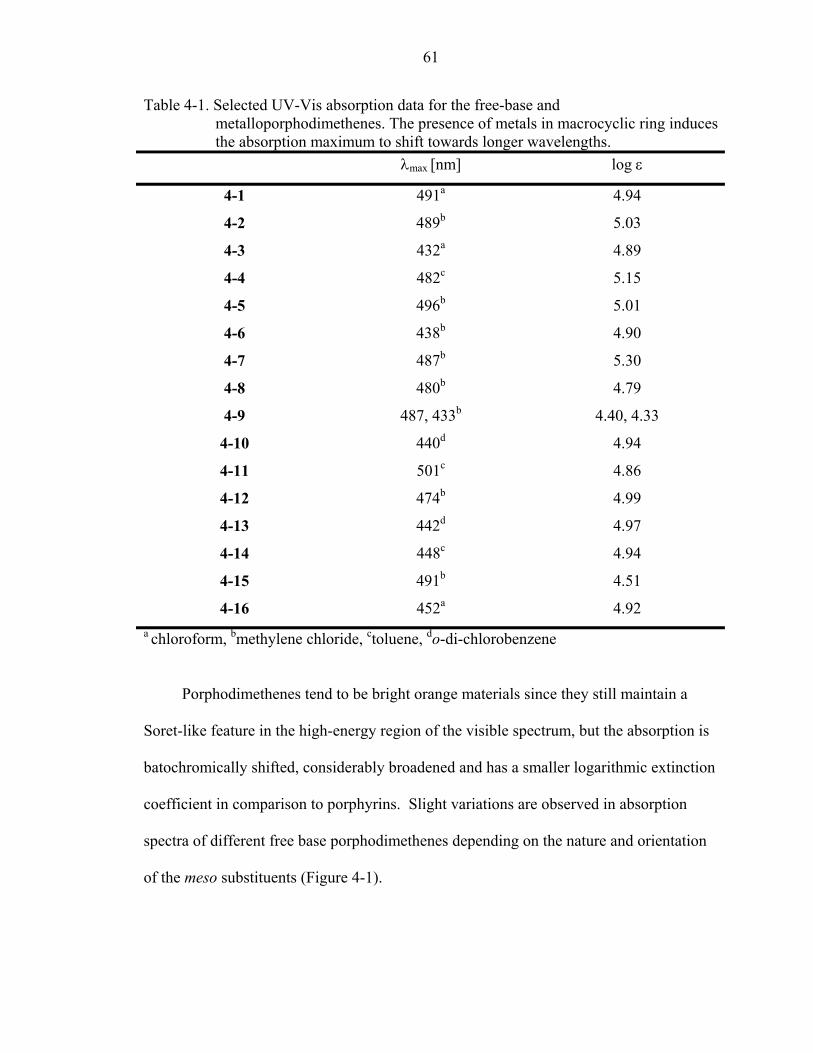
61
Table 4-1. Selected UV-Vis absorption data for the free-base and metalloporphodimethenes. The presence of metals in macrocyclic ring induces the absorption maximum to shift towards longer wavelengths.
λmax [nm] log ε
4-1 491a 4.94
4-2 489b 5.03
4-3 432a 4.89
4-4 482c 5.15
4-5 496b 5.01
4-6 438b 4.90
4-7 487b 5.30
4-8 480b 4.79
4-9 487, 433b 4.40, 4.33
4-10 440d 4.94
4-11 501c 4.86
4-12 474b 4.99
4-13 442d 4.97
4-14 448c 4.94
4-15 491b 4.51
4-16 452a 4.92 a chloroform, bmethylene chloride, ctoluene, do-di-chlorobenzene
Porphodimethenes tend to be bright orange materials since they still maintain a
Soret-like feature in the high-energy region of the visible spectrum, but the absorption is
batochromically shifted, considerably broadened and has a smaller logarithmic extinction
coefficient in comparison to porphyrins. Slight variations are observed in absorption
spectra of different free base porphodimethenes depending on the nature and orientation
of the meso substituents (Figure 4-1).

62
Wavelength / nm350 400 450 500 550 600 650
Abs
orba
nce 4-3
4-104-14
Figure 4-1: Illustration of the UV-Vis absorption spectra of free base porphodimethenes.
The meso substituents do not significantly influence spectral characteristics of the free-base spiro-tricyclic porphodimethenes.
Wavelength / nm
350 400 450 500 550 600 650
Abso
rban
ce
4-74-84-9
Figure 4-2. Illustration of the absorption spectra of metalloporphodimethenes. Spectral
properties depend on the nature of central atom.

63
Insertion of metals into the porphodimethene core causes the absorption maximum
to shift toward longer wavelengths, and the magnitude of this shift varies with the nature
of the metal (Figure 4-2).
With palladium incorporated into the macrocycle, in porphodimethenes 4-1 and 4-2
depicted in Scheme 4-1, main absorption band shifts towards longer wavelengths in
comparison to the free base macrocycles: from 432 nm to 489 nm and from437 nm to 491
nm, respectively for 4-1 and 4-2. Despite numerous crystallization attempts, compound
4-1 consistently formed small needle-like crystals which were unsuitable for diffraction
experiments, but a related porphodimethene 4-1b was structurally characterized.42 This
species differs from 4-1 in that it bears p-tolyl groups at two meso-positions rather than
mesityls and lacks the t-butyl groups on the naphthyl substituents. As can be seen in the
solid-state structure of 4-1b presented in Figure 4-3, the palladium resides in a square
planar arrangement, but the metal center is situated slightly above the mean plane of the
four pyrrole nitrogens by 0.068 Å. The two spiro-locks, defined by the five-membered
ring of the acenaphthenone, are trans to each other in an anti-configuration, and these
saturated carbon atoms force the tetrapyrrolic macrocycle to adopt a roof-like fold with
an interplanar roof angle of 135 °. In solution, however, the macrocycle flexes about the
saturated carbons, equilibrating the resonances in the 1H NMR of the two p-tolyl groups
and the two naphthyl moieties, and this tendency to flex about the sp3 carbons appears to
be general for most metalloporphodimethenes.
In contrast to the naphthenone substituted porphodimethene 4-1, the increased
flexibility afforded by the two phenanthrenone moieties and the six-membered rings at
the spiro-lock in 4-2 allow the angle between the two dipyrromethene units to flatten out
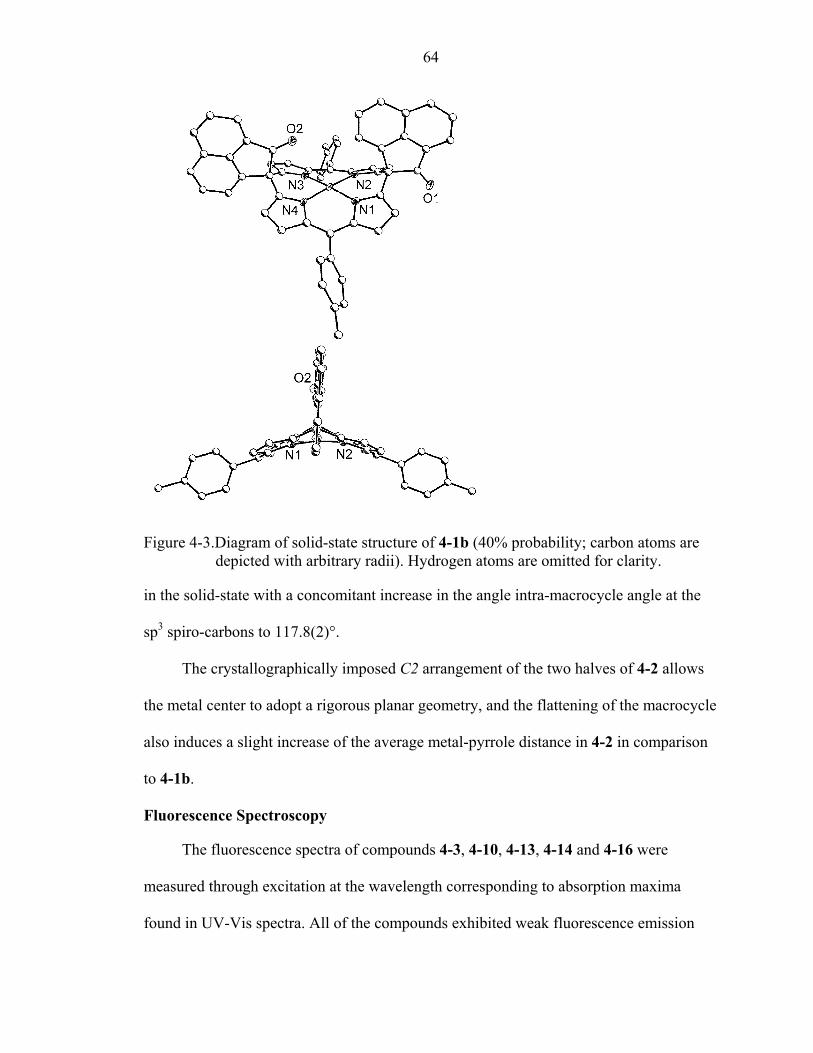
64
Figure 4-3.Diagram of solid-state structure of 4-1b (40% probability; carbon atoms are depicted with arbitrary radii). Hydrogen atoms are omitted for clarity.
in the solid-state with a concomitant increase in the angle intra-macrocycle angle at the
sp3 spiro-carbons to 117.8(2)°.
The crystallographically imposed C2 arrangement of the two halves of 4-2 allows
the metal center to adopt a rigorous planar geometry, and the flattening of the macrocycle
also induces a slight increase of the average metal-pyrrole distance in 4-2 in comparison
to 4-1b.
Fluorescence Spectroscopy
The fluorescence spectra of compounds 4-3, 4-10, 4-13, 4-14 and 4-16 were
measured through excitation at the wavelength corresponding to absorption maxima
found in UV-Vis spectra. All of the compounds exhibited weak fluorescence emission

65
between 592 nm and 688 nm (Table 4-2). Quantum yields range between 1.02x10-4 and
8.9x10-3 depending on the substituents and the isomer (syn or anti), which is low in
comparison to quantum yields of porphyrins.42 No trend was observed among different
macrocycles with regard to their fluorescence, although isomer pairs exhibited
comparable quantum yields.
Table 4-2: The values of fluorescence emission maxima and quantum yields for selected free-base porphodimethenes. The fluorescence is very weak.
Compound λexc [nm] λem [nm] Φf
4-3 433 592 7.94 x10-4
4-10 440 624 8.70 x10-3
4-13 442 624 1.41 x10-3
4-14 452 688 1.13 x10-4
4-16 448 615 1.02x10-4
Phosphorescence Emission
As described above, the absence of a porphyrin-like 18-annulene ring system in
porphodimethenes simplifies the absorption spectrum of the macrocycle, and given the
paucity of data concerning the photophysical characteristics of porphodimethenes, we
instigated a study of the emission properties of both 4-1 and 4-2. While free base
porphodimethenes do exhibit weak fluorescence emission, the insertion of a heavy atom
(palladium) should influence the spin-orbit coupling within the macrocycle and thus
enhance the singlet-triplet intersystem crossing at the expense of the fluorescence
emission from the singlet state. Indeed, although emission from the triplet-excited states
of 4-1 and 4-2 (phosphorescence) is not observed at room temperature, strong bands in
the visible region are evident upon cooling the samples. The highest intensity emission
maximum (at 703 nm) for compound 4-2 was observed at 100 K (the solvent glass

66
transition temperature), and as the temperature of the sample was increased; the maxima
of this feature in the spectra shifted towards longer wavelengths, and the signal
disappeared completely when the solution reached room temperature (Figure 4-4). The
identity of the substituents at the spiro-locks on the macrocycle (4-1-naphthenone, 4-2-
phenanthrenone) appears to have negligible influence on the energy of triplet state,
presumably since the groups are perpendicular to the macrocyclic ring. The emission
maxima of compound 4-1 (at 708 nm) also reached maximum at 100 K. Unlike in 4-2,
this feature was more sensitive to temperature changes and was completely quenched at
200 K (Figure 4-4). Given that 4-1 has been shown to undergo a light induced radical
rearrangement at room temperature (vide infra), the rapid decrease in the intensity of the
spectral feature is likely a result of activation of the macrocycle by the light source. The
respective triplet state energies for compounds 4-1 and 4-2 are 40 kcal/mol and 41
kcal/mol.
Transient Absorption
Transient absorption spectroscopy was used to probe the lifetime of the triplet
states, and after irradiation at 355 nm, both 4-1 and 4-2 exhibit sharp transients at 520 nm
(Figure 4-5). The disappearance of transient absorption for 4-1 was fit with a two-
component decay (lifetimes 2.2 µs and 360 ns), likely corresponding to the fading of the
porphodimethene triplet state absorbance in addition to the decay of a radical formed
upon laser irradiation of the porphodimethene. For compound 4-2, on the other hand, the
decay of the transient absorption could be fitted to single component pathway with a
triplet lifetime of 2.7 µs, as expected given that 4-2 is not nearly as sensitive to light as 4-
1.

67
4-1
Wavelength / nm
660 680 700 720 740 760 780 800
Rel
. I100 K140 K200 K
4-2
660 680 700 720 740 760 780 800
Rel
. I
100 K140 K180 K220 K298 K
Figure 4-4. Depiction of phosphorescence emission for 4-1 and 4-2. The triplet state
energies are similar, but the phosphorescence of light-sensitive 4-1 disappears at lower temperature than the phosphorescence of 4-2.

68
Wavelength / nm500 600 700 800
∆ A
0.0
0.1
0.2
0.3
0.4
4-24-14
Figure 4-5. Depiction of transient absorption of 4-1 and 4-2. Both molecules feature sharp transients at 520 nm.
Even though the lifetimes of their triplet states are short, both 4-1 and 4-2 exhibit
effective energy transfer to dioxygen molecules resulting in generation of singlet oxygen
with quantum yields of 0.85 and 0.87 (respectively for 4-1 and 4-2).
Conclusions
Insertion of metals in tetrapyrrolic ring induces red-shifts in the UV-Vis spectra of
both the porphodimethenes with the alkyl20 and the aryl substituents.21 The spectra
presented here are in good agreement with this trend, showing that the nature of the
substituents on macrocycle does not influence the spectral properties significantly.
The presence of the central metal atom, on the other hand, changes both the
absorption and the emission spectra of the porphodimethenes. While the free-base
porphodimethenes exhibit weak fluorescence, the first detailed investigation of the
photophysical properties of porphodimethenes revealed that the fluorescence emission in
palladium derivatives 4-1 and 4-2, and these compounds show low temperature

69
phosphorescence instead. Porphodimethenes 4-1 and 4-2 have short triplet state lifetimes
and sharp triplet absorptions and can generate singlet oxygen in high quantum yields.
Experimental
Porphodimethenes were synthesized as described in chapters 2 and 3. All solvents
were used as received from commercial sources, unless otherwise specified. UV-Vis
spectra were recorded with a Varian Cary 50 spectrophotometer. Spectroscopic
experiments were carried out either in tetrahydrofuran or in distilled 2-
methyltetrahydrofuran (2-MeTHF) for variable temperature emission. Spectroscopy
carried out at room temperature was performed using samples that were degassed by a 20
min argon purge. Low-temperature spectroscopic experiments were conducted on
samples that were degassed by three repeated freeze-pump thaw cycles on a high-vacuum
line. Steady-state photoluminescence spectroscopy and singlet oxygen quantum yield
measurements were carried out using a SPEX Fluorolog 2 instrument. For steady-state
spectroscopy measurements, the samples were irradiated at the absorption maxima and
emission response in the UV-Vis region was monitored. Sample concentrations were
sufficiently low such that the absorbance at all wavelengths was less than 0.2. Integrity
of the samples was confirmed by taking excitation spectra. Quantum yield of singlet
oxygen is defined as the ratio of the number of generated singlet oxygen molecules to the
number of light-excited photosensitizer molecules. The standard used for quantum yield
calculations was tetraphenylporphyrin (Φ = 0.68).43 Samples 4-1 and 4-2 were dissolved
in THF, and the concentration was adjusted so that their absorbance at 420 nm matched
that of a standard (0.2). The solutions were then irradiated at 420 nm, the emission
response at 1270 nm (emission of the singlet oxygen) was monitored, and the resulting
spectra integrated to obtain the quantum yields by comparison with the standard. The

70
instrument used for transient absorption spectroscopy has previously been described in
the literature.44 Samples were contained in a cell that holds a total volume of 10 ml, and
the contents were continuously recirculated through the pump-probe region of the cell.
Samples were degassed by argon purging for 30 min. Excitation was provided by the
third harmonic output of a Nd:YAG laser (355 nm, Spectra Physics, GCR-14). Typical
pulse energies were 5 mJ‚ which corresponded to irradiance in the pump-probe region of
20 mJ/cm2. The samples were dissolved in THF with absorbance of 0.6 at 355 nm.
Transient absorption decay lifetimes were determined from the multiwavelength
difference-absorption data by using the SPECFIT/32 factor analysis software.

CHAPTER 5 SYNTHESES OF PORPHYRINS WITH EXOCYCLIC RING SYSTEMS
Introduction
The spectroscopic properties of terapyrolic chromophores can be altered by
distortions from planarity or the presence of aromatic systems fused to the periphery of
the macrocycle. These modifications induce red-shifts in the porphyrin absorption
spectra, which is of particular interest for optical or therapeutic materials.48 The
placement of one meso substituent in the plane of the porphyrin by fusing it at the β
position causes a batochromic shift in the visible absorption by approximately 100 nm.49
The inclusion of additional aryl moiety on another pyrrole β position in a porphyrin
should cause further change in the optical properties, but only a few examples of
porphyrins with two meso, β fused conjugated rings had been reported50-52 (prior to the
first synthesis of porphyrins with exocyclic rings in our group53). Herein, we present a
synthetic pathway for the preparation of unprecedented palladium porphyrins with
exocyclic eight-membered keto systems, as well as the modification of existing
methodology54 resulting in novel palladium porphyrins with six-membered exocycling
rings.
Results and Discussion
In work with the metallated porphodimethenes such as 5-1, solutions of the
compound were found to be susceptible to a light induced oxidative rearrangement
presumably provoked by the their inclination to form the fully aromatic, 2 electron
oxidized porphyrin macrocycles. Through the use of a combination of light and an
71

72
excess of the oxidant DDQ under anhydrous conditions, a preparative method was
developed for the formation of the non-planar porphyrins depicted in Scheme 5-1, likely
via a Norrish Type I pathway53. The reaction produces two structural isomers, cis-5-3
and trans-5-3, and in each, the porphyrin contains two naphthyl groups fused to the
macrocycle meso- and β-positions by cyloheptanone moieties. The compounds are easily
separated by column chromatography and can be isolated in high combined yield as
crystalline solids.53
O
O
R
R
R
R
N N
N N
Ar
Ar
Pd
N
N
N
N
Pd
OO
Ar
ArR
R
R
R
N
N
N
N
Pd
O
O
Ar
ArR
RR
R
hν, DDQ
CH2Cl2
Ar = Mesityl, R = t-Bu
5-1
cis-5-2
trans-5-2
Scheme 5-1. Illustration of the cycloheptanone porphyrin synthesis. The reaction proceeds at room temperature in 90 minutes.
Due to a steric clash between the naphthyl hydrogens and the β hydrogens on the
porphyrin ring in cis 5-2 and trans 5-2, these macrocycles are easily oxidized to sheet-
like porphyrins with exocyclic azulenone moieties (Scheme 5-2).

73
N
N
N
NPd
OO
Ar
ArR
R
R
R
N
N
N
NPd
O
O
Ar
ArR
RR
R
cis-5-2
trans-5-2
N
N
N
NPd
OO
Ar
ArR
R
R
R
N
N
N
NPd
O
O
Ar
ArR
R R
R
cis-5-3
trans-5-3
Ar = Mesityl, R = t-Bu
FeCl3, DDQ
∆, CH2Cl2
FeCl3, DDQ
∆, CH2Cl2
Scheme 5-2. Depiction of oxidation of heptanone porphyrins to azulenone porphyrins.
Steric clash of naphthyl and β hydrogens favors this oxidative coupling.
Cyclooctanone Porphyrins
In view of the tendency of naphthenone porphodimethene 5-1 to form porphyrins,
phenanthrenone porphodimethene 5-4 was subjected to similar reaction conditions, but
the macrocycle was particularly robust. Even in refluxing benzonitrile with one
equivalent of Pd(PhCN)2Cl2, complete conversion of the porphodimethene took seven
days. As with 5-4, two porphyhrin isomers are formed in the reaction, cis-5-5 and trans-
5-5 (Scheme 5-2). The reaction was followed spectroscopically, by monitorig the
disappearance of the porphodimethene absorption at 489 nm, and the emergence of the
porhyrin Soret band at 438 nm, as illustrated in Figure 5-1.
Despite numerous attempts, all efforts to separate the isomers by column
chromatography have been unsuccessful, but the material can be obtained as analytically
pure mixture of isomers cis-5-4 and trans-5-4. The physical measurements have been

74
performed on the mixture, which, based on NMR integrations, contained approximately
40% of cis-5-5 and 60% of trans-5-5.
Wavelength / nm400 500 600 700
Abs
orba
nce
20'6 h6 days
Figure 5-1. Illustration of the reaction progress for synthesis of 5-4. The
porphodimethene absorption at 489 nm disappears as the porphyrin Soret band grows in.
N N
N N
Ar
Ar
O
Pd
O
∆, C6H5CN
Pd(C6H5CN)2Cl2
N
N
ArO
N
N
O
Ar
N
N
ArO
N
N
OAr
Pd
Pd
Ar = Mesityl5-4
cis 5-5
trans 5-5 Scheme 5-3. Diagram of octanone porphyrin formation. The reaction takes 7 days in
refluxing benzonitrile.

75
Apart from the different exocyclic ring size, both cis-5-5 and trans-5-5 contain
biphenyl moieties in place of the naphtyl groups found in cis-5-2 and trans-5-2. As a
consequence, the increased flexibility of exocyclic substituents in cis-5-5 and trans-5-5
compared to cis-5-2 and trans-5-2 may decrease the polarity difference between the
isomer pair, making separation of cis-5-5 and trans-5-5 difficult under different
chromatographic conditions. Due to our inability to resolve the two isomers, all the
photophysical measurements for 5-5 described in Chapter 6, were conducted on the
mixture of cis and trans porphyrins.
In an effort to improve the chromatographic properties of cis- 5-4 and trans- 5-4,
the various metals including platinum, copper and nickel were incorporated into the
macrocyclic ring, and the meso aryl subtituents were replaced with 3,5-di-t-butyl phenyl
groups. Unfortunately, all attempts to separate the isomers were still unsuccessful.
Nevertheless, single crystals of trans-5-5 can be obtained after diffusion of pentane
into a concentrated toluene solution containing the two isomers. This molecule
crystallizes in the point group C2 and adopts anti configuration with respect to the
carbonyl groups of cyclooctanone moieties. As can be seen from the solid-state structure
(Figure 5-2), the presence of an exocyclic octanone moiety allows for the phenyl
substituents to assume a nonplanar conformation with respect to the porphyrin ring. The
angle between phenyl groups adjacent to the porphyrin ring and the ring itself is 70.2(1)º,
while the angle between the two phenyls within biphenyl moiety is 60.6(1)º. The
flexibility of the octanone rings relieves the strain imposed on the porphyrin by the
presence of heptanone rings in cis-5-2 and trans-5-2, and the core of trans-5-5 is nearly
planar with mean plane deviation of 0.125 Å. Palladium adopts a square-planar geometry,

76
with N-Pd-N angles of 89.3(1)º and 90.8(1)º. Selected bond lengths are listed in Table 5-
1.
Figure 5-2. Diagrams (side view on the bottom) of the solid-state structure of trans-5-5
(40% probability; carbon atoms depicted with arbitrary radii). Hydrogen atoms omitted for clarity. Primed and non-primed atoms related by 2-fold symmetry.
Without the steric clash between pyrrolic hydrogens of the porphyrin ring and the
aromatic hydrogens on the substituents, that is highly pronounced in 5-2, porphyrins cis-
and trans-5-5 did not exhibit reactivity towards further oxidation that would result in
forming highly conjugated counterparts of cis-5-3 and trans-5-3.

77
Table 5-1. Selected bond lengths for trans-5-5
trans-5-5·C7H8
Pd1-N1 2.003(2)
Pd1-N1’ 2.003(2)
Pd1-N2 2.013(2)
Pd1-N2’ 2.013(2)
N1-C4 1.368(4)
N1-C1 1.382(4)
N2-C9 1.378(4)
N2-C6 1.380(4)
O1-C11 1.207(5)
C11-C7 1.489(5)

78
Cyclohexannone Porphyrins
NNAr
N N
O
ArPd
Ar
NNAr
O
N N
O
ArNiNN
Ar
O
N N
O
ArNi
5-6 cis-5-7 trans-5-7Ar =
t-But-Bu Figure 5-3. Diagram of porphyrins with exocyclic rings synthesized in Callot’s lab.
With palladium porphyrins containing seven and eight-membered rings in hand, we
were interested in synthesizing analogues macrocycles with six-membered exocyclic
rings in order to explore the photophysical properties of the whole series of the
compounds. Palladium porphyrin with one six-membered ring 5-6 and nickel porphyrins
with two six-membered rings 5-7 were reported in the literature (Figure 5-3)54, 55 and the
published synthetic procedures were modified to obtain cis and trans palladium
cyclohexanone porphyrins (Scheme 5-4). Callot and coworkers isolated α,α and α,β
porphyrins 5-9 from the condensation reaction of α-methoxybenzaldehyde with 3,5-di-t-
butylphenyl aldehyde and pyrrrole. In this work, we synthesized 2-methoxyphenyl
dipyrromethane and condensed it with 3,5-di-t-butylphenyl aldehyde under Lindsey
conditions to obtain α,α and α,β 5-9 in combined yield of 34%. Porphyrin 5-9 was
metallated with palladium, diesters hydrolyzed and transformed to acid chlorides that

79
HNNAr
MeO2C
NH N
CO2Me
Ar
Ar =
t-Bu t-Bu
NNAr
O
N N
O
ArPd
NH HN
CO2Me+
i
i) TFA, DDQ
ii) LiOH, dioxane, H2Oiii) oxallylchloride, SnCl4, benzene
2ArCHO2
NNAr
MeO2C
N N
CO2Me
ArPdNN
Ar
O
N N
O
ArPdii, iii
5-8 5-9
5-10 cis-5-11 trans-5-11
Scheme 5-4. Diagram of cyclohexanone porphyrin formation.

80
were then subjected to intramolecular Friedel-Craft’s acylation to form porphyrins cis-5-
11 and trans-5-11 with exocyclic six-membered rings.
The first step in the synthesis of porphyrins with exocyclic six-membered rings
designed by Callot and coworkers55 involves condensation of 3,5-di-t-butylphenyl
aldehyde with pyrrole and 2-methoxy-bezaldehyde, resulting in a statistical mixture of a
number of porphyrins. This approach, while useful for obtaining starting material for
different mono and bi exocyclic porphyrins, produces individual porphyrins in low to
moderate yields ( 2 – 10%). By using 2+2 condensation of 3,5-di-t-butylphenyl aldehyde
and 2-methoxy-phenyl dipyrromethene, we increased the yield of 5-9 to 34% (combined,
for α,α and α,β) and minimized the formation of porphyrin side products. Metallation of
5-9 with palladium in the second step and carrying on the reaction sequence with
palladium instead of nickel as a central atom eliminated the demetallation and
trasmetallation steps required for synthesis of 5-6.55
In addition to obtaining palladium cyclohexanoneporphyrins, this methodology
allowed for isolation and structural characterization of a new palladium porphyrin
bearing two methyl ester groups, as illustrated by the solid-state structure diagram in
Figure 5-4. The esters are on benzylic carbons α to the two trans meso positions of the
porphyrin ring, and they are oriented away from each other. Phenyl rings bearing
functional groups are rotated 85.3(2) and 86.7(2) degrees with respect to the mean plane
of the porphyrin ring defined by 20 carbon atoms and four nitrogens of the tetrapyrrolic
core. The average deviation of the 24 atoms from the mean plane is 0.033 Å, and
palladium is situated in a square planar arrangement 0.014(3)Å below the mean plane.

81
The angles between the 3,5-di-t-butylphenyl substituents and the porphyrin core are
73.3(2)° and 72.2(2)°.
Figure 5-4. Diagram of the solid-state structure of 5-10 (40% probability; carbon atoms are depicted with arbitrary radii). Hydrogen atoms are omitted for clarity.
Conclusions
A synthetic pathway for unprecedented palladium porphyrins contaning eight-
membered exocyclic keto-rings was devised. Even though the isomers could not be
resolved chromatographically, a single crystal of trans-5-5 was obtained and the
compound was structurally characterized. Biphenyl moeties in this structure are rotated
away from the porphyrin plane, and the flexibility of the eight-memberd rings allows the
aryl and β pyrrolic hydrogens to point away from each other. In agreement with the lack
of steric hindrance observed for compounds 5-4 compared to the cycloheptanone
porphyrins 5-2, in which naphthyl and β hydrogens are in intimate proximity, compounds
5-4 showed no reactivity in further oxidative coupling to form analogues of 5-3.

82
A literature procedure for synthesis of porphyrins with exocyclic six-memberd
rings54, 55 was modified to give cis-5-11 and trans-5-11 that have not been previously
reported. The porphyrins were synthesized in fewer steps and with higher overall yields
than the literature analogues 5-6 and 5-7. Cyclohexanone porphyrins 5-11 show
significant red-shifts in their UV-Vis absorption spectra in comaparison to typical
tetraarylporpyrins, due to the extended π-conjugation outside of the porphyrin ring.
Palladium porphyrins with six and eight-memebered exocyclic rings were used for
photophysical studies described in Chapter 6.
Experimental
General Procedures
NMR spectra were recorded on Varian Mercury or VXR 300 MHz
spectrometers. UV-Vis spectra were recorded with a Varian Cary 50 spectrophotometer.
High resolution mass spec analyses were performed by University of Florida Mass Spec
services using FAB or ESI as ionization method. All solvents were used as purchased,
unless otherwise specified.
Chromatography
Absorption column chromatography was performed using neutral alumina
(Aldrich, Brockman I ~ 158 mesh, 58 Ǻ) or chromatographic silica gel (Fisher, 200 – 425
mesh). HPL chromatography was performed on Waters Breeze HPLC system equipped
with the dual wavelength detector, using Alltech Econosphere 10µm silica column (10
mm x 250 mm).
Synthesis of cis-5-5 and trans-5-5
A sample of 0.050 g (0.05 mmol) of 5-4 was dissolved in 30 ml of benzonitrile and
0.021 g (0.05 mmol) of Pd(PhCN)2Cl2 was added. The solution was heated to reflux with

83
stirring, and the progress of the reaction was monitored by UV-Vis spectroscopy. After 7
days, the reaction was complete and the solvent was removed under vacuum. The solid
residue was redissolved in a minimal amount of methylene chloride and purified by
column chromatography with a 1:1 mixture of methylene chloride/toluene as the eluant.
The second, purple fraction was collected, and the solvent evaporated to dryness to afford
0.024 g (48%) of product as a mixture of cis-5-5 and trans-5-5 isomers. HRMS (FAB)
calcd. for MH+ (C64H45O2N4Pd): 1006.2498. Found 1006.2518. Anal.Calcd. for
C64H44N4O2Pd: C, 76.30; H, 4.40; N, 5.56. Found: C, 76.36; H, 4.39; N, 5.63. 1H NMR
indicated the presence of both the cis and the trans isomer in a ratio of 2:3 respectively,
and to date, all efforts to separate the two isomers using either regular silica column or
HPL chromatography have been unsuccessful. X-ray quality crystals of trans-5-5 were
grown from pentane/toluene diffusion.
Synthesis of 5-8
Dipyromethene 5-8 was synthesized using standard literature procedure.34 A
sample of 5.490 g (33.5 mmol) of 2-formyl methylbezoate was dissolved in 100.0 ml of
freshly distilled pyrole and 1.26 ml of BF3·Et2O was added. The mixture was stirred for
30 minutes in the dark and diluted with 170 ml of methylene chloride. It was then
washed with 170 ml of 0.1 M NaOH and water, dried over Na2SO4 and methylene
chloride was removed on the rotary evaporator. Excess pyrolle was distilled off on the
vacuum line; solid residue was redissolved in minimal amount of methylene chloride and
passed through alumina column. The first yellowish fraction was collected, solvent
evaporated and solid recrystalized from hot hexanes to yield 2.556 g (25 %) of white
solid. 1H NMR (300 MHz, CDCl3) d= 8.49 bs 2H, 7.77 (dd, 1H, J1 =8.1 Hz, J2 =1.5 Hz),

84
7.45-7.24 (m, 3H), 6.70 (dd, 2H, J1 = 3.9 Hz, J2 = 2.4 Hz), 6.31 (s, 1H), 6.14 (dd 2H, J1 =
5.7 Hz, J2 = 3.0 Hz), 5.92-5.90 (m, 2H), 3.81 (s, 3H).
Synthesis of 5-9
A portion of 1.000 g (3.56 mmol) of 5-8 and 0.779 g (3.56 mmol) of 3,5-di-t-butyl
benzaldehyde were dissolved in 360 ml of methylene chloride and 0.48 ml of TFA was
added dropwise. The mixture was stirred at room temperature for 30 minutes and 0.811 g
(3.56 mmol) of DDQ was added and stirred for another hour. The excess DDQ was
filtered off, two drops of triethylamine added to the filtrate, and the solution volume was
reduced to 10 %. The mixture was purified over neutral alumina, and the porphyrin
products were separated on a silica column using toluene as eluent. The third, purple
band was collected, and the solvent was evaporated to yield 0.310 g (18 %) of purple α, β
isomer. The fifth, purple band from this column was collected, and the solvent was
evaporated to give 0.276 g (16 %) of the α, α isomer of 5-9. α, α :1H NMR (300 MHz,
CDCl3) d= 8.72 (d, 4H, J = 4.8 Hz), 8.62 ( J = 4.5 Hz), 8.40 (d, 1 H, J = 2.1), 8.38 (d, 1H,
J = 1.8 Hz ), 8.18-8.16 (m, 4H), 7.97 (dd, J1 = J2 = 1.5 Hz), 7.87 – 7.77 (m, 6H). 2.87 (s,
6H), 1.54 (s, 18H),1.49 (s, 18 H) - 2.55 (s, 2H).
α, β: 1H NMR (300 MHz, CDCl3) δ = 8.81 (d, 4H), 8.59 (d, 4H, J= 4.8 Hz), 8.39
(d, 1H, J= 1.8 Hz), 8.36 (d, 1H, J = 1.5 Hz), 8.12 (d, 1H, J = 1.2 Hz), 8.10 (d,1H, J = 2.1
Hz), 8.04 (d, 4H, J= 1.8 Hz), 7.88 – 7.76 (m, 6H), 2.70 (s, 6H), 1.51 (s, 36H), - 2.52 (s,
2H).
Synthesis of 5-10
A sample of 0.310 g (0.325 mmol) of 5-9 (α, β isomer) was dissolved in 100.0 ml
of toluene and 0.155 g (0.692 mmol) of Pd(OAc)2 was added. The solution was kept at

85
reflux overnight. The reaction mixture was purified through a short silica gel column and
the first bright orange fraction containing both was collected, and the solvent was
evaporated to yield 0.284 g (83 %)of 5-10. The product was obtained as a mixture of
α,α and α,β isomers due to interconversion of 2-methoxy phenyl substituents in refluxing
toluene. Analytical sample of α,β isomer was purified on a silica column with toluene.
1H NMR (300 MHz, CDCl3) δ= 8.81 (d, 4H, J = 4.8 Hz), 8.59 (d, 4H, J = 4.8 Hz) 8.39 (d,
1H, J = 2.1 Hz), 8.36 (d, 1.2 Hz), 8.12 (d, 1H, J = 1.2 Hz), 8.10 (d, 1H), 8.04 (d, 4H, J =
2.1 Hz), 7.87 – 7.76 (m, 6H), 2.80 (s, 6H), 1.51 (s, 36H). Slow diffusion of pentane into
the chloroform saturated solution of α,β isomer of 5-10 afforded single crystals suitable
for X-ray diffraction studies.
Synthesis of cis-5-11 and trans-5-11
A portion of 0.204 g (0.193 mmol) of 5-10 was dissolved in 210 ml of dioxane and
a solution of LiOH (6.000 g) in 25 ml of water was added. The solution was kept at
reflux for 48 hours and it was the filtered. The precipitate was washed with 5% acetic
acid in methylene chloride. The filtrate was washed with water, organic layer separated
and dried over sodium sulfate. The solvent was removed under vacuum to yield 0.170 g
of orange solid (prphyrin diacid), and the compound was used without further
purification for the subsequent reaction.
A sample of 0.170 (0.165 mmol) g of the diacid was dissolved in 210 ml of
benzene and 6.3 ml of oxallyl chloride was added. The reaction mixture was stirred at
room teperature for 4 hours. Distillation of 20 ml of solvent (to remove the excess oxalyl
chloride) was followed by the addition of 4.2 ml of SnCl4. The reaction mixture was
stirred at room temperature for 75 minutes, diluted with 200 ml of methylene chloride,

86
neutralized with aqueous sodium hydroxide, washed with water and dried over sodium
sulfate. The solvent was evaporated, and the solid was redissolved in methylene chloride
and percipitated by addition of methanol. Yield:0.088 g (55 % overall for the two steps)
of product as a mixture of cis and trans isomers. A sample of the solid was redissolved
in CHCl3 and the two isomers were separated on semi-preparative HPLC column using
toluene as an eluant.
The trans isomer eluted first with retention time of 23 minutes. 1H NMR (300
MHz, CDCl3) δ= 9.07 (d, 2H, J = 5.1 Hz ), 9.02 (s, 2H), 8.54 (d, 2H, J = 5.1 Hz ), 8.47
(d, 2H, J = 5.1 Hz ), 7.82-7.76 (m, 8H), 7.51 (dd, 2H, J1 = J2 = 3.9 Hz ), 1.52 (s, 36 H).
UV-Vis [methylene chloride, λmax (log ε)] 765 nm (3.69), 681 nm (3.83), 466 nm (4.55).
HRMS (FAB) calcd. for M+ (C62H56O2N4Pd) 994.3437. Found: 994.3444.
The cis isomer eluted second, after 40 minutes. 1H NMR (300 MHz, CDCl3) δ=
9.06 (d, 2H, J = 5.1 Hz), 9.00 (s, 2H), 8.54 (d, 2H, J = 5.1 Hz), 8.47 (d, 2H, J = 7.5 Hz),
8.22 (d, 2H, J = 8.1 Hz), 7.83 – 7.72 (m, 6H), 7.28 (s, 4H, under the solvent peak), 1.52
(s, 18H), 1.50 (s, 18H). UV-Vis [methylene chloride, λmax (log ε)] 718 nm (4.18), 547
nm (4.05), 484 nm (4.83), 411 nm (4.32).
X-ray Crystallography
Unit cell dimensions were obtained and intensity data collected by Prof. Michael
Scott on a Siemens CCD SMART diffractometer at low temperature, with
monochromatic Mo-Kα X-rays (λ = 0.71073 Å). The data collections nominally covered
over a hemisphere of reciprocal space, by a combination of three sets of exposures; each
set had a different φ angle for the crystal and each exposure covered 0.3° in ω. The

87
crystal to detector distance was 5.0 cm. The data sets were corrected empirically for
absorption using SADABS.37
Table 5-2. Crystallographic data for trans-5-5 and 5-10 trans-5-5·2C7H8 5-10
Formula C78H60N4 O2Pd C66H54N4O4Pd
Formula weight 1191.70 967.13
Crystal system Monoclinic Triclinic
Space group C2 P1
Z 2 2
Temp, K 173(2) 193(2)
Dcalc/ gcm-3 1.329 1.313
a Å 25.2799(12) 11.1639(5)
b Å 9.5071(4) 11.9487(6)
c Å 12.7357(6) 12.5807(6)
a, deg - 116.497(1)
β, deg 103.398(1) 106.317(1)
γ, deg - 101.036(1)
V Å3 2977.6(2) 1339.8(1)
µ, mm-1 0.37 0.399
Uniq. data coll./obs. 5537/9702 7039/7596
R1[I > 2σ(I)data]a 0.0434 0.0370
wR2[I > 2σ(I)data]b 0.1179 0.0929 a R1 = Σ||Fo| - |Fc||/ Σ| Fo| bwR2 = { Σ[w (Fo
2 – Fc2)2/ Σ[w ( Fo
2)2}
The structure was solved using the Bruker SHELXTL software package for the
PC, by direct method option of SHELXS. The space group was determined from an
examination of the systematic absences in the data, and the successful solution and
refinement of the structure confirmed these assignments. All hydrogen atoms were

88
assigned idealized locations and were given a thermal parameter equivalent to 1.2 or 1.5
times the thermal parameter of the carbon atom to which it were attached. For the methyl
groups, where the location of the hydrogen atoms was uncertain, the AFIX 137 card was
used to allow the hydrogen atoms to rotate to the maximum area of residual density,
while fixing their geometry. Relevant crystallographic data are listed in table 5-2.

CHAPTER 6 PHOTOPHYSICAL PROPERTIES OF PORPHYRINS WITH EXOCYCLIC RING
SYSTEMS
Introduction
Artificial porphyrins have potential use in diverse applications ranging from
chemotherapy to material science, since they often exhibit unusual photochemical,56
magnetic,57 and electronic33 properties. For example, enhanced intersystem crossing in
free base and metalloporphyrins can be exploited for the photo-induced generation of
singlet oxygen within cancer cells resulting in cell death.58 Moreover, porphyrin species
with highly absorbing transients represent good candidates for optical limiters.59 Finally,
phosphorescent porphyrins are used to improve the efficiency of light-emitting devices.59
The ability of porphyrins to trap and transfer energy when organized in the arrays is
utilized for energy-transporting antennae in e.g. dye-sensitized TiO2 solar cells.60
Regardless of what the new artificial porphyrins could be used for, understanding their
physical properties is essential for the potential application.
Herein, we report detailed photophysical studies of palladium porphyrins with
unprecedented cyclooctanone exocyclic rings and related porphyrins with exocyclic
cycloheptanone and cyclohexanone systems.
Both cis and trans 6-1 are bright green due to a significant red shift of the
porphyrin Soret and Q-bands (Figure 6-2). Palladium porphyrins typically exhibit Soret
absorptions between 380-412 nm,24 but cis-6-1 has an absorption maximum at 470 nm
(log ε = 5.4) while the analogous absorption for trans-6-1 occurs at 486 nm (log ε = 5.4).
89

90
Along with shifting to lower energy (652 nm for cis-6-1 and 674 nm for trans-6-1), the
Q-bands for these green porphyrins are unusually broad and intense (log ε = 4.6).
N
N
N
N
Pd
OO
Ar
ArR
R
R
R
N
N
N
N
Pd
O
O
Ar
ArR
R R
R
Ar = Mesityl, R = t-Bucis 6-1 trans 6-1
N
N
ArO
N
N
O
Ar
N
N
ArO
N
N
OAr
PdPd
cis 6-2 trans 6-2
N
N
N
N
Pd
OO
Ar
Ar
N
N
N
N
Pd
O
O
Ar
Ar
Ar = 3,5-di-t-butylphenylcis 6-3 trans 6-3
Ar = Mesityl
N
N
N
N
Pd
OO
Ar
ArR
R
R
R
N
N
N
N
Pd
O
O
Ar
ArR
RR
R
cis 6-4 trans 6-4 Ar = Mesityl, R = t-Bu Figure 6-1. Diagram of the porphyrins with exocyclic rings used for the photophysical
measurements reported herein.

91
Wavelength / nm400 500 600 700 800
Eps
ilon
/ M-1
cm-1
0.0
5.0e+4
1.0e+5
1.5e+5
2.0e+5
2.5e+5
cistrans
Figure 6-2. The cycloheptanone porphyrins exhibit red-shifts in the absorption spectra.
In the solid-state, trans-6-1 adopts an anti configuration with respect to the
carbonyl groups of the cycloheptanone moieties. Due to a steric clash between
hydrogens on the napthyl group and the β-pyrrolic positions, the macrocycle assumes a
classic, ruffled B1u deformation with the meso-carbon atoms displaced alternately above
and below the plane formed by the four pyrrole nitrogens. Both the non-planar
distortion61 and the delocalization of electron density through the fused
naphthocycloheptanone ring systems likely contribute to the bathochromic shifts of the
Soret bands.
The rigidity of the seven-membered keto ring enables the electronic delocalization
by forcing a 30.1° angle between the mean plane of the porphyrin (defined by 20 carbon
atoms and 4 nitrogens of the core) and the naphthyl substituents. Moreover, the naphthyl
groups are only displaced by 15.3° from the pyrrole rings to which they are connected via
the carbonyl carbon. In view of the long wavelength absorption of these porphyrins, the
emission would be expected to occur in the low-energy region of the spectrum, and
indeed, both cis-6-1 and trans-6-1 exhibited low temperature phosphorescence emissions

92
in the IR region that disappear rapidly with increasing temperature (Figure 6-2). Triplet
state energies for cis-6-1 and trans-6-1 are 30 kcal/mol and 28 kcal/mol, respectively.
Wavelength / nm900 1000 1100 1200
Rel
. I
cistrans
Figure 6-3. Depiction of the phosphorescence emission of cis-6-1 and trans-6-1. Both
isomers emit in the IR region.
The local symmetry of the two isomers in solution is entirely consistent with their
solid-state structures. As expected for C2 symmetry, trans-6-1 exhibits only two peaks
for the mesityl o-hydrogens and 3 peaks for the mesityl methyls in the 1H NMR
spectrum, while the four one-proton singlets corresponding to mesityl o-hydrogens and
six peaks for the mesityl methyls are evident in the spectrum of the Cs symmetric cis-
isomer. The cores of both cycloheptanone porphyrins are distorted from the mean plane
defined by the 20 carbon and 4 nitrogen atoms in the macrocycle, and the distortion is
somewhat more pronounced in trans-6-1, as compared to cis-6-1 (mean plane deviation
of 0.27 Å vs. 0.24 Å), possibly explaining the red shift of the lowest energy band in the
former.62
The low-energy absorption in 6-1 is accompanied by the low- energy emission, and
the phosphorescence feature of trans-6-1 at 1017 nm is, to the best of our knowledge, the

93
longest wavelength emission measured for the monomeric palladium porphyrin species.
The triplet states in cis-6-1 and trans-6-1 have the absorption maxima at 520 nm and the
lifetimes of 10 µs and 4.3 µs, respectively (Figure 6-4). The absorption peaks in TA
spectra of these porphyrins are very broad and extend into IR region. The shape of the
TA spectra, together with the low energies and short lifetimes of the triplet states
suggests a high degree of electronic delocalization in cis-6-1 and trans-6-1.63
In the triplet excited state the cycloheptanone porhyrins will induce the excitation
of dioxygen molecules into the singlet state via energy transfer, and both compounds cis-
6-1 and trans-6-1 generate singlet oxygen with quantum yields of 1. Quantitative singlet
oxygen production makes these molecules particularly interesting oxygen sensitizing.
The strong absorption of 6-1 in far red region of the visible spectrum along with the
ability to tune chemical properties of the macrocycle through changes in the meso-aryl
groups makes these molecules particularly interesting for applications in photodynamic
therapy. The cycloheptanone porphyrins also exhibit significant triplet-state absorption
in the low-energy region, where their ground states do not absorb, a key feature for
materials used as optical limiters.64
Cyclooctanone Porphyrins.
Much like solutions of cis-6-1 and trans-6-1, the Soret bands in the UV- Vis
spectrum of mixture of cis- and trans-6-2 are red-shifted with the respect to simple
palladium porphyrins and are coincident at 438 nm (Figure 6-5). The spectrum also
features two bands in the low energy region at 544 nm and 584 nm, but both the Q-band
and the Soret bands are blue-shifted in comparison to 6-1, presumably due to a smaller
degree of delocalization of the π-system over the two exocyclic octanone rings.

94
Wavelength / nm
500 550 600 650 700 750 800
∆A
-0.3
-0.2
-0.1
0.0
0.1
0.2 cistrans
Figure 6-4. Illustration of transient absorption of cis-6-1 and trans-6-1. Broad transients
indicate high delocalization of electron density.
Table 6-1. Summary of photophysical data
Compound Abs [nm], ε [M-1cm-1]
Phosphorescence [nm] (kcal/mol) Φ 1O2 τ [ µs ]
cis-6-1 486, 250000 949 (30) a 1.0 10.2
trans-6-1 470, 250000 1017 (28) a 1.0 4.3
cis-6-2, trans-6-2 438 820 (35) b 1.0 12 c, 43 d
cis-6-3 484, 68000 1015 (28) a 2.3
trans-6-3 466, 35000 1010 (28) b 0.9
cis-6-4 553, 79000 - 0 -
trans-6-4 567, 79000 - 0 - a Low temperature. b Room temperature. c τ1. d τ2.

95
The conjugation outside the porphyrin ring in 6-2 extends only to the carbonyl groups,
which are displaced from the plane of the core by 30.3º, and any further electronic
communication is lost due to the rotation of the phenyl groups.
Wavelength /nm350 400 450 500 550 600
Abs
orba
nce
Figure 6-5. Diagram of the absorption spectrum of the mixture of cis-6-2 and trans-6-2
highlights the coincidence of their Soret bands at 438 nm.
The rotation of the phenyl substituents out of the plane of the porphyrin, causes a
substantial difference between the electronic absorption spectra of 6-2 and 6-1, and can
account for different shapes of their transient absorptions.
In this study only cis-6-2 and trans-6-2 exhibited room temperature
phosphorescence, and the emission maximum occurred at 820 nm (Fig. 6-6). Saturation
of the solution with air quenched this emission due to energy transfer to dioxygen
molecules.
In comparison to palladium tetraphenylporphyrin (PdTPP), the triplet state energy
of 6-2 is somewhat lower (40 kcal/mol vs. 35 kcal/mol), perhaps due to a slightly
extended π-conjugation to exocyclic carbonyl groups in the cyclooctanone porphyrins.

96
Wavelength / nm600 650 700 750 800 850
Rel
. I Argon degassedOxygen saturated
Figure 6-6. Diagram of the phosphorescence emission of the mxture of cis-6-2 and trans-
6-2. The emission is quenched by saturation with air.
The mixture of cis-6-2 and trans-6-2 exhibits a strong transient around 480 nm
(Figure 6-7). When the TA spectrum of the isomer mixture was deconvoluted in
SPECFIT, two spectra of nearly identical shape were identified, but the signals had
different ∆A intensities. The isomer with more intense absorption exhibited λmax at 575
nm with the lifetime of 43 µs, while the λmax for the isomer with less intense absorption
occurred at 585 nm with the lifetime of 12 µs. If the extinction coefficients of cis-6-2
and trans-6-2 are identical, the transient at 475 nm would correspond to the trans isomer,
while the one at 485 nm can be assigned to cis, based on the isomer ratio calculated from
the NMR spectrum of the isomer mixture. The position of the transient absorption in cis-
6-2 and trans-6-2 are comparable to PdTPP (λmax = 475 nm)63, but the lifetimes of the
triplet states are significantly shorter than the 250 µs lifetime of PdTPP.46, 63, 65 The
broadening of the transient absorption of 6-2 compared to the PdTPP spectrum is
consistent with a higher degree of delocalization due to the presence of carbonyl groups,
and the triplet-state absorptions of cis-6-1 and trans-6-1 are much broader than the

97
signals in TA spectra of cis-6-2 and trans-6-2, providing further evidence for higher
electron delocalization in the former isomer pair compared to the later.
Wavelength / nm500 600 700 800
∆A
-0.1
0.0
0.1
0.2
Figure 6-7. Depiction of transient absorption of cyclooctanone porphyrins. The spectrum
can be deconvoluted into two spectra with transients at 475 nm and 485 nm.
Cyclohexanone Porphyrins
The UV-Vis spectra of cis-6-3 and trans-6-3 (Figure 6-8) exhibit red-shifts in both
the Soret and the Q-bands with respect to the regular meso aryl substituted porphyrins.
The Soret absorptions at 484 nm and 466 nm respectively for cis and trans , as well as the
Q-absorptions at 681 nm, 765 nm (cis) and 718 nm (trans) are indicative of significant
electronic delocalization outside the porphyrin ring. The steric clash between the aryl
and β hydrogens in 6-3 is smaller than in 6-1, due to the smaller exocyclic ring-size in the
former. This reduced steric hindrance can allow the phenyl substituents on the six-
membered keto-rings to get closer to the porphyrin plane, thus enabling a higher degree
of electronic delocalization.

98
Porphyrin trans-6-3 exhibits room temperature phosphorescence emission at 1010
nm (Figure 6-9), while the cis isomer has a very weak room temperature phosphoresce at
1015 nm.
Wavelength / nm400 500 600 700 800
Abs
orba
nce
transcis
Figure 6-8. Diagram of the UV-Vis spectra of trans-6-3 and cis-6-3. Both the Soret and
the Q absorptions are red-shifted in comparison to simple meso-substituted porphyrins.
The triplet state absorption reaches maximum at 520 nm for trans-6-3 and at 760
nm for cis-6-3. The triplet-state lifetimes are 0.9 µs and 2.4 µs for the cis and the trans
isomer, respectively. The short triplet-state lifetimes and the broad features in the
absorption spectra of 6-3 give further support for extended electronic delocalization in
porphyrins with exocyclic six-membered rings.
Azulenone Porphyrins
Oxidative dehydrogenation of compounds cis-6-1 and trans-6-1 results in highly
conjugated porphyrins cis-6-4 and trans-6-4, that absorb light throughout the whole
visible (and a part of the IR) spectrum (Figure 6-11). The Soret bands for cis-6-4 and
trans-6-4 occur at 553 nm and 567 nm, respectively, while the lowest energy absorptions

99
Wavelength / nm
900 1000 1100 1200 1300 1400 1500
Rel
. I trans
Figure 6-9. Illustration of the room temperature phosphorescence emission of trans-6-3.
The low-energy emission is due to electronic delocalization outside the porphyrin ring.
Wavelength / nm
500 550 600 650 700 750 800
∆A
-0.2
-0.1
0.0
0.1
0.2
0.3
cistrans
Figure 6-10. Transient absorption of cis-6-3 and trans-6-3. Triplet states absorb
throughout the visible region.

100
are at 850 nm for the cis isomer and at 1145 nm for the trans. The Q-band of trans-6-4 at
1145 nm is, to the best of our knowledge, the furthest red-shifted absorption observed for
any monomeric porphyrin reported in the literature. The extended conjugation and
higher degree of electron delocalization in these fully planar macrocycles induce an
extreme shift to lower energy in the absorption spectra of cis-6-4 and trans-6-4 compared
to cis-6-1 and trans-6-1.
Wavelength /nm400 600 800 1000 1200
Eps
ilon
/ M-1
cm-1
0.0
2.0e+4
4.0e+4
6.0e+4
8.0e+4
cis trans
Figure 6-11. Diagram of the electronic absorption of cis-6-4 and trans-6-4. The
absorption of the trans isomer extends to 1145 nm.
Porphyrins cis-6-4 and trans-6-4 exhibit no detectable low-temperature emission,
or transient absorption. The lack of phosphorescence in these molecules is in agreement
with the energy gap law,66-68 considering that both compounds absorb in the low energy
region of the spectrum (850 nm and 1145 nm for cis-6-4 and trans-6-4, respectively).
Azulenone porphyrins did not create singlet oxygen since the energies of the triplet states
in these molecules are lower than the energy needed for the excitation of oxygen into the
singlet state.

101
Conclusions
Both the size of and the nature of the substituents on exocyclic rings influence the
photophysical properties of porphyrins. The greater degree of electronic delocalization in
cyclohexanone and cycloheptanone porphyrins causes a decrease in the triplet state
energy, and the compounds only phosphoresce in the IR region, while the less
delocalized cyclooctanone porphyrins 6-2 exhibit phosphorescence at room temperature
in the visible region. Increasing the amount of aromaticty also induces shorter triplet
lifetimes and broader TA spectra for 6-1 and 6-3 in comparison to 6-2. Literature data on
photophysical properties of simple PdTPP46, 63, 65 are in agreement with the trends found
in this work. PdTPP has virtually no electronic delocaliztion outside of the porphyrin
ring, exhibits higher energy absorption and phosphorescence emission, displays a sharper
transient absorption spectrum,63 and has a longer triplet state lifetime than the porphyrins
reported herein.46, 69
Considering the difference between the ground state absorption and TA spectra for
both 6-1 and 6-3, these porphyrins could be tested as optical limiters. For applications
such as oxygen sensing, room temperature phosphorescence observed in 6-2 is
particularly usefull.66, 67 Porphyrins cis-6-1, trans-6-1, cis-6-2 and trans-6-2 generate
singlet oxygen with quantum yield of 1, which is higher than any palladium porphyrin
studied thus far. Quantitative singlet oxygen generation is promising for potential
application of these molecules as photosensitizers. The quantum yield of 1 for singlet
oxygen generation in 6-1 and 6-2 indicates that the triplet states are fully populated, due
to enhanced intersystem crossing induced by the presence of palladium within the
macrocycles.

102
Experimental
All solvents were used as received from commercial sources, unless otherwise
specified. UV-Vis spectra were recorded with a Varian Cary 50 spectrophotometer (for
compounds 6-1 through 6-3) or Varian Cary 500 spectrophotometer (for compounds 6-4).
Spectroscopic experiments were carried out either in tetrahydrofuran or in distilled 2-
methyltetrahydrofuran (2-MeTHF) for variable temperature emission. Spectroscopy
carried out at room temperature was performed using samples that were degassed by a 20
min argon purge. Low-temperature spectroscopic experiments were conducted on
samples that were degassed by three repeated freeze-pump thaw cycles on a high-vacuum
line. Steady-state photoluminescence spectroscopy and singlet oxygen quantum yield
measurements were carried using a SPEX Fluorolog 2 instrument. For steady-state
spectroscopy measurements, the samples were irradiated at the absorption maxima and
emission response in the UV-Vis and IR regions was monitored. Sample concentrations
were sufficiently low such that the absorbance at all wavelengths was less than 0.2.
Integrity of the samples was confirmed by taking excitation spectra. Quantum yield of
singlet oxygen is defined as the ratio of the number of generated singlet oxygen
molecules to the number of light-excited photosensitizer molecules. The standard used
for quantum yield calculations was tetraphenylporphyrin (Φ = 0.68).46 Samples 6-1 – 6-3
were dissolved in THF, so that their absorbance at 420 nm matched that of a standard
(0.2). The solutions were then irradiated at 420 nm, the emission response at 1270 nm
(emission of the singlet oxygen) was monitored, and the resulting spectra integrated to
obtain the quantum yields by comparison with the standard. The instrument used for
transient absorption spectroscopy has previously been described in the literature.47
Samples were contained in a cell that holds a total volume of 10 ml, and the contents

103
were continuously recirculated through the pump-probe region of the cell. Samples were
degassed by argon purging for 30 min. Excitation was provided by the third harmonic
output of a Nd:YAG laser (355 nm, Spectra Physics,GCR-14). Typical pulse energies
were 5 mJ‚ which corresponded to irradiance in the pump-probe region of 20 mJ/cm2.
The samples were dissolved in THF with absorbance of 0.6 at 355 nm. Transient
absorption decay lifetimes were determined from the multiwavelength difference-
absorption data by using the SPECFIT/32 factor analysis software.

CHAPTER 7 SUMMARY
This work demonstrates the utility of aromatic vicinal diketones containing both
five- and six-membered rings for the preparation of spiro-tricyclic porphodimethenes.
Depending on the diketone used, porphodimethenes capable of or resistant to ring
opening at the spiro-lock can be prepared. All porphodimethenes have been successfully
metallated, and the reactivity of compounds susceptible to ring opening was studied,
revealing a synthetic pathway for unprecedented bis-cyclooctanone porphyrins. The
solid state-structures of different metalloporphodimethenes illustrate the influence of the
metal and the meso substituents on the geometry of the macrocycle.
The first extensive studies of photophysical properties of porphodimethenes were
conducted, and low temperature phosphorescence emission in the UV-Vis region was
detected. The rate of disappearance of the phosphorescence with increasing temperature
in two different porphodimethenes is in agreement with different light reactivity of these
compounds. The triplet-state lifetimes and the singlet oxygen quantum yields were
determined for two palladium porphodimethenes.
In order to investigate the differences in photophysical properties of porphyrins
with various exocyclic ring systems, palladium porphyrins bearing 6 and 8-membered
exocyclic rings were prepared, and their spectroscopic features were compared to the
porphyrins bearing 7-membered rings whose synthesis had been previously reported.
We have demonstrated that the size of exocyclic keto-rings fused to the porphyrin
periphery has a profound impact on the overall geometrical features of the macrocycle.
104

105
Smaller exocyclic rings allow for a higher degree of coplanarity between the aryl
moieties and the porphyrin core, inducing a higher degree of electronic delocalization
outside of the core. In the series of porphyrins with cyclooctanone, cycloheptanone and
cyclohexanone keto-rings the increase in the electronic delocalization is accompanied by
decrease in the absorption and emission energies, decrease in the triplet-state lifetimes,
and broadening of the transient absorption spectra.
Oxidative biaryl coupling of cycloheptanone porphyrins flattens out the
macrocycle, which further increases the delocalization, and the resulting azulenone
porphyrins absorb light throughout the whole visible and a part of the IR region of the
spectrum. Due to the low-energy absorption, no emission features were observed for
these macrocycles.
The ease of syntheses and the photophysical properties of cycloheptanone and
cyclooctanone porphyrins suggest that these molecules could be further modified to find
application in areas of oxygen sensing, optical limiting and photosensitizing.

LIST OF REFERENCES
1. Gouterman, M. ed, Porphyrins.; Ameican Chemical Society: Washington DC, 1986;.
2. Buchler, J. W.; Puppe, L., Annalen Der Chemie-Justus Liebig 1974 (7), 1046-1062.
3. Buchler, J. W.; Dreher, C.; Lay, K. L.; Lee, Y. J.; Scheidt, W. R., Inorganic Chemistry 1983, 22, 888-891.
4. Botulinski, A.; Buchler, J. W.; Tonn, B.; Wicholas, M., Inorganic Chemistry 1985, 24 (20), 3240-3243.
5. Botulinski, A.; Buchler, J. W.; Wicholas, M., Inorganic Chemistry 1987, 26, 1540-1543.
6. Botulinski, A.; Buchler, J. W.; Lee, Y. J.; Scheidt, W. R.; Wicholas, M., Inorganic Chemistry 1988, 27 (5), 927-931.
7. Buchler, J. W.; Lay, K. L.; Smith, P. D.; Scheidt, W. R.; Rupprecht, G. A.; Kenny, J. E., J. Organometal. Chem. 1976, 110, 109-120.
8. Fontecave, M.; Battioni, J. P.; Mansuy, D., Journal of the American Chemical Society. 1984, 106 (18), 5217-5222.
9. Benech, J. M.; Bonomo, L.; Solari, E.; Scopelliti, R.; Floriani, C., Angewandte Chemie-International Edition 1999, 38 (13-14), 1957-1959.
10. Harmjanz, M.; Scott, M. J., Chemical Communications 2000, 5, 397-398.
11. Kral, V.; Sessler, J. L.; Zimmerman, R. S.; Seidel, D.; Lynch, V.; Andrioletti, B., Angewandte Chemie-International Edition 2000, 39 (6), 1055-1057.
12. Senge, M. O.; Kalisch, W. W.; Bischoff, I., Chemistry-a European Journal 2000, 6 (15), 2721-2738.
13. Senge, M. O.; Runge, S.; Speck, M.; Ruhlandt-Senge, K., Tetrahedron 2000, 56 (45), 8927-8932.
14. Rhee, S. W.; Na, Y. H.; Do, Y.; Kim, J., Inorganica Chimica Acta 2000, 309, 49-56.
106

107
15. Dwyer, P. N.; Buchler, J. W.; Scheidt, W. R., Journal of the Chemical Society 1974, 96 (9), 2789-2784.
16. Buchler, J. W.; Lay, K. L.; Lee, Y. J.; Scheidt, W. R., Angewandte Chemie-International Edition 1982, 21 (6), 432-435.
17. Dwyer, P. N.; Puppe, L.; Buchler, J. W.; Scheidt, W. R., Inorganic Chemistry 1975, 14 (8), 1782-1785.
18. Botulinski, A.; Buchler, J. W.; Abbes, N. E.; Scheidt, W. R., Liebigs Annalen 1987, 4, 305-309.
19. Botulinski, A.; Buchler, J. W.; Lay, K. L.; Stoppa, H., Liebigs Annalen Chemie 1984, 7, 1259-1266.
20. Re, N.; Bonomo, L.; Da Silva, C.; Solari, E.; Scopelliti, R.; Floriani, C., Chemistry-a European Journal 2001 7 (12), 2536-2546.
21. Harmjanz, M.; Gill, H. S.; Scott, M. J., Journal of Organic Chemistry 2001, 66 (16), 5374-5383.
22. Harmjanz, V.; Bozidarevic, I.; Scott, M. J., Organic Letters 2001, 3 (15), 2281-2284.
23. Bonomo, L.; Solari, E.; Scopelliti, R.; Floriani, C.; Re, N., Journal of the American Chemical Society 2000, 122 (22), 5312-5326.
24. Gouterman, M., Optical Spectra and Electronic structure of Porphyrins and Related Rings. In The porphyrins; Dolphin, D.,' Academic Press: New York, 1978; Vol. III.
25. Anderson, H. L., Chemical Communications 1999, 21, 2323-2330.
26. Renner, M. W.; Buchler, J. W., Journal of Physical Chemistry 1995, 99, 8045-8049.
27. Botulinski, A.; Buchler, J. W.; Lay, K. L.; Ensling, J.; Twilfer, H.; Billecke, J.; Leuken, H.; Tonn, B., Advances in Chemistry Series 1982, 201, 253-277.
28. Bonomo, L.; Toraman, G.; Solari, E.; Scopelliti, R.; Floriani, C., Organometallics 1999, 18 (25), 5198-5200.
29. Bonomo, L.; Solari, E.; Scopelliti, R.; Latronico, M.; Floriani, C., Chemical Communications 1999, 21, 2227-2228.
30. Lindsey, J. S.; Wagner, R. W., Journal of Organic Chemistry 1989, 54 (4), 828-836.
31. Chang, C. K.; Kondylis, M. P., Chemical Communications 1986, 4, 316-318.

108
32. Harmjanz, M.; Scott, M. J., Inorganic Chemistry 2000, 39 (24), 5428-5429.
33. Harmjanz, M.; Gill, H. S.; Scott, M. J., Journal of the American Chemical Society 2000, 122, 10476-10477.
34. Chang-Hee, L.; Lindsey, J. S., Tetrahedron 1994, 50, 11427-11440.
35. Young, E. R.; Funk, R. L., Journal of Organic Chemistry 1998, 63, 9995-9996.
36. Beavington, R.; Burn, P. L., Journal of the Chemical Society Perkin Transactions I 1999, 4, 383-385.
37. Blessing, R. H., Acta Crystallographica Section A 1995, 51, 33-38.
38. Borovkov, V. V.; Lintuluoto, J. M.; Inoue, Y., Synlett 1999, 1, 61-62.
39. Buchler, J. W.; Dreher, C.; Kunzel, F. M., Synthesis and coordination chemistry of noble metal porphyrins. In Metal Complexes with Tetrapyrrole Ligands III, 1995; Vol. 84, pp 1-69.
40. Bischoff, I.; Feng, X. D.; Senge, M. O., Tetrahedron 2001, 57 (26), 5573-5583.
41. Bucher, C.; Seidel, D.; Lynch, V.; Kral, V.; Sessler, J. L., Organic Letters 2000, 2 (20), 3103-3106.
42. Belanzoni, P.; Rosi, M.; Sgamellotti, A.; Bonomo, L.; Floriani, C., Journal of the Chemical Society-Dalton Transactions 2001, 9, 1492-1497.
43. Da Silva, C.; Bonomo, L.; Solari, E.; Scopelliti, R.; Floriani, C.; Re, N., Chemistry-a European Journal 2000 6, (24), 4518-4531.
44. Hayashi, T.; Miyahara, T.; Koide, N.; Kato, Y.; Masuda, H.; Ogoshi, H., Journal of the American Chemical Society. 1997, 119, 7281-7290.
45. Wiehe, A.; Stollberg, H.; Runge, S.; Paul, A.; Senge, M. O.; Roder, B., Journal of Porphyrins and Phthalocyanines 2001 5 (12), 853-860.
46. Wang, Y. S.; Schanze, K. S., Chemical Physics 1993, 176 (2-3), 305-319.
47. Chou, J. H.; Nalwa, H. S.; Kosal, M. E.; Rakow, N. A.; Suslick, S. S., The Porphyrin Handbook. In The Porphyrin Handbook, ed.; Kadish, K. M.; Smith, K. M.; Guilard, R.,' Academic Press: San Diego, 2000; Vol. 6, pp 43-132.
48. Callot, H. J.; Schaeffer, E.; Cromer, R.; Metz, F., Tetrahedron 1990, 46 (15), 5253-5262.
49. Nath, M.; Huffman, J. C.; Zaleski, J. M., Journal of the American Chemical Society 2003, 125 (38), 11484-11485.

109
50. Aihara, H.; Jaquinod, L.; Nurco, D. J.; Smith, K. M., Angewandte Chemie-International Edition 2001, 40 (18), 3439-3444.
51. Richeter, S.; Jeandon, C.; Kyritsakas, N.; Ruppert, R.; Callot, H. J., Journal of Organic Chemistry 2003, 68 (24), 9200-9208.
52. Gill, H. S.; Harmjanz, M.; Santamaria, J.; Finger, I.; Scott, M. J., Angewandte Chemie-International Edition 2004, 43 (4), 485-490.
53. Richeter, S.; Jeandon, C.; Gisselbrecht, J. P.; Graff, R.; Ruppert, R.; Callot, H. J., Inorganic Chemistry 2004, 43 (1), 251-263.
54. Richeter, S.; Jeandon, C.; Gisselbrecht, J. P.; Ruppert, R.; Callot, H. J., Journal of the American Chemical Society 2002, 124 (21), 6168-6179.
55. Kyrychenko, A.; Andrasson, J.; Martensson, J.; Albinsson, B., Journal of Physical Chemistry B 2002, 106 (48), 12613-12622.
56. Mascarenhas, F.; Falk, K.; Klavins, P.; Schilling, J. S.; Tomkowicz, Z.; Haase, W., Journal of Magnetism and Magnetic Materials 2001, 231 (2-3), 172-178.
57. Bonnett, R., Chemical Society Reviews 1995, 24 (1), 19-33.
58. Armstrong, N. R., Journal of Porphyrins and Phthalocyanines 2000, 4 (4), 414-417.
59. Odobel, F.; Blart, E.; Lagree, M.; Villieras, M.; Boujtita, H.; El Murr, N.; Caramori, S.; Bignozzi, C. A., Journal of Materials Chemistry 2003, 13 (3), 502-510.
60. Shelnutt, J. A., Journal of Porphyrins and Phthalocyanines 2001, 5 (3), 300-311.
61. Haddad, R. E.; Gazeau, S.; Pecaut, J.; Marchon, J. C.; Medforth, C. J.; Shelnutt, J. A., Journal of the American Chemical Society 2003, 125 (5), 1253-1268.
62. Rogers, J. E.; Nguyen, K. A.; Hufnagle, D. C.; McLean, D. G.; Su, W. J.; Gossett, K. M.; Burke, A. R.; Vinogradov, S. A.; Pachter, R.; Fleitz, P. A., Journal of Physical Chemistry A 2003, 107 (51), 11331-11339.
63. Tutt, L. W.; Boggess, T. F., Progress in Quantum Electronics 1993, 17 (4), 299-338.
64. Rozhkov, V. V.; Khajehpour, M.; Vinogradov, S. A., Inorganic Chemistry 2003, 42 (14), 4253-4255.
65. Amao, Y.; Miyashita, T.; Okura, I., Reactive & Functional Polymers 2001, 47 (1), 49-54.

110
66. Amao, Y.; Tabuchi, Y.; Yamashita, Y.; Kimura, K., European Polymer Journal 2002, 38 (4), 675-681.
67. Englman, R.; Jortner, J., Molecular Physics 1970, 18 (2), 145-168.
68. Shinozaki, K.; Hotta, Y.; Yasue, R., Inorganica Chimica Acta 2002, 328, 229-231.

BIOGRAPHICAL SKETCH
Ivana Božidarević was born in 1975 in Belgrade, Serbia, where she finished
elementary school and high school. She decided to study chemistry when she was
thirteen, because she had the best chemistry teacher in the world. During high school,
she won a couple of awards for young researchers and was highly ranked at all the
chemistry competitions on the federal level. She spent her summers doing research in the
labs of Petnica Science Center for high school students. Ivana entered The Faculty of
Chemistry at Belgrade University in 1994 and defended her BS Thesis in June 1999,
being the second t to graduate from her class of 150 students. She started her Ph.D.
studies at the University of Florida in 1999 by joining Prof. Scott’s group, and she will be
graduating in August 2004 ready for new challenges in the world of chemistry.
111





























