Embed Size (px)
Citation preview

www.pnas.org/cgi/doi/10.1073/pnas. 115
1
Supplementary Information for Two-step pathway for isoprenoid synthesis A.O. Chatzivasileioua, V. Warda,b, S. Edgara,1, G. Stephanopoulosa aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA bDepartment of Chemical Engineering, University of Waterloo, 200 University Avenue West, Waterloo, ON N2L 3G1, Canada
1: Present address: Zymergen, 5980 Horton St #105, Emeryville, CA 94608, USA Corresponding author: G. Stephanopoulos Email: [email protected] This PDF file includes:
Supplementary text Figs. S1 to S7 Tables S1 to S4 References for SI reference citations
1 129358

2
Supplementary Information Text
Supplemental Materials and Methods
Routine Cloning Protocol. A standard protocol was used for the cloning of all plasmids described in this work. Primers were designed for Gibson Assembly using the NEBuilder online tool (NEB), and primers were purchased from Sigma-Aldrich. PCR reactions were performed in a Bio-rad C1000 Touch Dual Block thermocycler using 2x Q5 polymerase master mix (NEB) according to manufacturer’s recommendations. The products were digested with DpnI (NEB) enzyme for 1h at 37°C to digest the template DNA. The digested PCR products were then run on a 0.8% agarose gel using a Mini or Sub Cell and a Powerpac Basic power supply (Bio-rad). Fragments were gel-extracted using a Zymoclean Gel DNA recovery kit (Zymo Research) according to the manufacturer’s recommendation. Fragments were ligated using the Gibson Assembly Master Mix (NEB) for 1h at 50°C and transformed into DH5α (NEB) high efficiency chemical competent cells (NEB) according to standard protocol. Transformants were screened by PCR using colonies boiled in water for 10 min as the template. Two to three positive transformants were cultured overnight in LB media and the plasmid was purified using a Mini-prep kit (Qiagen). Overlapping regions of the new construct were sequenced (Quintara Biosciences, Boston) to confirm the sequence of the new plasmid. Plasmids with confirmed sequences for protein purification were transformed into BL21(DE3) using heat-shock, otherwise plasmids were transformed into MG1655(DE3) for further study by electroporation using a MicroPulser (Bio-rad). Electrocompetent cells were made by a standard glycerol washing of mid-log phase cells (Bio-rad) and stored at -80oC until future use. For electroporation, 1 μL of purified plasmid in water was added to 50 μL of electrocompetent cells using 1 kV and electroporated in 1 mm path-length cuvettes (Bulldog Bio).
Construction of enzyme expression vectors. All enzyme expression vectors were based on the pET-28 vector, into which the genes for enzyme expression were inserted. Backbone fragments were amplified from pET-28 a (+) vector using the primer pair GB pET28-HisT-vec_f/r. The insert fragments were amplified as follows: the fragments containing the genes for ScCK or ScMK expression were amplified from S. cerevisiae genomic DNA using the primer pairs GB-pET28-CHOLKIN_f/r or GB-pET28-MEVKIN_f/r respectively, the fragments containing the gene for EcGK or EcHK expression were amplified from E. coli genomic DNA using the primer pairs GB-pET28-GLYCKIN_f/r or GB-pET28-HSERKIN_f/r respectively, whereas the fragments the genes for HvIPK, MtIPK, MjIPK, TaIPK or TaIPK-3m expression were amplified from custom synthesized, and codon optimized DNA using the primer pairs GB-pET28-HV_f/r, GB-pET28-MTH_f/r, GB-pET28-MJ _f/r, GB-pET28-THA_f/r, or GB-pET28-THA3m_f/r respectively. The backbone and insert fragments were then assembled to give the respective plasmids. Sequences for the primers used are listed in Table S 3.
Construction of the IUP expression vectors. The plasmid pSEVA228-proDIUPi was generated by amplifying the backbone pSEVA228 with the primers GB-SEVA228_f/r and inserting a custom-synthesized promoter sequence, shown in Table S 4, which incorporates the proD promoter system (1), which was amplified using the primer pairs GB-proD_f/r, along with the IUP operon, consisting of the genes Scck, ipk and idi. Each of the three operon elements was amplified from custom synthesized DNA fragments (IDT) using the primer pairs GB-chk_f/r, GB-atipk_f/r and GB-iditerm_f/r respectively. In all three cases, the codon-optimized gene coding sequence was preceded by a corresponding optimized RBS (shown in Table S 4) and in the case of idi, it was followed by a T7 terminator derived from pET-28(+) (shown in Table S 4). The RBSs were optimized using the Salis lab RBS optimization tool (2, 3). The PCR fragments were assembled to give pSEVA228-proDIUPi. The plasmid pSEVA228-pro4IUPi was created by

3
replacing the 6 nucleotides in the proD promoter sequence of pSEVA228-proDIUPi with the primer pairs GB-pro4_Mut_f/GB-ProLibrary_Mut_r to amplify the whole plasmid and then subsequently assembling the amplification product. The plasmid pSEVA228pro4-ck-idi, which carries a reduced version of the IUP operon, lacking ipk, was created by amplifying pSEVA228-pro4IUPi using the primer pair GB-IUPnoIPK_f/r and then subsequently assembling the amplification product. The pTET-IUPi plasmid was created as follows: The pTET backbone was created by replacing the T7/lac promoter region of pET-28a carrying a methanol utilization operon (pETMeOH500) with the androtetracycline repressor and promoter region of pBbS2k-rfp by Gibson assembly using primers GB-pETMeOH500-f/r and GB-pBbS2k-rfp-f/r. Then, the methanol utilization operon was replaced with the IUP operon from pSEVA228-pro4IUPi by Gibson assembly using the primers pTet-IUP-ins_f/r, and pTet-IUP-ins_f/r, to produce plasmid pTET-IUPi. pSEVA228 was a gift from Jason King. pETMeOH500 was a gift from Benjamin Woolston. pBbS2k-RFP was a gift from Jay Keasling (Addgene plasmid # 35330). Sequences for the primers used are listed in Table S 3.
Construction of downstream isoprenoid pathways. The copy number of the lycopene plasmids were varied by first amplification of the genes crtE crtI, crtB and ipi as well as the endogenous lycopene promoter using primers GB_pAC-LYCipi_f/r from the pAC-LYCipi plasmid. The fragment containing the gene aadA1 (spectinomycin resistance) was amplified using GB_aadA1_f/r from p5T7tds-ggpps and the origins pUC19 and pBR322 were amplified using GB_pUC19_f/r and GB_pBR322_f/r from pUC19 and pET28a respectively. These fragments were assembled with the appropriate origin to create pUC-LYCipi and p20-LYCipi. To create p5T7-LYCipi, the backbone of p5T7tds-ggpps was amplified using GB_p5T7_f/r and the lycopene synthesis genes were amplified from p20-LYCipi using the primers GB_p20-LYCipi_f/r and then assembled. To make p5T7-LYCipi-ggpps, the backbone of p5T7-LYCipi was amplified using primers p5T7Lyc-back_f/r and the ggpps was amplified from p5T7tds-ggpps using p5T7Lyc-ggpps_f/r and then assembled. p5T7gpps-ls and p5T7ispA-ads were created by PCR amplification of the p5T7tds-ggpps vector using primers GB_p5t7ggppstds_f/r to create the backbone from the T7 terminator to the T7 promoter. The primers GB_gpps_ls_f/r were used to amplify the gpps-ls operon from JBEI-6409 for Gibson Assembly in this backbone created the p5T7-gpps-ls vector. Primers GB_ispA_f/r and GB_ads_f/r (with RBS encoded on the primer) were used to amplify ispA from p5T7vs-ispA and ads from pADS respectively. These two fragments were assembled into the same backbone as gpps-ls to create the p5T7-ispA-ads vector. pAC-LYCipi was a gift from Francis X Cunningham Jr (Addgene plasmid # 53279) pADS was a gift from Jay Keasling (Addgene plasmid # 19040). JBEI-6409 was a gift from Taek Soon Lee (Addgene plasmid # 47048). Sequences for the primers used are listed in Table S 3.
Enzyme expression and purification. BL21 harboring a pET-28 vector for the expression of proteins in Fig. 1B was revived from a glycerol stock by inoculating into LB media and growing at 37C overnight. Two hundred milliliters of SOB media (Amresco) in a baffled 1L flask was inoculated at 1% with the overnight culture and grown until an OD of 0.5 at 30°C. The culturere then induced with IPTG at a final concentration of 100 uM. Cultures were incubated for 3-4 h at 30°C for protein synthesis after which they were centrifuged in an Allegra X12R centrifuge (Beckman-Coulter) at 3273 x g for 15 min. The supernatant was removed, and the cell pellets were stored at -20°C until purification. Proteins were purified using the following protocol and at all stages proteins were keep on ice. First, cells were lysed using 5 mL of ice-cold NPI-10 buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) using a gas driven high-pressure homogenizer, the EmulsiFlex-C5 (Avestin). After disruption, 100 μM PMSF was added to the lysate. The lysate was centrifuged at 15 000 x g for 10 min at 4°C. Ni-NTA resin purchased from Qiagen was equilibrated using 10 column volumes (CV) of NPI-10 buffer in gravity column (Fisher-Scientific). The clarified supernatant was loaded to the column and allowed to drip

4
through by gravity. After all of the lysate was loaded, the column was washed with 10 CV of NPI-20 buffer (20 mM imidazole). Then the protein was eluted from the column using 3 CV of NPI-250 buffer (250 mM imidazole). Protein purification was confirmed by protein gel electrophoresis using a Mini-protean system (Bio-rad) using precast 4-20% acrylamide gels (Bio-rad), Kalidescope Prestained Protein Ladder (Bio-rad), and Tris-glycine, SDS buffer (Bio-rad) at 200V for 20 min. Gels were stained with InstantBlue (Expedeon).
IUP flux estimation through metabolite measurements. In order to have a first-order estimate of IPP flux through the IUP a simple model was developed that utilizes the results of the pulse labeling experiment (see Fig 3 in main text). The basis of the model lies on Eq 1, which states that in our experiment, IPP is being produced through either the MEP pathway (rMEP) or through the IUP (rIUP) and is consumed at a rate rC.
IPP can be either labeled or unlabeled, with labeled IPP being produced from the MEP pathway and unlabeled IPP being produced from the IUP, that is:
d[IPPUL]d𝑡𝑡
= 𝑟𝑟IUP − (1 − 𝜆𝜆)𝑟𝑟C (𝟑𝟑)
In the above λ indicated the fraction of IPP that is labeled, i.e:
𝜆𝜆 = [IPPL][IPPTOT]
⇒ 1 − 𝜆𝜆 = [IPPUL]
[IPPTOT] (𝟒𝟒)
Using the definition of λ, we can rework Eq. 3 as follows:
(1 − λ)d[IPPΤΟΤ]
d𝑡𝑡=
d𝜆𝜆d𝑡𝑡
[IPPΤΟΤ] + 𝑟𝑟IUP − (1 − 𝜆𝜆)𝑟𝑟C (𝟓𝟓)
We then make the following assumptions. First we assume that in the cell, as an aggregate, IPP consumption follows a 1st order rate law. Secondly, we assume that the fraction of labeled IPP is very small, something that is corroborated by our data (Fig 3A & B). Therefore:
𝑟𝑟C = 𝑘𝑘[IPPΤΟΤ]
1 − 𝜆𝜆 ≈ 1
This leads to Eq. 5 being transformed as follows:
d[IPPΤΟΤ]d𝑡𝑡
= �d𝜆𝜆d𝑡𝑡
− 𝑘𝑘� [IPPΤΟΤ] + 𝑟𝑟IUP (𝟔𝟔)
d[IPPΤΟΤ]d𝑡𝑡
= 𝑟𝑟MEP + 𝑟𝑟IUP − 𝑟𝑟C (𝟏𝟏)
d[IPPL]d𝑡𝑡
= 𝑟𝑟MEP

5
If it is assumed that for at least the first 30 mins of the experiment we have a quasi-steady state, meaning that the terms �d𝜆𝜆
d𝑡𝑡− 𝑘𝑘� and rIUP will remain relatively constant. Thus, Eq. 6 can be
integrated. Using IPPTOT,0=IPP0 (IPP at t=0, which is measured) as its initial condition, we get:
[IPPΤΟΤ] =eA𝑡𝑡�B + A IPP0� − B
A (𝟕𝟕)
By least-square fitting our data on total IPP concentration (which is calculated by summing the measured values of IPPL and IPPUL against Eq. 7, we can then estimate a value for rIUP.
Synthesis of IP and DMAP. Isopentenyl monophosphate (IP) and dimethylallyl monophosphate (DMAP) were chemically synthesized using isoprenol or prenol respectively as they are not commercially available. The synthesis process was adapted from (4, 5). All chemicals and solvents were used as supplied without further purification. Trichloroacetonitrile (2.26 equiv.) tetrabutylammonium phosphate (1.66 equiv.) was added to a solution of the appropriate isopentenol (1 equiv.) in acetonitrile and stirred for 4h. The acetonitrile was evaporated, and the synthesized isopentenyl phosphate was resuspended in water and cooled for 6h at 4°C. This caused the crystallization and precipitation of the trichloroamide which was removed by filtration. The isopentenyl phosphate was purified by flash chromatography using a DOWEX 50WX8 ion-exchange column by percolation using NH4HCO3 (0.025 M) then eluted using isopropanol/NH4OH/H2O 7:2:1. The resulting product was recovered as a solid by lyophilization.
The structure of the resulting IP and DMAP were confirmed by 1H and 31P-NMR. Spectra were recorded on a Varian Mercury-300 NMR Spectrometer in deuterated water (Sigma-Aldrich) at 300 MHz and chemical shifts (δ) are reported in parts per million (ppm) downfield from the internal standard, tetramethylsilane (TMS). The resulting spectra for IP were 1H NMR (300MHz, D2O): δ: 4.71 (s, 1H), 3.80 (q, 2H), 2.21 (t, 2H), 1.61 (s, 3H) and 31P NMR (300MHz, D2O): δ 2.38. The resulting spectra for DMAP were 1H NMR (300MHz, D2O): δ 5.26 (t, 1H), 4.17 (t, 2H), 1.61 (s, 3H), 1.56 (s, 3H) and 31P NMR (300MHz, D2O): δ 2.96.
Quantification of metabolites. IP/DMAP, IPP/DMAPP, MEC, PEP and 3PG were quantified by LC-MS/MS by comparison to synthetic IP/DMAP made in house according to the procedure described above and IPP, DMAPP, MEP, PEP and 3PG standards purchased from Sigma-Aldrich. Liquid Chromatography was conducted using an Agilent 1100 Series HPLC (Agilent Technologies) and the MS/MS was conducted using an API 4000 triple quadrupole mass spectrometer (SCIEX) with ESI running in negative MRM mode. Mobile phases consisted of LCMS grade 10 mM tributylammonium (TBA) (Sigma-Aldrich), 15 mM acetic acid (Sigma) in water (A) and 100% acetonitrile (B). A EC18 column (2.7 μm, 2.1 mm, 50 mm length) (Agilent) was used to separate 20 μL of sample with a flow rate of 0.3 mL/min and linear gradient program: 0-3 min 0% B, 3-10 min 0-50% B, 10-12 min 50-100% B, 12-18 min 100% B, 18-18.5 min 100-0% B, 0% B until 25 min. Metabolite specific ionization and fragmentation voltages were obtained from a 1 μM standard solution of each metabolite using the Analyst software (v 1.6) and monitored during the chromatography. Peaks were integrated using the Analyst software and compared to a standard curve generated for each metabolite.
GPP, FPP, and GGPP were quantified using an alternative method using the same instrumentation described above. An Xbridge C18 column (150mm, 3.5 μm, 2.1 mm) (Waters) was operated with a mobile phase of 0.1% v/v TBA, 0.12% v/v acetic acid, and titrated with ~0.5% v/v 5N NH4OH until a pH of 8.5 was reached (A). The elutant was 100% acetonitrile (B). A series of linear gradients: 0-5 min 0% B, 5-20 min 0-65% B, 20-25 min 65% B, 25-30 min

6
100% B, 30-35 min 100% B, 35-36 min 100-0% B, 0% B until 45 min, was used to separate these analytes which were then compared to standard curves generated using standards purchased from Sigma-Aldrich and/or Cayman Chemicals. Samples were resuspended in the aqueous mobile phases described above.
Quantification of lycopene. Lycopene content was assessed by UV-Vis spectroscopy. First, 1 mL of cells was transferred to an amber microtube and centrifuged at 16 000 x g for 2 min. The cell pellet was then resuspended in 1mL of a 50% ethanol, 50% acetone solution and vortexed for 30 min (VWR). The solution was centrifuged to remove particulates and 200 μL was transferred to a microplate and the absorbance at 475 nm was recorded. This was compared to a standard curve generated using a standard freshly purified in-house as the commercial standards purchased from three different companies (Indofine Chemical Company, Inc., Carbosynth, Santa Cruz Biotechnology) were found to be overestimating the lycopene content by about 10-fold, presumably due to degradation and bleaching of these standards during storage (6). Therefore, a fresh standard was prepared according to a standard protocol (7) from E. coli biomass expressing the lycopene synthesis genes. First, E. coli biomass was centrifuged, then resuspended in acetone in the dark and left to stir for 1h. This solution was then filtered, and a small amount of acetone was added to wash residual lycopene from the cells. The filtrate was then chilled at -20°C to induce crystallization of the lycopene. Crystals were recovered by filtration and the crystallization process was repeated twice to purify the lycopene. The resulting lycopene was dried, weighed and subjected to UV/VIS spectroscopy to confirm its authenticity. It was compared to a commercially available standard from Indofine in Fig S 5. It was then resuspended in 50% ethanol, 50% acetone solution to create a standard curve for quantification. Lycopene content was calculated using the cell density of the culture calculated from the optical density at 600 nm using a correlation of 0.33 g/A.U.
Quantification of volatile isoprenoids. Volatile isoprenoids were quantified using GC-MS using ultra-pure helium as the carrier gas. First, the C18 resin used to capture the isoprenoids was vacuum-filtered from the cells and culture media using BioSpin columns (Bio-rad). The resin was then spun at 1000 x g to remove residual water, then eluted in ethyl acetate containing 36 mg/L caryophyllene as an internal standard which allowed a 20-fold concentration of the isoprenoid for quantification. The 1 μL of the eluted isoprenoid was quantified on a HP-5 MS UI capillary column (30m, 250 μm, 0.25μm) (Agilent Technologies) using a 7890B Series GC and a 5977B MS. Chromatography was performed under the following conditions: splitless injection, inlet temperature 280°C, constant inlet pressure 115.8 kPa, valve temperature 300°C, and MS transfer line 300°C. A oven program of 100°C, hold 1min, 15°C/min until 200°C, hold 2 min, 30°C/min until 250°C, hold 1 min, and 30°C/min until 290°C, hold 2 min was used for determination of taxadiene, miltiradiene, valencene, and amorphadiene. Limonene was separated using an oven program of 80°C, hold 3min, 10°C/min until 140°C, hold 2 min, 45°C/min until 290°C, hold 1 min. The MS was operated at an ion source temperature of 280°C, and a quadrupole temperature of 180°C. Ions were scanned between a mass of 40 to 400 at 1.562 u/s. Taxadiene was quantified using a standard curve based on the m/z 122 ion which has the greatest abundance in unlabeled taxadiene. The 131 m/z ion was used to quantify labeled taxadiene using the same standard curve generated from purified unlabeled taxadiene. Taxadiene was purified using a semi-preparative HPLC using a Supelco Discovery C18 (25 cm, 10 mm, 5 um) column under isocratic conditions, 89% acetonitrile in water at 8 mL/min on a Shimadzu LC-2AD HPLC (8) equipped with a SPD-M20A diode array set at 210 nm. The fractions containing taxadiene as confirmed by GCMS were collected using a fraction collector, pooled and recovered by rotary evaporation on a Buchi Rotavapor R-210. The purified taxadiene was weighed and resuspended for generation of the standard curve.
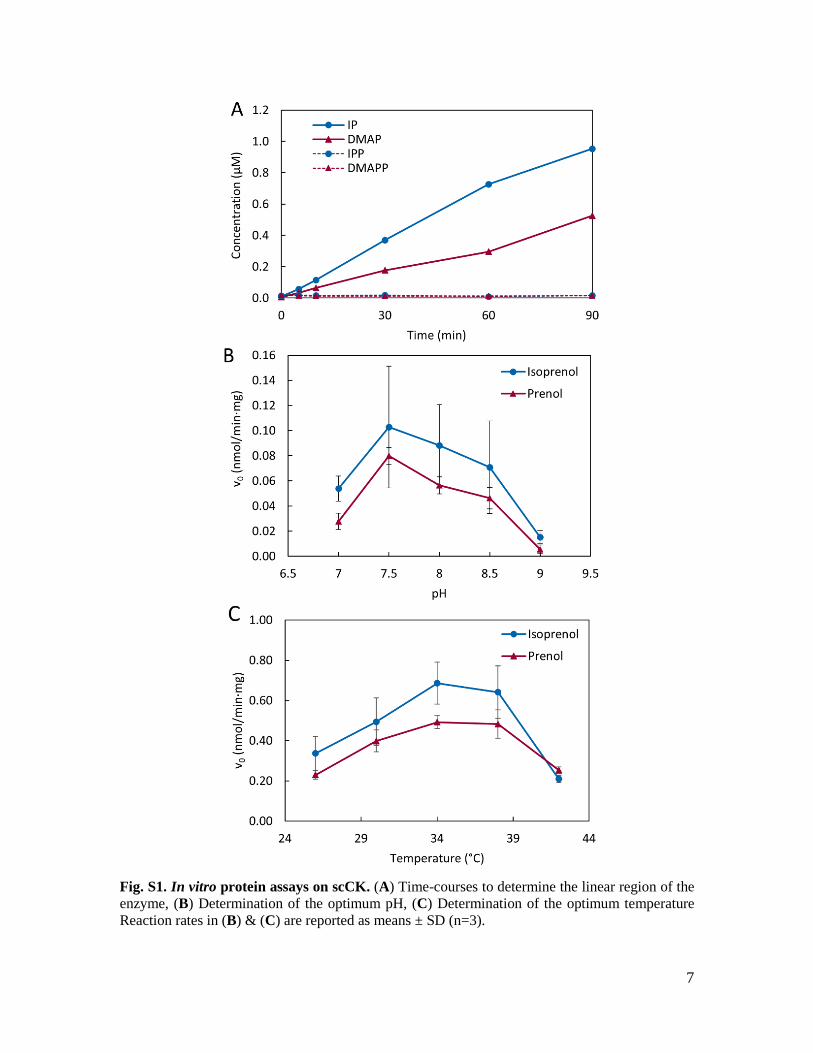
7
Fig. S1. In vitro protein assays on scCK. (A) Time-courses to determine the linear region of the enzyme, (B) Determination of the optimum pH, (C) Determination of the optimum temperature Reaction rates in (B) & (C) are reported as means ± SD (n=3).
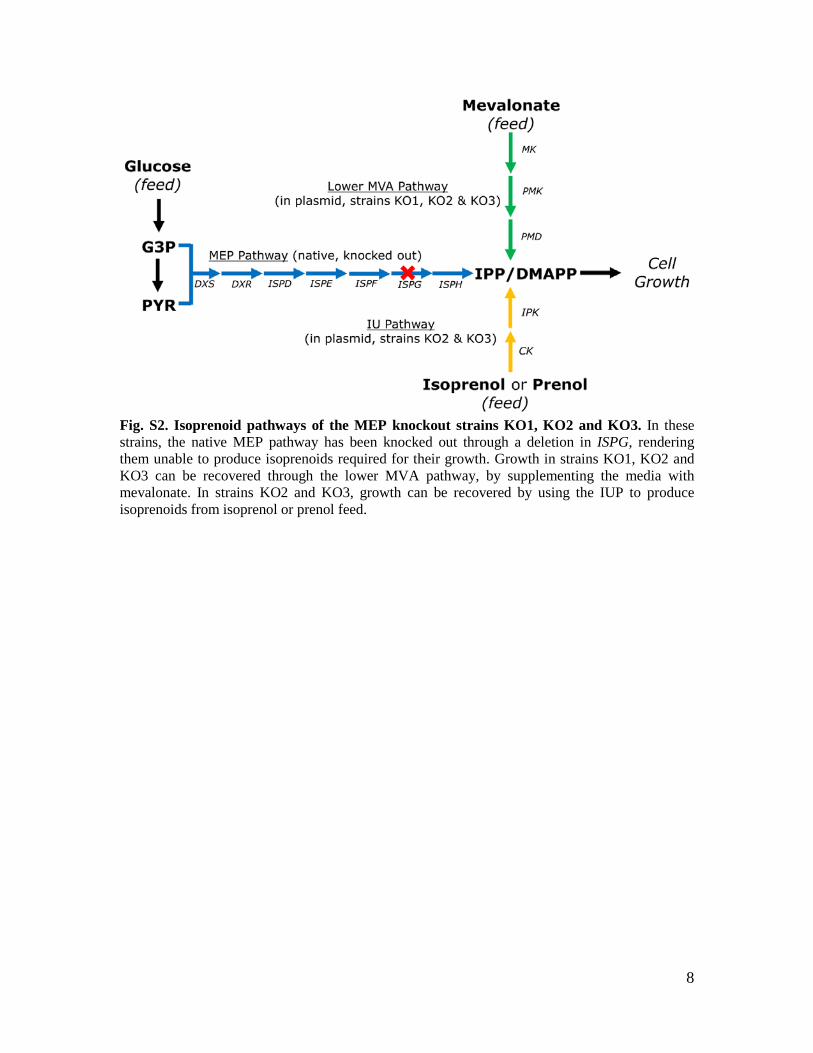
8
Fig. S2. Isoprenoid pathways of the MEP knockout strains KO1, KO2 and KO3. In these strains, the native MEP pathway has been knocked out through a deletion in ISPG, rendering them unable to produce isoprenoids required for their growth. Growth in strains KO1, KO2 and KO3 can be recovered through the lower MVA pathway, by supplementing the media with mevalonate. In strains KO2 and KO3, growth can be recovered by using the IUP to produce isoprenoids from isoprenol or prenol feed.

9
Fig. S3. Initial characterization of expression system strength. Characterization of expression plasmids using GFP as a reporter protein, with either the pSEVA228pro4-gfp plasmid (pro4), the pTET-gfp plasmid (pTET) induced with 10 or 20 ng/mL aTC, or no plasmid (Control). All values represent the means ± SD of 3 biological replicates.
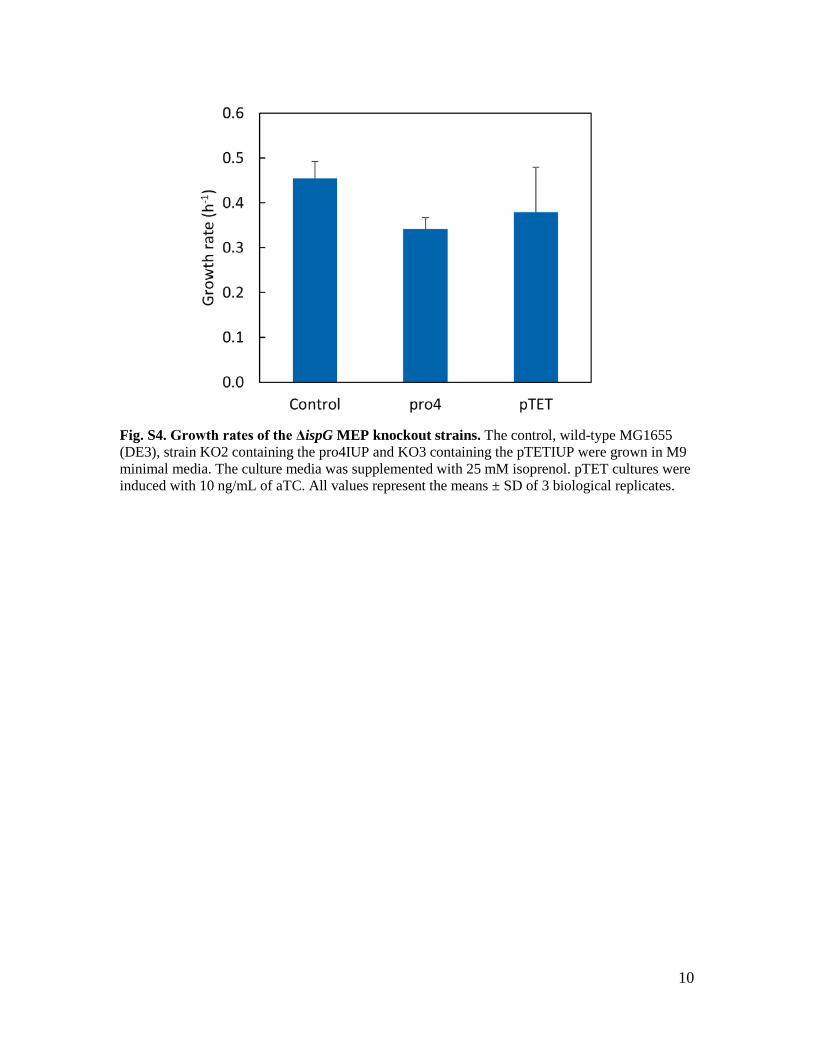
10
Fig. S4. Growth rates of the ΔispG MEP knockout strains. The control, wild-type MG1655 (DE3), strain KO2 containing the pro4IUP and KO3 containing the pTETIUP were grown in M9 minimal media. The culture media was supplemented with 25 mM isoprenol. pTET cultures were induced with 10 ng/mL of aTC. All values represent the means ± SD of 3 biological replicates.

11
Fig. S5. Growth Rate Comparison. Exponential growth rates of strains containing the pSEVA228pro4-gfp plasmid (pro4), the pTET-gfp plasmid (pTET) or no plasmid (control), grown in M9 minimal media supplemented with either 0 or 25 mM isoprenol. All values represent the means ± SD of 3 biological replicates.

12
Fig. S6. Standard Curve for lycopene quantification. Lycopene purchased from a commercial source is compared to lycopene freshly purified in-house. All values represent the means ± SD of 3 replicates.
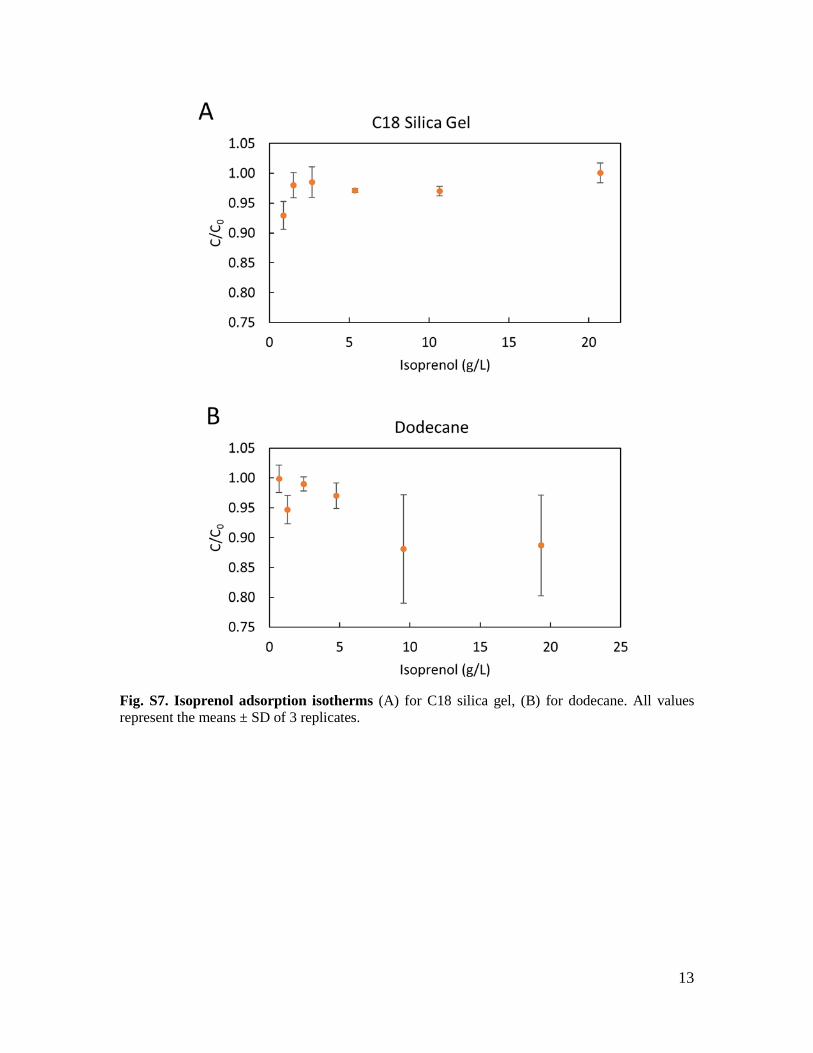
13
Fig. S7. Isoprenol adsorption isotherms (A) for C18 silica gel, (B) for dodecane. All values represent the means ± SD of 3 replicates.

14
Table S1. List of strains and plasmids Host/Strain Description Reference MG1655 (DE3) ΔendA ΔrecA (λ DE3) DH5α fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15
gyrA96 recA1 relA1 endA1 thi-1 hsdR17 NEB
BL21 (DE3) fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5
NEB
KO1 MG1655 (DE3), ΔispG, pBAD33-proA-MEVI This study KO2 MG1655 (DE3), ΔispG, pBAD33-proA-MEVI,
pSEVA228pro4IUPi This study
KO3 MG1655 (DE3), ΔispG, pBAD33-proA-MEVI, pTETIUPi This study Plasmids Description (origin, antibiotic marker, promoter, operon) Reference pET28a(+) pBR322, KnR, PT7lacUV, enzymes from Table 1 with N
terminal 6 x his tag Novagen
pADS pTrc99A derivative containing the ADS gene; ApR (9) pJBEI-6409 p15A, CmR, PlacUV5, atoB, hmgs, hmgr, PlacUV5, mvk, pmk,
pmd, idi, Ptrc, trGPPS, ls (10)
pAC-LYCipi p15A, CmR, crtE, ipi, crtI, crtB, endogenous promoter (11) pSEVA228 RK2, KnR, xlyS-Pm (12) pBbS2k-RFP SC101, KnR, PTET
, rfp (13) pETMEOH500 pBR322, KnR, PT7lacUV, mdh This study pTETmdh pBR322, KnR, PTET, mdh This study pMBIS RK2, TcR, Ptrc, erg12, erg8, mvd1, idi, ispA (9) pBAD33-proA-MEVI p15A, CmR, PproA, erg12, erg8, mvd1 This study pCas9 pSC101 ori, RepA101ts, KnR, ParaC, cas9 (14) pTargetF pij23119, pMB1, SpR (14) pTargetF-ispG pij23119, pMB1, SpR This study p20-LYCipi pBR322, SpR, crtE, ipi, crtI, crtB, endogenous promoter This study pUC-LYCipi pUC19, SpR, crtE, ipi, crtI, crtB, endogenous promoter This study p5T7-LYCipi pSC101, SpR, PT7lacUV, crtE, ipi, crtI, crtB This study p5T7-LYCipi-ggpps pSC101, SpR, PT7lacUV, ggpps, ipi, crtI, crtB This study p5T7tds-ggpps pSC101, SpR, PT7lacUV, tds, ggpps (15) p5T7ksl-ggpps pSC101, SpR, PT7lacUV, ksl, ggpps (16) p5T7vs-ispA pSC101, SpR, PT7lacUV, vs, ispA (16) p5T7ggps-ls pSC101, SpR, PT7lacUV, gpps, ls This study p5T7ispA-ads pSC101, SpR, PT7lacUV, ispA, ads This study pSEVA228-pro4IUPi RK2, KnR, Ppro4, ck, ipk, idi This study pSEVA228-proDIUPi RK2, KnR, PproD, ck, ipk, idi This study pTET-IUPi pBR322, KnR, PTET, ck, ipk, idi This study pTrcsGFP pBR322, AmpR, Ptrc, sgfp (17) pSEVA228pro4-gfp RK2, KnR, Ppro4, sgfp This study pTET-gfp pBR322, KnR, PTET, sgfp This study pSEVA228pro4-ck-idi RK2, KnR, Ppro4, ck, idi This study ApR = ampicillin, KnR = kanamycin, TcR = Tetracyclin, SpR = Spectinomycin

15
Table S2. List of genes used in this study and their origins Genes Origin (Accession Number) tds Taxus brevifolia (AAC49310.1), codon optimized, truncated first 60 amino
acids, methionine added ggpps Taxus canadensis (AAD16018.1), codon optimized, truncated first 98 amino
acids, methionine added crtE, crtI, crtB,
ipi Pantoea agglomerans, crtE (AAA21260.1), crtB (AFZ89043.1), crtI
(AFZ89042.1), ipi (AAA64978.1) ksl Salvia miltiorrhiza, codon optimized, methionine added, (ABV08817.1) vs Callitropsis nootkatensis, codon optimized, methionine added
(AFN21429.1) ls Mentha spicata (AAC37366.1), codon optimized gpps Abies grandis (AAN01134.1), codon optimized ads Artemisia annua (AEQ63683.1), codon optimized ispA E. coli (WP_097750737.1) ipk Arabidopsis thaliana (AAN12957.1), codon optimized Scck S. cerevisiae (AAA34499.1), codon optimized Hvipk Haloferax volcanii (ADE04091.1), codon optimized Mtipk Methanothermobacter thermautotrophicus (AAB84554.1), codon optimized Mjipk Methanocaldococcus jannaschii (AAB98024.1), codon optimized Taipk Thermoplasma acidophilum (CAC11251.1), codon optimized Taipk-3m Thermoplasma acidophilum (CAC11251.1), codon optimized, three
mutations (V72I , Y140V, K203G) Ecgk E. coli (AAA23913.1) erg12/Scmk S. cerevisiae (CAA29487.1) Echk E. coli (AAC73114.1) idi E. coli (AAD26812.1)

16
Table S3. List of primers used in this study Name Sequence GB_p5t7tds-ggpps_r ATGGTATATCTCCTTATTAAAGTTAAAC GB_p5t7tds-ggpps_f TATTAGTTAAGTATAAGAAGGAGATATAC GB_gpps_ls_f TAATAAGGAGATATACCATATGGAATTTGACTTCAACAAATAC GB_gpps_ls_r CTTCTTATACTTAACTAATACGAGGAAGCGGAATATATC GB_ispA_f TAATAAGGAGATATACCATATGGACTTTCCGCAGCAAC GB_ispA_r CTCCTTCTTAAAAGATCCTTTATTTATTACGCTGGATGATGTAGTC GB_ads_f GTAATAAATAAAGGATCTTTTAAGAAGGAGATATACATGGCCCTGACCGAAGAG GB_ads_r CTTCTTATACTTAACTAATATCAGATGGACATCGGGTAAAC GB_pAC-LYCipi_r CAGTTATTGGTGCCCTTAAACG GB_pAC-LYCipi_f TAAGCTTTAATGCGGTAGTTTATCAC GB_aadA1_f AGGGCACCAATAACTGGGTGAACACTATCCCATATC GB_aadA1_r TAACCGTATAATCATGGCAATTCTGGAAG GB_pUC19_f GCCATGATTATACGGTTATCCACAGAATC GB_pUC19_r CTACCGCATTAAAGCTTAAGGATCTAGGTGAAGATC GB_pBR322_f ATTGCCATGATTCCCCTTGTATTACTGTTTATG GB_pBR322_r CTACCGCATTAAAGCTTAACTCAAAGGCGGTAATAC GB_p5T7_r ATGGTATATCTCCTTATTAAAGTTAAACAAAATTATTTCTACAGGG GB_ p5T7 _f TTAATAAGGAGATATACCATATGGTGAGTGGCAGTAAAGC GB_p20-LYCipi_f CTCCTTCTTATACTTAACTAATACTGCGTGAACGTCATGGC GB_p20-LYCipi_r TATTAGTTAAGTATAAGAAGGAGATATAC GB-pET28-HisT-vec f CACCACCACCACCACCAC GB-pET28-HisT-vec r CGGTATATCTCCTTCTTAAAGTTAAACAAAATTATTTC GB-pET28-ScCK_f AAGAAGGAGATATACCGATGGTACAAGAATCACGTC GB-pET28-ScCK_r TCAGTGGTGGTGGTGGTGGTGCAAATAACTAGTATCGAGGAAC GB-pET28-EcGK f AAGAAGGAGATATACCGATGACTGAAAAAAAATATATCGTTGC GB-pET28-EcGK r TCAGTGGTGGTGGTGGTGGTGTTCGTCGTGTTCTTCCCAC GB-pET28-EcHK f AAGAAGGAGATATACCGATGGTTAAAGTTTATGCCCC GB-pET28-EcHK r TCAGTGGTGGTGGTGGTGGTGGTTTTCCAGTACTCGTGC GB-pET28-TaIPK-3m f AAGAAGGAGATATACCGATGATGATTCTGAAAATCGGAG GB-pET28-TaIPK-3m r TCAGTGGTGGTGGTGGTGGTGTCGAATGACAGTACCGATG GB-pET28-MjIPK_f AAGAAGGAGATATACCGATGCTGACCATCCTGAAATTAG GB-pET28-MjIPK_r TCAGTGGTGGTGGTGGTGGTGTTCGCTAAAGTCGATCTC GB-pET28-TaIPK f AAGAAGGAGATATACCGATGATGATTCTTAAGATAGGGGG GB-pET28-TaIPK r TCAGTGGTGGTGGTGGTGGTGACGAATGACGGTTCCGATG GB-pET28-Mtipk_f AAGAAGGAGATATACCGATGATCATTCTGAAACTGGG GB-pET28-Mtipk_r TCAGTGGTGGTGGTGGTGGTGATGTTTTCCTGTGATACGC GB-pET28-HvIPK_f AAGAAGGAGATATACCGATGTCCCTGGTGGTCCTTAAA GB-pET28-HvIPK_r TCAGTGGTGGTGGTGGTGGTGTTCCCCGCGAATGACTGT GB-pET28-ScMK_f TTTAAGAAGGAGATATACCGATGTCATTACCGTTCTTAAC GB-pET28-ScMK_r CAGTGGTGGTGGTGGTGGTGCTATGAAGTCCATGGTAAATTC GB-pETMeOH500_f ATGACCCACCTGAACATC GB-pETMeOH500_r GCGCAACGCAATTAATGTAAG GB-pBBS2k-rfp_f TTACATTAATTGCGTTGCGCTTAAGACCCACTTTCACATTTAAG GB-pBBS2k-rfp _r GCGATGTTCAGGTGGGTCATATGTATATCTCCTTCTTAAAAGATC GB-pTet-IUP-Ins_f TTTAAGAAGGAGATATACATATGGTGCAGGAGTCCCGC GB-pTet-IUP-Ins_r GTCGACGGAGCTCGAATTCGTTATTTGCTGAAGCGGATGATGGTC

17
GB-pTet-Vec_f CGAATTCGAGCTCCGTCG GB-pTet-Vec_r ATGTATATCTCCTTCTTAAAAGATCTTTTGAATTC Pro4 Mut_f GGGCATGCATAAGGCTCGGATGATATATTCAGGGAGACC ProLibrary_Mut_r CGAGCCTTATGCATGCCC GB-SEVA228_f GGGTCCCCAATAATTACG GB-SEVA228_r CAGCTGGGCGCGCCGTAG GB-proD_f TTCTACGGCGCGCCCAGCTGTTCTAGAGCACAGCTAACAC GB-proD_r TCCTTGCGTTGAAACCGTTGTGGTCTCC GB-chk_f CAACGGTTTCAACGCAAGGAAACACATTAAG GB-chk_r TTTCTTGTACTTACAGGTAGCTGGTGTC GB-atipk_f CTACCTGTAAGTACAAGAAAAGTCAGTAGTC GB-atipk_r CTCCTTAGTTTTATTTGCTGAAGCGGATG GB-iditerm_f CAGCAAATAAAACTAAGGAGGTCTATATGC GB-iditerm_r ATCGTAATTATTGGGGACCCGATATAGTTCCTCCTTTCAG GB-IUPnoIPK_f CTACCTGTAAAACTAAGGAGGTCTATATGC GB-IUPnoIPK_r CTCCTTAGTTTTACAGGTAGCTGGTGTC pCas9-ispG_f GCGACATTGAAGAAGATAAGG pCas9-ispG_r GTTTACGGTGTAAGCGATCC pCas9-ispG-seq_f GATTGCTGGCTGGAGGTCAC GB-pTargetF-ispGN20_f
GTCCTAGGTATAATACTAGTCGCTGCGTATCCGTTCGCGAGTTTTAGAGCTAGAAATAGC
GB-ptargetF-N20_r ACTAGTATTATACCTAGGACTGAG GB-pTargetF-vec_f CACCACCGACTATTTGCAAC GB-pTargetF-vec_r CTCGAGTAGGGATAACAGGGTA GB-ispG-H1_f CCCTGTTATCCCTACTCGAGCCAGCGTCTGTGGATACTACC GB-ispG-H1_r TCCCATCACGTCTCCCGCGTTACCCGTC GB-ispG-H2_f ACGCGGGAGACGTGATGGGAAGCGCCTC GB-ispG-H2_r GTTGCAAATAGTCGGTGGTGCTTCGCAGCCCAACTGATG p5T7Lyc-ggpps_f TTAATAAGGAGATATACCATATGTTCGACTTCAACGAG p5T7Lyc-ggpps_r TTGAACCCAAAAGGGCGGTATTAGTTTTGACGAAAGGC p5T7Lyc-back_f TACCGCCCTTTTGGGTTC p5T7Lyc-back_r ATGGTATATCTCCTTATTAAAGTTAAAC GB-sGFP-pSEVA F GAAAGAGGAGAAATACTAGTATGAGCAAGGGCGAAGAG GB-sGFP-pSEVA R CAAGCTTGTCGACGGAGCTCTTACTTATAGAGTTCATCCATGCC GB-pSEVA-back F GAGCTCCGTCGACAAGCTTG GB-pSEVA-back R ACTAGTATTTCTCCTCTTTCTCTAGTAAAAGTTAAAC GB-sGFP-pTET F TTTAAGAAGGAGATATACATATGAGCAAGGGCGAAGAG GB-sGFP-pTET R GTCGACGGAGCTCGAATTCGTTACTTATAGAGTTCATCCATGCC GB-pTET-back F CGAATTCGAGCTCCGTCG GB-pTET-back R ATGTATATCTCCTTCTTAAAAGATCTTTTGAATTC GB-pBro IAI Vec F GAGCTCCGTCGACAAGCT GB-pBro IAI Vec R ACTAGTATTTCTCCTCTTTCTCTAGTAAAAG GB-proX-Mevi Ins F CTAGAGAAAGAGGAGAAATACTAGTATGTCATTACCGTTCTTAACTTC GB-proX-Mevi Ins R CAAGCTTGTCGACGGAGCTCTTATTCCTTTGGTAGACCAG

18
Table S4. Custom elements used for the creation of vector pSEVA228-proDIUPi Promoter sequence TTCTAGAGCACAGCTAACACCACGTCGTCCCTATCTGCTGCCCTAGGTCTATGAGTGGTTGCTGGATAACTTTACGGGCATGCATAAGGCTCGTATAATATATTCAGGGAGACCACAACGGTTTC RBS for ck AACGCAAGGAAACACATTAAGGAGGTTTAA RBS for ipk GTACAAGAAAAGTCAGTAGTCTAAGGAGGTAAGC RBS for idi AACTAAGGAGGTCTAT T7 terminator region GCTAACAAAGCCCGAAAGGAAGCTGAGTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGCTGAAAGGAGGAACTATATC

19
References 1. Davis JH, Rubin AJ, Sauer RT (2011) Design, construction and characterization of a set
of insulated bacterial promoters. Nucleic Acids Res 39(3):1131–1141. 2. Salis HM, Mirsky EA, Voigt CA (2010) Automated Design of Synthetic Ribosome
Binding Sites to Precisely Control Protein Expression. Nat Biotechnol 27(10):946–950. 3. Espah Borujeni A, Channarasappa AS, Salis HM (2014) Translation rate is controlled by
coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42(4):2646–2659.
4. Lira LM, Vasilev D, Pilli RA, Wessjohann LA (2013) One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett 54(13):1690–1692.
5. Wang Y, Xu H, Jones MK, White RH (2015) Identification of the final two genes functioning in methanofuran biosynthesis in Methanocaldococcus jannaschii. J Bacteriol 197(17):2850–2858.
6. Srivastava S, Srivastava AK (2015) Lycopene; chemistry, biosynthesis, metabolism and degradation under various abiotic parameters. J Food Sci Technol 52(1):41–53.
7. Davis WB (1949) Preparation of Lycopene from Tomato Paste for Use as a Spectrophotometric Standard. Anal Chem 21(10):1226–1228.
8. Edgar S, et al. (2016) Mechanistic Insights into Taxadiene Epoxidation by Taxadiene-5α-Hydroxylase. ACS Chem Biol 11(2):460–469.
9. Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21(7):796–802.
10. Alonso-Gutierrez J, et al. (2013) Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng 19:33–41.
11. Cunningham FX, Lee H, Gantt E (2007) Carotenoid biosynthesis in the primitive red alga Cyanidioschyzon merolae. Eukaryot Cell 6(3):533–545.
12. Silva-Rocha R, et al. (2013) The Standard European Vector Architecture (SEVA): A coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 41(D1):666–675.
13. Lee TS, et al. (2011) BglBrick vectors and datasheets: A synthetic biology platform for gene expression. J Biol Eng 5(1):12.
14. Jiang Y, et al. (2015) Multigene editing in the Escherichia coli genome using the CRISPR-Cas9 system. Appl Environ Microbiol 81(7):2506–2514.
15. Ajikumar PK, et al. (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330(2010):70–74.
16. Zhou K, Qiao K, Edgar S, Stephanopoulos G (2015) Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat Biotechnol 33(4):377–383.
17. Santos CNS, Koffas M, Stephanopoulos G (2011) Optimization of a heterologous pathway for the production of flavonoids from glucose. Metab Eng 13(4):392–400.
















![Hirsutism (androgen excess) warda [compatibility mode]](https://img.dokumen.tips/doc/110x75/559d189d1a28ab64558b469c/hirsutism-androgen-excess-warda-compatibility-mode.jpg)

![Anemias during pregnancy warda [compatibility mode]](https://img.dokumen.tips/doc/110x75/554b248ab4c905da088b45c1/anemias-during-pregnancy-warda-compatibility-mode.jpg)










