Embed Size (px)
Citation preview

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 1
STUDY PROCEDURES MANUAL (SPM)
Proton Collaborative Group Protocol GU003-10
Phase III Study of Image Guided Radiation Therapy with or without Androgen Suppression for
Intermediate Risk Adenocarcinoma of the Prostate
Current Version: 2016Oct14
Revison History:
2015Apr16 2014Dec24 2012Feb17 2012Feb02 2012Jan05
Initial Issue: 2011Nov01
CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 2
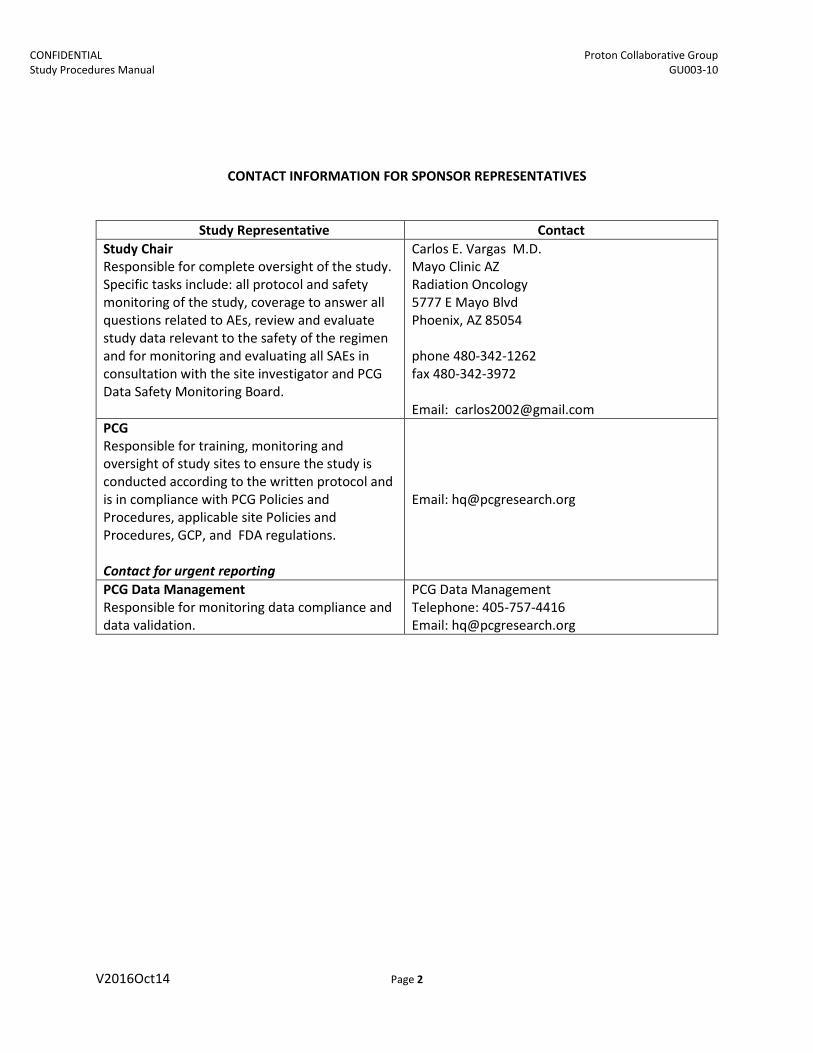
CONTACT INFORMATION FOR SPONSOR REPRESENTATIVES
Study Representative Contact Study Chair Responsible for complete oversight of the study. Specific tasks include: all protocol and safety monitoring of the study, coverage to answer all questions related to AEs, review and evaluate study data relevant to the safety of the regimen and for monitoring and evaluating all SAEs in consultation with the site investigator and PCG Data Safety Monitoring Board.
Carlos E. Vargas M.D. Mayo Clinic AZ Radiation Oncology 5777 E Mayo Blvd Phoenix, AZ 85054
phone 480-342-1262 fax 480-342-3972
Email: [email protected] PCG Responsible for training, monitoring and oversight of study sites to ensure the study is conducted according to the written protocol and is in compliance with PCG Policies and Procedures, applicable site Policies and Procedures, GCP, and FDA regulations.
Contact for urgent reporting
Email: [email protected]
PCG Data Management Responsible for monitoring data compliance and data validation.
PCG Data Management Telephone: 405-757-4416 Email: [email protected]

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 3
TABLE OF CONTENTS
1.0 GENERAL INFORMATION ............................................................................................. 4 2.0 ELIGIBILITY AND ENROLLMENT .................................................................................... 6 3.0 VISIT WINDOWS AND SCHEDULE OF TIME AND EVENTS ............................................... 8 4.0 PROTOCOL EXCEPTIONS AND DEVIATIONS .................................................................. 8 5.0 DATA SAFETY MONITORING ........................................................................................ 8 6.0 SUBJECT WITHDRAWAL AND LOST TO FOLLOW-UP GUIDELINES .................................. 9 7.0 TRANSFER SUBJECT TO ANOTHER FACILITY………………………………………………………………..10 8.0 DATA ENTRY.............................................................................................................. 10 9.0 ADVERSE EVENT AND UNANTICIPATED PROBLEM REPORTS...……………………………………12 10.0 CENTRAL REVIEW OF TREATMENT PLANS .................................................................. 13 11.0 NONCOMPLIANCE ..………………………………………………………………………………………………..…13
APPENDIX I - FORMS ……………………………………………………………………………………………………………14

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 4
1.0 GENERAL INFORMATION
1.1 OVERVIEW OF MANUAL
This document is confidential. It is to be used by PCG members only in conjunction with the PCG protocol. No other use or reproduction is authorized by the Study Chair nor does the Study Chair assume any responsibility for unauthorized use of this document.
This Study Procedures Manual (SPM) complements Protocol PCG GU003-10 by providing additional information and clarification about the following: how the clinical and administrative aspects of the study should be conducted to aid in ensuring compliance with the protocol, the principals of Good Clinical Practice (GCP), all applicable federal regulations and Proton Collaborative Group (PCG) requirements.
All individuals who are responsible for conducting Protocol PCG GU003-10 should refer to this SPM in conjunction with the protocol.
1.2 MANUAL REVISIONS
A copy of the original SPM will be issued to the study site at or prior to the site initiation. If revisions to the SPM become necessary, the revised SPM, with the revision date on the front cover will be distributed to the investigative site.
The investigative site study coordinator will be responsible for placing the revised SPM in the investigator’s study files and utilizing the current SPM.
1.3 REGULATORY PROCEDURES
This study must be conducted according to applicable regulations stated in title 21 of the United States Code of Federal Regulations (CFR) and the ICH Good Clinical Practice (GCP): Consolidated Guideline (ICH E6). Additional requirements are also outlined in protocol.
As specified in ICH E6, sections 4.9 and 8, the investigator is responsible for retaining all study-related documentation.
The study site’s regulatory documents must be current with PCG HQ prior to participation in this study. This study will be conducted only at institutions who are members of PCG. The respective local institutional review board (IRB), independent ethics committee (IEC), or central IRB must be used during this study to grant approval for research conduct at each site. The GU003-10 protocol has been approved as a multi-site study through Western IRB (WIRB). Sites that are not obligated to use a local IRB may submit the study to WIRB using their short form process. The study monitor can assist those sites with the WIRB submission but the site is still ultimately responsible for assuring compliance with all IRB related regulations and policies.
If needed per site or IRB requirements, a complete CRF template packet is available by contacting [email protected].

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 5
Samples of forms to meet regulatory requirements, such as a Delegation Log and Subject Identification List are available at: http://pcgresearch.org/login/. When utilizing these templates, the original should be maintained, and updates should be made to the original to maintain legibility of the document after multiple changes.
1.4 INVESTIGATOR RESPONSIBILITIES
The site Principal Investigator (PI) is responsible for all aspects of the clinical research protocol being conducted at their site. For those protocols that involve chemotherapy and/or surgical intervention, the PI must work closely with their designated medical oncologist or surgeon to assure protocol compliance and protection of human research subjects. It is highly recommended that the site PI host a meeting with designated surgeon(s) or medical oncologist(s) to review the protocol prior to enrollment of subjects.
Protocol changes or amendments that may affect the modality must be circulated to all participating surgeons and/or medical oncologists at the institution.
If patient protocol compliance appears to be a problem, or if the protocol regimen will not be followed by the attending physician, the patient should not be entered into a study. Problems with treatment compliance or surgical or medical oncology data submission will be referred to the site PI for resolution. Problems that persist will be referred to the Study Chair.
1.5 GENERAL RESEARCH CONSIDERATIONS/EXPECTATIONS
Revisions to paper or electronic documents (source, regulatory and/or CRF) must follow acceptable guidelines:
• Original electronic or paper documentation must be maintained.• Be sure the revisions are clearly identifiable in either paper or electronicrecords. On paper documents, use a single line to cross through the informationbeing changed and initialize the revision. You may circle the question or writeon top of the page specific items that have been revised.• Do not use whiteout or highlighter.
Laboratory results must be reported in US equivalents unless otherwise specified.
All source documents including required reports (e.g., pathology, surgery) must be in English.
1.6 MONITORING PROCEDURES
In order to assure data integrity for the GU003-10 protocol, sites will be monitored following the PCG clinical trial monitoring SOP by the study monitor or other applicable PCG staff.
Prior to the first subject enrollment, the study monitor or other assigned PCG staff will conduct a site initiation visit. This may be done in person or via teleconference. It is expected that the site research staff will have read the protocol and this SPM prior to

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 6
this meeting. The goal of this meeting is to assure the staff are qualified and prepared to comply with the protocol and all related study procedures.
Each site participating in this study will be subject to remote monitoring every three months. Monitoring will be conducted by a member of the PCG staff and will include the following: • A full source verification of 10% of all active study patients at that site• A 100% source verification for critical data as defined by the August 2013 Food and
Drug Administration Guidance for Industry: Oversight of Clinical Investigations- ARisk-Based Approach to Monitoring.
o Verification that informed consent was obtained properlyo Adherence to protocol eligibilityo Source verification and review of all SAEs and any grade 3 or higher AEo Source verification for any treatment failures or deaths
• Review of the site regulatory files• Review of any outstanding queries
Sites will also be subject to on-site monitoring or more frequent remote monitoring if issues are identified that could disrupt the integrity of the trial or compromise patient safety.
Sites will be notified by email at least one week in advance of each remote monitoring visit. Reports of all findings will be sent to the site’s study staff, including the Principal Investigator if requested, within one week of monitoring completion. The site will be responsible for addressing all findings prior to the start of the next monitoring visit for this study.
2.0 ELIGIBILITY AND ENROLLMENT
2.1 INFORMED CONSENT
Informed consent must be obtained from subjects before they can be enrolled in the study (and entered in Velos) as specified in 21 CFR 50 (Protection of Human Subjects) and ICH E6 Section 4.8. The subject must sign the IRB approved consent form. In the Food and Drug Administration Information Sheets dated September 1998, the following specifications are made and adopted by PCG as policy:
Procedures that are to be performed as part of the practice of medicine and which would be done whether or not study entry was contemplated, such as diagnosis or treatment of disease or medical condition, may be performed and the results subsequently used for determining study eligibility without first obtaining consent. On the other hand, informed consent must be obtained prior to initiation of any clinical screening procedure that is performed solely for the purpose of determining eligibility for research. When a doctor-patient relationship exists, prospective subjects may not realize that clinical tests performed solely for determining eligibility for research enrollment are not

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 7
required for their medical care. Physician-Investigators should take extra care to clarify with their patient-subjects why certain tests are being conducted.
In addition, in the United States each subject must sign and date the authorization for use and disclosure of Protected Health Information (PHI) under the Health Insurance Portability and Accountability Act (HIPAA) before the investigational site can transmit any PHI. Note that it is acceptable to combine the informed consent with the HIPAA authorization; in this case, only a single signature and date is required.
2.2 SUBJECT REGISTRATION
Refer to the Eligibility Worksheet (available either at our website (www.pcgresearch.org) or in Appendix I) for information required prior to patient registration. Once the investigator verifies a subject’s eligibility, the subject will be registered in the Proton Collaborative Group’s Electronic Data Capture (EDC) system. Please see the EDC Manual (section 1.4) for detailed instructions on registering a subject in the EDC system.
• Upon entering a new eligible subject into Velos eResearch, a unique patientidentification number (Pt Study ID) will be assigned. The first three digitsrepresent the site number. The last four digits are assigned sequentially forpatients enrolled at the site. The Pt Study ID (i.e. 002-0001) will be used for theremainder of the study to identify the enrolled subject.
• Once randomization questions are completed and submitted, an e-mail will besent to the site assigning the treatment arm. It is recommended that sitesmaintain all randomization e-mails for regulatory purposes.
• Notes:o Radiation treatment must begin within 56 days after randomization
for Arm 1 and 56 days after starting LHRH agonist treatment in arm 2.o Per protocol section 5.5, the first 3 treatment plans at each site must
be reviewed and approved prior to starting treatment. See section 10.0for additional information regarding treatment plan reviews.
2.3 INVESTIGATOR REFUSAL
An investigator should not enroll patients in a study unless s/he has reviewed the protocol and agrees to accept all of the study treatment options. Failure to accept an assigned option may result in suspension of the investigator from further enrollment in the study.
2.4 PATIENT REFUSAL
All treatment options in a study must be explained to potential study participants and the role of randomization explained. If a patient changes his mind and is unwilling to start or continue with the assigned option, PCG should be notified in writing. Please see section 6.0 for additional information on subject withdrawal.

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 8
3.0 VISIT WINDOWS AND SCHEDULE OF TIME AND EVENTS
The study will be conducted on an outpatient basis. The study must be conducted according to the schedule of time and events provided in the GU003-10 protocol. See protocol Section 4.0 for study specific patient assessments.
In order to assure subject safety and integrity of the study, follow-up visits should be completed per the Study Parameters table in the GU003-10 protocol (Section 4.1). However, PCG allows windows for the completion of follow up visits, as indicated below. If follow up visits are missed or completed outside of these windows, you will be required to complete a Deviation Report Form (see section 4.2). Please see the EDC Manual for instructions regarding how to enter a Missed or Out of Window visit.
• For the 3 and 6 month follow-ups, +/- 45 day window• For 12, 18, and 24 month follow-ups, +/-60 day window• For 36 month and greater follow ups, +/-90 day window
4.0 PROTOCOL EXCEPTIONS AND DEVIATIONS
4.1 PROTOCOL EXCEPTIONS
No protocol exceptions will be granted for this study. Contact the PCG Study Monitor for any questions regarding eligibility.
4.2 PROTOCOL DEVIATIONS
It is expected that the protocol will be followed without variation. However, if a protocol procedure is not completed according to the protocol, a Deviation Report Form must be completed upon discovery and emailed to [email protected] (form available at www.pcgesearch.org). All deviations will be reported to the Study Chair by the Study Monitor who will determine if further action is warranted.
If the site is aware that a deviation will occur, for example if a patient refuses to return to the office for follow-up but is willing to complete visit assessments by phone, the site can request that the Study Chair grant a planned deviation (deviation waver) Note to File (NTF). This will be kept in the site’s regulatory file as well in PCG’s Trial Master File. In certain circumstances, the subject may be changed to Follow-Up Off Protocol status after discussion with PCG staff (please see the EDC Manual for status details).
5.0 DATA SAFETY MONITORING
Data will be reviewed by the PCG Data Safety Monitoring Board, as required per the protocol. A summary of this report will be sent to participating sites for submission to their IRB, if required.

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 9
6.0 SUBJECT WITHDRAWAL AND LOST TO FOLLOW-UP GUIDELINES
6.1 EARLY WITHDRAWAL
All treatment options in a study must be explained to potential study participants and the role of randomization explained, if applicable. If a patient changes his/her mind and is unwilling to start or continue with the assigned option, notify the Lead Study Monitor and/or PCG headquarters (HQ) in writing. Instructions regarding collection of future data will be given on a case by case basis in collaboration with the Study Chair and Statistician.
Subjects may choose to withdraw from a study at any time. There are two scenarios for withdrawal: 1) withdrawal from study only or 2) withdrawal from study AND withdrawal of consent. The medical and study record must reflect the specific wishes of the subject and the following procedures should be followed:
a) Subject withdraws from study but does NOT withdraw consent. For example, thesubject may not want to continue prescribed treatment, he/she does not wish to followall protocol assessments, or he/she does not want to participate in follow-up visits.However, consent remains in place to allow for continued use of data collection and usefor research. If the patient decides to discontinue follow-up with the PCG investigator,this too is acceptable; however, a process to obtain information from other sourcesshould be discussed, i.e. release of information by other sources, etc. If this is notacceptable to the patient, the investigator should encourage the patient and requestpermission to submit survival status data. Although the patient has the right to refusesubmission of all data, she/he should be informed that failure to provide survival statusand information about treatment toxicity may adversely affect the study results. A“release of information” document may need to be signed by the patient. The policy andduration of the release may differ among institutions.
b) Patient withdraws from the study AND withdraws consent. This must be documentedin writing by the patient to the site PI. This document must then be forwarded to PCG HQ.Per regulations, information that has previously been collected may still be used but nonew information will be gathered or used.
6.2 LOST TO FOLLOW-UP
If absolutely no information is available regarding the status of the patient, a follow-up form should still be submitted per the protocol follow-up schedule listing all efforts made to obtain patient information. Examples:
o Call patient – if patient refuses to come to the clinic for a visit, conduct a phoneinterview to obtain as much information for the protocol as possible
o Send certified letter- requesting patient to contact the siteo Check Social Security Death Indexo Contact Emergency Contacto Contact other medical providers for information on patient statuso Contact local tumor registry for update on patient status

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 10
Note: If the only information available is that the patient is alive, this must be reported on the follow-up form noting the date and patient status. The remainder of the form can be filled out by marking Unknown.
In the event that there is no information regarding the patient status over a period of 2 consecutive years past the last missed follow up visit (and above attempts made at least every 6 months), the patient will be deemed “Lost to Follow-up” with the status being appropriately changed in the Electronic Data Capture System (EDC) to “Lost to F/U”. At this point no further follow-up forms will be expected but collected data will remain in the database and utilized for analysis. If the patient is located after being deemed “Lost to Follow-up”, complete a Follow-up Form with the information collected and contact PCG regarding revising status.
The principal investigator of an institution with frequent occurrences of lost patients may be requested to submit in writing a Corrective Action Plan to avoid additional lost to follow-up occurrences.
7.0 TRANSFER SUBJECT TO ANOTHER FACILITY
To transfer a protocol patient to another PCG institution with protocol approval, the investigator who originally enrolled the patient must submit a written request to the Study Monitor and/or PCG HQ via the Patient Transfer Form, available at www.pcgresearch.org.
Issues related to medical insurance are the responsibility of the investigators involved in the case transfer. All delinquent data through the date of transfer should be resolved before the transfer. Subsequent to case transfer, responsibility for all data requests and data submission is transferred to the recipient investigator including institutional requirements.
Transfer of cases from a PCG member institution to a member of a different cooperative group or transfer of a case from another cooperative group to a PCG member institution cannot be made.
8.0 DATA ENTRY
8.1 PCG ELECTRONIC DATA CAPTURE SYSTEM
For instructional guidance on how to log in, update password, register patients and enter data utilizing the PCG EDC System, please access the EDC Manual, which is available on the PCG website (www.pcgresearch.org).
8.1 QUALITY OF LIFE FORMS DATA
Quality of Life Forms (QoL) information will be collected and entered utilizing the PCG EDC system. The survey being used is The Expanded Prostate Cancer Index Composite (EPIC v 2.2002), which is available on the PCG website (www.pcgresearch.org). For sites utilizing VTOC, a pdf of the form can be uploaded into the Appendix. For sites not utilizing VTOC, responses need to be entered into the Patient Surveys section. Contact PCG headquarters (HQ) for further questions and details.

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 11
If the patient cannot complete the form on his own, he should be given assistance to complete the form electronically either at the time of his clinic visit or over the phone.
If the patient is unable to enter the information himself, staff may assist but should limit assistance as much as possible in order to avoid any bias. Patients should be instructed to read the brief directions at the top of the page. After it has been confirmed that the patient understands the directions, patients should be encouraged to answer all items, in order, without skipping any. If the patient feels an item does not apply to him, he should be encouraged to choose the response that is the most applicable. Patients should complete the questionnaires without coaching by staff. Once completed, research staff should verify that every item has been answered and remind the patient to answer any questions that were missed. If the patient declines to respond to any item, or the entire form, staff should document an explanatory response in the patient’s medical/source record.
It is important to note that the QoL forms are utilized to collect patient reported outcomes whereby physician and nurse assessments and documentation are a separate research data source. Staff should not try to cross reference the information given on the QoL forms with their own assessment of the patient’s condition. The answers on the QoL forms should not be used to “quiz” the patient but rather kept as confidential as possible in order to avoid influencing the patient responses. The information provided by the patient in the completed questionnaires is confidential and should not be discussed with, or shown to, anyone who is not a member of the study team.
8.3 DATA SUBMISSION SCHEDULE
The below electronic case reports forms must be completed for each patient within the timeframes indicated below via PCG’s EDC System. Data should be submitted in a timely manner to assure subject safety and meet high data integrity standards. As per PCG Policy, sites will be periodically evaluated for continued membership; timely data submissions are part of that evaluation.
Category Form Due for Completion Baseline Demographics Within 72 hours of study entry
OnStudy 1- Initial Diagnosis Within 2 weeks of study entry OnStudy 2 – Prior Treatment Within 2 weeks of study entry OnStudy 3 – Baseline Parameters Within 2 weeks of study entry
Adverse Event Log Adverse Events Within 2 weeks of treatment completion & 1 week of each follow up visit, if applicable
Treatment Summary Androgen Suppression Treatment Within 2 weeks of treatment completion Dosimetry Form 003 Within 2 weeks of treatment completion Meds at Prostate EOT Within 2 weeks of treatment completion Radiation Treatment Within 2 weeks of treatment completion
Follow-Up* Follow-up - Prostate Within 1 week of each follow-up visit Failure Failure Form – Prostate Within 2 weeks of event Patient Surveys EPIC form Within 2 weeks of study entry & 1 week of each
follow up visit, if applicable

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 12
* Patient follow-up visits will be completed as outlined in the protocol at 3 months;at 6 months and every 6 months x 3; yearly up to year 5; every 2 years thereafter.
In addition to the above eCRFs, paper forms may need to be completed during the study. These forms are referenced throughout this SPM in pertinent sections, and are available either at our website (www.pcgresearch.org) or in Appendix I.
9.0 ADVERSE EVENT AND UNANTICPATED PROBLEM REPORTS
9.1 ADVERSE EVENT REPORTING
All adverse event information related to protocol therapy will be collected in the EDC system according to protocol specifications. The severity of the AEs and SAES will be graded using the CTCAE v4 criteria.
For the GU003-10 protocol, only possibly, probably or definitely related (to radiation) adverse events will be collected. Sites are strongly encouraged to use the GU Study Adverse Events Assessment form (available either at our website (www.pcgresearch.org) or in Appendix I).
9.2 SERIOUS ADVERSE EVENT REPORTING
If the subject experiences a Serious Adverse Event as defined by the protocol, a Serious Adverse Event Report Form must be completed and submitted to PCG HQ via e-mail within one business day of when the investigator or site becomes aware of the event. The SAE form must be updated and resubmitted when any new information regarding the event is known.
In addition, adverse events meeting all of the following criteria must be reported to PCG by contacting PCG HQ within 1 business day of the time the investigator becomes aware of them:
• Unexpected (in terms of nature, severity, or frequency) given (a) the researchprocedures that are described in the protocol-related documents, such as the IRB-approved research protocol and informed consent document, or the InvestigatorBrochure; and (b) the characteristics of the subject population being studied; and
• Related or possibly related to participation in the research (possibly related meansthere is a reasonable possibility that the incident, experience, or outcome may havebeen caused by the drugs, devices or procedures involved in the research); and
• Suggests that the research places subjects or others at a greater risk of harm(including physical, psychological, economic, or social harm) than was previouslyknown or recognized.
The Study Monitor or other designated staff will determine additional steps and work with the site to complete the applicable documentation. As a reminder, sites are responsible for following the reporting requirements of AEs, SAEs, and unanticipated problems set forth per their own IRB policies and procedures since requirements may vary among IRBs. These reporting requirements may differ from those set forth by PCG.

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 13
9.3 UNANTICIPATED PROBLEMS
Reporting Unanticipated Problems that are not Adverse Events Investigators are to report Unanticipated Problems that fit the following criteria within 5 working days of becoming aware of them by contacting the Study Monitor or PCG HQ: • Unanticipated problems that do not fit the definition of an adverse event, but which
may, in the opinion of the investigator, involve risk to the subject, affect others inthe research study, or significantly impact the integrity of research data. Forexample, report occurrences of breaches of confidentiality, or accidentaldestruction of study records.
• Unplanned protocol deviations/violations that have already occurred, that mayadversely affect the rights, safety or welfare of subjects or the integrity of theresearch data, AND that meet the definition of an unanticipated problem (i.e., itinvolves risk to subjects or other people).
10.0 CENTRAL REVIEW OF TREATMENT PLANS
Per section 5.5 of the GU003-10 protocol, the first 3 cases enrolled into the study at each site must be reviewed by a PCG Medical Physicist, the site Dosimetrist who completed the treatment plan and the Study Chair or designee, prior to start of treatment. These reviews are typically performed locally or via web conference and arranged by the Study Monitor. However, if any treatment plan documents are submitted to PCG, they should be de-identified and the subject number indicated. The Dosimetry Form (available at www.pcgresearch.org and in Appendix I) should be completed to verify and document protocol compliance for treatment planning. This meeting, held at the earliest convenience of all parties, is to review the treatment plan, resolve issues and approve to begin treatment. Additional cases will undergo the same review process as determined by PCG.
Once the treatment plan is approved, the Dosimetry Form will be completed and signed by the site Dosimetrist, and provided to the Study Monitor, who will then obtain the signatures of the PCG Physicist and Study Chair. The PCG Monitor will return the completed Dosimetry Form to the site for their files to document the plan was approved and will maintain a copy for PCG files. Approval by the PCG Physicist representative and Study Chair or Designee will be documented.
The Dosimetry Form should be completed and provided to PCG for all subjects treated on PCG protocol GU003-10 (via uploading in the Velos Appendix). However, if a central review of the treatment plan is not required for that subject, the PCG Study Chair and PCG Physicist signatures are not required. The lack of a Dosimetry Form will be consider being non-compliant with the protocol. In the event of non-compliance, a Deviation Report Form will need to be sent to PCG HQ.
11.0 NONCOMPLIANCE Research sites that fail to comply with protocol, PCG policies/procedures, GCP and/or FDA regulations, may be subjected to develop and comply with a Corrective Action Plan to prevent future issues.
Continued or chronic noncompliance with protocol, policies/procedures, regulations or corrective action plans could result in study closure at the research center.

CONFIDENTIAL Proton Collaborative Group Study Procedures Manual GU003-10
V2016Oct14 Page 14
Appendix I – Forms
GU003-10 Eligibility Worksheet………………………………………………………….………………………………………………15
GU003-10 Dosimetry Form………………………………………………………………………………………………………………..17
GU Study Adverse Events Assessment……………………………………………………………………………………………….19

GU003-10 Eligibility Worksheet
Gu003-10 Eligibility Worksheet v5 Page 1 of 2
This form may be used as a tool when assessing patient eligibility and to assure all pretreatment evaluations are completed per protocol. This form is NOT a protocol requirement – it is for optional use. It does NOT replace referencing the current protocol as changes may be made that are not included here in a timely manner.
Subject Initials: Treating Investigator:
Form Completed By: Date Form Completed:
Inclusion Criteria
Yes
3.1.1 Histologically confirmed prostate adenocarcinoma( within 365 days prior to randomization) at intermediate risk for reoccurrence determined by at least one of the following:
Gleason Score 7 PSA 10- 20T stage T2b-T2c
Biopsy Date: Pathology Report Date:
Yes 3.1.2 Clinical stages T1-T2c N0 M0 (AJCC Criteria, 7th Ed.) as staged by treating investigator. T N M
Yes 3.1.3 Histological evaluation of prostate biopsy with assignment of Gleason score to the biopsy
material; Gleason Score must be in the range of 2-7. >6 cores are strongly recommended. Gleason Score:
Yes 3.1.4 PSA values < 20 ng/ml within 90 days prior to randomization. Obtained prior to biopsy or
at least 21 days after prostate biopsy. Date: Result:
Yes 3.1.5 ECOG performance status 0-1 (appendix I) assessed within 90 days prior to randomization. Date: Result:
Yes 3.1.6 Patients must sign IRB approved study specific informed consent. Date ICF Signed:
Yes
3.1.7 Patients must complete all required pre-entry tests listed in section 4.0 within the specified time frames.
History assessment with DRE Medication assessment Adverse Events assessment CTCAE v4 ( Baseline) CT or MRI pelvis (planning) must be done prior to treatment. CT or MRI pelvis (diagnostic) must be done prior to treatment. Fiducial marker placement must be done prior to treatment.
Yes 3.1.8 Patients must be able to start treatment within 56 days of randomization.
Page 15

GU003-10 Eligibility Worksheet
Gu003-10 Eligibility Worksheet v5 Page 2 of 2
Yes 3.1.9 Patients must be at least 18 years old. Date of Birth: Age:
Yes NA
3.1.10 For brachytherapy, an IPSS ≤ 21, or ≤ 17 if the patient is on medications to improve urination. EPIC/QOL completed within 90 days prior to randomization. Date: Result:
Yes NA
3.1.11 For brachytherapy, prostate volume must be less than 55cc prior to AS. Volume (cc):
Exclusion Criteria
No 3.2.1 Pelvic lymph nodes > 1.5cm in greatest dimension, unless the enlarged lymph node is biopsied and negative.
No 3.2.2 Previous prostate cancer surgery to include: prostatectomy, hyperthermia and cryosurgery.
No 3.2.3 Prior pelvic radiation for prostate cancer.
No 3.2.4 Prior androgen suppression therapy for prostate cancer.
No 3.2.5 Active rectal diverticulitis, Crohn’s disease affecting the rectum or ulcerative colitis (non-active diverticulitis and Crohn’s disease not affecting the rectum are allowed).
No 3.2.6 Prior systemic chemotherapy for prostate cancer.
No 3.2.7 History of proximal urethral structure requiring dilatation.
No 3.2.8 Current and continuing anticoagulation therapy with warfarin sodium (Coumadin),
heparin, low molecular weight heparin, Clopidogrel bisulfate (Plavix), or equivalent (unless it can be stopped to manage treatment related toxicity or to have a biopsy is needed).
No 3.2.9 Major medical, addictive or psychiatric illness which in the investigators opinion, will
prevent the consent process, completion of the treatment and/or interfere with follow-up. (Consent by legal authorized representative is not permitted for this study).
No 3.2.10 Evidence of any other cancer within the past 5 years AND < 50% probability of a 5-year
survival. Prior or concurrent diagnosis of basal cell or non-invasive squamous cell cancer of the skin is allowed.
No 3.2.11 History of myocardial infarction with the last 6 months.
Additional information: Central Treatment Plan review completed (if required, initial 3 cases from each institution) Documentation of consenting process that meets all applicable GCP requirements Bone scan (highly recommended) Urethrogram (recommended at time of CT Sim if no MRI is available) Start androgen suppression (for Arm II only, must start 8-10 weeks prior to the start of radiation therapy)
I certify that I have reviewed the eligibility and all questions have been answered accurately.
Investigator’s Signature: __________________________________ Date: ________________________
Page 16

Dosimetry Form
Dosimetry Form GU003-10 v2
GU003-10 Dosimetry Form
Patient Study ID # Institution# Date Form Complete
Treating Investigator
ARM I or II
Proton Therapy:
Structure Constraint (Major Deviation) Value
Rectum V44 <35% (V44 ≥ 40%) %
V60 <15% (V60 ≥ 20%) %
Bladder V71 <8cc (V71 ≥ 12cc) cc
Femoral heads V40 <1 cc (V40 ≥ 2cc) cc
PTV Min dose PTV * %
PTV eval PTV Coverage ** Gy (RBE)
IMRT/Photon Therapy Alone:
Structure Constraint (Major Deviation) Value
Rectum V70 <10% (V70 >20%) %
Max <V105% (V106 > 1 cc) cc
Bladder V70 <10% (V70 >20%) %
Max <V105% (V106 > 1 cc) cc
Femoral heads Max < V55 Gy (>V55 max point) Gy
PTV Min dose PTV ≥90% (Min dose PTV ≤90%) %
PTV V100 95-97% (V100 <94% or ≥100%) %
PTV V100 <110% (V100 >110%) %
IMRT/Photon Therapy and Brachytherapy:
Structure Constraint (Major Deviation) Value
Rectum V40 <10% (V40 >20%) %
Max <V105% (V106 > 1 cc) cc
Bladder V40 <10% (V40 > 20%) %
V105% <1cc (V106 > 1 cc) cc
Femoral heads Max < V55 Gy (>V55 max point) Gy
PTV Min dose PTV ≥90% (Min dose PTV ≤90%) %
PTV V100 95-97% (V100 <94% or ≥100%) %
PTV V100 <110% (V100 >110%) %
Page 17

Dosimetry Form
Dosimetry Form GU003-10 v2
Real Time LDR Brachytherapy:
Structure Constraint (Major Deviation) Value
Rectum V100 <1cc (V100 >2cc) cc
Prostate D90 >90% (D90<85%) %
Dosimetrist’s Signature: _________________________________________________________
PCG Physicist’s Signature: ________________________________________________________
Study Chair’s Signature: ___________________________________________________________
* Volume of the PTV receiving 95% of the prescribed dose (66.5 Gy(RBE)); Goal = 99.5% of PTV volume.** Minimum dose to 95% of the PTV eval volume; Goal = prescribed dose of 70 Gy (RBE).
Completed signed copies to be filed in patient’s medical record and PCG EDC database.
Page 18

GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
Please assess and record all adverse events previously reported or currently ongoing for study subjects. This form is provided as an optional worksheet tool and is NOT required by protocol.
Subject Name: Treatment Week :
Subject ID: -OR- Follow/up Period
Assessed by: Assessment Date:
ADVERSE EVENT ASSESSMENT *Instrumental ADL refers to preparing meals, shopping for groceries or clothes, using the telephone, managing money, etc. **Self-care ADL refers to bathing, dressing and undressing, feeding self, using the toilet, taking medications, and not bedridden.A semi-colon indicates ‘or’ within the description of the grade.
Fatigue Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Fatigue relieved by rest
2 Fatigue not relieved by rest; limiting instrumental ADL
3 Fatigue not relieved by rest, limiting self-care ADL
Bone Pain (HIP) Relationship to
Protocol Therapy 0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Mild pain
2 Moderate pain; limiting instrumental ADL
3 Severe pain; limiting self careADL
Anorexia Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Loss of appetite without alteration in eating habits
2 Oral intake altered without significant weight loss or malnutritional supplements indicated
3 Associated with significant weight loss or malnutrition (e.g., inadequate oral caloric and/or fluid intake); tube feeding or TPN indicated.
4 Life-threatening consequences; urgent intervention indicated
5 Death
Nausea Relationship to
Protocol Therapy
0 None Definitely related
Probably related 1 Loss of appetite alteration in eating habits
2 Oral intake decreased without significant weight loss, dehydration or malnutrition
Page 19
GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
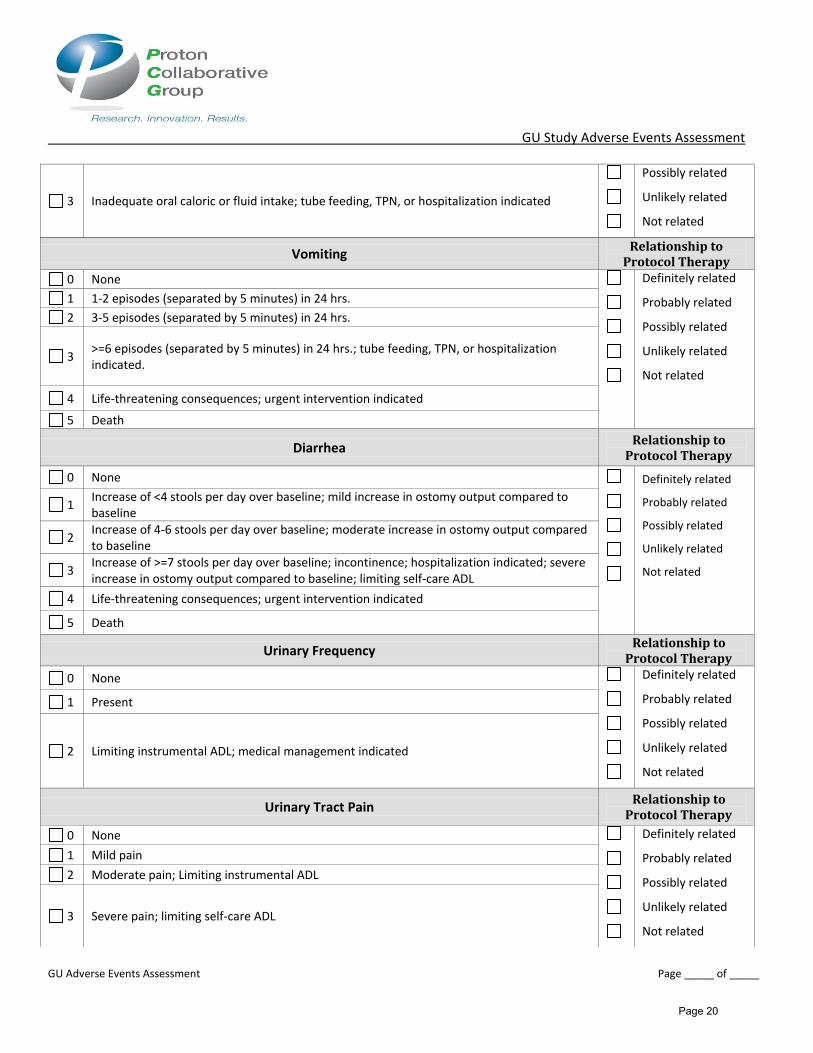
3 Inadequate oral caloric or fluid intake; tube feeding, TPN, or hospitalization indicated
Possibly related
Unlikely related
Not related
Vomiting Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 1-2 episodes (separated by 5 minutes) in 24 hrs.
2 3-5 episodes (separated by 5 minutes) in 24 hrs.
3 >=6 episodes (separated by 5 minutes) in 24 hrs.; tube feeding, TPN, or hospitalization indicated.
4 Life-threatening consequences; urgent intervention indicated
5 Death
Diarrhea Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Increase of <4 stools per day over baseline; mild increase in ostomy output compared to baseline
2 Increase of 4-6 stools per day over baseline; moderate increase in ostomy output compared to baseline
3 Increase of >=7 stools per day over baseline; incontinence; hospitalization indicated; severe increase in ostomy output compared to baseline; limiting self-care ADL
4 Life-threatening consequences; urgent intervention indicated
5 Death
Urinary Frequency Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Present
2 Limiting instrumental ADL; medical management indicated
Urinary Tract Pain Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Mild pain
2 Moderate pain; Limiting instrumental ADL
3 Severe pain; limiting self-care ADL
Page 20
GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
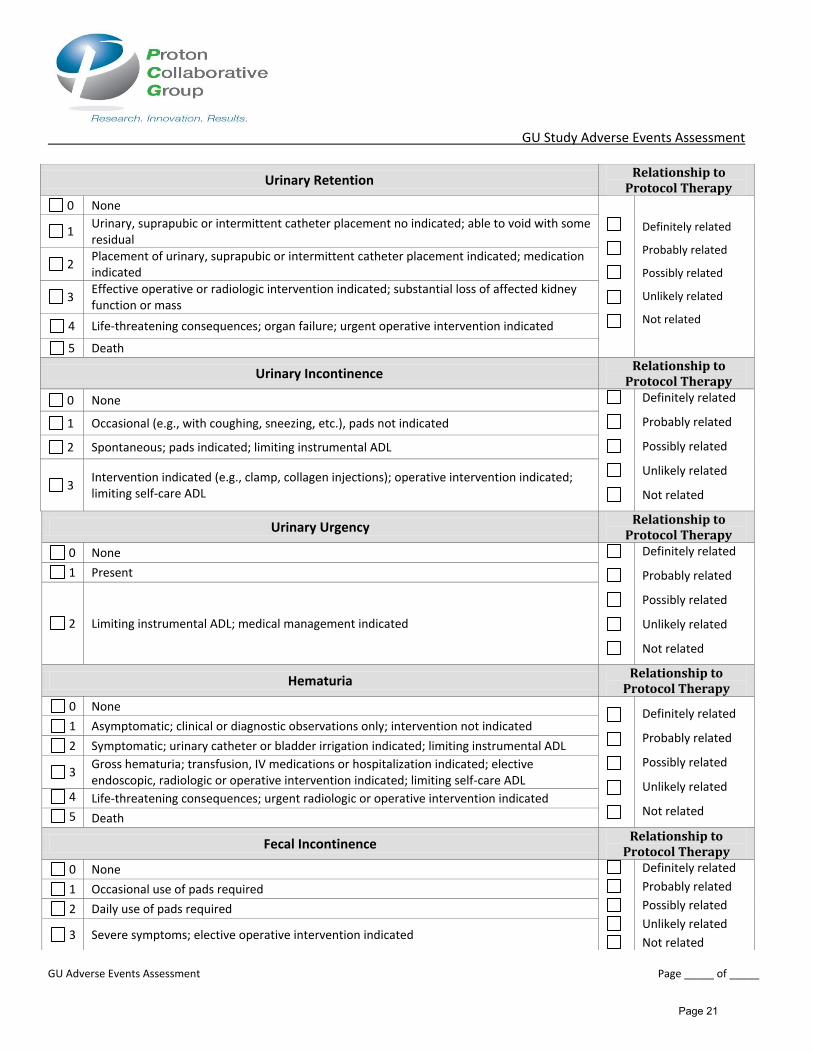
Urinary Retention Relationship to
Protocol Therapy
0 None
Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Urinary, suprapubic or intermittent catheter placement no indicated; able to void with some residual
2 Placement of urinary, suprapubic or intermittent catheter placement indicated; medication indicated
3 Effective operative or radiologic intervention indicated; substantial loss of affected kidney function or mass
4 Life-threatening consequences; organ failure; urgent operative intervention indicated
5 Death
Urinary Incontinence Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Occasional (e.g., with coughing, sneezing, etc.), pads not indicated
2 Spontaneous; pads indicated; limiting instrumental ADL
3 Intervention indicated (e.g., clamp, collagen injections); operative intervention indicated; limiting self-care ADL
Urinary Urgency Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Present
2 Limiting instrumental ADL; medical management indicated
Hematuria Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Asymptomatic; clinical or diagnostic observations only; intervention not indicated
2 Symptomatic; urinary catheter or bladder irrigation indicated; limiting instrumental ADL
3 Gross hematuria; transfusion, IV medications or hospitalization indicated; elective endoscopic, radiologic or operative intervention indicated; limiting self-care ADL
4 Life-threatening consequences; urgent radiologic or operative intervention indicated
5 Death
Fecal Incontinence Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Occasional use of pads required
2 Daily use of pads required
3 Severe symptoms; elective operative intervention indicated
Page 21

GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
SIGNATURES
Assessed by (Signature) Date
Rectal Hemorrhage Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Mild; intervention not indicated
2 Moderate symptoms; medical intervention or minor cauterization indication
3 Transfusion, radiologic, endoscopic, or elective operative intervention indicated
4 Life-threatening consequences; urgent intervention indicated
5 Death
Dermatitis Radiation Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Faint erythema or dry desquamation
2 Moderate to brisk erythema; patchy moist desquamation, mostly confined to skin folds and creases; moderate edema
3 Moist desquamation in areas other than skin folds and creases; bleeding induced by minor trauma or abrasion
4 Life-threatening consequences; skin necrosis or ulceration of full thickness dermis; spontaneous bleeding from involved site; skin graft indicated
5 Death
Erectile Dysfunction Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Decrease in erectile function (frequency or rigidity of erections) but intervention not indicated (e.g., medication or use of mechanical device penile pump
2 Decrease in erectile function (frequency/rigidity of erections), erectile intervention indicated, (e.g., medication or mechanical devices such as penile pump)
3 Decrease in erectile function (frequency/rigidity of erections) but erectile intervention not helpful (e.g., medication or mechanical devices such as penile pump); placement of permanent penile prosthesis indicated (not previously present)
Proctitis Relationship to
Protocol Therapy
0 None Definitely related
Probably related
Possibly related
Unlikely related
Not related
1 Rectal discomfort, intervention not indicated
2 Symptoms (e.g., rectal discomfort, passing blood or mucous); medical intervention indicated; limiting instrumental ADL
3 Severe symptoms; fecal urgency or stool incontinence; limiting self-care ADL
4 Life-threatening consequences; urgent intervention indicated
5 Death
Page 22

GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
I have reviewed and approved the above assessments.
Investigator Signature Date
Use this page to assess and record additional adverse events not specifically listed.
Subject Name: Treatment Week :
Subject Study ID: -OR- Follow/up Period
Assessed by: Assessment Date:
OTHER AEs NOT SPECIFCALLY LISTED Relationship to
Radiation Therapy CTCAE
v4 Grade
CTCAEv4 Term Description (i.e. fever, allergic reaction etc.)
Definitely related Probably related Possibly related Unlikely related Not related
Definitely related Probably related Possibly related Unlikely related Not related
Definitely related Probably related Possibly related Unlikely related Not related
Definitely related Probably related Possibly related Unlikely related Not related
Definitely related Probably related Possibly related Unlikely related Not related
Definitely related Probably related Possibly related Unlikely related Not related
Page 23

GU Study Adverse Events Assessment
GU Adverse Events Assessment Page _____ of _____
SIGNATURES
Assessed by (Signature) Date
I have reviewed and approved the above assessments.
Investigator Signature Date
Page 24





























