Embed Size (px)
Citation preview

Studies on Fluorinated PyrimidinesXVI. Metabolism of 5-Fluorouracil-2-C4 and 5-Fluoro-2'-
deoxyuridine-2-Cu in Cancer Patients*
KANAILAL MuKHERjEE,tJUDITHBOOHAR,DOROTHYWENTLAND,FREDJ. ANSFIELD,ANDCHARLESHEIDELBERGER^
(McArdle Memorial Laboratory, University of Wisconsin, Madison, Wisconsin)
SUMMARY5-Fluorouracil-2-C14 (FU) and 5-fluoro-2'-deoxyuridine-2-C14 (FUDR) were admin
istered to cancer patients by oral, intravenous, intramuscular, and intraperitonealroutes. The radioactivity in the plasma, respiratory CCh, and urine was measured, andfractionation of radioactive materials on ion-exchange columns into the unchangeddrug and the various degradation products was carried out with urines and some acid-soluble extracts of plasma. The level of radioactivity in the plasma was higher for thefirst 2 hours after intravenous injections of FU-2-C14 and FUDR-2-C14 than after administration by any other route. The unchanged drugs persisted longer in urine afterintravenous injections than after oral, intramuscular, or intraperitoneal administrations. The excretion of radioactivity as respiratory CÛ2was least after the intravenousinjection of FUDR. It was concluded, therefore, that intravenous administration ofboth drugs is the method of choice in the clinical use of these drugs. There was moredegradation of FU injected as a continuous infusion than following rapid intravenousinjection, and the opposite was true for FUDR.
One patient, who tolerated large doses of FUDR without toxic manifestations, degraded the drug rapidly ; whereas a second patient, who rapidly became toxic to FUDR,degraded the drug at a much slower rate. When the degradation of 3',5'-diacetyl-5-fluoro-2'-deoxyrudine-2-C14 and FUDR-2-C14, both given orally to the same patient,was compared, it was found that 5'-monoacetyl-FUDR was present in the urine 24 hours
after administration of the diacetyl compound, whereas no unchanged drug was foundin the urine after administration of FUDR. After intravenous injections of FU-2-C14and FUDR-2-C14, radioactivity was found in the cerebrospinal fluid. Fractionationof the radioactive compounds demonstrated the presence of unchanged drug in thecerebrospinal fluid.
The metabolism of 5-fluorouracill and 5-fluoro- the catabolic pathway has been shown to involve2'-deoxyuridine has been worked out in mice, and degradation of the compounds through DHFU,
* This investigationwassupported in part by grant, T-157, FUPA, FGPA, Urea, Carbon dioxide, and FBAL,from the AmericanCancer Society. as shown in Chart 1 (4). The anabolism of these
t Fellow of the Damon Runyon Memorial Fund for Cancer compounds proceeds through FUR to FURP andResearch- thence to FUDRP, or, in the case of FUDR, di-
t American Cancer Society Professorof Oncology. ^ t(J FUDRp (Chart j). FUDRp has been'Abbreviations used in this paper are as follows: rU = . 1111 i -11 i
5-fluorouracil; FUDR = 5-fluoro-2'-deoxyuridine;DHFU = shown to block the enzyme thymidylate synthe-5,6-dihydro-S-fluorouracil;FUPA = o-fluoro-(3-ureidopropion- tase (9), which catalyzes the methylation of deoxy-ic acid; FGPA = a-fluoro-/3-guanidopropionicacid; FBAL = uridylic acid to thymidylic acid. The blockage ofa-fluoro-/3-alanine; FUR = 5-fluorouridine; FURP = 5-flu- ., . . • i j Ì•u-u-o.- t r»vrt iU •orouridine-S'-monophosphate; FUDRP = 5-fluoro-2'-deoxy- thls ^action leads to inhibition of DNA synthesis,uridine-5'-monophosphate; UDRP=2'-deoxyuridine-5'-mono- and, therefore, this effect is considered to be of pri-phosphate;TDRP = thyinidine-S'-monophosphate. mary importance in the carcinostatic action of
Receivedfor publication June il, lose. these compounds (16). The degradative products
49
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

50 Cancer Research Vol. 23, January 1963
of fluorinated pyrimidines are inactive as carci-nostatic agents.
FU and FUDR are currently undergoing extensive clinical trial as tumor-inhibitory compounds in patients suffering from various kinds ofcancer. Evidence is steadily accumulating that inpatients, particularly with carcinomas of thebreast and colon, both drugs produce clinical improvement (2) in about one-third of cases (19).FUDR presents a definite clinical advantage overFU in the treatment of patients with cancer of therectum or colon, whereas in patients with carcinoma of the breast both drugs are effective to the
times the type of response induced by a drug (6).Thus, it has been shown by Heidelberger et al.(11) that FU was much less effective against Sarcoma 180 in mice when it was administered by thestomach tube than when it was given intraperi-toneally. It was, therefore, considered desirableto study the metabolism of FU and FUDR inhuman patients with various kinds of cancer whenthe drugs were administered by different routes.The present paper describes the absorption, excretion, and degradation of FU-2-C14 and FUDR-2-Cu in cancer patients by oral, intravenous, intramuscular, and intraperitoneal administrations.
OH2N-CH2CHF COOH + H^CNHg +
FBAL*
UREA
o i. HgN-C-NH-CHg CHF COOH •
VvPUPA
CHF COOH
iOF•FURP-
•FUDRP
F-RNA
•FURPPP
UDRP
P
TTP
DMA
CIIAKT1.-—Metabolismof 5-fluorouracil
same extent (1). In mice, Heidelberger et al. (10)had found FUDR to be more effective againstsome transplanted tumors and less toxic than FU.It is desirable, therefore, to ascertain why certaintypes of cancer respond to either FU or FUDRand others do not, and also why only a certain proportion of patients suffering from the same type ofcancer derived benefit from the use of the drug. Aprobable reason for differential responses of cancerpatients to FU and FUDR may be the extent andrapidity of degradation of the compounds in individual cases. It appeared necessary, therefore,to study the degradation of FU and FUDR inpatients with various kinds of cancer.
It is well known that the route of administrationoften modifies the extent, duration, and some-
Lemon (13) reported that the toxicity from theusual chemotherapeutic dose of FU (15 mg/kg)was markedly reduced in patients who were giventhe drug by continuous intravenous infusion,whereas Sullivan et al. (20) reported that the toxicity of FUDR was greatly increased when administered by continuous infusion. Thus, the presentpaper also reports on the degradation of FU-2-C14and FUDR-2-C14 in cancer patients when thedrugs were given by a continuous intravenous infusion.
Many compounds when injected intravenouslyfail to appear in the cerebrospinal fluid (7). Thishas been attributed to the existence of a blood-brain barrier, which prevents the free equilibration of compounds between systemic and intra-
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

MuKHERJEE et al.—Metabolism of FU and FU DR in Patients 51
cerebral circulation. It was considered desirableto discover whether such a barrier existed in thecase of FU or FUDR injected intravenously. Consequently, the present paper reports observationson the levels of radioactivity in various samples ofcerebrospinal fluid and blood of patients giveninjections of FU-2-C14 or FUDR-2-C14 intravenously.
MATERIALS AND METHODSPatients suffering from various kinds of cancer,
and whose life expectancies were presumed not toexceed a few years, served as volunteers for thesetests. It was explained to them that the amount ofradioactivity they would receive during the testswould not be harmful. Usually the patients wereadmitted to the hospital for the period of investigation. In some cases the patients went home afterthe first 12 hours and collected their urine according to instructions.
The mode of administration of the drugs variedin different cases. The single intravenous dose wasinjected through a No. 22 needle. The continuousintravenous infusion was given by the drip method. The intramuscular doses were administeredin the gluteal region, and the oral doses were givenafter breakfast, usually dissolved in lemonade or incapsules. The drugs were injected intraperitoneallyin patients who had ascites from peritoneal involvement by the tumor; the fluid was drained, and afterthe site of puncture was sealed the drugs were injected in the peritoneal cavity above the level oftapping. The patients had their usual diets and remained ambulatory during the tests.
Venous blood was collected under aseptic conditions through an indwelling Rochester needle;after each withdrawal of blood the stylet of theneedle was moistened with a drop of sterile hepa-rin (1 in 10,000 solution) to prevent clotting. Thesamples of respiratory air were collected inDouglas bags. The urines were collected in separate bottles under toluene as voided for the first24 hours and in a single bottle for the next 24hours. The cerebrospinal fluids were obtainedeither by repeated lumbar punctures or by meansof a catheter introduced through the lumbar puncture needle. No stools were collected, since no significant amount of the drug or its metabolites hadbeen found in the stools in our previous studies(4). The blood was allowed to clot, and the serumwas separated by centrifugation. A sample of theserum was plated on aluminum planchets under acurrent of cold air for the determination of radioactivity. In the earlier part of these investigationsblood was collected in oxalated tubes, and plasmawas separated and used for determination of radio
activity and fractionation of acid-soluble extracton Dowex-1 (formate). In the present studies, theterms serum and plasma are used synonymously.The remaining serum was cooled in ice, andperchloric acid was added to a final concentrationof 4 per cent. The precipitated protein was centri-fuged in the cold, and the supernatant fraction wascollected. The protein was washed with an equalvolume of 4 per cent perchloric acid, and the combined acid-soluble extracts were neutralized with20 per cent KOH and left in the cold overnight.The precipitated KC1Ü4was centrifuged, and analiquot of the supernatant fraction was platedand its radioactivity was determined. If the totalradioactivity exceeded 1000 counts/min, the extracts were fractionated on Dowex-1 (formate)columns.
The urine volumes were measured, and a samplewas diluted and plated and the radioactivity wasdetermined. Samples of urine containing 40,000-50,000 counts/min were brought to pH 11. Almostall the urine showed some flocculent precipitate,which was removed by centrifugation, and the supernatant fraction was used for the column separations. The Dowex-1 (formate) column fractionation of the metabolites of FU in the urines andserum acid-soluble extracts was carried out by theprocedure described previously (4). The metabolites of FU or FUDR that were separated on thecolumns were urea, FU or FUDR, FUTA, FGPA,and nucleotides of FU. The urea was eluted fromthe column with water at pH 8, FU or FUDRwith 0.05 M formic acid, and FUTA with 1.5 Mformic acid. The nucleotides were eluted with0.5 N HC1. The FGPA formed a second peaktoward the latter part of the 0.05 Mformate eluate.A sample from each tube was plated, and theradioactivity was measured to locate the peaks indifferent fractions. The contents of the tubesshowing the radioactive peaks were pooled andplated, and the radioactivity was determined ineach fraction. The percentages of urea, FU orFUDR, FUPA, and the nucleotides in the originalspecimen were then calculated. In the patientstreated with FU, the 0.05 M formic acid fractiondid not contain any detectable amount of FUDR;however, in the patients treated with FUDR, the0.05 Mformic acid fraction contained both FU andFUDR. In the latter case this fraction was ly-ophilized and acetylated according to the methodof Kaldor and Heidelberger (12), and FU andactylated FUDR were separated by paper chro-matography. The identity of the individual compounds obtained by fractionation on the ion-exchange column was frequently checked by paperchr omatography.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

52 Cancer Research Vol. 23, January 1963
The C14O2in the respiratory air, collected at different time intervals, was precipitated as bariumcarbonate. In our previous studies (4) we had usedthe basal metabolic rate to calculate the total COzexpired in a given amount of time. However, itwas soon found that such a calculation did notgive an accurate estimation of total CCs The procedure employed in the present studies for measurement of total CÛ2expired per minute was asfollows. The patient was asked to relax andbreathe in through the nose and out through themouth into the mouthpiece of the Douglas bag.After the patient became accustomed to the procedure, respiratory air was collected in the Douglas bag for exactly 2 minutes. Two hundred cc. ofthe respiratory air was introduced into a calibrated volumetric gas buret by displacement ofmercury. This volume of air was then bubbledthrough carbonate-free NaOH, and the COz wasprecipitated as barium carbonate. The procedurewas done in duplicate. The remaining air in theDouglas bag was quantitatively transferred bywater displacement to a chain-compensated gasometer (manufactured by Warren E. Collins, Boston, Mass.). The total volume of air was thencalculated from the displacement of the drum, andto it was added the 400 cc. which was taken out ofthe bag and another 300 cc. of dead space air.Thus from the total volume of air breathed overthe 2-minute period and the weight of the bariumcarbonate given by the C02 in 400 cc. of air, thetotal volume of CO2expired in 1 minute was calculated. The average specific activity of CÛ2 incounts/min/mg of barium carbonate was calculated from the average of the counts obtained intwo succeeding periods; it was then multiplied byweight of the barium carbonate that was equivalent to the total CO2 expired during the intervalbetween the samples. Thus the total amount ofC14Oathat was excreted during the appropriatetime interval was calculated.
The radioactivity was usually measured inwindowless gas-flow counters and corrected forself-absorption of barium carbonate or the dryweights of the sera. In some specimens the radioactivity was too low to be measured with thesecounters, and, therefore, they were determined in aguarded automatic counter (Nuclear Chicago)which has a background of 1.5 counts/min. Thespecimens were counted for a minimum of 600counts. Sometimes the samples containing ureawere too hygroscopic to be measured in the gas-flow counters; so a sample was dissolved in 10 ml.of a mixture containing 100 gm. of naphthalene,10 gm. of 2,5-diphenyloxazole, 250 mg. of 2,2'-paraphenylene-bis-5-phenyloxazoIe, and 1000 ml.
of dioxane and determined in the Tri-Carb Scintillation counter.
FU and FUDR were administered in the usualchemotherapeutic doses—i.e., 15 mg/kg for FU,up to a total of 1 gm. of FU, and 30 mg/kg forFUDR, up to a total of 2 gm. of FUDR. Theradioactive FU and FUDR were transferredaseptically from sterile solutions with a pipetteinto the corresponding solution containing non-radioactive FU or FUDR. Usually 100-200 ¿ic.ofFU-2-C14 or FUDR-2-C14 were administered.
Preparation of 5-fluoro-3',5'-diacetyl-S'-deoxyuri-dine (diacetyl-FUDR)-2-Clt.—To about 2 mg. ofFUDR-2-C14 containing 200 X IO6 counts/minwere added 0.5 ml. of freshly distilled aceticanhydride and a drop of pyridine, and the solutionwas shaken at 50°C. for 15 minutes. The acetic
anhydride was removed by distillation in vacuo,and the acetylated material was spotted on stripsof Whatman No. 1 filter paper. The paper chro-matogram was developed with the upper phase (8)of ethyl acetate :formic acid: water (60:5:35) andthen was scanned through the automatic stripscanner (14). There was a single radioactive peakwith an RF of 0.94, which corresponded exactlywith authentic nonradioactive diacetyl-FUDR.FUDR in this system has an RF of 0.6. To the required amount of nonradioactive diacetyl-FUDR(30 mg/kg) was added the radioactive diacetyl-FUDR (94 X IO6 counts/min in one patient,R.L., and 42 X IO6counts/min in another, A.I.),and an intimate mixture was made with a pestleand mortar. The drug was then put in five or sixcapsules. FUDR-2-C14 was similarly mixed withnonradioactive FUDR and put in capsules. Thepatients swallowed the capsules containing eitherFUDR or diacetyl-FUDR with warm coffee afterbreakfast.
FUDR-2-C14 was prepared enzymatically byDr. D. B. Koechlin of Hoffman-LaRoche fromFU-2-C14 purchased from California Foundationfor Biochemical Research. FU-2-C14 was alsokindly supplied by Dr. Koechlin of Hoffman-LaRoche.
RESULTS
Levels of radioactivity in the plasma after administration of FU-2-Cu and FUDR-2-Cli.—Five-mi, samples of blood were collected 10, 20, 30, and45 minutes and l, 1J, 2, 4, 8, 12, 24, and 48 hoursafter administration of the drug. The plasma orserum was separated, and a sample was plated fordetermination of radioactivity. The radioactivitieswere then expressed as /ig. of drug equivalent perml. of plasma by dividing the counts per minuteper ml. by the specific activity of the drug. The
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

MuKHERJEEet al.—Metabolism of FU and FUDR in Patients 53
acid-soluble extracts of most of the samples ofplasma did not contain enough radioactivity topermit separation on Dowex-1 (formate) columns.In the specimens with which such separation couldbe carried out it was found that the radioactivitywas present mostly as one or more of the degradation products. Although it was recognized that theproportion of unchanged FU-2-C14 or FUDR-2-C14 was not known in most of the samples, forconvenience the radioactivity was neverthelessexpressed as ng. drug equivalent per ml. When thelevels in the plasma of such drug equivalents afteroral, intraperitoneal, intramuscular, and intravenous administrations of FU-2-C14 were plottedagainst the log of time, the results shown inCharts 2-4 were obtained. Two patients werestudied with each mode of administration. Thedata show that the radioactivity was highest between \ and 1 hour after intramuscular, intraperitoneal, and oral routes, and thereafter itgradually decreased. After the intravenous injections, the level of radioactivity in the plasma declined in almost a straight line on a log-log plot.Four hours after administration of the drug theamounts of radioactivity in the plasma were almost the same after all modes of administration.There was considerable individual variation in thelevels of radioactivity between the two patientsinvestigated with each route of administrationexcept with the intravenous route, in which caseboth patients had comparable levels of drugequivalent in the plasma.
When the pg. drug equivalents per ml. ofplasma after the administration of FUDR-2-C14 byoral, intramuscular, intraperitoneal, and intravenous routes were plotted in the same way, theresults shown in Charts 5 and 6 were obtained. Thehighest level of radioactivity in the two oral andintramuscular cases and in one intraperitonealcase was reached in 1-1 \ hours, after which theradioactivity gradually declined. In one patientgiven the drug intraperitoneally the radioactivityin the plasma increased up to 10 hours and thendecreased. Three patients were studied by theintravenous route of administration. In all threepatients the radioactivity in the plasma declinedin a straight line, and the plasma samples hadcomparable radioactivities. The radioactivity washighest after intravenous administrations of bothFU-2-C14 and FUDR-2-C14 for the first 2 hours.
In most of the patients studied, the total radioactivity in the acid-soluble extract of the plasmadid not exceed 1000 counts/min, so that fractiona-tion of the radioactive materials on Dowex-1(formate) could not be undertaken. However, itwas possible to carry out the fractionations in
I£
JO
S'
10-
05
01
«INTRAMUSCULARO INTRAMUSCULARA INTRAFERITONEAL«INTRAPERITONEAL
0.5 1.0 10 24
HOURS
CHART2
I
50
ID
S'
1.0-
0.5-
0.1 0.5 1.0 4
HOURS
10 24
CHABTS
5Q
IO
5-
10
05-
0.10.5 IO
HOURS
IO 24 OO
CHABT4
CHARTS2—4.—Levelsof plasma drug equivalent after administration of FU-2-C14.Chart 8, oral; Chart 4, intravenous.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

54 Cancer Research Vol. 23, January 1963
plasmas obtained from three patients after oral,intravenous, and intraperitoneal administrationsof FU-2-C14, and the results are shown in Chart 7.The solid lines show the amounts of FU in theacid-soluble extracts of the plasma. The patientwho had been given injections intravenously wasfound to have the greatest amount of the unchanged drug in the 1-hour specimen of plasma(50 per cent as compared with 28 per cent afterthe oral and 18 per cent after the intraperitonealadministration). The plasma urea and FUPA,which are catabolic products of FU, were foundto be proportionally less following the intravenousadministration than by oral and intraperitonealroutes (48 per cent as compared with 70 per cent
PLASMA LEVEL OF RADIOACTIVITY,
FUDR NTRAVENOUS
SC
IO-
5-
10-
Q5-
OJ
and 80 per cent after the oral and intraperitonealadministrations, respectively, in the 1-hour specimens). We do not have corresponding data withFUDR-2-C14.
Excretion of CU02 after administrations of FU-2-C"4 and FUDR-2-C1*.—Samples of respiratoryair were collected at the same times at which thespecimens of blood were withdrawn. The CÛ2ofthe respiratory air was precipitated as BaCO3, theradioactivity of which was determined. When thespecific activity of respiratory COj as counts/min/mg of BaCOs was plotted against time on asemi-log plot, the results shown in Charts 8 and 9were obtained after the administration of FU-2-C14. The specific activity was highest between §
PLASMA LEVEL OF RADIOACTIVITY, FUDR
O-
5\ oORAL
05 IO 4 IO 24
0 0.5 IO 4 IO 24 100HOURS
Caini
CHART6CHARTS5, 6.—Levelsof radioactivity in the plasma after administration of FUDR-2-C14 by various routes
fiCD
300
250
200
150
100
SO
SPECIFIC ACTIVITY OFRESPIRATORY .CO2
FU INTRAMUSCULAR
O-OFU0-0 »
INTRAPERITONEAL
Ql 0.5 1 2 4HOURS
8 12 24 40 100
CHA*T7.-Fractionation of the acid-soluble extract ofplasma on Dowex-1 (formate) after administration of FU-2-C'4.
and 1 hour after oral, intravenous, intramuscular,and intraperitoneal administrations of the drug.There was considerable individual variation in thelevels of the specific activity of C14O2between thetwo patients receiving the drug by the same route.When the total amount of respiratory C14O2,in percent of the amount of radioactivity of the totaldose, was plotted against the log of time, the results shown in Charts 10 and 11 were obtained.When administered by the intravenous and oralroutes, 60-90 per cent of FU was excreted as COzin 12 hours. When the drug was injected intramuscularly 40-50 per cent was excreted as CÜ2inthe same period of time. In the patients to whomFU-2-C14 was given intraperitoneally there was
considerable variation in the excretion of the drugas CO2. One patient excreted 25 per cent and an-
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

et al.—Metabolismof FU and FUDR in Patients 55
other 84 per cent of the administered drug as CÛ2in 12 hours.
The specific activities of respiratory COi afterintravenous, oral, intramuscular, and intraperi-toneal administrations of FUDR-2-C14 are shown
in Charts 12 and 13. After oral, intravenous, andintramuscular doses of FUDR-2-C14, the specific
activity was highest between 1 and 2 hours. Whenthe drug was injected intraperitoneally the specificactivity reached the highest level If hours afterinjection of the drug in one patient and 8 hoursafter administration of the drug in the other. Thetotal excretion of C14U2 in percentage of the dose
administered by different routes is shown in Charts14-16. It was found that, when FUDR-2-C14 was
injected intravenously, 38, 44, and 63 per cent of
SPECIFIC ACTIVITY OFRESPIRATORY CO,
300
250
200
150
100
50
FU INTRAMUSCULAR
0-OFU INTRAPERITONEALO-O -
CI 0.5 i 4HOURS
CHART8
300
260
200
ISO
100
50
SPECIFIC ACTIVITY OFRESPIRATORY
)t
8 12 24 4O 100
FU INTRAVENOUS
O—O FU ORAL
O—O •'
0.5 2 4HOURS
CHART9
8 12 24 40 100
CHARTS8, 9.—Specificactivity of respiratory CO2 afteradministration of FUDR-2-C14 by various routes.
the administered drug was excreted as CÛ2in threeindividual patients. After oral and intramuscularadministrations, 58-95 per cent of FUDR appeared in the respiratory CO2. When FUDR-2-C14was injected intraperitoneally, 34 per cent wasexcreted as CO-¡in one patient and 65 per cent inthe other.
Excretion of radioactivity in urine after administration ofFU-2-Cli and FUDR-2-C™.—Samples of
urine were collected in separate bottles as voidedduring the first 24 hours and then in a single bottlefor the next 24 hours. The total amounts of radioactivity in the individual specimens were measuredand plotted against the log of time. In Charts 17 and18 are shown the results after administration of FU-2-C14 by different routes. The total radioactivity
in urine in per cent of the administered dose
TOTAL EXCRETIONIN RESPIRATORY C0?x ORALO ORA
DO
83
60
40.
20-
0
AMUSCULARv INTRAMUSCULAR
24 40 100
CHAUT10
TOTAL EXCRETION IN RESPIRATORY CO
Q!} INTRAVENOUS
'D-INTRAPERITONEAL100-
80
60
4O
2O
I 2 4HOURS
CHABT11
CHARTS10, 11.—Totalexcretion of respiratory C'H^ afteradministration of FU-2-C" by various routes.
reached 7-14 per cent in 48 hours after adminis
tration of FU by oral, intramuscular, intravenous,and intraperitoneal routes. In the case of FUDR,the total radioactivity in urine amounted to 7-21per cent of the administered dose in 48 hours indifferent patients (Chart 19). In the patient inwhom the radioactivity in the plasma was highest,10 hours after the intraperitoneal injection ofFUDR-2-C14, the total radioactivity in the urine
amounted to 34 per cent of the injected dose in 48hours.
Individual urine samples containing 40,000-50,000 counts/min were fractionated on Dowex-1
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

56 Cancer Research Vol. 23, January 1963
(formate) columns. The unchanged drug in thespecimens of urine after administration of FU,expressed as per cent of the total radioactivity inthe urine, is shown in Charts 20 and 21. Whereasthe first samples of urine voided after oral, intramuscular, and intraperitoneal administration contained 25-64 per cent of the unchanged drug, thelater specimens were found to have little or nounchanged FU. One of the two patients who wasgiven injections intravenously of FU-2-C14 wasfound to have 75 per cent of the total radioactivity
3001
250
200
ISO
100
50
SPECIFIC ACTIVITY OF
RESPIRATORY CO,
£^| FUDR INTRAMUSCULAR
O—O FUDR INTRAPERITONEALO—O -
05 I 2 4HOURS
CHABT12
8 12 24 4O 100
the drug was injected intravenously, urea andFUPA constituted 51 per cent and 66 per cent ofthe total radioactivity in the urine passed 24hours after the injection.
The urine collected from patients after the administration of FUDR-2-C14 by different routes
fractionated similarly, and the resultswas art-
TOTAL RADIOACTIVITYRESPIRATORYCOg, FUDR
g0. 40
20-
X ORALO ORAL4 INTRAMUSCULAR7 INTRAMUSCULAR
TOTAL RADIOACTIVITY IN RESHRATORY COg,
FUCR INTRAVENOUS
300
250
200
ISO
100
50
SPECIFIC ACTIVITY OFRESPIRATORY CO0
X * FUDR INTRAVENOUSX— --K
O—O FUDR ORAL0-0 "
O.5 2 4HOURS
CHART13
8 12 24 4O 100
CHARTS12, 13.—Specificactivity of respiratory C14O2afteradministration of FUDR-2-C14 by various routes.
in the urine as unchanged drug in the specimenpassed at 2 hours, and 50 per cent at 24 hours.The other patient, however, had 62 per cent unchanged FU in the urine voided at 2j hours butonly 3 per cent in the urine passed at 24 hours. Theamounts of urea and FUPA, the catabolic products of FU in the urine, were plotted against thelog of time. The results in each of two patientsafter oral and intravenous administrations of FU-2-C14are shown in Chart 22. It was found that thedegradation products constituted 93-95 per centof the radioactivity in the urine samples 24 hoursafter the oral administration of FU-2-C14. When
KO
80
60
40
20-
10 24 DO
CHABT15
TOTAL RADtOACTMTY OF RESPIRATORY C02,
FLOR INTRAPERITONEAL
K»
80.
60
40
20
05HOURS
CllART*16
ICO
CHARTS14-16.—Total excretion in respiratory CO»ofFUDR-2-C14 admimsterc<Tby¡variousroutes.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

MUKHERJEËet al.—Metabolism of FU and FU DR in Patients 57
shown in Table 1. In the urine of the two patients given injections intravenously the fractionscontaining both FU and FUDR constituted 72and 82 per cent of the total radioactivity at0-3 hours after the injections. When this fractionwas separated into FU and FUDR by acetylationand paper chromatography (12), it was found that
oa:UJCL
12-
D-
8-
6-
4-
2-
I
12-
D-
8
4-
2-
RADIOACTMTY IN URINE
x ORALO ORAL4 INTRAMUSCULARv INTRAMUSCULAR
4 10 24 48 100HOURS
CHABT17
% RADIOACTIVITY IN URINE
x INTRAVENOUSO INTRAVENOUS
INTRAPERITOMEAL»INTRAPERITONE/t
4HOURS
10 24 48 00
CHART18
CHARTS17, 18.—Excretion of radioactivity in urine afteradministration of FU-2-C14expressed as % of dose.
unchanged FUDR was present to the extent of 38and 28 per cent of the total radioactivities in thisurine. However, after oral administration of thedrug, no unchanged FUDR could be found in theurine. When the drug was given intramuscularly,FUDR amounted to 32 per cent of the total radioactivity in the urine passed at 10.5 hours. Afterintraperitoneal injections, unchanged FUDR wasfound to constitute 48, 14, and 8 per cent of the
total radioactivity in the urine passed 24 hoursafter administration. Urea and FUPA accountedfor 61-98 per cent of the total radioactivity in theurine passed 12-24 hours after the administrationof FUDR-2-C14 by any of these routes.
Metabolism of FU-2-C" and FUDR-2-C1* inpatients receiving the drugs by continuous intravenous drip method.—Thetoxicity of FU is diminished and of FUDR increased when the drugsare injected intravenously by continuous intravenous drip method (13, 20). It was, therefore,considered advisable to study the degradation ofthese two drugs in patients who received continuous intravenous administrations of FU or FUDR.Three patients were studied with FU-2-C14 and
TOTAL RADIOACTIVITY IN URINE FUDR
30-
20-
10-
INTRAPERITONEAL
i111uKUJO.
20-INTRAMUSCULAR
XO ORAL
INTRAVENOUS
4 IOHOURS
CHART19.—Excretion of radioactivity in urine after administration of FUDR-2-C14 expressed as percentage of dose.
one with FUDR-2-C14. The doses administeredwere 3 mg FU/kg and 6 mg FUDR/kg in 1 literof normal saline. The infusions were given at therate of 6-8 drops/min, and the total dose was administered in 24 hours. Frequent samples of bloodand respiratory air were collected. Charts 23 and24 show the results obtained in two patients aftercontinuous intravenous infusion of FU-2-C14. Thelevels of radioactivity in the plasma remained approximately the same during the period of infusion except during the last 4 hours, when the flowrate was increased so that the total infusion couldbe finished in 24 hours, which led to an increase ofradioactivity in the plasma. After the infusionwas stopped the radioactivity in the plasmagradually decreased. The average specific activity of the respiratory CO»reached a maximum at 6hours and remained approximately constantthroughout the period of infusion except for a
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

58 Cancer Research Vol. 23, January 1963
slight increase during the last 4 hours. After theinfusion was stopped, the spécifieactivity of therespiratory CO2 gradually decreased. The totalexcretion of C02, in percentage of the dose administered, showed that in 24 hours about 70 percent of the injected FU was excreted as CO2 andin 48 hours about 90 per cent. The urine of thispatient showed an excretion of only 0.1 per cent
% UNCHANGED
IOOH
FU IN URINE SAMPLESORALORALINTRAMUSCULARINTRAMUSCULAR
CHABT20
% UNCHANGED FU IN URINE SAMPLES
x INTRAVENOUSO INTRAVENOUSA INTRAPERITONEAL' INTRAPERITONEAL
124 10
HOURS
CBABT«1
CHARTS20, 21.—Percentage of unchanged FU in samplesof urine after administration of FU-2-C14by various routes.
of the total dose. Three specimens of urine fromanother patient who also had continuous intravenous infusion of FU-2-C14 for 24 hours werefractionated on Dowex-1 (formate) columns, andthe results are shown in Chart 24. It was foundthat the percentages of unchanged FU (19-25per cent) and urea (1-3 per cent) remained approximately constant in all three specimens, andthere was a slight increase of FUPA in the later
specimens (from 58 per cent in the 4-hour sampleto 74 per cent in the ll^-hour sample).
Similar investigations were carried out on apatient who was treated with FUDR-2-C14 bycontinuous intravenous drip. The results areshown in Chart 25. The levels of radioactivity inthe plasma were approximately the same for thefirst 7 hours, after which the flow rate of the intravenous drip was increased from 6 drops/min toabout 10 drops/min. The radioactivity in theplasma increased concomitantly with the increasein flow rate and then stayed at an approximatelyconstant level up to 23 hours when the flow ratewas again increased to finish the total dose. Thelevel of radioactivity in the plasma also showed aslight increase in the 24th hour. The average
% OF UREA 8. FUPA IN URINE
IOO
80
60
40
20
4 8 12 16 20 24HOURS
CHART 22.—Percentages of urea and FUPA in samples ofurine after administration of FU-2-C14.
X X, Urea + FUPA; intravenous.O O, Urea + FUPA; intravenous.X X, Urea + FUPA; oral.O O, Urea + FUPA; oral.
specific activity of the respiratory CO»reached apeak at about 10 hours after the beginning of theinfusion. However, in contrast to the cases inwhich FU-2-C14 was administered by intravenousdrip, the specific activity of respiratory C02 decreased after it reached the highest level, althoughthe infusion was continued for another 14 hours.The total excretion of C14O2during the 24 hoursof infusion amounted to only 45 per cent of thetotal injected dose. The excretion in the urinevoided during the 24 hours of infusion constituted10 per cent of the total dose. Three specimens ofurine, excreted at 3, 6, and 11 hours after the beginning of the infusion, were fractionated on Dowex-1 (formate) columns. The results show that thefractions containing FU and FUDR amounted toabout 38 per cent of the total radioactivity in theurine. When these fractions were further separated
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

TABLE1FRACTIONATIONOFURINEONDOWEX-I(FORMATE)AFTERADMINISTRATIONOFFUDR
PatientDO.12345678Time(hours)0-33-55-7la
-240-33-712
-240-22-39
-120
-2.52.5-712-240-55
-10.512-240-22-412
-240
-0.70.7-22
-3.83.8-5.90
-2.82.8-99
-24Mode
of administrationI.V.I.V.I.V.I.V.I.V.I.V.I.V.OralOralOralOralOralOralI.M.I.M.I.M.I.M.I.M.I.M.IP.IP.IP.IP.IP.IP.IP.%Dose4.15.12.10.812.51.70.90.91.00.31.51.60.80.71.21.410.52.60.41.31.41.41.60.66.026.1%FU+FUDR726652138228174029IS2175645454692645823192222149%FUDR385260543217481480%Urea0256832954394615216825202.512500727344%FUPA141923201941165165376860727354128713532496757798994
100
80
6O
40
20
AVERAGE SPECIFICACTIVITY OF CO,
PLASMA LEVEL U9/n'ml
PERIOD OF INFUSIONI
8 12 16 20 24HOURS
32 40 48
Ç
00
80
60f
40 §
20
CHART23.—Levelof drug equivalent in the plasma, average specific activity of respiratory C02, and total excretion ofC14O2in a patient who'received FU-2-C14by continuous intra
venous drip for 24 hours.
100
80
60
40
20
URINE FRACTIONATION
UREA X XFUPA ®—®FU *—y
URINE EXCRETION
12 16HOURS
24
CHART24.—Excretion of radioactivity in urine and frac-tionation of the radioactive compounds in samples of urineafter continuous infusion of FU-2-C14.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

60 Cancer Research Vol. 23, January 1963
into FU and FUDR by acetylation and paper chro-matography (12), it was found that FUDR constituted 8-10 per cent of the radioactivity in the
urine.Metabolism of FUDR-2-Cu: comparison of a pa
tient who tolerated the drug well with one who did not.—Clinical evidence (20) has suggested that there
is a relationship between toxicity of FUDR andthe rate at which the drug is degraded in vivo.Hence, the metabolism of FUDR-2-C14 was
studied in two patients, one of whom (Patient #9,carcinoma of breast) tolerated large doses ofFUDR without exhibiting toxicity; the other patient (Patient #10, carcinoma of colon) exhibitedtoxicity with small doses of FUDR.
Both patients received an intravenous injection of FUDR (30 mg/kg) containing 150 /jc. of
FUDR-2-C14 in 10 ml. of normal saline. Samples
of respiratory air, blood, and urine were collectedat various time intervals and assayed for radioactivity. The results summarized in Table 2 showthat the plasma drug level decreased more rapidlyin the case of the patient who tolerated the drugwell (Patient #9) than in that of the patient whorapidly became toxic to FUDR (Patient #10).However, no significant difference in the rates atwhich the label was excreted as respiratory C14Oa
was observed as shown in Table 3.Similarly, the amount of radioactivity excreted
in the urine did not differ significantly betweenthe two patients (Table 2). However, fractionationof the urine samples on Dowex-1 (formate) columns indicated that Patient #9 had degraded thedrug much more rapidly than Patient #10 (Table
LU100
80
60
040
IO
LUOOC(PDL
URINE FRACTIONATION
FUPA
EXCRETION IN URINE
I
LU
8 12 16 20 24HOURS
8 12 16 20 24HOURS
CHART25.—Levelof drug equivalent in the plasma, average specific activity of respiratory COo,and total excretion of CltO¡,excretion of radioactivity in the urine and fractionation of radioactivity in the urine of a patient who received FUDR-2-C14 in acontinuous intravenous drip.
TABLE 2DEGRADATIONOFFUDR-2-C14IN PATIENTSSUSCEPTIBLEANDRESISTANTTOITSTOXICITY
Patientno.910SamplePlasmaUrinePlasmaUrineTime
of sampleafter injection30
min.2hr.
8hr!4
hr. 10min.7hr. 30min.12-24hr.30
min.2hr.4
hr.8hr.4
hr. 15min.7hr. 25min.12-24hr.Drug
dig/ml)93.4]
1.6/0.87.84.92.31.4%
of doseaccumulated11.611.912.910.114.315.9%Urea017.50.244.256.214.112.11.24.566.5%FU+FUDR25.83.376.86.05.931.45.177.466.04.5%FUDRinMiit.76.677.374.7%FUPA49.529.120.355.237.855.032.621.328.029.0
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

MuKHERJEE et al.—Metabolism of FU and FUDR in Patients 61
2). Only 6 per cent of the radioactivity in the urinesample collected from Patient #9 (7j hours afteradministration of the drug) was present as FU orFUDR, whereas 49 per cent of the radioactivityin the corresponding urine sample from Patient#10 was present as unchanged FUDR and an additional 17 per cent was FU.
Metabolism of 5-fluoro-3',5'-diacetyl-2'-deoxyuri-dine (diacetyl-FUDR).—Diacetyl-FUDR, whenadministered to mice by stomach tube, was foundto be inhibitory toward some transplanted tumors,whereas FUDR given in the same way was ineffective.2 We had found previously (4), and also in ourpresent studies, that FUDR was rapidly degradedwhen it was administered by mouth. Therefore, inthe clinic the drug is usually injected intravenously. It would be a clinical advantage, however, if aderivative of FUDR could be found which wouldnot be degraded so rapidly as FUDR but would beinhibitory toward the cancer when administeredorally. It was of interest, therefore, to study thedegradation of diacetyl-FUDR given orally andto compare it with that of FUDR administered inthe same way. These investigations were carriedout on two patients. The level of radioactivity inthe plasma, excretion of C14Oain the respiratoryair, and the excretion of the unchanged drug andits metabolites in the urine were followed for 48hours after oral administration of FUDR-2-C14.After an interval of 1 week the patients returnedto the hospital, and the same procedure wasrepeated after oral administration of diacetyl-FUDR-2-C14. The results are shown in Charts 26and 27 and Table 4.
The solid lines show the data obtained after theoral administration of FUDR-2-C14 and the brokenlines after that of diacetyl-FUDR-2-C14. In one patient (Patient #11), no difference was found in theamount of respiratory COi excreted in 12 hours orin the total excretion of radioactivity in the urineat 24 hours. The specimens of urine were fractionated on Dowex-1 (formate) columns, and the fractions containing FU and FUDR were further separated into FU and FUDR by acetylation andpaper chromatography (12). It was found that 5'-monoacetyl-FUDR constituted 26 per cent of thetotal radioactivity in the urine at 9 hours afteradministration of diacetyl-FUDR, whereas FUDRamounted to only 1.6 per cent of the radioactivityat 10 hours after the ingestion of FUDR (Table 4).
In the other patient (Patient #12), no differencecould be found in the amount of C14Oiexcreted at48 hours after the administration of either FUDR-2-C14or diacetyl-FUDR-2-C14 (Table 4). The levelof radioactivity in the plasma and the total radio-
2Unpublished observations.
activity excreted in the urine was higher after theadministration of diacetyl-FUDR-2-C14 than afterFUDR-2-C14. When the urines were fractionated,it was found that, after the administration ofFUDR, unchanged drug could not be detected inany of the urine samples. However, after the administration of diacetyl-FUDR, 5'-monoacetyl-
FUDR was found to constitute 25 per cent of thetotal radioactivity in the urine passed 12 hoursafter ingestion of the drug.
Radioactivity in the cerebrospinal fluid after intravenous injection of FU-2-Cu and FUDR-2-C14.—The fluorinated pyrimidines have been reportedto be clinically ineffective in patients having eitherprimary or metastatic tumors of the brain.3 The
TABLE 3
EXCRETIONOFC14OîIN PATIENTSSUSCEPTIBLEANDRESISTANTTOTHE
TOXICITYOFFUDR
TIME10
rain.20rain.30min.45min.60min.Uhr.2hr.è
ir.8hr.24
hr.PERCENTAGE
OFDOSEACCUMULATEDABC^OjPatient
i»0.82.44.78.812.518.623.134.546.7PatientÕIO0.72.54.88.512.017.722.234.548.956.5
failure of these patients to respond may result fromthe capacity of the drugs to pass through thechoroid plexuses after intravenous injection. Itwas desirable, therefore, to study the distributionof FU-2-C14 and FUDR-2-C14 in the cerebrospinalfluid after intravenous injection.
Three patients were injected with FU-2-C14 andone with FUDR-2-C14. The drugs were injectedintravenously, and samples of blood and cerebrospinal fluid were withdrawn at different time intervals. A portion of each sample was plated, and theradioactivity was determined. The counts/min/mlof the plasma and cerebrospinal fluid were converted to ¡jig.drug equivalent/ml and plottedagainst the log of time. Chart 28 shows the levelof radioactivity in cerebrospinal fluid and plasmain one patient who had an intravenous injection ofFU-2-C14. Ten minutes after injection of the drugthe cerebrospinal fluid was found to contain verylittle radioactivity. However, the level of radio-
3Personal communication from Dr. F. J. Ansfield.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

100
080
I 60
040
S 20û.
LU
0Q.
108642
IONATION
FUDR
EXCRETION IN URINE
RESPIRATORYC02
'TOTAL AS COg
4 8 12 16 20 24HOURS
AVERAGE SPECIFICACTIVITY OF CO2
I !
200 n
160 8120 m
80 I
0
8 12 1620 24HOURS
CHART26.—Levelof drug equivalent in the plasma, average specific activity of respiratory CO-<and total excretion ofexcretion of radioactivity in the urine and fractionation of radioactivity in the urine of a patient who received FUDR-2-C" and3',5'-diacetyl-FUDR-2-C14 by mouth.
, FUDR. ,3', 5'-diacetyl-FUDR.
8 12 16 20 24HOURS
8 12 16 20 24HOURS
CHART27.—Levelof drug equivalent in the plasma, average specific activity of respiratory COo,and total excretion of C14O2,excretion of radioactivity in the urine and fractionation of radioactivity in the urine of a patient who received FUDR-2-C14 and3',5'-diacetyl-FUDR-2-C14 by mouth.
, FUDR. , 3',5'-diacetyl-FUDR.
TABLE 4
METABOLISMOFFUDR ANDDIACETYL-FUDR,ADMINISTEREDORALLYIN Two PATIENTS,#11 AND#12
Dose(mg/kg)Dose
(totalcounts/min)Per
cent of dose inCO¡Per
cent of dose inurineMaximum
/¿gequivalents/mlplasmaPer
cent of total radioactivity in urine as:diOAc-FUDRFU
FUDR or 5'-monoacetyl-FUDRPATIENT
illFUDR3092X10"83
(12hr.)6.35.6
(45min.)016.6
( 1 hr.)11.5 ( 3hr.)6.3 (10 hr.)
16.6 ( 1 hr.)1.6 ( 3hr.)1.6 (10 hr.)diOAc
FUDR3094X10«84
(12hr.)6.89.3
(30min.)031.5
( 3hr.)4.3 ( 9hr.)10.7
( 3hr.)26. 7 ( 9hr.)PATIENT
fitFUDR31100X10"723301470
06
(48hr.)85
(lihr.)(
7 hr.)5 (12hr.)(
7 hr.)(12 hr.)diOAc
FUDR3242X10«72.8(48hr.)7.76.9
( 4hr.)046
( 5 hr.)27 ( 9 hr.)35 (12 hr.)31 ( 5 hr.)13 ( 9 hr.)25 (12 hr.)
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
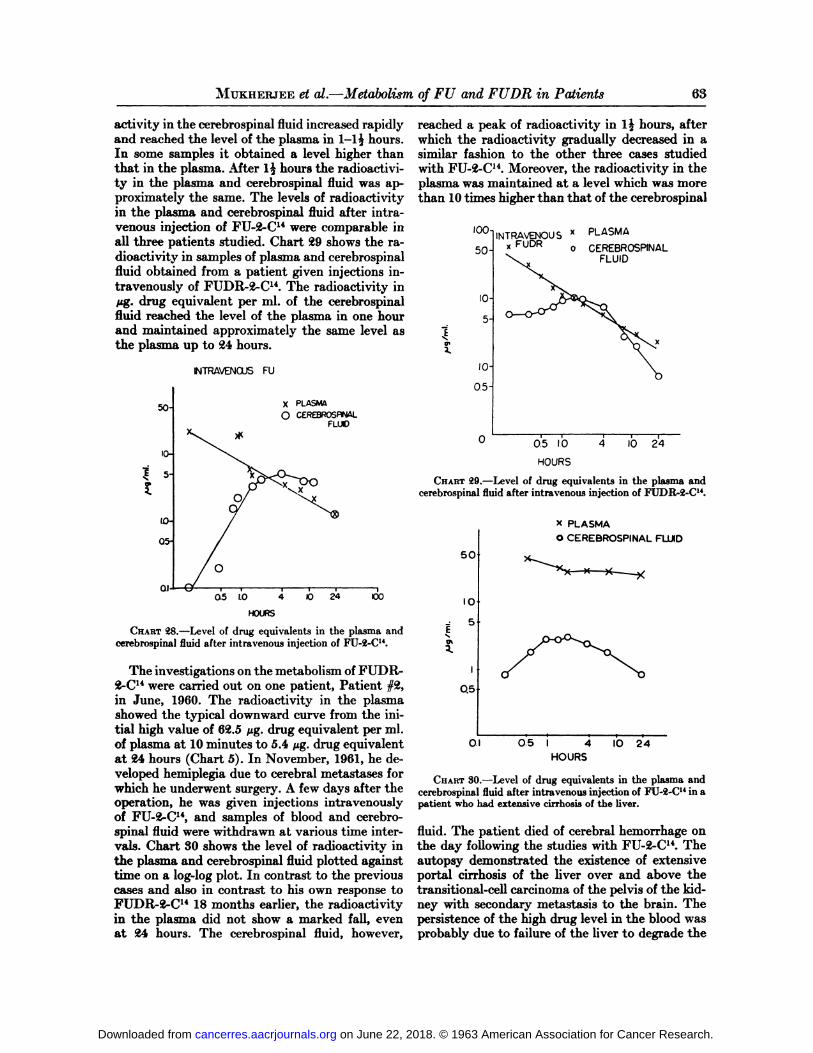
MuKHERJEEet al.—Metabolismof FU and FU DR in Patients 63
activity in the cerebrospinal fluid increased rapidlyand reached the level of the plasma in 1-15 hours.In some samples it obtained a level higher thanthat in the plasma. After 1^ hours the radioactivity in the plasma and cerebrospinal fluid was approximately the same. The levels of radioactivityin the plasma and cerebrospinal fluid after intravenous injection of FU-2-C14 were comparable inall three patients studied. Chart 29 shows the radioactivity in samples of plasma and cerebrospinalfluid obtained from a patient given injections intravenously of FUDR-2-C14. The radioactivity inHg. drug equivalent per ml. of the cerebrospinalfluid reached the level of the plasma in one hourand maintained approximately the same level asthe plasma up to 24 hours.
INTRAVENOUS FU
o*
so
10-
5-
X PLASMA
O CEREBROSPNALFLUO
01J—005 1.0 O 24 OC
HOURS
CHAUT28.—Levelof drug equivalents in the plasma andcerebrospinal fluid after intravenous injection of FU-2-C14.
The investigations on the metabolism of FUDR-2-C14were carried out on one patient, Patient #2,in June, 1960. The radioactivity in the plasmashowed the typical downward curve from the initial high value of 62.5 jug-drug equivalent per ml.of plasma at 10 minutes to 5.4 fig. drug equivalentat 24 hours (Chart 5). In November, 1961, he developed hemiplegia due to cerebral métastasesforwhich he underwent surgery. A few days after theoperation, he was given injections intravenouslyof FU-2-C14, and samples of blood and cerebrospinal fluid were withdrawn at various time intervals. Chart 30 shows the level of radioactivity inthe plasma and cerebrospinal fluid plotted againsttime on a log-log plot. In contrast to the previouscases and also in contrast to his own response toFUDR-2-C14 18 months earlier, the radioactivityin the plasma did not show a marked fall, evenat 24 hours. The cerebrospinal fluid, however,
reached a peak of radioactivity in lj hours, afterwhich the radioactivity gradually decreased in asimilar fashion to the other three cases studiedwith FU-2-C14. Moreover, the radioactivity in theplasma was maintained at a level which was morethan 10 times higher than that of the cerebrospinal
100
10
5-
10
0.5H
INTRAVENOU SFUDR
x PLASMA
CEREBROSPINALFLUID
0.5 10
HOURS
10 24
CHART29.—Levelof drug equivalents in the plasma andcerebrospinal fluid after intravenous injection of FUDR-2-C14.
X PLASMA
O CEREBROSPINAL FLUID
50
Em
IO
5
I
Q5
01 05 I 4 IO 24
HOURS
CHAUT80.—Levelof drug equivalents in the plasma andcerebrospinal fluid after intravenous injection of FU-2-C14in apatient who had extensive cirrhosis of the liver.
fluid. The patient died of cerebral hemorrhage onthe day following the studies with FU-2-C14. Theautopsy demonstrated the existence of extensiveportal cirrhosis of the liver over and above thetransitional-cell carcinoma of the pelvis of the kidney with secondary metastasis to the brain. Thepersistence of the high drug level in the blood wasprobably due to failure of the liver to degrade the
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Cancer Research Vol. 23, January 1963
drug. The acid-soluble extracts of the samples ofplasma and cerebrospinal fluid obtained at 1^-2and 16 hours after injection of the FU-2-C14 werefractionated on Dowex-1 (formate), and the resultsare shown in Table 5. It was found that the proportions of urea, FU, and FUPA in the acid-soluble extracts of the plasma obtained at 1| hoursafter injection were almost the same as in those ofthe cerebrospinal fluid withdrawn at about thesame time. The identity of the FU eluted from theDowex-1 (formate) column was confirmed by paper chromatography. The proportion of FU in theplasma obtained 16 hours after injection was muchlower than at If hours while the proportions ofurea and FUPA were higher. The radioactivityfrom the 16-hour specimen of the cerebrospinalfluid, when fractionated on a Dowex-1 (formate)column, was not recovered quantitatively.
TABLE5DOWEX-1 (FORMATE) FRACTIONATION OF ACID-
soLUBLEMATERIALOFTHEPLASMAANDCEREBROSPINALFLUIDOFPATIENT#2
SpecimenPlasmaCerebrospinal
fluidPlasmaCerebrospinal
fluidTime(hours)1.521616Urea(per
cent)5330FU(percent)597019FUPA(percent)691835
DISCUSSIONThe appearance of radioactivity in the plasma
at 10 minutes and the attainment of the highestpeak at 5-! 5 hours after oral and intramuscularadministrations of FU and FUDR indicate rapidabsorption of the drugs by these routes. The erratic absorption, which was observed by the intra-peritoneal route, does not seem surprising, sincethese patients had ascites and involvement of theperitoneum. Although the ascites fluid was drainedprior to the intraperitoneal injection, the extent ofthe peritoneal adhesions probably influenced theamount of drainage as well as the rate and degreeof absorption by this route.
The rates of absorption of FU and FUDR afteroral, intramuscular, and intraperitoneal administrations were different in individual patients, asshown by the variation in the amount of radioactivity in the plasma between the two patientstreated in the same manner. A difference in therate of degradation and elimination may also account for the variation. However, in the two patients who received FU orally there was a difference in the drug level in the plasma, even thoughthe urine fractionation studies showed that degradation was rapid in both cases. Moreover, when
the drugs were injected intravenously, where thefactor of absorption was eliminated, the individualvariations were found to be minimal. Therefore,differences in the rates of absorption, rather thanelimination, presumably account for the differencein the plasma levels.
Fractionation of the acid-soluble extracts of theplasmas after administration of FU by oral, intraperitoneal, and intravenous routes showed thatdegradation was comparatively slower after theintravenous than after oral or intraperitoneal administrations. Fractionation could not be undertaken in samples of plasma later than 2 hours because of the low radioactivity, and so the correspondence between the radioactive compounds inthe plasma and urine could not be studied.
The degradation of the drug occurs primarily inthe liver (4). In the present series of investigationsextensive liver cirrhosis was found in one patientto inhibit the decrease of drug level in the plasma,presumably because of failure of the liver to degrade the injected FU. The circulating drug musttraverse the hepatic circulation for some time inorder that the successive stages of degradationthrough FU, DHFU, and FUPA to CO2may takeplace. Thus, although the drug level is at its highest at the beginning of intravenous injection, thespecific activity of CÛ2takes about 5 hour to reachthe maximum. However, if the absorption takesplace more gradually as in the case of oral, intramuscular, and intraperitoneal administrations, theliver presumably is not overloaded and hence candegrade the drug more efficiently. Moreover, whenthe drugs were administered by mouth, the bacterial flora of the gastrointestinal tract probablydegrade the drug to some extent. The specific activity of respiratory COi, therefore, reaches thehighest peak at about the time when the amountof radioactivity is also highest after oral, intramuscular, and intraperitoneal administration. Thetotal excretion of C140a was least in the patientswho were given injections intramuscularly of FU-2-C14.However, among the patients who receivedFUDR, those who were injected intravenouslyexcreted the least in the expired air.
The total excretion of radioactivity in the urinevaried in individual patients. The patients whoexcreted more radioactivity in the CÜ2usually excreted less in the urine and vice versa. If the proportion of the different radioactive compounds present in the urine can be considered as representativeof those in the blood, it appears that intravenousadministration of FU should be the method ofchoice, because unchanged FU persisted in theurine even 24 hours after intravenous injection.After administration of FUDR, the urine wasfound to contain unchanged FUDR when the drug
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

MuKHERJEE et al.—Metabolism of FU and FUDR in Patients 65
was given by intravenous, intramuscular, and in-traperitoneal injections; no unchanged FUDR wasfound to be present in the urine after oral administration of the drug. However, excretion in the respiratory air was least when FUDR was given intravenously. Therefore, with FUDR also, intravenous injection appears to be preferable to the othermethods of administration. In the clinic the drugsare usually injected intravenously.
The fluorinated pyrimidines have to be given tothe point of toxicity before they are clinically effective (5). The toxicity of these drugs depends on,among other factors, the extent of degradation ofFU and FUDR. When FU was injected intravenously in a single dose, 63 per cent was excretedin the respiratory CÛ2and 11 per cent in the urinein 24 hours, whereas, when it was injected by continuous drip, 89.6 per cent was excreted in CO2and4 per cent in the urine. It appears, therefore, thatthere was more degradation of FU when it wasgiven as a continuous drip than as a single dose,which probably explains the report of Lemon (13),who found that, when FU was administered in acontinuous intravenous drip, it was less toxic thanwhen given in a single rapid injection. The chemo-therapeutic effectiveness of the fluorinated pyrimidines is considered to be primarily the result of theformation in vivoof FUDRP (16). However, onlya very small fraction of the administered dose (afew m/ig/mg) is actually present as nucleotides(17) in the tumor and other tissues. This tinyamount may be too small to be affected by an increase of the degradation of the drug which is normally extensive. However, the toxicity is diminished by an increase in degradation. When thedegradation of FUDR, administered as a singledose or as continuous intravenous drip, was compared in the same way it was found that, after asingle intravenous injection, about 67 per cent ofFUDR was excreted as CÛ2and 25 per cent in theurine in 12 hours. However, when it was injectedas a continuous drip, 45 per cent was excreted asCOo in 24 hours and 9 per cent in the urine. Itappears, therefore, that there was less degradationafter continuous intravenous infusion of FUDRthan after a single intravenous dose. The toxicityof FUDR has been reported to be greatly increasedwhen it was injected as a continuous intravenousdrip (20). There appears, therefore, to be an inverserelation between the amount of degradation andtoxicity produced by these drugs.
This conclusion was borne out by the comparisonof the rates at which FUDR was degraded by twopatients, one of whom tolerated the drug well andthe other of whom rapidly exhibited toxicity tothe drug. The former patient degraded the drugrapidly, whereas the latter patient did not, thus
maintaining a high level of FUDR in the circulatory system for a longer time.
The chemotherapeutic effectiveness of diacetyl-FUDR when administered by stomach tube tomice, in contrast to the inactivity of FUDR givenin the same way,4 led to a study of degradation ofthe two compounds in human patients under comparable conditions. The extent of degradation ofboth drugs to respiratory COj was the same, butin the case of diacetyl-FUDR it was found thatS'-monoacetyl-FUDR persisted in the unchanged
form for a long time in the urine. When Ehrlichascites cells were incubated in vitro with diacetyl-FUDR, the incorporation of formate-C14 intoDNA thymine was not inhibited to a significantextent, whereas FUDR was a powerful inhibitor ofthis reaction (16). It was suggested that the tumorcells probably could not easily hydrolyze thediacetyl-ester, and hence FUDR was not set freeto be phosphorylated to FUDRP. Casida andHeidelberger5 have found that liver of mice cande-esterify diacetyl-FUDR, so that when diacetyl-FUDR is given by mouth it is absorbed and transported by the portal circulation to the liver, wherethe compound is deacetylated and FUDR is setfree. The administration of FUDR by mouth, onthe other hand, would lead to the transport of thecompound in the portal circulation to the liverwhere it would be rapidly broken down. The administration of diacetyl-FUDR by mouth becomes, therefore, almost equivalent to FUDRintravenously. Deacetylation in the liver can bepresumed to take place in the same way in humanpatients. A clinical trial is, therefore, under way inwhich the administration of diacetyl-FUDR bymouth is being compared with intravenous injection of FUDR.
The slow appearance and gradual increase ofradioactivity in the cerebrospinal fluid after theintravenous injection of FU-2-C14 or FUDR-2-C14indicates that either the drugs or their metabolicproducts are entering the cerebrospinal fluid. Itwas only in one patient that fractionation of radioactivity in simultaneous specimens of plasma andcerebrospinal fluid could be carried out. The datashowed that unchanged FU could be found in thecerebrospinal fluid, and the proportion of the unchanged drug was roughly equal to that in thecorresponding sample of plasma. However, the radioactivity in the plasma was maintained at amuch higher level than the cerebrospinal fluid inthis particular patient. It appears, therefore, thatthere is no free diffusion of radioactive compoundacross the blood-brain barrier after intravenous injection of FU-2-C14.
4Dr. F. J. Ansfield, personal communication.5Unpublished data.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

Gü Cancer Research Vol. 23, January 1963
It is well recognized by clinicians that the biology of human cancer differs very considerablyamong individual patients. It is not surprising,therefore, to find wide variations in the extent andtype of response elicited in cancer patients by atumor-inhibitory drug, which undergoes varioustransformations in vivo of synthetic and degrada-tive nature. Hence, the carcinostatic action will bedependent on the balance between the two processes. The tumor-inhibitory action of the fluori-nated pyrimidines is the result of what Peters calls"lethal synthesis" (18), in which the fluoropyrim-
idine is converted in vivoto the lethal compound,FUDRP, which is the inhibitor of thymidylatesynthetase (9). The biosynthesis of nucleo tides after administration of FU and FUDR to cancer patients is described in the next paper of this series(17). The present paper demonstrates that thereis a considerable individual variation in the extentand rate of degradation of FU and FUDR. It ishoped that the eventual clinical response of thesepatients may be correlated with the distributionand the extent of degradation of the drugs. Thepresent studies also demonstrate that the degradation of these drugs is very rapid, especially afteradministration by certain routes. From the slopeof the decline of the drug level in the plasma afterintravenous injection we can extrapolate the levelof the drug to zero time; thus, after the administration of FUDR, the drug level at zero time wasfound to be about 100 ¿ig/ml.This corresponds toa space of 20 liters. Therefore, the drug was probably rapidly distributed over a larger volume thanthat of either plasma or extracellular fluid.
The biological activity of a drug is determinedby the following factors: (a) the capacity to beabsorbed from the site of administration, (6) excretion, (c) the metabolic transformations in thebody, (d) the distribution in the tissues, and (è)thenature of the interaction of the active form of thedrug with the target enzyme. The present investigations shed light on the first three of these factors.The distribution of the drug in the tissues and conversion to the nucleotides are dealt with in anaccompanying paper (17).
ACKNOWLEDGMENTSThe authors wish to express their deep gratitude to Drs.
A. R. Curreri and M. J. Ja vid for making the patients availablefor the studies. We particularly express our appreciation of theco-operation of all the patients who voluntarily submitted toextra discomfort to make these studies possible. We acknowledge with affection and gratitude the expert technical assistance of the late Miss Laura Davis, who worked devotedly formore than two years while receiving monthly courses of one ofthese drugs.
REFERENCES1. ANSFIELD,F. J. Further Clinical Comparison between
5-Fluoro-2'-deoxyuridine and 5-FluorouraciI. A Compari
son of the Clinical Effectiveness of 5-FUDR Administeredby the Continuous Infusion and the Single Daily Injection. Proc. Am. Assoc. Cancer Res., 3:205, 1961.
2. ANSFIELD,F. J., and CURRERI,A. R. Clinical Studies with5-Fluoro-2'-deoxyuridine. Cancer Chem. Rep. #6, 21-25,
1960.3. BHENNAN,M. J., and VAITKEVICIUS,V. K. 5-Fluorouraeil
in Clinical Cancer Experience with 155 Patients. CancerChem. Rep. #6, 8-11, 1960.
4. CHAUDHURI,N. K.; MUKHERJEE,K. L.; and HEIDELBERGER,C. Studies on Fluorinated Pyrimidines VII. TheDegradative Pathway. Biochem. Pharmacol., 1:328-41,1959.
5. CURRERI,A. R.; ANSFIELD,F. J.; MclvER, F. A.; WAIB-MAN,H. A.; and HEIDELBERGER,C. Clinical Studies with5-Fluorouracil. Cancer Res., 18:478-84, 1958.
6. DRILL, V. A. Pharmacology in Medicine, p. 9. McGraw-Hill, 1954.
7. DOBBING,J. Blood-Brain Barrier. Physiol. Rev., 41:130-88, 1961.
8. FINK, K.; CLINE, R. E.; HENDERSON,R. B.; and FINK,R. M. Metabolism of Thymine (Methyl-C14 or -2-C14)byRat Liver in Vitro. 3. Biol. Chem., 221:4-25-33, 1956.
9. HARTMANN,K-U., and HEIDELBERGER,C. Studies onFluorinated Pyrimidines XIII. Inhibition of ThymidylateSynthetase. J. Biol. Chem., 236:3006-13, 1961.
10. HEIDELBERGER,C.; GRIESBACH,L.; CRUZ,O.; SCHNITZER,R. J.; and GRTTNBERG,E. Fluorinated Pyrimidines. VI. Effects of 5-Fluorouridine and S-Fluoro-a'-deoxyuridine onTransplanted Tumors. Proc. Soc. Exp. Biol. Med., 97:470-75, 1958.
11. HEIDELBERGER,C.; GHIESBACH,L.; MONTAG,B. J.;MOOREN, D.; and CRUZ, O. Studies on FluorinatedPyrimidines. II. Effects on Transplanted Tumors. CancerRes., 18:305-17, 1958.
12. KALDOR,G., and HEIDELBERGER,C. A New Paper Chromatographie Method for Separation of Various Pyrimidines and Their Deoxyribonucleosides. Biochim. Biophys.Acta, 36:249-50, 1959.
13. LEMON,H. N. Reduction of 5-Fluorouracil Toxicity inMan with Retention of Anticancer Effects by ProlongedIntravenous Administration in 5% Dextrose. CancerChem. Rep. #8, pp. 97-101, 1960.
14. LUDWIG,H.; POTTER,V. R.; HEIDELBERGER,C.; andDEVERDIER,C. H. Automatic Direct Quantitation ofRadioactivity on Paper Chromatograms. Biochim. Biophys. Acta, 37:525-27, 1960.
15. MUKHERJEE,K. L., and HEIDELBERGER,C. Studies onFluorinated Pyrimidines IX. The Degradation of 5-Fluorouracil-6-C14. J. Biol. Chem., 236:433-37, 1960.
16. . Studies on Fluorinated Pyrimidines. XV. Inhibition of the Incorporation of Formate-C" into DNA Thymine of Ehrlich Ascites Cells in Vitro. Cancer Res., 22:815-22, 1962.
17. MUKHERJEE,K. L.; CURRERI,A. R.; JAVID, M.; andHEIDELBERGER,C. Studies on Fluorinated Pyrimidines.XVII. Tissue Distribution of 5-Fluorouracil-2-C14 and5-Fluoro-2'-deoxyuridine in Cancer Patients. Cancer Res.,23:67-77, 1963.
18. PETERS,R. A. In: R. J. C. HARRIS(ed.), Biological Approaches to Cancer Chemotherapy, pp. 11-19. New York:Academic Press, 1961.
19. SCHNEIDERMAN,M. A. The Clinical Excursion into 5-Fluorouracil. Cancer Chem. Rep. #16, pp. 107-18, 1962.
20. SULLIVAN,R. D.; YOUNG,C. W.; MILLER,E.; GLATSTEIN,N.; CLARKSON,B.; and BURCHENAL,J. H. The ClinicalEffects of the Continuous Administration of FluorinatedPyrimidines (5-Fluorouracil and 5-Fluoro-L2'-deoxyuri-dine). Cancer Chem. Rep. #8, pp. 77-83, 1960.
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1963;23:49-66. Cancer Res Kanai Lal Mukherjee, Judith Boohar, Dorothy Wentland, et al. Cancer Patients
-deoxyuridine-2-C14 in′5-Fluorouracil-2-C14 and 5-Fluoro-2Studies on Fluorinated Pyrimidines: XVI. Metabolism of
Updated version
http://cancerres.aacrjournals.org/content/23/1/49
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/23/1/49To request permission to re-use all or part of this article, use this link
on June 22, 2018. © 1963 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
![Electrochemica l studies of two pyrrolo[1,2 -c]pyrimidines · 2018. 6. 1. · M.-L. Tatu: Electrochemical studies of two pyrrolo[1,2-c]pyrimidines … 28 . Table 1. Investigated pyrrolo[1,2-c]pyrimidines](https://img.dokumen.tips/doc/110x75/6126a41ecf6a744fa06703e9/electrochemica-l-studies-of-two-pyrrolo12-c-2018-6-1-m-l-tatu-electrochemical.jpg)
![Supplementary Information · S1 Supplementary Information Efficient synthesis and preliminary biological evaluations of trifluoromethylated imidazo[1,2-a]pyrimidines and benzimidazo[1,2-a]pyrimidines](https://img.dokumen.tips/doc/110x75/5f8a85792012cc11f3416f3f/supplementary-s1-supplementary-information-efficient-synthesis-and-preliminary-biological.jpg)



























