Embed Size (px)
Citation preview

Published on Web Date: November 10, 2009
r 2009 American Chemical Society 136 DOI: 10.1021/jz900080q |J. Phys. Chem. Lett. 2010, 1, 136–140
pubs.acs.org/JPCL
Structures and Electronic Properties of Si-SubstitutedBenzenes and Their Transition-Metal ComplexesVaisakh Mohan† and Ayan Datta*,‡
†National Institute of Science Education and Research, Institute of Physics Campus, Bhubaneswar-751005, Orissa, India and‡School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, CET Campus,Thiruvananthapuram-695016, Kerala, India
ABSTRACT Structural and electronic properties for a series of silicon-substitutedbenzenes (CnSimH6, where n=0-6,m=0-6, and nþm=6) are studied throughdensity functional theory calculations. Benzene is found to sustain its planarityup to two Si substitutions for all isomers. For three Si substitutions, only the 1,3,5-alternate structure (6) is planar, while for four Si substitution, only the 2,3,5,6structure (10) is planar. Further Si substitution makes all the isomers for the ringsnonplanar, which eventually leads to the fully puckered C3v structure for hexasi-labenzene (13). The reorganization energies for these molecules are sufficientlylow to be favorably utilized for hole conduction. All themolecules form very stablefull-sandwich and half-sandwich complexes of the type η6-(CnSimH6)2Cr andη6-(CnSimH6)Cr(CO)3. The binding energies for these complexes increase withincrease in the number of Si atoms in the rings. Strategies are proposed forexperimental design of extended sheets of silicenes and mixed C/Si graphenesthrough transition-metal complexation of the six-membered rings.
SECTION Dynamics, Clusters, Excited States
T he chemistry and physics of new formsof carbonhavebeen in the frontline of research in nanotechnologyfor the last two decades.1,2 Interest in carbon-based
materials arises primarily because of their easy tailoribility,rational structural design, and versatile state of aggregationat various length and time scales.3,4 Contrary to the hugeinterest in carbon-based nanomaterials, the chemistry ofsilicon, belonging to the same group in the periodic table, isnot very well studied.5 The lack of interest in Si-basednanomaterials maybe attributed to the unavailability of pla-nar sp2-like bonding environment. Si prefers an sp3 bondingenvironment. Hence, ideal two-dimensional sheets of sili-cenes undergo puckering in the six-membered rings to formordered ripples (unpublished results). Thus, the basic buildingblock of fullerenes, graphenes, and nanotubes, namely, ben-zene, is mostly confined to the carbon family.
However, the past few years has witnessed a renewedinterest in Si-based nanomaterials for applications as electro-nic and optical materials. All-Si fullerenes and quantum dotsof various nuclearities and shapes have been synthesized.6,7
Many new structures of all-Si clusters have been proposedbased on computational studies mainly through densityfunctional theory (DFT) calculations.8,9 However, a rationaldesign for truly all-Si-based nanomaterials requires a bottom-up approach starting from a fundamental unit like benzene.The all-Si analogue of benzene, hexasilabenzene, is wellcharacterized through computational studies.10,11 Unlike ben-zene, it has a puckeredC3v geometry. The puckering distortionis a consequence of the weak Si-Si π-bonding, which results
in the electron-electron interactions being overcome by theelectron-nuclear interactions and a strong Jahn-Teller dis-tortion. It is interesting to study the trade-off between theplanarity of the six-membered ringand the ratio ofC andSi formixed carbo-sila benzenes. In this manuscript, we criticallystudy the effect of substitution of C by Si in the six-memberedπ-framework to their structural and electronic properties. Wealso proposemechanisms for stabilizing such partially Si/all-Sirings through complexation to transition metals such as Cr.We predict the structures and stabilities of all the Si analoguesof silico-transition-metal complexes.
The structures forall the clusterswere optimizedat theDFTlevel using the hybrid B3LYP functional at the 6-31þG(d) basisset level. All the calculations are performedusing theGaussian03 set of codes.12 Frequency calculations were performed forremoval of vibrational instabilities in all the structures. InFigure 1, the structures for the various clusters considered inthe study are shown. Benzene (1) with a D6h symmetry stillretains it planarity with a single substitution of C by a Si atom(2). A search for such a molecule in the Cambridge Crystal-lographic Database (CCDC)13 leads to five structures.14 Inter-estingly, all the structures have a planar six-memberedmotif,suggesting reliability in the level of our calculations. Thecomputed (experimental)15 C-Si bond-lengths are found to
Received Date: October 2, 2009Accepted Date: November 2, 2009

r 2009 American Chemical Society 137 DOI: 10.1021/jz900080q |J. Phys. Chem. Lett. 2010, 1, 136–140
pubs.acs.org/JPCL
be 1.774 Å (1.765(4) Å and 1.770(4) Å), indicating equalC-Si bonds in even the experimental molecules. Also, abond-length of∼1.77 Å indicates that the C-Si bonds essen-tially have an intermediate character between a double bondand single bond (∼CdSi= 1.70 Å and∼C-Si= 1.89 Å) andtheπ electrons are fairlywell delocalized along the ring. This isalso supported by almost all equal computed (experimental)C-C bond lengths of 1.402 Å and 1.405 Å (1.381(6)-1.399(6) Å). Previous computational study of the nucleus-independent chemical shift (NICS) (at the GIAO-SCF/6-31þG(d) level) in 2 are also indicative of its strong aromaticbehavior.16,17
Substituting the second silicon in benzene leads to threedifferent isomers, namely, the ortho (3), meta (4), and para(5) forms. Interestingly, all the three isomers are planar,and the stabilities follow the order: 4 (0.0 kcal/mol) > 3(þ0.9 kcal/mol)> 5 (þ11.4 kcal/mol). Such a variation in thestability for the isomers is understoodon thebasis of the lowerstability of conjugation along a SidSi bond compared to thatof a CdC bond. For the meta isomer, no-conjugation existsbetween the two Si atoms, while for both the ortho and paraforms, π-conjugation is allowed, the extent of conjugationbeing more for the para isomer.
For the trisubstituted benzene, C3Si3H6, various structuresare considered, and the structures 6, 7, and 8 are the lowest
energy isomers. Of them, only the 1,3,5-alternately substi-tuted structure (6) is found to be planar. The origin for thenonplanarity of the 1,2,3-substituted isomer (7) and the1,2,4-substituted isomer (8) is traced to the stronger steric repulsionbetween the bulkier Si atoms, when nearest neighbor to eachother. This is also supported by the larger mean puckeringangle, j = 9.31� in 7 (two nearest-neighbor Si-Si inter-actions) compared to j = 5.41� in 8 (one nearest-neighborSi-Si interaction). The stabilities of the isomers decrease inthe following order: 6 (0.0 kcal/mol)> 7 (þ4.8 kcal/mol)> 8(þ11.5 kcal/mol). The greater stability of 6 is understood onthe basis of its planar structure, which allows delocalizationssimilar to borazine, albeit lesser than benzene.
For the tetra-substituted benzenes, the stabilities of theisomers decrease in the following order: 11 (0.0 kcal/mol,j=15.3�)>9 (þ4.2 kcal/mol,j=18.03�)> 10 (þ9.5 kcal/mol,j = 0�). Interestingly, even though 10 has a perfectly planarstructure, it is the least stable of all the isomers. This suggeststhat in tetrasubstituted isomers, delocalization of π electronsacross the ring is minimal, and even a planar structure isinsufficient to ensure stability through aromaticity. The lack ofaromaticity 10 is easily understood on the basis of the verylong Si 3 3 3 Si bond of 2.22 Å, which breaks π conjugationacross the verticle mean plane of the ring. The penta-silicon-substituted benzene (12) is nonplanar with j = 26.0�. Thehexa-Si-substituted benzene (13) has chairlike distorted struc-ture with j = 37.1�.
It is interesting to study the effect of Si substitution inbenzene on its reorganization energies (λ).18 The hole andelectron reorganization energies (λhole and λelectron) are de-fined as λhole [λelectron] = ionization energy (vertical) - ioni-zation energy (adiabatic) and [|electron affinity (vertical) -electron affinity (adiabatic)|], respectively. λhole (λelectron) forthe molecules are 1 (0.15 eV, 0.11 eV), 2 (0.08 eV, 0.18 eV), 3(0.09 eV, 1.10 eV), 4 (0.12 eV, 2.53 eV), 5 (0.06 eV, 1.24 eV), 6(0.17 eV, 0.54 eV), 7 (0.18 eV, 0.79 eV), 8 (0.10 eV, 2.45 eV), 9(0.09 eV, 0.61 eV), 10 (0.12 eV, 0.38 eV), 11 (0.29 eV, 0.53 eV),12 (0.16 eV, 0.48 eV), and 13 (3.44 eV, 0.43 eV), respectively.Benzene, 1, has an amphiphilic behavior with comparableλhole and λelectron. However, most of the Si-substututed ben-zenes (with the exception of hexasilabenzene, 13) havesmaller λhole, suggesting that partial doping of benzene withSi makes the molecules predominatingly hole conductors. 13is a poor hole conductor but a moderate electron conductor.The origin of the large (small) cation (anion) reorganizationenergies in hexasilabenzene is understood on the basis of thedelocalization of the π-electrons in the rings. For the cation,the C3v puckering distortions lead to poor overlap between the3pz orbitals in comparison to that for benzene (as seen inFigure 2), which increases the λhole. However, for the anion,the additional electron is delocalized over five Si atoms,whichstabilizes the anion and thus reduces the λelectron.
One of the well-established strategies to stabilize newmolecules is through the coordination of the molecules asligands to transition metals through the formation of organo-metallic complexes. Benzene forms stable full-sandwich andhalf-sandwich organometallic complexes like η6- (C6H6)2Crand η6-(C6H6)Cr(CO)3.
19 The exceptional stability of thesecomplexes is understood on the basis of the 18-electron rule,
Figure 1. Minimum energy structures for six-membered rings forhomogeneous and heterogeneous C/Si nuclearities.

r 2009 American Chemical Society 138 DOI: 10.1021/jz900080q |J. Phys. Chem. Lett. 2010, 1, 136–140
pubs.acs.org/JPCL
leading to the formation of a closed shell configuration.20 Wehave modeled structures for similar complexes using themixed C-Si rings and the all-Si-rings. All the possible con-formations for the Si-substituted rings in complexes wereconsidered. Those reported inFigure 3 andFigure 4 for the fulland half-sandwich complexes, respectively, are the lowestenergy geometries. The binding energies for the complexesare calculated as ΔE (full-sandwich) = Ecomplex - 2Eligand -ECr(0) and ΔE (half-sandwich)= Ecomplex - Eligand - ECr(CO)3.The lowest energy spin-state for the Cr(0) atom correspondsto a septet state (S= 3), while Cr(CO)3 is diamagnetic in theground state. All the calculations for the transition-metalcomplexes are performed at the B3PW91/LANL2DZ level of
theory, which has been shown to be quite satisfactory forcalculationofmetal complexes.21 Thebindingenergies for14,15, 16, 17, 18, 19, and 20 are -12.5, -29.2, -41.3, -50.0,-51.9, -53.1, and -58.2 kcal/mol, respectively. Interest-ingly, in all the complexes, the most stable isomers corre-spond to nearest-neighbor presence of the Si atoms. Forexample, the ortho-isomer complex (16) is more stable thanthe meta or the para isomers. Also, Si atoms on the top andbottom rings of the full-sandwich complexes prefer closerproximity, and thus, in all the complexes other than 18, fullyeclipsed orientation of the Si-substituted rings is preferred. Forexample, the eclipsed conformer (15) is 3.0 kcal/mol morestable than its staggered form. The exceptional structure of18with twisted alternate rings arises as a result of its skewedstructure that leads to steric repulsion in the fully eclipsedconformer. For the all-Si-substituted full-sandwich complex(20), the chairlike structure of the Si6H6 is conserved.
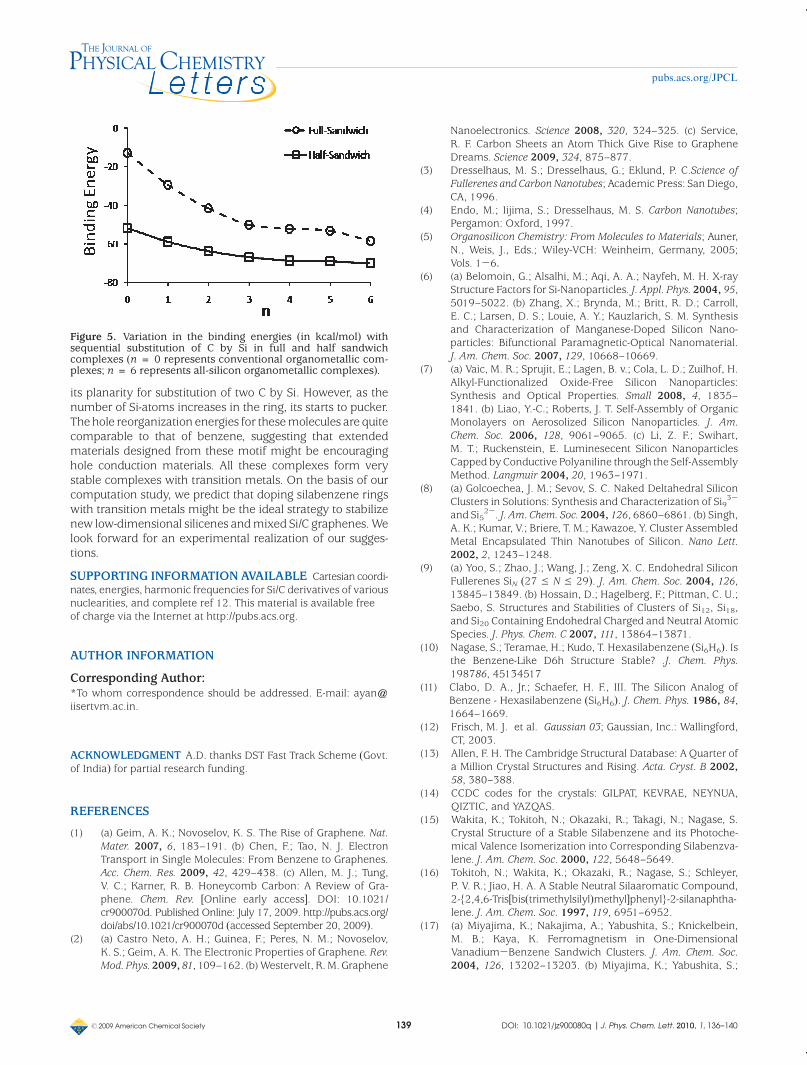
For the half-sandwich complexes, the binding energies for21, 22, 23, 24, 25, 26 and 27 are -51.8, -58.8, -63.6,-66.6, -68.5, -69.0, and -69.7 kcal/mol, respectively.Similar to the full-sandwich complexes, the chair-type struc-ture of Si6H6 is also maintained in (Si6H6)Cr(CO)3 (27). InFigure 5, the profiles for the variation in the binding energiesin the full and half-sandwich complexes with the sequentialsubstitution of C by Si in the rings are shown. The half-sandwich complexes are more stable than the full-sandwichcomplexes for the same degree of substitution. This is under-stood based on the fact that metal (Cr(0)) to ligand (CnSimH6)backbonding increases in the half-sandwich complexes as aresult of the strong σ-donating and π-accepting abilities of thethree carbonyl groups. Also, the sequential substitution of Cby Si stabilize the complexes. Thus, Si-substituted benzenesform more stable complexes with the transition metals thantheir organic counterparts.
In conclusion, we have for the first time, performed anextensive search for various possible structures of sila-sub-stituted benzenes. Interestingly, benzene can easily maintain
Figure 2. HOMO of benzene (I) and SOMOof (II) hexasilabenzenecation and (III) hexasilabenzene anion.
Figure 3. Ground-state minimum energy structures for thefull-sandwich complexes for (CnSimH6)2Cr (n þ m = 6; n = 0-6;m = 0-6).
Figure 4. Ground-state minimum energy structures for the half-sandwich complexes for (CnSimH6)Cr(CO)3 (n þm= 6; n= 0-6;m = 0-6).
r 2009 American Chemical Society 139 DOI: 10.1021/jz900080q |J. Phys. Chem. Lett. 2010, 1, 136–140
pubs.acs.org/JPCL
its planarity for substitution of two C by Si. However, as thenumber of Si-atoms increases in the ring, its starts to pucker.The hole reorganization energies for thesemolecules are quitecomparable to that of benzene, suggesting that extendedmaterials designed from these motif might be encouraginghole conduction materials. All these complexes form verystable complexes with transition metals. On the basis of ourcomputation study, we predict that doping silabenzene ringswith transition metals might be the ideal strategy to stabilizenew low-dimensional silicenes andmixed Si/C graphenes.Welook forward for an experimental realization of our sugges-tions.
SUPPORTING INFORMATION AVAILABLE Cartesian coordi-nates, energies, harmonic frequencies for Si/C derivatives of variousnuclearities, and complete ref 12. This material is available freeof charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author:*To whom correspondence should be addressed. E-mail: [email protected].
ACKNOWLEDGMENT A.D. thanks DST Fast Track Scheme (Govt.of India) for partial research funding.
REFERENCES
(1) (a) Geim, A. K.; Novoselov, K. S. The Rise of Graphene. Nat.Mater. 2007, 6, 183–191. (b) Chen, F.; Tao, N. J. ElectronTransport in Single Molecules: From Benzene to Graphenes.Acc. Chem. Res. 2009, 42, 429–438. (c) Allen, M. J.; Tung,V. C.; Karner, R. B. Honeycomb Carbon: A Review of Gra-phene. Chem. Rev. [Online early access]. DOI: 10.1021/cr900070d. Published Online: July 17, 2009. http://pubs.acs.org/doi/abs/10.1021/cr900070d (accessed September 20, 2009).
(2) (a) Castro Neto, A. H.; Guinea, F.; Peres, N. M.; Novoselov,K. S.; Geim, A. K. The Electronic Properties of Graphene. Rev.Mod. Phys.2009, 81, 109–162. (b)Westervelt, R.M. Graphene
Nanoelectronics. Science 2008, 320, 324–325. (c) Service,R. F. Carbon Sheets an Atom Thick Give Rise to GrapheneDreams. Science 2009, 324, 875–877.
(3) Dresselhaus, M. S.; Dresselhaus, G.; Eklund, P. C.Science ofFullerenes and Carbon Nanotubes; Academic Press: SanDiego,CA, 1996.
(4) Endo, M.; Iijima, S.; Dresselhaus, M. S. Carbon Nanotubes;Pergamon: Oxford, 1997.
(5) Organosilicon Chemistry: From Molecules to Materials; Auner,N., Weis, J., Eds.; Wiley-VCH: Weinheim, Germany, 2005;Vols. 1-6.
(6) (a) Belomoin, G.; Alsalhi, M.; Aqi, A. A.; Nayfeh, M. H. X-rayStructure Factors for Si-Nanoparticles. J. Appl. Phys. 2004, 95,5019–5022. (b) Zhang, X.; Brynda, M.; Britt, R. D.; Carroll,E. C.; Larsen, D. S.; Louie, A. Y.; Kauzlarich, S. M. Synthesisand Characterization of Manganese-Doped Silicon Nano-particles: Bifunctional Paramagnetic-Optical Nanomaterial.J. Am. Chem. Soc. 2007, 129, 10668–10669.
(7) (a) Vaic, M. R.; Sprujit, E.; Lagen, B. v.; Cola, L. D.; Zuilhof, H.Alkyl-Functionalized Oxide-Free Silicon Nanoparticles:Synthesis and Optical Properties. Small 2008, 4, 1835–1841. (b) Liao, Y.-C.; Roberts, J. T. Self-Assembly of OrganicMonolayers on Aerosolized Silicon Nanoparticles. J. Am.Chem. Soc. 2006, 128, 9061–9065. (c) Li, Z. F.; Swihart,M. T.; Ruckenstein, E. Luminesecent Silicon NanoparticlesCapped byConductive Polyaniline through the Self-AssemblyMethod. Langmuir 2004, 20, 1963–1971.
(8) (a) Golcoechea, J. M.; Sevov, S. C. Naked Deltahedral SiliconClusters in Solutions: Synthesis and Characterization of Si9
3-
and Si52-. J. Am. Chem. Soc. 2004, 126, 6860–6861. (b) Singh,
A. K.; Kumar, V.; Briere, T. M.; Kawazoe, Y. Cluster AssembledMetal Encapsulated Thin Nanotubes of Silicon. Nano Lett.2002, 2, 1243–1248.
(9) (a) Yoo, S.; Zhao, J.; Wang, J.; Zeng, X. C. Endohedral SiliconFullerenes SiN (27 e N e 29). J. Am. Chem. Soc. 2004, 126,13845–13849. (b) Hossain, D.; Hagelberg, F.; Pittman, C. U.;Saebo, S. Structures and Stabilities of Clusters of Si12, Si18,and Si20 Containing Endohedral Charged and Neutral AtomicSpecies. J. Phys. Chem. C 2007, 111, 13864–13871.
(10) Nagase, S.; Teramae, H.; Kudo, T. Hexasilabenzene (Si6H6). Isthe Benzene-Like D6h Structure Stable? .J. Chem. Phys.198786, 45134517
(11) Clabo, D. A., Jr.; Schaefer, H. F., III. The Silicon Analog ofBenzene - Hexasilabenzene (Si6H6). J. Chem. Phys. 1986, 84,1664–1669.
(12) Frisch, M. J. et al. Gaussian 03; Gaussian, Inc.: Wallingford,CT, 2003.
(13) Allen, F. H. The Cambridge Structural Database: AQuarter ofa Million Crystal Structures and Rising. Acta. Cryst. B 2002,58, 380–388.
(14) CCDC codes for the crystals: GILPAT, KEVRAE, NEYNUA,QIZTIC, and YAZQAS.
(15) Wakita, K.; Tokitoh, N.; Okazaki, R.; Takagi, N.; Nagase, S.Crystal Structure of a Stable Silabenzene and its Photoche-mical Valence Isomerization into Corresponding Silabenzva-lene. J. Am. Chem. Soc. 2000, 122, 5648–5649.
(16) Tokitoh, N.; Wakita, K.; Okazaki, R.; Nagase, S.; Schleyer,P. V. R.; Jiao, H. A. A Stable Neutral Silaaromatic Compound,2-{2,4,6-Tris[bis(trimethylsilyl)methyl]phenyl}-2-silanaphtha-lene. J. Am. Chem. Soc. 1997, 119, 6951–6952.
(17) (a) Miyajima, K.; Nakajima, A.; Yabushita, S.; Knickelbein,M. B.; Kaya, K. Ferromagnetism in One-DimensionalVanadium-Benzene Sandwich Clusters. J. Am. Chem. Soc.2004, 126, 13202–13203. (b) Miyajima, K.; Yabushita, S.;
Figure 5. Variation in the binding energies (in kcal/mol) withsequential substitution of C by Si in full and half sandwichcomplexes (n = 0 represents conventional organometallic com-plexes; n = 6 represents all-silicon organometallic complexes).

r 2009 American Chemical Society 140 DOI: 10.1021/jz900080q |J. Phys. Chem. Lett. 2010, 1, 136–140
pubs.acs.org/JPCL
Knickelbein, M. B.; Miyajima, K. Stern-Gerlach Experimentsof One-Dimensional Metal-Benzene Sandwich Clusters: Mn-(C6H6)m (M= Al, Sc, Ti, and V). J. Am. Chem. Soc. 2007, 129,8473–8480. (c) Rehaman, A.; Datta, A.; Mallajosyula, S. S.;Pati, S. K. Quantifying Aromaticity at the Molecular andSupramolecular Limits: Comparing Homonuclear, Hetero-nuclear, and H-Bonded Systems. J. Chem. Theory Comput.2006, 2, 30–36. (d) Datta, A.; Mallajosyula, S. S.; Pati, S. K.Nonlocal Electronic Distribution in Metallic Clusters: A Criti-cal Examination of Aromatic Stabilization. Acc. Chem. Res.2007, 40, 213–221. (e) Datta, A.; Pati, S. K. New Examples ofMetalloaromatic Al-Clusters: (Al4M4)Fe(CO)3 (M=Li, Na andK). Chem. Commun. 2005, 5032–5034. (f) Datta, A.; Pati, S. K.Limit to Puckering of Benzene with Sterically CrowdedMolecules: Hexaferrocenylbenzene. Chem. Phys. Lett. 2006,433, 67–70.
(18) (a) Deng, W. -Q; Goddard, W. A., III. Prediction of HoleMobilities in Oligoacene Organic Semiconductor fromQuan-tum Mechanical Calculations. J. Phys. Chem. B 2004, 108,8614–8621. (b) Datta, A.; Mohakud, S.; Pati, S. K. Electronand Hole Mobilities in Polymorphs of Benzene and Naphtha-lene: Role of Intermolecular Interactions. J. Chem. Phys. 2007,126, 144710–144717.
(19) Crabtree, R. H. The Organometallic Chemistry of the TransitionMetals, 5th ed.; Wiley: New York, 2009.
(20) Huheey, J. E.; Keiter, E.; Keiter, R. L. Inorganic Chemistry:Principles of Structure and Reactivity, 4th ed.; Prentice Hall:Upper Saddle River, NJ, 1997.
(21) Szabo, K. J.; Hupe, E.; Larsson, A. L. E. StereoelectronicControl on the Kinetic Stability of β-Acetoxy-Substituted(η3-Allyl)palladium Complexes in a Mild Acidic Medium.Organometallics 1997, 16, 3779–3785.






















![Chapter 19: Benzene and Aromatic Substitution Reactions...Chapter 19: Benzene and Aromatic Substitution Reactions [Sections: 18.2, 18.6; 19.1-19.12] Nomenclature of Substituted Benzenes](https://img.dokumen.tips/doc/110x75/610b07013e4c45556a417ac5/chapter-19-benzene-and-aromatic-substitution-reactions-chapter-19-benzene.jpg)






