Embed Size (px)
Citation preview

Structure and Dynamics of Elastin Cross-linking Domains
by
Aditi Ramesh
A thesis submitted in conformity with the requirementsfor the degree of Master of Science
Graduate Department of BiochemistryUniversity of Toronto
Copyright c© 2015 by Aditi Ramesh

Abstract
Structure and Dynamics of Elastin Cross-linking Domains
Aditi Ramesh
Master of Science
Graduate Department of Biochemistry
University of Toronto
2015
The secondary structure of elastin cross-linking domains has been shown to be sequence
and context dependent, but the role of these domains in the function of elastomeric pro-
teins remains unclear. We use molecular dynamics simulations (MD), circular dichroism
spectroscopy (CD), and nuclear magnetic resonance to probe the conformational equilib-
ria of model elastin-like cross-linking peptides. We tested four recently developed force
fields using MD to select the one that best reproduces the amount of alpha-helix seen in
CD. Simulation studies of the aggregative properties of the cross-linking domains found
that they occasionally interact, but not in any specific way. Additionally, multifaceted
studies of biphasic systems show that these domains do not partition preferentially into
or on the interface of a hydrophobic surface. Further experiments on constructs of cross-
linking and hydrophobic domains will help elucidate how cross-linking modulates the
self-assembly and mechanical properties of elastomeric proteins.
ii

Dedication
To my parents.
iii

Acknowledgements
I would like to thank my supervisors Dr. Simon Sharpe and Dr. Regis Pomes for their
constant guidance and advice throughout my graduate work. They have shaped the
scientist I am today and fostered my deep passion for the biological sciences. I also wish
to thank the members of my supervisory committee, Drs. Hue Sun Chan, Fred Keeley,
and Julie Forman-Kay, for their critical analysis of my work and suggestions along the
way.
I wish to thank the members, both past and present, of both labs for their constant
advice, help, and, most importantly, moral support. I would especially like to thank Drs.
Chris Neale, Loan Huynh and Sarah Rauscher for their invaluable help in getting me
started in the simulation work and teaching me the ropes. I would like to thank Zhuyi
Xue for our daily discussions about elastin and Chris Ing for his scripting help in times
of distress. My deepest, most heartfelt thanks to Dr. Grace Li, Kethika Kulleperuma,
and Dr. Nilu Chakrabarti for their advice about everything in life.
I thank Dr. Patrick Walsh and Jason Yau for leading the way in the peptide work in
the Sharpe lab and teaching me the ins and outs of working with peptides for the first
time. Greg Cole and Dave Davidson are thanked for their constant help and support in
the lab. I have Karen Simonetti to thank for her patience, support and encouragement
as I struggled in the wet lab and tried not to break equipment.
I wish to thank my amazing friends Tracy Stone, Noor Alnabelseya, and countless
other friends I have made over the years from my labs and the rest of the department in
various labs who have kept me sane inside and outside the lab. These friends have heard
me vent and cry through the years and I treasure their patience, love and support. They
kept me going with their encouragement and optimism during the rough and turbulent
times and have become a second family for me in Toronto. I also wish to thank Daniel
Schep for his support and patience during the long months of my thesis writing.
I thank with all my heart my family, especially my parents, for supporting me in all
my endeavours and being the loving, encouraging people they have always been and for
always having faith in my abilities, even though my own belief sometimes faltered.
iv

Contents
List of Tables vii
List of Figures viii
List of Acronyms and Symbols x
1 Introduction 1
1.1 Elastin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.2 Elastin cross-linking . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.3 Elastin structure and mechanism . . . . . . . . . . . . . . . . . . . . . . 8
1.4 Peptide studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.5 Recombinant elastin-like polypeptides . . . . . . . . . . . . . . . . . . . . 10
1.6 Rationale and aims . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2 Methods 14
2.1 Molecular Dynamics Simulations . . . . . . . . . . . . . . . . . . . . . . 14
2.1.1 Molecular mechanics . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.1.2 Force fields . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
OPLS force fields . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
CHARMM force fields . . . . . . . . . . . . . . . . . . . . . . . . 18
CHARMM27 (CHARMM22/CMAP) . . . . . . . . . . . . 18
CHARMM36 . . . . . . . . . . . . . . . . . . . . . . . . . 19
CHARMM22* . . . . . . . . . . . . . . . . . . . . . . . . . 19
AMBER force fields . . . . . . . . . . . . . . . . . . . . . . . . . . 20
v

AMBER ff03w . . . . . . . . . . . . . . . . . . . . . . . . . 20
AMBER ff99sb*-ildn . . . . . . . . . . . . . . . . . . . . . 20
Water models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Periodic boundary conditions . . . . . . . . . . . . . . . . . . . . 21
Temperature and pressure coupling . . . . . . . . . . . . . . . . . 22
2.1.3 System setup . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.2 Biophysical techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.2.1 Peptide synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.2.2 Peptide sample preparation . . . . . . . . . . . . . . . . . . . . . 25
2.2.3 Circular dichroism . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.2.4 Partitioning and analytical RP-HPLC . . . . . . . . . . . . . . . . 25
2.2.5 NMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.3 Data Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3 Results 28
3.1 Choice of force field . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
3.2 Spectroscopic characterization of the monomeric cross-linking domains . 32
3.3 Aggregative properties of the cross-linking domains - a simulation perspective 37
3.4 Tying biophysical results back to simulation . . . . . . . . . . . . . . . . 58
3.4.1 Solution NMR of the model peptides . . . . . . . . . . . . . . . . 58
3.4.2 Circular dichroism spectra calculated helicity of the model peptides 60
3.5 Biphasic systems as a way to model the coacervate . . . . . . . . . . . . 64
4 Discussion 66
5 Future Directions 74
Bibliography 76
vi

List of Tables
2.1 Summary of force fields and water models used. . . . . . . . . . . . . . . 24
vii

List of Figures
1.1 Domain architecture of the tropoelastin monomer . . . . . . . . . . . . . 3
1.2 Cross-linking domain sequences in natural elastin . . . . . . . . . . . . . 3
1.3 Pseudo-periodic hydrophobic domain sequences in natural elastin . . . . 3
1.4 Molecular view of how cross linking is achieved in different types of elas-
tomeric proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.5 Mechanism of cross-linking . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.6 List of model peptides . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
1.7 Position of lysines in the model peptides in a perfect α-helix . . . . . . . 13
2.1 Schematic illustrating the different energy terms of the potential energy
formula for a force field . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.1 PMFs of backbone dihedral angles of A2 in the force fields tested . . . . 30
3.2 Time evolution of α-helix in KA16K and A7KAAKA7 . . . . . . . . . . . 31
3.3 Time evolution of secondary structure in molecular dynamics . . . . . . . 33
3.4 Average α-helix percentages in the A2 peptide for the four force fields tested 34
3.5 Circular dichroism spectra of model peptides in TFE . . . . . . . . . . . 35
3.6 Circular dichroism spectra of model peptides in NaF . . . . . . . . . . . 36
3.7 Circular dichroism spectra of model peptides in MeOH . . . . . . . . . . 37
3.8 Average peptide-peptide distance in dimer simulations . . . . . . . . . . 38
3.9 Histograms of the end-to-end distance of the model peptides in the monomer,
dimer, and tetramer simulations . . . . . . . . . . . . . . . . . . . . . . . 40
3.10 Histograms of the radius of gyration of the model peptides in the monomer,
dimer, and tetramer simulations . . . . . . . . . . . . . . . . . . . . . . . 41
viii

3.11 Histograms of the probability of having 0 through 18 helical residues . . 42
3.12 Time evolution of the radius of gyration of A8KKA8 in CHARMM22* . . 44
3.13 Time evolution of the radius of gyration for a representative A0 dimer
simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.14 Sample contact maps for the A0 dimer system . . . . . . . . . . . . . . . 47
3.15 Snapshots at 100ns of six A0 dimer replicas . . . . . . . . . . . . . . . . 48
3.16 Sample contact maps for the A0 tetramer system . . . . . . . . . . . . . 49
3.17 Snapshots at 100ns of six A0 tetramer replicas . . . . . . . . . . . . . . . 50
3.18 Dimer contact maps for the six model peptides averaged over all replicas 52
3.19 Tetramer contact maps for the six model peptides averaged over all replicas 53
3.20 Comparison of total fraction helix formed by all peptides over the 1, 2,
and 4 peptide simulation systems . . . . . . . . . . . . . . . . . . . . . . 54
3.21 Histograms of pairwise distances between the centers of mass of all peptides 54
3.22 Average fraction helicity for dimer simulations of all peptides with and
without formation intermolecular contacts . . . . . . . . . . . . . . . . . 55
3.23 Average fraction helicity for tetramer simulations of all peptides with and
without formation intermolecular contacts . . . . . . . . . . . . . . . . . 56
3.24 Solution NMR of the A2 peptide . . . . . . . . . . . . . . . . . . . . . . 59
3.25 Secondary chemical shifts of Cα (red) and Cβ (blue) atoms for the A2,
A3, and A16 peptides and A3 at different temperatures . . . . . . . . . . 61
3.26 Comparison of helicity calculated from MD and CD . . . . . . . . . . . . 62
3.27 Average helicity per residue for all the model peptides in CHARMM22* . 63
3.28 Integrated peptide peak areas from RP-HPLC chromatograms for each
phase in octane and octanol partitioning experiments . . . . . . . . . . . 65
4.1 Position of lysines in the helical aggregated multimer simulations . . . . . 71
4.2 Schematic of the proposed cross-linking mechanism in elastin . . . . . . . 72
ix

List of Acronyms and Symbols
A Angstrom
AMBER assisted model building and energy refinement
CD Circular dichroism
DIEA N,N-Diisopropylethylamine
DMF Dimethylformamide
DSSP Dictionary of protein secondary structure
EBP elastin-binding protein
ELP elastin-like peptide
EM electron microscopy
Fmoc Fluorenylmethyloxycarbonyl
fs femtosecond
GROMACS Groningen machine for chemical simulations
HFIP 1,1,1,3,3,3-Hexafluoro-2-propanol
HPLC High-performance liquid chromatography
K Kelvin
MD molecular dynamics
x

nm nanometer
NMR Nuclear magnetic resonance
ns nanosecond
OPLS optimized potentials for liquid simulations
TIP3P transferable intermolecular potential function, three point model
xi

Chapter 1
Introduction
1.1 Elastin
Elastic proteins, which are found in many animal species [1], include abductin (which is
found in the flexible hinge ligament of a scallop’s shell) [2], resilin (found in the cuticle
of many insects) [3], spider silks [4], and elastin [5]. These proteins are found to fulfill
a diverse set of functions and showcase a wide range of properties, with some spider
silks demonstrating incredible rigidity while others are more elastic and resilient. The
mechanical properties of elastomeric proteins motivates the study of these proteins from
a biomaterials and bioengineering perspective.
Elastin is an extracellular matrix protein found in a number of tissues including skin,
blood vessels, and lungs. Elastic fibers are characterized by their ability to undergo
repetitive cycles of stretching and relaxation, properties which are integral to the phys-
iological function of these tissues. This function is achieved with very little turnover
(with the exception of the uterine wall [6]) of the elastin protein, which is an insoluble
biopolymer with a half-life of about 70 years [7], and is thus able to undergo billions
of cycles of stretching and recoil over a lifetime [8]. Elastin confers vital function to
numerous tissues, thus many diseases are associated with abnormalities of elastin pro-
duction and deposition. Fragmentation of elastin or an overall decrease in the amount
of elastin produced leads to diseases such as atherosclerosis [9], pulmonary emphysema
1

Chapter 1. Introduction 2
[10, 11, 12], cutis laxa [13, 11], which is characterized by wrinkled and sagging skin, and
Menkes syndrome, a disease resulting in the inability to absorb copper in the intestines
and distribute it to other cells in the body [14]. Excessive accumulation of elastin leads
to scleroderma, elastomas, and endocardial fibroelastosis, which is characterized by a
thickened lining in the heart [15].
Modern biological and biochemical techniques have made it possible to generate arti-
ficial mimics of elastin, which self-assemble into fibrils and membranes with properties
resembling those of human elastin. Gaining structural insight into the molecular basis of
tissue elasticity will help in the design of both biomimetic materials with application to
medicine (such as grafts for skin and heart tissues) and non-immunogenic materials that
absorb and release drugs with defined binding constants [16, 17, 18, 19].
Tropoelastin, the soluble, monomeric precursor of elastin, is composed of alternating
hydrophobic and cross-linking domains. Figure 1.1 depicts the domain architecture of
the tropoelastin monomer. Self-association of these monomers in vitro occurs by an in-
verse temperature transition called coacervation. There are two types of cross-linking
domains in elastin, the KA-type and the KP-type. The KA-type domains are composed
primarily of stretches of alanine residues containing two or three lysines spaced three or
four residues apart, while the KP-type domains resemble hydrophobic domains in amino
acid sequence with the addition of precisely spaced lysine pairs [20]. The hydrophobic
domains are comprised largely of glycine (G), valine (V), proline (P), and alanine (A)
residues arranged in pseudo-repetitive motifs, or tandem repeats, with typical motifs such
as PGV, GVA, GV, and GGV [21]. Figure 1.2 shows a couple of sequences of each of
the two types of cross-linking domains. Figure 1.3 shows the sequences of some elastin
hydrophobic domain exons in human and chicken, highlighting the key repetitive motifs.
Previous studies indicate that the aggregation of the hydrophobic domains is modulated
by their combined proline and glycine content, whereby elastomeric proteins have a higher
content of proline and glycine than amyloid-forming proteins and peptides [21]. Amyloid
forming proteins, as compared to elastomers, switch from disordered or unfolded states

Chapter 1. Introduction 3
Figure 1.1: Domain architecture of the tropoelastin monomer (adapted from [7]). White
boxes represent hydrophobic domains, yellow represents KA-type cross-linking domains,
maroon boxes are KP-type cross-linking domains.
Figure 1.2: Cross-linking domain sequences in natural elastin. The lysine residues avail-
able for cross-linking are highlighted in red.
Figure 1.3: Pseudo-periodic hydrophobic domain sequences in natural elastin. The
coloured parts of the sequences indicate the various periodic motifs. PGV is shown
in red, GGV in blue, GVA in green and GV in orange.

Chapter 1. Introduction 4
into rigid β-sheet assemblies [22] - a fate that is avoided by elastin through its unique
amino acid composition. A higher content of proline and glycine results in greater struc-
tural disorder [21, 23]. Tropoelastin molecules are thought to aggregate by interactions
between hydrophobic domains, which confer the properties of self-aggregation and exten-
sibility to elastin [24] and cause the protein to deposit in ordered, fibrous structures. In
vitro, the self-association of hydrophobic domains, driven by the hydrophobic effect, is
thought to drive coacervation and, because elastomeric chains do not fold into an ordered,
native structure, their aggregates are amorphous and hydrated and able to undergo en-
tropically driven extension and recoil. The temperature at which coacervation occurs
depends on a number of factors, including ionic strength, pH, protein concentration and
the relative proportions of hydrophobic and hydrophilic residues [25]. Upon coacervation,
the turbidity of the solution increases (as measured by a drop in light intensity in light
scattering experiments). Birefringence is seen at the surface of coacervate droplets, as
shown by dye binding studies [23], implying there is some level of ordering at the surface
of coacervate droplets.
Coacervation is a reversible process; upon cooling a coacervated solution, tropoelastin
monomers go back into solution. However, if a coacervated solution is left to mature,
the coacervate droplets can settle and form an organized fibrillar structure. There are
a specific set of requirements and controlling factors that govern coacervation. As such,
coacervation is thought to occur not by nonspecific aggregation of monomers but by
an increase in the secondary structure and specific intermolecular contacts.. Below the
transition temperature, tropoelastin monomers look like polyamorphous unstructured el-
ements while above this temperature, coacervates of tropoelastin and recombinant pep-
tides with the ability to coacervate take on a fibrillar structure with a diameter of ∼
5nm. These fibres are similar in structure to mature elastin fibres. Occasionally, lateral
association of the fibres or 100-150nm wide banded filaments, representative of the elastin
network, are also formed [26]. Coacervation is an important step in ordering tropoelastin
monomers and coacervation produces the assembled elastin state, but how this process
occurs is largely unknown.

Chapter 1. Introduction 5
Elastin fibres have several other components apart from elastin, including fibrillins, fibu-
lins, and glycoproteins, collectively referred to as microfibrils [20]. Tropoelastin mRNA is
translated at the surface of the rough endoplasmic reticulum in a number of different types
of cells, including the smooth muscle, endothelial, and fibroblast cells [5]. The approxi-
mately 70kDa precursor protein is transported as a nascent chain to the ER lumen where
its signal peptide is cleaved cotranslationally. Tropoelastin is bound by the chaperone
FKBP65 once it is synthesized in the endoplasmic reticulum (ER) and is then trans-
ported to the cell surface via the Golgi apparatus (where it is bound by elastin-finding
protein (EBP)) and is then released and secreted from the cell [5, 27]. It is thought to
deposit on microfibril scaffolds [24] and that alignment of tropoelastin monomers occurs
via coacervation, which results in ordering and alignment for subsequent cross-linking
[21, 1].
1.2 Elastin cross-linking
Elastomeric proteins achieve cross-linking either covalently, as in elastin, or non-covalently,
as seen in spider silks. Covalent cross-links are largely formed by lysine residues, though
some proteins, such as resilin, have covalent cross-links between tyrosine residues. Non-
covalent cross-links are found in many types of spider silks, which achieve cross-linking by
the formation of β-sheets between domains consisting largely of alanine residues. Figure
1.4 depicts different types of cross-linking schemes in elastomeric proteins.
Cross-linking of collagen and elastin occurs through the action of the copper-dependent
amine oxidase lysyl oxidase. It catalyzes oxidative deamination of the ε-amino group in
the lysine side-chain, forming peptidyl α-aminoadipic-δ-semialdehyde (allysine) [29] us-
ing a ping-pong kinetic mechanism [30]. Subsequent spontaneous aldol condensation and
Schiff base reactions with nearby aldehydes or ε-amino groups result in the formation
of di-, tri-, and tetra-functional cross-links such as desmosine and isodesmosine. Figure
1.5 depicts where the lysine residues are catalysed by lysyl oxidase and the subsequent
reactions to form the desmosine linkages.

Chapter 1. Introduction 6
desmosine
di- and tri-tyrosine
polyalanine beta-sheetlysinonorleucine
Figure 1.4: Molecular view of how cross linking is achieved in different types of elastomeric
proteins (cross-linked filament network diagram obtained from [28]). Desmosine linkages
are found in elastin and are tetrafunctional, pyridinium ring-containing cross-links derived
from four lysine residues. The lysinorleucine bivalent elastin cross-link is formed between
two lysine residues. Many spider silks form a polyalanine β-sheet, with β-sheet crystalline
domains surrounded by semi-amorphous domains. Di- and tri-tyrosine linkages are found
in resilin and formed between tyrosine residues interspersed through the elastic repeat
motifs (not in specific cross-linking or hydrophobic domains, which are not found in
resilin).

Chapter 1. Introduction 7
Figure 1.5: Molecular mechanism of cross-linking and the role of lysyl oxidase (figure
obtained and modified from [31]). Lysyl oxidase initiates oxidative deamination of the
lysine side chains, releasing oxygen and ammonia. The aldehyde formed by this reaction
condenses with other aldehydes to form a bivalent aldol condensation product (ACP).
The aldehyde can also react with an unmodified lysine side chain and form a dehydrolysi-
nonorleucine (dLNL). Spontaneous condensation reactions between ACP and dLNL form
tetra-functional cross-links desmosine or isodesmosine.

Chapter 1. Introduction 8
1.3 Elastin structure and mechanism
NMR studies of elastin show that most of the backbone carbonyl carbon atoms are highly
mobile in polar solvents and that hydrated elastin lacks a defined tertiary structure due to
a highly disordered backbone [20, 32, 33, 34, 35, 36]. Previous studies of the hydrophobic
domains have concluded that elastin and elastomeric proteins remain disordered upon
aggregation [21] and that point mutations in these domains suppress phase separation
and promote amyloid-fibril formation [23]. The cross-linking domains provide stability
and mechanical integrity, as without these domains, pulling on elastin would cause the
polymer to fall apart when extended. They have long been predicted to form α-helices
since this would position the lysines on the same side of the helix, allowing the formation
of cross-links between two pairs of lysine residues from adjacent tropoelastin monomers
[37]. While the cross-linking domains of elastin are α-helical in the polymerized, or
cross-linked, state [38], the cross-linking domains of other elastomeric proteins, such as
the alanine-rich domains of spider silk, are made of β-sheets [39]. Recent studies have
shown that single-point mutations (K to Y or K to A) can switch the conformation of
crosslinking domains from α-helical to β-sheet [40]. Furthermore, SSNMR studies have
shown that the cross-linking domains of recombinant elastomeric peptides are unstable
α-helices in the monomeric state, form β-sheet upon coacervation, but are found to be
very stable α-helices when they are cross-linked after coacervation [40]. Thus it is clear
that the cross-linking domains of elastin have the potential to affect the aggregation
propensity of elastin-like recombinant peptides, as well as the mechanical properties of
the assembled protein.
1.4 Peptide studies
The cross-linking domains of elastin and spider silks are rich in alanine, which is re-
ported to be a strong helix former by secondary structure propensity scales [41, 42, 43].
However, experimental [44, 45, 46] and computational work [47, 48, 49] show that polyala-
nine peptides adopt largely random-coil structures in aqueous solution. Most polyalanine

Chapter 1. Introduction 9
peptides, except the very shortest, are insoluble. Solvation of these peptides has been
achieved by inserting polar or charged resides at the ends [50]. A a result, a number
of studies have been conducted with polyalanine peptides interspersed with amino acids
such as lysine, glutamine, and arginine. Some studies have shown that the helicity of
alanine-rich peptides can be increased by introducing charged or polar residues such as
lysine, arginine, or glutamine into the sequence [51], while other studies show that these
residues decrease the helicity of alanine-rich peptides [52]. Yet another study corrobo-
rates the helix stabilizing effect of charged residues, but shows that increasing the number
of these solubilizing residues is a factor in decreasing helix stability [53]. Thus environ-
ment (sequence and context) plays an important role in modulating secondary structure.
Polypeptide sequences have been the subject of much study over the last few decades
[54, 46, 55]. The secondary structure of small peptides is highly dependent on environ-
ment and sequence. In particular, polyalanine peptides have been extensively studied in
different solvents and shown to be highly sensitive to environment and sequence length,
as well as the guest residues [43, 44, 53]. Chakrabartty and coworkers [43] studied guest
amino acids in a series of alanine-based peptides without helix-stabilizing interactions.
Using circular dichroism, they found that the helix propensities of all residues, except
alanine, leucine, and arginine, oppose folding. Marqusee and coworkers [53] found that
16-residue alanine-based peptides containing between 3 and 6 lysines and glutamates
formed stable helices, also determined by circular dichroism. Sung [48] corroborated
these findings by simulation methods (Monte Carlo simulations) on the same peptides.
The 3K peptides formed 60-80% helix while the 6K peptides formed significantly less
helix (only 8-14%). Polyalanine peptides have been studied in aqueous environments,
SDS, TFE, and in the presence of hydrophobic interfaces by both experiment [56, 57]
and molecular dynamics simulations [58, 59, 60] yielding different percentages of the three
major secondary structure types - α-helix, β-sheet and random coil. Best and coworkers
[58] tested various MD force fields and gauged the extent of different secondary struc-
tures formed in the peptides Ala5 and Ac-(AAQAA)3-NH2. They found anywhere from
15 to 30% helix in the Ala5 peptide and as high as 94% helix in the ff03 force field
for Ac-(AAQAA)3-NH2. These studies are of note because the cross-linking domains in

Chapter 1. Introduction 10
tropoelastin are alanine-rich and have been long proposed to act as alpha-helical linkers
between tropoelastin monomers, thus positioning the lysines for cross-linking and con-
ferring strength and stability to the elastin fiber. Notably, the method used to gauge
helicity also affects the percent helicity measured in the studies cited above.
1.5 Recombinant elastin-like polypeptides
Difficulty in obtaining detailed structural information for full length tropoelastin has
prompted the study of synthetic elastin-like peptides (ELPs). Elastin is characterized
by its ability to undergo repetitive stretching and relaxation and return to its original
shape after large deformations. Additionally, elastomeric proteins are characterized by
their ability to self-assemble into a polymeric matrix. Previous studies have shown that
recombinant elastin polypeptides based on repetitive motifs found in human elastin have
physical and mechanical properties that are similar to full length elastin. These peptides
encapsulate key features of the entire protein, such as the ability to self-assemble and
organize into fibrillar structures and form lysine derived cross-links [8, 23, 26]. They also
recapitulate the local structural propensities found in full-length elastin. The means to
recombinantly express proteins has allowed the study of homogenous protein prepara-
tions to study structural characteristics.
Studies of recombinant elastin-like polypeptides have shown that the hydrophobic do-
mains are required for coacervation (the cross-linking domains do not coacervate on their
own) [8]. Electron microscopy (EM) of coacervates of EP 20-24 and EP 20-24-24, where
exons 20 and 24 are hydrophobic domains and the hyphen represents the two cross-linking
exons 21 and 23, show fibrillar structures similar to tropoelastin upon self-aggregation [8].
Proline-rich hydrophobic domains influence coacervation by lowering the temperature at
which it occurs [61]. Helix-breaking proline residues in the hydrophobic domains imply
that the small abount of helix observed is confined to the alanine-rich cross-linking do-
mains [62, 63, 64, 65]. The EP 20-24-24[21Y/A] mutant shows no changes in the amount
of α-helix compared to wild type but has a coacervation temperature that is 7◦C higher

Chapter 1. Introduction 11
[37]. Thus the biophysical properties of the cross-linking sequences are highly susceptible
to point mutations and these domains also play an important role in the mechanical
properties of elastin. Essentially, mutations and domain rearrangements [66] affect the
properties of the materials formed by recombinant elastin-like polypeptides.
1.6 Rationale and aims
The elastic properties of self-assembled elastomeric proteins depend on cross-linking:
how do the cross-linking domains modulate the structure, self-assembly, and mechanical
properties of elastomeric proteins? To answer this broad question, I aim to examine
and characterize how the cross-linking of hydrophobic domains modulates the structure,
self-assembly, and mechanical properties of elastomeric proteins using molecular dynam-
ics (MD) simulations and experiments on model peptides. We adopted a reductionist
approach to study the cross-linking domains. I first studied the cross-linking domains
separately to characterize their inherent structural and self-assembly properties. I then
investigated the effects of lysine spacing on the conformational equilibrium and secondary
structure characteristics of the peptides, as well as their aggregation properties in silico.
Figure 1.6 lists the peptides I have studied. The A0, A1, A2, A4, and A16 peptides have
an alanine background and are 18 resides in length, akin to real cross-linking domains
found in elastin. The lysines are centrally placed so as to study lysine spacing (from
zero through four alanines apart and at the ends) but not register. The A2Y and A3A
peptides are designed to be more like actually cross-linking domains, where the lysines
are C-terminally located and the second lysine sometimes followed by a tyrosine residue
instead of an alanine residue. Substrate recognition by lysyl oxidase is said to be partly
dependent on local conformation, leading to the hypothesis that there must be prior
alignment of the cross-linking domains before cross-linking can occur [26]. Coacervation
has been found to promote the formations of ordered filaments and thus play a role in the
alignment of monomers [23]. As a result, we wanted to probe whether the cross-linking
domains order at the surface of coacervate droplets. The model peptides were studied in
the presence of a hydrophobic phase to determine whether they partition preferentially

Chapter 1. Introduction 12
Ace-AAAAAAAAKKAAAAAAAA-NH2
Ace-AAAAAAAAKAKAAAAAAA-NH2
Ace-AAAAAAAKAAKAAAAAAA-NH2
Ace-AAAAAAAKAAAKAAAAAA-NH2
Ace-AAAAAAKAAAAKAAAAAA-NH2
Ace-KAAAAAAAAAAAAAAAAK-NH2
A0
A1
A2
A3
A4
A16
Ace-AAAAAKAAKYGA-NH2
Ace-AAAAAKAAAKAA-NH2
A2Y
A3A
Figure 1.6: List of model peptides. The A0, A1, A2, A4, and A16 peptides have an
alanine background and are 18 resides in length, akin to real cross-linking domains found
in elastin. The lysines are centrally placed so as to study lysine spacing (from zero
through four alanines apart and at the ends) but not register. The A2Y and A3A
peptides are designed to be more like actually cross-linking domains, where the lysines
are C-terminally located and the second lysine sometimes followed by a tyrosine residue
instead of an alanine residue.
into this phase (mimicking a coacervate droplet surface).
Figure 1.7 shows the position of lysines in an idealized helix. It is of note that the
A2 and A3 peptides, which have spacing akin to those found in natural elastin, position
the lysine residues on the same side of the helix. The other peptides do not position the
lysines as distinctly to one side of the helix.

Chapter 1. Introduction 13
A16
A0 A1 A2
A3 A4
Figure 1.7: Position of lysines in the model peptides in a perfect α-helix. Helical wheels
generated from http://kael.net/helical.htm.

Chapter 2
Methods
2.1 Molecular Dynamics Simulations
The molecular structure and interactions of biological macromolecules can be predicted
in computer simulations from first principles using quantum mechanics [67]. However,
quantum mechanical calculations are computationally expensive. This necessitates a
simplification of the method to calculate the structure and dynamics of biological macro-
molecules in simulation.
2.1.1 Molecular mechanics
Molecular mechanics uses classical mechanics to model atomic interactions and the poten-
tial energy of the system is calculated with force fields. Essentially, the electronic degrees
of freedom are ignored and separated from the nuclear motions, which are the only mo-
tions considered in all calculations (atoms move in the Born-Oppenheimer ground-state
energy surface [68]). A force field is the form and set of parameters of the function used
to describe the potential energy of the particles in the system. Classical force fields have
terms associated with the potential energy of five physically interpretable entities:
1. stretching and compression of bonds
2. bending of angles
3. rotation about torsion angles
4. electrostatic interactions
14

Chapter 2. Methods 15
5. van der Waals forces
These terms can be expressed in the following formula for the potential energy of a
molecular system, V(r):
V (r) =∑
bonds(i)
kd2
(di − d0)2 +
∑angles(i)
kθ2
(θi − θ0)2 +
∑dihedrals(i)
kφ2
(1 + cos(nφi − φ0))
+∑
impropers(i)
kψ2
(ψi − ψ0)2 +
∑non−bondedpairs(i,j)
4εij[(σijrij
)12 − (σijrij
)6] +∑
non−bondedpairs(i,j)
qiqjεDrij
(2.1)
The first term in equation 2.1 is the bond stretching term, where each bond is approx-
imated as a spring, so the potential energy becomes the harmonic potential as determined
by Hooke’s law. This potential is suitable for small deviations from the initial bond
length. The second term is the potential energy upon deformation of angles. The third
term is the torsional term, and represents the potential energy of the system in terms of
rotations about the dihedrals. The fourth term considers the planarity of geometrically
flat groups and chirality [67]. The last two terms consider the non-bonded components
of the potential energy. They are the van der Waals interactions Lennard-Jones potential
and the electrostatic potential respectively, where rij is the distance between nuclei i and
j. These interactions are shown in Figure 2.1.

Chapter 2. Methods 16
rijφ
θ
d
Figure 2.1: Schematic illustrating the different energy terms of the potential energy
formula for a force field. d is the bond length, θ is the angle between topological triples
of atoms, φ is the dihedral angle and rij is the distance between nuclei i and j.

Chapter 2. Methods 17
2.1.2 Force fields
The formula for the potential energies is one component of a force field. The other is a set
of parameters for each atom type, including the partial charges for individual atoms (q),
van der Waals radius (σ), atomic mass, spring constant values for each potential energy
term (kd, kθ, kφ, kψ), and equilibrium values of various bond lengths, bond angles, and
dihedral angles (d0, θ0, φ0, ψ0).
The parametrization or re-parametrization of force fields is a complex process as there
are an endless array of parameter combinations where one subset of parameters can
compensate for another subset in order to reproduce experimentally observed structural
and energetic data. Each potential energy term needs to be calibrated relative to quan-
tum mechanical data, vibrational spectra, crystal information and other experimental
data. Recent advances in computer hardware and software have allowed long all-atom
molecular dynamic simulations on the tens of µs to ms timescale. These studies have en-
abled detailed understanding of protein folding and conformational dynamics on a longer
timescale than ever before. However, these long-scale simulations show inaccuracies in
the physical models on which the force fields are based and inconsistencies with experi-
mental data [69].
There are a number of systematic projects underway in many academic labs to refine
the parametrization of various force fields. Each force field is parametrized with re-
spect to experimental values and optimized for different systems. There are three pop-
ular force fields that are commonly used in current molecular dynamics work. Below, I
briefly discuss the major force field developments pertinent to my project and the key
re-parametrizations and empirical comparisons for recent force fields. It is important to
consider and validate different force fields, as they are parametrized in different ways and
in reference to different empirical data. The same simulation system may adopt different
conformational ensembles in different force fields. It is therefore useful to consider how
each force field in my validation studies was parametrized.

Chapter 2. Methods 18
OPLS force fields
The OPLS (Optimized Potentials for Liquid Simulations) force fields were first developed
in the the early 1980s [70]. The potentials in this set of force fields were developed for
simulating liquid state properties (initially water, but later more than 40 other organic
liquids) [68]. The emphasis was on non bonded interactions and these were compared
to liquid-state thermodynamics and optimizing charges and van der Waals parameters
from simulations of pure liquids. The weights for each fitting point were based on the
magnitudes of the potential-energy gradient.
Quantum chemical data was used to evaluate the current OPLS-AA force field (back-
bone and side chain torsional parameters were refit to QM data) and the transferability
of parameters was demonstrated using the same alanine dipeptide-fitted backbone tor-
sional parameters for all other dipeptides (with appropriate side-chain refitting) and the
alanine tetra-peptide. This re-parametrization of Coulombic charges and van der Waals
interactions was validated by reproducing gas-phase energies of complex formation of
heats of vaporization and densities of pure model liquids. [71, 72].
CHARMM force fields
The CHARMM (Chemistry at HARvard using Molecular Mechanics) force fields were
also initially developed in the early 1980s [73]. Parametrization was initially achieved
using model compounds such as form amide and N-methylacetamide and aimed to get
balanced interactions between solute-water and water-water interactions. The Lennard-
Jones parameters were refined to reproduce densities and heats of vaporization of liquids.
CHARMM27 (CHARMM22/CMAP) This particular force field was developed by
the MacKerell lab in 2004 [74]. In addition to the preceding versions of the CHARMM
force field, MacKerell’s group performed additional parameter optimization via Monte
Carlo simulated annealing. The potential energy function was extended to contain pep-
tide backbone φ, ψ dihedral cross terms or φ, ψ grid-based energy correction terms.
Empirical adjustments to grid-based corrections for alanine and glycine were applied to
account for their systematic differences in the helical and sheet regions.

Chapter 2. Methods 19
QM and MM calculations on alanine, glycine, and proline dipeptides were combined
with MD simulations of proteins in crystal and aqueous environments. Monte Carlo sim-
ulated annealing was used to optimize parameters and MD simulations of seven proteins
in crystalline environments were used to validate these parameters.
CHARMM36 The parametrization of the CHARMM36 force field involved a refine-
ment of the backbone CMAP potential for non-Gly, non-Pro residues and compared to
solution NMR data for weakly structured peptides [75]. This resulted in a force field that
was intended to rebalance the α-helix and extended regions of the Ramachandran map,
correcting the overwhelming helical bias seen in CHARMM22/CMAP.
Re-parametrization was performed using simulation of Ala3 and other short peptides,
as well as replica exchange simulations of Ac-(AAQAA)3-NH2, solute tempering simula-
tion of unfolded proteins in urea, and crystal structure simulations. Quantum mechanical
calculations of glycine and proline dipeptides were performed and 2D CMAP potentials
were compared to NMR 3J scalar couplings (Ala5) and carbonyl chemical shifts (Ac-
(AAQAA)3-NH2) data to optimize backbone parameters - comparisons were made to
calculated NMR chemical shifts and J couplings from SPARTA+ [76]. Additionally, un-
folded ubiquitin and GB1, a 19-residue disordered fragment of hen lysozyme and dimeric
coiled-coil 1U0I were used as test systems.
CHARMM22* This force field is based on the CHARMM22 force field [77]. The
details of the re-parametrization can be found in [78], though generally CHARMM22* is
CHARMM22 with newly modified backbone torsions potentials. The CMAP corrections
were replaced with new backbone torsions terms for all residues, except proline and
glycine. Partial charges for asparagine, glutamate, and arginine side chains were modified
to get a better description of salt-bridge interactions as well as χ1 and χ2 torsion terms
for asparagine side chains as done for AMBER ff99SB [79].
The backbone torsions parameters for non-proline, non-glycine residues were opti-
mized to match the φ-ψ energy map of di-alanine and NMR data on polyalanine peptides
in water. Additionally, simulations were conducted by the Shaw lab on the villin head-

Chapter 2. Methods 20
piece and Cα RMSDs from PDB structures, the order of helix formation, and various
kinetic and thermodynamics properties were evaluated. Of note is that simulations were
conducted with CHARMM-modified TIP3P (a flexible water model)[80]. Each force field
discussed has been parametrized with a different water model. A brief discussion of the
different water models used is found in the following section.
AMBER force fields
The AMBER (Assisted Model Building and Energy Refinement) force field was initially
developed in the early 1980s in Peter Kollman’s group [81, 82].
AMBER ff03w The AMBER ff03w force field [83] was re-parametrized from previous
versions of the force field with small backbone modifications to match the population
of helical states obtained with a new water model, a highly optimized TIP4P/2005,
to experiment. Experimental data was used to re-parametrize the backbone dihedral
potential correction for AMBER ff03* so that the fraction helix for the 15 residue pep-
tide Ac-(AAQAA)3-NH2 was correctly reproduced in optimized TIP4P/2005 water. To
compare to experiment, SPARTA was used to compute temperature-dependent carbonyl
chemical shifts for the same peptide.
AMBER ff99sb*-ildn The Shaw lab optimized side-chain torsion potentials of the I,
L, D, and N residues to parametrize AMBER ff99sb*-ildn [79]. The re-parametrization
was done to match new quantum mechanical calculations. Millisecond scale molecular
dynamics simulations were performed in explicit solvent to validate the resulting force
field against experimental NMR measurements. Problematic residue types were identified
by comparing the distribution of χ1 dihedrals in simulations of short helical peptides with
statistics for residues in helices in the PDB. The water model used in this work was TIP3P
or TIP4P-Ew (depending on the system used for validation).

Chapter 2. Methods 21
Water models
Water models are used to simulate hydrogen bonding and aqueous solutions. These mod-
els are derived from quantum mechanical calculations and comparisons to experiments
(as with the parametrization of any force field or computational model). The number
of interaction points, the rigid or flexible nature of the model, and whether polarization
effects characterize different types of water models.
In the work that follows, I use a three-site water model (TIP3P) and a four-site water
model (TIP4P), depending on the force field used. A three-site model is characterized by
three interactions points, which correspond to the three atoms of a water molecule. The
TIP3P model [84] is a three-site, rigid model, with a 104.5◦ HOH angle. This water model
is rigid, implying that only non-bonded interactions are considered. That is, holonomic
constraints, which are constraints on coordinates, are applied on all bonding interactions.
The TIPS3P is a flexible version of this water model. TIP4P is a four-site water model,
whereupon a dummy atom with a negative charge is used to improve the electrostatic
distribution around the entire water molecule. The TIP4P/2005 model extended the
TIP4P model to simulate the entire phase diagram of condensed water.
Periodic boundary conditions
Periodic boundary conditions (PBC) are used to approximate a much larger/infinite
system and minimize the artifacts from phase boundaries by replicating a unit cell, or
simulation box, along its axes. Net neutrality of the system is important in order to
avoid summing to an infinite charge. Interactions between nearest neighbours are the
only ones counted, so as to avoid duplication of interactions. Care must also be taken
to ensure a large enough simulation box so that artifacts from unphysical interactions
do not arise. For example, if the box is too small, a molecule can interact with its own
image in a neighboring box. That is, the ’head’ of a molecule could ostensibly interact
with its own ’tail’, leading to an unphysical interaction.

Chapter 2. Methods 22
Temperature and pressure coupling
The canonical, or NVT, ensemble ensures conservation of the number of particles (N),
volume (V), and temperature (T) in the system. The energy of endothermic and exother-
mic processes are exchanged with a thermostat. Velocity rescaling considers the velocities
at each step and rescales them so that the kinetic energy yields the target temperature.
The Nose-Hoover thermostat allows temperature fluctuation about an average value and
this oscillation is minimized by the use of a damping factor that controls the oscillation.
The canonical ensemble is produced with this thermostat [85, 86]. The Berendsen ther-
mostat ensures fast equilibration by allowing exponential decay of temperatures to the
target value [87].
The isothermal-isobaric, or NPT, ensemble ensures conservation of particles (N), pres-
sure (P), and temperature (T). In addition to a thermostat, a barostat is required to con-
serve pressure. This equilibration setup can be likened to an open flask equilibrated to
ambient temperature and pressure. As with thermostats, there are a couple of schemes
to pressure-couple the system to the environment. Depending on the type of integra-
tion used (leap-frog and velocity Verlet are two common methods), the pressure coupling
method will vary. For leap-frog, the Berendsen or Parinello-Rahman [88] barostats can be
used, whereas the Martyna-Tuckerman-Tobias-Klein barostat [89] can be used in com-
bination with the Nose-Hoover thermostat for velocity Verlet integrated (a numerical
method used to integrate Newton’s equations of motion) systems [90].
2.1.3 System setup
A significant part of my initial molecular dynamics simulations involved testing various
force fields on my systems and comparing to experiments I performed on the same pep-
tides. I needed a force field that would recapitulate the same average secondary structure
I saw in my biophysical experiments. To this end, of the the force fields discussed above,
I tested OPLS-AA with TIP4P (the initial force field I intended to use before I noticed
significant deviations in secondary structure and dynamics in this force field as compared
to experiment), CHARMM22* with TIPS3P, AMBER ff03w with TIP4P/2005, and AM-

Chapter 2. Methods 23
BER ff99SB*-ILDN with TIP3P. Table 2.1 summarizes the four force fields tested in this
work.
All peptides were built in PyMOL and solvated in water as terminally capped pep-
tides (N-terminal acetylation, C-terminal amidation). A cubic box was used with size
and number of waters varying depending on the size of the system. Protonation states of
lysines in the systems simulated were proposed to be that found at neutral pH. The sys-
tem was equilibrated at 300K and 1atm for 100ps in the NVT ensemble with the velocity
rescaled, modified Berendsen thermostat. Another 100ps of equilibration was performed
in the NPT ensemble with the Berendsen thermostat and the Parinello-Rahman baro-
stat. All bonds involving hydrogen atoms were constrained using a fourth-order LINCS
algorithm. A 10A cutoff was used for Lennard-Jones interactions and short-range electro-
static interactions. Electrostatic interactions were calculated using Particle Mesh Ewald
(PME) summation fourth-order interpolation with a grid size of 0.16nm and pair lists
were updated every 10fs with a 10nm cutoff. Covalent bonds on hydrogen atoms were
constrained using the LINCS algorithm.
2.2 Biophysical techniques
2.2.1 Peptide synthesis
Peptides were synthesized by solid-phase Fmoc synthesis [91] using either PAL-PEG-
PS resin or Fmoc-alanine-Rink amide-MBHA resin. Both of these resins are amidated,
meaning the first, or C-terminal, residue coupled to the resin will be amidated at the C-
terminus. Peptides were acetylated on the resin with 96:1:3 mixture of DMF:DIEA:acetic
anhydride and cleaved with an 88:2:5:5 mixture of TFA:TIPS:phenol:water and ether
precipitated. The peptides were lyophilized for storage and subsequent purification. The
peptides were dissolved in 21% acetonitrile in water and purified by C18 reverse-phase
HPLC in a 10% to 90% acetonitrile gradient. Peptide identity was confirmed by MALDI-
TOF mass spectrometry.

Chapter 2. Methods 24
Force&Field&
Water&
Mod
el&
Orig
in&
Refin
emen
t/Va
lidation&
OPLS%AA
'TIP4
P'Jorgen
sen'
lab'
Reprod
ucing'gas%ph
ase'en
ergies'of'com
plex'heats'of'
vapo
rization'and'de
nsities'of'p
ure'mod
el'liqu
ids'
Ambe
r'ff03w
'TIP4
P/2005
'Be
st'lab'
Helix%coil'transition
'in'alanine
%based
'helical'pep
tides'
Ambe
r'ff99SB*
%ILDN
'TIP3
P'Shaw
'lab'
Optim
ized'sid
e%chain'torsion'po
tentials'of're
sidue
s'that'd
iffered
'from
'PDB
'statistics'
CHAR
MM22*'
TIP3
P'Shaw
'lab'
Alph
a/be
ta'balance'
!
Tab
le2.
1:Sum
mar
yof
forc
efiel
ds
and
wat
erm
odel
suse
d.

Chapter 2. Methods 25
2.2.2 Peptide sample preparation
5-10mg of lyophilized peptide were solubilized in 200 µl of HFIP and sonicated for 10
minutes. HFIP was subsequently dried under N2(g) and peptide HFIP films were sol-
ubilized in 500 µl water and sonicated for 10 minutes. All samples were centrifuged at
13000rpm and the top 400 µl of sample was extracted for peptide stocks. Peptide concen-
trations were determined using the Waddell method [92]. Equation 2.2 shows how the
concentration of peptides was calculated based on a cuvette with a 1cm path length.
144µg/ml(A215 − A225) (2.2)
Peptide concentrations were measured on a nanophotometer (MBI Lab Equipment).
2.2.3 Circular dichroism
Peptide stocks, prepared as in section 2.2.2, were diluted to 50 µM for CD samples.
Circular dichroism measurements were made using a JASCO J-810 spectropolarimeter
in a 1.0-mm quartz cell. Single spectrum measurements were performed at 10C and
temperature melts were performed starting at 10◦C and melting at 1◦C/min until 80◦C
with a Jasco PFD-425S Peltier temperature controller. Measurements were performed
at a scan rate of 1nm/sec in 0.1nm steps. Each reported spectrum is the mean of three
stepwise scans between 250nm and 190nm averaged for 1s at each wavelength.
2.2.4 Partitioning and analytical RP-HPLC
Partitioning experiments in vitro [93, 94] were performed in 1.5ml glass vials. Peptide
stocks (made as described in section 2.2.2) were diluted to 50 µM in 750 µl. This aqueous
peptide sample was added to glass vials and 750 µl of the hydrophobic solvent (either
octane or octanol) was added on top. Vials were capped and inverted a few times and
then placed on a nutator overnight for equilibration. Phases were separated by extracting
650 µl of the top layer (the hydrophobic phase), 650 µl of the bottom layer (the aqueous
phase) and the remaining 200 µl was considered the interface. Quantitative HPLC was
performed with a Waters HPLC system whereby 200 µl of each phase were injected into

Chapter 2. Methods 26
a 200 µl loop and run on an Xbridge BEH130 C18 analytical column equilibrated with
10% acetonitrile in water. All phases were run on a 10% to 90% acetonitrile gradient.
All octane samples were dried down after separating the three phases and re-solubilized
in 400 µl water. Peptide was quantitated by integrating the area under the peptide peak
and areas were normalized by volume injected relative to volume in the initial phase.
The interface was assumed to be of negligible volume.
If A, I, and H are the normalized aqueous, interface, and hydrophobic peak areas and a,
i, and h are the raw integrated peak areas, the following equations show how normalized
areas were calculated for octanol samples:
A = a750200
H = h750200
I = i-(A100750
+H100750
)
The equations below show how normalized areas were calculated for octane samples:
A = a400200
H = h400200
I = i400200
-(A100750
+H100750
)
2.2.5 NMR
I performed a series of Correlation Spectroscopy (COSY), Total Correlated Spectroscopy
(TOCSY), Heteronuclear Single Quantum Coherence (HSQC), and HSQC-TOCSY ex-
periments on the A2, A3, and A16 peptides at temperates at, below, and above their
melting temperatures.
A COSY experiment allows identification of spins that are coupled to each other.
TOCSY spectra show through bond correlations through spin-spin coupling. Both the
COSY and TOCSY experiments are homonuclear. A 1H-13C HSQC experiment yields
correlations between aliphatic carbons and their attached protons. All the unique protons
attached to the heteronucleus considered, in this case 13C, are seen. This allows us to
track the chemical shifts of various atoms (or types of atoms) in the peptide as a function
of position and temperature.

Chapter 2. Methods 27
Solution NMR samples were prepared by dissolving lyophilized peptides in HFIP and
drying as described previously. Samples were run in a Bruker Avance III spectrometer
with a 1H frequency of 600MHz. Samples were approximately 500µM peptide, 20mM
sodium phosphate buffer, and 10% D2O. Temperature was controlled using a variable
temperature unit and a high flow rate of dry air for below room temperatures. All
samples were run in a 5mm PATXI 1H/D-13C/15N Z-GRD probe. Binomial water
suppression was applied to all pulse sequences and isotropic mixing was achieved using a
DIPSI sequence in the TOCSY and HSQC-TOCSY experiments. Mixing times were 0.08
seconds for all TOCSY experiments and 0.06 seconds for all HSQC-TOCSY experiments
and the relaxation delay of 2 seconds was used for all experiments.
2.3 Data Analysis
Molecular dynamics simulations were all performed in GROMACS version 4.5.5 [95, 96]
on the SciNet [97] or MP2 computing clusters using precompiled GROMACS on the
cluster. Most analyses were performed using GROMACS analysis tools or using Python
scripts written for that purpose in conjunction with Matplotlib for graphing analyzed
data. NMR data was processed in NMRPipe [98] and subsequently analyzed in CCPNMR
[99].

Chapter 3
Results
Molecular dynamics simulations yield information about low-population intermediate
states and conformational dynamics that many other biophysical methods involving en-
semble averaging cannot provide. Quantitative comparison of MD studies with exper-
iments will give us insight into protein biophysics at the atomic level and give us the
ability to devise more accurate force fields by using empirical evidence [58].
The following chapter delves into validation of a few recent force fields for the elastin
cross-linking peptides studied by comparing molecular dynamics computational results
to biophysical observables in vitro.
3.1 Choice of force field
As summarized in the above Methods section, the choice of force field is an important
consideration when performing biomolecular simulations. The peptide systems that I
have simulated have the ability to sample a diverse set of conformations and, as such, a
force field that best recapitulates these properties is optimal for our studies. The force
fields OPLS-AA, CHARMM22*, AMBER ff99sb*-ildn, and AMBER ff03w were used
to conduct simulations of the A2 peptide with an acetylated N-terminus and amidated
C-terminus as well as amidated and acetylated A18. The Methods section describes the
techniques and simulation protocol used. Each system was run 100 times for a 100ns
each run, with the monomeric peptide starting in the extended state. The first 50ns was
28

Chapter 3. Results 29
discarded before analysis.
The cross-linking peptides have historically been hypothesized to be α-helical. This
is because this would facilitate cross-linking by bringing the lysine residues of one cross-
linking domains onto the same helical face [40]. We thus performed molecular dynamics
simulations and investigated the secondary structure of the peptides. More specifically,
we measured the extent of α-helicity over the course of the simulation. To compare the
conformational space explored by the peptides, we plotted the potentials of mean force
(PMFs) of all the backbone dihedral angles in the peptide for each of the four force fields
tested. Figure 3.1 shows the results of this analysis.
The predominant energetic basin in CHARMM22* is in the α-helical region of the
Ramachandran plot. The OPLS and AMBER ff99sb*-ildn force fields have a much more
extended basin in the same part of the plot, extending past the canonical α-helical bounds
(roughly φ and ψ angles of -64 +/- 7, -41 +/- 7). The ff03w force field has a significant
basin in the polyproline/β-sheet region (roughly φ and ψ of -120, +120).
Additionally, I used the dictionary of protein secondary structure (DSSP) [100] in-
cluded in Gromacs as an analysis tool to compute secondary structure. DSSP identifies
intra-backbone hydrogen bonds with a purely electrostatic definition, where a +0.20e
partial charge is assigned to amide hydrogens, -0.42e to carbonyl oxygens and the op-
posites to the amide nitrogens and carbonyl carbons respectively. A hydrogen bond is
defined as an energy E less than -0.5 kcal/mol where
E = 0.084[1
rON+
1
rCH− 1
rOH− 1
rCN] · 332kcal/mol (3.1)
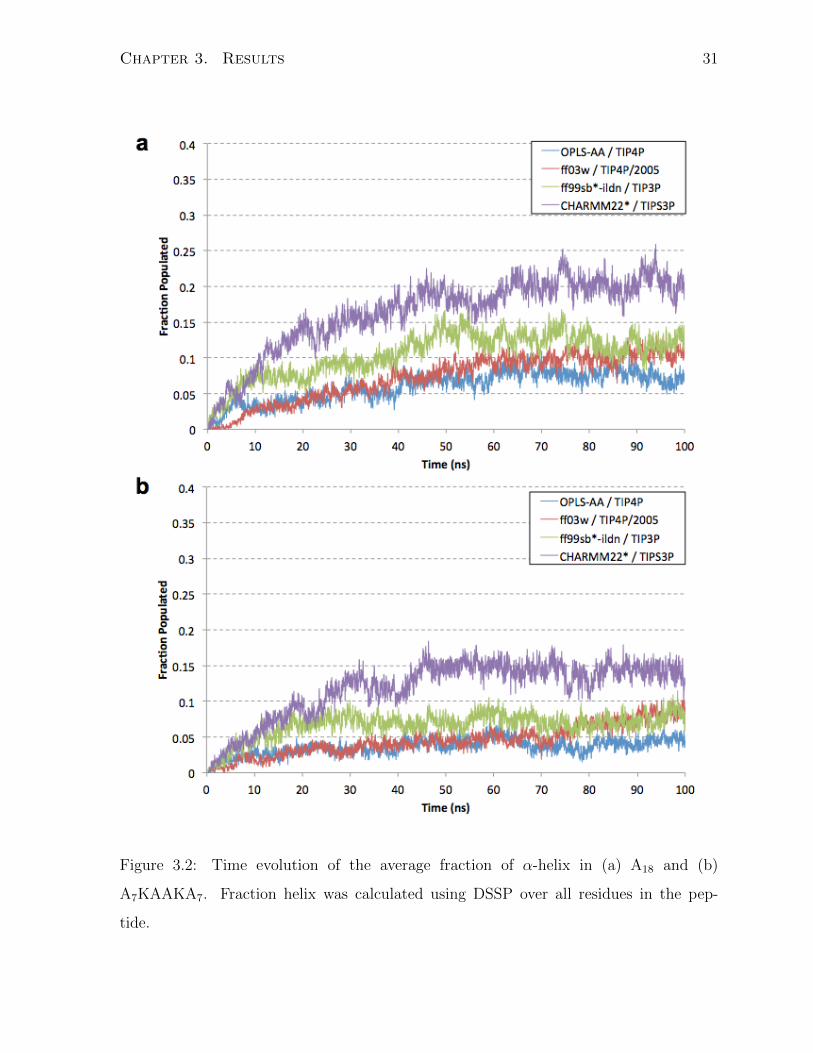
The computed fraction of α-helix by DSSP are plotted in Figure 3.2 for the acetylated
and amidated A18 peptide as well as the A2 peptide from my set of model peptides.
The graphs show the fractional amount of helix as a function of time. DSSP calculated
values yield significant amount of helix in the CHARMM22* force field, which increases
over the course of the simulations since the peptides start from an extended state in
the monomer simulations. Comparatively, the other force fields do not yield very helical
peptides at all, with significantly less than 15% helix in the AMBER ff03w, ff99sb*-ildn,
and OPLS-AA force fields.

Chapter 3. Results 30
Figure 3.1: Potentials of mean force of the φ and ψ angles of the A2 peptide (over all
residues) for the four force fields tested. The PMF is given by W(φ,ψ) = -RTlnρ(φ,ψ),
where R is the gas constant (8.3145 JK−1mol−1), T is the temperature in Kelvin (K),
and ρ(φ,ψ) is the probability distribution of φ and ψ.
Chapter 3. Results 31
Figure 3.2: Time evolution of the average fraction of α-helix in (a) A18 and (b)
A7KAAKA7. Fraction helix was calculated using DSSP over all residues in the pep-
tide.

Chapter 3. Results 32
Since CHARMM22* produces the most α-helix, I investigated this force field further.
I wanted to see to what extent the other secondary structures were formed. The amount
of β-sheet and β-turns formed were negligible, as seen in Figure 3.3. Essentially, the
predominant secondary structure is α-helix. This is of note because the cross-linking
domains have long been hypothesized to be α-helical and we know that the secondary
structure of the cross linking domains in the coacervate is actually β-sheet [40].
Figure 3.4 shows the average helicity (using DSSP constraints) for the A2 and A18
peptides in the four force fields tested. As shown in the time trajectories above, the
overall helicity is notably higher in CHARMM22* than for the other force fields.
The goal of the force field validation was to obtain an amount of α-helix in simula-
tion that was comparable to results obtained in biophysical experiments in vitro. The
following section details the results from some of these experiments.
3.2 Spectroscopic characterization of the monomeric
cross-linking domains
The analysis in the previous section focused on the secondary structure of the monomeric
peptides in water. Comparing the above simulation results with in vitro spectroscopic
secondary structure information yields one metric of determining force field quality - or
at least a force field that will recapitulate the secondary structure properties seen in
vitro. To this end, I performed a series of circular dichroism experiments to probe the
secondary structure of the model peptides in various conditions.
I first studied these peptides in TFE, a known helix stabilizer, to determine the effect
of this solvent on the helicity of the model peptides. As expected, TFE increased helicity
in all the peptides tested, as seen in Figure 3.5. This is denoted by a more negative [θ]222.
The A2Y and A3A peptides have a stronger random coil component in water (0% TFE)
that disappears upon addition of TFE.
However, fitting the melting curves shown on the lefthand panels in Figure 3.5 proved
difficult since there is no folded baseline in the curves. We see that TFE stabilizes the

Chapter 3. Results 33
Figure 3.3: Time evolution of the average fraction of α-helix, β-sheet and random coil in
(a) A2 and (b) A16. Fraction of secondary structure was calculated using DSSP over all
residues in the peptide.

Chapter 3. Results 34
Figure 3.4: Average α-helix percentages in the A2 peptide for the four force fields tested.
Fraction helix was calculated using DSSP over all residues in the peptide.
helix, but these peptides are never strongly helical and ‘fully folded’. The more ‘realistic’
cross-linking domains, A2Y and A3A, retain the same global properties as the other
model peptides, except that they are overall less helical as monomers.
Coacervation is promoted by an increase in salt concentration in vitro (among other
factors). Therefore, I wanted to monitor the helicity of these peptides in increased sodium
fluoride concentration. Although sodium chloride is ordinarily used to represent a phys-
iological salt, circular dichroism experiments preclude the use of large concentrations of
chloride because it absorbs strongly below 200nm. Anions such as sulphate or fluoride do
not absorb significantly in this range [101] and allow us to increase the ionic concentra-
tion to levels used in vitro for coacervation. Figure 3.6 shows that an increase in [NaF]
does not noticeably change the intensity of the α-helical minimum at 222nm.
Previous simulation studies in the Pomes lab have indicated that the hydrophobic
domains of elastin have a greater propensity to form β strands in methanol versus water
[102]. A higher secondary structure propensity (namely, β-sheet) is seen for amyloido-
genic sequences like (GV)18, which is similar to the cross-linking poly(GA) sequences in

Chapter 3. Results 35
Figure 3.5: Circular dichroism spectra of model peptides in (a,d) 0% TFE, (b,e) 20%TFE,
and (c,f) 50%TFE. The left-hand panels show temperature melts following the ellipticity
at 222nm as a function of temperature while the right-hand panels show far-UV CD
spectra at 10 ◦C.

Chapter 3. Results 36
Figure 3.6: Mean residue ellipticity at 222nm from CD in varying [NaF] for the A2, A3,
A16, A2Y, and A3A peptides (∼ 66.5 µg/ml) in water and sodium fluoride at 10 ◦C for
each of the model peptides.
spider silks, than in elastomeric sequences.
Methanol is a poorer solvent of the peptide backbone than water and I wanted to
see if the cross-linking domains showed any differences in CD spectra upon altering
the concentration of methanol in the sample. If methanol preferentially solvated the
side chains relative to water, then marked differences in sidechain hydrophobicity would
impact solvation. Figure 3.7 shows that the A2Y peptide has a strongly enhanced helicity
upon an increase in the concentration of methanol. However, the A2Y peptide has
a strong random coil component in its CD spectrum, so this drastic increase in the
ellipticity at 222nm simply means that the conformational ensemble favoured is more
helical at higher concentrations of methanol. Methanol has a smaller impact on the
other two peptides tested, A2 and A3A, but helicity is still slightly increased upon an
increase in methanol concentration.

Chapter 3. Results 37
Figure 3.7: Mean residue ellipticity at 222nm from CD in varying [MeOH] for the A2,
A2Y, and A3A peptides (∼ 66.5 µg/ml) in water and methanol at 10 ◦C.
3.3 Aggregative properties of the cross-linking do-
mains - a simulation perspective
A large part of the data in the previous sections details the properties of the monomeric
cross-linking domains. In simulations, this means one peptide solvated in a box of wa-
ter. In biophysical experiments, one can consider the peptide to be monomeric if it is
solubilized by water and not aggregating in solution.
In order for cross-linking to occur, two cross-linking domains from different tropoe-
lastin monomers must come together such that a desmosine or isodesmosine linkage can
be formed. The monomer molecular dynamics simulations show the intrinsic properties
of the peptides in an aqueous environment. However, studying the structure and dy-
namics of the peptides in the presence of each other better recapitulates cross-linking
conditions. To this end, I performed simulations of both two peptides and four peptides
in a box of water.
We hypothesize that the cross-linking domains have a role in the assembly and order-

Chapter 3. Results 38
ing of the elastin fiber. Having characterized these domains as monomers, we wanted to
then investigate the aggregation of these domains. MD simulations were conducted for
all six peptides in the lysine spacing table where two peptides were solvated in a box of
water and run for 100ns starting from 100 different starting conformations (which were
equilibrated conformations from monomer simulations of the same peptide). Figure 3.8
shows that, for all peptides, the two monomers come closer together as the simulation
progresses.
Figure 3.8: Average peptide-peptide distance in dimer simulations. This was calculated
by finding the distance between the centres of mass of each peptide chain in the simula-
tion.
Figure 3.9 and Figure 3.10 encapsulate two macromolecular properties of the peptides
over the course of the simulation: the end-to-end distance and the radius of gyration.

Chapter 3. Results 39
The end-to-end distance is the straight-line distance between the ends of a polymer while
the radius of gyration is the root-mean-square distance of the segments of a polymer from
its centre of mass [103]. Both of these metrics provide a measure of peptide size.
The end-to-end distance is largely unchanged, both in terms of average value and
distribution, over all peptides. The A16 peptide has a slightly narrower distribution over
all system sizes. This correlates with a peptide that has a tendency to be more helical
than the rest, and perhaps more compact as a result. The radius of gyration, histograms
of which are shown in figure 3.10, is roughly the same for all peptides as well. The average
peak is around 1.0nm. The only anomaly is the higher peak/narrower distribution of the
A16 dimer. The dashed lines in all panels show the normal distributions calculated from
the mean and standard deviation of each data set. We can see that the distribution
of the data in each case tends to have a similar shape on its right side to the normal
distribution. Also, there is a shift of the maximum peak, which lies to the right of the
normal distribution maximum in all cases. In general, the radius of gyration and end-
to-end distance, indicators of overall peptide size and compactness, do not vary between
peptides and over system size.
Figure 3.11 shows the number of residues in the helical conformation over the course
of the simulation for the monomer, dimer, and tetramer simulations. The peptides have
no helical residues roughly 60% of the time. Additionally, there are no significant differ-
ences between the monomer, dimer, and tetramer systems for each model peptide. All
peptides have a similar spread in the number of helical residues, tapering off at 16 helical
residues. Overall, there are no major differences between peptides for the same system
size (monomer, dimer, or tetramer).
We don’t see significant differences in the amount of helicity between peptides or
system size. We also don’t see any substantial differences in the types of contacts made
between peptides and between peptide and water when comparing contacts over all sizes
of systems over the entire set of replicas. However, there are a few qualitative observations
that can be made for a few specific trajectories. Looking back at the helix histograms in
figure 3.10, there is a smaller spread (standard deviation) and mean for the A16 dimer
radius of gyration. As a result, we looked at specific trajectories from this system. A few

Chapter 3. Results 40
Figure 3.9: Histograms of the end-to-end distance of the model peptides in the monomer,
dimer, and tetramer simulations, calculated between the carbon of the N-terminal acetyl
group and the oxygen of the C-terminal residue. The end-to-end distance during the last
50ns of the simulation (the equilibrated portion) was divided into 50 bins of the same
size for each of the monomer, dimer, and tetramer systems for each of the six peptides.
The dashed lines are normal distributions based on the mean and standard deviation
calculated in each data set.

Chapter 3. Results 41
Figure 3.10: Histograms of the radius of gyration of the model peptides in the monomer,
dimer, and tetramer simulations. The radius of gyration during the last 50ns of the
simulation (the equilibrated portion) was divided into 50 bins of the same size for each
of the monomer, dimer, and tetramer systems for each of the six peptides. The dashed
lines are normal distributions based on the mean and standard deviation calculated in
each data set.

Chapter 3. Results 42
Figure 3.11: Histograms of the probability of having 0 through 18 helical residues (DSSP
calculations) in each of the peptides for the monomer, dimer, and tetramer systems.

Chapter 3. Results 43
of the peptides were almost completely helical by the end of the trajectory but did not
interact. A few formed sporadic peptide-peptide interactions (something that was seen
in the other multimer simulations as well), but these interactions were transient and,
moving through the simulation, we saw that these peptides then drifted away from each
other at a later time point. Some simulations started out completely helical and retained
most of this helicity while other were predominantly random coil but formed intermittent
β-sheets. A small number formed peptide-peptide interactions. In one instance one of
the peptides was largely helical throughout while the other chain sampled all secondary
structures. Some inter-strand hydrogen bonding occurred. In certain cases, the peptides
actually move apart over the course of the trajectory. In fact, all multimer simulations
had very few instances of hydrogen-bonded peptides for large portions of the trajectory.
Additionally, helicity within each peptide, if helix was indeed formed, was transient.
These trends were also observed in the other systems studied. Although each replica had
slightly different behaviour, the entire ensemble of replicas for each system showed no
statistically significant differences in bonding and interaction when compared to the other
systems, both between peptides and between monomer, dimer, and tetramer simulations.
The macromolecular polymer properties of radius of gyration and end-to-end distance
plateaued after 50ns of simulation and this, along with block-averaging, was used to
determine that the simulations had converged. One question that arose over the course
of my studies was whether the secondary structure was correlated to these properties.
That is, is the overall size of the monomers correlated to helicity and is this modulated
by interactions between peptides in the multimer simulations?
Figure 3.12 shows the evolution of the radius of gyration over time for a single replica
of the A0 peptide. Snapshots of the peptide conformation, with helix highlighted in
magenta, are shown for the 10ns, 20ns, 30ns, 50ns, 80ns, 90ns, and 100ns time points.
In this replica, more extended conformations (larger Rg) are largely random coil while
more helical conformations have a smaller radius of gyration.
In the dimer simulations, we studied whether peptide-peptide interactions played a
role in modulating the radius of gyration and/or end-to-end distances as well as secondary
structure. As with the monomer simulations, the peptides are largely random coil. As

Chapter 3. Results 44
Figure 3.12: Time evolution of the radius of gyration of A8KKA8 in CHARMM22*. The
graph shows the radius of gyration of the peptide monomer at a given point in time.
Selected snapshots of the peptide showing secondary structure are shown along with
the timepoint at which the snapshot was taken. The radius of gyration for the entire
production run (after NVT and NPT equilibration) is depicted in the plot.

Chapter 3. Results 45
seen in figure 3.13, the two peptides each have a slightly different radius of gyration but
increase and decrease at similar times by the same amount on average. The peptides
also form similar amounts of helix at the same time, though this could also be attributed
to the fact that both peptides begin from the same starting conformation (albeit far
apart). The question here is whether interactions between peptides causes both peptides
to adopt the same structure and if helix is preferentially formed when there are contacts.
To answer this question, a closer look at the number and type of contacts formed between
peptides (if they are formed at all) is important.
0ns 20ns 40ns 60ns 80ns 100ns
0ns 20ns 40ns 60ns 80ns 100ns
Figure 3.13: Time evolution of the radius of gyration of A8KKA8 in CHARMM22*. The
blue plot is the Rg of one peptide chain in one replica of the dimer simulation, shown
above the graph is a snapshot of the peptide every 20ns over the course of the simulation
with secondary structure highlighted. The green plot is the Rg of the other peptide chain,
with representative snapshots shown below the graph.

Chapter 3. Results 46
We first investigated if the peptides in the multimer simulations interact at all. Figure
3.14 shows contact maps for six different replicas in the A0 dimer simulations. Each map
shows all interpeptide and intrapeptide interactions between non polar atoms of each
residue in the two peptides in each simulation. Figure 3.15 shows the structure, with
secondary structure elements highlighted in different colours, for each peptide and shows
the distance of approach between peptides in each replica.
We see, for example, that replica 12 (top right panel of figure 3.14) has many i,i+4
contacts within peptide 1. These i+4 contacts suggest that the peptide is forming a
significant amount of helix. Furthermore, peptide 2 is forming a diffuse array of contacts,
suggesting a largely random coil peptide 2 in this replica. Additionally, the top left and
bottom right quadrants of this contact map are mostly black. These quadrants depict
intermolecular contacts and thus show that the two peptides hardly interact over the
course of the simulation in this replica. Looking at figure 3.15, which shows a snapshot
of the simulation at the 100ns time point, we see these descriptions of the system hold
true. Replica 83 (bottom right panel of figure 3.14) shows a lot of i,i+4 contacts in
the off diagonal quadrants and also a lot of intermolecular contacts in the on diagonal
quadrants. This shows two peptides that form helix down nearly their entire length that
interact. Other maps show helix-turn-helix motifs and a diverse, heterogeneous, array of
intermolecular interactions.
Figure 3.17 shows snapshots of the six A0 tetramer replicas shown in figure 3.16. We
see that replicas 2, 41, and 47 have formed helical bundles. This is seen by the extended
i,i+4 contacts as well as a scattered, nonadjacent, pattern of intermolecular contacts,
showing that the peptides interact via specific faces of the helix, essentially disallowing
adjacent residues from forming contacts with the same residue (as they will be positioned
on different faces of the helix).
The contact maps have been averaged over all six two-peptide combinations to yield
two-peptide by two-peptide contact maps.
As previously discussed for the monomer simulations, the dimer and tetramer sim-
ulations also show a lot of heterogeneity in the type of contacts they form. Although
clear patterns in inter- and intramolecular contacts are seen in individual replicas, fig-

Chapter 3. Results 47
Figure 3.14: Sample contact maps for six replicas of the A0 dimer system showing in-
termolecular and intramolecular contacts between nonpolar atoms of all residues. The
top left and bottom right quadrants identically show intermolecular contacts between
peptides 1 and 2. The top right and bottom left quadrants show intramolecular contacts
for peptide 2 and 1 respectively.

Chapter 3. Results 48
Figure 3.15: Snapshots at 100ns of six A0 dimer replicas. These replicas are the same as
those shown in figure 3.14.

Chapter 3. Results 49
Figure 3.16: Sample contact maps for six replicas of the A0 tetramer system showing
intermolecular and intramolecular contacts between nonpolar atoms of all residues. The
top left and bottom right quadrants identically show intermolecular contacts between
peptides 1 and 2 (averaged over all six possible pairs of peptides). The top right and
bottom left quadrants show intramolecular contacts for peptide 2 and 1 respectively
(again averaged over all six pairs).

Chapter 3. Results 50
Figure 3.17: Snapshots at 100ns of six A0 tetramer replicas. These replicas are the same
as those shown in figure 3.16.

Chapter 3. Results 51
ures 3.18 and 3.19 show that averaging the contacts over all replicas washes out specific
interactions. In fact, the only average interactions seen with large propensity are the
i+1 interactions as well as i+2 and i+3 turns. A small proportion of i,i+4 turns, denot-
ing helical contacts, are also seen. Otherwise, all intermolecular contacts and all other
intramolecular contacts are of lower propensity. This underscores the structural hetero-
geneity of the cross-linking peptides and shows that there is no preferential arrangement
or aggregation state.
However, we do see a significant amount of helical structure in these simulations and
also see interpeptide interactions. We know from previous work in our labs that the cross-
linking peptides are helical in the cross-linked state and perhaps their helicity pushes the
peptides to interact or vice versa. Namely, is interaction between chains correlated to
helicity?
Figure 3.20 shows average fraction helix formed using DSSP calculations for the
monomer, dimer, and tetramer simulations for all six lysine-spacing model peptides.
On average, the systems with two or four peptides formed more helix, with only the A16
peptide forming significantly more helix in the dimer and tetramer simulations relative to
the monomer case. Additionally, figure 3.21 shows a distribution of the distance between
the centers of mass (COM) of all pairs of peptides. Panel a shows the distribution for
the dimer systems, with two clear distances of approach seen by the two peaks in the
distribution. The first peak is also seen in panel b for the tetramer systems. The second
peak is a more diffuse plateau, which makes sense because there are many more confor-
mations that can be sampled in the comparatively larger tetramer system. Additionally,
only the first peak is indicative of any interactions between peptides.
We see two distances of approach in the COM plots (figure 3.21) as well as an increase
in helix in the multimer simulations. Figures 3.22 and 3.23 show the fraction helix formed
on average for each peptide, divided into two cases: when the peptides are forming
intermolecular contacts and when they are not.
When the peptide is helical, it is more frequently not in contact with another peptide
(greater than factor of 2 in most cases for dimer, still significantly more for the tetramer).
However, considering the fraction of time they are in contact, they are largely helical

Chapter 3. Results 52
Figure 3.18: Dimer contact maps for the six model peptides averaged over all replicas.
Intramolecular contacts are shown above the diagonal and intermolecular contacts are
shown below the diagonal for the(a) A0, (b) A1, (c) A2, (d)A3, (e) A4, and (f) A16
peptides.

Chapter 3. Results 53
Figure 3.19: Tetramer contact maps for the six model peptides averaged over all replicas.
Intramolecular contacts are shown above the diagonal and intermolecular contacts are
shown below the diagonal for the(a) A0, (b) A1, (c) A2, (d)A3, (e) A4, and (f) A16
peptides.

Chapter 3. Results 54
Figure 3.20: Comparison of total fraction helix formed by all peptides over the 1, 2, and
4 peptide simulation systems. Fraction helix was computed using DSSP and normalized
by the total number of frames.
Figure 3.21: Histograms of pairwise distances between the centers of mass of all peptides
in the (a) dimer and (b) tetramer systems.

Chapter 3. Results 55
Figure 3.22: Average fraction helicity for dimer simulations of all peptides with and with-
out formation intermolecular contacts. The dark grey bar denotes fraction helix formed
by the peptides when they are making intermolecular contacts with other peptides, the
hatched bar denotes fraction helix formed by the peptides when they are not making any
contacts with other peptides.

Chapter 3. Results 56
Figure 3.23: Average fraction helicity for tetramer simulations of all peptides with and
without formation intermolecular contacts. The dark grey bar denotes fraction helix
formed by the peptides when they are making intermolecular contacts with other pep-
tides, the hatched bar denotes fraction helix formed by the peptides when they are not
making any contacts with other peptides.

Chapter 3. Results 57
when they are in contact with another peptide (results not graphed). That is to say, the
peptides in contact are helical but helicity does not imply they are in contact. However,
we do see from the previous figure 3.20 that the multimeric peptides form more overall
helix, suggesting that increasing peptide concentration in the box yields more helix,
regardless of the fraction of time they spend in contact (since the difference in helicity
between system sizes when the peptides are in contact is negligible). The monomer
simulations were conducted in a 6 nm x 6 nm x 6 nm box, yielding a total volume of
216nm3 available to one peptide. The dimer box also had the same dimensions, thus
crowding each peptide to an available volume of 108nm3. The tetramer simulations were
conducted in a 12 nm x 6 nm x 6 nm box, also yielding 108nm3 in volume per peptide.
Thus the increased helicity in the multimer simulations could be due to a crowding of
their environment, as they have two-fold less space available per peptide, even if they are
not in contact. There is enough steric hindrance and charge repulsion between lysines to
prevent long-lasting contact formation but molecular crowding could cause the peptides
to form more compact structures and, if these structures are compact enough, they have
backbone hydrogen bonds that need to be fulfilled amongst themselves due to exclusion
of water. These bonds are most easily fulfilled by a helical structure.
We can conclude that in the dimer and tetramer simulations, both secondary structure
and peptide-peptide associations (not necessarily hydrogen bonding) are transient. Per-
haps if there is any ordering in the domains, it requires the entire tropoelastin monomer.
A study of tropoelastin by Baldock and coworkers [104] used small angle X-ray scatter-
ing (SAXS) and neutron scattering and presented a head-to-tail model of tropoelastin
assembly, where propagation of the fiber occurred through a stacked spring design. This
type of ordered alignment and assembly may only occur when the cross-linking domains
are placed in the context of the entire tropoelastin molecule. Previous studies from our
labs have shown that the hydrophobic domains lack a defined order on their own as well
[21]. Additionally, tropoelastin is notoriously difficult to work with in vitro, leading to
study of simplified domains that are chemically synthesized or expressed recombinantly
in E. coli. We may thus not see the possible ordered alignment of tropoelastin monomers
by studying either the cross-linking or hydrophobic domains in isolation or together but

Chapter 3. Results 58
as simplified constructs. This motivates the development of methods that would allow
the study of tropoelastin in greater resolution.
3.4 Tying biophysical results back to simulation
Molecular dynamics simulations give us atomistic level detail but comparison to circular
dichroism spectra, which only yields average secondary structure of the peptide, is diffi-
cult. The model peptides used in my work have highly repetitive sequences, which yields
high signal to noise in NMR experiments. The peptides are small and soluble enough
for study in solution NMR experiments, which are able to resolve differences between
populations of lysines and alanines in the peptides.
3.4.1 Solution NMR of the model peptides
NMR samples were prepared as described in section 2.2.5. The A2, A3, and A16 peptides
were used to create three different samples. COSY, TOCSY, HSQC, and HSQC-TOCSY
spectra were acquired for each peptide starting at 7◦C, followed by 15◦C, and then 37◦C.
The samples were stable for 1-2 weeks, allowing data collection for all experiments. Figure
3.24 shows the HSQC and TOCSY spectra for the A2 peptide at 15◦C. The peaks for
various Cα and Cβ atoms are shown and labelled on both spectra. As seen in the
HSQC, the alanine Cα peaks overlap in the same general area but do not populate one
homogeneous state. They appear to populate two major states, one centered further
upfield in the proton dimension. Also of note is that the two lysines, although centered
in the model peptide sequence, have different chemical shifts, denoting that they are in
slightly different environments. The HSQC and TOCSY spectra allow us to distinguish
the different populations of alanine and lysine residues in the peptide and thus allow
comparison between peptides and across temperatures.
The secondary structure and dihedral angles of the residues in a peptide can be
determined by their chemical shifts. One way to determine the secondary structure of a
particular residue is to calculate the secondary chemical shift. Equation 3.2 shows that
the calculation to determine the secondary chemical shift. δobserved is the chemical shift of

Chapter 3. Results 59
Figure 3.24: Solution NMR on unlabelled A2 peptide. The top panel shows a 1H-13C
HSQC with the carbon chemical shifts labelled according to atoms in the peptide. The
bottom panel shows the correlated peaks from a TOCSY spectra collected on the same
sample.

Chapter 3. Results 60
the peak observed in the spectrum, and δrandom coil is the published random coil chemical
shift value. Wishart et al. [105] have published random coil chemical shift values for
C=O, Cα, Cβ, NH, Hα, and Hβ for the 20 amino acids.
∆δ = δobserved - δrandom coil (3.2)
In α-helices, Cα atoms will have positive secondary chemical shifts while Cβ atoms
will have negative secondary chemical shifts. The opposite holds for β-strands (i.e. Cα
atoms will have negative secondary chemical shifts while Cβ atoms will have positive
secondary chemical shifts). The Cα atoms are most strongly correlated to secondary
structure of all the types of atoms that can be observed in an experiment. Figure 3.25a
shows the secondary chemical shifts for the A2, A3, and A16 peptides at 15◦C. As
denoted by the positive secondary chemical shifts for the Cα residues and the negative
secondary chemical shifts for the Cβ residues, they are all shifted towards helix, with two
populations of roughly equally helical lysine Cαs in A2, only one population of lysine
Cα in A3, and two populations of lysine Cα, one of which is much more shifted to helix
than the other, in A16. Overall, all residues are helical, and to roughly the same degree
over all peptides. In figure 3.25b, we see a decrease in the magnitude of the secondary
chemical shift with increasing temperature for the A3 peptide, denoting loss of helix with
increasing temperature, which corroborates our CD melt data (which shows gradual loss
of helix, i.e. unfolding, with increasing temperature).
3.4.2 Circular dichroism spectra calculated helicity of the model
peptides
Going back to the circular dichroism data, we can calculate percent helicity from the
mean residue molar ellipticity at 222nm. It can be calculated as follows:
Fraction helix = −40, 000(1 − 2.5/n) (3.3)
where n is the number of amino acid residues in the peptide.
Converting calculated fraction helix into percentages, we can compare the helicity

Chapter 3. Results 61
Figure 3.25: Secondary chemical shifts of the Cα (red) and Cβ (blue) atoms for (a) the
A2, A3, and A16 peptides at 15 ◦C and (b) the A3 peptide at three different temperatures.
Positive Cα and negative Cβ secondary chemical shifts denote helical structure.

Chapter 3. Results 62
calculated from CD experiments to what was calculated from DSSP analysis of the MD
data. Figure 3.26 summarizes the comparison. We obtain significantly more amounts of
helix in CD versus MD for all peptides (A2, A3, A16, A2Y, and A3A). However, if we
look at the inset graph of figure 3.26, we see a linear relationship between the CD and
MD percent helix, though MD underestimates helicity by a factor of 3 relative to CD
(the slope of the graph is about 0.3).
Figure 3.26: Comparison of helicity calculated from molecular dynamics simulations
(DSSP constraints) and circular dichroism (molar ellipticity at 222nm) for the model
peptides.
Although some residue specific information can be obtained from NMR experiments,
the degeneracy of the sequences of the model peptides does not yield more than general
populations of the alanine and lysine residues present in the peptides. To this end, we
can use molecular dynamics simulations, which give us atomistic level detail and insight

Chapter 3. Results 63
into the system.
Figure 3.27 shows the DSSP calculated secondary structure on a per residue basis,
averaged over the equilibrated portion of the trajectory (the last 50ns) for the monomer
simulations in CHARMM22*. We can see the the helicity is lowest at either end of the
peptide, where the helix cannot be hydrogen bonded on both sides. helicity increases
towards the middle of the peptide and dips slightly about the lysine residues. This
dip is seen in most of the model peptides. This is most probably due to unfavourable
helix elongation by the lysine residues, which are long, charged, and bulkier than alanine
residues. Furthermore, the helicity is markedly less at the C-terminal end than at the
N-terminal end (with the exception of the A1 peptide), which might be due to the high
N-cap propensity of the acetyl group [43].
Figure 3.27: Average helicity per residue for all the model peptides in CHARMM22*.
Helicity was calculated based on structure assignment to each residue from DSSP criteria.

Chapter 3. Results 64
3.5 Biphasic systems as a way to model the coacer-
vate
Coacervation is a process whereby an increase in temperature causes a phase separation of
a solution into protein rich droplets and a surrounding, protein depleted, solution. It has
already been shown that the hydrophobic domains on their own are able to coacervate.
They form a partially-hydrated aggregate, as shown by molecular dynamics simulations
[21, 106]. We wanted to probe the behaviour of the cross-linking domains at the interface
of a coacervate droplet. To simulate this, we performed molecular dynamics simulations
in the presence of an octane slab [107]. Additionally, RP-HPLC was used to analyze par-
titioning experiments in vitro. We hypothesized that the cross-linking domains partition
at the interface of a coacervate droplet and, due to positively charged lysine residues,
do not insert into the droplets. Rather, we predicted that they would have the ability
to order at the surface of a droplet and drive subsequent cross-linking, as cross-linking
domains need to come together in some way so that lysyl oxidase can convert lysines
to allysines and the spontaneous condensation reaction can happen immediately. The
reaction would be most specific and efficient if the cross-linking domains were already
near each other after coacervation in preparation for cross-linking.
The overarching conclusions of this work were that the cross-linking domains do not
partition at the interface. They neither move towards the octane slab by the end of
simulation nor do they preferentially partition at or insert into the octane slab. Addi-
tionally, HPLC results, shown in figure 3.28, show that peak areas for the interface and
hydrophobic phases are negligible and that, with the exception of the A16 peptide in
octane, the peptides do not partition into the hydrophobic phase. It is worth noting that
the A16 peptide has the lysines at either end of the sequence, which is not representa-
tive of the lysine spacing and register in natural cross-linking domains (these lysines are
found at the C-terminal end and spaced 2 or 3 residues apart). Essentially, the cross-
linking domains in isolation (that is, without the hydrophobic domains) do not partition
in a biphasic system. Further experiments will need to be performed on more complex
systems involving both the hydrophobic and cross-linking domains.

Chapter 3. Results 65
!0.6%
!0.4%
!0.2%
0%
0.2%
0.4%
0.6%
0.8%
1%
1.2%
1.4%
1.6%
A2% A3% A16% A2Y% A3A%
Peak%Area%(A.U)%
Octanol%Par22oning%
Aqueous%
Interface%
Hydrophobic%
!0.6%
!0.4%
!0.2%
0%
0.2%
0.4%
0.6%
0.8%
1%
1.2%
1.4%
1.6%
A2% A3% A16%
Peak%Area%(A.U)%
Octane%Par00oning%
Aqueous%
Interface%
Hydrophobic%
a b
Figure 3.28: Integrated peptide peak areas from RP-HPLC chromatograms for each
phase in octane and octanol partitioning experiments. The peptide peak was integrated
for each of the three phases run separately in triplicate on an analytical C18 column.
The hypothesis based on current results is that the cross-linking domains are ex-
cluded by coacervation when the hydrophobic domains form the interior of the droplet.
The hydrophobic environment is unfavourable for the positive residues in the cross-linking
domains. They may be positioned, by exclusion, at the surface of the droplet and are
oriented for subsequent cross-linking. We know from other studies that the sequence
of the cross-linking domains has an effect on their aggregation properties and that ly-
sine residues contribute to preventing aggregation [40]. My results have shown that the
cross-linking domains are partially helix in solution as monomers. This helicity is melted
below physiological temperatures. These results are corroborated in recent elastin liter-
ature [40]. Additionally, Reichheld and coworkers found that the cross-linking domains
are β-sheet in the coacervate and predominantly helix in the cross-linked state. Thus
modulation of secondary structure is a phenomenon of the state of assembly of the recom-
binant peptides. We can think of the cross-linking domains as floating on the coacervate
droplet surface, rather than inserting into the droplet. They are heavily influenced by
their environment but do not have an intrinsic ability to drive coacervation or self-order.

Chapter 4
Discussion
The development of molecular mechanics force fields began 40 years ago [108] and the
first molecular dynamics simulation of a protein was also conducted at this time. With
advances in computational power, the sampling of the energy landscape becomes a lesser
concern as longer simulations are being conducted and making it clear that accurate
energy functions are required for reliable results. Quantum mechanical calculations are
the most accurate way to computationally track atomic movement, but are also com-
putationally costly. Molecular mechanics energy functions are much simpler and more
computationally efficient but pre-existing force fields have conformational biases. Choos-
ing an appropriate force field for a study is a challenge since different force fields have
specific biases towards certain secondary structures. These biases are usually only evident
with thorough validation against experimental data, which isn’t always possible [109] due
to the lack of experimental data or difficulty in calculating experimental observables from
simulation data [78] and the accuracy of the experimental observables themselves.
Many force fields are parametrized for peptide and protein simulation studies, how-
ever these force fields all have different parametrization and have been validated against
different sets of experimental data. As previously mentioned, four force fields were tested
for their ability to recapitulate biophysical data for the alanine-rich peptides studied.
Alanine-based peptides form varying levels of helix in solution. However, this helicity is
in balance with the tendency for these peptides to sample a diverse set of random coil
conformations. Thus the force field most suitable for my studies necessitates the right
66

Chapter 4. Discussion 67
balance of helix and random coil in my simulations.
Recent developments in computer hardware and simulation methods have allowed
simulations on the millisecond timescale [110]. Sampling representative conformations
for peptides and small proteins is thus a lesser concern than the reliability and accuracy of
force field parametrization. As computational power increases, the development of force
field parameters that accurately recapitulate and predict biophysical properties becomes
a more important topic of study.
Re-parametrization of force fields is done most effectively when simulation data is
compared to experimental results, such as NMR J-coupling constants and other biophys-
ical data. Ultimately, the future use of data like what I have obtained over the course of
my project can be used to further validate or re-parametrize force fields. Direct compar-
ison between experimental and computational results on the same systems and similar
metrics between techniques will enable a more thorough validation of simulation results.
In my study, I used the same set of peptides in both simulation and in biophysical exper-
iments, along with similar methods of calculating secondary structure. This led to direct
comparison and validation of the best force field for the systems in my study.
My work has shown that CHARMM22* leads to the largest amount of helix formation
in the tested peptides. This compares most favourably to experimentally determined
percent helicity from CD and NMR and also compares best to pre-existing literature
results. Robert Best and coworkers [58] found anywhere from 15 to 30% helix in the Ala5
peptide and as high as 94% helix in the ff03w force field for Ac-(AAQAA)3-NH2. Many
other peptide studies, as mentioned in Section 1.4, use various simulation and biophysical
techniques to quantify the amount of helix in poly-alanine peptides. Ultimately, it is
important to select a force field for the system of relevance that compares most favourably
to experimental data for the same system. Consequently, the force field to use might be
different for a stable membrane protein embedded in a lipid bilayer (where the study
might involve rare ion channel events) versus a disordered peptide that forms very little
secondary structure and needs to sample a great variety of conformations (such as elastin-
like peptides).
Force field validation is important for a number of reasons, including achieving the

Chapter 4. Discussion 68
proper balance of secondary structure in a particular system, adequately sampling con-
formational space over the course of a simulation, and being able to compare and validate
computational simulation results against experimental findings. In this context, Best and
coworkers looked at residue specific α-helix propensities in alanine-based peptides [111]
in comparison to experimental Lifson-Roig parameters. This yielded a picture of he-
lix nucleation and propagation propensities for the 20 amino acids within alanine-based
peptides for various AMBER force fields, including two force fields that I have tested -
AMBER ff03w and AMBER ff99sb*-ildn. Temperature dependent Lifson-Roig parame-
ters from experiment [112, 43] nearly overlapped for the AMBER ff99SB force field with
backbone and ILDN side-chain corrections - that is, the AMBER ff99sb*-ildn force field.
The agreement for ff03w was also very good.
Another important consideration is the quality of the solvent model used with a par-
ticular force field. I have used different solvent models for each of the force fields I
tested. Previous studies [113] looked at the stability and conformational ensemble of
disordered tripeptides as a function of the solvent model used. Careful consideration of
protein-solvent interactions is especially important for simulations of intrinsically disor-
dered peptides and proteins, since the energy differences between different conformations
of these systems is minimal and interactions with water are significant in these systems.
It is precisely for this reason that I used the CHARMM-modified TIP3P water model
for my simulations in CHARMM22*, as it is essential to match the solvent and the force
field, as they are parametrized together and experimental comparisons are made with
respect to this specific pairing of water model and force field.
Additionally, comparison to experimental measurements yields a benchmark of force
field accuracy. My studies looked at the comparative secondary structure (more specif-
ically, helicity) of my model peptides in silico and in vitro. Other studies have pointed
out the limitations of current force fields by comparing simulation data to NMR measure-
ments [114] to benchmark current force fields and gain an idea of force field dependent
dynamics and the accuracy of potential energy parameters in penta-peptide simulations
[115]. The robustness of protein folding with respect to the force field (i.e is the path of
folding the same) is also an important consideration [78] and transferability of force fields

Chapter 4. Discussion 69
is important if simulations are used to elucidate folding mechanism [116]. In the context
of these, and other, molecular dynamics simulation studies, force field validation is an
important consideration for any system of study. Now that temporally longer and spa-
cially larger simulations are being conducted, force field accuracy becomes the dominant
concern as issues with convergence due to insufficient sampling become less important.
The force field validation I performed is important in the context of simulation litera-
ture and other peptide studies. In this regard, not only are my model peptides represen-
tative cross-linking domains in terms of their sequence composition, but also a peptide
set that can be studied within the framework of polyalanine peptide studies. Intrinsic
α-helix and β-sheet preferences were studied by Cabellero and coworkers [117] and found
to be different for different force fields. Polyalanine peptides have long been found to
be α-helical [53, 43, 48] and lysines within these peptides decrease the amount of helix
formed. This validates the work presented in my thesis regarding peptide helicity, as
I find local decreases in helix about lysine residues and at the termini of each peptide,
but find that helix is the dominant secondary structure overall. Similar comparisons be-
tween CD and simulation data have been made before [52] to determine the equilibrium
structure and folding of helix-forming peptides.
Investigation of the formation and stabilization of α-helices in short peptides is in
itself a worthwhile study since the α-helix is a dominant secondary structure in proteins
and small helical peptides are also finding a role in anti-microbial applications. How-
ever, the context in which such peptides are found is also of note. My model peptides,
specifically, have been designed to resemble elastin cross-linking domains. It is thus also
useful to look at these peptides in the framework of elastin and elastin-based peptide
studies. Conformational disorder is integral to elastin function, requiring a force field
that achieves the right balance of structure and disorder in an elastin-based peptide
[20]. Previous studies on elastin have shown that tropoelastin, the soluble monomer that
cross-links and deposits into insoluble fibers to form mature elastin, has an α-helical
component in CD measurements [65]. This reinforces the relevance of focusing on the
cross-linking domains, since the hydrophobic domains are not α-helical in isolation and
show significant structural disorder that is essential to their function in elastin [21]. Mu-

Chapter 4. Discussion 70
tations in the hinge region of the cross-linking domain encoded by exons 21 and 23 of
human tropoelastin affect the stability of the α-helix formed by this region and found to
be important in the function of elastin [37, 118]. We know that short elastin-like polypep-
tides (ELPs) exhibit the same temperature-induced structural transitions as full-length
elastin polymers. This motivates continued study of shorter, representative recombinant
constructs that resemble full length elastin.
The key structural features of the peptides are similar within the model set. The
A2 and A3 peptides, both of which have lysine spacings found in natural elastin, adopt
similar amounts of helix as the other model peptides. On their own, these peptides do not
show significant difference in secondary structure or interactions. The only exception is
the A16 peptide, which adopts significantly more helix in vitro. However, this is a more
canonical polyalanine peptide with a longer, uninterrupted stretch of alanine residues
and serves as a sequence composition control for our model peptide set. My results also
suggest that these peptides do not partition or assemble in a meaningful way on their own.
They are perhaps passive in the coacervation process and cross-linking after coacervation
may occur simply by exclusion of the cross-linking domains from the interior of the
coacervate droplet. Analogous to this hypothesis is the association of charged detergent
micelles, where the association of hydrophobic tails brings charged headgroups in close
proximity to one another despite coulombic repulsion between the like charges on these
headgroups. The exclusion of cross-linking domains from hydrophobic solvents suggests
that they are pushed to the surface of these droplets, increasing their local concentration
and potentially aiding in cross-link formation. The following chapter proposes a few
experiments to probe this hypothesis.
The cross-linking domains aggregate in water without the hydrophobic domains and
are preferentially helical in their aggregated state. Charge repulsion and steric hindrance
between lysines lead to the lysines preferentially pointing away from each other on the
outside of the helix dimer. Figure 4.1 shows lysines pointing away from packed helices
in a snapshot of (a) an A0 dimer simulation, and (b) an A0 tetramer simulation. Fig-
ure 4.2 shows this interaction, where (a) shows a sideview of interacting helices and (b)
shows a top down view of the same helix dimer interaction. Figure 4.2 (b) also shows

Chapter 4. Discussion 71
the hydrophobic helix interaction face (shaded). Favourable interaction between the hy-
drophobic faces is what drives association of these helices. Simulation results show similar
helix packing but differences in the exact face of the helix forming the interaction (see
figure 4.1). This rotational freedom in inter-helix interactions is indicated by the arrows
in part (b). This same interaction face is hypothesized to be responsible for interaction
with the surface of the coacervate droplet. Figure 4.2 (c) shows the proposed interaction
of the aggregated cross-linking domains with the hydrophobic droplet interface, where
the hydrophobic interface between the helices and the droplet is similar to the interface
between helices in part (b).
Figure 4.1: Position of lysines in the helical aggregated multimer simulations. (a) Helical
dimer formed in an A0 two peptide simulation, with lysines (green stick representation)
pointing away from the hydrophobic helical interface, (b) Helical trimer formed in an
A0 four peptide simulation, again with lysines (green stick representation) pointing away
from the helix bundle.
We know that four lysines from two different tropoelatin monomers must come to-
gether for cross-linking to occur. The model proposed in figure 4.2 is one hypothesis
for how these lysines might come together in the early stages of coacervation. As yet
unresolved is the alteration of secondary structure in the cross-linking domains in the
coacervate, which is rich in β-sheet [40]. Our experimental data only considers the cross-

Chapter 4. Discussion 72
Figure 4.2: Schematic of the proposed cross-linking mechanism in elastin. (a) Helix dimer
formed between cross-linking peptides, with lysines pointing away from the hydropho-
bic helical interface, (b) top down view of a, showing the hydrophobic interaction face
between helices and the dynamic nature of this face (arrows show the helices can rotate
about their axis), and (c) proposed cross-linking domain interaction with the hydropho-
bic droplet interface - association between cross-linking domains is as in (b), but rotated
such that the hydrophobic helical face is at the surface of the droplet.

Chapter 4. Discussion 73
linking domains in isolation. Further experiments with the hydrophobic and cross-linking
domains together will be required to probe what type of ordering occurs in the cross-
linking domains at the surface of a coacervate droplet. Furthermore, simulation studies
of the cross-linking peptides considered two or four peptides in the same box. In vivo,
cross-linking in the extracellular matrix occurs in a more crowded environment and the
local concentration of elastin is much higher prior to cross-linking. The conformational
equilibrium of the cross-linking domains may switch to favour β-sheet or other more
extended structures when a sufficiently high concentration of cross-linking domains is
achieved locally and minimization of steric and charge repulsions needs to occur.
That being said, we have considered the early stages of aggregation of the cross-
linking domains in our simulation results and have a working hypothesis for the assembly
mechanism of elastin that will lend us insight for larger scale studies.

Chapter 5
Future Directions
The model cross-linking peptides have been studied thoroughly in the monomer and ag-
gregative states. RP-HPLC data and initial simulation data suggest that the cross-linking
domains on their own, whether monomeric or somehow assembled, do not partition pref-
erentially at the water-octane interface. We can extrapolate these results to say that
they do not drive coacervation by partitioning preferentially to the outside of coacervate
droplets.
Once it has been conclusively determined whether these domains partition prefer-
entially at the interface of a biphasic system, it is possible to devise experiments to
determine whether the cross-linking domains are ordering at this interface. One method
is to label the peptides with tryptophan and monitor whether their structure is affected
by the presence of other cross-linking domains or a biphasic environment. This can be
done by tracking tryptophan fluorescence and monitoring whether there is any quenching
of these residues. Alternatively, fluorescence anisotropy measurements can be performed
to determine mobility of these peptides. NMR PFG diffusion experiments can also be
performed to track changes in peptide oligomeric state and possible aggregation and mo-
bility changes. We can also spin label the lysines and gather more long range distance
information than can be obtained by NMR by doing electron paramagnetic resonance
(EPR) if we have access to a magnet and even probe partitioning with the use of appro-
priate paramagnetic compounds that partition into either phase.
Eventually, we will want to study larger constructs involving the cross-linking domains
74

Chapter 5. Future Directions 75
in the context of the hydrophobic domains. Such studies have already been conducted
by our lab in conjunction with the Keeley lab [40]. Essentially, we want to study the
assembly of these larger constructs and cross-link them to get samples that mimic elastin-
like aggregates. This will involve coarse-grained methods for simulation due to the size
of these constructs.
Molecular dynamics simulations are a valuable tool when performed at the atomistic
scale, but require large computational resources when performed on large systems. To
study these systems atomistically, the simplest system of relevance to study coacervation
would be two hydrophobic domains linked by a cross-linking domain. This system can
be used to probe whether the cross-linking domains can be forced to lie at the interface
of a hydrophobic droplet if multimers of this construct are simulated in the same system.
Coarse-graining, in the broadest sense, allows groups of atoms to be lumped together
and re-parametrized, thus reducing the number of degrees of freedom and thus lowering
computational cost. This enables greater sampling of conformational space for larger sys-
tems. One particular coarse-grained force field that can be used for the aforementioned
studies is MARTINI [119, 120]. Additionally, coacervated and cross-linked constructs
form insoluble aggregates that are not amenable to study by solution NMR. They can be
studied by SSNMR of selectively and uniformly 13C/15N labelled constructs with magic
angle spinning (MAS) to average orientation dependent parameters. A number of differ-
ent experiments can be performed to assign chemical shifts and determine internuclear
distances. Dihedral angle information can be obtained and compared to results from
molecular dynamics simulations. Additionally, information about the mobility of specific
residues can be obtained from these experiments.

Bibliography
[1] S. Rauscher and R. Pomes, “Structural disorder and protein elasticity,” in Fuzzi-
ness, pp. 159–183, Springer, 2012.
[2] B. Bochicchio, A. Pepe, and A. M. Tamburro, “Circular dichroism studies on re-
peating polypeptide sequences of abductin,” Chirality, vol. 17, no. 7, pp. 364–372,
2005.
[3] J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp, and K. Savage, “Elas-
tic proteins: biological roles and mechanical properties,” Philosophical Transactions
of the Royal Society of London. Series B: Biological Sciences, vol. 357, no. 1418,
pp. 121–132, 2002.
[4] J. M. Gosline, M. E. DeMont, and M. W. Denny, “The structure and properties of
spider silk,” Endeavour, vol. 10, no. 1, pp. 37–43, 1986.
[5] M. B. van Eldijk, C. L. McGann, K. L. Kiick, and J. C. van Hest, “Elastomeric
polypeptides,” in Peptide-Based Materials, pp. 71–116, Springer, 2012.
[6] B. Starcher and S. Percival, “Elastin turnover in the rat uterus.,” Connect. Tissue
Res., vol. 13, no. 3, pp. 207–15, 1985.
[7] S. M. Mithieux and A. S. Weiss, “Elastin,” Advances in Protein Chemistry, vol. 70,
pp. 437–461, 2005.
[8] F. W. Keeley, C. M. Bellingham, and K. A. Woodhouse, “Elastin as a self–
organizing biomaterial: use of recombinantly expressed human elastin polypeptides
76

Bibliography 77
as a model for investigations of structure and self–assembly of elastin,” Philosoph-
ical Transactions of the Royal Society of London. Series B: Biological Sciences,
vol. 357, no. 1418, pp. 185–189, 2002.
[9] L. B. Sandberg, N. T. Soskel, and J. G. Leslie, “Elastin structure, biosynthesis,
and relation to disease states.,” N. Engl. J. Med., vol. 304, pp. 566–79, Mar 1981.
[10] R. M. S. Richard A. Pierce, Thomas J. Mariani, “Elastin in lung development and
disease,” Ciba Foundation Symposium 192 - The Molecular Biology and Pathology
of Elastic Tissues, 1995.
[11] B. Levinson, J. Gitschier, C. Vulpe, S. Whitney, S. Yang, and S. Packman, “Are
X-linked cutis laxa and Menkes disease allelic?,” Nat. Genet., vol. 3, p. 6, Jan 1993.
[12] S.-H. Lee, S. Goswami, A. Grudo, L.-Z. Song, V. Bandi, S. Goodnight-White,
L. Green, J. Hacken-Bitar, J. Huh, F. Bakaeen, H. O. Coxson, S. Cogswell,
C. Storness-Bliss, D. B. Corry, and F. Kheradmand, “Antielastin autoimmunity
in tobacco smoking-induced emphysema.,” Nat. Med., vol. 13, pp. 567–9, May
2007.
[13] M. C. Zhang, M. Giro, D. Quaglino, and J. M. Davidson, “Transforming growth
factor-beta reverses a posttranscriptional defect in elastin synthesis in a cutis laxa
skin fibroblast strain.,” J. Clin. Invest., vol. 95, pp. 986–94, Mar 1995.
[14] C. Vulpe, B. Levinson, S. Whitney, S. Packman, and J. Gitschier, “Isolation
of a candidate gene for Menkes disease and evidence that it encodes a copper-
transporting ATPase.,” Nat. Genet., vol. 3, pp. 7–13, Jan 1993.
[15] L. Bader, “Disorders of elastic tissue: a review.,” Pathology, vol. 5, pp. 269–89, Oct
1973.
[16] Y. Wu, J. A. MacKay, J. R. McDaniel, A. Chilkoti, and R. L. Clark, “Fabrica-
tion of elastin-like polypeptide nanoparticles for drug delivery by electrospraying.,”
Biomacromolecules, vol. 10, pp. 19–24, Jan 2009.

Bibliography 78
[17] J. R. McDaniel, D. J. Callahan, and A. Chilkoti, “Drug delivery to solid tumors by
elastin-like polypeptides.,” Adv. Drug Deliv. Rev., vol. 62, pp. 1456–67, Dec 2010.
[18] S. S. Amruthwar and A. V. Janorkar, “Preparation and characterization of elastin-
like polypeptide scaffolds for local delivery of antibiotics and proteins.,” J Mater
Sci Mater Med, vol. 23, pp. 2903–12, Dec 2012.
[19] M. S. Kim, D.-W. Lee, K. Park, S.-J. Park, E.-J. Choi, E. S. Park, and H. R.
Kim, “Temperature-triggered tumor-specific delivery of anticancer agents by crgd-
conjugated thermosensitive liposomes.,” Colloids Surf B Biointerfaces, vol. 116,
pp. 17–25, Apr 2014.
[20] L. D. Muiznieks, A. S. Weiss, and F. W. Keeley, “Structural disorder and dynamics
of elastin,” Biochemistry and Cell Biology, vol. 88, no. 2, pp. 239–250, 2010.
[21] S. Rauscher, S. Baud, M. Miao, F. W. Keeley, and R. Pomes, “Proline and glycine
control protein self-organization into elastomeric or amyloid fibrils,” Structure,
vol. 14, no. 11, pp. 1667–1676, 2006.
[22] T. P. J. Knowles, M. Vendruscolo, and C. M. Dobson, “The amyloid state and its
association with protein misfolding diseases.,” Nat. Rev. Mol. Cell Biol., vol. 15,
pp. 384–96, Jun 2014.
[23] M. Miao, C. M. Bellingham, R. J. Stahl, E. E. Sitarz, C. J. Lane, and F. W.
Keeley, “Sequence and structure determinants for the self-aggregation of recombi-
nant polypeptides modeled after human elastin,” Journal of Biological Chemistry,
vol. 278, no. 49, pp. 48553–48562, 2003.
[24] B. Vrhovski and A. S. Weiss, “Biochemistry of tropoelastin,” European Journal of
Biochemistry, vol. 258, no. 1, pp. 1–18, 1998.
[25] C. Bellingham, K. Woodhouse, P. Robson, S. Rothstein, and F. Keeley, “Self-
aggregation characteristics of recombinantly expressed human elastin polypep-
tides,” Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular En-
zymology, vol. 1550, no. 1, pp. 6–19, 2001.

Bibliography 79
[26] G. C. Yeo, F. W. Keeley, and A. S. Weiss, “Coacervation of tropoelastin,” Advances
in colloid and interface science, vol. 167, no. 1, pp. 94–103, 2011.
[27] M. Miao, S. E. Reichheld, L. D. Muiznieks, Y. Huang, and F. W. Keeley, “Elastin
binding protein and FKBP65 modulate in vitro self-assembly of human tropoe-
lastin.,” Biochemistry, vol. 52, pp. 7731–41, Nov 2013.
[28] G. Bratzel and M. J. Buehler, “Sequence-structure correlations in silk: Poly-ala
repeat of N. clavipes MaSp1 is naturally optimized at a critical length scale,”
Journal of the Mechanical Behavior of Biomedical Materials, vol. 7, pp. 30–40,
2012.
[29] A. Sethi, R. J. Wordinger, and A. F. Clark, “Focus on molecules: lysyl oxidase.,”
Exp. Eye Res., Nov 2012.
[30] H. M. Kagan and W. Li, “Lysyl oxidase: properties, specificity, and biological roles
inside and outside of the cell.,” J. Cell. Biochem., vol. 88, pp. 660–72, Mar 2003.
[31] J. E. Wagenseil and R. P. Mecham, “Vascular extracellular matrix and arterial
mechanics.,” Physiol. Rev., vol. 89, pp. 957–89, Jul 2009.
[32] D. Torchia and K. Piez, “Mobility of elastin chains as determined by 13C nuclear
magnetic resonance,” Journal of Molecular Biology, vol. 76, no. 3, pp. 419–424,
1973.
[33] J. Lyerla Jr and D. Torchia, “Molecular mobility and structure of elastin deduced
from the solvent and temperature dependence of 13C magnetic resonance relaxation
data,” Biochemistry, vol. 14, no. 23, pp. 5175–5183, 1975.
[34] A. Perry, M. P. Stypa, B. K. Tenn, and K. K. Kumashiro, “Solid-state 13C NMR re-
veals effects of temperature and hydration on elastin,” Biophysical Journal, vol. 82,
no. 2, pp. 1086–1095, 2002.
[35] M. S. Pometun, E. Y. Chekmenev, and R. J. Wittebort, “Quantitative observation
of backbone disorder in native elastin,” Journal of Biological Chemistry, vol. 279,
no. 9, pp. 7982–7987, 2004.

Bibliography 80
[36] X. Yao and M. Hong, “Structure distribution in an elastin-mimetic peptide
(VPGVG)3 investigated by solid-state NMR,” Journal of the American Chemical
Society, vol. 126, no. 13, pp. 4199–4210, 2004.
[37] M. Miao, J. T. Cirulis, S. Lee, and F. W. Keeley, “Structural determinants of cross-
linking and hydrophobic domains for self-assembly of elastin-like polypeptides,”
Biochemistry, vol. 44, no. 43, pp. 14367–14375, 2005.
[38] L. D. Muiznieks, S. A. Jensen, and A. S. Weiss, “Structural changes and facilitated
association of tropoelastin,” Archives of Biochemistry and Biophysics, vol. 410,
no. 2, pp. 317–323, 2003.
[39] A. H. Simmons, C. A. Michal, and L. W. Jelinski, “Molecular orientation and
two-component nature of the crystalline fraction of spider dragline silk,” Science,
vol. 271, no. 5245, pp. 84–87, 1996.
[40] S. Reichheld, L. Muiznieks, R. Stahl, K. Simonetti, S. Sharpe, and F. Keeley,
“Conformational transitions of the cross-linking domains of elastin during self-
assembly,” Journal of Biological Chemistry, vol. 289, no. 14, pp. 10057–10068,
2014.
[41] P. Y. Chou and G. D. Fasman, “Empirical predictions of protein conformation,”
Annual Review of Biochemistry, vol. 47, no. 1, pp. 251–276, 1978.
[42] C. Nick Pace and J. Martin Scholtz, “A helix propensity scale based on experimen-
tal studies of peptides and proteins,” Biophysical Journal, vol. 75, no. 1, pp. 422–
427, 1998.
[43] A. Chakrabartty, T. Kortemme, and R. L. Baldwin, “Helix propensities of the
amino acids measured in alanine-based peptides without helix-stabilizing side-chain
interactions,” Protein Science, vol. 3, no. 5, pp. 843–852, 1994.
[44] R. Ingwall, H. Scheraga, N. Lotan, A. Berger, and E. Katchalski, “Conformational
studies of poly-L-alanine in water,” Biopolymers, vol. 6, no. 3, pp. 331–368, 1968.

Bibliography 81
[45] K. Platzer, V. Ananthanarayanan, R. Andreatta, and H. Scheraga, “Helix-coil
stability constants for the naturally occurring amino acids in water. IV. Alanine
parameters from random poly (hydroxypropylglutamine-co-L-alanine),” Macro-
molecules, vol. 5, no. 2, pp. 177–187, 1972.
[46] S. E. Blondelle, B. Forood, R. A. Houghten, and E. Perez-Paya, “Polyalanine-based
peptides as models for self-associated β-pleated-sheet complexes,” Biochemistry,
vol. 36, no. 27, pp. 8393–8400, 1997.
[47] V. Daggett, P. A. Kollman, and I. D. Kuntz, “A molecular dynamics simulation of
polyalanine: an analysis of equilibrium motions and helix–coil transitions,” Biopoly-
mers, vol. 31, no. 9, pp. 1115–1134, 1991.
[48] S.-S. Sung, “Folding simulations of alanine-based peptides with lysine residues,”
Biophysical Journal, vol. 68, no. 3, pp. 826–834, 1995.
[49] A.-S. Yang, B. Honig, et al., “Free energy determinants of secondary structure
formation: I. Alpha-helices.,” Journal of Molecular Biology, vol. 252, no. 3, p. 351,
1995.
[50] J. S. Miller, R. J. Kennedy, and D. Kemp, “Short, solubilized polyalanines are
conformational chameleons: Exceptionally helical if N-and C-capped with helix
stabilizers, weakly to moderately helical if capped with rigid spacers,” Biochemistry,
vol. 40, no. 2, pp. 305–309, 2001.
[51] J. A. Vila, D. R. Ripoll, and H. Scheraga, “Physical reasons for the unusual α-helix
stabilization afforded by charged or neutral polar residues in alanine-rich peptides,”
Proceedings of the National Academy of Sciences, vol. 97, no. 24, pp. 13075–13079,
2000.
[52] J. M. Stewart, J. C. Lin, and N. H. Andersen, “Lysine and arginine residues do not
increase the helicity of alanine-rich peptide helices,” Chemical Communications,
vol. 39, pp. 4765–4767, 2008.

Bibliography 82
[53] S. Marqusee, V. H. Robbins, and R. L. Baldwin, “Unusually stable helix formation
in short alanine-based peptides,” Proceedings of the National Academy of Sciences,
vol. 86, no. 14, pp. 5286–5290, 1989.
[54] L. Zhong and W. Johnson, “Environment affects amino acid preference for sec-
ondary structure,” Proceedings of the National Academy of Sciences of the United
States of America, vol. 89, no. 10, pp. 4462–4465, 1992.
[55] D. Roccatano, M. Fioroni, M. Zacharias, and G. Colombo, “Effect of hexafluoroiso-
propanol alcohol on the structure of melittin : A molecular dynamics simulation
study,” Protein Science, vol. 14, no. 10, pp. 2582–2589, 2005.
[56] C. Huang, Z. Getahun, Y. Zhu, J. Klemke, W. DeGrado, and F. Gai, “Helix for-
mation via conformation diffusion search,” Proceedings of the National Academy of
Sciences of the United States of America, vol. 99, no. 5, pp. 2788–2793, 2002.
[57] P. Thompson, V. Munoz, G. Jas, E. Henry, W. Eaton, and J. Hofrichter, “The
helix-coil kinetics of a heteropeptide,” Journal of Physical Chemistry B, vol. 104,
no. 2, pp. 378–389, 2000.
[58] R. B. Best and G. Hummer, “Optimized Molecular Dynamics Force Fields Applied
to the Helix-Coil Transition of Polypeptides,” The Journal of Physical Chemistry
B, vol. 113, no. 26, pp. 9004–9015, 2009.
[59] G. Hummer, A. Garcıa, and S. Garde, “Conformational diffusion and helix forma-
tion kinetics,” Physical Review Letters, vol. 85, no. 12, pp. 2637–2640, 2000.
[60] A. E. Garcıa and K. Y. Sanbonmatsu, “α-helical stabilization by side chain shield-
ing of backbone hydrogen bonds.,” Proc. Natl. Acad. Sci. U.S.A., vol. 99, pp. 2782–
7, Mar 2002.
[61] A. Pepe, D. Guerra, B. Bochicchio, D. Quaglino, D. Gheduzzi,
I. Pasquali Ronchetti, and A. M. Tamburro, “Dissection of human tropoe-
lastin: supramolecular organization of polypeptide sequences coded by particular
exons.,” Matrix Biol., vol. 24, pp. 96–109, Apr 2005.

Bibliography 83
[62] L. B. Sandberg, N. Weissman, and W. R. Gray, “Structural features of tropoelastin
related to the sites of cross-links in aortic elastin,” Biochemistry, vol. 10, no. 1,
pp. 52–56, 1971.
[63] D. P. Varadi, “Studies on the chemistry and fine structure of elastic fibers from
normal adult skin,” Journal of Investigative Dermatology, vol. 59, no. 3, pp. 238–
246, 1972.
[64] W. R. Gray, “Molecular model for elastin structure and function,” Nature, vol. 246,
pp. 461–466, 1973.
[65] A. M. Tamburro, A. Pepe, and B. Bochicchio, “Localizing α-helices in human
tropoelastin: assembly of the elastin puzzle,” Biochemistry, vol. 45, no. 31,
pp. 9518–9530, 2006.
[66] P. Toonkool, S. A. Jensen, A. L. Maxwell, and A. S. Weiss, “Hydrophobic domains
of human tropoelastin interact in a context-dependent manner.,” J. Biol. Chem.,
vol. 276, pp. 44575–80, Nov 2001.
[67] L. Monticelli and D. P. Tieleman, “Force fields for classical molecular dynamics.,”
Methods Mol. Biol., vol. 924, pp. 197–213, 2013.
[68] J. W. Ponder and D. A. Case, “Force fields for protein simulations.,” Adv. Protein
Chem., vol. 66, pp. 27–85, 2003.
[69] S. Piana, J. L. Klepeis, and D. E. Shaw, “Assessing the accuracy of physical mod-
els used in protein-folding simulations: quantitative evidence from long molecular
dynamics simulations,” Current opinion in structural biology, vol. 24, pp. 98–105,
2014.
[70] W. L. Jorgensen and J. Tirado-Rives, “The OPLS [optimized potentials for liquid
simulations] potential functions for proteins, energy minimizations for crystals of
cyclic peptides and crambin,” Journal of the American Chemical Society, vol. 110,
no. 6, pp. 1657–1666, 1988.

Bibliography 84
[71] G. A. Kaminski, R. A. Friesner, J. Tirado-Rives, and W. L. Jorgensen, “Evaluation
and reparametrization of the OPLS-AA force field for proteins via comparison
with accurate quantum chemical calculations on peptides,” The Journal of Physical
Chemistry B, vol. 105, no. 28, pp. 6474–6487, 2001.
[72] W. L. Jorgensen, D. S. Maxwell, and J. Tirado-Rives, “Development and testing of
the OPLS all-atom force field on conformational energetics and properties of organic
liquids,” Journal of the American Chemical Society, vol. 118, no. 45, pp. 11225–
11236, 1996.
[73] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan,
and M. Karplus, “Charmm: A program for macromolecular energy, minimization,
and dynamics calculations,” Journal of Computational Chemistry, vol. 4, no. 2,
pp. 187–217, 1983.
[74] A. D. MacKerell, M. Feig, and C. L. Brooks, “Extending the treatment of back-
bone energetics in protein force fields: Limitations of gas-phase quantum mechanics
in reproducing protein conformational distributions in molecular dynamics simula-
tions,” Journal of Computational Chemistry, vol. 25, no. 11, pp. 1400–1415, 2004.
[75] R. B. Best, X. Zhu, J. Shim, P. E. Lopes, J. Mittal, M. Feig, and A. D. MacK-
erell Jr, “Optimization of the additive charmm all-atom protein force field targeting
improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles,”
Journal of Chemical Theory and Computation, vol. 8, no. 9, pp. 3257–3273, 2012.
[76] Y. Shen and A. Bax, “SPARTA+: a modest improvement in empirical NMR chem-
ical shift prediction by means of an artificial neural network.,” J. Biomol. NMR,
vol. 48, pp. 13–22, Sep 2010.
[77] A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J.
Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kucz-
era, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E.
Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe,

Bibliography 85
J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus, “All-atom empirical potential for
molecular modeling and dynamics studies of proteins.,” J Phys Chem B, vol. 102,
pp. 3586–616, Apr 1998.
[78] S. Piana, K. Lindorff-Larsen, and D. E. Shaw, “How robust are protein folding
simulations with respect to force field parameterization?,” Biophysical Journal,
vol. 100, no. 9, p. L47, 2011.
[79] K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror,
and D. E. Shaw, “Improved side-chain torsion potentials for the Amber ff99SB
protein force field,” Proteins: Structure, Function, and Bioinformatics, vol. 78,
no. 8, pp. 1950–1958, 2010.
[80] A. D. MacKerell, J. Wiorkiewicz-Kuczera, and M. Karplus, “An all-atom empiri-
cal energy function for the simulation of nucleic acids,” Journal of the American
Chemical Society, vol. 117, no. 48, pp. 11946–11975, 1995.
[81] S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona,
S. Profeta, and P. Weiner, “A new force field for molecular mechanical simulation
of nucleic acids and proteins,” Journal of the American Chemical Society, vol. 106,
no. 3, pp. 765–784, 1984.
[82] W. Cornell, P. Cieplak, C. Bayly, I. Gould, K. Merz Jr, D. Ferguson, D. Spellmeyer,
T. Fox, J. Caldwell, and P. Kollman, “A new force field for molecular mechanical
simulation of nucleic acids and proteins,” J. Am. Chem. Soc, vol. 117, pp. 5179–
5197, 1995.
[83] R. B. Best and J. Mittal, “Protein simulations with an optimized water model:
cooperative helix formation and temperature-induced unfolded state collapse,” The
Journal of Physical Chemistry B, vol. 114, no. 46, pp. 14916–14923, 2010.
[84] W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein,
“Comparison of simple potential functions for simulating liquid water,” The Journal
of Chemical Physics, vol. 79, no. 2, pp. 926–935, 1983.

Bibliography 86
[85] S. Nose, “A molecular dynamics method for simulations in the canonical ensemble,”
Molecular Physics, vol. 52, no. 2, pp. 255–268, 1984.
[86] W. G. Hoover, “Canonical dynamics: equilibrium phase-space distributions,” Phys-
ical Review A, vol. 31, no. 3, pp. 1695–1697, 1985.
[87] H. J. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. Haak,
“Molecular dynamics with coupling to an external bath,” The Journal of Chemical
Physics, vol. 81, no. 8, pp. 3684–3690, 1984.
[88] M. Parrinello and A. Rahman, “Polymorphic transitions in single crystals: A new
molecular dynamics method,” Journal of Applied Physics, vol. 52, no. 12, pp. 7182–
7190, 1981.
[89] G. J. Martyna, M. E. Tuckerman, D. J. Tobias, and M. L. Klein, “Explicit reversible
integrators for extended systems dynamics,” Molecular Physics, vol. 87, no. 5,
pp. 1117–1157, 1996.
[90] W. C. Swope, H. C. Andersen, P. H. Berens, and K. R. Wilson, “A computer
simulation method for the calculation of equilibrium constants for the formation of
physical clusters of molecules: Application to small water clusters,” The Journal
of Chemical Physics, vol. 76, no. 1, pp. 637–649, 1982.
[91] E. Atherton and R. Sheppard, “Solid phase peptide synthesis,” 1989.
[92] W. J. Waddell, “A simple ultraviolet spectrophotometric method for the determi-
nation of protein.,” J. Lab. Clin. Med., vol. 48, pp. 311–4, Aug 1956.
[93] W. C. Wimley and S. H. White, “Quantitation of electrostatic and hydrophobic
membrane interactions by equilibrium dialysis and reverse-phase HPLC.,” Anal.
Biochem., vol. 213, pp. 213–7, Sep 1993.
[94] W. C. Wimley, T. P. Creamer, and S. H. White, “Solvation energies of amino acid
side chains and backbone in a family of host-guest pentapeptides.,” Biochemistry,
vol. 35, pp. 5109–24, Apr 1996.

Bibliography 87
[95] S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts,
J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess, and E. Lindahl, “GRO-
MACS 4.5: a high-throughput and highly parallel open source molecular simulation
toolkit.,” Bioinformatics, vol. 29, pp. 845–54, Apr 2013.
[96] D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark, and H. J. C.
Berendsen, “GROMACS: fast, flexible, and free.,” J Comput Chem, vol. 26,
pp. 1701–18, Dec 2005.
[97] C. Loken, D. Gruner, L. Groer, R. Peltier, N. Bunn, M. Craig, T. Henriques,
J. Dempsey, C.-H. Yu, J. Chen, L. J. Dursi, J. Chong, S. Northrup, J. Pinto,
N. Knecht, and R. V. Zon, “SciNet: Lessons Learned from Building a Power-
efficient Top-20 System and Data Centre,” Journal of Physics: Conference Series,
vol. 256, p. 012026, 2010.
[98] F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax, “NMRPipe:
a multidimensional spectral processing system based on UNIX pipes.,” J. Biomol.
NMR, vol. 6, pp. 277–93, Nov 1995.
[99] W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L.
Ulrich, J. L. Markley, J. Ionides, and E. D. Laue, “The CCPN data model for NMR
spectroscopy: development of a software pipeline.,” Proteins, vol. 59, pp. 687–96,
Jun 2005.
[100] W. Kabsch and C. Sander, “Dictionary of protein secondary structure: pattern
recognition of hydrogen-bonded and geometrical features.,” Biopolymers, vol. 22,
pp. 2577–637, Dec 1983.
[101] S. M. Kelly, T. J. Jess, and N. C. Price, “How to study proteins by circular dichro-
ism.,” Biochim. Biophys. Acta, vol. 1751, pp. 119–39, Aug 2005.
[102] Z. Xue, “Understanding the molecular mechanism of elasticity in elastin from a
solvation perspective,” Master’s thesis, University of Toronto, 2013.
[103] A. Rudin and P. Choi, “The elements of polymer science and engineering,” 2012.

Bibliography 88
[104] C. Baldock, A. F. Oberhauser, L. Ma, D. Lammie, V. Siegler, S. M. Mithieux,
Y. Tu, J. Y. H. Chow, F. Suleman, M. Malfois, S. Rogers, L. Guo, T. C. Irving,
T. J. Wess, and A. S. Weiss, “Shape of tropoelastin, the highly extensible protein
that controls human tissue elasticity.,” Proc. Natl. Acad. Sci. U.S.A., vol. 108,
pp. 4322–7, Mar 2011.
[105] D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges, and B. D. Sykes, “(1)H,
(13)C and (15)N random coil NMR chemical shifts of the common amino acids. i.
investigations of nearest-neighbor effects.,” J. Biomol. NMR, vol. 5, pp. 67–81, Jan
1995.
[106] S. Rauscher, Protein Non-Folding: A Molecular Simulation Study of the Structure
and Self-Aggregation of Elastin. PhD thesis, University of Toronto, 2012.
[107] A. Nikolic, S. Baud, S. Rauscher, and R. Pomes, “Molecular mechanism of β-sheet
self-organization at water-hydrophobic interfaces,” Proteins: Structure, Function,
and Bioinformatics, vol. 79, no. 1, pp. 1–22, 2011.
[108] M. Bixon and S. Lifson, “Potential functions and conformations in cycloalkanes,”
Tetrahedron, vol. 23, pp. 769–784, Jan 1967.
[109] E. A. Cino, W.-Y. Choy, and M. Karttunen, “Comparison of Secondary Structure
Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics
Simulations.,” J Chem Theory Comput, vol. 8, pp. 2725–2740, Aug 2012.
[110] K. Lindorff-Larsen, P. Maragakis, S. Piana, M. P. Eastwood, R. O. Dror, and D. E.
Shaw, “Systematic validation of protein force fields against experimental data,”
PLoS One, vol. 7, no. 2, pp. 3787–3791, 2012.
[111] R. B. Best, D. de Sancho, and J. Mittal, “Residue-specific -helix propensities from
molecular simulation,” Biophysical Journal, vol. 102, no. 6, pp. 1462–1467, 2012.
[112] R. J. Moreau, C. R. Schubert, K. A. Nasr, M. Torok, J. S. Miller, R. J. Kennedy,
and D. S. Kemp, “Context-independent, temperature-dependent helical propensi-

Bibliography 89
ties for amino acid residues.,” J. Am. Chem. Soc., vol. 131, no. 36, pp. 13107–16,
2009.
[113] P. S. Nerenberg and T. Head-Gordon, “Optimizing Protein-Solvent Force Fields
to Reproduce Intrinsic Conformational Preferences of Model Peptides,” Journal of
Chemical Theory and Computation, vol. 7, no. 4, pp. 1220–1230, 2011.
[114] K. A. Beauchamp, Y.-S. Lin, R. Das, and V. S. Pande, “Are protein force fields
getting better? A systematic benchmark on 524 diverse NMR measurements,”
Journal of Chemical Theory and Computation, vol. 8, no. 4, pp. 1409–1414, 2012.
[115] W. A. Hegefeld, S.-E. Chen, K. Y. DeLeon, K. Kuczera, and G. S. Jas, “Helix
formation in a pentapeptide: experiment and force-field dependent dynamics.,” J
Phys Chem A, vol. 114, pp. 12391–402, Dec 2010.
[116] J. Mittal and R. B. Best, “Tackling force-field bias in protein folding simulations:
folding of Villin HP35 and Pin WW domains in explicit water.,” Biophys. J., vol. 99,
pp. L26–8, Aug 2010.
[117] D. Caballero, J. Maatta, A. Q. Zhou, M. Sammalkorpi, C. S. O’Hern, and L. Regan,
“Intrinsic α-helical and β-sheet conformational preferences: a computational case
study of alanine.,” Protein Science, vol. 23, pp. 970–80, Jul 2014.
[118] J. Djajamuliadi, T. F. Kagawa, K. Ohgo, and K. K. Kumashiro, “Insights into
a putative hinge region in elastin using molecular dynamics simulations.,” Matrix
Biol., vol. 28, pp. 92–100, Mar 2009.
[119] M. Seo, S. Rauscher, R. Pomes, and D. P. Tieleman, “Improving Internal Peptide
Dynamics in the Coarse-Grained MARTINI Model: Toward Large-Scale Simula-
tions of Amyloid- and Elastin-like Peptides.,” J Chem Theory Comput, vol. 8,
pp. 1774–1785, May 2012.
[120] J. Barnoud and L. Monticelli, “Coarse-grained force fields for molecular simula-
tions.,” Methods Mol. Biol., vol. 1215, pp. 125–49, 2015.








![Surface and adsorption characteristics of three elastin ... · PDF fileand ELP20-244 [ELP4] contains five hydrophobic domains flanked by four cross-linking domains [18, 19]. The](https://img.dokumen.tips/doc/110x75/5a88daa47f8b9a9f1b8e99a2/surface-and-adsorption-characteristics-of-three-elastin-elp20-244-elp4-contains.jpg)




















