Embed Size (px)
Citation preview

Starting Material Selection for Type II Drug Master Files
Ronald S. Michalak Quality Assessment Lead (Acting),
Division of Lifecycle API Office of New Drug Products,
OPQ/CDER/ FDA

CDER Reorganization
Office of Pharmaceutical Quality In Operation since Jan 11, 2015
Combines Former Office of Pharmaceutical Science (OPS), Office of Compliance and DMF Review Staff
from Office of Generic Drugs
Immediate Office PMAS Staff
Office of Biotechnology
Products Office of New Drug
Products Office of Lifecycle
Drug Products Office of Testing
and Research
Office of Surveillance
Office of Process and Facilities
Office of Program and Regulatory
Operations
Office of Policy for Pharmaceutical
Quality
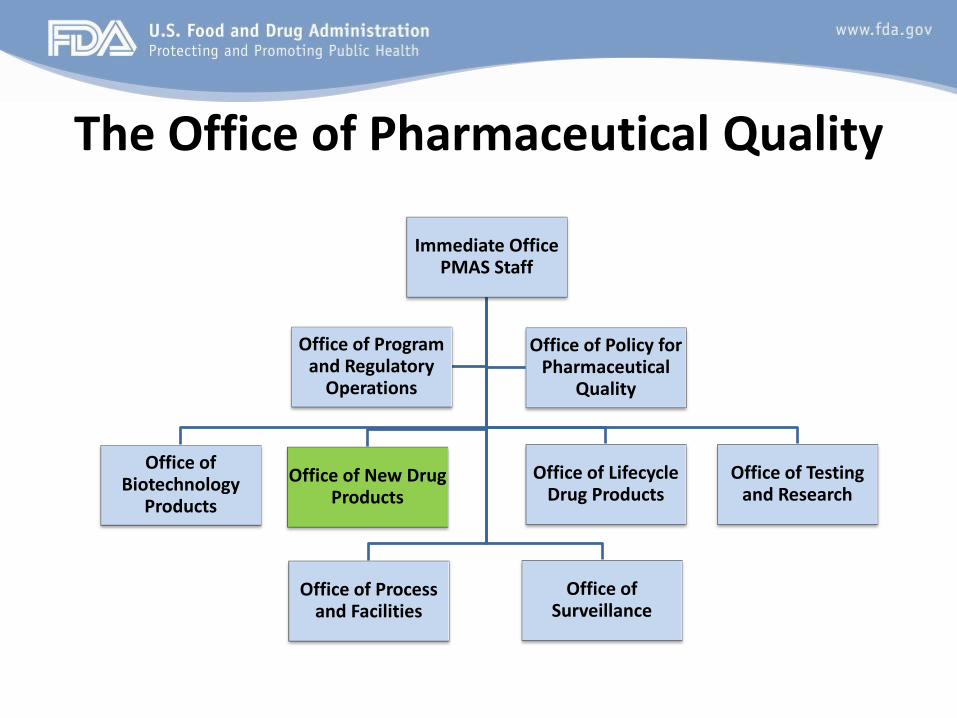
The Office of Pharmaceutical Quality

Office of New Drug Products
• Office of the Director
• Division of New Drug API: NDA/IND Drug Substance Review
• Division of Lifecycle API: DMF Staff/ANDA Drug Substance Review
• Division of Biopharmaceutics
• Division of New Drug Products I
• Division of New Drug Products II

ONDP Drug Substance Review • Division of New Drug API
– 2 Branches each aligned with the OND Clinical Divisions
– Review of Drug Substance sections of NDAs, INDs and Meeting Packages
• Division of Lifecycle API – 3 Branches – Review of DMFs primarily in support of ANDAs
(generics)

• Reviewers trained in organic/analytical/process chemistry, many staff with industrial experience – Drug substance reviews are performed by approximately 80
reviewers (20 for New Drugs and 60 for Lifecycle)
– One Office improves communication regarding starting material selection between RLD and subsequent lifecycle submissions
• DMF Scientific Review Process: – Primary review
– Secondary review
– Tertiary review (if 1st generic or high risk application)
– DMF is then adequate, inadequate, or adequate with information request(s)
ONDP Drug Substance Review

What is a Regulatory Starting Material? – High level definition in ICH Q7 and Q11
– Starting point for cGMP manufacturing process
– Starting point for the detailed description of the synthetic process in a marketing application
– One or more starting materials is possible depending on linear or convergent syntheses
– Can be a commercially available (commodity) chemical manufactured mainly for the non-pharmaceutical market
– Can be synthesized in-house or outsourced
– Has defined chemical structure

Dilemma: How do we balance starting material selection that impacts industry concern for supply chain flexibility
and cost with adequately low risk to quality and patient?

Why do Regulators Request Longer GMP Synthetic Routes?
• Information on earlier synthetic steps is necessary in order to understand the risk of impurity carryover and to demonstrate that the proposed control strategy sufficiently mitigates this risk
• A sufficient number of purification steps need to be documented so that the fate and purge of impurities can be understood
• Recrystallizations and salt formation are not considered chemical transformation steps
• Multiple synthetic transformations carried out in one vessel without intermediate isolations (sometimes referred to as telescoped steps or “one pot reactions”) provide fewer opportunities for purification than if isolation of intermediates were carried out
• For these scientific reasons, short synthetic routes will not normally be accepted

Regulatory Perspective On considerations for starting material selection In Lifecycle API: (DMF scientific review)
• Q7 and Q11 principles • Commercial availability • Information known to the Agency. • Is the starting material complex?
– How is stereochemistry or regiochemistry established – Where in the process is the core of the structure formed?
• Was information provided on how the starting material is made so that the impurity profile can be adequately assessed?
• What is the justification for the SM specification? – Justified limits based on observed impurity values during process
validation (retrospective) – Spike and purge studies at impurity limits (prospective)

Starting Material Specification Example In-House Specification for Starting Material 1 Test / Method Acceptance Criteria
Description White to Off White Solid ID / HPLC RT of main peak matches RS
Water / KF NMT 0.50% Purity / HPLC NLT 90.0%
Any Single Unspecified Impurity/HPLC
NMT 2.0%
Total Impurities/HPLC NMT 5.0%
• No specific ID test • Are impurity limits justified • Does purity limit reflect observed values in COAs • Is test omission for residual solvents justified

Guidance Documents for Selection of Starting Materials
– ICH Q11 • Guidance on Drug Substances with a sub-section on
Starting materials
– ICH Q11, Q & A (in progress) • Companion document to Q11 providing additional
clarification
– ICH Q7A • Guidance for GMP from the starting material to API

ICH Q11 A multi-step GMP process is clearly implied
• Process risk inherently higher the closer the regulatory starting material is to the final API
GMP should be applied to critical process steps which impact the quality of the API • Steps that introduce key structural features • Steps where careful control of stoichiometry,
temperature, other process variables is crucial for API quality
• Complex chemical transformations
Overview of Q11 Principles

Overview of Current Guidances ICH Q11 – Seven General Principles • The manufacturing route begins from the starting material and GMPs apply from there
onwards; • The changes in material attributes and process conditions at the beginning of the
manufacturing process have a lower potential to impact API quality; • Manufacturing steps that impact the impurity profile of the API should be included in
the process description; • Enough of the manufacturing process must be disclosed so impurity fate and purge can
be understood; • Starting materials should be a substance of defined chemical properties, and non-
isolated intermediates are not appropriate; • Starting materials should be a significant structural fragment and should be
distinguished from reagents and solvents; and • Commercially available commodity chemicals need not be justified as a starting
material as long as these are available in the pre-existing non-pharmaceutical market.

Clarification of Q11 Principles • ICH Q11
– “Starting materials should be a significant structural fragment ..”
• Should not be interpreted to imply close structural similarity between the proposed starting material and the API
• Intention was to distinguish between raw materials and starting materials
– Commercially available commodity chemicals need not be justified as starting materials …
• Provide supporting documentation regarding the non-pharmaceutical market for these chemicals
• Procuring intermediates from vendors is not commercial availability

Clarification of Q11 Principles - “Manufacturing steps that impact the impurity profile of the API should be included in the process description”
- “Enough of the manufacturing process must be disclosed so impurity fate and purge can be understood”
Is enough of the process covered under GMP that we are confident that the process can purge all impurities resulting from the starting material to the specification levels in the final API? Tip: Include a narrative description of the generation, fate, and purge of impurities in the process
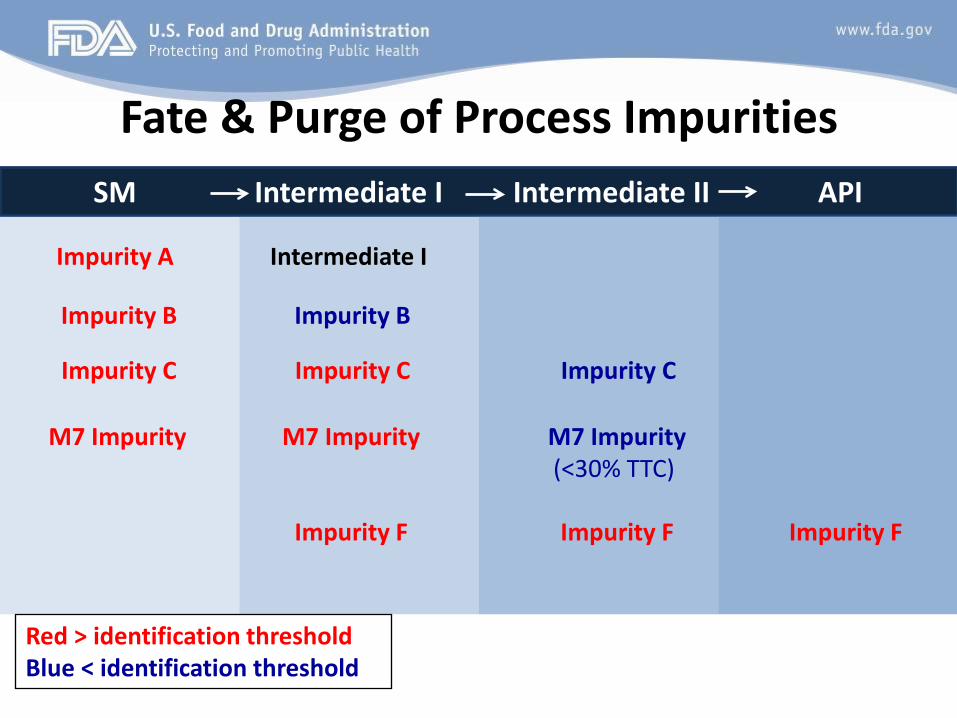
Fate & Purge of Process Impurities SM Intermediate I Intermediate II API
Impurity A Intermediate I
Impurity B Impurity B
Impurity C Impurity C Impurity C
M7 Impurity M7 Impurity M7 Impurity (<30% TTC)
Impurity F Impurity F Impurity F
Red > identification threshold Blue < identification threshold
18
[H] FI DS
G
C A + B D
DS (salt) J F E
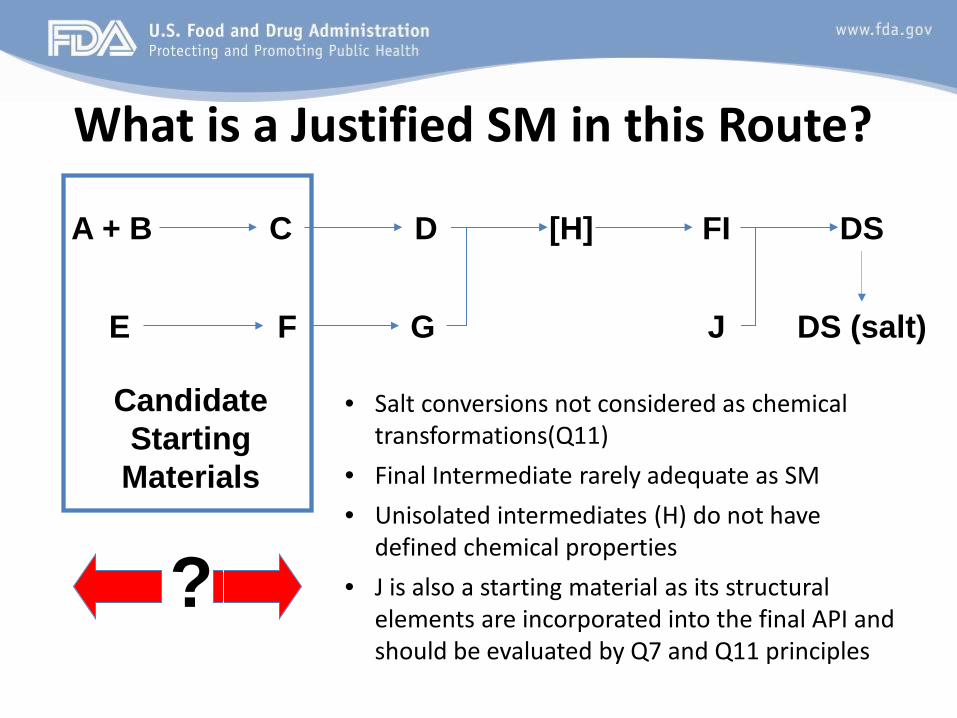
What is a Justified SM in this Route?
Candidate Starting
Materials
?
Temporary slide to justify late stage SM’s
• Salt conversions not considered as chemical transformations(Q11)
• Final Intermediate rarely adequate as SM • Unisolated intermediates (H) do not have
defined chemical properties • J is also a starting material as its structural
elements are incorporated into the final API and should be evaluated by Q7 and Q11 principles

FDA Concerns Regarding Starting Material Designation
• Starting material is not manufactured under GMP • Change Control and Vendor Qualification
– Outsourcing is common – Cost benefit to industry / High risk for regulators
• Changes to manufacturing process of starting material and impact on drug substance – Risk to impurity control strategy (genotoxic, regioisomers,
chirality, heavy metals, etc.) – Applicant should have a strong Quality Management
System (vendor qualification) (not in scope of review)

Common Issues with Starting Material Designation by Industry
• Proposing starting materials only one or two steps from the API in a multi-step synthesis – Justified by starting material contributing a “significant structural
fragment”; by this logic even the final intermediate could be a starting material. Contradicts all other Q11 principles and Q7A.
• Proposing starting materials multiple steps from the API, however: – The steps are simple chemical manipulations e.g., esterification,
removal of protecting groups. – The “starting material” itself is synthesized by complex reactions,
e.g., stereospecific reactions, complex ring systems, etc. – The process is telescoped: few or no isolated intermediates

Common Issues with Starting Material Designation by Industry
• Proposal of an advanced intermediate as a starting material and making a commitment not to change the manufacturing process for it without FDA notification. – The ‘advanced intermediate’ should still meet all criteria for SM
outlined in Q11 and Q7A – According to interpretation of regulations 21 CFR 314.70 and 21
CFR 314.420 (c), ‘any change’ could include starting material vendors with a new route of synthesis which results in the addition of tests to the starting material specification.
– These types of proposals are evaluated on a case-by-case basis

Other Recommendations Justification why steps prior to the starting material, which are not carried out under GMP, are not likely to affect the quality of the API should always be provided.
It is strongly recommended that industry seek FDA input on SM selection: • For Generics/DMF holders by email at

Starting Material Selection: Bottom Line • All the general principles in Q11 and Q7A for selection of starting
materials should be considered rather than applying any individual one in isolation • Commercial availability • Complexity of the process and structure • Impurity fate and purge • Significant structural fragment • Adequate SM specification
• Deficiencies for inadequate SM are common during DMF reviews (both CA and full scientific review) and can be costly: • Extra review cycles • More work (validation/method development) • Risk of RTR for the referencing ANDA






























