Embed Size (px)
Citation preview

Irish College of General Practitioners
Research Ethics Committee
4/5 Lincoln Place Dublin 2
Standard Operating Procedures
♦ SOP Title: Irish College of General Practitioners Ethics Committee standard operating
Procedures
PROCEDURES ♦ SOP Author(s. REC Committee Members
♦ SOP Status: Current
♦ Version No: 2 December 2003
Approval 1: Signed: Dr. Niall Maguire
Chairman
Approval 2: Signed: Pauline Tierney Administrator
December 2003 ____________________________________________________________________________________

2
CONTENTS 1.0 OBJECTIVE _______________________________________________________________________ 2.0 POLICY ________________________________________________________________________ 3.0 PROCESS FLOW 3.1 Application Procedure. 3.2. Pre Meeting Procedure. 3.3. Meeting Procedure. 3.4. Post Meeting Procedure. _______________________________________________________________________ 4.0 ICGP/ APPLICABLE TO 4.1 All Members of Ethics Committee. 4.2. All personnel of the Ethics Office. 4.3. All personnel submitting an application. ________________________________________________________________________ 5.0 DEFINITIONS ________________________________________________________________________ 6.0 PROCEDURES 6.1. Composion of Committee. 6.2. Functions of Committee. 6.3. Responsibilities of Committee. 6.4. Meeting Procedures. 6.5. Committee Review Procedure. 6.6. Record Keeping. ________________________________________________________________________ 7.0 COMPANY/INVESTIGATORS 7.1. Notification of Amendments. 7.2. Notification of Adverse Events. 7.3. Annual Review and Renewal. 7.4. Patient Information Leaflet.
7.5. Patient Consent. 7.6. Final Report
________________________________________________________________________ 8.0 APPENDICES 8.0.1 Appendix A List of meeting dates 2002 8.0.2 Appendix B Constitution of Ethics Committee 8.0.3 Appendix C ICH GCP Guidelines section 2 & 3 8.0.4 Appendix D The Clinical Trials Act 1987 (1990)
________________________________________________________________________

3
1.0 OBJECTIVE To ensure that the content of the clinical trial documentation is relevant and accurate, the submissions are prepared and progressed in a consistent manner, and are subject to appropriate ethical review and approval procedures. ________________________________________________________________________ 2.0 POLICY The Irish College of General Practitioners Ethics Committee will safeguard the rights safety and well being of all trial subjects. Special attention will be paid to trials that may include vulnerable subjects. The Committee will ensure that clinical trials are conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with GCP and the Clinical Trials act 87/1990. 3.0 PROCESS FLOW
3.1 APPLICATION PROCEDURE Post Meetin
Inquiry to Ethics Office re submission procedures. See ICGP Website: www.icgp.ie for further information.
Ethics Office to forward 1. Application Form. 2. Submission Check list Available in both Hard copy or disc
Submission must be received by Ethics Office at least three weeks prior to next review meeting.
All details of 1. Application form x13 2. Patient consent x13 3. Patient Information Leaflets x13 4. Questionnaires x13 5. Protocol x3 6. Investigator Brochure x3 Where applicable Processed by Ethics Office.
When all the above requirements are processed. 1. Log Proposal on Database. 2. Include on Agenda for next Committee Meeting.
4
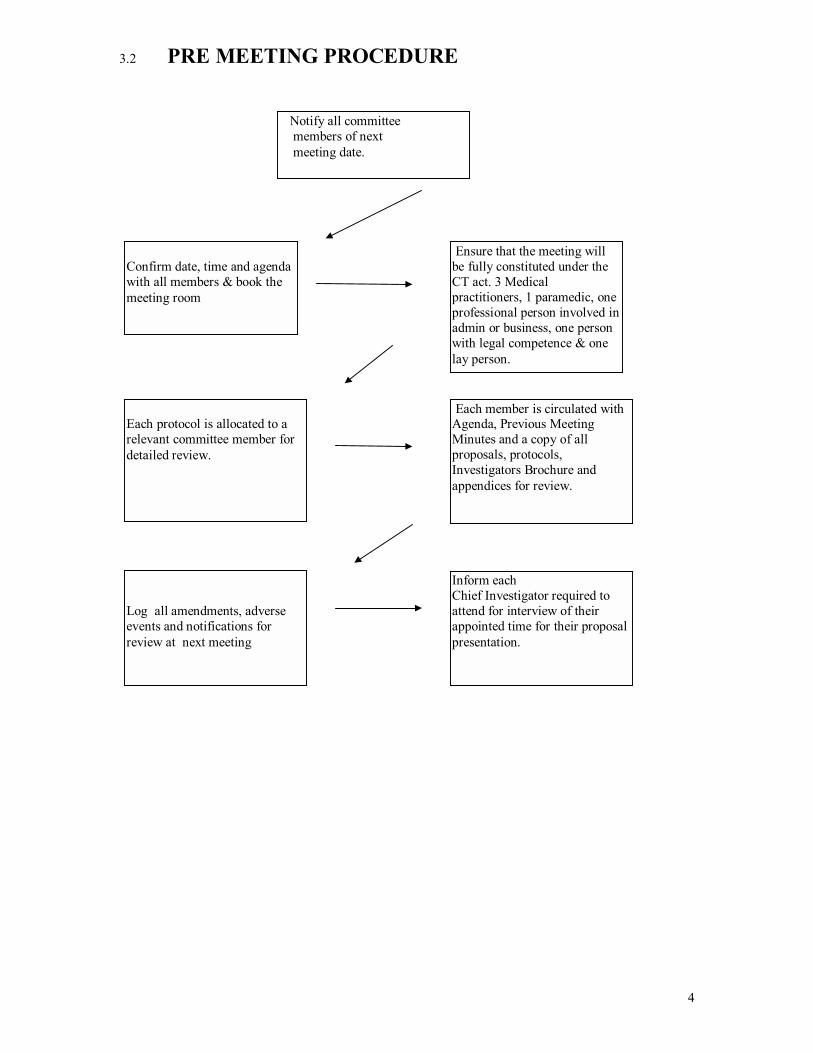
3.2 PRE MEETING PROCEDURE
Confirm date, time and agenda with all members & book the meeting room
Each protocol is allocated to a relevant committee member for detailed review.
Log all amendments, adverse events and notifications for review at next meeting
Each member is circulated with Agenda, Previous Meeting Minutes and a copy of all proposals, protocols, Investigators Brochure and appendices for review.
Inform each Chief Investigator required to attend for interview of their appointed time for their proposal presentation.
Notify all committee members of next meeting date.
Ensure that the meeting will be fully constituted under the CT act. 3 Medical practitioners, 1 paramedic, one professional person involved in admin or business, one person with legal competence & one lay person.

5
3.3 COMMITTEE MEETING PROCEDURE
Previous Minutes and Matters arising are discussed.
Each submission is discussed in detail and a list of queries is compiled
A quorum of at least 5 members must be present Hon. Sec. may act as Chairman in his/her absence
Each Company/Investigator in turn answers any queries The committee may have.
A vote on each submission is taken and recorded on the minutes of the meeting. If any of the members are participating in a trial that is under review by the Committee, he/she may not participate in the discussion on that submission and must absent him/herself from the vote.

6
3.4. POST MEETING PROCEDURE
Recorded Minutes are Kept on file.
All Companies/Investigators of protocols submitted for review are informed of the committee�s decision in writing and any recommendations.
Investigators/Companies who submitted Amendments or Adverse events for review will be notified of the committee�s decision in writing.
All correspondence must be signed by either the Administrator or Chairman
All Ethically Approved Trials Must Include: Copy of protocol. * Copy of Investigator Brochure. * Copy of Pt Information Sheet. Copy of Patient Consent Form. Copy of Ethics Proposal Form Copy of Ethics Approval Letter Copy of IMB approval * Copy of Signed Indemnity.* * = Where Applicable.

7
4.0 APPLICABLE TO 4.1 All members of the Ethics Committee. 4.2 All personnel of the Ethics Office. 4.3 All investigators / Companies submitting an application. _______________________________________________________________________ 5.0 DEFINITIONS IMB : Irish Medicine Board.
ICH GCP : International Conference on Harmonisation of Good Clinical Practice. (Harmonized Tripartite Guidelines for Good Clinical Practice.)
CT :Clinical Trials Act ________________________________________________________________________ 6.0 PROCEDURES 6.1. Composition of Committee. The Ethics Committee will comprise of a minimum seven members and up to twenty members. A quorum of at least five members must be present at each meeting. See Appendix A for full list of Members and qualifications. The term of office of each member is three years (renewable for one term). Any change to membership of the Ethics Committee must be notified to the Irish medicines Board in writing. 6.2 FUNCTIONS 6.2.1. To consider the justification for conducting the proposed trial and the circumstances under which proposed trial is to be conducted and, where the committee considers that the proposed trial is justified and it is satisfied with those circumstances, it shall give its approval to the conducting of the proposed trials and the person who is arranging for the conducting of the proposed trial. in compliance with the Clinical Trials Act 1987/1990 and ICH GCP guidelines. 6.2.2. The committee shall not consider the proposed clinical trial justified unless it is satisfied that the risks to be incurred by participants would be commensurate with the objectives of the trial. 6.3. Committee Responsibilities.
6.3.1. To safeguard the rights, safety and well being of all trial subjects. 6.3.2 Obtain the following documents · Trial Protocol. * · Protocol Amendments * · Investigator Brochure. * · Patient Information Sheet. · Patient consent form. · Available Safety reports
· Investigator Details � Name, Address, Medical Council Registration No, Medical Insurance Name, Number & expiry date
· Copy of IMB approval * · Copy of signed Indemnity Form. * * = Where applicable.

8
6.3.3. To review proposed trial with regard to the following; • The objectives of the proposed trial and its planning and organisational structure. • The qualifications and competence of each person who would conduct the trial and where appropriate
the resources available to them. • The criteria to be used for the recruitment and the selection of participants. • The extent and nature of the medical examination that persons selected as participants are to undergo
before participating in the trial. • The extent to which the health of the participants is proposed to be monitored during and after the trial. • Whether or not the persons selected as participants are to undergo independent medical examination
before, during or after the clinical trial. • Details of the proposed method or methods by which participants are to be recruited. • Details of any inducements or rewards, monetary or otherwise to be made for becoming or being a
participant. • Any payments whether monetary or otherwise to be made to a person for conducting the clinical trial or
any part of the trial. • The criteria to be used to ensure that the identity of each participant remains confidential. • Any payments whether monetary or otherwise to be made to any person for facilities used for the
purposes of the trial. A fee of �1269.74 will be charged for Protocol review, there will be no further fee changed for review of any Amendments to the Protocol. This fee is to cover administrative expenses, where the protocol is supported by a Pharmaceutical company. 6.4. MEETING PROCEDURES • A Maximum of five meetings to be held per annum. • All members to be notified of meetings. • Agenda, Previous Minutes and Application forms to be circulated to the committee two weeks prior to the meeting. • Minutes of the meeting to be recorded. • All meetings must consist of at least a quorum. • Not more than 8 protocols will be reviewed at any one meeting. 6.5. COMMITTEE REVIEW PROCEDURE: 6.5.1. When protocols involve more than minimal risk to human subjects, meaning a greater risk than that found in ordinary daily life, they must be reviewed by the Ethics Committee. A review of the Ethics Committee is defined as a regular meeting of a quorum (5 or more) of the committee members 6.5.2. The Committee will meet on the third/fourth Wednesday of the month at the discretion of the
Chairman. Project applications must be submitted at least three weeks prior to each meeting. If a large volume of applications are received for a meeting, the applications may not all be dealt with and some may have to be postponed to a subsequent meeting. The applications will be taken on a first come, first served basis or at the discretion of the Chairman. As part of the review process, an individual on the committee will be assigned the task of acting as the primary reviewer.

9
6.5.3. Each committee member will be circulated with the protocol submission form, patient information leaflet and patient consent form. 6.5.4. The Company/ Investigator is invited to attend the meeting to answer any questions on the protocol. The protocol is then discussed by the committee until a consensus is reached and then a vote on whether to approve the protocol is taken. Following each Ethics committee meeting the company/investigator will be informed of the protocol status. 6.5.5 Approval: If full ethical approval is granted, the company/investigator may only begin the research proposed in the protocol when it has been outlined in a letter. 6.5.6. Conditional Approval: Conditional approval of a protocol may be granted, requiring modifications in the protocol and/or patient information sheet and/or patient consent form before initiation or responses to specific inquiries by the Ethics Committee. In this case, the company/ investigator should submit a cover letter along with a modified submission form, protocol, consent form or supplemental information as requested with the changes highlighted. No research may be started until all conditions have been met and full approval has been obtained. Where conditional approval is granted the Chairman and Chief Executive may review the modified submissions and grant approval subject to the affirmation of the committee at its following meeting. 6.5.7. Deferral: A deferred protocol must be revised and resubmitted to the Ethics committee 6.5.8. Rejection: Protocols may be rejected by the Ethics Committee. This may occur if a protocol has been deferred several times and the Ethics Committee feels that the proposed research is not justified and/or poses severe or unnecessary risk to the subjects. The Ethics Committee will not accept any further revisions to a rejected protocol. 6.5.9 Amendments: Amendments to approved trials may be submitted and reviewed by the Chairman and Chief Executive and approval issued as appropriate subject to affirmation by the committee at its following meeting. 6.6. RECORD KEEPING. • Written Procedures. • Membership List. • List of membership qualifications. • Agendas and Minutes of meetings. • All Correspondence. • All submitted documentation must be retained for at least three years after completion of trial 7.0. COMPANY/INVESTIGATORS RESPONSIBILITIES The Company Representative or Investigator of the trial must attend the Ethics Committee review. It is the responsibility of the company/ Investigator to provide the Ethics Committee with the appropriate information on the research protocol including initial information, notification of subsequent modifications, terminations and adverse reactions. It is the responsibility of the Company/ Investigator to ensure that all equipment used in trials has certification of compliance for the Year 2004.

10
7.1 NOTIFICATION OF AMENDMENTS When any revision to an approved research protocol or written consent form is proposed the amendment must be brought to the attention of the ethics committee for approval. Amendments to approved protocols may not be initiated until Ethics Committee approval has been obtained. 7.2 NOTIFICATION OF ADVERSE EVENTS All adverse events which may be related to the trial drug must be brought to the attention of the ethics committee. These events must be accompanied by an explanatory letter from the Company or Principal Investigator involved. 7.3. ANNUAL REVIEW AND RENEWAL Update reports of all trials must be forwarded to the ethics committee on an annual basis. The committee must be notified when trials are completed or terminated. 7.4. PATIENT INFORMATION LEAFLET Patient information leaflet must include the following elements. • A statement that the protocol involves research, an explanation of the purposes of the research and the expected duration of the subject�s participation, a description of the procedures to be followed and identification of any procedures, drugs or devices which are experimental. • Description of any reasonably foreseeable risks or discomforts to the subject. • Description of any benefits to the subject or to others that may reasonably be expected from the research, including payment or free treatment. • A disclosure of appropriate alternative procedures or courses of treatment, if applicable, that might be advantageous to the subject. • A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained. • Have the title of the project/trial and the names of the investigators clearly printed on the top of the
form. • Have space for signature/name/date of both the patient and witness/guardian and the investigator (or investigator�s nominee). • Include the statement �I�ve read and understand the attached patient information leaflet.� 7.5. PATIENT CONSENT The following points must be included in the written patient informed consent. • That the trial involves research. • The purpose of the trial. • The trial treatment(s) and the probability for random assignment to each treatment. • The trial procedures to be followed including all invasive procedures. • The subject�s responsibilities. • Those aspects of the trial that are experimental. • The reasonably foreseeable risks. • The reasonably expected benefits. • The alternative procedures/treatments available. • Compensation/ treatment available in event of trial related injury. • Payment to subject for participating. • Anticipated expenses to subject for participating. • That the subject�s participation is voluntary and may withdraw at any time. • That the monitor/auditor/regulatory authority will be granted direct access to the patient�s medical records.

11
• That records identifying the subject will be kept confidential. • That the subject will be informed in a timely manner if information becomes available that may be relevant to the subjects willingness to participate. • The name of the person to contact for further information re the trial. • The foreseeable circumstances/reasons under which the subject�s participation may be terminated. • The expected duration of the subject�s participation in the trial. • The approximate number of subjects involved in the trial. _________________ 7.6. Final Report A copy of the final report should be sent to the Committee as soon as it is available. 8.0 APPENDICES
8.0.1 Appendix A List of meeting dates 2004 8.0.2 Appendix B Constitution of Ethics Committee 8.0.3 Appendix C ICH GCP Guidelines section 2 & 3 8.0.4 Appendix D The Clinical Trials Act 1987 (1990)

12
Appendix A
Research Ethics Committee
Meeting Dates for 2004
Meeting Date Submission Deadline 1) 4th February 14th January 2) 21st April 31st March 3) 16th June 27th May 4) 15th September 25th August 5) 17th November 7th October Please note these Dates may be subject to change. See the ICGP Web page: www.icgp.ie

13
Irish College of General Practioners Independent Ethics Committee The Ethics Committee is constituted according to the ICH GCP guidelines & the Clinical Trials act 1987 (amended 1990). This Ethics Committee operates to the standards laid down in the ICH GCP guidelines published 1997 and implemented January 1998.
Constitution Name Position on Committee Experience &
Qualifications Address
Dr. Niall Maguire Ordinary member Medical Practitioner
MB, MICGP 2 Bedford Place Navan Co. Meath
Dr. John Mason Ordinary Member Medical Practioner
MB, BCh, FRCGP, MICGP
22 Killiney Avenue Ballybrack Co. Dublin
Dr. Thomas Maher Ordinary member Medical Practitioner
MB. MICGP, MRCGP 1 Whitworth Road Drumcondra Dublin 9
Ms. Marie Faughey Ordinary Member Acting Director of Public Health Nursing South Western Health Authority 21/25 Lord Edward St Dublin 2
Dr. Walter Cullen Ordinary y Member Medical Practitioner
MRCGP, MICGP, DCH, BObs
U.C.D Dept of General Practice Coombe Healthcare Centre Dolphins Barn Dublin 8
Dr. David Smith Ordinary Member Ethicist
Ethicist Woodview 34 Mount Merrion Avenue Blackrock Co. Dublin
Dr. Deirdre Madden Ordinary Member Lawyer
BCL, LLM, BL, PH.D Dept Law UCC Cork
Dr. Teresa Maguire Ordinary Member PhD Head of Division Research and Development for Health Division HRB 73 Lower Baggot Street Dublin 2
Dr. Deirdre Murphy Ordinary Member
MD, MICGP Merrion Medical Centre 240 Dublin Road Dublin 4
Prof. Colin Bradley Ordinary Member Prof. of General Practice
MD, MICGP, FRCGP Dept. of General Practice Distillery Road University College Cork
Mr. Michael Griffith Ordinary Member Chief Executive Fighting Blindness 1 Christchurch Hall High Street Dublin 8
Mr. Fionan O� Cuinneagain Ordinary Member Chief Executive ICGP 4/5 Lincoln Place Dublin 2
Ms Pauline Tierney Administrator ICGP 4/5 Lincoln Place Dublin 2

14
Appendix C
ICH GCP GUIDELINES
Section 2 & 3
Section 2 Statement of Compliance with International Conference on Harmonisation/Good Clinical Practice (ICH) Guideline for the conduct of Trials involving the Participation of Human Subjects. This Ethics Committee is fully compliant with the above guidelines as they relate to the responsibilities, composition, function, operations and records of an Independent Ethics Committee. The Principles of ICH GCP 2.1 Clinical trials should be conducted in accordance with the ethical principles that have their origin
in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s).
2.2 Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the
anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
2.3 The rights, safety, and well being of the trial subjects are the most important considerations and
should prevail over interests of science and society. 2.4 The available non-clinical and clinical information on an investigational product
should be adequate to support the proposed clinical trial. 2.5 Clinical trials should be scientifically sound, and described in a clear detailed protocol. 2.6 A trial should be conducted in compliance with the protocol that has received prior institutional
review board (IRB)/independent ethics committee (IEC) approval/favourable opinion. 2.7 The medical care given to, and medical decisions made on behalf of, subjects should always be the
responsibility of a qualified physician or, when appropriate, of a qualified dentist. 2.8 Each individual involved in conducting a trial should be qualified by education, training and
experience to perform his or her respective task(s). 2.9 Freely given informed consent should be obtained from every subject prior to clinical trial
participation. 2.10 All clinical trial information should be recorded, handled, and stored in a way that allows its
accurate reporting, interpretation and verification. 2.11 The confidentiality of records that could identify subjects should be protected, respecting the
privacy and confidentiality rules in accordance with the applicable regulatory requirement(s). 2.12 Investigational products should be manufactured, handled, and stored in accordance with
applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol.
2.13 Systems with procedures that assure the quality of every aspect of the trial should be implemented.

15
Section 3 Institutional Review Board/Independent Ethics Committee (IRB/IEC) 3.1 Responsibilities 3.1.1 An IRB/IEC should safeguard the rights, safety, and well being of all trial subjects. Special
attention should be paid to trials that may include vulnerable subjects 3.1.2 The IRB/IEC should obtain the following documents: trial protocol(s)/amendment(s), written
informed consent form(s) and consent form updates that the investigator proposes for use in the trial, subject recruitment procedures (eg. Advertisements), written information to be provided to subjects, Investigator�s Brochure (IB), available safety information, information about payments and compensation available to subjects, the investigator�s current curriculum vitae and/or other documentation evidencing qualifications, and any other documents that the IRB/IEC may need to fulfill its responsibilities.
The IRB/IEC should review a proposed clinical trial within a reasonable time and document its views in writing, clearly identifying the trial, the documents reviewed and the dates for the following: - approval/favourable opinion - modifications required prior to it�s approval/favourable opinion - disapproval/negative opinion; and termination/suspension of any prior
approval/favourable opinion. 3.1.3 The IRB/IEC should consider the qualifications of the investigator for the proposed trial, as
documented by a current curriculum vitae and/or by any other relevant documentation the IR/IEC requests.
3.1.4 The IRB/IEC should conduct continuing review of each ongoing trial at intervals appropriate to
the degree of risk to human subjects, but at least once per year. 3.1.5 The IRB/IEC may request more information than is outlined in 4.8.10 be given to subjects when,
in the judgment of the IRB/IEC, the additional information would add meaningfully to the protection of the rights, safety and/or well being of the subjects.
3.1.6 When a non-therapeutic trial is to be carried out with the consent of the subject�s legally
acceptable representative (see 4.8.12, 4.8.14), the IRB/IEC should determine that the proposed protocol and /or other document(s) adequately address relevant ethical concerns and meets applicable regulatory requirements for such trials.
3.1.7 Where the protocol indicates that prior consent of the trial subject or the subject�s legally
acceptable representative is not possible, the IRB/IEC should determine that the proposed protocol and/or other document(s) adequately address relevant ethical concerns and meets applicable regulatory requirements for such trials (ie in emergency situations).
3.1.8 The IRB/IEC should review both the amount and method of payment to subjects to assure that
neither presents problems of coercion or undue influence on the trial subject should be prorated and not wholly contingent on completion of the trial by the subject.
3.1.9 The IRB/IEC should ensure that information regarding payment to subjects, including the
methods, amounts, and schedule of payment to trial subjects, is set forth in the written informed consent form and any other written information to be provided to subjects. The way payment will be prorated be specified.

16
3.2 Composition, Functions and Operations 3.2.1 The IRB/IEC should consist of a reasonable number of members, who collectively have the
qualifications and experience to review and evaluate the science, medical aspects, and ethics of the proposed trial. It is recommended that the IRB/IEC should include:
(a) at least five members (b) at least one member whose primary are of interest is in a non-scientific area (c) at least one member who is independent of the institution/trial site
Only those IRB/IEC members who are independent of the investigator and the sponsor of the trial should vote/provide opinion on a trial-related matter.
A list of IRB/IEC members and their qualifications should be maintained. 3.2.2 The IRB/IEC should perform it functions according to written operating procedures, should
maintain written records of its activities and minutes of its meetings, and should comply with GCP and with the applicable regulatory requirement(s)
3.2.3 An IRB/IEC should make its decisions at announced meetings at which at least a quorum, as
stipulated in its written operating procedures, is present. 3.2.4 Only members who participate in the IRB/IEC review and discussion should vote/provide their
opinion and/or advise. 3.2.5 The investigator may provide information on any aspect of the trial, but should not participate in
the deliberations of the IRB/IEC or in the vote/opinion of the IRB/IEC 3.2.6 An IRB/IEC may invite non-members with expertise in special areas for assistance.

17
3.3 Procedures The IRB/IEC should establish, document in writing and follow it procedures, which should include: 3.3.1 Determining its composition (names and qualifications of the members) and the authority under
which it is established. 3.3.2 Scheduling, notifying its members of, and conducting its meetings. 3.3.3 Conducting initial and continuing review of trials. 3.3.4 Determining the frequency of continuing review, as appropriate. 3.3.5 Providing, according to the applicable regulatory requirements, expedited review and
approval/favorable opinion of minor change(s) in ongoing trials that have the approval/favorable opinion of the IRB/IEC.
3.3.6 Specifying that no subject should be admitted to a trial before the IRB/IEC issues its written
approval/favorable opinion of the trial. 3.3.7 Specifying that no deviations form, or changes of, the protocol should be initiated without prior
written IRB/IEC approval/favorable opinion of an appropriate amendment, except when necessary to eliminate immediate hazards to the subjects or when the change(s) involves only logistical or administrative aspects of the trial (eg. Change of monitor(s), telephone number(s).
3.3.8 Specifying that the investigator should promptly report to the IRB/IEC:
(a) Deviations from, or changes of, the protocol to eliminate immediate hazards to the trial subjects.
(b) Changes increasing the risk to subjects and/or affecting significantly the conduct of the trial (c) All adverse drug reactions (ADRs) that are both serious and unexpected. (d) New information that may affect adversely the safety of the subjects or the conduct of the trial
3.3.9 Ensuring that the IRB/IEC promptly notify in writing the investigator/institution concerning:
(a) its trial-related decisions/opinions (b) the reasons for its decisions/opinions (c) procedures for appeal of its decisions/opinions
3.4 Records The IRB/IEC should retain all relevant records (e.g. written procedures, membership lists, lists of occupations/affiliations of members, submitted documents, minutes of meetings, and correspondence) for a period of at least three years after completion of the trial and make them available upon request from the regulatory authority (ies). The IRB/IEC may be asked by investigators, sponsors, or regulatory authorities to provide its written procedures and membership lists.

18

19
Appendix D Clinical Trials Act 1987 and 1990 (1) Where the Minister is satisfied that a proposed ethics committee for a clinical trial is competent to
consider the justification for conducting the proposed clinical trial and the circumstances under which it is to be conducted, he shall give his approval of the proposed committee, which shall thereupon become the ethics committee for the proposed clinical trial and, where the ethics committee gives its approval in accordance with subsection (2), for the clinical trial.
(2) The ethics committee for a proposed clinical trial shall consider the justification for conducting the
proposed trial and the circumstances under which it is proposed to be conducted and, where the committee considers the proposed trial is justified an it is satisfied with those circumstances, it shall give its approval to the conducted and, where the committee considers he proposed trial is justified and it is satisfied with those circumstances, it shall give its approval to the conducting of the proposed trial and the person who is arranging for the conducting of the proposed trial shall communicate such approval to the minister in writing.
(3) For the purposes of subsection (2), the ethics committee shall not consider the proposed clinical
trial justified unless it is satisfied that the risks to be incurred by participants would be consumerate with the objectives of the trial.
(4) Without prejudice to the generality of subsection (2), the ethic committee for a proposed clinical
trial shall, in considering the circumstances under which the proposed trial is to be conducted, have regard to the following matters:
(a) the objectives of the proposed trial and its planning and organizational structure (b) the qualification and competence of each person who could conduct the clinical trial and,
where appropriate, the resources available to him; (c) the criteria to be used for the recruitment and the selection of participants (d) the procedures proposed for compliance with section 9 (e) the extent and nature of the medical examination that persons selected as participants are
to undergo before participating in the clinical trial; (f) the extent to which the health of participants is proposed to be monitored during and after
the clinical trial (g) whether or not the persons selected as participants are to undergo independent medical
examinations before, during or after the clinical trial (h) details of the proposed method or methods by which participants are to be reduced (i) details of the proposed inducements or rewards, whether monetary or otherwise, to be
made for becoming or being a participants; (j) any payments, whether monetary or otherwise, to be made to a person conducting the
clinical trial or any part of the trial (k) the criteria to be used to ensure that the identity of each participant remains confidential (l) any payments, whether monetary or otherwise, to be made to any person for facilities
used for the purposes of the clinical trial; (m) such other matters as may be prescribed by regulations made by the Minister under this
Act (5) The composition of the ethics committee may at any time be changed with the approval of the
Minister. (6) (1) A person shall not be a participant in a clinical trial unless a consent to such participation
has been given in accordance with this section
(2) Subject to subsection (70, a consent to participate in a clinical trial shall not be valid unless given in writing and signed by the person who is to be the participant in the trial.

20
(3) Any consent given for the purposes of this section will not be valid unless -
(a) the person so consenting is capable of comprehending the nature, significance and scope of his consent, and
(b) it is obtained by or on behalf of the person conducting the clinical trial (4) The person conducting the clinical trial shall ensure that every person shall, before giving his
consent in accordance with this section, be made aware of the following matters-
(a) the objectives of the trial (b) the manner in which the substance or preparation is to be administered (c) the risk of any discomfort involved in, and the possible side-effects of the trial. (d) Whether or not a pharmacologically inactive substance or preparation is to be
administered to some persons in respect of each of whom a consent has been given to being a participant in the trial in accordance with this section.
(e) Such other matters (if any) as may be �
(i) prescribed by regulations made by the Minister under this Act, or (ii) specified in the permission granted by the Minister under section 4.
(5) Unless otherwise provided for by virtue of the permission granted by the minister under section 4
or of any amendment to that permission agreed to by him under section 5, a clinical trial shall not be conducted on any person within the period of 6 days after the day on which the provisions of section 9 (4) have been compiled with in respect of such person.
(6) Any person who has given his consent in accordance with this section may withdraw it at any time
and no contractual liability shall be incurred by such person from such withdrawal. (7) Where it is proposed to conduct a clinical trial on any person suffering from an illness, the remedy
for or alleviation of which constitutes an objective of the trial, the following provisions shall apply:
(a) where such a person is capable of comprehending the nature, significance and scope of a
consent to be given for the purposes of this section but is physically unable to give such consent in accordance with subsection 920, his consent clearly given in any other manner shall be sufficient where it is so given, in the presences of two witnesses present at the same time, to a registered medical practitioner who is treating him for that illness and where the consent is expressed in writing and is attested by the signature of both witnesses.
(b) Where such a person is incapable of comprehending the nature, significance and scope of a consent to be given for the purposes of this section, that person may be a participant in a clinical trial only if a written and signed consent is given for such a participation by a person or persons, independent of the person who applied to undertake or is conducting the trial, who I the opinion of the ethics committee is or are competent to give a decision on such a participation.
Provided that a clinical trial to which this subsection relates may be conducted only if the substance or preparation under trial is to be administered for the purpose of saving the life of such a person, restoring his health, alleviating his condition or relieving his suffering.
(8) No person shall offer or cause to be offered to a person for becoming or being a participant in a
clinical trial any inducement or reward (whether monetary or otherwise) unless provided for by the permission to undertake the clinical trial.

21
(10) 1. A person shall not -
(a) conduct a clinical trial or, (b) administer any substance or preparation for the purpose of a clinical trial, unless such
person establishes to the satisfaction of the ethics committee that he can provide sufficient security to ensure that adequate funds are available to provide appropriate compensation for each participant who may suffer injury or loss as a result of the trial.
2. It shall be a good defence for a person prosecuted for contravening subsection (1) to show that he
reasonably believed that the provisions of that subsection had been complied with.







![Operating Procedures 1 G2 - OPERATING PROCEDURES [6 Exam Questions - 6 Groups] G2APhone operating procedures; USB/LSB utilization conventions; procedural](https://img.dokumen.tips/doc/110x75/56649e4d5503460f94b4351a/operating-procedures-1-g2-operating-procedures-6-exam-questions-6-groups.jpg)




















