Embed Size (px)
Citation preview

IMMUNOBIOLOGY
Regulation of Fas-mediated immune homeostasis by an activation-inducedprotein, CyclonShella Saint Fleur,1 Akemi Hoshino,1 Kimie Kondo,1 Takeshi Egawa,2 and Hodaka Fujii1,3
1Department of Pathology and 2Molecular Pathogenesis Program, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, NewYork, NY; and 3Combined Program on Microbiology and Immunology, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan
Activation-induced cell death (AICD) playsan essential role in the contraction ofactivated T cells after eradication of patho-gen. Fas (APO-1/CD95) is one of the keycell surface proteins that mediate AICD inCD4� and CD8� T cells. Despite its primeimportance in cell death, regulation ofFas expression in T cells is poorly under-stood. Here we show that Cyclon, a newly
identified cytokine-inducible protein, isinduced in T cells on T-cell receptor liga-tion and important for immune homeosta-sis. Transgenic expression of Cyclon ame-liorated autoimmune phenotype in micelacking subunits of IL-2R. Transgenic ex-pression of Cyclon markedly enhancedAICD through increased expression ofFas whose expression is essential for
Cyclon action. Finally, we demonstratedthat activated but not resting CD4� T cellswith targeted deletion of a Cyclon alleleshow reduced AICD and expression ofFas, indicating a critical role of Cyclon inFas expression in activated T cells. Wethink that our data provide insight intoexpression regulation of Fas in T cells.(Blood. 2009;114:1355-1365)
Introduction
Activation of acquired immunity induces rapid expansion ofantigen-specific CD4� and CD8� T cells to eradicate the patho-gens. After eradication of the pathogens, however, most activatedT cells are removed.1,2 The elimination of activated T cells islargely mediated by apoptotic cell death.2 Activated T-cell autono-mous death and activation-induced cell death (AICD) are the2 mechanisms by which programmed death of activated T cellsoccurs.3-7 Activated T-cell autonomous death is mediated by theintrinsic death pathway,8-10 which is regulated by the B-celllymphoma 2 (Bcl-2) family proteins. In contrast, the other mecha-nism of activated T-cell death, AICD, requires antigen restimula-tion of activated effector T cells and is triggered by the extrinsicdeath pathways regulated by signals of death receptors (DRs) of thetumor necrosis factor receptor (TNFR) family.1,11-13 Members ofthe DR subgroup include type I TNFR (TNFRI)/DR1, Fas/CD95/Apo1/DR2, DR3, TNF-related apoptosis-inducing ligand receptor1 (TRAIL-R1)/DR4, TRAIL-R2/DR5/Apo2, and DR6. These recep-tors contain extracellular cysteine-rich domains and an intracellulardeath domain, which is necessary for transducing death signals.14-16
Binding of ligands to DRs triggers trimerization of the receptors,which is required for the recruitment of the adaptor proteinFas-associated death domain (FADD),16 and subsequently pro-caspase 8, also known as FLICE, or procaspase 10. The ensembleconstitutes what is known as the death-inducing signaling complex,leading to auto-processing of initiator procaspases into activecaspases, which activate downstream effector caspases.14,17,18 Otherinhibitors of the pathway include the FLICE inhibitory protein(FLIP), which competes with procaspase 8 (FLICE) for binding tothe DED of FADD.19-23
Fas ligand (FasL) and TNF are the key death ligands thatmediate AICD in mature T cells.1,7,24-29 Importance of the Fas/FasLsystem in regulation of immune homeostasis has been established
by genetic studies showing that lpr mice, which have a loss-of-function mutation in the Fas gene,30-32 and gld mice, which have aloss-of-function mutation in the Fasl gene,33-35 develop severelymphadenopathy caused by defects in Fas-mediated cell death.36,37
In humans, defects in Fas and FasL cause similar abnormalities inthe autoimmune lymphoproliferative syndrome type Ia and Ib,respectively.38-40
Expression levels of Fas or FasL markedly impact T-cell death.Regulation of FasL expression in primary T cells has been exten-sively examined.41-43 In contrast, surprisingly little is known aboutregulation of Fas expression in primary T cells. T-cell death-associated gene 51 (TDAG51) was shown to be required for T-cellreceptor (TCR)–mediated expression of Fas in a T-cell hybrid-oma.44 However, it was later shown that TDAG51 is not essentialfor Fas regulation and apoptosis of primary mouse T cells in vivo.45
NF-�B was also implicated in Fas expression in a human T-cellline, Jurkat.46 However, TCR-mediated Fas up-regulation andAICD were normal in c-Rel/p50 doubly deficient primary T cells,47
suggesting that NF-�B is not essential for TCR-mediated Fasup-regulation in primary T cells.
Cytokines also play a role in the regulation of Fas-mediatedactivated T-cell death. Sensitivity to AICD is chiefly the result ofthe effect of interleukin-2 (IL-2) in inducing cell-cycle progressioninto late G1 to S phases, which confers susceptibility to death.1,7 Ithas been reported that the IL-2/IL-2 receptor (IL-2R) system isessential for AICD. IL-2R consists of IL-2R�, IL-2R�, and thecommon cytokine-receptor �-chain (�c).48-50 Mice deficient inIL-2, IL-2R�, or IL-2R� gene develop massive enlargement ofperipheral lymphoid organs and multiple autoimmune diseases,including hemolytic anemia and inflammatory bowel disease.51-54 Ithas also been reported that peripheral T cells in �c-deficient (�c�)mice are spontaneously activated.55 Defective AICD in mice lacking
Submitted November 11, 2008; accepted June 6, 2009. Prepublished online asBlood First Edition paper, June 15, 2009; DOI 10.1182/blood-2008-11-189118.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 USC section 1734.
© 2009 by The American Society of Hematology
1355BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

IL-2 signaling is at least partially responsible for the autoimmunephenotype of these animals.56 IL-2 was shown to increase transcrip-tion and surface expression of Fas and FasL and suppress transcrip-tion and expression of FLIP. However, it remains unknown howIL-2 receptor signals induce Fas up-regulation.
We have recently identified a novel nuclear protein, Cyclon,encoded by Ccdc86, which is induced on cytokine stimulation inthe Ba/F3 cell line.57 Cyclon is a phosphorylated nuclear proteinconsisting of repetitive sequences in the amino-terminus and acoiled-coil domain in the carboxyl-terminus.57 Here, we examinedin vivo function of Cyclon in T cells using Cyclon transgenic miceand Cyclon-deficient mice. In T cells, expression of Cyclon wasalso induced by TCR ligation. T cell–specific transgenic expressionof Cyclon normalized the number of activated T cells, suppressedenlargement of peripheral lymphoid organs, and decreased serumIL-17 levels in IL-2R�-deficient (IL-2R��) mice. Transgenicexpression of Cyclon also normalized splenomegaly in the �c�
background. Attenuation of these autoimmune phenotypes byCyclon overexpression was correlated with increased Fas expres-sion and enhanced AICD. Finally, AICD and Fas expression inactivated but not in resting CD4� T cells was substantially reducedin mice with targeted deletion of a Cyclon allele. These datacollectively indicate a critical role of Cyclon in Fas expression andAICD in activated T cells. We think our data provide a clue tounderstanding of Fas expression regulation in primary T cells.
Methods
Mice
Generation of Cyclon-transgenic and Cyclon-deficient mice is described insupplemental data (available on the Blood website; see the SupplementalMaterials link at the top of the online article). IL-2R��, �c�, and Fas� lprmice were purchased from The Jackson Laboratory. All animal experimentswere approved by the Institutional Animal Care and Use Committee at NewYork University School of Medicine.
Isolation of cells and cell culture
CD4� and CD8� T cells were isolated from splenocytes by magneticsorting (Miltenyi Biotec). Cells were cultured in RPMI complete medium.
Reverse-transcription polymerase chain reaction
Reverse-transcription polymerase chain reaction (RT-PCR) was performedwith BD TITANIUM One-Step RT-PCR kit (Clontech) as describedbefore.57 Real-time RT-PCR was performed with iScript One-Step RT-PCRkit with SYBR Green (Bio-Rad). Primers for real-time RT-PCR weredescribed previously.57,58
Immunoblot analysis
Preparation of nuclear extracts and whole cell lysates and immunoblotanalysis were performed as described before.59,60 Scanned images werequantified with ImageJ software.
Cell staining and flow cytometry
Cells were stained for 30 minutes at 4°C with fluorochrome-conjugatedantibodies (Abs). Abs used for surface staining were fluorescein isothiocya-nate (FITC)–, phycoerythrin (PE)�, or PE-Cy7–conjugated CD4 (cloneGK1.5), FITC- or Cychrome-conjugated CD8� (clone 53-6.7), PE-conjugated CD44 (clone IM7), PE-conjugated FasL (clone MFL3), andPE-conjugated Fas (clone Jo2; BD Biosciences). For T-reg staining, splenocytes were stained with FITC-labeled anti-CD4 inflow cytometry buffer, followed by staining with PE-conjugated anti-Foxp3
Ab in the Foxp3 staining kit (eBioscience) according to the manufacturer’sprotocol. Flow cytometric analysis was performed on FACSCalibur (BDBiosciences) and analyzed with FlowJo software (TreeStar).
IL-17 measurement
IL-17 in sera was analyzed by enzyme-linked immunosorbent assay withmouse IL-17 Quantikine assay kits (R&D Systems) according to themanufacturer’s protocol.
AICD assay
We followed an authentic AICD protocol.61 Briefly, CD4� and CD8�
T cells were purified from spleens by magnetic sorting and activated withanti-CD3 Ab (2 �g/mL) � anti-CD28 Ab (2 �g/mL) for 2 or 3 days. Livecells were purified by density centrifugation with Lymphocyte-M (Cedar-lane) and restimulated with different concentrations of anti-CD3 Ab in thepresence of IL-2 (5 ng/mL). Cells were incubated for 1 to 3 days, andviability was examined by propidium iodide (PI) staining and flowcytometry.
For analysis of Fas-mediated cell death, CD4� and CD8� T cells werepurified from spleens by magnetic sorting and activated with anti-CD3 Ab(2 �g/mL) � anti-CD28 Ab (2 �g/mL) for 2 or 3 days. Live cells werepurified by density centrifugation with Lymphocyte-M (Cederlane) andrecultured for 1 day with the indicated concentrations of superFasL (Alexis)in RPMI complete media in the presence of IL-2 (5 ng/mL).
Statistical analysis
Statistical analysis was performed with the Prism software (GraphPad)using t test or 1-way analysis of variance.
Results
Cyclon mRNA expression is induced in both CD4� and CD8�
T cells on activation
We found that Cyclon mRNA is induced in splenocytes byanti-CD3 stimulation (data not shown). To determine whichsubpopulations of T cells express Cyclon, purified splenic CD4�
and CD8� T cells were stimulated with anti-CD3 and anti-CD28Abs, and expression of Cyclon mRNA was examined (Figure 1A).TCR stimulation-dependent up-regulation of Cyclon was moreevident in CD4� T cells. Real-time PCR analysis showed thatCyclon mRNA was up-regulated by 30-fold 8 hours after stimula-tion in CD4� T cells, whereas Cyclon mRNA was up-regulated by13-fold in CD8� T cells (Figure 1B). Immunoblot analysis withanti-Cyclon Ab57 revealed that Cyclon protein expression isinduced more robustly in CD4� than in CD8� splenic T cells byanti-CD3 and anti-CD28 stimulation (Figure 1C). These resultssuggest a potential role of Cyclon in the regulation of function ofactivated T cells.
To analyze function of Cyclon in T cells in vivo, we generatedtransgenic mice overexpressing Cyclon in the T-cell lineages(Cyclon-Tg mice) under the control of the VA-hCD2 cassette(Figure 1D),62 which drives transgene expression in virtually allthymocytes as well as peripheral T cells.62,63 mRNA expression ofthe Cyclon transgene and its protein expression in splenocytes wereconfirmed by RT-PCR (Figure 1E) and by immunoblot analysisusing anti-HA Ab (Figure 1F). In addition, immunoblot analysiswith anti-Cyclon Ab revealed that expression levels of total Cyclon(endogenous and transgenic) were 1.70- and 1.49-fold higher inCD4� and CD8� T cells, respectively, from Cyclon-Tg mice thanin those cells from WT mice (Figure 1G). These mice werebackcrossed with C57BL/6 mice at least 6 times for further
1356 SAINT FLEUR et al BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom
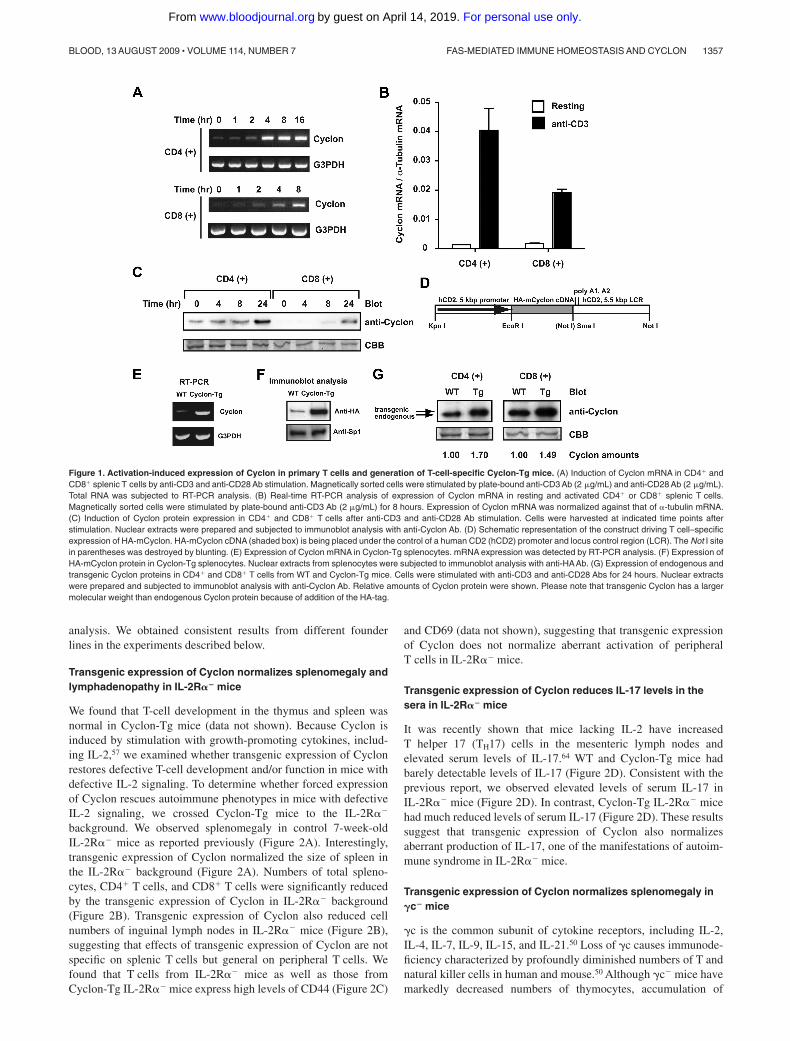
analysis. We obtained consistent results from different founderlines in the experiments described below.
Transgenic expression of Cyclon normalizes splenomegaly andlymphadenopathy in IL-2R�� mice
We found that T-cell development in the thymus and spleen wasnormal in Cyclon-Tg mice (data not shown). Because Cyclon isinduced by stimulation with growth-promoting cytokines, includ-ing IL-2,57 we examined whether transgenic expression of Cyclonrestores defective T-cell development and/or function in mice withdefective IL-2 signaling. To determine whether forced expressionof Cyclon rescues autoimmune phenotypes in mice with defectiveIL-2 signaling, we crossed Cyclon-Tg mice to the IL-2R��
background. We observed splenomegaly in control 7-week-oldIL-2R�� mice as reported previously (Figure 2A). Interestingly,transgenic expression of Cyclon normalized the size of spleen inthe IL-2R�� background (Figure 2A). Numbers of total spleno-cytes, CD4� T cells, and CD8� T cells were significantly reducedby the transgenic expression of Cyclon in IL-2R�� background(Figure 2B). Transgenic expression of Cyclon also reduced cellnumbers of inguinal lymph nodes in IL-2R�� mice (Figure 2B),suggesting that effects of transgenic expression of Cyclon are notspecific on splenic T cells but general on peripheral T cells. Wefound that T cells from IL-2R�� mice as well as those fromCyclon-Tg IL-2R�� mice express high levels of CD44 (Figure 2C)
and CD69 (data not shown), suggesting that transgenic expressionof Cyclon does not normalize aberrant activation of peripheralT cells in IL-2R�� mice.
Transgenic expression of Cyclon reduces IL-17 levels in thesera in IL-2R�� mice
It was recently shown that mice lacking IL-2 have increasedT helper 17 (TH17) cells in the mesenteric lymph nodes andelevated serum levels of IL-17.64 WT and Cyclon-Tg mice hadbarely detectable levels of IL-17 (Figure 2D). Consistent with theprevious report, we observed elevated levels of serum IL-17 inIL-2R�� mice (Figure 2D). In contrast, Cyclon-Tg IL-2R�� micehad much reduced levels of serum IL-17 (Figure 2D). These resultssuggest that transgenic expression of Cyclon also normalizesaberrant production of IL-17, one of the manifestations of autoim-mune syndrome in IL-2R�� mice.
Transgenic expression of Cyclon normalizes splenomegaly in�c� mice
�c is the common subunit of cytokine receptors, including IL-2,IL-4, IL-7, IL-9, IL-15, and IL-21.50 Loss of �c causes immunode-ficiency characterized by profoundly diminished numbers of T andnatural killer cells in human and mouse.50 Although �c� mice havemarkedly decreased numbers of thymocytes, accumulation of
Figure 1. Activation-induced expression of Cyclon in primary T cells and generation of T-cell-specific Cyclon-Tg mice. (A) Induction of Cyclon mRNA in CD4� andCD8� splenic T cells by anti-CD3 and anti-CD28 Ab stimulation. Magnetically sorted cells were stimulated by plate-bound anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL).Total RNA was subjected to RT-PCR analysis. (B) Real-time RT-PCR analysis of expression of Cyclon mRNA in resting and activated CD4� or CD8� splenic T cells.Magnetically sorted cells were stimulated by plate-bound anti-CD3 Ab (2 �g/mL) for 8 hours. Expression of Cyclon mRNA was normalized against that of �-tubulin mRNA.(C) Induction of Cyclon protein expression in CD4� and CD8� T cells after anti-CD3 and anti-CD28 Ab stimulation. Cells were harvested at indicated time points afterstimulation. Nuclear extracts were prepared and subjected to immunoblot analysis with anti-Cyclon Ab. (D) Schematic representation of the construct driving T cell–specificexpression of HA-mCyclon. HA-mCyclon cDNA (shaded box) is being placed under the control of a human CD2 (hCD2) promoter and locus control region (LCR). The Not I sitein parentheses was destroyed by blunting. (E) Expression of Cyclon mRNA in Cyclon-Tg splenocytes. mRNA expression was detected by RT-PCR analysis. (F) Expression ofHA-mCyclon protein in Cyclon-Tg splenocytes. Nuclear extracts from splenocytes were subjected to immunoblot analysis with anti-HAAb. (G) Expression of endogenous andtransgenic Cyclon proteins in CD4� and CD8� T cells from WT and Cyclon-Tg mice. Cells were stimulated with anti-CD3 and anti-CD28 Abs for 24 hours. Nuclear extractswere prepared and subjected to immunoblot analysis with anti-Cyclon Ab. Relative amounts of Cyclon protein were shown. Please note that transgenic Cyclon has a largermolecular weight than endogenous Cyclon protein because of addition of the HA-tag.
FAS-MEDIATED IMMUNE HOMEOSTASIS AND CYCLON 1357BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

activated T cells in the periphery occurs as mice age.55 Accumula-tion of activated T cells can be caused at least partly by the lack ofFoxp3�CD4� regulatory T cells (T-regs) in �c� mice65 and bydefects in AICD in �c� T cells.56 When we examined thymocytedevelopment of �c� mice and Cyclon-Tg �c� mice, thymocytedevelopment of �c� mice was not restored by transgenic expres-sion of Cyclon (data not shown). As shown in Figure 3A,8-week-old �c� mice had enlarged spleens compared with �c
�and
Cyclon-Tg �c� mice. As was observed in the Cyclon-Tg IL-2R��
mice, Cyclon-Tg �c� mice had markedly smaller spleens thanlittermate �c� mice. Numbers of total splenocytes, CD4� T cells,and CD8� T cells were significantly reduced by the transgenicexpression of Cyclon in �c� background (Figure 3B). PeripheralT cells in �c� mice and Cyclon-Tg �c� mice had activatedphenotype shown by high expression levels of CD44 (Figure 3C)and CD69 (data not shown), suggesting that transgenic expres-sion of Cyclon does not normalize aberrant activation ofperipheral T cells in �c� mice. The size of T cells from �c� micewas much larger than that of T cells from �c� mice (larger
forward scatter values in flow cytometry, data not shown)because of their activated phenotype. This resulted in theincrease in the size of spleens from �c� mice compared with �c�
mice (Figure 3A), although splenocyte numbers of �c� micewere slightly lower than those of �c� mice (Figure 3B). Theseresults collectively suggest that transgenic expression of Cyclonnormalizes autoimmune-associated splenomegaly/lymphad-enopathy and other autoimmune symptoms in mice lackingIL-2 signaling without normalizing activated phenotype of per-ipheral T cells.
Transgenic expression of Cyclon does not restoredevelopment of T-regs
Autoimmunity in mice lacking IL-2 signaling can be caused at leastpartly by reduced numbers of T-regs in these mice65,66 and bydefects in AICD. IL-2 signaling has been shown to be important fordevelopment and maintenance of Foxp3-expressing T-regs. Recent
Figure 2. Normalization of autoimmune phenotype of IL-2R�� mice by transgenic expression of Cyclon. (A) Spleens from 7-week-old IL-2R��, Cyclon-Tg IL-2R��,IL-2R��, and Cyclon-Tg IL-2R�� mice. (B) Cell numbers of splenocytes, CD4�, and CD8� splenic T cells, and cells in inguinal lymph nodes from IL-2R�
�, Cyclon-Tg IL-2R��,IL-2R��, and Cyclon-Tg IL-2R�� mice. (C) Expression of CD44 on CD4� and CD8� splenic T cells from IL-2R��, Cyclon-Tg IL-2R��, IL-2R��, and Cyclon-Tg IL-2R�� mice.(D) Transgenic expression of Cyclon decreases IL-17 in serum of IL-2R�� mice. Serum concentrations of IL-17 were analyzed in 7-week-old IL-2R��, Cyclon Tg IL-2R��,IL-2R��, and Cyclon TgIL-2R�� mice by enzyme-linked immunosorbent assay.
1358 SAINT FLEUR et al BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

studies showed that IL-2R� and IL-2R� are necessary for expan-sion of T-regs,65,66 whereas �c is essential for generation of T-regsin the thymus.65 To examine whether transgenic expression ofCyclon normalizes autoimmunity-associated splenomegaly throughrestoration of T-reg development in IL-2R�� mice and �c� mice,we performed intracellular staining of Foxp3 in splenic CD4�
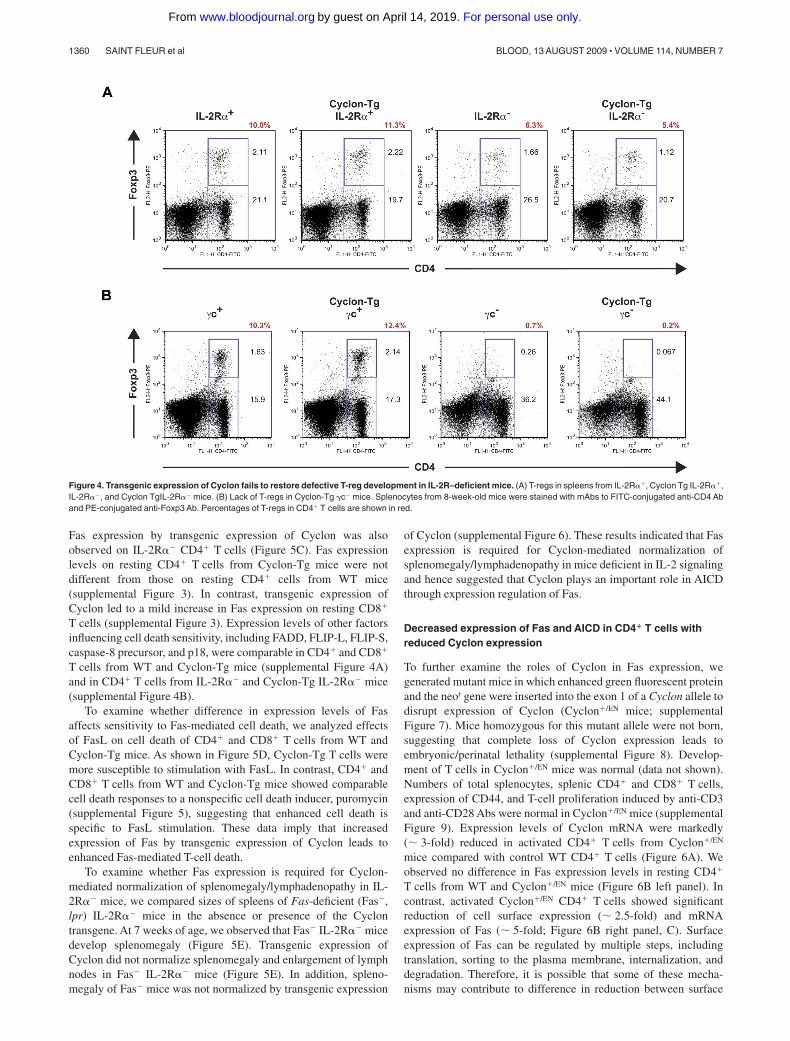
T cells. As shown in Figure 4A, IL-2R�� mice had reducedpercentages of T-regs. Transgenic expression of Cyclon failed tonormalize numbers of T-regs in IL-2R�� mice (Figure 4A). �c�
and Cyclon-Tg �c� splenocytes contained sizable CD4�Foxp3�
T-reg populations (Figure 4B). In contrast, CD4�Foxp3� cells wereobserved in neither �c� mice nor Cyclon-Tg �c� mice (Figure 4B),indicating that transgenic expression of Cyclon is unable to restoreT-reg development in the absence of �c expression. These resultsindicate that Cyclon-mediated normalization of splenomegaly andlymphadenopathy in mice with defective IL-2 signaling is indepen-dent of function of T-regs.
Transgenic expression of Cyclon enhances AICD
Decreased T-cell numbers in peripheral lymphoid organs bytransgenic expression of Cyclon in IL-2R-deficient mice could becaused by either reduced cell proliferation or increased cell death,or both. In this regard, proliferation of CD4� and CD8� T cells wasindistinguishable between WT and Cyclon-Tg (supplemental Fig-ure 1). In addition, no difference was observed in proliferation of
T cells in IL-2R-deficient background (data not shown), suggestingthat Cyclon does not affect cell proliferation. AICD is an importantmechanism to limit lymphocyte expansion after eradication ofpathogens. Both IL-2R�� and �c� mice have defects in AICD.56 Todetermine whether transgenic expression of Cyclon affects AICD,we purified splenic CD4� or CD8� T cells from WT or Cyclon-Tgmice and examined cell death after secondary stimulation in thepresence of IL-2. As shown in Figure 5A, Cyclon-Tg CD4� orCD8� T cells showed markedly increased AICD activity comparedwith WT CD4� or CD8� T cells. These data suggest that enhancedAICD by transgenic expression of Cyclon may be one of themechanisms by which transgenic expression of Cyclon normalizesautoimmune phenotypes in mice deficient in IL-2 signaling.
Cyclon positively regulates Fas expression, which is essentialfor reduction of splenomegaly by transgenic expression ofCyclon
It has been shown that the Fas/FasL system is critical forIL-2–dependent AICD.67,68 We examined whether enhancement ofAICD by transgenic expression of Cyclon is the result of alteredexpression levels of Fas or FasL. We detected no changes inexpression levels of FasL between WT and Cyclon-Tg T cells(supplemental Figure 2). In contrast, transgenic expression ofCyclon significantly enhanced expression levels of Fas both inactivated CD4� and CD8� T cells (Figure 5B). Enhancement of
Figure 3. Normalization of splenomegaly of �c� mice by transgenic expression of Cyclon. (A) Spleens from 8-week-old �c�, Cyclon-Tg �c�, �c�, and Cyclon-Tg �c�
mice. (B) Cell numbers of splenocytes, CD4� splenic T cells, and CD8� splenic T cells from �c�, Cyclon-Tg �c�, �c�, and Cyclon-Tg �c� mice. (C) Expression of CD44 onCD4� and CD8� splenic T cells from �c�, Cyclon-Tg �c�, �c�, and Cyclon-Tg �c� mice.
FAS-MEDIATED IMMUNE HOMEOSTASIS AND CYCLON 1359BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom
Fas expression by transgenic expression of Cyclon was alsoobserved on IL-2R�� CD4� T cells (Figure 5C). Fas expressionlevels on resting CD4� T cells from Cyclon-Tg mice were notdifferent from those on resting CD4� cells from WT mice(supplemental Figure 3). In contrast, transgenic expression ofCyclon led to a mild increase in Fas expression on resting CD8�
T cells (supplemental Figure 3). Expression levels of other factorsinfluencing cell death sensitivity, including FADD, FLIP-L, FLIP-S,caspase-8 precursor, and p18, were comparable in CD4� and CD8�
T cells from WT and Cyclon-Tg mice (supplemental Figure 4A)and in CD4� T cells from IL-2R�� and Cyclon-Tg IL-2R�� mice(supplemental Figure 4B).
To examine whether difference in expression levels of Fasaffects sensitivity to Fas-mediated cell death, we analyzed effectsof FasL on cell death of CD4� and CD8� T cells from WT andCyclon-Tg mice. As shown in Figure 5D, Cyclon-Tg T cells weremore susceptible to stimulation with FasL. In contrast, CD4� andCD8� T cells from WT and Cyclon-Tg mice showed comparablecell death responses to a nonspecific cell death inducer, puromycin(supplemental Figure 5), suggesting that enhanced cell death isspecific to FasL stimulation. These data imply that increasedexpression of Fas by transgenic expression of Cyclon leads toenhanced Fas-mediated T-cell death.
To examine whether Fas expression is required for Cyclon-mediated normalization of splenomegaly/lymphadenopathy in IL-2R�� mice, we compared sizes of spleens of Fas-deficient (Fas�,lpr) IL-2R�� mice in the absence or presence of the Cyclontransgene. At 7 weeks of age, we observed that Fas� IL-2R�� micedevelop splenomegaly (Figure 5E). Transgenic expression ofCyclon did not normalize splenomegaly and enlargement of lymphnodes in Fas� IL-2R�� mice (Figure 5E). In addition, spleno-megaly of Fas� mice was not normalized by transgenic expression
of Cyclon (supplemental Figure 6). These results indicated that Fasexpression is required for Cyclon-mediated normalization ofsplenomegaly/lymphadenopathy in mice deficient in IL-2 signalingand hence suggested that Cyclon plays an important role in AICDthrough expression regulation of Fas.
Decreased expression of Fas and AICD in CD4� T cells withreduced Cyclon expression
To further examine the roles of Cyclon in Fas expression, wegenerated mutant mice in which enhanced green fluorescent proteinand the neor gene were inserted into the exon 1 of a Cyclon allele todisrupt expression of Cyclon (Cyclon�/EN mice; supplementalFigure 7). Mice homozygous for this mutant allele were not born,suggesting that complete loss of Cyclon expression leads toembryonic/perinatal lethality (supplemental Figure 8). Develop-ment of T cells in Cyclon�/EN mice was normal (data not shown).Numbers of total splenocytes, splenic CD4� and CD8� T cells,expression of CD44, and T-cell proliferation induced by anti-CD3and anti-CD28 Abs were normal in Cyclon�/EN mice (supplementalFigure 9). Expression levels of Cyclon mRNA were markedly(� 3-fold) reduced in activated CD4� T cells from Cyclon�/EN
mice compared with control WT CD4� T cells (Figure 6A). Weobserved no difference in Fas expression levels in resting CD4�
T cells from WT and Cyclon�/EN mice (Figure 6B left panel). Incontrast, activated Cyclon�/EN CD4� T cells showed significantreduction of cell surface expression (� 2.5-fold) and mRNAexpression of Fas (� 5-fold; Figure 6B right panel, C). Surfaceexpression of Fas can be regulated by multiple steps, includingtranslation, sorting to the plasma membrane, internalization, anddegradation. Therefore, it is possible that some of these mecha-nisms may contribute to difference in reduction between surface
Figure 4. Transgenic expression of Cyclon fails to restore defective T-reg development in IL-2R–deficient mice. (A) T-regs in spleens from IL-2R��, Cyclon Tg IL-2R��,IL-2R��, and Cyclon TgIL-2R�� mice. (B) Lack of T-regs in Cyclon-Tg �c� mice. Splenocytes from 8-week-old mice were stained with mAbs to FITC-conjugated anti-CD4 Aband PE-conjugated anti-Foxp3 Ab. Percentages of T-regs in CD4� T cells are shown in red.
1360 SAINT FLEUR et al BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

Figure 5. Cyclon positively regulates AICD through up-regulation of Fas expression. (A) Increased AICD of T cells by transgenic expression of Cyclon. CD4� or CD8�
T cells sorted magnetically were activated with anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for 2 days. Live cells were purified by density centrifugation andrestimulated with different concentrations of anti-CD3 in the presence of IL-2 (5 ng/mL). Cells were incubated for 2 days (left panel, CD4� T cells) or 3 days (right panel, CD8�
T cells), and viability was examined by PI staining and flow cytometry. Indicated are representative data using WT and Cyclon-Tg littermates from 4 independent experiments.(B) Transgenic expression of Cyclon enhances Fas expression. CD4� or CD8� T cells were activated with anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for 2 days. Livecells were purified by density centrifugation and restimulated with anti-CD3 Ab (1 �g/mL) in the presence of IL-2 (5 ng/mL). Cells were incubated for 16 hours and stained withPE-conjugated anti-Fas Ab. (C) Transgenic expression of Cyclon enhances Fas expression on IL-2R�� CD4� T cells. CD4� T cells sorted magnetically were activated withanti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) in the presence of IL-15 (100 ng/mL) for 2 days. Cells were stained with PE-conjugated anti-Fas Ab. (D) Increased celldeath by FasL stimulation of T cells by transgenic expression of Cyclon. CD4� or CD8� T cells were activated with anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for2 days. Live cells were purified by density centrifugation and restimulated with different concentrations of FasL in the presence of IL-2 (5 ng/mL). Cells were incubated for24 hours, and viability was examined by PI staining and flow cytometry. Indicated are representative data using WT and Cyclon-Tg littermates from 4 independent experiments.(E) Cyclon transgenic expression fails to suppress splenomegaly and lymphadenopathy in Fas� IL-2R�� mice. Spleens and numbers of splenocytes and inguinal lymph nodesfrom 7-week-old Fas� IL-2R��, Fas� IL-2R��, and Cyclon-Tg Fas� IL-2R�� mice are shown.
FAS-MEDIATED IMMUNE HOMEOSTASIS AND CYCLON 1361BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom
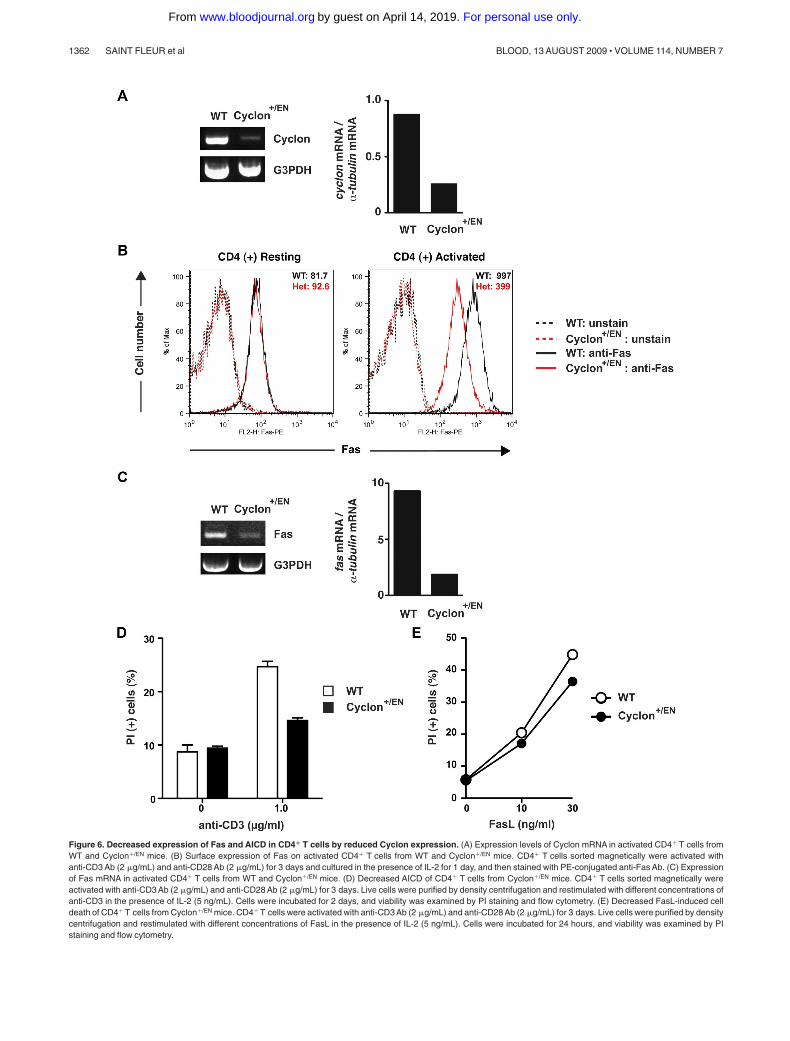
Figure 6. Decreased expression of Fas and AICD in CD4� T cells by reduced Cyclon expression. (A) Expression levels of Cyclon mRNA in activated CD4� T cells fromWT and Cyclon�/EN mice. (B) Surface expression of Fas on activated CD4� T cells from WT and Cyclon�/EN mice. CD4� T cells sorted magnetically were activated withanti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for 3 days and cultured in the presence of IL-2 for 1 day, and then stained with PE-conjugated anti-Fas Ab. (C) Expressionof Fas mRNA in activated CD4� T cells from WT and Cyclon�/EN mice. (D) Decreased AICD of CD4� T cells from Cyclon�/EN mice. CD4� T cells sorted magnetically wereactivated with anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for 3 days. Live cells were purified by density centrifugation and restimulated with different concentrations ofanti-CD3 in the presence of IL-2 (5 ng/mL). Cells were incubated for 2 days, and viability was examined by PI staining and flow cytometry. (E) Decreased FasL-induced celldeath of CD4� T cells from Cyclon�/EN mice. CD4� T cells were activated with anti-CD3 Ab (2 �g/mL) and anti-CD28 Ab (2 �g/mL) for 3 days. Live cells were purified by densitycentrifugation and restimulated with different concentrations of FasL in the presence of IL-2 (5 ng/mL). Cells were incubated for 24 hours, and viability was examined by PIstaining and flow cytometry.
1362 SAINT FLEUR et al BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

Fas protein and Fas mRNA. We did not detect change in Fasexpression levels in resting and activated CD8� T cells fromCyclon�/EN mice (supplemental Figure 10A). Because Cyclonexpression levels were similarly decreased in activated CD8�
T cells (supplemental Figure 10B), Fas expression may be differen-tially regulated in CD4� and CD8� T cells. Expression levels ofother factors influencing cell death sensitivity, including FADD,FLIP-L, FLIP-S, caspase-8 precursor, and p18, were comparable inCD4� T cells from WT and Cyclon�/EN mice (supplemental Figure11). Anti-CD3-mediated AICD of CD4� T cells from Cyclon�/EN
mice was markedly lower than that of T cells from WT mice(Figure 6D). Furthermore, CD4� T cells from Cyclon�/EN miceshowed significantly reduced cell death in response to differentconcentrations of FasL (Figure 6E). CD4� T cells from WT andCyclon-Tg mice had comparable cell death responses to a nonspe-cific cell death inducer, puromycin (supplemental Figure 5),indicating that enhanced cell death is specific to FasL stimulation.These data clearly indicate that Cyclon plays a critical role inup-regulation of Fas expression and AICD in CD4� T cells onactivation.
Discussion
In this study, we showed that T-cell activation induces expressionof Cyclon, a novel cytokine-inducible nuclear protein (Figure 1).Initially, Cyclon was identified as an immediately early geneinduced by growth-promoting cytokines, including IL-3 and IL-2.57
Because TCR-mediated expression of Cyclon mRNA is promptlyinduced, we think that TCR stimulation directly induces Cyclonexpression but not indirectly, such as by IL-2 produced by activatedT cells. In other words, there may be different mechanisms ofCyclon induction by different stimuli, such as growth-promotingcytokine (eg, IL-3, IL-2) and TCR stimulation. At this stage, we donot know the significance of IL-2� or IL-3�mediated induction ofCyclon in T cells and other cell lineages.
Next, we examined roles of Cyclon in the regulation of T-cellfunction in vivo. Transgenic expression of Cyclon specifically inthe T-cell lineages suppressed development of splenomegaly,lymphadenopathy, and aberrant production of IL-17 in micelacking IL-2 signaling (Figures 2,3). Because IL-17 has beenlinked to autoimmunity,69 reduction of IL-17 expression bytransgenic expression of Cyclon may also contribute to normal-ization of autoimmune phenotype of IL-2R�� mice. Autoimmu-nity in mice with defective IL-2 signaling is considered to becaused at least partly by the defects in T-regs65,66 and AICD.56
We showed that transgenic expression of Cyclon neither restoresdevelopment of T-reg in mice (Figure 4) nor corrects aberrantactivation of peripheral T cells in mice deficient in IL-2signaling (Figure 2C,3C). These results are consistent with theinterpretation that Cyclon-mediated normalization of autoim-mune symptoms in these mice is independent of function ofT-regs. In contrast, transgenic expression of Cyclon enhancedAICD both in CD4� and CD8� T cells (Figure 5A), whereasCD4� T cells from Cyclon�/EN mice showed reduced AICD(Figure 6D), indicating that enhancement of AICD is one of themechanisms of Cyclon action.
The Fas-dependent death pathway is well characterized, and itsimportance in immune homeostasis is firmly established.37 IL-2was shown to increase transcription and surface expression of FasLand suppress transcription and protein expression of FLIP, an
inhibitor of apoptosis. We showed that transgenic expression ofCyclon significantly enhances up-regulation of Fas on activatedCD4� and CD8� T cells in WT and IL-2R�� background (Figure5B-C). Furthermore, Fas up-regulation and AICD were attenuatedin activated CD4� T cells from Cyclon�/EN mice (Figure 6B-D). Incontrast, expression of other proteins involved in the Fas pathwaywas not affected (supplemental Figures 2,4,11). Furthermore,expression levels of Fas were directly correlated with sensitivity toFasL-mediated cell death (Figures 5D,6E). In addition, our geneticanalysis showed that Cyclon-mediated normalization of spleno-megaly and lymphadenopathy of IL-2R�� mice requires expres-sion of Fas (Figure 5D), collectively suggesting that Cyclon plays acritical role in AICD presumably through regulation of Fasexpression after TCR stimulation.
Apoptosis is regulated by the extrinsic stimuli, such as signalsthrough the FasL/Fas pathway as well as the intrinsic pathwayinvolving Bcl-2 family members.1,2 It was recently shown thatT-cell death after acute infection is mainly regulated by the intrinsicpathway in which Bim plays a critical role, whereas the Faspathway regulates T-cell death in chronic infection and autoimmu-nity.3-5 Consistent with this notion, there was no difference incontraction of T cells between WT and Cyclon-Tg mice afterinjection of staphylococcal enterotoxin B (data not shown), furtherconfirming that Cyclon regulates Fas expression but not expressionor activities of regulators of the intrinsic pathway.
Expression of Fas on CD4� and CD8� T cells was differentiallyaffected by an increase or decrease in expression levels of Cyclon.In CD4� T cells, the effects of an increase or decrease in Cyclonexpression on Fas expression were observed in activated but notresting cells (Figures 5-6, supplemental Figure 3). These resultssuggest an essential role of Cyclon in Fas up-regulation onactivation in CD4� T cells. Because Cyclon expression was low inresting T cells and induced by T-cell activation, it may bereasonable to assume that Cyclon is involved in enhancement ofFas expression on T-cell activation. On the other hand, transgenicexpression of Cyclon increased expression of Fas in activated aswell as resting CD8� T cells (Figure 5, supplemental Figure 3),suggesting that forced expression of Cyclon is sufficient forup-regulation of Fas in CD8� T cells. In contrast, reduction ofCyclon expression in CD8� T cells did not affect Fas expressionlevels (supplemental Figure 10). These results suggest that enhance-ment of Fas expression in CD8� T cells requires less Cyclonprotein than in CD4� T cells. Analysis of conditional deletion ofCyclon in T cells would be an interesting future issue to clarifyroles of Cyclon in Fas expression in CD8� T cells.
Understanding of regulation mechanisms of Fas expression inT cells is important for development of novel therapeutic ap-proaches for autoimmune diseases and lymphoma/leukemia. How-ever, surprisingly little is known about regulation mechanisms ofFas expression in primary T cells. It has been shown that TDAG51is important for Fas induction in a protein kinase C�dependentmanner in a T-cell hybridoma.44 Transcription factors, includingp53,41 c-myc single-strand binding protein,70 Sp3,71 NF-�B,46,47 andAP-1,72 have been suggested to be involved in Fas expressionregulation in various cell lines. However, involvement of theseproteins in regulation of Fas expression in primary T cells has notbeen clearly demonstrated. In this regard, it was shown that neitherTDAG51 nor NF-�B is essential for Fas expression regulation andapoptosis of primary mouse T cells in vivo.45,47 Information oncritical elements in promoter/enhancer of the Fas gene in primaryT cells is also very limited. Our preliminary data showed that the6-kbp fragment upstream of the start codon of mouse Fas gene is
FAS-MEDIATED IMMUNE HOMEOSTASIS AND CYCLON 1363BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

not sufficient to support expression of a reporter gene in primarymouse CD4� T cells (S.S.F. and H.F., unpublished observation,February 2008), suggesting that elements outside of this fragmentmight be important for expression of the Fas gene in primaryT cells. Our present study revealed a nuclear modifier of Fasexpression in primary T cells in vivo for the first time. Cyclon is aphosphorylated nuclear protein with a coiled-coil domain at theC-terminus.57 Coiled coils can be involved in signal-inducingevents, molecular recognition, mechanical stability of cells, andmovement processes.73 Therefore, it would be reasonable toassume that Cyclon interacts with Cyclon itself or other proteinsvia its coiled-coil domain to regulate expression levels of Fas.Considering the fact that Cyclon is localized in the nucleus57 andFas mRNA levels were decreased by the reduction of Cyclonexpression (Figure 6C), candidate Cyclon-interacting proteinswould be transcription factors involved in the regulation ofexpression levels of Fas. We think that our data provide animportant clue to elucidation of Fas expression regulation in T cellsand that future investigation of mechanisms of Cyclon action willbe an important step for understanding of molecular mechanisms ofFas expression regulation. Furthermore, our data suggest thatpharmacologic regulation of Cyclon expression levels may be apotential clinical intervention of Fas expression levels in thetreatment of autoimmune diseases and leukemia/lymphoma.
Acknowledgments
The authors thank Dan R. Littman for his kind and generous help,the Transgenic Core Facility of New York University CancerInstitute for generation of transgenic mice, Lina Kozhara andDerya Unutmaz for flow cytometric sorting, and Adriana Arita andQuazi Al-Tariq for technical assistance.
This work was supported by National Institutes of Health grantR01 AI059315 (H.F.).
Authorship
Contribution: S.S.F. designed and performed research and wrotethe paper; A.H. and K.K. performed research; T.E. contributed togeneration of Cyclon-deficient mice; and H.F. directed, designed,and performed research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Hodaka Fujii, Combined Program on Microbi-ology and Immunology, Research Institute for Microbial Diseases,Osaka University, 3-1 Yamadaoka, Suita-shi, Osaka 565-0871,Japan; e-mail: [email protected].
References
1. Chan FK-M, Lenardo MJ. Programmed celldeath. In: Paul WE, ed. Fundamental Immunol-ogy (5th Ed). Philadelphia, PA: Lippincott Wil-liams & Wilkins; 2003:841-864.
2. Tough DF, Sprent J. Immunological memory. In:Paul WE, ed. Fundamental Immunology (5th Ed).Philadelphia, PA: Lippincott Williams & Wilkins;2003:865-899.
3. Weant AE, Michalek RD, Khan IU, Holbrook BC,Wilingham MC, Grayson JM. Apoptosis regula-tors Bim and Fas function concurrently to controlautoimmunity and CD8� T cell contraction. Immu-nity. 2008;28:218-230.
4. Hutchenson J, Scatizzi JC, Siddiqui AM, et al.Combined deficiency of proapoptotic regulatorsBim and Fas results in the early onset of systemicautoimmunity. Immunity. 2008;28:206-217.
5. Hughes PD, Belz GT, Fortner KA, Budd RC,Strasser A, Bouillet P. Apoptosis regulators Fasand Bim cooperate in shutdown of chronic im-mune responses and prevention of autoimmunity.Immunity. 2008;28:197-205.
6. Brenner D, Krammer PH, Arnold R. Concepts ofactivated T cell death. Crit Rev Oncol Hematol.2008;66:52-64.
7. Krammer PH, Arnold R, Lavrik IN. Life and deathin peripheral T cells. Nat Rev Immunol. 2007;7:532-542.
8. Hildeman DA, Zhu Y, Mitchell TC, Kappler J,Marrack P. Molecular mechanisms of activated Tcell death in vivo. Curr Opin Immunol. 2002;14:354-359.
9. Hildeman DA, Zhu Y, Mitchell TC, et al. ActivatedT cell death in vivo mediated by proapoptotic bcl-2-family member bim. Immunity. 2002;16:759-767.
10. Marsden VS, Strasser A. Control of apoptosis inthe immune system: Bcl-2, BH3-only proteins andmore. Annu Rev Immunol. 2003;21:71-105.
11. Ashwell JD, Cunningham RE, Noguchi PD,Hernandez D. Cell growth cycle block of T cellhybridomas upon activation with antigen. J ExpMed. 1987;165:173-194.
12. Shi YF, Sahai BM, Green DR. Cyclosporin A in-hibits activation-induced cell death in T cell hy-
bridomas and thyocytes. Nature. 1989;339:625-626.
13. Strasser A, O’Connor L, Dixit VM. Apoptosis sig-naling. Annu Rev Biochem. 2000;69:217-245.
14. Contassot E, Gaide O, French L. Death receptorsand apoptosis. Dermatol Clin. 2007;25:487-501.
15. Thornburn A. Death receptor-induced cell killing.Cell Signal. 2004;16:139-144.
16. Schulze-Osthoff K, Ferrari D, Los M, WesselborgS, Peter ME. Apoptosis signaling by death recep-tors. Eur J Biochem. 1998;254:439-459.
17. Baud V, Karin M. Signal transduction by tumornecrosis factor and its relatives. Trends Cell Biol.2001;11:372-376.
18. Krueger A, Baumann S, Krammer PH, KirchhoffS. FLICE-inhibitory proteins: regulators of deathreceptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247-8254.
19. Hu S, Vincenz C, Ni J, Gentz R, Dixit VM.I-FLICE, a novel inhibitor of tumor necrosis factorreceptor-1- and CD95-induced apoptosis. J BiolChem. 1997;272:17255-17257.
20. Inohara N, Koseki T, Hu Y, Chen S, Nunez G.CLARP, a death effector domain-containing pro-tein interacts with caspase 8 and regulates apo-ptosis. Proc Natl Acad Sci U S A. 1997;94:10717-10722.
21. Irmler M, Thome M, Hahne M, et al. Inhibition ofdeath receptor signals by cellular FLIP. Nature.1997;388:190-195.
22. Rasper DM, Vaillancourt JP, Hadano S, et al. Celldeath attenuation by “Usurpin,” a mammalianDED-caspase homologue that precludes caspase8 recruitment and activation by the CD-95 (Fas,Apo-1) receptor complex. Cell Death Differ. 1998;5:271-288.
23. Srinivasula SM, Ahmad M, Ottilie S, et al.FLAME-1, a novel FADD-like antiapoptotic mole-cule that regulates Fas/TNFR1-induced apopto-sis. J Biol Chem. 1997;272:18542-18545.
24. Dhein J, Walczak H, Baumler C, Debatin KM,Krammer PH. Autocrine T-cell suicide mediatedby APO-1/(Fas/CD95). Nature. 1995;373:438-441.
25. Brunner T, Mogil RJ, LaFace D, et al. Cell-
autonomous Fas (CD95)/Fas-ligand interactionmediates activation-induced apoptosis in T-cellhybridomas. Nature. 1995;373:441-444.
26. Ju ST, Panka DJ, Cui H, et al. Fas(CD95)/FasLinteractions required for programmed cell deathafter T-cell activation. Nature. 1995;373:444-448.
27. Zheng L, Fisher G, Miller RE, Peschon J, LynchDH, Lenardo MJ. Induction of apoptosis in matureT cells by tumour necrosis factor. Nature. 1995;377:348-351.
28. Lenardo M, Chan KM, Hornung F, et al. Mature Tlymphocyte apoptosis: immune regulation in adynamic and unpredictable antigenic environ-ment. Annu Rev Immunol. 1999;17:221-253.
29. Algeciras-Schimnich A, Griffith TS, Lynch DH,Paya CV. Cell cycle-dependent regulation of FLIPlevels and susceptibility to Fas-mediated apopto-sis. J Immunol. 1999;162:5205-5211.
30. Watanabe-Fukunaga R, Brannan CI, CopelandNG, Jenkins NA, Nagata S. Lymphoproliferationdisorder in mice explained by defects in Fas anti-gen that mediates apoptosis. Nature. 1992;356:314-317.
31. Chu JL, Drappa J, Parnassa A, Elkon KB. Thedefect in Fas mRNA expression in MRL/lpr miceis associated with insertion of the retrotranspo-son, ETn. J Exp Med. 1993;178:723-730.
32. Adachi M, Suematsu S, Kondo T, et al. Targetedmutation in the fas gene causes hyperplasia inperipheral lymphoid organs and liver. Nat Genet.1995;11:294-300.
33. Takahashi T, Tanaka M, Brannan CI, et al. Gener-alized lymphoproliferative disease in mice,caused by a point mutation in the Fas ligand.Cell. 1994;76:969-976.
34. Ramsdell F, Seaman MS, Miller RE, Tough TW,Alderson MR, Lynch DH. gld/gld mice are unableto express a functional ligand for Fas. Eur J Im-munol. 1994;24:928-933.
35. Lynch DH, Watson ML, Alderson MR, et al. Themouse Fas-ligand gene is mutated in gld miceand is part of a TNF family gene cluster. Immu-nity. 1994;1:131-136.
36. Siegel RM, Chan FK, Chun HJ, Lenardo M. Themultifaceted role of Fas signaling in immune cell
1364 SAINT FLEUR et al BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

homeostasis and autoimmunity. Nat Immunol.2000;1:469-474.
37. Wajant H. The Fas signaling pathway: more thana paradigm. Science. 2002;296:1635-1636.
38. Rieux-Laucat F, Le Deist F, Hivroz C, et al. Muta-tions in Fas associated with human lymphoprolif-erative syndrome and autoimmunity. Science.1995;268:1347-1349.
39. Fisher GH, Rosenberg FJ, Straus SE, et al.Dominant interfering Fas gene mutations impairapoptosis in a human autoimmune lymphoprolif-erative syndrome. Cell. 1995;81:935-946.
40. Straus SE, Sneller M, Lenardo MJ, Puck JM,Strober W. An inherited disorder of lymphocyteapoptosis: the autoimmune lymphoproliferativesyndrome. Ann Intern Med. 1999;130:591-601.
41. Sharma K, Wang RX, Zhang LY, et al. Death theFas way: regulation and pathophysiology ofCD95 and its ligand. Pharmacol Ther. 2000;88:333-347.
42. Kavurma MM, Khachigian LM. Signaling andtranscriptional control of Fas ligand gene expres-sion. Cell Death Differ. 2003;10:36-44.
43. Zhang J, Xu X, Liu Y. Activation-induced celldeath in T cells and autoimmunity. Cell Mol Im-munol. 2004;1:186-192.
44. Park CG, Lee SY, Kandala G, Lee SY, Choi Y. Anovel gene product that couples TCR signaling toFas (CD95) expression in activation-induced celldeath. Immunity. 1996;4:583-591.
45. Rho J, Gong S, Kim N, Choi Y. TDAG51 is notessential for Fas/CD95 regulation and apoptosisin vivo. Mol Cell Biol. 2001;21:8365-8370.
46. Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the hu-man Fas promoter requires NFkB p50-p65 re-cruitment. Mol Cell Biol. 1999;19:2098-2108.
47. Zheng Y, Ouaaz F, Bruzzo P, Singh V, GerondakisS, Beg AA. NF-kB RelA (p65) is essential forTNF-alpha-induced fas expression but dispens-able for both TCR-induced expression and acti-vaion-induced cell death. J Immunol. 2001;166:4949-4957.
48. Taniguchi T. Cytokine signaling through nonre-ceptor protein tyrosine kinases. Science. 1995;268:251-255.
49. Sugamura K, Asao H, Kondo M, et al. Theinterleukin-2 receptor gamma chain: its role in themultiple cytokine receptor complexes and T celldevelopment in XSCID. Annu Rev Immunol.1996;14:179-205.
50. Leonard WJ. Type I Cytokines and Interferonsand their receptors. In: Paul WE, ed. Fundamen-tal Immunology (5th Ed). Philadelphia, PA: Lippin-cott Williams & Wilkins; 2003:701-747.
51. Horak I, Lohler J, Ma A, Smith KA. Interleukin-2deficient mice: a new model to study autoimmu-nity and self-tolerance. Immunol Rev. 1995;148:35-44.
52. Kramer S, Schimpl A, Hunig T. Immunopathologyof interleukin (IL) 2-deficient mice: thymus depen-dence and suppression by thymus-dependentcells with an intact IL-2 gene. J Exp Med. 1995;182:1769-1776.
53. Suzuki H, Kundig TM, Furlonger C, et al. Deregu-lated T cell activation and autoimmunity in micelacking interleukin-2 receptor beta. Science.1995;268:1472-1476.
54. Willerford DM, Chen J, Ferry JA, Davidson L, MaA, Alt FW. Interleukin-2 receptor alpha chainregulates the size and content of the peripherallymphoid compartment. Immunity. 1995;3:521-530.
55. Cao X, Shores EW, Hu-Li J, et al. Defective lym-phoid development in mice lacking expression ofthe common cytokine receptor gamma chain. Im-munity. 1995;2:223-238.
56. Nakajima H, Leonard WJ. Impaired peripheraldeletion of activated T cells in mice lacking thecommon cytokine receptor gamma-chain. J Im-munol. 1997;159:4737-4744.
57. Hoshino A, Fujii H. Redundant promoter elementsmediate IL-3-induced expression of a novel cyto-kine-inducible gene, cyclon. FEBS Lett. 2007;581:975-980.
58. Wang Y, Zhu W, Levy DE. Nuclear and cytoplas-mic mRNA quantification by SYBR green basedreal-time RT-PCR. Methods. 2006;39:356-362.
59. Hoshino A, Matsumura S, Kondo K, Hirst JA, FujiiH. Inducible translocation trap: a system for de-tecting inducible nuclear translocation. Mol Cell.2004;15:153-159.
60. Hoshino A, Hirst JA, Fujii H. Regulation of cellproliferation by interleukin-3-induced nucleartranslocation of pyruvate kinase. J Biol Chem.2007;282:17706-17711.
61. Zhang J, Gao J-X, Salojin K, et al. Regulation ofFas ligand expression during activation-inducedcell death in T cells by p38 mitogen-activated pro-tein kinase and c-jun NH2-terminal kinase. J ExpMed. 2000;191:1017-1030.
62. Zhumabekov T, Corbella P, Tolaini M, Kioussis D.
Improved version of a human CD2 minigenebased vector for T cell-specific expression intransgenic mice. J Immunol Methods. 1995;185:133-140.
63. Festenstein R, Tolaini M, Corbella P, et al. Locuscontrol region function and heterochromatin-in-duced position effect variegation. Science. 1996;271:1123-1125.
64. Laurence A, Tato CM, Davidson TS, et al.Interleukin-2 signaling via STAT5 constrains Thelper 17 cell generation. Immunity. 2007;26:371-381.
65. Fontenot JD, Rasmussen JP, Gavin MA,Rudensky AY. A function for interleukin 2 inFoxp3-expressing regulatory T cells. Nat Immu-nol. 2005;6:1142-1151.
66. D’Cruz LM, Klein L. Development and function ofagonist-induced CD25�Foxp3� regulatory T cellsin the absence of interleukin 2 signaling. Nat Im-munol. 2005;6:1152-1159.
67. Jaleco S, Swainson L, Dardalhon V, BurjanadzeM, Kinet S, Taylor N. Homeostasis of naive andmemory CD4� T cells: IL-2 and IL-7 differentiallyregulate the balance between proliferation andFas-mediated apoptosis. J Immunol. 2003;171:61-68.
68. Yang J, Epling-Burnette P-K, Painter JS, et al.Antigen activation and impaired Fas-induceddeath-inducing signaling complex formation inT-large-granular lymphocyte leukemia. Blood.2008;111:1610-1616.
69. Bettelli E, Oukka M, Kuchroo VK. Th-17 cells inthe circle of immunity and autoimmunity. Nat Im-munol. 2007;8:345-350.
70. Numura J, Matsumoto K, Iguchi-Ariga SM, ArigaH. Positive regulation of Fas gene expression byMSSP and abrogation of Fas-mediated apoptosisinduction in MSSP-deficient mice. Exp Cell Res.2005;305:324-332.
71. Pang H, Miranda K, Fine A. Sp3 regulates fasexpression in lung epithelial cells. Biochem J.1998;333:209-213.
72. Lasham A, Lindridge E, Rudert F, Onrust R,Watson J. Regulation of the human fas promoterby YB-1, Puralpha and AP-1 transcription factors.Gene. 2000;252:1-13.
73. Burkhard P, Stetefeld J, Strelkov SV. Coiled coils:a highly versatile protein folding motif. Trends CellBiol. 2001;11:82-88.
FAS-MEDIATED IMMUNE HOMEOSTASIS AND CYCLON 1365BLOOD, 13 AUGUST 2009 � VOLUME 114, NUMBER 7
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom

online June 15, 2009 originally publisheddoi:10.1182/blood-2008-11-189118
2009 114: 1355-1365
Shella Saint Fleur, Akemi Hoshino, Kimie Kondo, Takeshi Egawa and Hodaka Fujii activation-induced protein, CyclonRegulation of Fas-mediated immune homeostasis by an
http://www.bloodjournal.org/content/114/7/1355.full.htmlUpdated information and services can be found at:
(5665 articles)Immunobiology and Immunotherapy Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on April 14, 2019. by guest www.bloodjournal.orgFrom






![Braille Module 63 Special Formats: Poetry, Columns, Tables, …profitt.gatech.edu/drupal/sites/default/files/curriculum... · 2012. 9. 24. · Lesson 18.3 [BF Rule 7] Lesson 18.4](https://img.dokumen.tips/doc/110x75/6046a2830d5010642b0d440b/braille-module-63-special-formats-poetry-columns-tables-2012-9-24-lesson.jpg)






















