Embed Size (px)
Citation preview

Some Gas Kinetic Studies □! Reactive Opganosilicon Intermediates
A Thesis presented by
IAN THOMAS WOOD
fo r the degree of
Doctor of Philosophy
of the
U nivers ity of Leicester
Department of ChemistryUniversity of Leicester November 1982

UMI Number: U334065
All rights reserved
INFO RM ATIO N TO ALL U SER S The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
DiSËürtâtion Publishing
UMI U334065Published by ProQuest LLC 2015. Copyright in the Dissertation held by the Author.
Microform Edition © ProQuest LLC.All rights reserved. This work is protected against
unauthorized copying under Title 17, United States Code.
P r o Q u e s t
ProQuest LLC 789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106-1346

yq-SO V et

STATEMENT
The acconpanying thesis submitted for the degree of Ph.D. entitled "Sane Gas Kinetic Studies of Reactive Organosilicon Intermediates" is based on work conducted by the author mainly in the Department of Chemistry of the University of Leicester during the period between October 1979 and September 1982.
All the work recorded in this thesis is original unless otherwise acknowledged in the text or by references. None of the work has been submitted for another degree in this or any other university.
Signed: ....................... Date:

ACKNCWLEaX3EMENTS
I should like to express my sincere thanks to my supervisor,Dr. I. M. T. Davidson, who gave me the opportunity to study for a Ph.D., and for his help and guidance throughout the course of this work.
I am also grateful to Drs. P. Potzinger and B. Reimann for their assistance during ny research at the Max-Planck- Institute, Milheim a.d. Ruhr, West Germany.
Finally, I should like to thank all members of our research group, past and present, Vicky Orson-Wright for typing the thesis and Ann Crane for tracing the diagrams.

LIST OF CONTENTS
CHAPTER 1 - REVIEWPage No.
1.1 Introduction 21.2.1 Silyl radicals 31.2.2 Intermediates containing pïï-pïï bonds to silicon 31.2.3 Silylenes 121.3.1 Allyltrimethylsilane 171.3.2 Other allylsilanes 181.3.3 Silirenes 211.4.1 Thermochemistry 241.4.2 Electron impact measurements 241.4.3 Benson's electrostatic model 261.4.4 Bell and Perkins' calculations 281.4.5 Gas-phase iodine atom reactions 281.4.6 Pyrolysis techniques 31
CHAPTER 2 - APPARATUS AND EXPERIMENTAL PROCEDURE2.1 Vacuum Line, Reaction Vessel and Furnace 382.2 Mass Spectrometer 392.3 Computer 412.4 A Kinetic Run 432.5 Data Processing 432.6 Cleaning the Q 801K 442.7 Pulsed Stirred-Flow Technique 45
CHAPTER 3 - PYROLYSIS OF 1. l-DIMETHYL-2,3-BIS (TRIMETHYLSILYL)- 1-SILIRENE
3.1 Cyclopropenes 483.2 Mechanism of Silirane and Silirene Pyrolysis 493.3 Low-Pressure Pyrolysis 533.4 Kinetics 553.5 Discussion 563.6 Conclusion 57
CHAPTER 4 - ALLYLTRIMETHYLSILANE PYROLYSIS4.1 Introduction 594.2 Pyrolysis Products 594.3 Pyrolysis at Lower Pressures 604.4 ATMS Decay Kinetic Measurements 664.5 Trapping of SE 694.5.1 Methanol 704.5.2 Methyltrimethylsilyl ether 724.5.3 Oxygen 724.5.4 Methyl chloride 734.6.1 Radical addition to allyltrimethylsilane 764.6.2 Benzyltrimethylsilane 7 84.6.3 Mercury dimethyl 804.6.4 Allyltrichlorosilane/mercury dimethyl 824.7 Discussion 834.8.1 Mercury Photosensitization 844.8.2 Apparatus 87

LIST OF CONTENTS (Continued)Page N
4. 8.3 Experimental 874. 8.4 Experiments HG4 894. 8.5 Experiments HG3 934. 8.6 Experiments HGl 934. 8.7 Experiments HG2 954. 8.8 Experiments HG5 974. 8.9 Discussion 974. 9.1 Introduction 994. 9.1 a) Kinetic measurements 100
b) Pyrolysis products 1014. 9.3 Discussion 1044.10.1 Introduction 1054.10.2 Experimental 1054.11 Discussion 106
CHAPTER 5 - PYROLYSIS OF VINYLDIMETHYLCARBINOXYDIMETHYLSILANEAND 1,1,3,3-TETRAMETHYL-1 -VINYLDISILOXANE
5.1.1 Introduction 1085.1.2 Pyrolysis of VCS 1085.2 Experimental 1085.3 LPP Products 1095.4 Kinetic Measurements 1105.5.1 Insertion into oxygen-hydrogen bonds 1115.5.2 Insertion into silicon-oxygen bonds 1125.5.3 Other insertion reactions 1135.5.4 Further ccmments 1145.6 Discussion 1145.7 Introduction 1155.8.1 Pyrolysis products 1165.8.2 Trapping experiments 1185.8.3 Mercury dimethyl 1195.8.4 LPP kinetic measurements 1215.9 Discussion 121
CHAPTER 6 - KINETICS OF REACTIONS OF 1,1-DIMETHYLSILAETHENE ANDDIMETHYLSILANONE
6.1 Introduction 1256.2 Experimental 1256.3 Kinetic Measurements 1276.4 Introduction 1306.5 Experimental 130
CHAPTER 7 - DISCUSSION7.1 Introduction 1337.2.1 Reaction of SE with unsaturated substrates 1337.2.2 Diels Alder reactions 1337.2.3 2tt + 2it reactions 1357.2.4 Hydrogen-transfer reactions 1377.3 Retroene Processes 1387.4 Reversibility of Retroene Reactions 140

LIST OF CONTENTS (Continued)Page No.
7.5 Entropy Calculations 1417.6 Strength of tt Bond in Me2Si=CH2 1457.7 Strength of the Silicon-Ally 1 Bond 1467.8 Heat of Formation of ATMS 1467.9 Heat of Formation of SE 1477.10 1,1,3-Trimethyl-1 -Silacyclobutane Formation 1487.11 Insertion of Di and SE into Methyl trimethylsilyl Ether 1487.12 Reversibility of Insertion Reactions 1517.13 Entropy Calculations 1527.14 Application of Results 1547.15 Summary 156REFERENCES 160APPENDICES 169

CHAPTER 1REVIEW

1.1 INTRODUCTION
Organosilicon chemistry is currently ccmtianding considerable attention. This is only partly due to its wide range of important applications in industry and organic synthesis : acadonic interest has been aroused by the recent discoveries that there exist several other reactive organosilicon intermediates besides silyl radicals, Wiich have been known for
2seme years. These species include silylenes #iich are analogous to carbenes, but the most interesting are compounds containing a silicon pTT-pTT double bond which have prompted studies of their spectroscopy and structure as well as their varied chemistry. Although much illuminating groundwork has been completed in investigating the fascinating field of these reactive intermediates, there remain many under-developed areas; studies are often handicapped by the scarcity of definitive thermochemical data, particularly bond dissociation energies and heats of formation of
3molecules and radicals.This thesis describes experimental work which was undertaken by the
author on the gas-phase studies (mainly kinetic) of the generation and reactions of some organosilicon reactive intermediates with a view to providing further valuable background information, and thermochemical data, vbich should lead to a better understanding of the chemistry of silicon-containing compounds in this area and others. The method used to produce these reactive intermediates was gas-phase pyrolysis reactions, of immense interest in their own right, mainly using a recently developed technique called low-pressure pyrolysis (LPP). The level of current knowledge is outlined in this chapter. Only thermal routes to the reactive intermediates are discussed; photochemical are omitted. Throughout this thesis first order rate constants and Arrhenius A factors are in s~ \ second order in dm^ mol" s" ; activation energies are in kJ mol"^.
— 2 —

REACTIVE INTERMEDIATES IN ORGANOSILICON CHEMISTRY
1.2.1 Silvl radicalsThese are species in which the radical centre is concentrated on a
silicon atan, e.g. trimethylsilyl, MesSi*, trichlorosilyl, ClaSi». The2 3 4chemistry of silyl radicals is well developed; ’ ’ more is known about
them than any of the other reactive intermediates. For example, silylradicals abstract a halogen atom from carbon-halogen bonds,^ chlorine
6 2 7from silicon-chlorine bonds, and hydrogen from carbon-hydrogen bonds. ’They also undergo rapid reccmbination^ or disproportionation^
14reactions and add readily to multiple bonds, e.g. tetrafluoroethene.
1.2.2 Intermediates containing pir-pTT bonds to siliconFor seme years this type of linkage to silicon was thought to be
impossible and, indeed, all early claims to have formulated such specieswere refuted.However, thermal decomposition studies on silacyclo-butanes in the late sixties^^ gave the first reliable evidence for theintermediacy of compounds containing pTT-pir silicon carbon bonds; by 1968this had been unequivocally confirmed in kinetic experiments by Flowers
17and Gusel'nikov. [It has now been established by direct detection mass18 19 20 21spectrcmetrically, ’ and by matrix isolation ’ that such compounds
can be generated.] Since then it has been shown that silicon can also2form weak pTr-pm bonds to oxygen, nitrogen or another silicon. These
compounds have been described in a recent review by Gusel'nikov and Nametkin;^^ very little is known about those containing silicon-nitrogen and silicon-silicon tt bonds.
(a) SilaalkenesSilaalkenes (also known as silenes) contain a silicon-carbon pTT-prr
bond, e.g. Me2Si=CH2, 1,1-dimethyl-1-silaethene (SE), the species usually
— 3 —

subjected to experimental and theoretical study. [A list of theabbreviations for chemical species used throughout this thesis is shownin Appendix 1.1.] The strength of the tt bond in SE has recently beenestimated to be 175 ± 25 kJ mol”^
The most frequently used precursors to silaalkenes are still sila-cyclobutanes since they are a clean and relatively low temperature sourceas well as being ccmmercially available. The kinetic measurements of
171,1-dimethyl-1-silacyclobutane (IMSCB) decaiposition vAiich gave: logio k/s” = (15.64 ±0.30) - (261.5 ±3.3 kJ mol"M/2.303 RT indicated that the mechanism of the reaction involves initial biradical formation:
M e a S Q 750J^ Me2S|. : Me2Si=CH2 + CH2=CH2 (1.1)
The Arrhenius parameters are identical to those for cyclobutanedecaiposition^^ - logio k/s"^ = (15.62 ±0.1) - (261.5 ±1.7 kJ mol"M/2.303 RT - vÆiich proceeds as above: the activation energy correspondsto the strained carbon-carbon bond hemolysis, and the size of the Afactor is in agreanent with a biradical intermediate formed by ring
25opening.Additional thermal routes to silaalkenes include reactions of the
26type:
,CF TOOK
r I + Me2Si=CH2 (1.2)
and the pyrolysis of allylsilanes, discussed in more detail later in this chapter.
— 4 —

Reactions of silaalkenesIn solution, polymerisation of SE usually occurs,but gas-phase
investigations have revealed several different reactions viiich can be broadly classified into two categories: additions and insertions. Thekinetics of a few of these reactions have been measured by Gusel'nikov
18 257 23et al., John et al., and workers in our laboratory. The27technique of John et al., that of conventional sealed tube pyrolyses,
is only suitable for measuring bimolecular rate constants for slower reactions than those measured by us - in the IPP apparatus - because their experiments are performed at higher pressures.
In the absence of a suitable trapping agent, cyclodimérisation by head-to-tail self-addition occurs to form 1,1,3,3-tetramethyl-l,3-disila- cyclobutane (DSCB):^^
2Me2Si=Œ2 — Me2Si— I (1.3)I— smez
Gusel'nikov has measured the kinetics of this process using a flowapparatus ccmprising a quartz tube with two zones of variable temperatureand length: one pyrolytic for SE generation frcm CMSCB, and the otherfor cyclodimérisation; this enabled the amount of dimérisation to becalculated at different tarperatures, giving : k = 10®’ dm^ mol"^ s”^
18with zero activation energy. For seme silaalkenes bulky substituents29effect head-to-head rather than head-to-tail addition.
The reaction of SE with ethene constitutes the reverse of CMSCB 17décomposition ; the reaction is therefore thought to proceed via a
30biradical intermediate:
Me2Si=CH2 + CH2—CH2 ^ Me2Si— : ^ Me2Si“ n (1.4)I : - 1
- 5 -
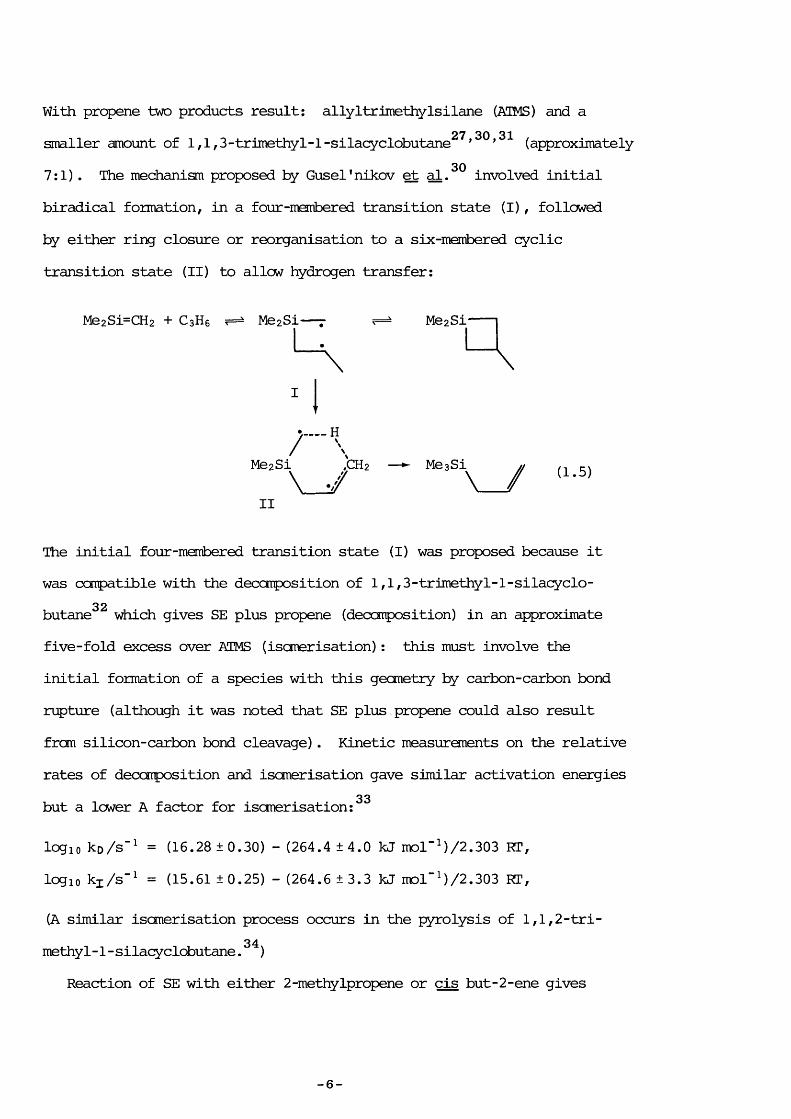
With propene two products result; allyltrimethylsilane (ATMS) and a27 30 31smaller amount of 1 ,1,3-trimethyl-1 -silacyclobutane ’ ’ (approximately307:1). The mechanism proposed by Gusel ' nikov et al. involved initial
biradical formation, in a four-member ed transition state (I), followed by either ring closure or reorganisation to a six-membered cyclic transition state (II) to allow hydrogen transfer:
Me2Si-CH2 + C 3H6 Me2Si ' —- Me 2 Si
I
•y H./ \Me2Si ,CH2 — ^ Me 3 Si ^v _ y v _ y
II(1.5)
The initial four-membered transition state (I) was proposed because itwas compatible with the decomposition of 1 ,1,3-trimethyl-1-silacyclo-
32butane which gives SE plus propene (decomposition) in an approximatefive-fold excess over ATMS (isomérisation): this must involve theinitial formation of a species with this geometry by carbon-carbon bondrupture (although it was noted that SE plus propene could also resultfrom silicon-carbon bond cleavage) . Kinetic measurements on the relativerates of decomposition and isomérisation gave similar activation energies
33but a lower A factor for isomérisation :
logio ko/s"^ = (16.28 ± 0.30) - (264.4 ± 4.0 kJ mol"M/2.303 RT, logic kj/s"^ = (15.61 ±0.25) - (264.6 ±3.3 kJ mol”M/2.303 RT,
(A similar isomérisation process occurs in the pyrolysis of 1,1,2-trimethyl-1 -silacyclobutane . )
Reaction of SE with either 2 -methyIpropene or cis but-2-ene gives
— 6 —

27 35entirely the hydrogen-transfer product. *
Conlin has tentatively proposed that the addition reaction with such alkenes could occur via an intennolecular concerted pericyclic process (an ene reaction) rather than via a biradical intermediate.^^ Similar solution reactions of a silaethene have been described in these
In the case of conjugated dienes a 27t + 47t Diels-Alder cycloadditionprocess, e.g. reaction 1.6,^^’ ^ v^ich occurs via a six-matiberedtransition state, is preferred over the hydrogen-transfer reaction whichis less important Wien the substrate is either 2-methyIbutadiene, 2,3-
35dimethyIbutadiene, penta-1,3-diene or cyclopentadiene, and does not35occur at all with butadiene, the Diels-Alder adduct being the only
product. No four-manbered ring products are formed with any of the dienes.
Me,Si=CH, Dlels-Alder
The reaction in solution between 1,1 -dimethyl-2,2-bis (trimethylsilyl) -1 -silaethene and 2,3-dimethyIbutadiene also gives both products: theDiels-Alder adduct is formed in four-fold excess over the product
38resulting frcm hydrogen transfer.Recently the relatively fast addition of SE to oxygen has been
28investigated kinetically using the LPP technique. The cyclic product is unstable and decomposes to form formaldehyde and dimethyIsilanone (Di) Wiich undergoes self - addition, in a similar fashion to SE, trimer is ing to form hexamethylcyclotrisiloxane (D3) because the dimer (D2 ) is unstable. [A ccmmon usage, adopted throughout this thesis, is to denote cyclic (Me2SiO)n by Dn] :
— 7 —
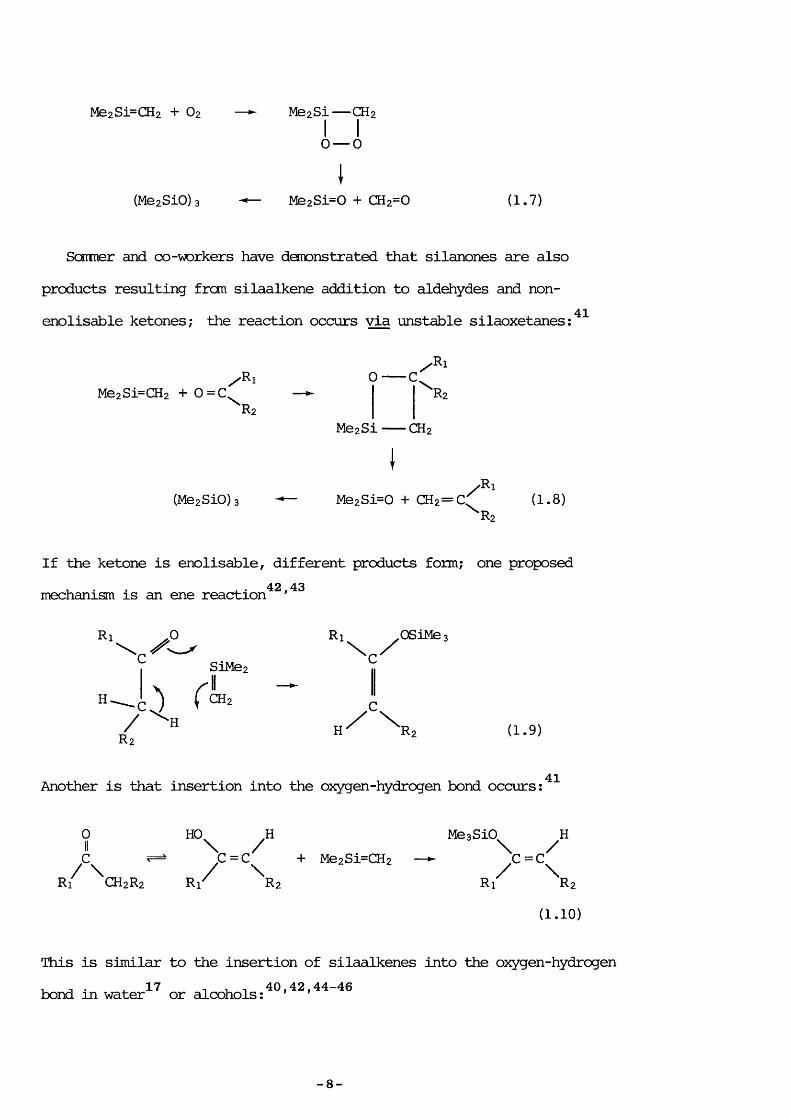
Me2Si—CH2 + O2 — Me2Si— CH2I I0 — 0
I(Me2SiO)3 Me2Si=0 + CH2=0 (1.7)
Santier and co-workers have demonstrated that silanones are also products resulting frcm silaalkene addition to aldehydes and non-
41enolisable ketones; the reaction occurs via unstable silaoxetanes:
Me2Si=Œ2 + 0 = CR2
/Ri O ---C/RiRz
Me2Si — ' ' CH2
I/Ri
(Me2SiO) 3 Me2Si=0 + CH2=C(' (1.8)
If the ketone is enolisable, different products form; one proposed, . 4 2 ,4 3mechanism is an ene reaction ’
Ri ^0 Ri ^0SiMe3
7 SiMe2
y ^ R2 (1.9)Kz
41Another is that insertion into the oxygen-hydrogen bond occurs:
0 HO^ H Me3SiO, HII \ / \ /C ^ C=C + Me2Si=CH2 — C =C
Ri CH2RZ Ri Rz Ri Rz(1.10)
This is similar to the insertion of silaalkenes into the oxygen-hydrogen, . . ^ 17 n T 4 0 ,4 2 ,4 4 -4 6bond in water or alcohols : ' ’
— 8 —
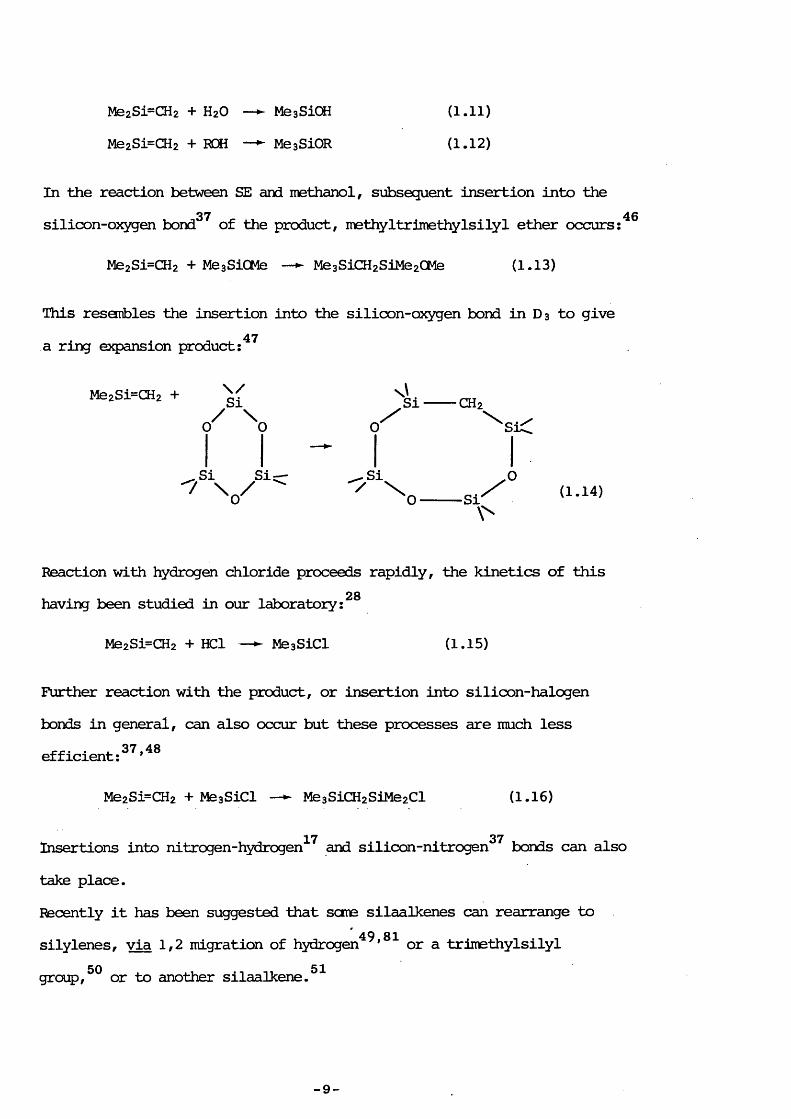
Me2Si=CH2 + H2O — MeaSiOi (1.11)Me2Si=CH2 + ROH — ^ MegSiOR (1.12)
In the reaction between SE and methanol, subsequent insertion into the37 46silicon-oxygen bond of the product, methyItrimethylsilyl ether occurs;
Me2Si=CH2 + MeaSiCMe — ► Me3SiCH2SiMe20Me (1.13)
This resembles the insertion into the silicon-oxygen bond in D 3 to give47a ring expansion product;
Me2Si=CH2 + n\.----
0^ '^0 o ' ^
^Si S i ^ ^ S i
Reaction with hydrogen chloride proceeds rapidly, the kinetics of this28having been studied in our laboratory:
Me2Si=CH2 + HCl — MeaSiCl (1.15)
Further reaction with the product, or insertion into silicon-halogen bonds in general, can also occur but these processes are much less efficient:
Me2Si=Œ2 + MeaSiCl — Me3SiCH2SiMe2Cl (1.16)
17 37Insertions into nitrogen-hydrogen and silicon-nitrogen bonds can alsotake place.Recently it has been suggested that sane silaalkenes can rearrange to silylenes, via 1,2 migration of hydrogen^^’®^ or a trimethylsilyl g r o u p , o r to another silaalkene.
- 9 -

(b) SilanonesSilanones contain a silicon-ojQ^en ïï bond, of approximate strength
160 kJ mol" . Routes to their production, Wiich are rarer and tend to be much less convenient than those for silaalkenes, include: thedecaiposition of unstable silaoxetanes, discussed earlier, which is unsatisfactory because of the necessary in situ preparation, and caiplications arising frcm other products; and the pyrolysis of cyclic siloxanes, difficult because of the ensuing reactions, e.g. octamethyl- cyclotetrasiloxane,
800 KDi| D 3 + Di (1.17)
Di + Dif D 5
Di + Ds ^ DeDe — ^ 2 D 3
Scheme 1.1
In 1979 a more facile route was discovered by Frye,^^ that of heating vinyldimethylcarbino2Q^imethylsilane (VCS) ; the mechanism was thought to be a retroene reaction:
Me ,Me
O O C H 950 K 9I — II I4- (1.18)
/ H Me' MeMe
Part of the research described in this thesis involved the kinetic study of the decaiposition of VCS to probe its usefulness as a Di source and to elucidate its mode of production, and also the study of a related catpound, ^ 1,1,3,3-tetramethyl-l-vinyldisiloxane (HMezSiOSiMe2CH=CH2) to investigate vhether it adopted similar decaiposition behaviour.
— 10 ”

Barton has also found another route^^ v^ich is less convenient than Frye ' s :
,56
300 K aCF3+ Me2Si=0
CF3 (1.19)
Barton has disputed the recent claims by Razuvaev et al. that a further silanone source, involving the thermolysis of silylperoxides, has been developed, e.g.:
HMe2SiOOCMe: 430 K Me2Si=mO! I
H— OCMe 3
Me2Si=0
+ H0CMe3 (1.20)
Reactions of silanonesStudies of reactions of silanones are less cannon than silaalkenes ;
one contributing factor has been the lack of a convenient source. Consequently, these recently discovered routes should allcw extended research in this area. Not surprisingly, the reactions of silanones kncwn so far bear a close resanblance to those of silaalkenes, e.g. the trimérisation of Di. No clear evidence of the nature of the adducts formed frcm silanone reaction with alkenes has been obtained, but it is known that D 5 production frcm pyrolysis (Scheme 1.1) is inhibited by addition of ccmpounds with carbon -carbon double bonds such as ethene, propene and butadiene. Reactions of this type should be moredifficult for silanones than silaalkenes because of the instability of silaoxetanes, probably the initial addition product.
The best-known reaction is that of insertion into a silicon-oxygen bond, e.g. in
— 11 “

Di + D; D, (1.21)
in other cyclics:60
RMeSi=0 + MegSi0.
WSiMez
MezSi^ ^SiMez (1 .22)
in dimethyIdirnethoxysilane^^ or in hexamethyldisiloxane.^^ Ring expansion products are also obtained by insertion into silirenes and siliranes.®^ Razuvaev et ad.^^ have claimed that silanes are the most effective traps, reaction occurring by insertion into the silicon-hydrogen bond, but Barton^^ doubts the validity of this reported process:
RiRzSi=0 + EtsSiH — EtaSiOSi (H)RiRz (1.23)
Insertion into silicon-chlorine bonds can also take place
MezSi=0 + MeaSiCl — ► MeaSiOSiMezCl (1.24)
Whilst there appears to be no evidence of insertion into an oxygen- hydrogen bond, products have been isolated which were thought to resultfrom a reaction sequence initiated by Di insertion into a carbon-oxygen, j 55 bond.
Seme of the work discussed in this thesis involved the investigation of several reactions of Di, including the first kinetic study, along with the corresponding SE process, so that comparisons could be made where appropriate.
1.2.3 SilvlenesThese reactive intermediates of silicon, analogous to carbenes, were
discovered in 1968 by the heating of readily available methoxydisilanes,64a-elimination occurring. This is still the most ccmmon thermal method
- 12 -

of generating silylenes:
Me MeI I 500 KMeO — Si Si QMe — MezSi: + (MeO)zSiMe2 (1.25)I IMe Me
Since then several other sources have been found, such as disilanes containing a silicon-hydrogen or silicon-halogen linkage;®^’®® these, and others, have been fully discussed in two recent reviews by Caspar One route worthy of note is the pyrolysis of sane siliranes:^^'^^
Me Me350 KMe Me _ >— ( + MezSi: (1.26)
SiMe^^ '^Me
/ \Me Me
Part of the author's research involved the pyrolysis of a silirene to investigate its usefulness as a silylene source. The indications, based on other studies of themolyses of such ccmpounds, discussed later in this chapter, were that silirenes are not good silylene generators.
Reactions of silvlenesMuch work has been done on the reactions of silylenes. The most
68important addition reactions include : reaction with alkenes and70acetylenes to give siliranes and silirenes, e.g.
R RMezSi: + RC ECR — >- = C (1.27)V
M e ^ '^Me
and reaction with dienesand ketones.Silylene dimérisation (vhich71is follcwed by rearrangement) can also occur:
- 13 -
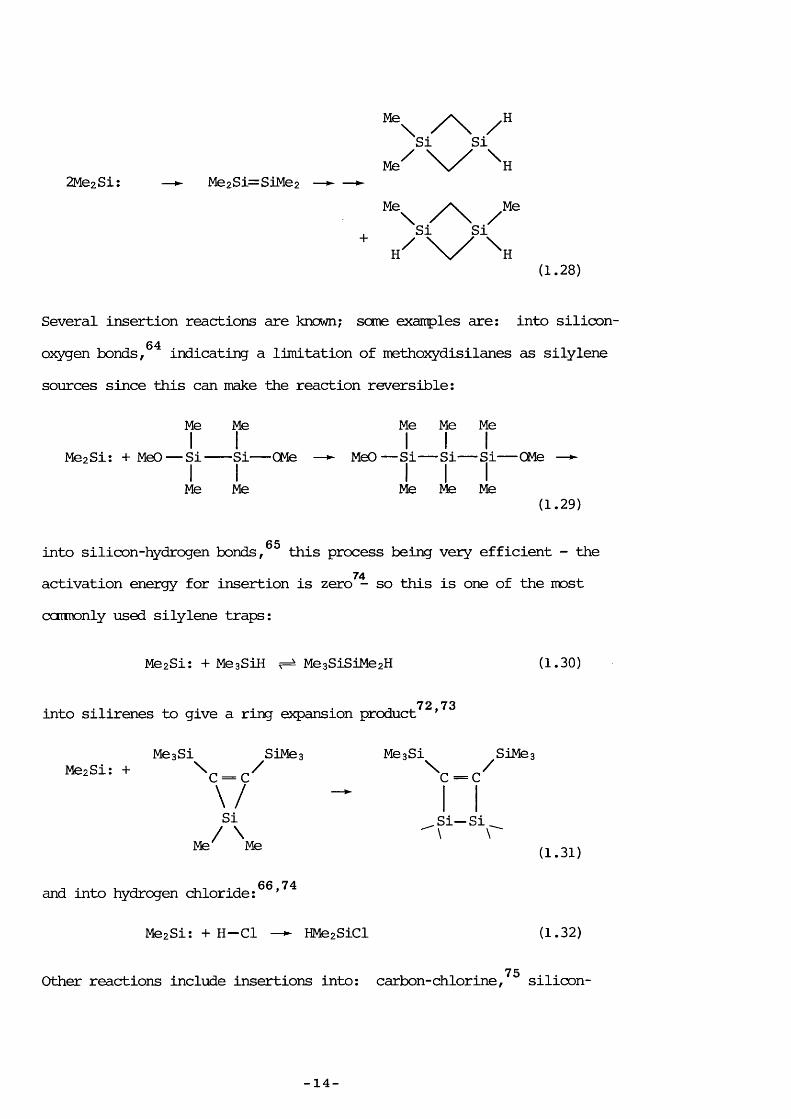
ZMezSi: Me 2 S i= SiMez
Me / HSi Si
M e ^
* H ^ \ / N(1.28)
Several insertion reactions are known; seme exairples are: into silicon-64oxygen bonds, indicating a limitation of methoxydisilanes as silylene
sources since this can make the reaction reversible:
Me MeI IMezSi: 4- MeO — Si--- Si QMeI IMe Me
Me Me MeI I IMeO — Si— Si— Si— OMeI I IMe Me Me
(1.29)
into silicon-hydrogen bonds,this process being very efficient - the activation energy for insertion is zero^- so this is one of the most ccmmonly used silylene traps:
MezSi: -H MegSiH ^ MegSiSiMezH (1.30)
into silirenes to give a ring expansion product72,73
Me 3 Si SiMe3MezSi: 4- ^ c = C ^
\/Si/ \Me Me
Me 3 Si ^SiMe3= C
and into hydrogen chloride ;
MezSi: 4- H— Cl HMezSiCl
Si— Si^ \ \
(1.31)
(1.32)
75Other reactions include insertions into: carbon-chlorine, silicon-
- 14 -

chlorine,nitrogen-hydrogen, oxygen-hydrogen, silicon-sulphur,78 77 79carbon-sulphur, sulphur-sulphur, sane carbon-hydrogen (rare) , and
strained silicon-silicon^^ and carbon-oxygen^^ bonds. Evidence hasrecently been presented that silylene to silaalkene rearrangements can
51occur.
- 15 -
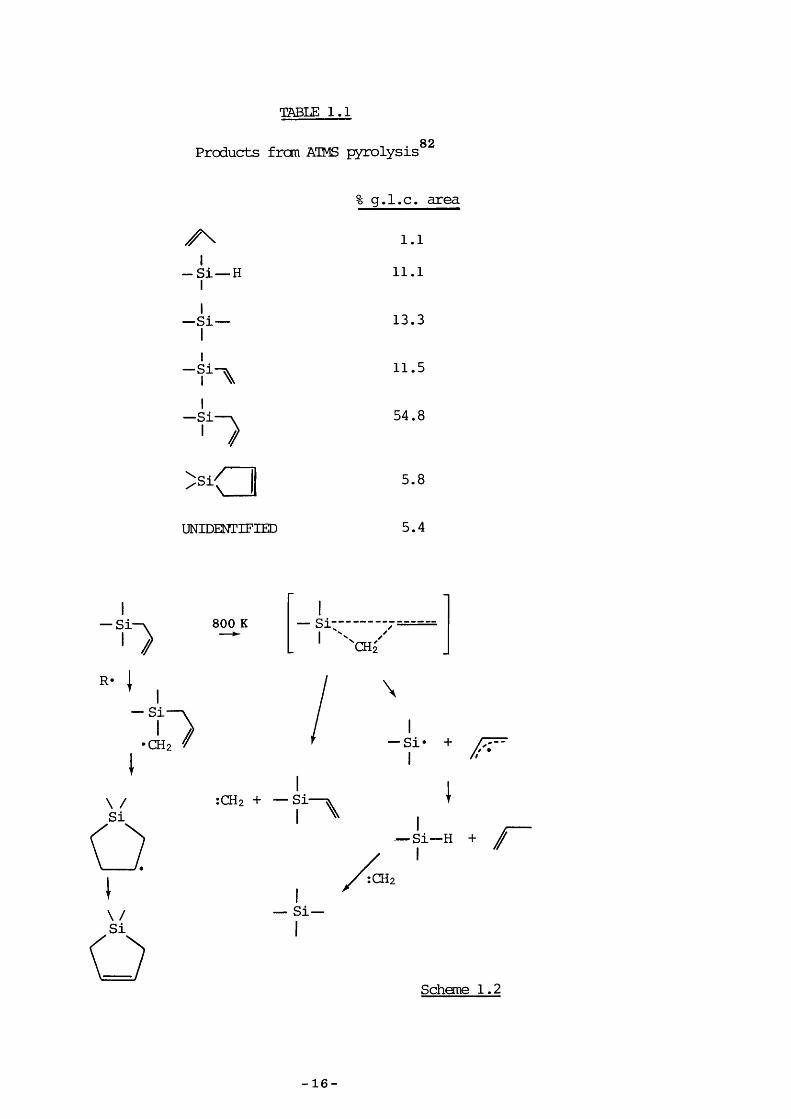
TABLE 1.182Products frcm ATMS pyrolysis
- S i — H
g.l.c. area
1.1
11.1
I— Si- 13.3
— Si
— SiI
A //
11.5
54.8
>< 5.8
UNIDENTIFIED 5.4
— Si 800 K
R«— Si
CHz
\— Si* +
\ /Si:CHz + — Si— ^
I ^■Si— H + r
îCHz
\/Si— Si—
Scheme 1.2
— 16 “

PYROLYSIS OF ORGANOSILICON CCMPOUNDS
Pyrolytic studies of organosilicon ccmpounds have revealed a large3variety of reactions; the pyrolysis of allylsilanes and silirenes alone
illustrates this diversity.
1.3.1 AllyltrimethvlsilaneIn 1968 Bailey found that allyltrimethylsilane (ATMS) was a thermal
39source of SE; a retroene mechanism was proposed:
Me^ ^^Me/Si 875 K
C H z / M — ^ MeCH=CH2 + Me2Si=CH2H
Me2Si^SiMe 2 (1.33)
82Sakurai et al. pyrolysed ATMS and reported results vhich appeared to conflict with those of Bailey since DSCB was not formed in large quantities. To explain the formation of the products, shewn in Table 1 .1, it was proposed that the décomposition mechanism was the concurrent extrusion of a methylene groip and silicon-ally 1 bond cleavage (Scheme 1.2).
83Kwart claims to have measured the kinetics of the retroene reaction, obtaining, logio k/s” = (12.55) - (234 kJ mol"^)/2.303 RT vhereas Gusel'nikov has postulated that the SE is formed by disproportionation of trimethylsilyl radicals.The issue was confused still further by Jones®^ vhose labelling studies seemed to have proved that vinyltri- methylsilane is not formed by methylene extrusion but is a result of silioon-methyl bond rupture (Scheme 1.3). Although the ensuing rearrangement sequence is entirely reasonable, the initial bond rupture is improbable since the veakest bond towards unimolecular hemolysis
— 17 “
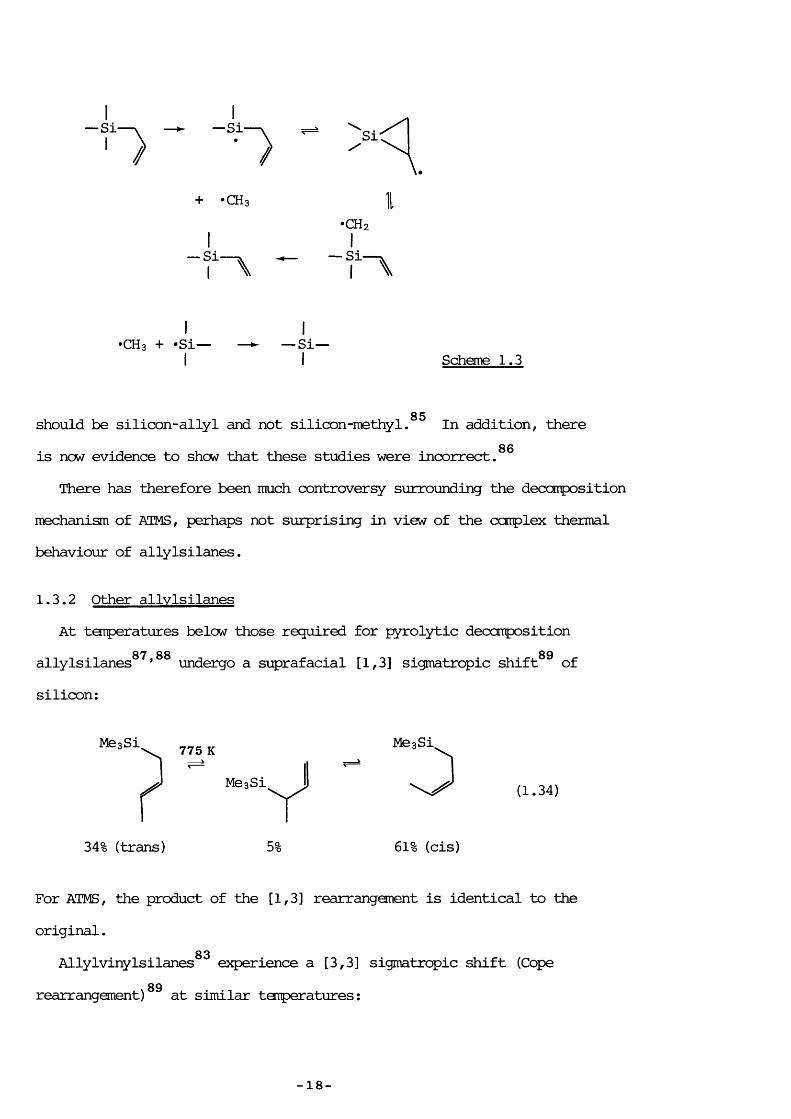
•Si— \ — ► — Si-/ ■ /
Si:
\.+ -CHs I
•CH2
~ f “ \ ^ ~ f " \
I I"CH 3 + "Si— — — Si—I I Scheme 1.3
should be silicon-allyl and not silicon-methyl. In addition, there86is now evidence to show that these studies were incorrect.
There has therefore been much controversy surrounding the décompositionmechanism of AIMS, perhaps not surprising in view of the catplex thermalbehaviour of allylsilanes.
1.3.2 Other allylsilanesAt toiperatures below those required for pyrolytic decarposition
8T 88 89allylsilanes ' undergo a suprafacial [1,3] sigmatropic shift ofsilicon:
775 K WesSi
(1.34)
34% (trans) 61% (cis)
For ATMS, the product of the [1,3] rearrangement is identical to the original.
83Allylvinylsilanes experience a [3,3] sigmatropic shift (Cope 89rearrangement) at similar temperatures:
MeaSi
- 18 -
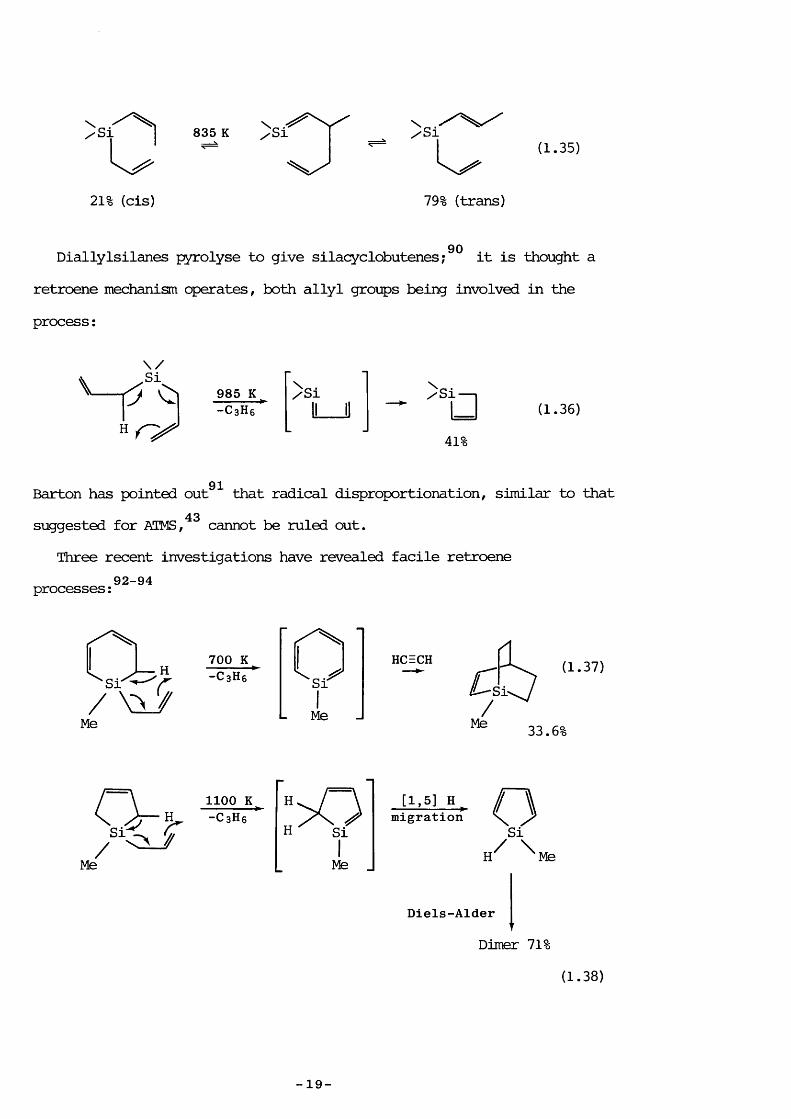
)si 835 K(1.35)
21% (cis) 79% (trans)
90Diallylsilanes pyrolyse to give silacyclobutenes; it is thought aretroene mechanism operates, both allyl groups being involved in the process :
\H
\ /Si985 K -C3H6
/Si1! I)
/Si□ (1.36)
91Barton has pointed out that radical disproportionation, similar to thatsuggested for ATMS,^^ cannot be ruled out.
Three recent investigations have revealed facile retroene 92-94processes:
/700 K -C3H6 sr
Me
HCECH (1.37)St
Me 33.6%
SiMe
1100-C3H6
SiIMe
[1,5]migration
HSi / \ Me
Diels-Alder
Dimer 71-(1.38)
— 19 —
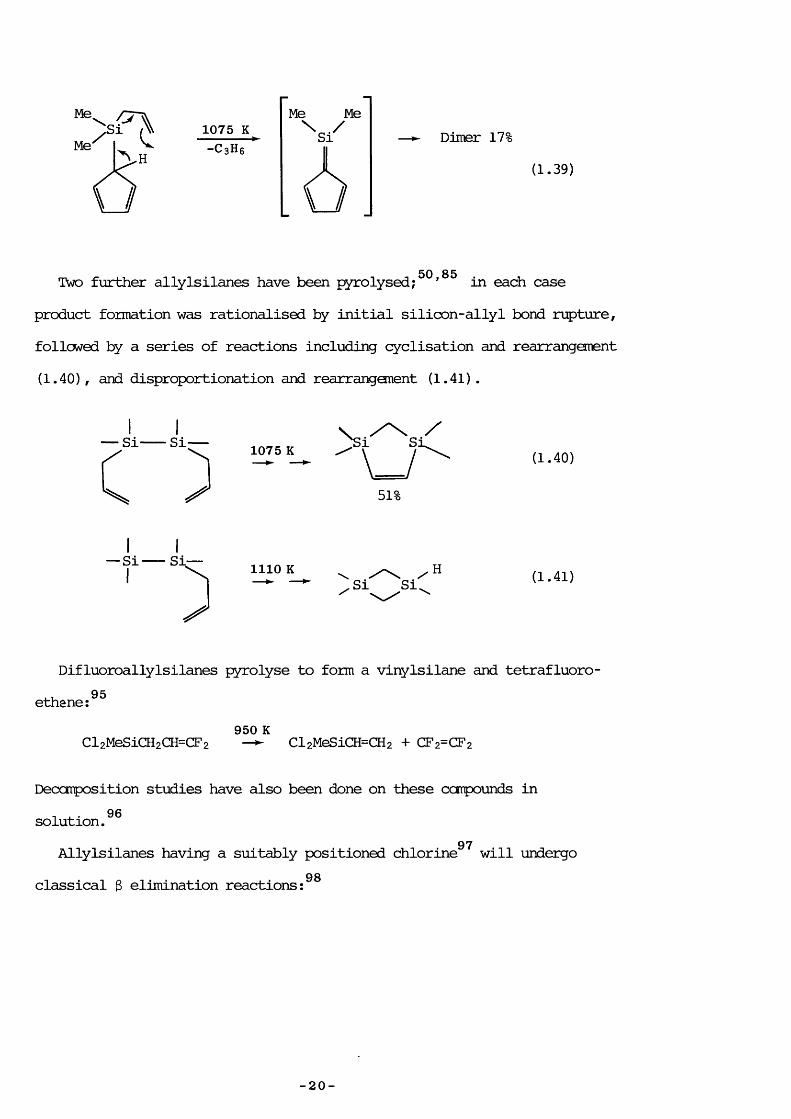
Me1075 K -C 3H6
\ //
Dimer 17‘(1.39)
Two further allylsilanes have been pyrolysed;^^’ ^ in each case product formation was rationalised by initial silicon-allyl bond rupture, followed by a series of reactions including cyclisation and rearrangement (1.40), and disproportionation and rearranganent (1.41) .
— Si Si- 1075 K )i Siw
51^
(1.40)
— Si Si- 1110 K HSi Si. (1.41)
DifluoroallyIsilanes pyrolyse to form a vinylsilane and tetrafluoro- 95ethene:
950 KCl2MeSiŒ2CH=CF2 — ^ Cl2MeSiCH=Œ2 + CF2=CF2
Deccmposition studies have also been done on these carpounds inI X . - 96solution.
97Allylsilanes having a suitably positioned chlorine will undergoclassical 3 elimination reactions: 98
— 2 0 —

RiRzSi „ — RiRzSi + CH2=CHMe
^ M e Cl
In addition a further decarposition pathway exists:Cl
A
(1.42)
Me(1.43)
73
A mechanism involving a transient silicon anion was proposed.The pyrolyses of ATMS and two related carpounds were studied by the author.
1.3.3 SilirenesAlthough the general chemistry of silirenes has been widely studied,
investigations of their thermal properties are much rarer. Whilst most siliranes extrude silylenes at relatively lew temperatures silirenesare thermally more stable (and usually give different products) . As ring strain in silirenes should be greater this is surprising; it can, in part, be rationalised by the stabilizing effect of pTr-dir overlap which leads to a small degree of aromatic character.
69Solution thennal deccmposition studies have been made by Seyferth(1.44), which suggested that sane silylene extrusion could have occurred,
70and Caspar (1.45) :
\ = C ^ ' Benzene Lin. % of\ / , ^ product ISSi t 2~60 hrs. MeaSiC^CSiMea
M e ^ ^ M e (1.44)
— 2 1 —
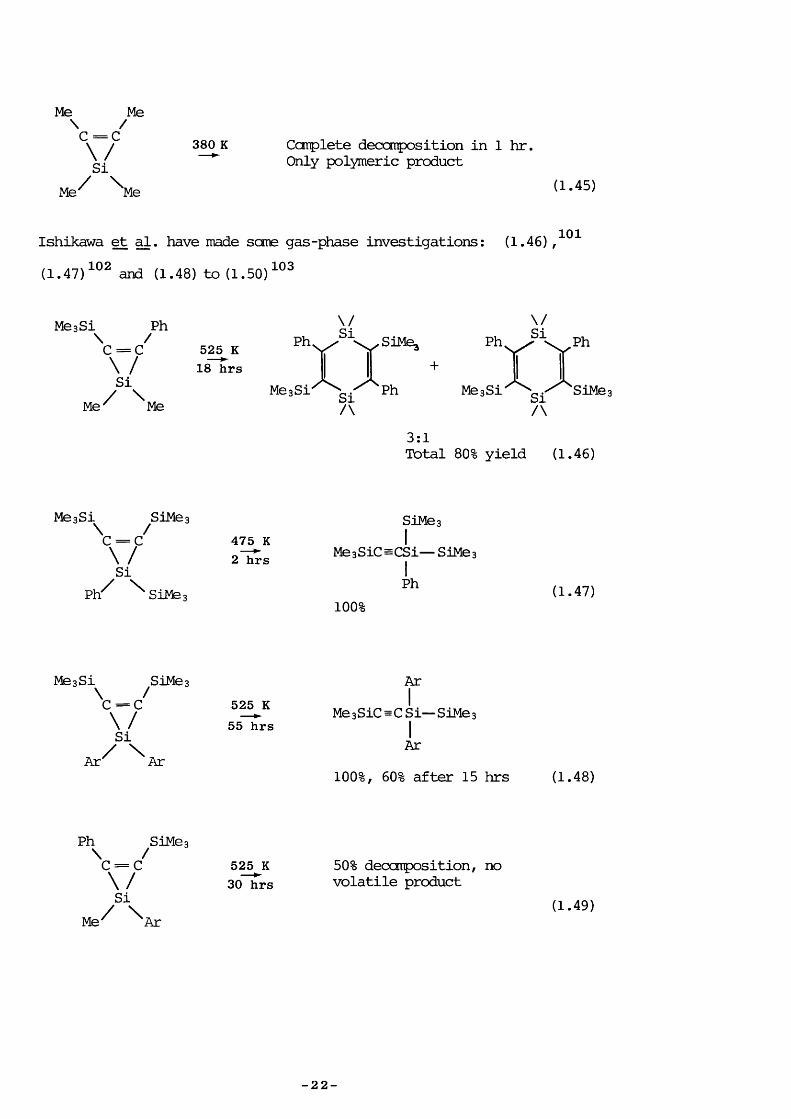
Me Me\ /C = C\ / 380 K Complété deccmpos it ion in 1 hr.g / Only polymeric product
Me^ '^Me (1-45)
Ishikawa et al. have made sane gas-phase investigations : (1.46)(1.47)^°^ and (1.48) to (1.50)^°^
525 K PhMe 3Si Ph
\ /Si 18 hrs +
\/SiA r^ Ar
1 1/ \ MesSi^^.-^Ph MesSi^c.-'^SiMeaMe^ Me /\ /\
3:1Total 80% yield (1.46)
MeaSi SiMea SiMea\ / IC = C 475 K I\ / 2 “h?s MeaSiC^CSi— SiMeaSi I
Ph^ '^SiMes (1.47)100%
MeaSi SiMea Ar\ / IC = C 525 K55 hrs MeaSiC=C Si— SiMea
IAr100%, 60% after 15 hrs (1.48)
Ph ^SiMeaC = C 525 K 50% décomposition, no\ / 3o”hrs volatile productSi
Me^ ^ Ar(1.49)
- 22 -
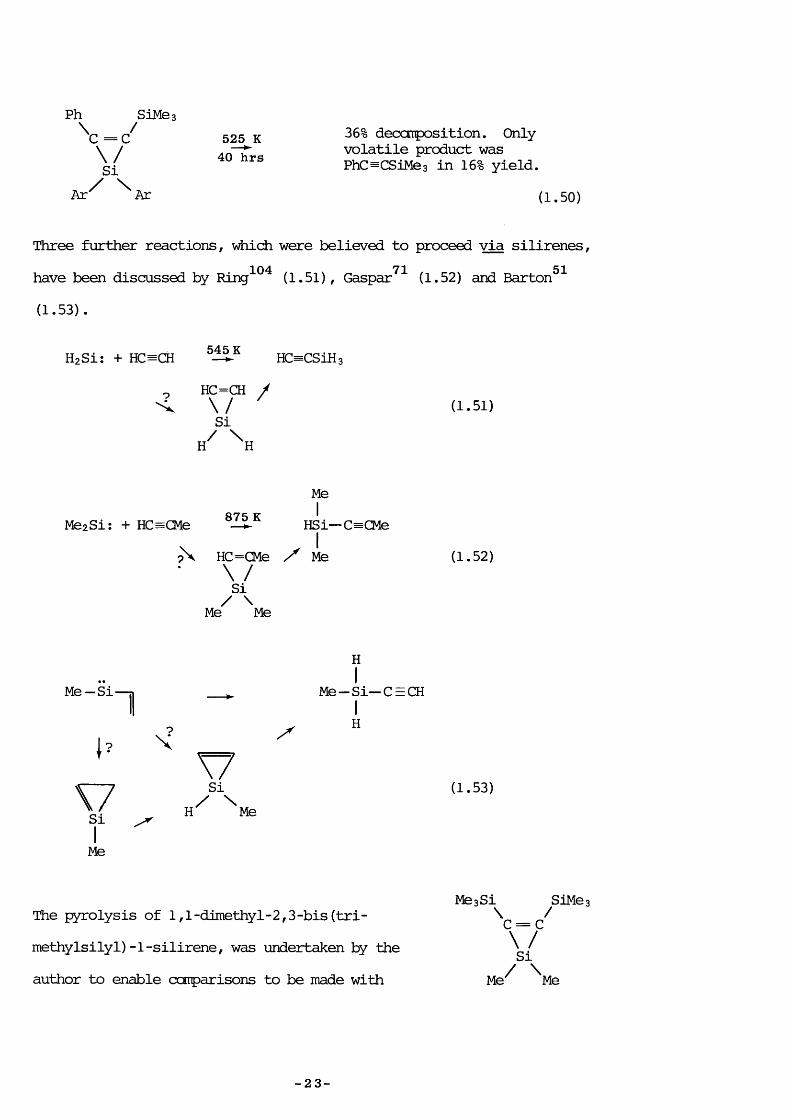
Ph SiMe;\ = Q\/SiA r ^ ^ Ar
525 K 40 hrs
36% deccmposition. Only volatile product was PhC^CSiMea in 16% yield.
(1.50)
Three further reactions, which were believed to proceed via silirenes, have been discussed by Ring^^^ (1.51), Caspar^^ (1.52) and Barton^^ (1.53).
HzSi: + HC=CH HC^CSiHaHC=CH /\/ ^Si
(1.51)
Me2Si: + HC=CMe 875 KMeIHSi— C=CMe
HC=CMe Me* \ /Si / \Me Me
(1.52)
HMe-Si1
SiIMe
SiH Me
Me-Si— CECHIH
(1.53)
The pyrolysis of 1,1-dimethyl-2,3-bis(tri- methylsilyl) -1 -silirene, was undertaken by the author to enable comparisons to be made with
MeaSi SiMea\ = C\/SiMe^ '^Me
- 23 -

these reactions, particularly Seyferth's solution study on the same ccmpound (Reaction 1.44) , and to attorpt to rationalise the differing products.
1.4.1 ThermochemistrySeme experimental techniques which have been successfully used to
measure thermochemical quantities in organic chemistry have been applied to organosilicon carpounds with varying success. The traditional calorimetric methods to find heats of canbustion, and hence heats of formation, have mainly proved unsuccessful owing to the involatility of silica, the formation of solid films over unccmbusted material, and a
4tendency to detonate. However, recently data which is considered to be more reliable^^^ has been acquired, largely by Pedley and co-workers, using hydrogen fluoride along with oxygen in the calorimeter vÆiich overccmes the problons of incarplete ccmbustion by converting the silicon dioxide quantitatively to HzSiFs. These data can be found in the CATCH T a b l e s , t h e most carprehensive collection of experimentally-determined thermochenical data with the best values being arrived at by ccmputer- aided statistical analysis. The CATCH Tables provide a valuable source of information since the heats of formation within are a result of independent experimental measuronents, in contrast to data frcm other channels v^ich are often calculated values reliant on an additivity scheme. Reliable heats of formation have been obtained for Me3Six, where ;x is Cl, Br and OH, largely frcm heats of hydrolysis.
1.4.2 Electron impact measurementsBond dissociation energies are obtainable frcm electron impact
imeasuronents in a mass spectrometer:
Ri — R 2 + e — ► Ri + R2* + 2e (1.54)
— 24 “

The appearance potential is related to the R 1-R2 bond strength and the ionization potential of Ri :
A (Ri") = D (R1-R2) + IP (Ri)
The bond strength therefore cannot be determined directly but depends on information frcm another source; this means that the technique is limited since additional errors can be introduced as well as those arising from experimental difficulties such as mis interpretation of ionization efficiency curves. Early determinations of bond strengths frcm appearance potentials gave several conflicting results * ^8,109 because oferroneous supporting d a t a . T h e s e problems led to the application of a modified version of Allen's bond - interaction schone by Potzinger and L a m p e , by Quane,^^^ and later by Potzinger, Ritter and Krause^(PRK).
Allen's bond-interaction schaneThis schene, for hydrocarbons and sulphanes, was developed by Allen
114in 1959 and is based on the interaction energies of atoms which are next-nearest neighbours; interaction energies involving hydrogen atoms and more distant neighbours are trivial and neglected since this involves unnecessary complications. These energies were calculated frcm thermodynamic data based on the strength of the carbon-carbon bond in ethane vhich has no next-nearest carbon atoms, and a high degree of agreement was found over a wide range of chain and branched hydrocarbons.
4PRK used as their basic information the heats of formation of
115silane (30.5 kJ m o l " ^ u p d a t e d to 34.3 kJ mol”^ ^ ) and disilane(71.5 kJ mol"^^^^’ ^ ) , measured calorimetrically by Gunn and Green,
113and those for several chlorosilanes. These were combined with aseries of equations relating appearance potential measuranents to heats
- 25 -

of formation, etc. For exarrple:
SiH4 + e“ — SiHz" + H2 + 2e" (1.55)AH55 = AP (55) = 11.9 eV
MeSiHs + e" — SiH2' + CH4 + 2e" (1.56)AH56 = AP (56) = 11.5 eV
CH4 + SiHit — ^ H2 + MeSiHa (1.57).*. AH (57) = AP (55) -AP (56) = 0.4eV
The PRK enthalpies were then additivity smoothed by the Allen schene, for example
AH" [Me(CH2)„SiH3] = Ahf (MeSiH3) -9.87 n - r (CCSi) - (n-1) r (CGC)
The 9.87n term arises frcm the -CH2- unit, and Y represents an interaction term between the three atoms in parentheses. Bond dissociation energies determined by PRK i n c l u d e : D ( M e 3Si-H) = 372 ±17,D(Me3Si-Me) = 355 ±17, D(Me3Si-SiMe3) = 313 ± 35 kJ mol"^. Heats of formation for several silanes as calculated by PRK or Quane and those in the CATCH Tables have been compared by Davidson^who applied Benson's electrostatic model for calculating enthalpies of formation.
1.4.3 Benson's electrostatic model117This model for hydrocarbons was developed recently by Benson and
visualizes differences in carbon-carbon bond strengths in different molecules as arising from coulcmbic interactions. For example, in ethane the carbon-hydrogen bond is polarised such that hydrogen is given a formal charge of +y, with each carbon therefore having a charge of -3y. The electrostatic energy, Eei is given by:
Eel = I qi qj / ri r j i<j
— 26 “

vÆiere qi, qj are charges and ri, rj are the distances between them. Assuming that the differences in heats of formation of structural hydrocarbon isaners arise entirely frcm the difference in electrostatic energies, AE@i, y was calculated, and then used to find the contribution of a -CH2- unit to the heat of formation of a hydrocarbon in a hcmologous series. This scheme is therefore similar to Allen's in that there are additive and interactive terms, the latter being calculated by coulcmbic interactions in this model.
Davidson has successfully demonstrated that Benson's electrostatic energy corrected bond additivity scheme (EECBA) could be applied to organosilicon c a r p o u n d s . T h e silicon-hydrogen and silicon-carbon bonds were assumed to be polarised and given formal charges which were estimated frcm dipole mcments, allowing heats of formation of several silanes to be calculated. It was concluded that the AH;^ values with vhich there was best agreenent were those of PRK rather than the ones in the CATCH Tables. One of the data bases used by Davidson was thesilicon-carbon bond strength (355 kJ mol"^) which has since been
118 22measured and estimated to be higher (ça. 370 kJ mol" ) . Using thisnew figure, better agreement is found with the CATCH values than was first thought.
O'Neal and Ring have developed the application of EECBA much furtherand used the heats of formation of silane, disilane, and trisilane
105(Gunn and Green) in their calculations. Heats of formation calculated agreed with the CATCH values much better than with PRK figures, and it was therefore suggested that the most recent calorimetric data is fairly accurate. A group additivity scheme, similar to that constructed by Benson (for carpounds containing hydrogen, carbon, oxygen, nitrogen, sulphur, phosphorus and the halogens) was subsequently created, vÈrich was
- 27 -

thought to be more reliable than the CATCH Tables themselves since any experimental errors are minimized. For example:
AH^ [(CH3) 3SiCH2SiH(CH3)2]= 5 [C- (H) 3 (Si) ] + [Si- (C) 4 ] + [C-(H) 2 (Si) 2 ] + [Si- (C) 3 (H) ]= 5 (-42.7) + (-76.6) + (-40.2) + (-47.3)= 377.6 kJ mol"^
1.4.4 Bell and Perkins' calculationsBell and Perkins have obtained heats of formation for a wide range of
107molecules and radicals, and several bond dissociation energies. Quantum-mechanical calculations at orbital level were performed using spatial co-ordinates evaluated frcm bond lengths and bond angles in the literature (called the MDBI method) . The bond indices calculated were used in conjunction with selected literature heats of formation to derive a bonding parameter for each type of atcm-pair bond. For exarrple, the molecule Si2H6 is considered as (H )3 Sia - Sib(Hb)3 ; the Sia - Ha bonding parameter is different frcm Sia - %. Using the CATCH Tables' value for AHf SiHit (+34.7 kJ mol"^) and Steele's figure for AHf SiMeit (-227.6 kJ mol"^ the heats of formation of several ccmpounds were calculated.The work of Bell and Perkins is, however, slightly controversial sincethe primary data were arbitrarily chosen.
Kinetic methods :1.4.5 Gas-phase iodine atcm reactions
This technique was developed by Benson in 1961; its subsequentextensive application to organic molecules has yielded extremely reliable
119carbon-hydrogen bond dissociation energies. More recently the provendependability of this method has been utilized to measure silicon-hydrogen
22bond strengths in a series of organosilicon ccmpounds. In its general
— 28 “

form, the technique involves the spectrophotanetric measurement of the kinetics of the gas-phase reaction between iodine and a hydrogen-containing species, XH:
I2 + XH — XI + HI (1.58)
In the majority of cases the mechanism can be described as an iodine atcm propagated chain reaction (Schene 1.4).
I2 + M ^ 21* + M K
I« + XH 4^ X' + HI
X* + I2 i XI + I'
Schene 1.4
The reactions of I2 with silanes are mechanistically and kinetically simpler than with hydrocarbons. When X is a carbon-centred radical step 4 is inportant, vhich leads to the iodination reaction being highly reversible (the strength of the silicon-iodine bond prevents this vhen X is silicon-centred) ; in addition the iodide XI is often unstable, undergoing the elimination reaction:
XI — ► alkene + HI (1.59)
(the weakness of the silicon-carbon tt bond makes this an unfavourable process).
In the early stages when reaction 4 is insignificant, the applicable rate equation - vÆiich can be further modified, if required, to allow for step 2 carpeting with step 3 - is:
-_d [I2] = ki [l2l^ [XH]dt
The experimental results are fitted to an integrated form of this expression to give ki Kj^ which allows ki to be determined (frcm the
— 2 9 —

known value of Ki^^) ; its tanperature dependence yields the activation energy, Ei. Since Ez is always very small and can be estimated with reasonable accuracy, Ei provides a good approximation of AH^z (= E 1-E2 ) ; adjustment to rocm temperature by standard thermodynamic formulae is also a minor correction. This enthalpy change allows D(X-H) to be determined because D(H-I) is known (298.49 ±0.21 kJ mol"M and
AHÎ2 = D(X-H) -D(H-I) .
Walsh has successfully used this technique to measure seme silicon- hydrogen bond strengths for seme of vAiich there has been a wide disparity of values, ,e.g, D(MegSi-H) = 378 ± 6 kJ mol“\^^^’ ^^ Re-interpretation^ of earlier data which gave a lower value (339 kJ mol"^ ) based on a determination of the activation energy for hydrogen formation in trimethylsilane pyrolysis, showed that the kinetics of the complex process was more consistent with a higher bond strength, so this supports Walsh's measurements.
A useful spin-off of bond strength determinations is the ability tocalculate radical heats of formation using the thermodynamic relation-, . 22ship:
AH^(R«) = AHf (RH) -AH^(H«) + D(R-H)
This can be used to generate other bond dissociation energies :D(R-X) = AH^(R-) + AH^(X«) - AH^ (RX)
These calculations are limited because additional information is required: AH]^ (H*) and AH^ (X*) values are available, but the familiar problem of unreliable AH^ (RH) and AH^ (RX) presents itself. An indication of the controversy surrounding current data is obtained frcm two recent publications by Davidson^and Walsh; AH^ (Mei+Si) is a key quantity since other heats of formation are derived frcm it.
— 3 0 —

1.4.6 Pyrolysis techniquesMeasurorient of the rate of dissociation of molecules into radicals is
useful since the energetics of radical formation may be deduced frcm the kinetic data. This can lead to the determination of bond strengths and heats of formation:
Ri R2 — ► Ri • + R2 • (1.60)AH = D(Ri"R2)
= AH^(Ri-) + AH^ (R2*) - AH^ (Ri R2 )
If the reverse reaction, i.e. that of radical reccmbination, is assumed to have zero activation energy, then the measured activation energy can be identified with the bond strength. This means that bond strengths can be determined directly and are independent of ancillary data. It is usually difficult to measure the rate of thermal dissociation of hydrocarbons and sinple organic molecules as the pyrolysis of these ccmpounds generally proceeds by a radical chain mechanism which is kinetically ccmplex: Rice-Herzfeld type mechanisms (Scheme 1.5) are often adopted;the alkene products often then change the course of the reaction:
C2H6 — ► 2CH3' initiation•CH3 + C2H6 — ► CHit + C2H5' transfer
C2H 5 ' — ► C2H1+ + H" propagationH* + C2H6 — ► H2 + C2H5 propagation
Schene 1.5
The chain is terminated by various radical reccmbination or disproportionation reactions. For the determination of the bond strength it is necessary to inhibit this chain; this is often done by performing the pyrolysis in an excess of a canpound containing a weakly-bound hydrogen.
"31 —

a) Very Icw-pressure pyrolysisThese problems have been largely circumvented in a flow technique
called very low-pressure pyrolysis (VLPP). This versatile technique iscapable of giving direct quantitative information on the rate ofelementary gas-phase chemical processes and, in particular, yieldinginformation on the nature of primary products of unimolecular pyrolytic
123 124reactions. ’ The principle behind VLPP is that a gas at very lewpressure (usually <10"^ Torr) is allowed to flow into a thermostattedreaction cell where it deccmposes molecularly since most collisions ofreactant molecules are with the walls of the reaction vessel and not inthe gas phase. Wall catalysis is relatively unimportant becauseheterogeneous reactions are usually slow, and secondary reactions areminimized since they are generally bimolecular. Products and undeccmposedreactant then flew molecularly (at large mean free paths) through arelatively large exit hole (e.g. 2-10 mm) into the ionization chamber ofa quadrupole mass spectremeter where they are analysed directly understeady-state conditions, enabling rate constants of deccmposition to bedetermined. Because of the lew pressures involved, most molecules are
125studied in their unimolecular fall-off region and the apparent unimolecular deccmposition rate constant, k, is well below the high- pressure limit rate constant, kœ, as energy transfer is rate controlling. The relationship between k and k^ is given by the PRK (M) theory and if the value of the Arrhenius A factor is known (and, in general, it can be estimated with considerable accuracy for an unambiguous unimolecular process) , the value of the activation energy may be determined frcm a fit of k vs. T to a cemputed curve.
Most of the problems associated with the investigation of pyrolyses of hydrocarbons and sirtple organic molecules are absent for silicon-
- 32 -

containing species; two factors are chiefly responsible: silicon-centred radicals are less likely to undergo hydrogen metathesis reactions
22cwing to the silicon-hydrogen bond being weaker than carbon-hydrogen,15and silicon only forms weak p-rr-pw bonds to other atans and itself so
that reaction schanes resembling the Rize-Herzfeld mechanism are much less likely to be adopted. This has promoted ideas that VLPP should be a technique of increased expedience in organosilicon chanistry. Unfortunately, these expectations have not been realised : a VLPP study of the
19thermolysis of DMSCB, for example, revealed ccmplex pyrolyticbehaviour which departed frcm the theoretical RRK calculations ; thiswas caused by significant heterogeneous reactions (even more important
126for ccmpounds containing silicon-hydrogen bonds) ; this also led to the ccmplete loss of the reactive silicon-containing pyrolysis product,SE, to the walls of the reaction vessel.
b) Lcw-pressure pvrolvsis126 127A technique has recently been devised in our laboratory ’ which
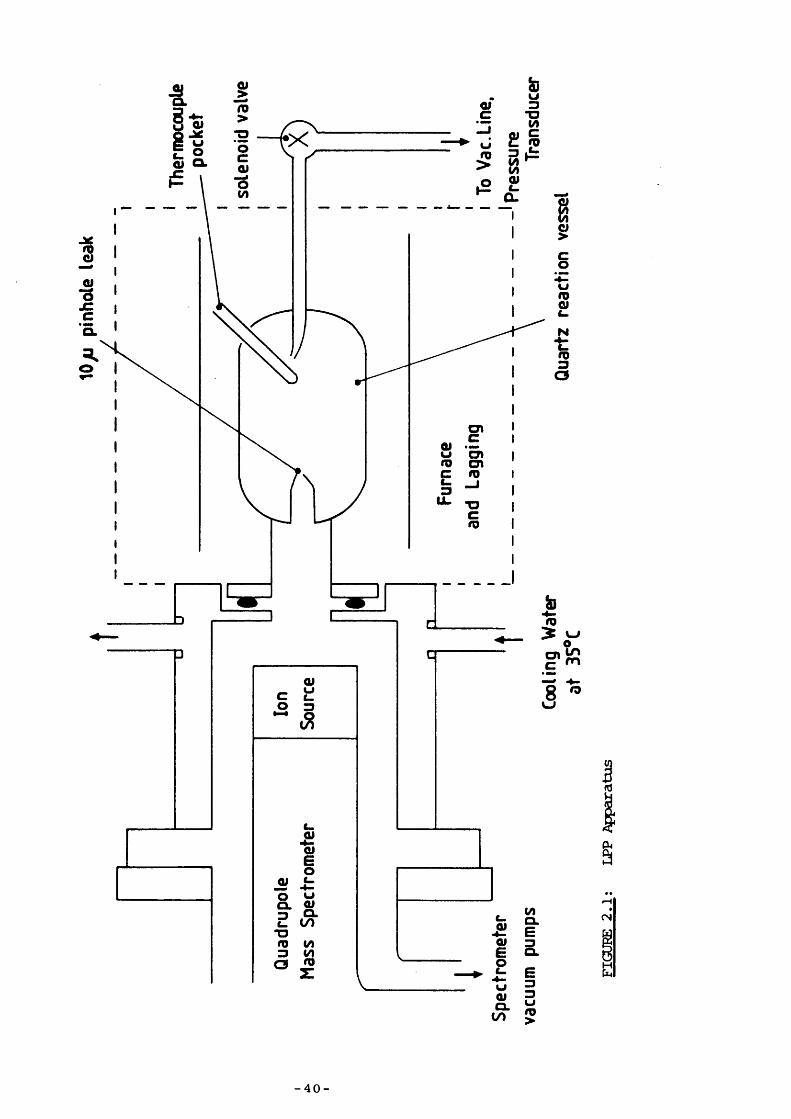
eliminates these problans and ccmbines the advantages of the determination of bond dissociation energies by pyrolytic methods with those involved in the thermolysis of organosilicon ccmpounds rather than hydrocarbons and organic molecules. This technique, called low-pressure pyrolysis (LPP) , has profited since it has been specifically designed to study thermolyses of organosilicon ccmpounds ; it has consequently developed into a powerful technique.
LPP is scmewhat similar to VLPP but there are two major differences : it is a static rather than a flow system; higher gas pressures are used - between 5x10"^ and 10"^ Torr, usually ça. 0.2 Torr. Pyrolyses are carried out in a quartz reaction vessel housed in an electrically heated furnace which is connected via a 10 y m pinhole leak to the ion
- 33 -

source of a fast-scanning quadrupole mass spectremeter to allow theprogress of the reaction to be monitored continually. These spectro-
128—130meters, developed in 1955 by Paul and co-workers, ” are much more useful here than the conventional magnetic deflection type. They work on the same principle except that the deflecting field for mass analysis is produced by four accurately machined and critically aligned cylindrical stainless steel rods. Opposite pairs are connected electrically; a ccmbination of superimposed RF and DC potentials of opposite polarii^ for each pair is applied, the resulting magnetic field being hyperbolic in nature. Ions produced in the ion source are injected along the longitudinal axis and are selected according to their m/e values, either passing straight through to be detected or colliding with the quadrupole rods. Advantages over conventional mass spectraneters include their ccmpactness and hence convenience, their fast response time, and their linear mass spectrum on the oscilloscope screen. Loss of material frcm the reaction vessel through the pinhole leak is small, so the apparatus functions essentially as a static rather than a flow system.
LPP's higher operating pressures (ccmpared with VLPP) mean that bulk collisions are more iirportant and the mean free path is reduced, whilst still operating in the region where pyrolysis mechanisms are dominated by unimolecular processes. Because wall reactions are less important (most radical abstraction processes take place at the walls however) , the silicon-containing products can be detected as they are rarely ccmpletely lost as in VLPP; elucidating the deccmposition mechanism is therefore easier. In addition, kinetic analysis in LPP is simpler as the deccmposition of most molecules can be investigated in their high pressure unimolecular decay region, i.e. above the fall-off transition pressure.
All theories, e.g. Lindenann-Hinshelwood, RRK(M) , Slater, predict that
- 34 -

the pressure for a given degree of fall-off depends on the number ofvibrational modes in the molecule. A plot of log (the pressure atvhich the experimental rate constant drops to half its high pressurevalue) against the number of atans in the molecule for several carpounds
131is approximately a straight line. This can be used to predict therange of carpounds capable of being investigated above their unimolecularfall-off region in. LPP: as a "rule of thumb" , if a molecule containsmore than 15 atans, then it should be in its high-pressure region attypical LPP operating pressures.
For exanple, the pyrolysis of methylsilane has recently been studied126by the LPP technique in its fall-off region, and was found to proceed
mainly by formation of hydrogen with a small amount of methane. These products resulted frcm two initial decarposition pathways, the other products being silylenes:
MeSiHs — ► HMeSi: + Hz (1.61)MeSiHa — ► HzSi: + CH^ (1.62)
c) Pulsed stirred-flew techniqueThis gas kinetic pyrolysis technique for organosilicon carpounds was
132 133developed recently in our laboratory. ’ The apparatus contains aquartz stirred-flow reactor, into which a pulse of reactant vapour in a carrier gas is injected, where it partially deccmposes before being carried into a gas chranatograph for analysis. The amount of deccmposition, and products formed, can be determined and the data treated kinetically. This systen has advantages over conventional flow techniquesin that it is much simpler and is relatively econonical in reactant. Inaddition, the risk of oxidation of ccmpounds is minimized since thesystem is run at a pressure above atmospheric. The chain-inhibited
- 35 -

133pyrolysis of hexamethyldisilane has been studied by this technique, allowing DdyieaSi-SiMes) to be determined as 337 kJ mol"S a figure which is thought to be reliable.
To summarize, there are seme reasonably reliable bond strengths,22e.g. MeaSi-SiMes, Me3Si-H, Me3Si-Me, in the literature (Walsh's review
contains the most recent measured and calculated figures); the most accurate heats of formation are obtained frcm the CATCH T a b l e s o r O'Neal and Ring's group additivity scheme.
— 36 “

CHAPTER 2APPARATUS AND EXPERIMENTAL PROCEDURE

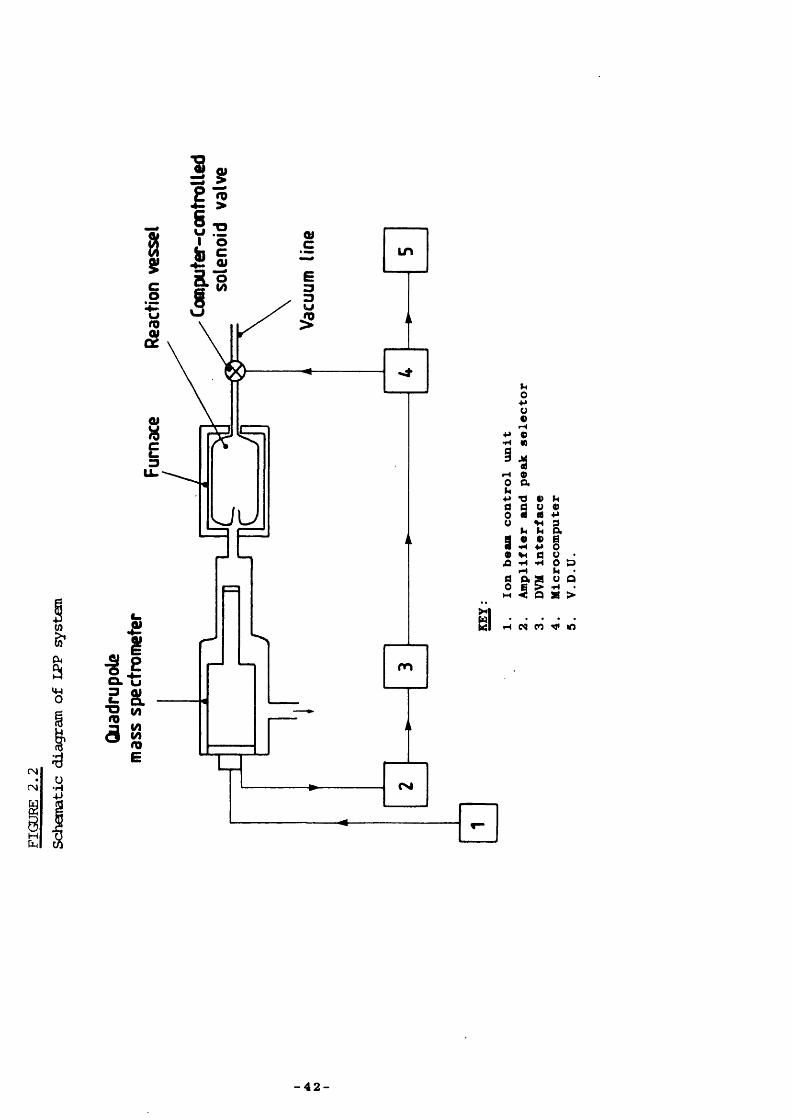
APPARATUS
The bulk of the experimental work, and all of the kinetic runs were126 127done using low-pressure pyrolysis (LPP) , ’ described in part in
Chapter 1. Whilst the overall systen underwent several modificationsduring the period of research covered in this thesis, the principal
129features renained unaltered, and can be divided into three sections :(i) Vacuum line, reaction vessel and furnace.(ii) Quadrupole mass spectremeter analyser.(iii) Computer for data processing.
2.1 VACUUM LINE, REACTION VESSEL AND FURNACE
The vacuum line, of conventional design, was fitted throughout with Young's greaseless stopcocks. Several ports were available for loading detachable sanple containers, using teflon collars, either standard 1 dm^ gas bulbs or 0.1 dm^ pear-shaped vessels for liquids. In addition to an entrance to the reaction vessel, the vacuum line was constructed so that vapour could be allowed directly into the ion source of the spectremeter via a metrosil leak, by-passing the reaction vessel.
Vacuum line pressure at the pumps was monitored using a Pirani gauge. Sanple pressures were measured using a Bell and Howell pressure transducer viiose resistance elements were wired into a Wheatstone bridge circuit; changes in pressure caused a deflection on a Kipp and Zonen Micrograph BD5 chart recorder. The transducer head was suitably positioned in a small region of line approximately equal in volume and adjacent to the reaction vessel. Connection was made via a solenoid
129valve - a later addition to the system, replacing a Young's stopcock viiich could be operated manually or by the computer, as shown later. The cylindrical quartz reaction vessel was of approximate volume 30 cm^ with
— 38 “

<4% dead space; temperatures were measured using a chrcmel-alumel thermocouple inserted into a small pocket. The furnace consisted of a cylindrical steel tube 10.5 cm in length and 5 cm in diameter around viiich was wrapped the heater winding (Kanthal A wire) in four parts with variable resistances in parallel with each section. This allowed the furnace temperature profile to be adjusted to give an approximately constant value over the length of the reaction vessel. Applied to the heater winding was a layer of fire-proof clay housed in a vessel constructed frcm asbestos board and aluminium. Heat losses were minimized at the open end of the furnace by asbestos packing. To maintain a vacuum at the reaction vessel/spectrcmeter interface a Viton rubber 'O' ring was tightened onto the quartz flange of the reaction vessel, and a flange and copper gasket were used at the spectrometer end. Cooling water at 35°C was circulated around this interface. These features are shown in Figure 2.1.
2.2 MASS SPECTROMETER
The quadrupole mass spectrometer used in our LPP apparatus is a VG Micrcmass Q801 K. Evacuation is achieved by an Edwards EDM2 direct drive rotary pump and an air-cooled Edwards E02 silicone oil diffusion pump. Situated between the two is a foreline trap containing molecular sieve.
The Q801 K can be operated in two ways :-( i ) As a conventional mass spectrometer, scanning the mass range
m/e 1'*' to m/e 300'*’.(ii) As a peak selector in kinetic experiments.
The oscilloscope (VG model 132) displays both types of output ; hard copy spectra are obtainable on light-sensitive paper by using the ultraviolet chart recorder. An 8-channel peak selector - replacing the original 4-
- 39 -
u
w5 a.
L_
W
Q.
O
cnw oi ro cn
LL
uW
L.
UCO
Ou
- 40 -

129channel - allows up to 8 different m/e values to be monitored during a kinetic run; usually peaks are duplicated on more than one channel.Peak heights, displayed as voltages on a Newport DVM, can be recorded at regular time intervals, depending on the cycle time used, viiich varies frcm Is to 30s. For example, for a 3s scan time readings are taken at time intervals of |s for each channel in turn; the time between successive readings for each channel is therefore 3s. The QBOl K is usually operated at an electron energy of 40 eV, lower than typical for mass spectrcmetry, because this enhances the size of the molecule ion peak (vÆiilst maintaining reasonable sensitivity), allowing easier kinetic measuraments: during kinetic runs, fragment ions vÈiich arecharacteristic of reactant and products are chosen to monitor pyrolysis; the molecule ion is favoured for the reactant.
2.3 COMPUTER
Seme kinetic runs were done using our old computing procedure:voltages were recorded on punched paper tape; these data were then
129analysed on the Leicester University CYBER 73 mainframe computer. Our present syston has been updated with a Research Machines 380Z microcomputer (56 K, having two disc-drives for double-sided floppy discs, high-resolution graphics, a real time clock, and using the BASIC programming language) . A machine code program has been written, by C. E. Dean, Leicester University, vÆiich collects peak height data via the DVM interface (and time using the 380Z clock) and opens the solenoid valve automatically to start a kinetic run. These features are shown in Figure 2 .2.
- 41 -
u
w
*»
c.u.
r4
<M
m
«> k O O « #
H> - H .Q a >
— 42 ”

2.4 A KINETIC RUN
A sample of reactant vapour is expanded into the region of line adjacent to the reaction vessel. The data collecting program is run, and the Q801 K peak selector cycling started. Four cycles are returned - these numbers constituting baseline readings which are averaged and subtracted from subsequent values during data processing - before the valve is automatically opened; it shuts again after a chosen time interval of 2s, determined by a separate control unit. A sample of vapour has now flowed into the reaction vessel and data points are collected until a suitable amount of decarposition has occurred; the Q801 K cycling is then switched off. The DVM interrupt button is pressed, this being the signal to the computer that data collecting is carplete; the software then instructs the 380Z to record the data on a floppy disc. The solenoid valve is opened manually to pump out the contents of the reaction vessel after v^ich another kinetic run can be performed.
2.5 DATA PROCESSING
Allowing for the leaking into (from the vacuum line) and leaking outof (through the pinhole leak) the reaction vessel, the processes occurringcan be described by the simple scheme:
ki kA — ► B — ► D
kobs = k + kL:L
A = vapour in line outside the reaction vesselB = vapour inside reaction vessel D = productki, k|_ = leak-in, leak-out rate constants ; both approximate
to first order
— 43 “

k = decomposition rate constant kobs - observed decay rate constant
Typical values of ki and kL are 1.5 s“ and 0.0005 s” and under normal conditions ki»k»k|_ and kgbs = k . In practice k[ is measured at temperatures below those required for pyrolysis and subtracted from kobs to obtain k, but this is usually only a minor correction. The lower limit of rate constant measurement is therefore dependent on the value of kL ; the highest decomposition rate constant that can be measured with confidence is ~0.3 s“ .
A BASIC program allows the data to be analysed by use of the high- resolution graphics on a VDU; individual users normally make small changes in the program to suit their own requirements. A In peak height against time plot, linear for first order decomposition, gives a least squares fitted slope - the best straight line through the points is chosen by the user - equal to k^bg» Other software routines have been written for different types of analysis, e.g. to measure the initial slope of a product formation curve and initial reactant concentration to determine the rate constant of formation, kf. The additional information of the relative sensitivity of the mass spectrcmeter to the m/e values in the authentic materials is also required in this case. Other routines include: a cracking pattern correction procedure (this allcws fragmentation contributions of a compound to more than one of the peaks monitored - determined using the authentic material) , and an Arrhenius plot frcm rate data. Full details of computer programs, etc., can be found in the Ph.D. theses of two members of our research g r o u p . *
2.6 CLEANING THE Q 801 K
About three times a year it is necessary to clean certain parts of the * Appendix 2.1 shows a typical data file and some plots.
- 44 -

Q 801 K analyser: the source plate, trap and shield acquire an electronb u m cover and the rods become dirty because ions not selected for analysis during deflection are lost there. The source must be completely dismantled before cleaning proceeds. The ceramics are boiled for 15 minutes in aqua regia to remove the metallic layers that are burnt on (if this fails this is followed by boiling in caustic soda) ; the nuts and washers are rubbed with a mild jewellers' rouge paper. All these parts are then boiled for 15 minutes in both isopropyl alcohol and acetone; the nuts and washers now undergo cleaning in an ultrasonic bath for 10 minutes in three successive solvents : i) Triklone, ii) distilledwater (followed by drying with a hot air blower), iii) Treble-One Chemiclene. The source, focus and earth plates, trap and shield, are rubbed with the jewellers' rouge; similar treatment on the rods would be too harsh so, instead, a diamond paste/glycerin mixture is used Wiich is then washed off with water ; all these parts now undergo ultrasonic cleaning alongside the nuts and washers.
2.7 PULSED STIRRED-FLCW TECHNIQUE132 133This technique, ' described in part in Chapter 1, was used to
supplement the LPP experiments: G.C. analysis indicated the number ofpyrolysis products and aided identification - particularly compounds with lew molecular weight - but was not used for any kinetic measurements
Reactants were stored on a greaseless vacuum line fitted with a pressure transducer vdiich enabled the pressure of vapour in the sampling volume of the loop (approximately 2 crn ) to be measured. The tap to the loop had two positions: "Fill" and "Inject". In the Fill position theloop was opened to the vacuum line; changing to Inject redirected the regulated stream of nitrogen carrier gas - which had been passed through
— 4 5 —

molecular sieve and an "Qxy Trap" - to sweep out the sample in the loop into the reaction vessel.
The design of the reactor (shown in Figure 2.3) allows rapid sample dispersion; the residence time is determined by the carrier gas flew rate. Reactants and products are carried into the gas chranatograph for analysis (Pye Unicam GCD Torperature Prograrrmable Chranatograph with a Flame Ionization Detector) ; separation was achi.eved using a 2.7m glass (4 mm i.d.) column packed with 10% ESDI on 100/120 mesh acid-washed Diatanite C treated with dimethyldichlorosilane, v^ch separated most catpounds well at 50°C, apart fron a few lew boiling point species, e.g. methane and ethene.
pyrex
graded seal
quartz
approx. vol. 9cm®
Carrier gas flow
thermocouple pocket
perforated sphere ~ 1 5 holes
FIGURE 2.3Reaction vessel in pulsed stirred-flew apparatus
— 46 —

CHAPTER 3PYROLYSIS OF 1,1 -DIMETHYL-2,3-BIS (TRIMETHYLSILYL) -1 -SILIRENE
nsiTRœucriON
The pyrolysis of silirenes can be carpared with the thermolysis of other three-membered cyclic corpounds.
3.1 CYCLOPROPENES
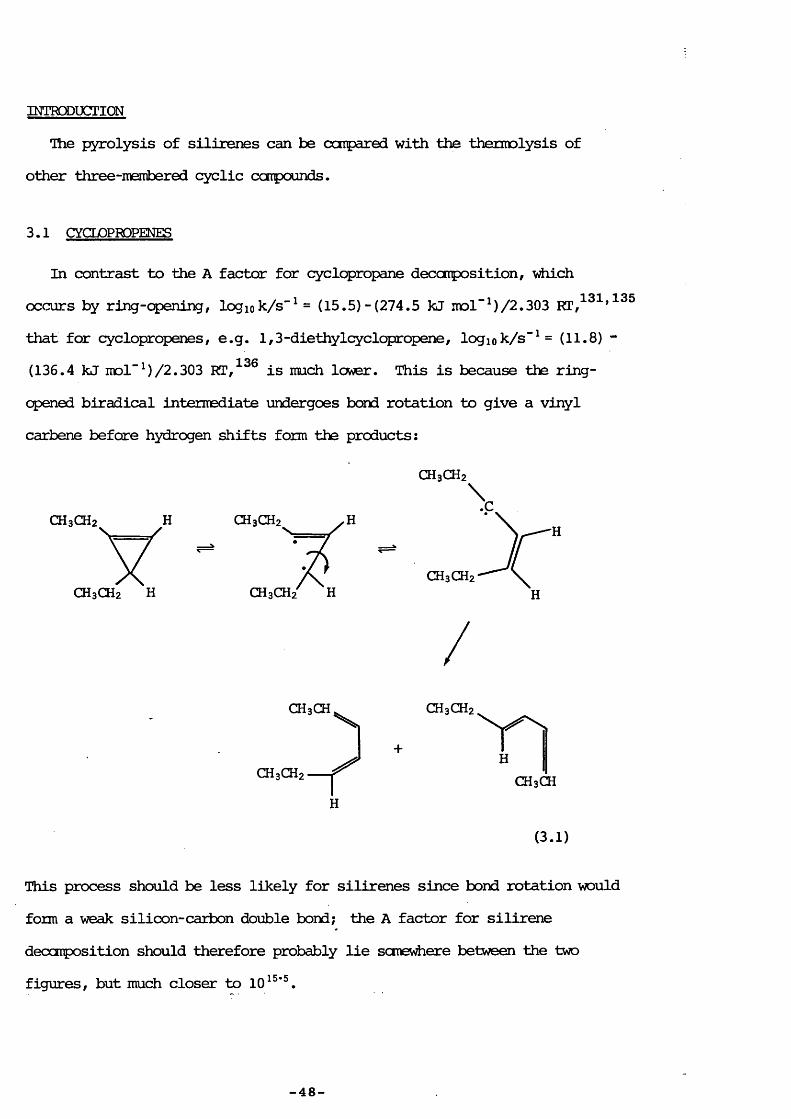
In contrast to the A factor for cyclopropane deccmposition, \^ch occurs by ring-opening, logiok/s"^ = (15.5) -(274.5 kJ mol"^)/2.303 that for cyclopropenes, e.g. 1,3-diethylcyclppropene, logiok/s"^= (11.8) - (136.4 kJ mol"M/2.303 is much lower. This is because ths ring-opened biradical intermediate undergoes bond rotation to give a vinyl carbene before hydrogen shifts form the products:
CH3CH2
CH3CH2
CH3CH2 H
CH3CH2
CH3CH2 H
H
CH3CH2
HCH3CH
(3.1)
This process should be less likely for silirenes since bond rotation would form a weak silicon-carbon double bond; the A factor for silirene decarposition should therefore probably lie sonevAere between the two figures, but much closer to 10^®’.
— 4 8 —

3.2 MECHANISM OF SILIRANE AND SILIRENE PYROLYSIS
Whereas the ground state for carbenes is usually a triplet, forsilylenes a singlet state is the more stable.
Carbene addition to olefins to give cyclopropanes is therefore normallya step-wise process via a biradical intermediate which ring closes afterspin inversion, but vhen the singlet state is of lower energy, for examplev\hen stabilized by a substituent containing a lone pair, e.g. :CClz,stereospecific addition occurs, probably by a pericyclic chelotropicreaction. Similar stereospecific addition behaviour has been demonstrated
137for the reaction of dimethylsilylene with but-2-ene. Orbital syirmetryrules require the adding species to have a lone pair (with a vacant porbital orthogonal to it) such as that found in singlet carbenes or
89molecules like sulphur dioxide. Consequently sane reactions Wiich areequivalent to the reverse process, e.g. sulphur dioxide extrusion frcm
■ n i_ 138episulphones:
HMeC -CMeH\ / — ^ HMeC =CMeH + SO2 (3.2)
SO 2
89are also stereospecific although the exact mechanism is unclear.Since the lowest energy form for most silylenes is a singlet, there
therefore arises the possibility that the decomposition to ground state silylenes of siliranes - or silirenes, should these be pyrolysis products - that a concerted mechanism operates, i.e. that a biradical is not necessarily formed in the course of the reaction. Results seen to be in disagreement with this possibility: heating hexamethylsilirane in thepresence of styrenes - vhich have a high reactivity towards radicals - did not give the expected siliranes which would result frcm reaction with the extruded silylene, but silacyclopentanes, indicating probable intermediate
- 49 -
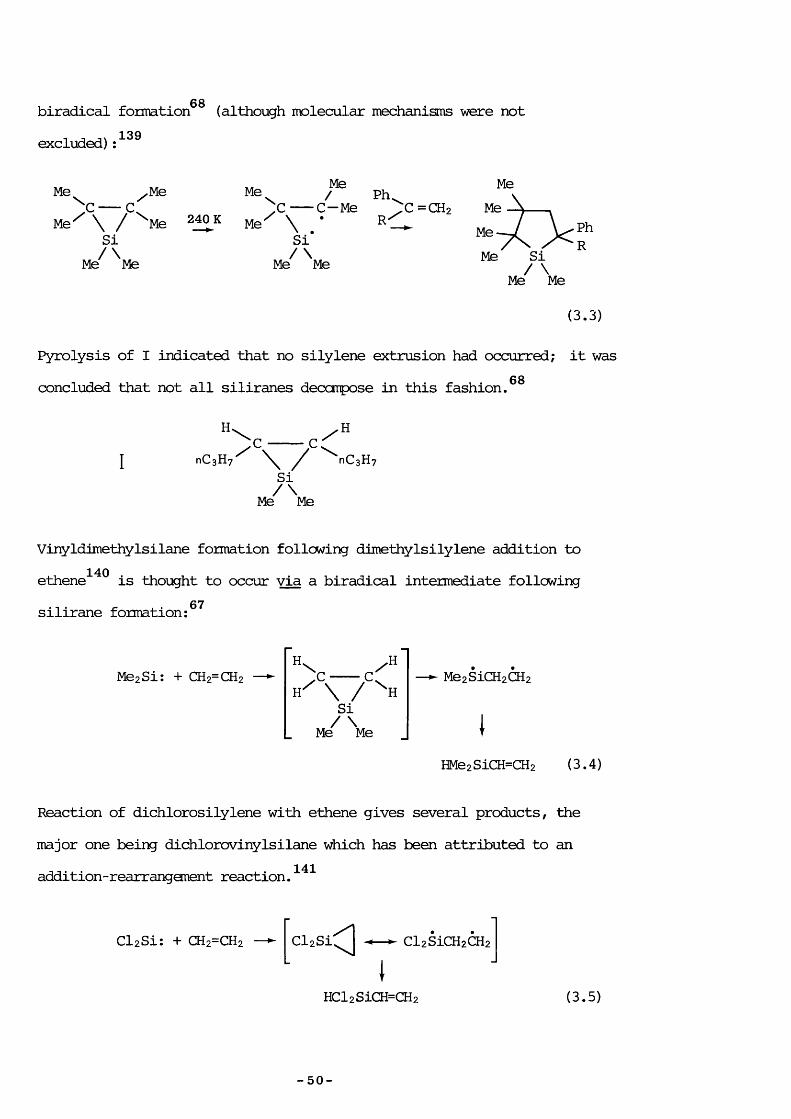
6 8biradical formation (although molecular mechanisms were not excluded)
Me /MeC C
Me^ \ / ^Me Si
Mé^ Me
Me240 K Me
Me/ Ph. C-Me %ZC=CH2/ \ •\
/ \Me MeMe Me
(3.3)
Pyrolysis of I indicated that no silylene extrusion had occurred; it was68concluded that not all siliranes decatpose in this fashion.
H. HnCsHy' ^ nCsHy
Si/\Me Me
Vinyldimethylsilane formation following dimethylsilylene addition to140
silirane formation:ethene is thought to occur via a biradical intermediate following
67
MezSi: + CH2=CH2 > x - < :Si
Me Me
Me2SiCH2CH2
HMe2SiCH=CH2 (3.4)
Reaction of dichlorosilylene with ethene gives several products, themajor one being dichlorovinylsilane which has been attributed to an
141addition-rearrangement reaction.
Cl2Si: + Œ2=CH2 Cl2Si Cl2SiCH2CH2
HCl2SiCH=CH2 (3.5)
- 50 -
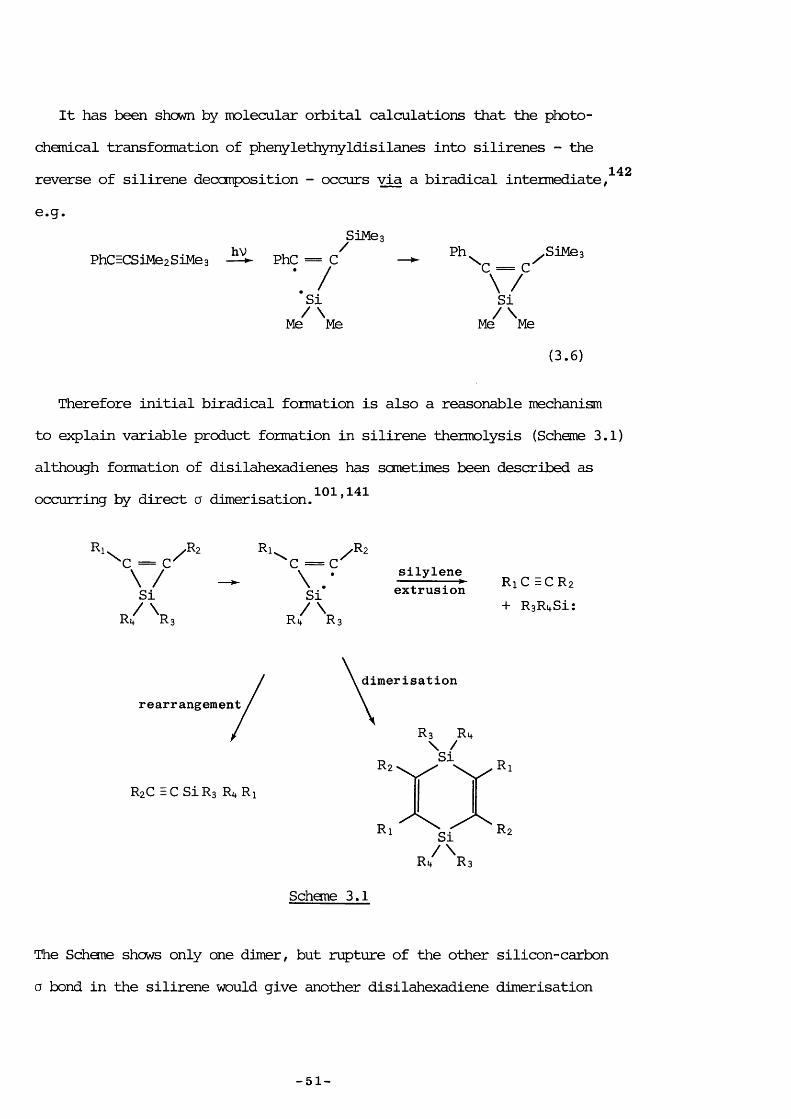
It has been shown by molecular orbital calculations that the photochemical transformation of phenylethynyldisilanes into silirenes - the reverse of silirene décomposition - occurs via a biradical intermediate, e.g.
142
hV /PhCECSiMezSiMes — PhC = CSiMe.
•s// \Me Me
Ph /SlMesC = C\ /SiMe^ ' Me
(3.6)
Therefore initial biradical formation is also a reasonable mechanism to explain variable product formation in silirene thermolysis (Schane 3.1) although formation of disilahexadienes has sometimes been described asoccurring by direct a dimérisation 101 ,14 1
\ /Sir /
Ri,C = C\ . •SiR i f Rs
Rzsilyleneextrusion RiC ECRz
+ RgRi+Si :
rearrangement
RzC EC SiRa R R]
dimérisation
Rz
R:
Si
Si/\Ri+ RsSchane 3.1
R
Rz
The Schane shows only one dimer, but rupture of the other silicon-carbon Q bond in the silirene would give another disilahexadiene dimérisation
-51-
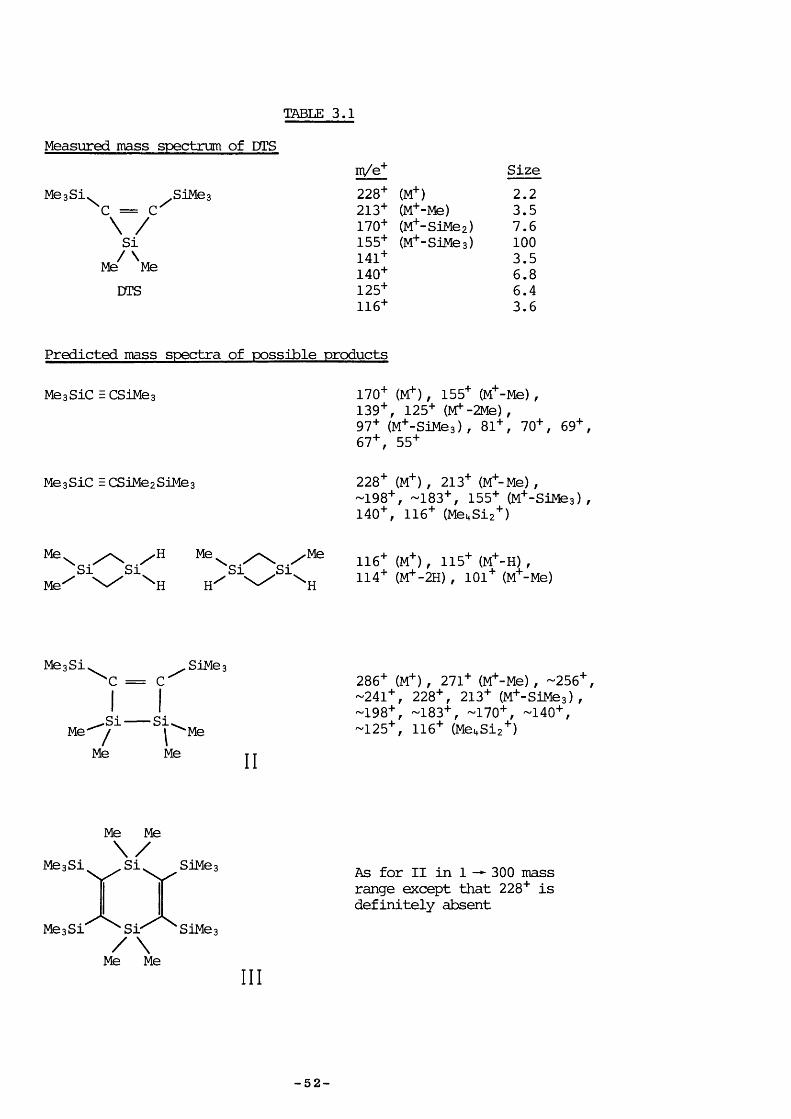
TABLE 3.1
Measured mass spectrum of DTS
MeaSi\m/e+ Size
.SiMeaC = C
228+ (M+) 2.2213+ (M+-Me) 3.5
\ / 170+ (M+'SiMez) 7.6Si 155+ (M+-SiMea) 100
141+ 3.5Me Me 140+ 6.8DTS 125+
116+6.43.6
Predicted mass spectra of possible products
MeaSiC E CSiMea 170+ (M+) , 155+ (M+-Me) ,139+, 125+ (M -2Me) ,97+ (M+-SiMea), 81+, 70+, 69+, 67+, 55+
MeaSic ECSiMezSiMea 228+ (M+) , 213+ (M+-Me) ,~198+, -183+, 155+ (M+-SiMea) , 140+, 116+ (Me4Siz+)
Me \ SiMe'
SiHH
Me.H
Si Si.Me
\ H116+ (M+) , 115+ (M+-H) , 114+ (M+-2H) , 101+ (M+-Me)
MeaSi^ SiMea*C = C
Me _^Si/MeSi. ‘MeMe II
286+ (M+) , 271+ (M+-Me) , -256+, -241+, 228+, 213+ (M+-SiMea), -198+, -183+, -170+, -140+, -125+, 116+ (Me4Siz+)
MeaSi
Me Me
Me a Si "‘Si"/ \Me Me
SiMe;
SiMe:
III
As for 11 in 1 — 300 mass range except that 228+ is definitely absent
— 5 2 —

prcxluct. Fran the results obtained by other workers, described in Chapter 1 (Reactions 1.46 to 1.53), the following observations can be made;-1. If R 3 or R4 is an aryl group then no dimérisation occurs - possibly
prevented by bulkiness in the biradical. [Disilahexadienes containing eight aryl groups have been synthesized, but not fran silirene pyrolysis.
2. For 1,2 rearrangement to occur R% or Rz must be a labile group, e.g. hydrogen or trimethylsilyl: for example, in Reactions 1.47 and 1.48 in vÆiich both Ri and Rz are trimethylsilyl groups, 100% rearrangement proceeds. This also seons to apply to silirane decarposition (Reactions 3.4 and 3.5); perhaps I pyrolysed to give the rearranged product.
On this basis, for the silirene studied by the author - 1,1 dimethyl- 2, 3-bis (trimethylsilyl) -1 -silirene (DTS) - which has Ri and Rz as trimethylsilyl and Rs and Ri+ methyl groups, a large percentage rearrangement was expected, with perhaps a small amount of dimérisation or silylene extrusion. The results obtained by Seyferth on the pyrolysis of the same ccmpound®^ (Reaction 1.44) - 30% bis(trimethylsilyl)acetylene formation - were surprising; the solvent (benzene) could have played a major rôle in the course of the reaction, possibly by interacting significantly with radical centres or the double bond.
EXPERIMENTAL3.3 im-PRESSURE PYROLYSIS
The DTS sample, a gift frcm Professor D. Seyferth, contained a quantity of bis(trimethylsilyl)acetylene impurity, estimated to be approximately 30%. The measured mass spectrum of DTS is shown in Table 3.1 along with
-53-

that for bis (trimethylsilyl) acetylene and the spectra expected for theother possible pyrolysis products. II could be formed fran dimethyl-
73silylene insertion into DTS (Reaction 1.31) and the two disilacyclo-71butanes fran dimethylsilylene dimérisation (Reaction 1.28). The two
important features of the Table are that the product of rearrangement,Me3Sic ECSiMezSiMes, should have no 170+ fragment (M+-SiMez) as this could only arise fran a rearrangement rather than a simple fragmentation process (cf. hexamethyldisilane, CH3SiMezSiMe3, viiich was found to have no m/e = 88+ : M+- SiMez) and that III, the DTS dimérisation product, has no 228+ peak. " ^
Investigation of pyrolysis products was made more difficult owing to the pulsed stirred-flow apparatus not being available at the time. DTS was pyrolysed, using LPP, between 420 and 550 K at initial pressures of ça. 0.2 Torr. The observed changes in the mass spectra were:-
1. No peaks disappeared.2. Four new peaks appeared:
286+, 271+ (both extremely small)197+ (final 228+/197+ intensity ratio ~3)183+ (final 228+/183+ -2)
3. Table 3.2 shows the effect of pyrolysis on the height of the observed mass spectral peaks.
These findings showed that the mass fragments present in bis(trimethylsilyl) acetylene all decreased; very little (if any) was therefore produced. Dimethylsilylene, formed concomitantly in this possible décomposition pathway (Schane 3.1) therefore did not play an important part in the reaction: the disilacyclobutanes were not formed: no m/e115+, 114+, 101+ were observed; nor was there any evidence for penta- methyldisilane (m/e 132+, 131+, 117+ : M+, M+-H, M+-Me) - the insertion product - in excess trimethylsilane (Reaction 1.30). In separate experi-
— 5 4 —

TABLE 3.2
Peak height changes during DTS pyrolysis
Before pyrolysis After228+ 1.0 Approx. constant213+ 1.6 3.0170+ 3.5 1.8140+ 6.8 7.1116+ 3.6 5.581+ 1.5 0.8
70+, 69+, 67+ all -9.0 all -1.555+ 3.0 1.2
ments dimethylsilylene, generated frcm pentamethylchlorodisilane^® at 770 K, gave small quantities of m/e = 101+, perhaps due to the disilacyclobutanes, and formed pentamethyldisilane in excess trimethylsilane.
Since this meant that dimethylsilylene was not a product of the DTS73reaction, then nor was II, v^ich is stable at these temperatures, a
ccmpound not reported as being formed during silirene thermolysis by any other research group. The remaining mass spectral features of the reaction product mixture are well explained by 80-90% rearrangement product. Me3Sic ECSiMezSiMe3, with the remainder being the product of DTS dimérisation. III. The approximate constancy of the 228+ fragment during pyrolysis, and the observed peak ratios showed that the major product was Me3SiC ECSiMe2SiMe3.
3.4 KTNETICS
No detailed kinetic measurements were made, but the following rate constants for DTS decomposition could be estimated
0.0001 s"^ (at 440 K), 0.05 s" (495 K), 0.5 s" (550 K), frcm Wiich the rather crude values for the Arrhenius parameters could be derived:
- 5 5 -

logic k/s"‘ = (14.9 ±3.3)-(158 ±31 kJ itcl"')/2.303 FT.
3.5 DISCTSSICTJ
The validity of these figures can be assessed by using the ring strain calculated by Gordon for silirene,equal to 198 ±8 kJ imol” . If pyrolysis proceeds by ring-opening, iy silicon-carbon bond rupture, and the estimated A factor (above) is in keeping with this, then the activation energy can be identified with the silioon-viryl bond strength minus the ring strain. This gives the unstrained silicon-viiyl bond energy to be equal to 198 + 158 = 356 ± 39 kJ mol” . Coiparisons with the carbon analogues may initially suggest that this figure is too lew;
tBu-Me = 342 kJ MeaSi-Me = 366^^®-374^^ kJ mol"^^Bu -CH=CH2 = 378 " Me3Si-CH=Œ2 ~ 400 " ?
Preliminary LPP experimental work on the pyrolysis of vinyltrimethyl-silane gave approximately equal yields of methane and ethene (resultingfrcm silioon-methyl and silicon-vinyl bond rupture) with apparently aslightly greater proportion of methane at higher temperatures! Theseresults agreed with those obtained for HMe2SiOSiMe2CH=CH2, discussed inChapter 5, in that significant silioon-viryl bond rupture occurred, andsuggest that the two bonds are approximately equal in strength. [Bondstrengths between silicon and another aton are essentially unaffected by
22the nature of the substituents on silicon. ] The reasons for this are unclear, but could be related to the shorter bond length required for a
linkage involving an sp^ hybridized carbon; this could be unfavourable vÆien bonding to a larger silicon atom. Since the resonance stabilization energy in radicals of the type (R)2Si-CH=CH2 is thought to be very lew
• 22 147(cf. H2Si-Ph ’ ), the silioon-methyl, and hence the silicon-vinyl.
— 5 6 —

bond strength should be -370 kJ mol"^, vhich resonbles the calculated value of 356 ± 39 kJ inol“ . This, and the measured A factor, is evidence in favour of the proposed ring-opening mechanism.
The 1,2 trimethylsilyl shift, vhich gives the rearranged product, is not unexpected in view of the known ability of silicon to migrate frcm silicon to carbon, e.g. as a step in the pyrolysis of hexamethyl- disilane:^®'^^®
MesSiSiMez — ► MeaSiCHzSiMez (3.7)•CHz
149or in the thermolysis of other organosilicon compounds. In our case, the rearrangement is frcm carbon to silicon, but there is the advantage of forming the carbon-carbon triple bond:
MeaSi^ SiMea“C =\ ’ — ► MeaSiSiMezC ECSlMea (3.8)Si */\Me Me
3.6 CaSfCLUSION
DTS pyrolyses between 440 K and 550 K to give mainly the rearranged product MeaSiSiMezC E cSiMea. No significant silylene extrusion occurred. Estimated rate constants indicate that pyrolysis probably proceeds via a biradical intermediate.
-57-

CHAPTER 4A3XYLTRIMETEÎYLSILANE PYROLYSIS

Allyltriraethylsilane pyrolysis4.1 INTRODUCTION
The heating of allylsilanes results in an extraordinary yariety of reactions; for many there is sane doubt about the processes involved; the one for viiich there is most confusion is allyltrimethylsilane (ATMS) . In view of the intrinsic interest in the thermolysis of this ccmpound - in particular because SE, an intermediate of considerable interest in this laboratory, is probably formed - it was investigated frcm several angles: to prove that SE is evolved and to decide on the mechanism; tofind the extent of silicon-ally 1 bond rupture; and to elucidate the mechanism for vinyltrimethylsilane production.
Experimental4.2 PYROLYSIS PRODUCTS
ATMS was pyrolysed at varying pressure between 860 and 960 K usinglow-pressure pyrolysis (LPP) with additional pulsed stirred-flowexperiments to aid product identification. Further pyrolyses in break-seal tubes were performed at the Max-Planck Institute, Miilheim a.d. Ruhr,West Germany, with analysis by gas chromatography (Carlo Erba 2900,having a flame ionization detector, product separation being achieved bya 77m glass capillary column coated with SF 96 methylsilicon oil) .
Typical results are shown in Tables 4.1 to 4.3; the last two Tables150use a shorthand notation (a methyl group attached to silicon, and a
hydrogen attached to carbon are emitted) and show only the major products, identified by comparison of retention times with authentic compounds or by performing suitable experiments (discussed later) . Approximate yield can be obtained by dividing % area by the number of carbon atoms in the ccmpound. Appendix 4.1 lists retention times with assigned compounds ;
— 5 9 —

Appendix 4.2 is a glossary of shorthand notation terms used. These39 82pyrolyses formed the products reported both by Bailey and Sakurai
in varying proportions; the results most strongly corroborating those ofSakurai were at an initial pressure of 1 Torr at 810 K (Table 4.2) : aninferior column would have separated the same number of peaks, as shownin Table 4.2. Under these conditions the seventh peak (unidentified bySakurai) is mainly hexamethyldisilane.
All LPP products were confirmed by pulsed stirred-flew experimentsusing authentic ccmpounds for identification, except 1,1-dimethyl-1-silacyclopent- 3 -ene vÆiich was not available ; the 97+ and 112+ fragmentswere taken as being indicative of its presence. This was probablyformed by endo cyclisation of the radical CHz (Me) z SiCHzCH=CHz as
82proposed by Sakurai, even though exo ring closure usually occurs forthis type of r a d i c a l . 1,1-Dimethyl-1-silacyclopentane was not aproduct because of the absence of m/e = 86+ (its base peak) the
153terrperature was also too low for its pyrolysis.
4.3 PYROLYSIS AT LCWER PRESSURES
Lowering the pressure simplified the reaction considerably: LPP,pulsed stirred-flow, and sealed tube experiments all showed that at initial ATMS pressures of 0.05 Torr the only products were: mainlypropene, trimethylsilane and 1,1,3,3-tetramethyl-l,3-disilacyclobutane (DSCB) , with ethene and methane in smaller quantities. The formation of DSCB indicated 1,1-dimethylsilaethene (SE) production; one of the two proposed mechanisms, that of trimethylsilyl radical disproportionation by Gusel ' nikov, can be eliminated because reccmbination competes withdisproportionation:
— 60“

TABLE 4.1
LPP products of ATMS pyrolysis at typical pressures(ca. 0.3 Torr)
Product % Yield Observed mass fragments
CH4 5-10 16+ (M+) , 15+ (M+-H)CH2=CH2 5-15 28+ (M+) , 27+ (M+-H) , 26+MeCH=CH2 85-100 42+ (M+), 41+ (M+-H), 40+, 39+,
27+MeaSiH 25-40 73+ (M+-H)MeitSi Up to 5 73+ (M+-Me)Me3SiCH=CH2 Up to 5 100+ (M+) , 85+ (M+-Me)
> S 1 ^ -5 112+ (M+) , 97+ (M+-Me)
5-20 144+ (M+), 129+ (M+-Me), 101+, 85+
- h114+ (M+), 99+ (M+-Me), 85+, 73+, 42+
— 61“
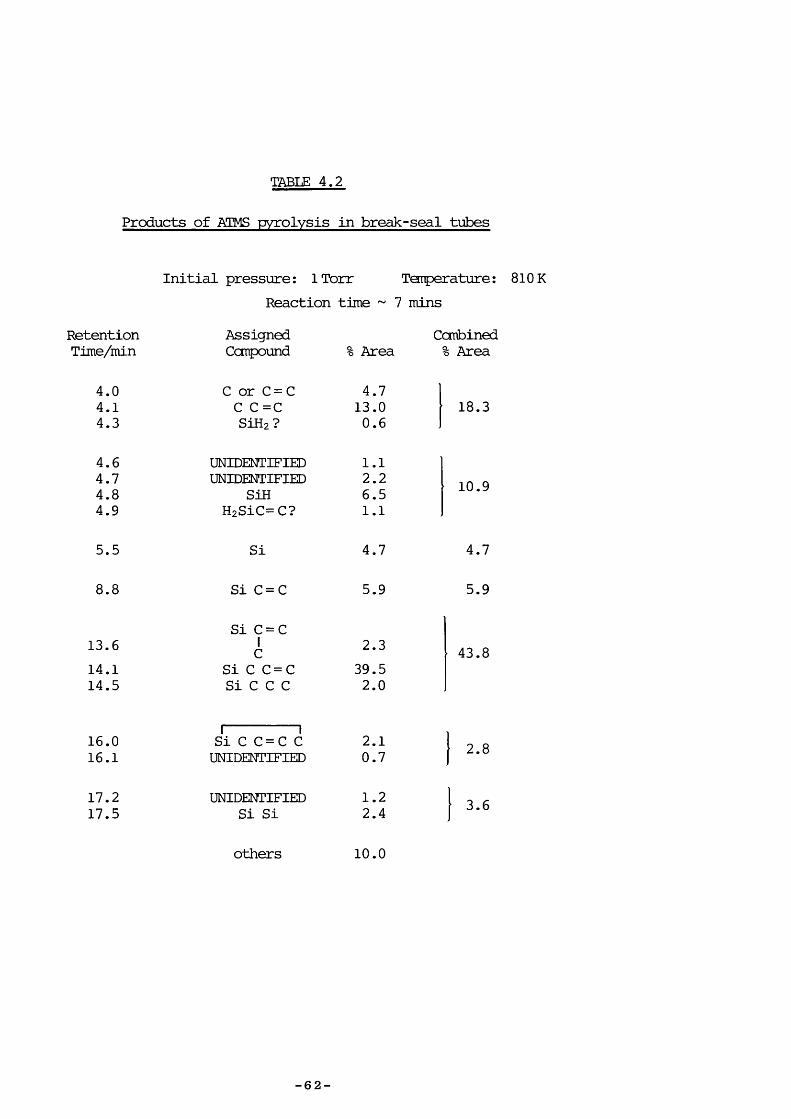
TABLE 4.2
Products of ATMS pyrolysis in break-seal tubes
Initial pressure: 1 Torr Tennperature: 810KReaction time - 7 mins
RetentionTime/min
AssignedCcmpound Area
Combined % Area
4.04.1 4.3
C or C = C C C=C SiHz?
4.713.00.6
18.3
4.64.74.84.9
UNIDENTIFIEDUNIDENTIFIED
SiHH2SiC=C?
1.12.26.51.1
10.9
5.5 Si 4.7 4.7
8.8 Si C=C 5.9 5.9
13.614.114.5
Si C = C ICSi C C=C Si C C C
2.339.52.0
43.8
16.016.1
Si C C=C C UNIDENTIFIED
2.10.7 2.8
17.217.5
UNIDENTIFIED Si Si
1.22.4 3.6
others 10.0
— 62 —
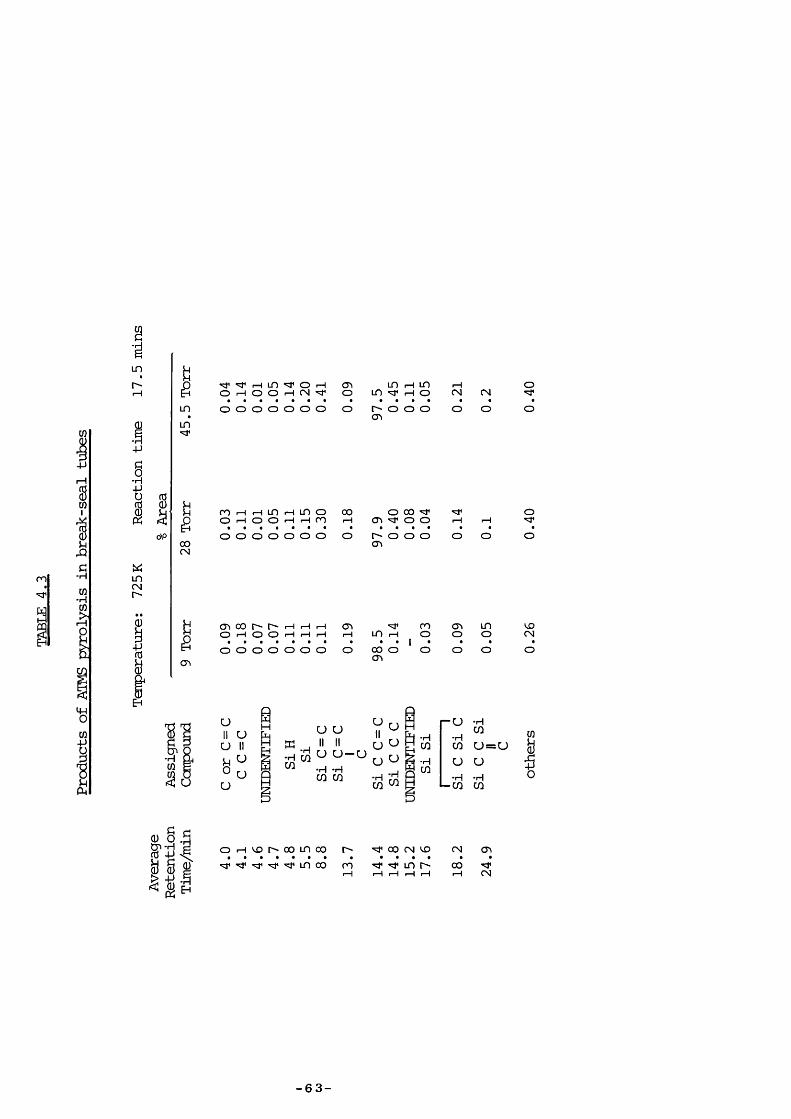
ro
I
0)
COI• Sin•H
in
I4JIintNI
E-iinin
I'0\0
g
00CN
g
(T\
r—1inTfo r—1 CT» in rH in <—1 Oo r—1o o 1—1CN O in 1—1o CN CNo o o o o O O O r~-
CTi o o o O O o
ro 1—1 rH in 1—1ino 00 o 00 oo rH o o 1—1 rH ro rH m o o rH rH 'Ifo o o o o o o o r"ono o o O o o
CT)00 r—1 rH rH "f ro cr> in kOO rH o o rH rH rH rH m rH o o o CN1O o o o o o o o 00 o cr» o o o o
01 uII u
U II
u
u u X , II II ■H w u o - u^ -rH -Hw œ
u u g
U •H
•HW
W U = U
U U 5•H •H 0
W
I o rH VOr"00in00 r-> 00 CN VO CN O'f in00 ro f in 00rH rH rH rH rH rH CN
— 0 3 —

Reccmbination :2M03Si* MeaSiSiMea (4.1)
Disproportionation: ^2MeaSi- — ► MeaSiH + M02Si=Œ2 (4.2)
Estimates for kd/kr range frcm 0.05 to 0.31.®"^® The absence of hexamethyldisilane (m/e = 131+: M+-Me) (rate ATMS deccmposition » rate
132hexamethyldisilane decomposition ) at low initial ATMS pressures whilst DSCB was a major product indicated that trimethylsilyl radical disproportionation was not the route to SE. The results showed that a unimole- cular process was responsible; kinetic experiments were necessary to confirm that Bailey’s retroene reaction occurred.
The absence at low pressure of 1,1 -dimethyl-1 -silacyclopent-3-eneindicated that its formation resulted frcm a bimolecular reaction, as
82expected; in addition, both tetramethylsilane and vinyltrimethylsilane,major products at higher pressure, were not formed below 0.05 Torr. This
82meant that vinyltrimethylsilane is not formed by methylene extrusion84or by a series of reactions following silicon-methyl bond cleavage
(since these are both unimolecular processes) but frcm reactions following an initial bimolecular step.
Whilst methane and ethene formation was reduced by lowering the pressure, they were still present in significant quantities. Seme was produced frcm the polymeric deposit on the sides of the reaction vessel, and, although a small amount was probably formed frcm head-to-head dimérisation of and unimolecular DSCB deccmposition, ®® themain route to their production was radical reactions with either SE or DSCB: pyrolysis in excess toluene (a radical trap) dramatically loweredtheir yields, whereas in excess DSCB their yields increased. One of the probable processes involved in ethene production is shown in Scheme 4.1; co-pyrolysis with hydrogen chloride gave dimethylsilylchloride
— 6 4 —

(m/e = 79+: M+-Me) , indicating dimethylsilylene formation.®®
/Si — CH 2 + — ► /Si— /Si1.i
Me2Si: CH2 =CH2 J \ ^ ^ ^ + CH2 =CH2
\ HCl
HMezSiClScheme 4.1
As well as traces of m/e = 95+ and 87+ (unidentified), a product in minor yield was probably 1,1-dimethyl-1-silacyclobut-2-ene (m/e = 83+: M+-Me®®), formed following hydrogen abstraction frcm the allyl group, vÆiich should have a Icwer activation energy than for abstraction frcm the methyl group (Scheme 4.2):
R' + -Si— \ — ► RH + -Si— /O ' 1
t/Si— j) /Si=n□ ^ J
Scheme 4.2
The resulting radical could then unimolecularly decompose to give a methylradical (subsequently forming methane) and a silabutadiene, a process
7viiich has a precedent :
Mo3S i Œ 2 — ^ 'Me + Me2Si=CH2 (4.3)
-65-

4.4 ATMS DECAY KINETIC MEASUREMENTS
Two primary processes could therefore be identified:
kiMe3SiCH2CH=CH2 — ► Me2Si=CH2 + MeCH=CH2 (4.4)
Me3SiCH2CH=CH2 ^ Me3Si* + CH2C H Π2 (4.5)
Reaction 4.4 is irreversible at the pressures used in LPP: SE, generatedby pyrolysing 1,1 -dimethyl-1 -silacyclobutane (EMSCB) at 800 K, did not react with added propene; no ATMS was formed. The radicals produced in Reaction 4.5 subsequently form trimethylsilane and propene by hydrogen abstraction processes. As the initial ATMS pressure was raised, several bimolecular reactions became increasingly important; this complex pyrolytic behaviour led to unclear kinetic data in our original work:^®® kinetic runs were carried out between 880.5 and 949.5 K at initial ATMS pressures of 0.2 Torr. [Each series of kinetic runs will be given a name, this series ATMS 1.] Decomposition data (Table 4.4) , obtained by monitoring the ATMS molecule ion (m/e = 114+) , gave (Figure 4.1) :-
logiok/s"^ = (16.01 ±0.37) - (304.63 ±6.52 kJ mol"M/2.303 RT
Direct quantitative measurements of product formation are difficult in LPP since a proportion of the product is often lost to the walls of the reaction vessel (forming a polymeric deposit), particularly silicon- containing species; in addition trimethylsilane has an indistinctive mass spectrum.
Further ccmplications arose before because the reaction vessel at that time contained a large amount of dead space v^ich is now thought to have affected the kinetics of the reaction. Subsequent experiments (ATMS 2 - ATMS 4) , after modification of the reaction vessel to decrease dead space to <4%, showed that SE formation had been underestimated, and gave
— 6 6 —
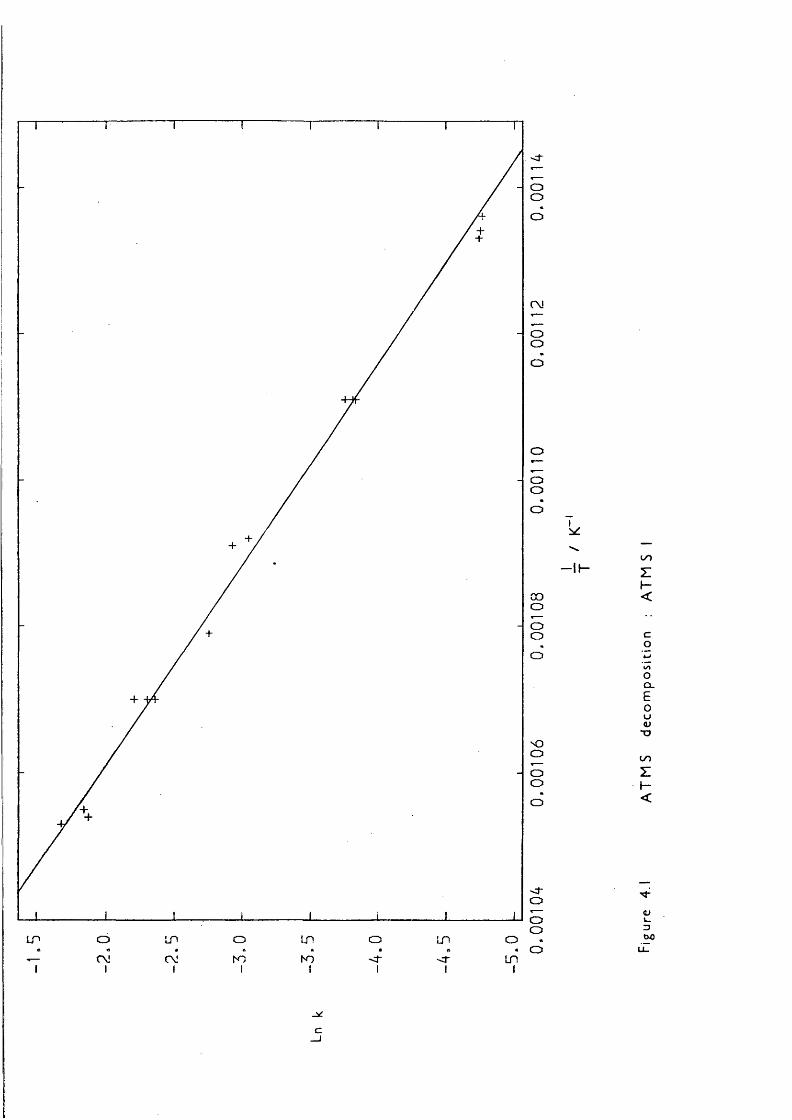
L:' s .
-IH LT)
zh-<
oQl£oui)*utoZI-<
LP O in O in o in o DO• o li.
-— OJ CM m m in
c

TABLE 4.4
Rate constants for ATMS deccmpos ition;experiments ATMS 1
Tarp/K k/s"i Tanp/K k/s-i880.5881.5882.5 900 900 900915.5 917
0.008550.008650.008750.02160.02200.02330.04740.0534
927935935935948949 949.5
0.06370.09450.09990.1100.1590.1540.187
TABLE 4.5
Rate constants for ATMS decomposition: experiments ATMS 2
Tarp/K k/s“^ Tarp/K k/s"^878 0.0121 921 0.0508879 0.0106 921 0.0527880 0.0122 921 0.0552881 0.0125 941 0.117899 0.0237 941.5 0.103899 0.0238 942 0.101900 0.0248 942 0.102901 0.0253 953 0.135909 0.0465 954 0.132910 0.0333 954 0.154910 0.0344 954 0.156911 0.0354921 0.0488
— 67”
TABLE 4.6
Rate constants for ATMS decorposition inATMS/N2 mixture; experiments ATMS 3
Tarp/K k/s-i Tamp/K k/s-i868 0.00915 919 0.0527869 0.00771 919 0.0546869.5 0.00885 937.5 0.0983883.5 0.0138 938 0.0929884 0.0133 938 0.0936884 0.0135 938 0.0960884 0.0136 956 0.180904 0.0295 957 0.184904.5 0.0334 957.5 0.175905 0.0332 958 0.182919 0.0523
TABLE 4.7
Rate constants for ATMS decorposition: experiments ATMS 4
Tarp/K k/s-i Tarp/K k/s"^867 0.00787 925 0.0608867 0.00810 925 0.0610891 0.0201 926 0.0611892 0.0202 934 0.0822892 0.0238 936 0.0835900 0.0255 936 0.0865901 0.0258 951 0.158909 0.0346 951 0.160909 0.0350 951.5 0.172909 0.0351 960 0.184917 0.0478 961 0.188918 0.0490 962 0.207919 0.0489
-68-

decaiposition parameters which differed from the original whilst ronaining mutually consistent.
ATMS decomposition obeyed first order kinetics; the rate constants obtained in these experiments (ATMS 2: 0.2 Torr ATMS; 878 to 954 K;Table 4.5; Figure 4.2, ATMS 3; 0.04 Torr ATMS in five-fold excess ofnitrogen; 868 to 958 K; Table 4.5, ATMS 4: 0.03 Torr ATMS; 867 to962 K; Table 4.7) gave:-
logiok/s"^ = (12.16 ±0.29) - (237.01 ±5.11 kJ mol"M/2.303 RT (ATMS 2) ,logiok/s"^ = (12.47 ±0.17) - (242.14 ±3.04 kJ inol"M/2.303 RT (ATMS 3),logiok/s’ = (12.20 ±0.20) - (237.40 ±3.48 kJ inol‘^)/2.303 RT (ATMS 4) .
A summary of decomposition kinetic data is shown in Table 4.13. Thesedata are considered to be much more reliable than the original^and
83therefore Kwart's reported Arrhenius parameters for the retroene reaction - v^ich give a larger rate constant than the above parameters - can be assumed to be decomposition data relating to the overall process at higher pressures which would increase the importance of bimolecular reactions and hence decomposition rate. Because of the difficulties involved in measuring product formation, to separate the two processes (4.4) and (4.5), it was necessary to trap out one of the species using a low ATMS partial pressure in excess of the trapping reagent; monitoring a peak characteristic of the trapped product would indicate the importance of that route and would give a rate constant identifiable with the appropriate ATMS decomposition reaction.
4.5 TRAPPING OF SE
Several unsuccessful attorpts to do this by trapping SE were made.This should give a higher percentage of trapped product at lower temperature, the activation energy for Reaction (4.4) being less than (4.5)
-69-
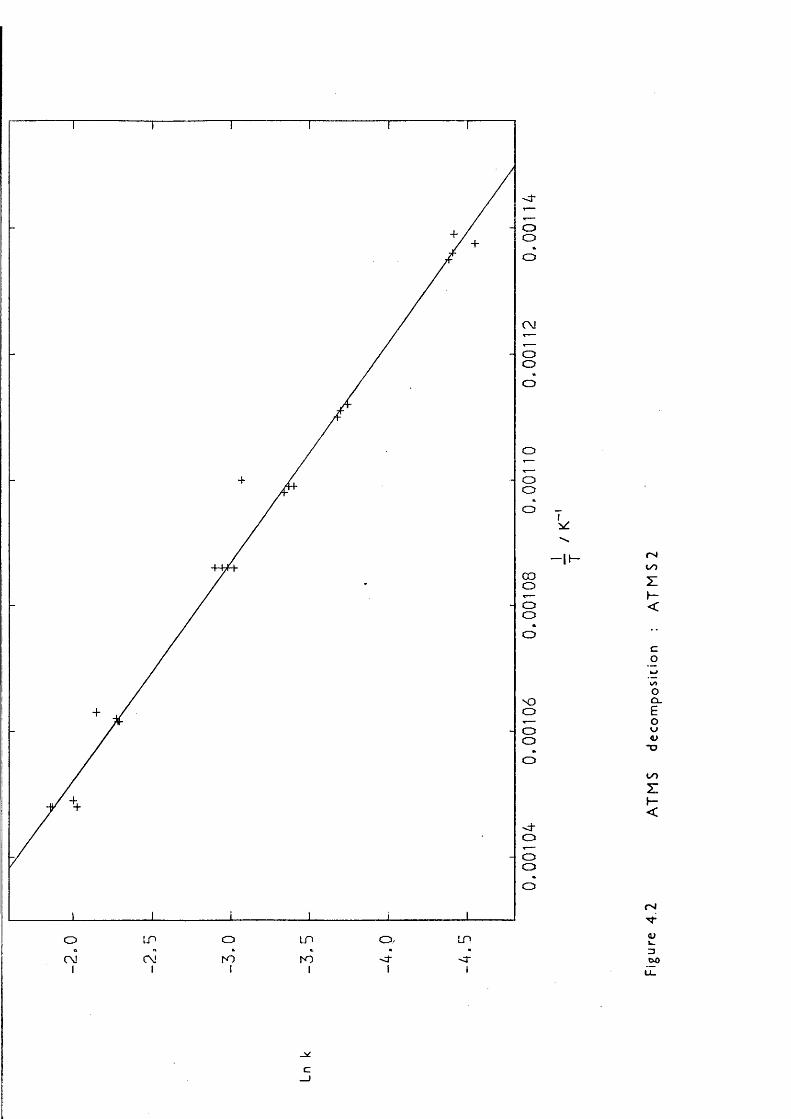
ooo
CM
OOO
oooo
COoooo
oooo
oooo
!_nLP o o.o
o
-lh- r>4ooZ\ -
<
oCLEoV
"OLOz
CM CMI
roI I <r
I
rMTf"OJ3&0
c

TABIiE 4.8
Rate constants in ATMS/methanol mixture:experiments ATMS 5
Tanp/K k/s-iMeasuredki/s"^
Calculatedk2/s"i
860 0.00412 0.000914 0.00321861 0.00426 0.000814 0.00345883 0.0101 0.00147 0.00863884 0.0102 0.00287 0.00733884 0.0104 0.00279 0.00761902 0.0188 0.00473 0.0141902 0.0195 0.00603 0.0135903 0.0208 0.00517 0.0156915 0.0307 0.00940 0.0213915 0.0312 0.00894 0.0223915 0.0315 0.00683 0.0247
4.5.1 MethanolATMS/methanol mixture kinetic runs (ATMS 5: 0.3 Torr ATMS in two-fold
excess of methanol; 860 to 915 K; Table 4.8) in which ki was measured by monitoring methyltrimethylsilyl ether production (m/e = 89'*’: M'"'-Me)(Reaction 4.6) and ka calculated by subtraction of ki frcm k, gave unsatisfactory ki data - an approximately constant percentage of trapped product was obtained over the entire temperature range.
Me2Si=CH2 + MeOH — ► MegSiOMe (4.6)
A modified technique v\Æiich involved placing a quantity of methanol in the reaction vessel to line the walls before adding a sample of the mixture gave increased methyltrimethylsilyl ether production, but still unsatisfactory ki data (ATMS 6: 0.02 Torr ATMS in six-fold excess ofmethanol; 834 to 938 K; Table 4.9) . Further runs using a greater excess of methanol were unsuccessful. The experiments did, however, confirm the ATMS decomposition data, givinglogiok/s"^ = (12.18 ±0.12) - (239.73 ± 2.07 kJ mol"M/2.303 RT (ATMS 5) and
— 7 0 —
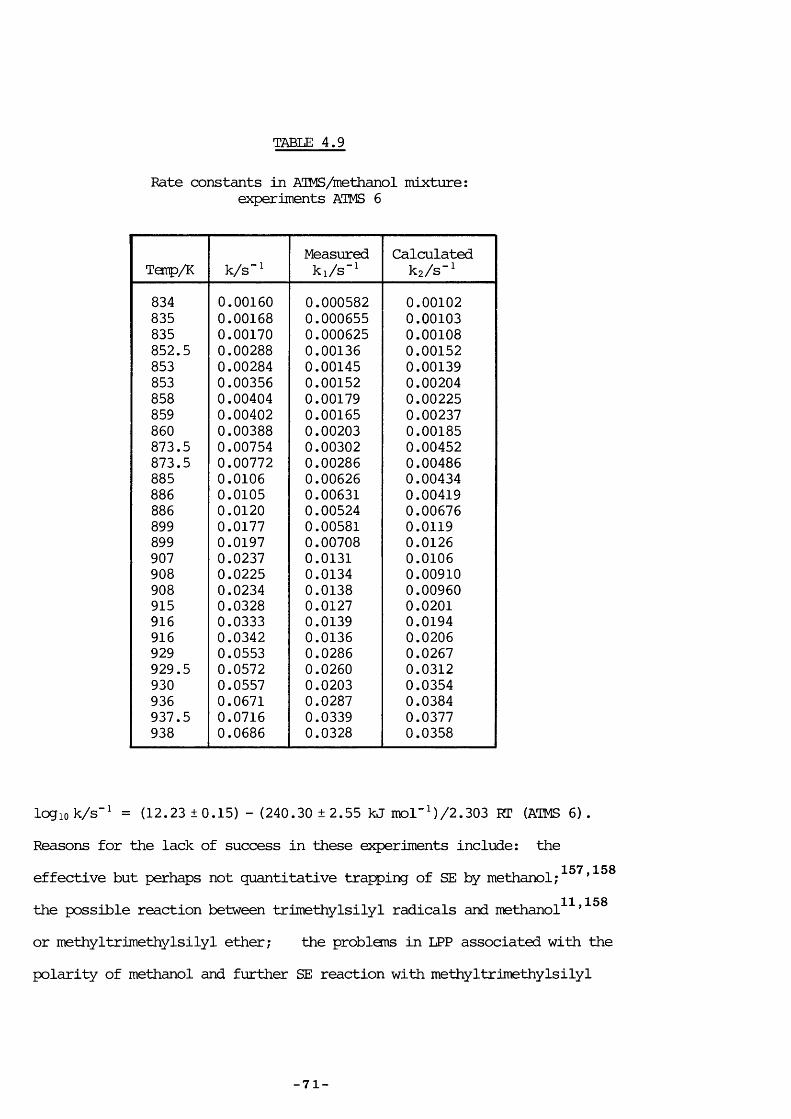
TABLE 4.9
Rate constants in ATMS/methanol mixture:experiments ATMS 6
Tarp/K k/s-iMeasuredki/s“^
Calculatedkz/s"!
834 0.00160 0.000582 0.00102835 0.00168 0.000655 0.00103835 0.00170 0.000625 0.00108852.5 0.00288 0.00136 0.00152853 0.00284 0.00145 0.00139853 0.00356 0.00152 0.00204858 0.00404 0.00179 0.00225859 0.00402 0.00165 0.00237860 0.00388 0.00203 0.00185873.5 0.00754 0.00302 0.00452873.5 0.00772 0.00286 0.00486885 0.0106 0.00626 0.00434886 0.0105 0.00631 0.00419886 0.0120 0.00524 0.00676899 0.0177 0.00581 0.0119899 0.0197 0.00708 0.0126907 0.0237 0.0131 0.0106908 0.0225 0.0134 0.00910908 0.0234 0.0138 0.00960915 0.0328 0.0127 0.0201916 0.0333 0.0139 0.0194916 0.0342 0.0136 0.0206929 0.0553 0.0286 0.0267929.5 0.0572 0.0260 0.0312930 0.0557 0.0203 0.0354936 0.0671 0.0287 0.0384937.5 0.0716 0.0339 0.0377938 0.0686 0.0328 0.0358
logiok/s"^ = (12.23 ±0.15) - (240.30 ±2.55 kJ mol"M/2.303 RT (ATMS 6) . Reasons for the lack of success in these experiments include: theeffective but perhaps not quantitative trapping of SE by methanol. the possible reaction between trimethylsilyl radicals and methanol^^ or methyltrimethylsilyl ether; the problems in LPP associated with the polarity of methanol and further SE reaction with methyltrimethylsilyl
— 7 1 "

ether (discussed in full in Chapter 6) :MezSi=CH2 + MesSiCMe — ^ MegSiCHzSiJMezOMe (4.7)
4.5.2 Methyltrimethylsilvl etherThis led to experiments using methyltrimethylsilyl ether itself as the
SE trap. This was unsuccessful because at the extranely low partialpressure of ATMS (in fifty-fold excess of methyltrimethylsilyl ether)required to give maximum MegSiCHzSiMezCMe (m/e = 163+: M' -Me) yield -SE reaction is less efficient than with methanol - the minor reactionsof SE with background oxpgen or water (also discussed in full in Chapter6) became significant. In addition, above 885 K, MeaSiCHzSiMezCMe
45began to deconpose rapidly.
4.5.3 Oxygen28The efficient reaction of SE with oxq gen - a four-fold oxygen
excess was used - to give formaldehyde (Reaction 1.7) (ATMS 7: 0.1 Torr;864 to 933 K; Table 4.10) gave unchanged ATMS decay rate constants : -
logiok/s'^ = (12.71 ± 0.42) - (245.82 ±7.23 kJ mol"M/2.303 RT Rate constants of formation of formaldehyde, k%, (m/e = 30^, M" ) were not corrected for sensitivity since they gave an activation energy of242.4 ±11.6 kJ mol"^, indicating an apparently constant percentage of SE formation frcm ATMS over the viiole temperature range. This erroneous solution is not surprising in view of the fact that other products resulting from SE reactions were observed: ethene and the cyclics(MezSiO)zMezSiCHz (m/e = 205+: M+-Me) , MezSiO(MezSiCHz)2 (m/e = 203+: M+-Me)
MezSi=CHz t CHz—0 — ^ MezSi O — ► MezSi=0 + CHz~CHzI ICHz GHz ISE
Schone 4.3 (MezSiO)zMezSiCHz, MezSiO(MezSiCHz)z
-72-
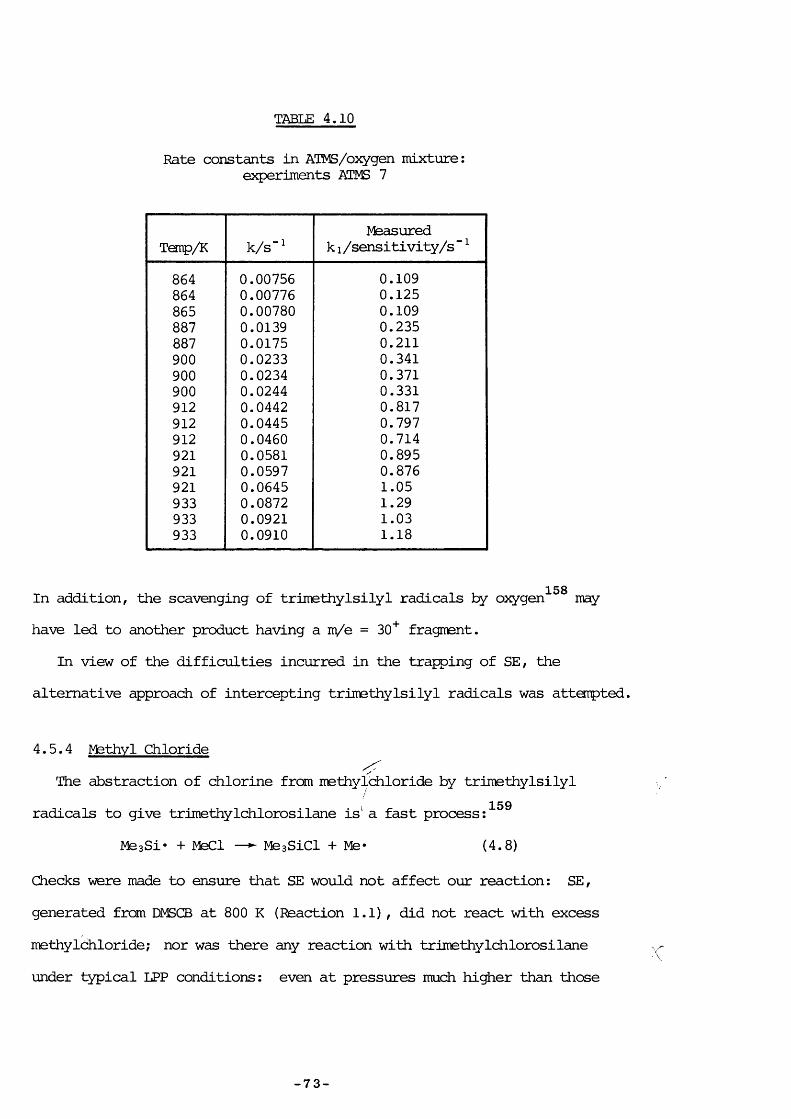
TABLE 4.10
Rate constants in ATMS/oxygen mixture;experiments ATJXG 7
Temp/K k/s-iMeasured
k 1/sensitivity/s"^864 0.00756 0.109864 0.00776 0.125865 0.00780 0.109887 0.0139 0.235887 0.0175 0.211900 0.0233 0.341900 0.0234 0.371900 0.0244 0.331912 0.0442 0.817912 0.0445 0.797912 0.0460 0.714921 0.0581 0.895921 0.0597 0.876921 0.0645 1.05933 0.0872 1.29933 0.0921 1.03933 0.0910 1.18
In addition, the scavenging of trimethylsilyl radicals by oxygen^may have led to another product having a m/e = 30+ fragment.
In view of the difficulties incurred in the trapping of SE, the alternative approach of intercepting trimethylsilyl radicals was attempted.
4.5.4 Methvl ChlorideThe abstraction of chlorine from methylchloride by trimethylsilyl
radicals to give trimethylchlorosilane is a fast processMssSi* + IfeCl — ► MeaSiCl + Me* (4.8)
Checks were made to ensure that SE would not affect our reaction: SE, generated from DMSCB at 800 K (Reaction 1.1) , did not react with excess methylchloride; nor was there any reaction with trimethylchlorosilane under typical LPP conditions : even at pressures much higher than those
-73-

used normally in LPP only a trace of MsgSiCHzSiMezCl (m/e 165+: M+-Me) ,the insertion product (Reaction 1.16), was formed.
Experiments showed that a five-fold excess of methylchloride over ATMS was sufficient to quantitatively trap the trimethylsilyl radicals; kinetic runs were performed in even greater excess, a quantity of methylchloride being initially placed in the reaction vessel. Trimethylchlorosilane formation, hence kz, was measured by monitoring m/e = 93+(M+-Me) ; methane was a product but no m/e = 165+ was formed, indicating that the reaction had proceeded as intended, i.e. only trimethylsilyl radicals had reacted with the methylchloride. In all, 77 runs were performed on three different days; combining all k2 rate constants gave a scattered Arrhenius plot because of small daily variations in the value of the relative sensitivity to m/e = 93+ in trimethylchlorosilane compared to 114+ in ATMS. The overall activation energy, however, and those for the results from each day were essentially the same. To decrease error limits, the k2 data from only one day have been taken (ATMS 8; Table 4.11) ; ki rate constants were calculated from k values which were unaffected by the presence of methylchloride. Between 859.5 and 919 K these data give:-
log A E/kJ mol“^ k890K/s”^ Figurek(=ki + k2) 12.51± 0.27 245.8± 4.6 1.2 X10"2 4.3
ki 10.55 ± 0.32 215.5 ±5.5 8 X 10-3 4.4kz 15.60 ± 0.50 306.6 ±8.5 4 X 10-3 4.5
No correction has been made for silicon-methyl bond rupture since this is insignificant; recent estimates for the silicon-methyl bond strength, (366 kJ mol"M^^^ and (374 kJ mol”M are thought to be more reliable than an earlier determination (355 kJ mol"^).^ Combined k values (ATMS 9; Table 4.12; Figure 4.6) give;-
-74-
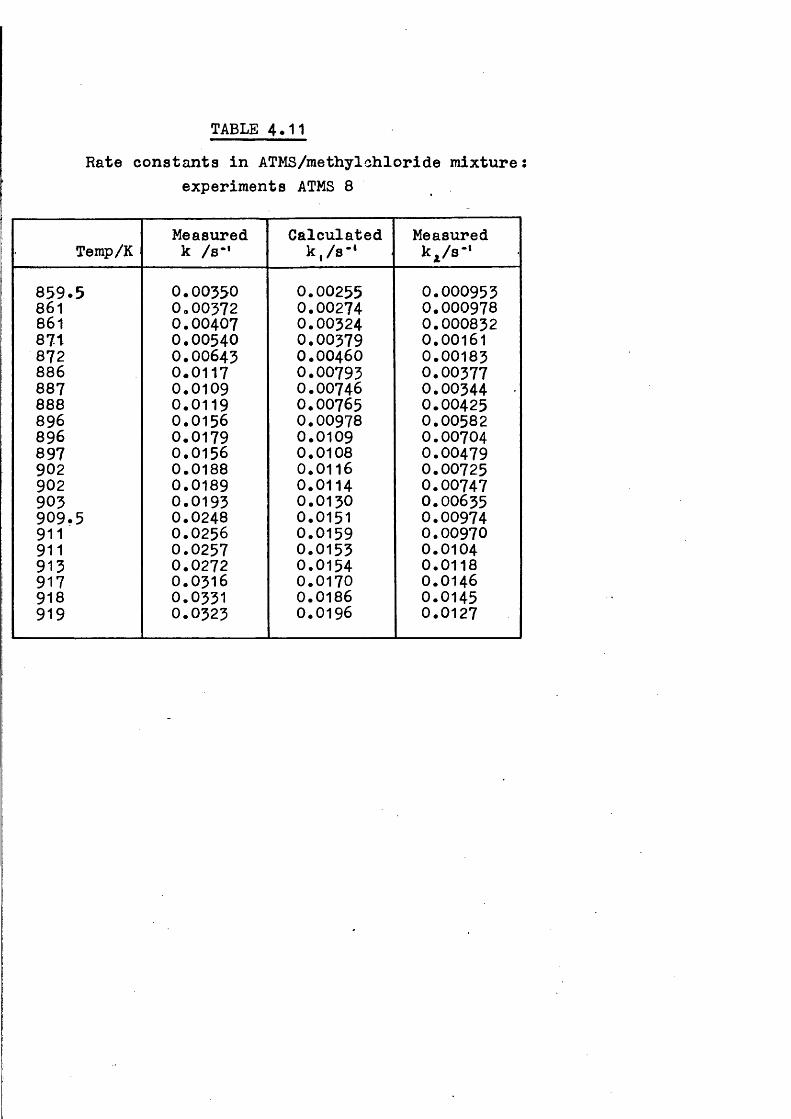
TABLE 4.11Rate constants in ATMS/methylchloride mixture :
experiments ATMS 8
M e a s u r e d C a l c u l a t e d M e a s u r e dT e m p / K k /s '* k , / s - ' k ^ / s - '
8 5 9 .5 0.00350 0.00255 0.000953861 0.00372 0.00274 0.000978861 0.00407 0.00324 0.000832871 0.00540 0.00379 0.00161872 0.00643 O.OO46O 0.00183886 0.0117 0.00793 0.00377887 0.0109 0.00746 0.00344888 0.0119 0.00765 0.00425896 0.0156 0.00978 0.00582896 0.0179 0.0109 0.00704897 0.0156 0.0108 0.00479902 0.0188 0.0116 0.00725902 0.0189 0.0114 0.00747903 0.0193 0.0130 0.00635909.5 0.0248 0.0151 0.00974911 0.0256 0.0159 0.00970911 0.0257 0.0153 0.0104913 0.0272 0.0154 0.0118917 0.0316 0.0170 0.0146918 0.0331 0.0186 0.0145919 0.0323 0.0196 0.0127
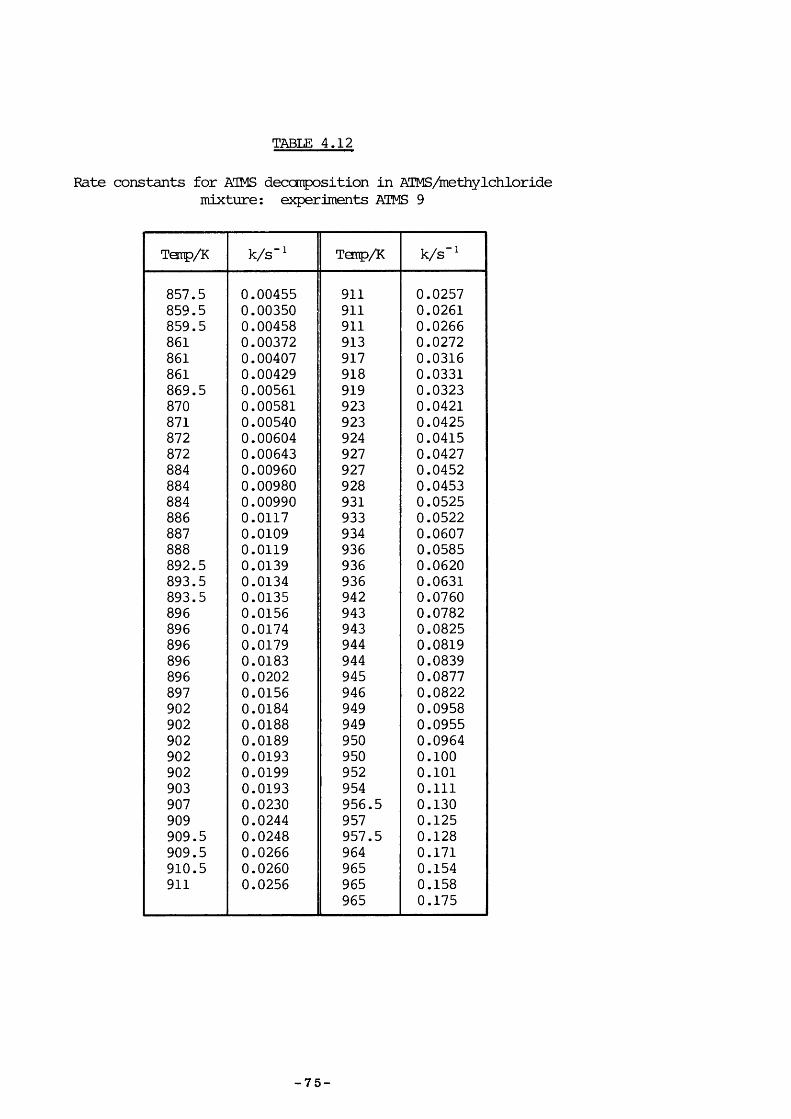
TABLE 4.12
Rate constants for AIMS decanposition in ATMS/methylchloride mixture: experiments ATMS 9
Temp/K k/s-i Temp/K k/s"'
857.5 0.00455 911 0.0257859.5 0.00350 911 0.0261859.5 0.00458 911 0.0266861 0.00372 913 0.0272861 0.00407 917 0.0316861 0.00429 918 0.0331869.5 0.00561 919 0.0323870 0.00581 923 0.0421871 0.00540 923 0.0425872 0.00604 924 0.0415872 0.00643 927 0.0427884 0.00960 927 0.0452884 0.00980 928 0.0453884 0.00990 931 0.0525886 0.0117 933 0.0522887 0.0109 934 0.0607888 0.0119 936 0.0585892.5 0.0139 936 0.0620893.5 0.0134 936 0.0631893.5 0.0135 942 0.0760896 0.0156 943 0.0782896 0.0174 943 0.0825896 0.0179 944 0.0819896 0.0183 944 0.0839896 0.0202 945 0.0877897 0.0156 946 0.0822902 0.0184 949 0.0958902 0.0188 949 0.0955902 0.0189 950 0.0964902 0.0193 950 0.100902 0.0199 952 0.101903 0.0193 954 0.111907 0.0230 956.5 0.130909 0.0244 957 0.125909.5 0.0248 957.5 0.128909.5 0.0266 964 0.171910.5 0.0260 965 0.154911 0.0256 965 0.158
965 0.175
-75-
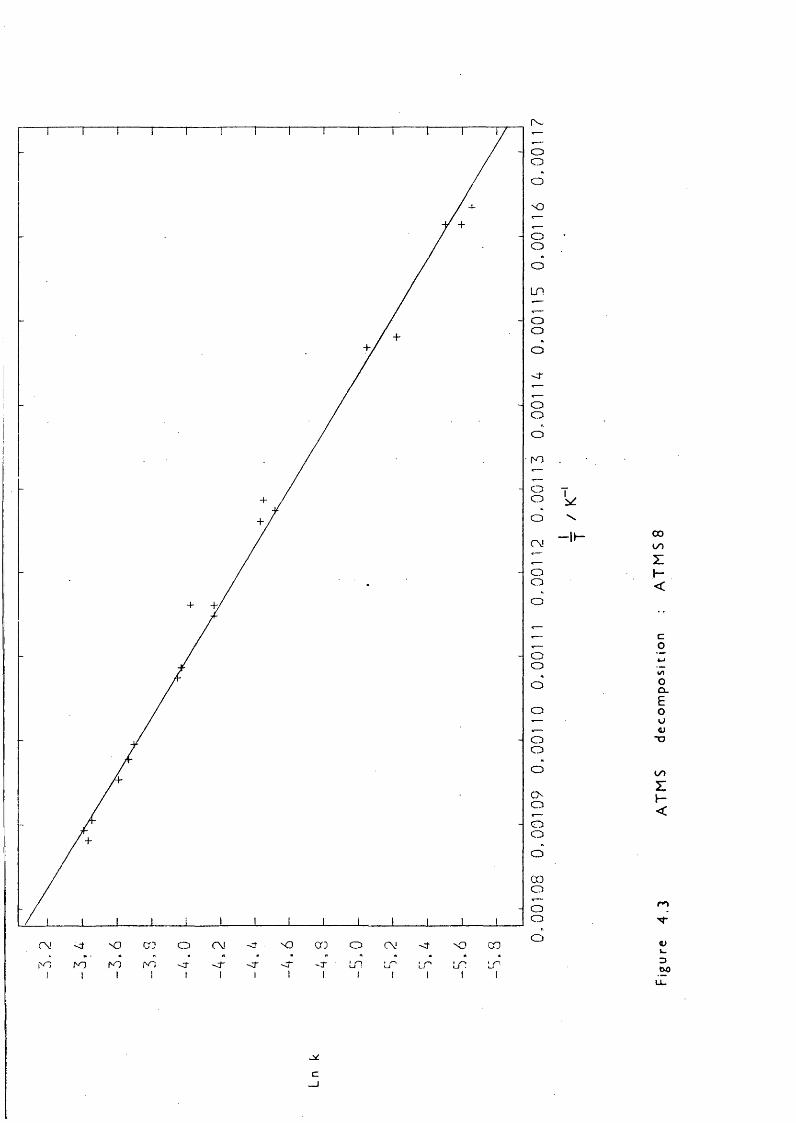
ooo
ooounooo
ooo
oooCM
ooo
oooooooooooo00ooooo 'Oo CM CM 00CM
o T\
-IH 00ooZh-<
001 £ o uCJ"O
toz<
ro
3(X)
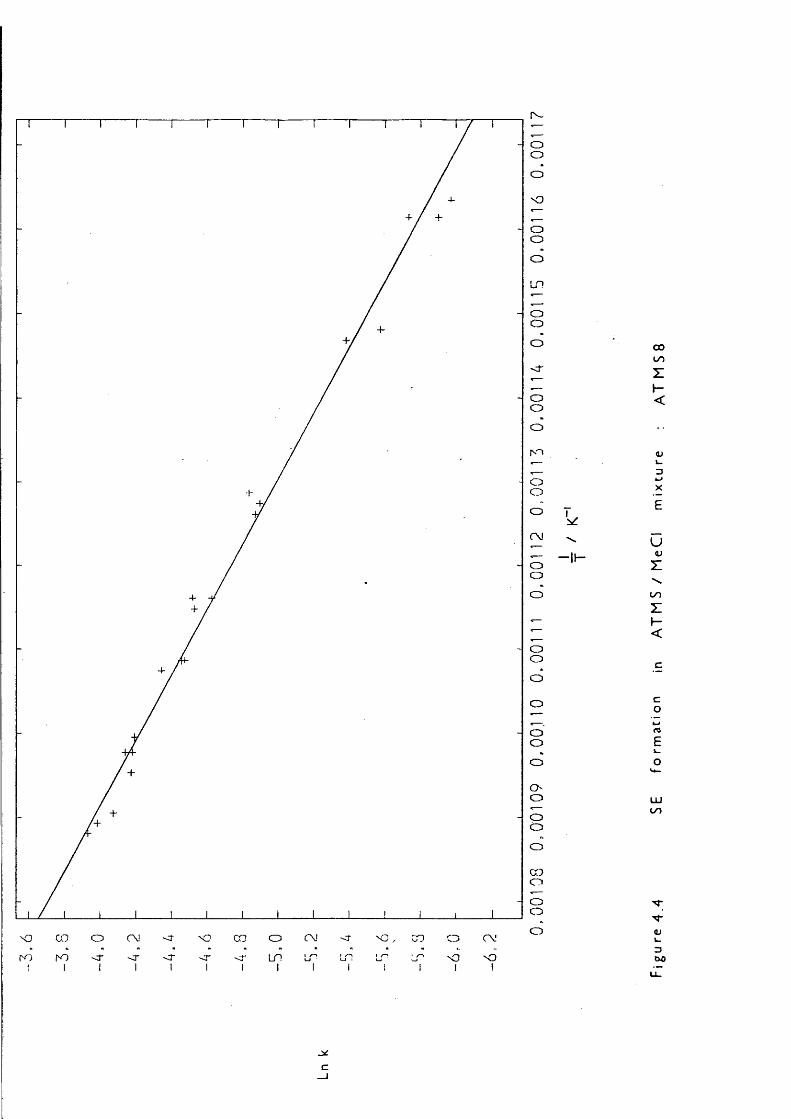
- o ooNDOOO
- O OO
ooo
oooOJ
ooo
oooooooooooo
oonO GO
roo C\!
<rI
<r<r <r
co oLO
I
OJ nO GO OnO
I
rvnd!
o
-IH
00LOzH<
U<ü
Z
L O
ZI-<
L O
Tf-0>3oo
c
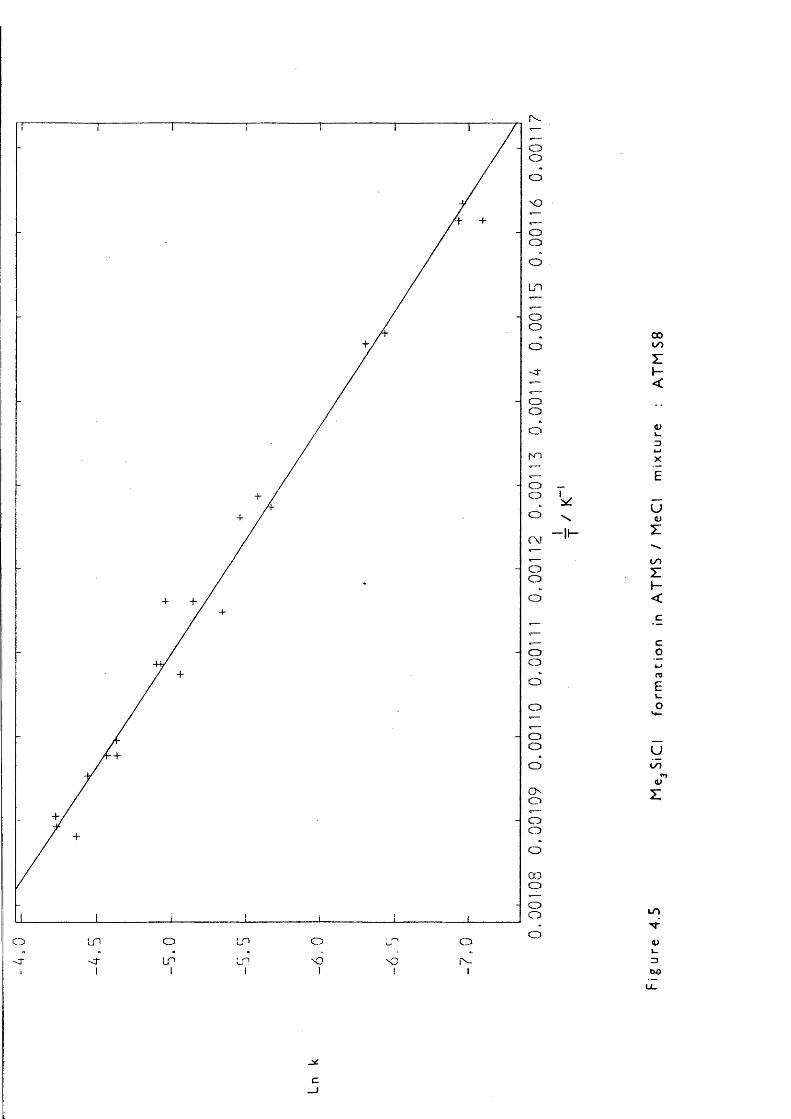
ooo
ooo
ooo
ooo
ooo
ooo
oooooooooooo00ooooooo i_n o
's- | H
00OOzl-<
uzunzK —<
uûooTz
un
3tX)
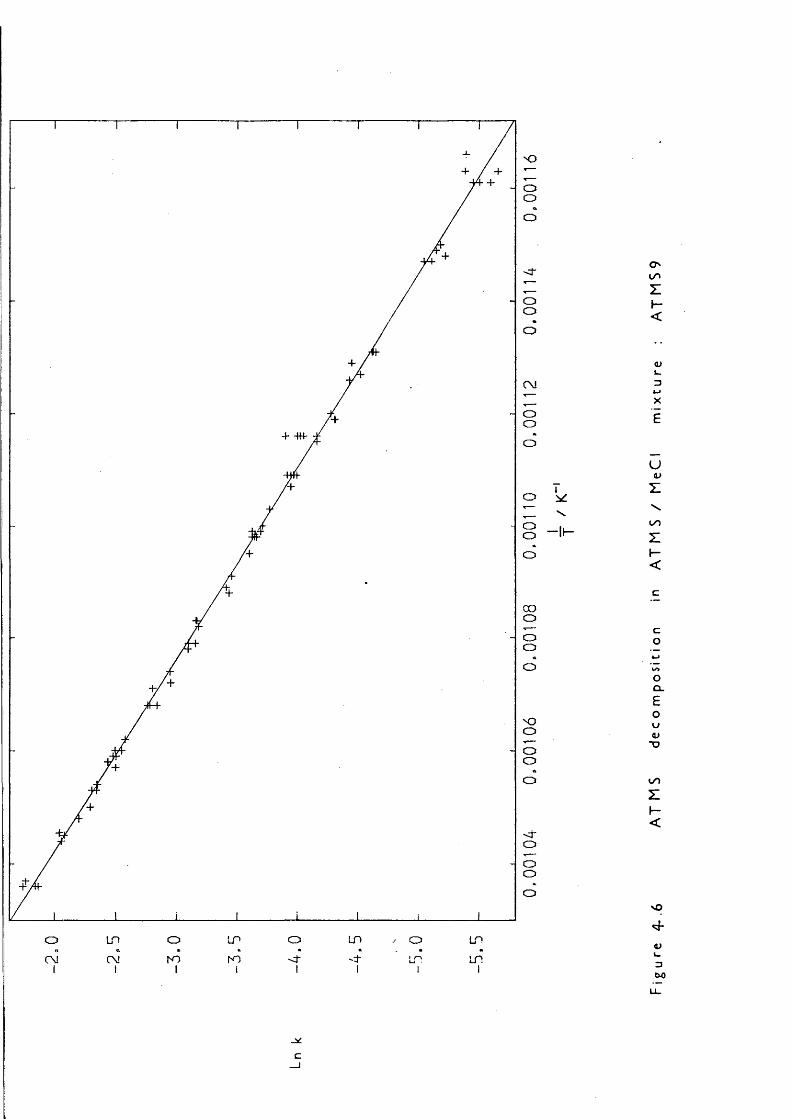
ooo
ooo
CM
OOO
oooo
cooooo
NOoooo
oooo
i_no LO o LPo oK) LPCM CM
L:V .
-IH
oIPzh-<
U( UZcozH<
OCL.Êou0>•U
coz
o
0)u( X )

logiok/s"^ = (12.30 ±0.10) - (241.93 ±1.79 kJ itiol"M/2.303 RT
4.6.1 Radical addition to allyltrimethylsilaneIn the radical addition to AIMS there are two sites of attack: terminal
or non-terminal. Studies both in the gas phase and in solution have shown that terminal addition is the dominant process in a favourable r e a c t i o n : a d d i t i o n occurs faster than with alkenes, an effect ascribed to 3 stabilization in the resulting radical^^^ vhich can decompose under suitable conditions. Migita et al.^^^ performed a series of reactions of phenyl radical additions to allyl compounds and found that there was a low yield of allylbenzene and trimethylsilyl radicals from ATMS:
Ph* • XMe3SiŒ2CH=CH2 — ► PhŒ2CHCH2 -4- SiMeg330 K >CH3CN solvent I
PhCH2CH=CH2 + *SiMe3 (4.9)
The allylbenzene yield depended on the strength of the bond broken: thiyl radicals were lost much more easily. All the above investigations were done at <400 K, but at ATMS pyrolysis temperatures, if terminal addition of trimethylsilyl radicals is a reversible process caused by bond rupture in the resulting radical, non-terminal addition would become important and provides an easy route to vinyItrimethyIs i lane and tetra- methylsilane vÆiich are always formed in comparable yields (Scheme 4.4) .
Our original proposal,based on Jones' labelling studies®" which require loss of methyl during the reaction sequence, was that vinyItri- methylsilane and tetramethylsilane resulted from terminal addition of a trimethylsilyl radical to ATMS followed by unimolecular elimination of tetramethylsilane to form an allyldimethylsilyl radical. This then re-
— 76“
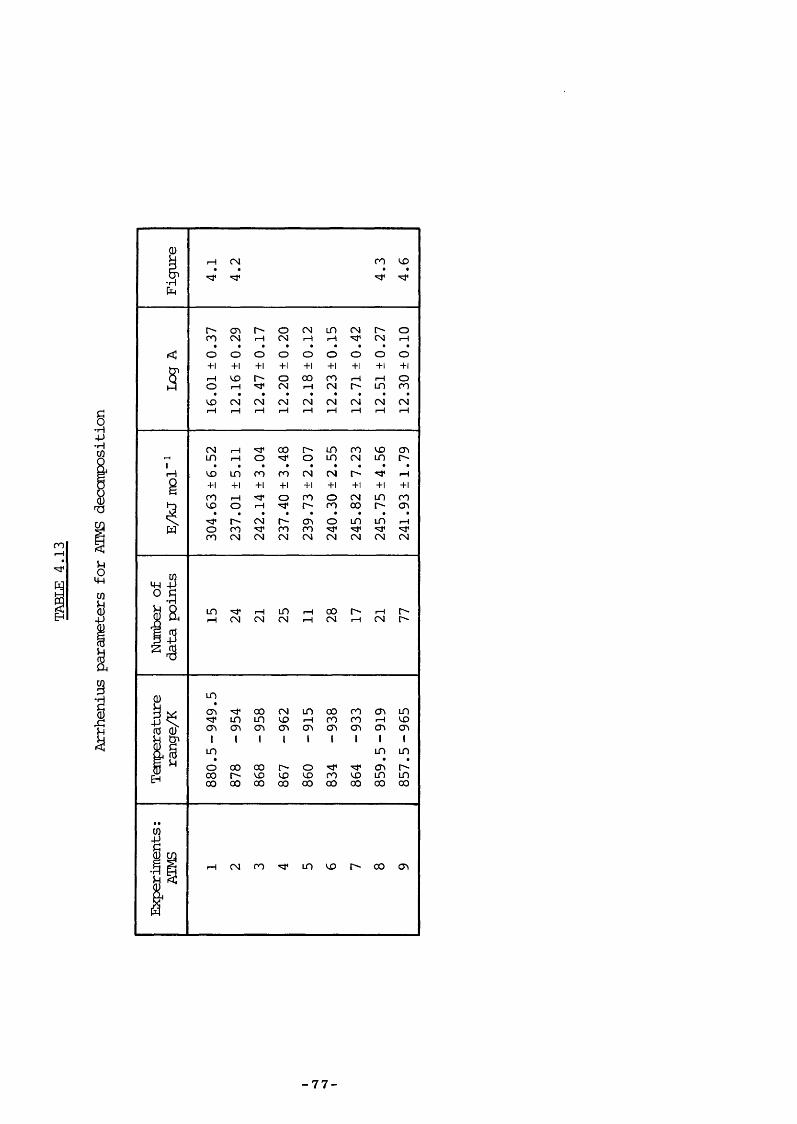
H
r H C N m VO
C7\ n- O CN m CN r~- oro CN 1-4 CN rH 1—4 TT CN iHo o O O O o O o O+1 +1 +1 +1 +1 +1 +1 +1 +11—1 VO o CO ro pH rH oo r—1 CN pH CN r- in COVO CN CN CN CN CN CN CN CN1—1 1—1 1-4 1-4 pH rH iH 1—1 iH
CN 1—1 CO in CO VO CTiin 1—4 o o in CN inVO in ro ro CN CN r~- iH+1 +1 +1 +1 +1 +1 +1 +1 +1ro 1—t TT o ro o CN m COVO o t-4 ro CO CTi
CN r- G\ o in m iHo ro ro CO TTro CN CN CN CN CN CN CN CN
LD >5* rHCM CN inCN C30CN CN
CN rn ^ in VO
mCO CN in 00 CO crv inm in VO iH CO CO pH VO
CTi (T\ CTi <Ti CTi cr> <Ti (Ti CTV
ino 00 CO r~- O
mC3>
inr»
CO VO vo VO CO VO m inCO CO 00 00 00 00 00 00 00
CO cr>
-77-

MesSi* + Mes Si— \ Me3Si—\ rA— siMe
Il l"
"■ r
I. •Me3SivT \ Mei+Si — — Si— Œ 2 + SiMe3
Me3Si^Scheme 4.4
arranged to viny Itr imethy Isilane (Scheme 1.3) . This apparently unlikely86mechanism can now be eliminated in the light of Barton's recent results
after repeating the same experiments as Jones.The plausibility of Schone 4.4 was investigated by a series of
experiments; an obvious check was to thermally generate trimethylsilyl radicals in the presence of ATMS below its pyrolysis temperatures to attempt to form viny Itr imethy Isilane and tetramethylsilane. A suitable precursor to trimethylsilyl radicals had to be found.
4.6.2 BenzyltrimethvlsilaneBenzyItrimethyIsilane was considered to be a likely candidate: its
silicon-carbon bond strength should be weaker than that in ATMS because of the slightly greater resonant stability of the benzyl radical compared to the allyl;perhaps allowing trimethylsilyl radical generation in the presence of unpyrolysed ATMS.
The pulsed stirred-flow technique showed that benzyItrimethyIsilane pyrolysed at about the same rate as ATMS to give several products : mainly five unidentified higher boiling point ccmpounds and, to a lesser extent, several other products including a low yield of trimethyIsilane.
82The pyrolysis of benzy Itr imethy Isilane has been studied by Sakurai et al. vho found that the principal product was:
-78-

SiMez
i.e. a carpound with a high boiling point, so this is therefore in agreement with our rudimentary observations. Our benzy Itr imethy Isilane contained two minor impurities vdiich ranained unaffected during the course of the reaction. Thus it was not possible to design an experiment in vhich ATMS itself did not pyrolyse, but it was nevertheless feasible to perform co-pyrolyses in vhich both ccmpounds decomposed.
A low pressure, ça. 0.2 Torr, of ATMS was chosen such that, vhen pyrolysed at 865 K with a reaction vessel residence time of ~13s vhich gave approximately 5% decomposition, it did not contain viny Itr imethy 1 - silane or tetramethylsilane amongst the products vhich were: methane,ethene (not separated by the column), propene, trimethyIsilane and DSCB. When the same pressure of ATMS was pyrolysed in the presence of an equal quantity of benzyItrimethyIsilane, it was found that vinyltrimethylsilane and tetramethylsilane had become major products : the relative yields ofviny Itr imethy Is i lane : tetramethylsilane : tr imethy Isilane : propene were 4:3:7:5. These relatively large quantities of vinyltrimethylsilane and tetramethylsilane could not be reproduced using neat ATMS whatever the pressure, and therefore must have been formed because of the presence of benzy Itrimethy Isilane, almost certainly a result of a bimolecular reaction between trimethylsilyl radicals and ATMS. The low yield of tr imethy 1- silane in the neat benzy Itrimethy Isilane experiments could be esq^lained by trimethylsilyl radicals favouring reaction with the aromatic ring, leading to the formation of higher molecular weight products. In the presence of ATMS there was a further addition reaction vhich the trimethylsilyl radicals could take part in. The higher boiling point products were still present in co-pyrolysis experiments.
-79-

4,6.3 Mercury dimethyl168Mercury dimethyl pyrolyses below 800 K giving methyl radicals vhich,
in the presence of tr imethy Isilane, undergo a metathesis reaction, efficiently generating trimethylsilyl radicals (Scheme 4.5).®’ ^^
HgMez — ► HgMe + "Me HgMe — ► Hg + "Me
•Me + MeaSiH — ► MegSi" + MeH
Schgne 4.5
Before this method was utilized it was necessary to investigate the effect of methyl radical reaction with ATMS.
ATMS/mercury dimethyl mixtures (from 3:1 to 1:1) were pyrolysed at 800 K using LPP and the pulsed stirred-flow technique. A large quantity of methane was produced; products formed following a hydrogen metathesis reaction were: mainly 1,l-dimethyl-l-silacyclopent-3-ene (m/e = 97'*’:M'*'-Me) (the absence of DSCB incidentally indicated that SE is not formed by Reaction 4.10; cyclisation is preferred) , m/e = 83''' (possibly 1,1-
I— Si— y Me2Si=CH2 + • }> (4.10)CH
dimethyl-1 -silacyclobut-2-ene) and traces of m/e OS’*" and 87^.Three additional minor products appeared in the pulsed stirred-flow
experiments, two of which were possibly hexamethyldisilane (Reaction 4.1) and 2,2,4,4-tetramethyl-2,4-disilapentane (Reaction 4.11); the third, unidentified, eluted just before 1,1-dimethy1-1-silacyclobut-2-ene.
MesSi" + MesSiCHz — ^ MesSiCHzSiMea (4.11)
The most important products, however, were: tr imethy Isilane, tetramethylsilane and vinyltrimethylsilane (in the ratio 4:2:1) and m/e = 56^, 55+,
-80-

3Ui
i
•HCO
Ia
I r•HCO
+
CO
s
I—cS OC •H•H -PB •HU "O0> •d4-> ' a
-f
• §CO
I
•HCO
i
i rH03a d
+ •H oa •H-P0)•HH->-ds 1 ■d1 a d1 §
g
— 81 —

42^, 41^, 40'*’, 39’*', 27'*’, 26'*' - mainly but-1-ene with sane propene, unseparated by the GC column along with methane, rationalised as resulting frcm methyl radical addition to ATMS both terminally and non-terminally (Schone 4.6) . Bond rupture in the radical resulting frcm terminal addition - vÈiich either leads to reaction reversibility or other products - is much more important at 800 K than at <400 K.
Repeating these experiments in the presence of a four-fold excess of trimethyIsilane gave results entirely consistent with this scheme. Preferential abstraction of hydrogen frcm tr imethy Isilane by methyl radicals (Scheme 4.5) increased the concentration of trimethylsilyl radicals; more vinyltrimethylsilane and less 1,1 -dimethyl-1 -silacyclo- pent-3-ene was fomed.
4.6.4 Allyltrichlorosilane/mercurv dimethvlLPP ej^Deriments at 800 K using a 1:1 mixture of allyltrichlorosilane
and mercury dimethyl gave mainly methane, but-1-ene and silicon tetrachloride (m/e = 168+, M+) (Schene 4.7).
ClsSi— f + Me*
CUSi
Scheme 4.7
Cl 3 Si
Cl,Si" + /
No evidence was found for any major products resulting frcm non-terminal addition of methyl radicals to ATMS. The same experiments in excess of tr imethy Isilane gave trimethylchlorosilane (m/e = 108+, 93+: M+,M+-Me) as the only major product, i.e. trimethylsilyl radicals abstracted chlorine frcm allyltrichlorosilane faster® than they added to the allyl group.
— 8 2 —

4.7 DISCUSSION
These experiments, and those with benzy Itr imethy Isilane, are consistent with our proposed mechanism for vinyltrimethylsilane and tetramethylsilane production in ATMS pyrolysis (Scheme 4.4) .
During ATMS pyrolysis, Wiilst products result from trimethylsilyl radical addition to ATMS, no product is formed following allyl radical addition: the LPP and sealed tube experiments showed no evidence foreither penta-1,4-diene (m/e = 68*, 67*, 53*: M*, M*-H, M*-Me) - frcmnon-terminal addition - or hexa-1,5-diene (m/e = 67+, 54+: M+-Me,M*-C2Hi+) - frcm terminal addition. The absence of hexa-1,5-diene also indicated that allyl radicals failed to dimerise.
To understand why trimethylsilyl radicals undergo two major concurrent processes (hydrogen abstraction and addition to AIMS) whilst allyl radicals have only one main result (propene formation) a ccmparison of bond strengths is helpful :
kJ mol-1MesSi-H 378 (Ref. 22)CH2=CHCH2- H 371 (Ref. 146)
MeaSi-C -374 (Ref. 22)CH2=CHCH2- C -303 (Ref. 146)
The abstraction of hydrogen by trimethylsilyl or allyl radicals shouldoccur at approximately the same rate, but trimethylsilyl radical additionto the carbon-carbon double bond in ATMS should be much faster than thecorresponding reaction for allyl radicals. The Arrhenius parameters for
170addition of trimethylsilyl radicals to ethene have been measured, giving: logio k/(dm^ mol” s"M = (7.0 ±0.2) -(10.50 ±0.84 kJ mol"^)/2.303 RT. A hydrogen metathesis reaction of trimethylsilyl radicals has been determined to occur approximately twenty times faster than the
— 8 3 —

corresponding reaction with methyl radicals but Walsh considered thisextranely unlikely and estimated the activation energy to be ça. 71 kJ
_ 22mol" . If log A for this process is 8.5, then at 900 K k(addtn) = 2.5x10® mol dm“ s"S and k(abst) = 2.3 x lO' mol dm"^ s"^. Therefore addition to ATMS should occur more readily than hydrogen abstraction, even without taking into account the approximate five-fold enhanced reactivity of AIMS to addition reactions.This agrees with the experimental observations.
In the mercury dimethyl experiments it was found that the rate of methyl radical addition to ATMS at 800 K occurs faster than abstraction, but both occur concanitantly. A ccmparison of Arrhenius parameters for methyl radical addition to ethene and a hydrogen abstraction reaction (frcm ethane) can be made:^® for addition, logio k/(dm^ mol"^ s"M =(8.5) - (32.2 kJ mol“M/2.303 RT; for abstraction, logio k/(dm^ mol"^ s" ) = (8.5) - (45.2 kJ mol"M/2.303 RT. At 800 K k(addtn) = 2.5x10® mol dm"^ s" and k(abst) = 3.5 x lO® mol dm"^ s” ; this means that both should occur simultaneously, agreeing with experimental observations.
Further evidence in support of our proposed mechanism for ATMS pyrolysis at higher pressures was obtained in experiments designed to investigate the temperature required for the radical resulting frcm terminal addition to decompose. This information was required to show that our ideas did not conflict with the results obtained by other workers { w h o found that terminal addition was much more important than non-terminal at <400 K). To facilitate these investigations a photochemical method was used to generate trimethylsilyl radicals: that ofmercury photosensitization.
4.8.1 Mercury Photosensitization171The mercury discharge lamp is the most extensively used source of
— 8 4 —

light in photochemistry. In high voltage, lew pressure mercury lanps adischarge at sane hundreds of volts is passed through a mixture of mercuryvapour (ça. 10” Torr) and a rare gas, e.g. helium, neon or argon, atroom tonperature. This produces excited mercury atoms vhich subsequentlyemit radiation consisting primarily of two bands - mainly (-90%) 253.7 nm,
172and 184.9 nm - as they return to their ground state:
Hg6(^Pi) — Hg6(^So) + hv (184.9nm) (4.12)Hg6(^Pi) — ^ Hg6(^So) + hv (253.7nm) (4.13)
The transition (4.13) is formally forbidden as the change in total spinangular momentum AS = 1, but this selection rule is relaxed for heavierelements. If the lamp is shone on a gaseous mixture of mercury andanother compound, this emitted radiation can be absorbed to give excitedmercury atoms which can then be quenched by the additive.
Quenching cross-sectional values for compounds containing silicon-hydrogen bonds are much higher than those with only carbon-hydrogenlinkages : the quenching efficiency of the silicon-hydrogen bond is much
173greater than the carbon-hydrogen bond. A consequence of this is that excited mercury atoms are selectively quenched by the silicon-hydrogen bond in trimethyIsilane: this gives a clean source of trimethylsilyl radicals vhich initiate a simple non-chain sequence vhich is obeyed ip to
173at least 673 K (Schane 4.8) .
Hg ('So) + hv (253.7 nm) — Hg (3pi) (4.14)Hg (3pi) + Me3SiH — ► tfeoSi* + H' + Hg ('So) (4.15)H* + M03;SiH — ► H2 + Me3Si* (4.16)H" + H* + M — ► H2 + M (4.17)
— Me3SiSiMe3 (4.18)
Scheme 4.8
— 8 5 —

Hydrogen atom abstraction from a silicon-hydrogen bond by atonic hydrogen(Reaction 4.16) occurs approximately 60 times faster at 485 K than the
174corresponding abstraction from a carbon-hydrogen bond. Similarly, dimethylsilane gives Hz and (jyfe2SiH)2 , v^le methylsilane gives Hz and (MeSiHz )2 as the main products of photosensitization. Although the main course of these reactions is determined by silyl radical reactions as in Schone 4.6, sane silylene formation may also occur by disproportionation reactions, e.g.:
2l4eSiHz — ► HMeSi: + MeSiHg (4.19)
The reaction of excited mercury atoms with trimethylsilane hassuccessfully been used as a source of trimethylsilyl radicals ininvestigations of their reactions with other species. B e c a u s e theonly silicon-hydrogen bonds in a mixture of ATMS and trimethylsilane arein the latter, mercury photozensitization should give trimethylsilylradicals whose subsequent reaction with ATMS could be studied. However,before applying this technique, it was necessary to stuc^ the reactionbetween hydrogen atoms and ATMS: during the course of the reactionsequence (Scheme 4.8) hydrogen atoms are produced which will inevitablyreact themselves with ATMS as well as trimethylsilane. The reaction withATMS should be similar to that with propene; hydrogen atom addition toethene coirpetes with hydrogen abstraction from a silicon-hydrogenbond^^^ (although no such problems were found with tetrafluoro-
14ethylene ) . The reaction between hydrogen atoms and ATMS could beinvestigated using molecular hydrogen and excited mercury atoms as a
178source:Hg(^Pi) + Hz — ^ Hg(^So) + 2H* (4.20)
The detailed mechanism is rather complex, involving species such as HgH,
-86-

also formed by the quenching of Hg ( Pi) :
Hg(^Pi) + Hz — HgH + H« (4.21)
Performing these experiments had the additional advantage that the tenperature required for the deccmposition of the radical resulting frcm terminal hydrogen atom addition to ATMS could be found.
All the photolytic experiments were done at Mülheim a.d. Ruhr, vAieresimilar work has recently been d o n e . A Icw-pressure mercuryarc from Grantzel, Karlsruhe was used, a vycor filter ronoving the189.4 nm band. Steady state photolysis was carried out in a 200 cm^cylindrical quartz cell having 5 cm diameter quartz suprasil windows.To allow for reactions to be performed at different torperatures, analuminium oven was used, positioned approximately 5 cm away frcm the larrpso that wire meshes to alter the intensity of light reaching the sanple
171could be interposed. Gaseous samples and mercury vapour were loaded into the reaction vessel on a separate vacuum line; product analysis was done by gas chromatography as described earlier in this chapter.
4.8.3 ExperimentalEach series of experiments was given a name to aid description. In
addition to t h e two series of experiments already described (HGl and HG2) , two others were carried out to help product identification (HG3 and HG4) ; extra experiments were done afterwards (HG5) ; the details of all are shown in Table 4.14. The argon used in experiments HG2, HG3 and HG5 was to maintain constant pressure. In each case the irradiation time was 3 minutes apart from the HG4 experiment which was only 2 minutes.
All discussions and Tables use the shorthand notation (described earlier in this chapter) for simplicity (Appendix 4.2) .
-87-
CN CN CN CN
g
a
-p
wIN
3I &I
m ro cnr~- r-in in inm m mr~ TT
..ro n mm cn m..o o oo o om m ro
5-Hœ m m
3cn
oon
s
CO
mm
ooro
H gœI k è i k a s s i ë x â l âH H H H h H H H H h H H H Hy 0 S 0 o 5 0 3 ^ y ^ 0 B BEh Eh Eh EH EH Eh EH EH Eh Eh Eh Eh Eh Eh
CN O in m o in in o ro CO o in in oCN O CN O CN O CN O CN
Ehr- CO CO in CTi1—1 i-H t—1 1—1 rH
Iz: a iin
— 88“

4.8.4 Experiments HG4Mercury photosensitization produced mainly two radicals: hydrogen (H*)
and trimethylsilyl (Si»)« H« radicals add to, rather than abstract a hydrogen from, CC = C;^^^ only about 6% addition occurs non - terminal ly : C G C is formed in high yield. The main products (Table 4.15) from these experiments therefore resulted from reactions of: H«, Si*, CGC (andG G C) , and were assigned on this basis, by making coiparisons with similar experiments,^^’ or by comparing retention times with authentic compounds :
G G G G G G G G G and Si Si66 ' 6
resulted from dimérisation reactions,SiG = G Si G G Si GG = G and SiGGG or Si G = G G
6 ' 6 '
(reactions of Si* with GG = G, GCG and GGC)Si G G Si /siGC + Si*
6 I 6
Si G G G G /SiGC + CG I I I IG G \ G G
As ATMS is particularly susceptible to radical attack, the other productswere perhaps:
Si G G G G G (SiGG = G + GCG, then + Si*)Si G
Si G G = G G G Si (cis and trans) or Si G = G G G G Si (c/t)G G
Si G G = G + * GGSiIG
— 89“
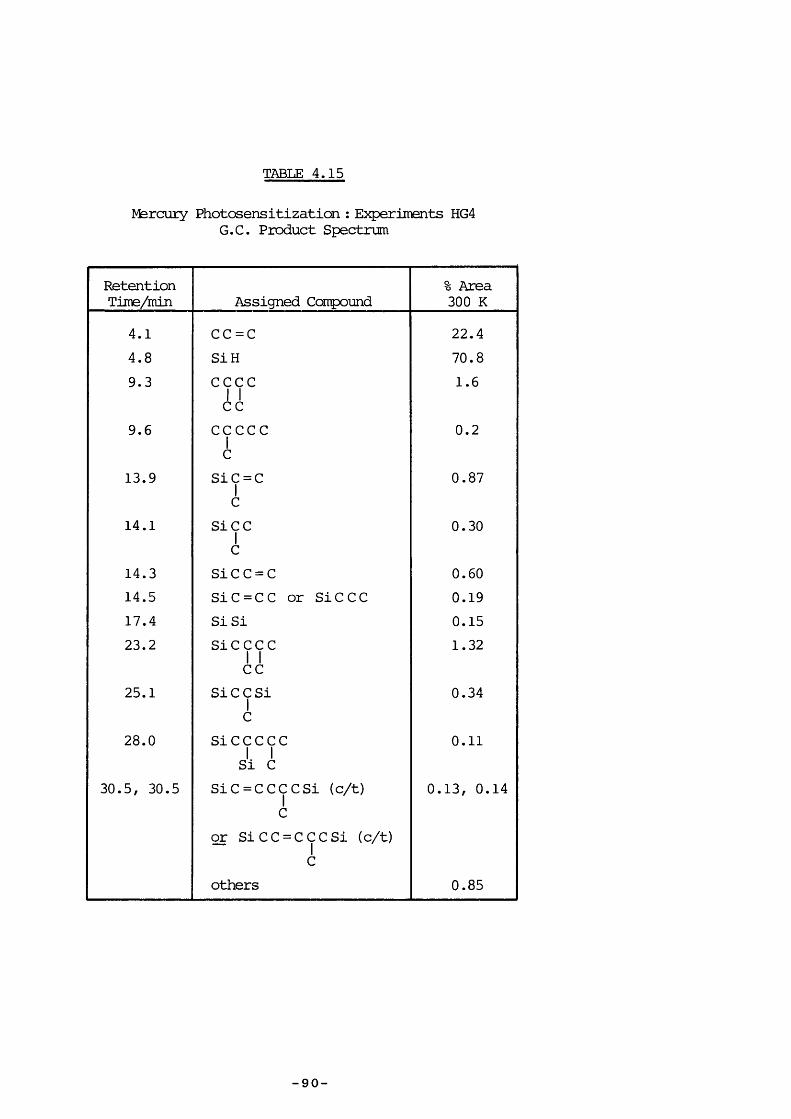
TABLE 4.15
Mercury Photosensitization ; Experiments HG4 G.C. Product Spectrum
RetentionTime/min Assigned Conpound
% Area 300 K
4.1 CC = C 22.44.8 SiH 70.89.3 C C C C
661.6
9.6 ccccc6
0.2
13.9 SiC = C 1C0.87
14.1 SiGC1G0.30
14.3 Si G G = G 0.6014.5 SiG = GG or SiGGG 0.1917.4 Si Si 0.1523.2 Si G G G G
661.32
25.1 Si G G Si 1G0.34
28.0 Si G G G G G Si G
0.11
30.5, 30.5 SiG=GGGGSi (c/t)G
or SiGG = GGGSi (c/t) G
0.13, 0.14
others 0.85
-90-
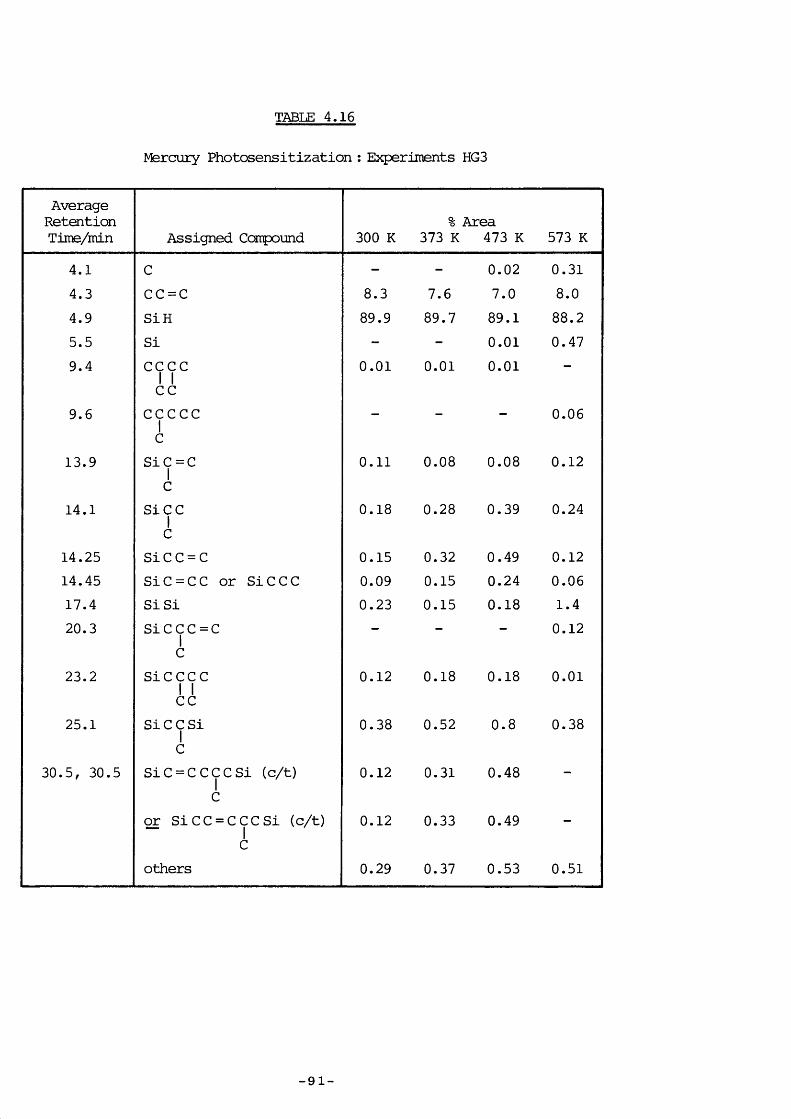
TABLE 4.16
Mercury Photosensitization : Experiments HG3
AverageRetentionTime/min Assigned Compound 300 K
%373 K
Area 473 K 573 K
4.1 C - - 0.02 0.314.3 CC = C 8.3 7.6 7.0 8.04.9 SiH 89.9 89.7 89.1 88.25.5 Si - - 0.01 0.479.4 CC C C
660.01 0.01 0.01
9.6 C C C C C1c0.06
13.9 SiC=C1c0.11 0.08 0.08 0.12
14.1 SiCC1C0.18 0.28 0.39 0.24
14.25 Si C C = C 0.15 0.32 0.49 0.1214.45 SiC = CC or SiCCC 0.09 0.15 0.24 0.0617.4 Si Si 0.23 0.15 0.18 1.420.3 SiCCC = C 1C
0.12
23.2 Si C C C C 1 1 cc0.12 0.18 0.18 0.01
25.1 Si C C Si C
0.38 0.52 0.8 0.38
30.5, 30.5 SiC = CCCCSi (c/t) C
0.12 0.31 0.48
or SiCC = CCCSi (c/t) C
0.12 0.33 0.49
others 0.29 0.37 0.53 0.51
— 9 1 —
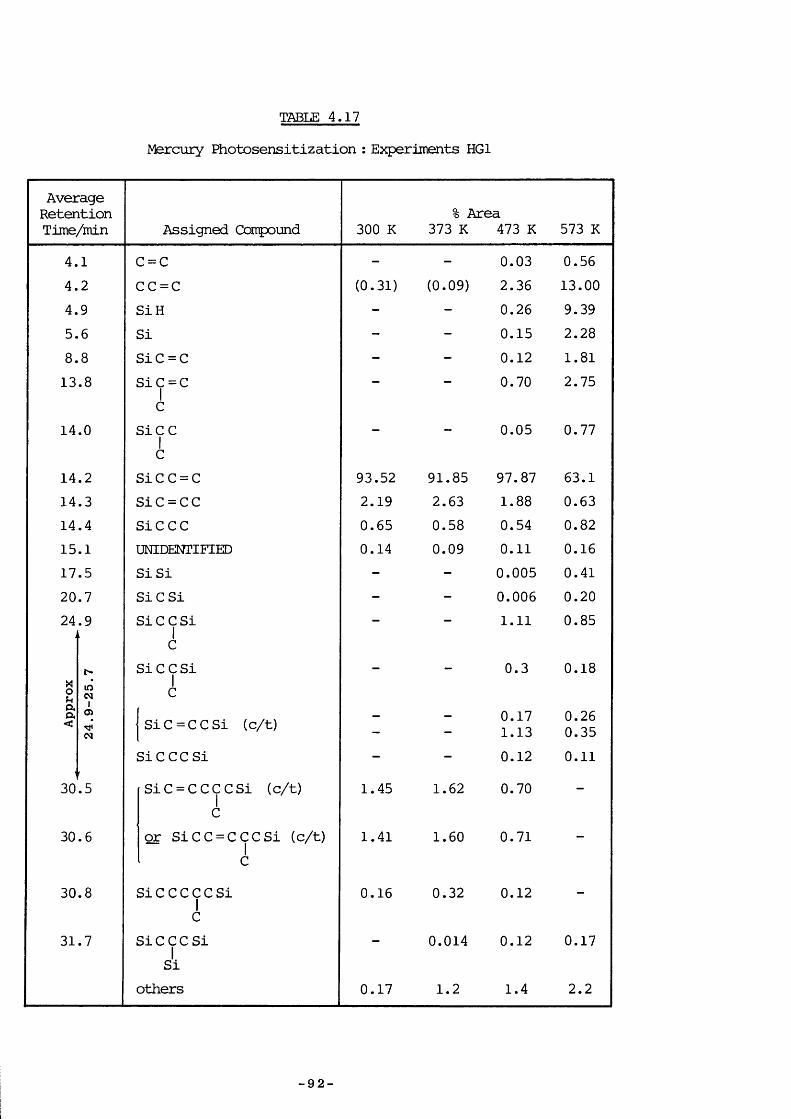
TABLE 4.17
]yfercury Photosensitization : Experiments HGl
AverageRetentionTime/min Assigned Compound 300 K
% Area 373 K 473 K 573 K
4.1 2 = C - - 0.03 0.564.2 CC = C (0.31) (0.09) 2.36 13.004.9 SiH - - 0.26 9.395.6 Si - - 0.15 2.288.8 SiC = C - - 0.12 1.8113.8 SiC = C - - 0.70 2.75
1C14.0 SiCC - - 0.05 0.77
1c14.2 Si C C = C 93.52 91.85 97.87 63.114.3 Si C = C C 2.19 2.63 1.88 0.6314.4 SiCCC 0.65 0.58 0.54 0.8215.1 UNIDENTIFIED 0.14 0.09 0.11 0.1617.5 Si Si - - 0.005 0.4120.7 Sic Si - - 0.006 0.2024.9 SiCCSi - - 1.11 0.85
1c
OPP.A<
i>inN1o>N
SiCCSi1CSi C = C C Si (c/t) - -
0.3
0.171.13
0.18
0.260.35
SiCCC Si - - 0.12 0.1130.5 SiC = CCCCSi (c/t) 1.45 1.62 0.70 -
C30.6 or SiCC = CCCSi (c/t) 1.41 1.60 0.71 -
C30.8 Si C C C C C Si 0.16 0.32 0.12 -
C31.7 SiCCC Si - 0.014 0.12 0.17
Siothers 0.17 1.2 1.4 2.2
— 9 2 —

4.8.5 Experiments HG3These experiraents alloved a check to be made on the conclusions drawn
already: the absence of hydrogen meant that products resulting fromreactions of CGC should be less important or absent. Peaks vhich met these requirements (Table 4.16) were:
C C CC, C C C C C , SiCC, Si C C C C and Si C C C C CI I I I I I I ICC C C cc S i c
At 573 K Schemes 4.9 and 4.10 may have operated:
SiCCC + CC = C — » - S i C C C C — ►C* + SiCCC = CI I ICC I C
CScheme 4.9
SiCCC + SiCC = C — ^ Si C C C C — ^ SiC* + Si C C C = CI I . ICCSi I C
SiScheme 4.10
Experiments HG3 and HG4 enabled several compounds' retention times to bedetermined, in particular Si Si and SiCCSi
C
4.8.6 Excperiments HGllyfercury photosensitization produced H* atons which subsequently
reacted with ATÜ4S in a similar fashion to their reaction with C C = C , as expected: the products (Table 4.17) resulted from reactions of theradicals in the concentration : SiCCC »SiCCC. Below 473 K, inaddition to seme C C = C (which appears to be an erroneous result) , the major products were:
-93-

SiC = CC, SiCCC, Si C = C C C C Si (c/t) or Si C C = C CC Si (c/t) ,C C
Sic C C C C SiC and an unidentified product.
Si C = C C and Si C C C were assumed to be the peaks at slightly longerretention time than SiCC = C, in the order of their boiling points(Appendix 4.1) . By 473 K there was a dramatic change in the product G.C.spectra: several more ccmpounds were formed, deduced to be:
large amounts of CC = C and SiH, C=C, Si, SiC = C,SiC = C, SiCC, Si Si , SiCSi, SiCCSi, SiCCSi,
C C C CSi C = C C Si (c/t), SiCCCSi, Si C C C Si
Si
These products are consistent with the reaction sequences in Scheme 4.11 (analogous to Scheme 4.6) in which significant terminal and non-terminal addition of hydrogen atoms to ATMS has occurred, followed by the breakdown of both the resulting radicals; secondary reaction of Si* radicals with ATMS ensued, the products indicating that both terminal and nonterminal addition had occurred. The peaks due to SiCCSi, SiCCSi ,
6 6
SiC=CCSi (c/t) and SiCCCSi could be assigned with confidence as fivepeaks were expected in this region of the spectrum, as observed, andbecause the retention time obtained for Si C C Si from experiments HG3 and
CHG4 coincided with the second peak in this group, again as expected (frcm boiling points) . Additional reactions also took place.
The relative importance of each H* atom addition route can be estimated at 573 K from the yields of C = C (0.56/2) and CC = C (13.00/3) from which it is calculated that non-terminal/terminal addition = 6.5%. This value
-94-

H- + SiCC = C — ^ SiCCC — ^ C C = C + Si- — ► SiH
\ / SiCC = C \
SiCCC SiCCC SiCCCSis'i
t IC = C + Sic Si Sic + SiC = C
Scheme 4.11
181agrees closely with that obtained (6%) by Cvetanovic and Falconer for the reaction of H- atons with propene.
4.8.7 Experiments HG2Mercury photosens itiz ation produced H- and Si- (Scheme 4.8) which both
reacted with ATMS, giving (Table 4.18) below 373 K:Si C = C C , SiCCC, SiC = CCCCSi (c/t) or Si CC = C C C Si (c/t)
C Cand Sic C C C C Si
C (frcm reactions of hydrogen atons) , with much largerquantities of:Si Si, SiCCSi, SiCCSi, Si C = CCSi (c/t) , SiCCCSi, SiCCCSi
C C Si(frcm reactions of Si-) .At 473 K and above the product spectra show a strong similarity with those for experiments HGl except that the reactions of Si- are much more important now because of its higher concentration : the SiC = C yield isincreased in relation to the extent of hydrogen atom reaction, indicated,for example, by the amount of C = C formed at 573 K:Experiments HGl: C = C yield = 0.28; SiC=C yield = 0.36;Experiments HG2: C = C yield =0.03; SiC = C yield = 0.18.
-95-
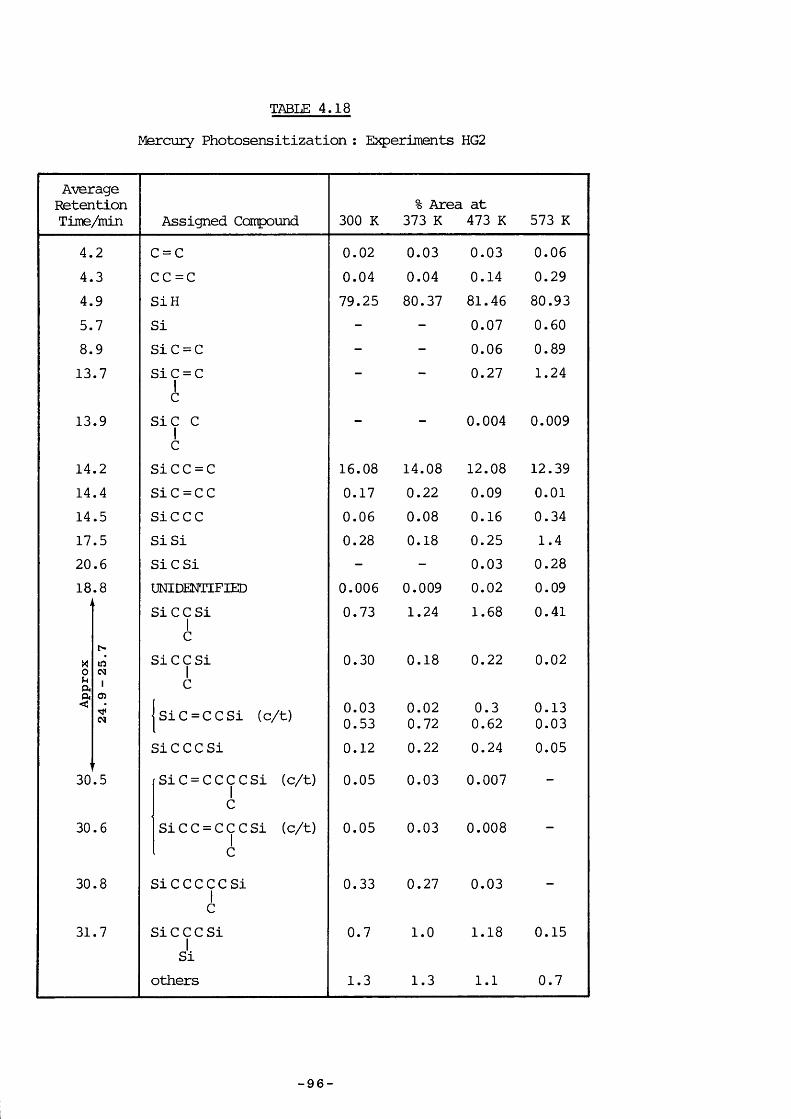
TABLE 4.18Mercury Photosensitization : Experiments HG2
AverageRetentionTime/min Assigned Compound 300 K
% Area 373 K
at473 K 573 K
4.2 : = c 0.02 0.03 0.03 0.064.3 C C = C 0.04 0.04 0.14 0.294.9 SiH 79.25 80.37 81.46 80.935.7 Si - - 0.07 0.608.9 SiC = C - - 0.06 0.8913.7 SiC = C - - 0.27 1.24
C13.9 SiC C - - 0.004 0.009
1C14.2 SiCC = C 16.08 14.08 12.08 12.3914.4 Si C = C C 0.17 0.22 0.09 0.0114.5 SiCCC 0.06 0.08 0.16 0.3417.5 Si Si 0.28 0.18 0.25 1.420 .6 Sic Si - - 0.03 0.2818.8 UNIDENTIFIED 0.006 0.009 0.02 0.09
Si C C Si 0.73 1.24 1.68 0.41
XokA5*
M10)M
cSiCCSi
CSiC = CCSi (c/t)
0.30
0.030.53
0.18
0.020.72
0.22
0.30.62
0.02
0.130.03
SiCCCSi 0.12 0.22 0.24 0.0530.5 SiC = CCCCSi (c/t)
C0.05 0.03 0.007 -
30 .6 Si C C = C C C Si (c/t) C
0.05 0.03 0.008
30.8 Si C C C C C Si 0.33 0.27 0.03 -
1c31.7 SiCCCSi 0.7 1.0 1.18 0.15
Siothers 1.3 1.3 1.1 0.7
-96-

4.8.8 Experiments HG5This series of experiments was performed in order to investigate
further radical addition processes. Mercury photosensitization produced H* and methylsilyl radicals, denoted by Hz Si*, (as described earlier in this chapter). The product spectra (Table 4.19) were very complex, giving several high boiling point products Wiich are not shown; peaks vere assigned as indicated. Two of the products formed at 573 K were thought to be Hz SiC = C and Hz SiCC = C. If this is correct then this WDuld show that Hz Si* reacted with ATMS (Scheme 4.12) .
Hz Si* + Si C C = C ^ SiCCC Si Hz — ► Hz SiCC = C + Si*
\ ^ SiHSiCCC HzSiC = C + Sic
I ^ ISiHz TSi
Scheme 4.12
These reaction sequences were superimposed on those in Scheme 4.11 vAiich provided further ccmplications. The formation of Si Hz was probably caused by radical addition to Hz SiCC = C (Scheme 4.13) .
Hz SiCC = C + R* — ► Hz SiCCC — ► Hz Si C + RC = C
« IHz Si
Schane 4.13
4.8.9 DiscussionThe mercury photosensitization experiments provided further evidence
in srpport of our proposed mechanism for ATD/B pyrolysis at high pressure (i.e. above ça. 0.3 Torr): non-terminal trimethylsilyl radical additionto ATMS gives a radical which decomposes at temperatures above 473 K,
“97 —
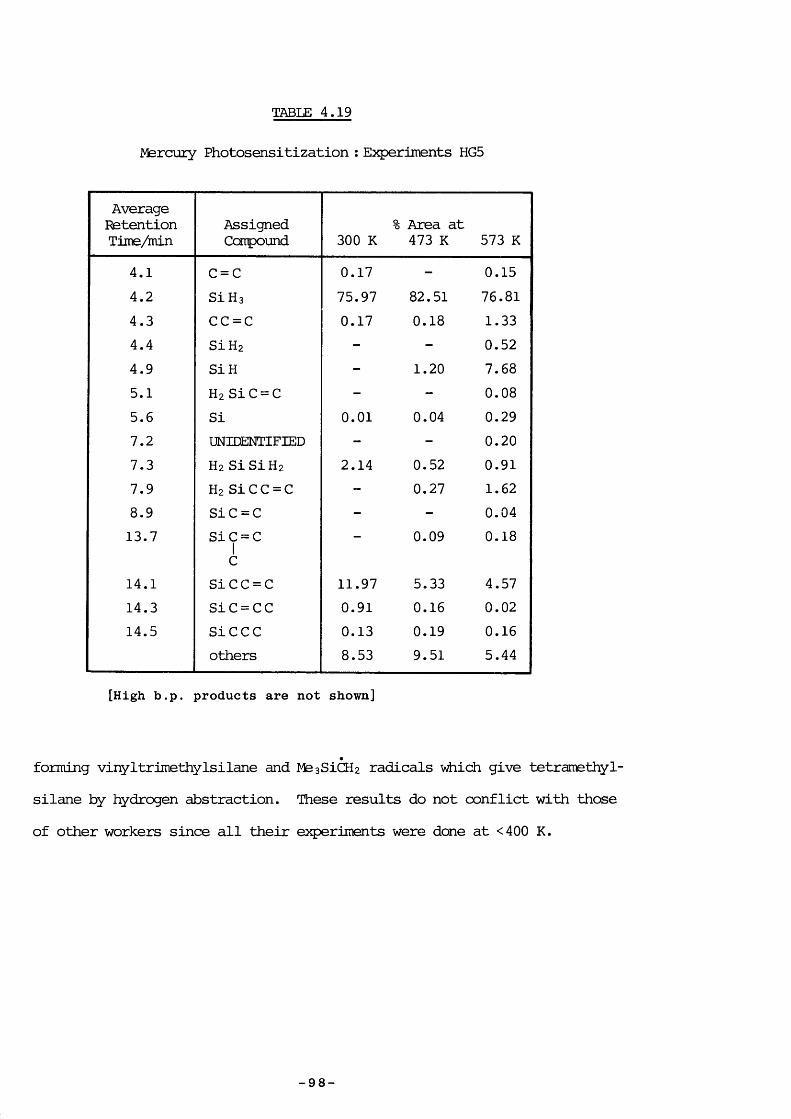
TABLE 4.19
Mercury Photosensitization : Experiments HG5
AverageRetentionTime/min
AssignedConpound 300 K
% Area at 473 K 573 K
4.1 C = C 0.17 - 0.154.2 SiHs 75.97 82.51 76.814.3 CC = C 0.17 0.18 1.334.4 SiHz - - 0.524.9 SiH - 1.20 7.685.1 Hz SiC = C - - 0.085.6 Si 0.01 0.04 0.297.2 UNICENTTFIED - - 0.207.3 Hz Si SiHz 2.14 0.52 0.917.9 Hz Si C C = C - 0.27 1.628.9 SiC = C - - 0.0413.7 SiC = C 1C
0.09 0.18
14.1 Si C C = C 11.97 5.33 4.5714.3 SiC = CC 0.91 0.16 0.0214.5 SiCCC 0.13 0.19 0.16
others 8.53 9.51 5.44
[High b.p. products are not shown]
forming vinyltrimethylsilane and î-feaSiCHz radicals viiich give tetramethyl- silane by hydrogen abstraction. These results do not conflict with those of other workers since all their experiments were done at <400 K.
— 98“
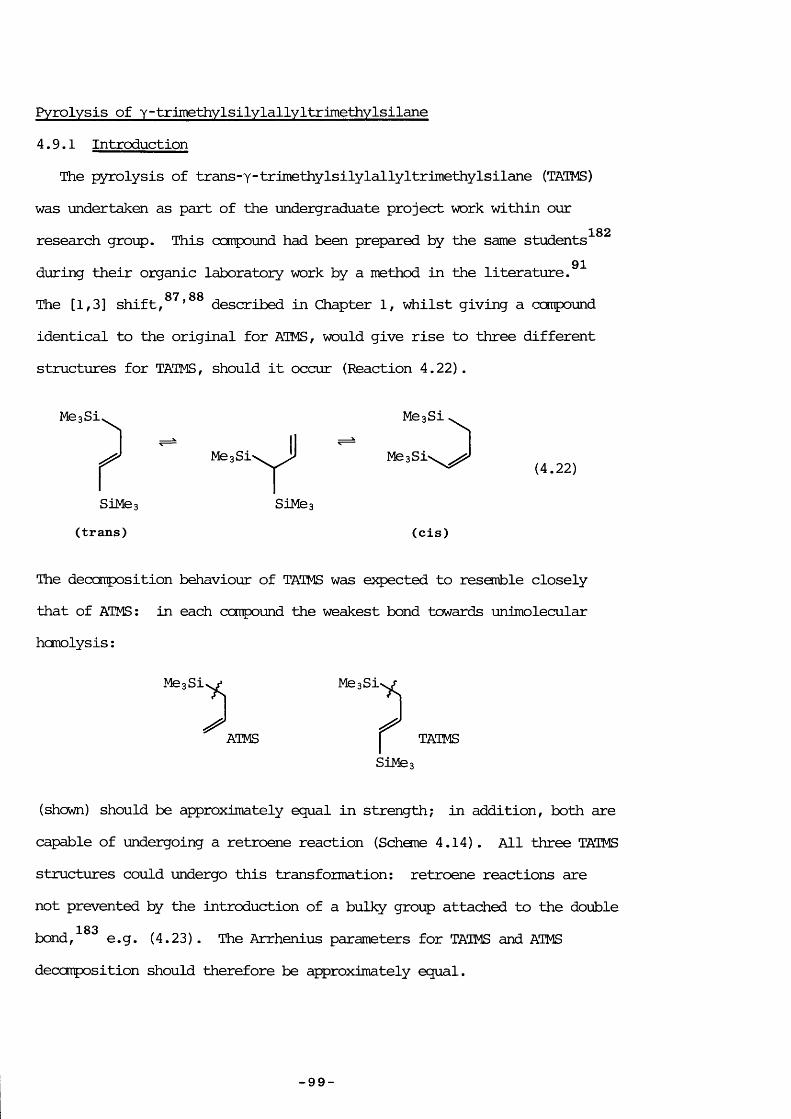
Pyrolysis of Y-tximethvlsilylallvltriinethvlsilane4.9.1 Introduction
The pyrolysis of trans-y-trimethyIsilylallyItrimethyIsilane (TATMS) was undertaken as part of the undergraduate project work within our
182research group. This conpound had been prepared by the same students91during their organic laboratory work by a method in the literature.
The [1,3] s h i f t , d e s c r i b e d in Chapter 1, whilst giving a conpound identical to the original for ATMS, would give rise to three different structures for TATMS, should it occur (Reaction 4.22).
MegSi^ Me 3 Si
MesSi\ Jj Me3Si (4.22)rSiMe3 SiMea
(trans) (cis)
The décomposition behaviour of TATMS was expected to resanble closely that of ATMS: in each compound the weakest bond towards unimolecularhomolysis :
MeaSi^y* MeaSi>
ATMS TATMSSiMea
(shown) should be approximately equal in strength; in addition, both arecapable of undergoing a retroene reaction (Scheme 4.14). All three TATMSstructures could undergo this transformation: retroene reactions arenot prevented by the introduction of a bulky group attached to the double
183bond, e.g. (4.23). The Arrhenius parameters for TATMS and ATMS decomposition should therefore be approximately equal.
-99-
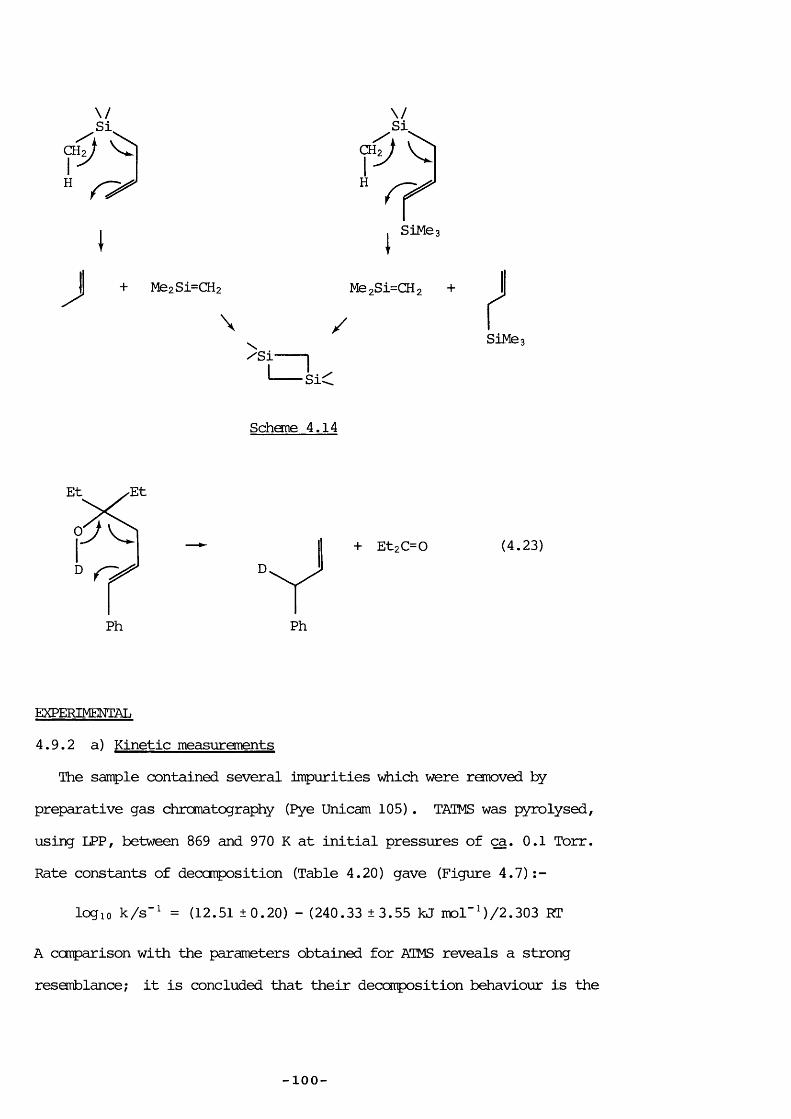
\/
+ Me2Si=Œ2
\/Si-
\/SiCH2H
SiMe.
Me2Si=CH2 +
/Sic
SiMe.
Scheme 4.14
Et
p c
Ph
Et
+ Et2C=0 (4.23)
EXPERIMENTAL4.9.2 a) Kinetic measurements
The sample contained several impurities vÆiich were ranoved by preparative gas chromatography (Pye Unicam 105) . TATMS was pyrolysed, using LPP, between 869 and 970 K at initial pressures of ça. 0.1 Torr. Rate constants of decomposition (Table 4.20) gave (Figure 4.7):-
logio k/s"^ = (12.51 ±0.20) - (240.33 ±3.55 kJ mol"^)/2.303 RT
A comparison with the parameters obtained for ATMS reveals a strong resemblance; it is concluded that their decomposition behaviour is the
— 10 0 —
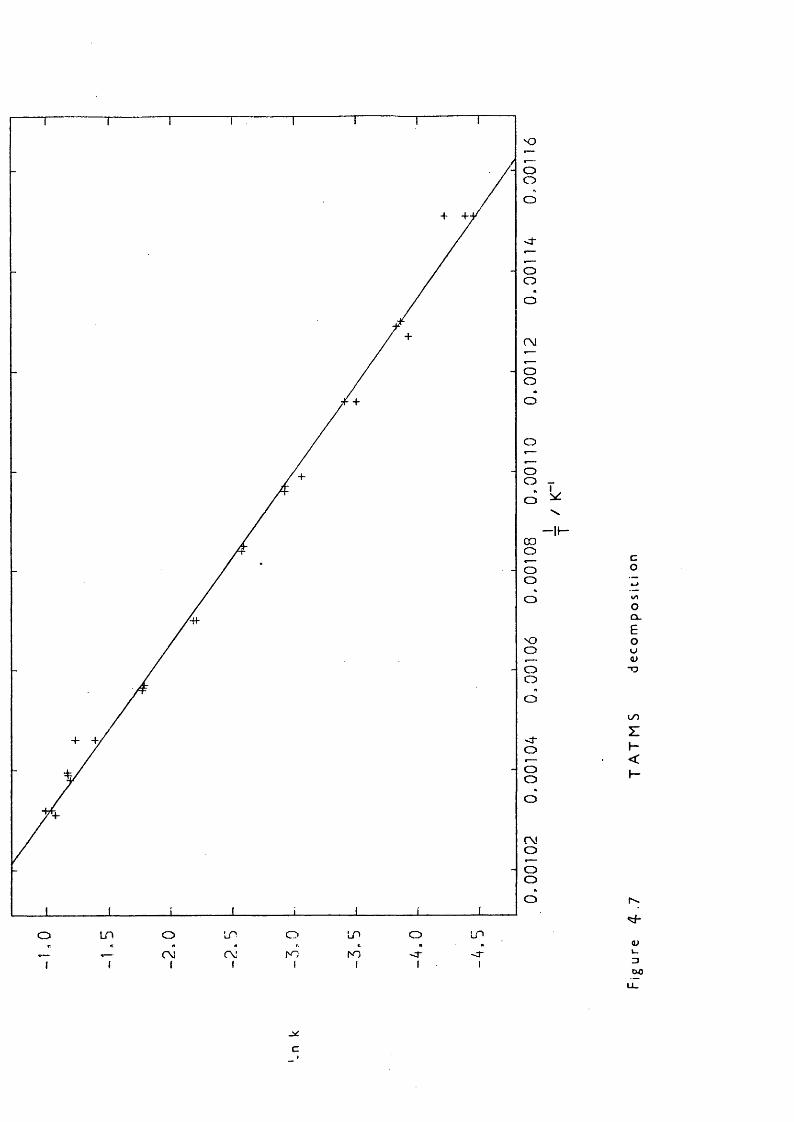
LP LO OO Omo
NDoo
oo
CM
OoC)
ooo
COoooo
nDoooo
ooo
oooo
-II-
oQl
Eou<u-O
zH<
CNJI CMI nI foI <rI <rI
V
3W)
c
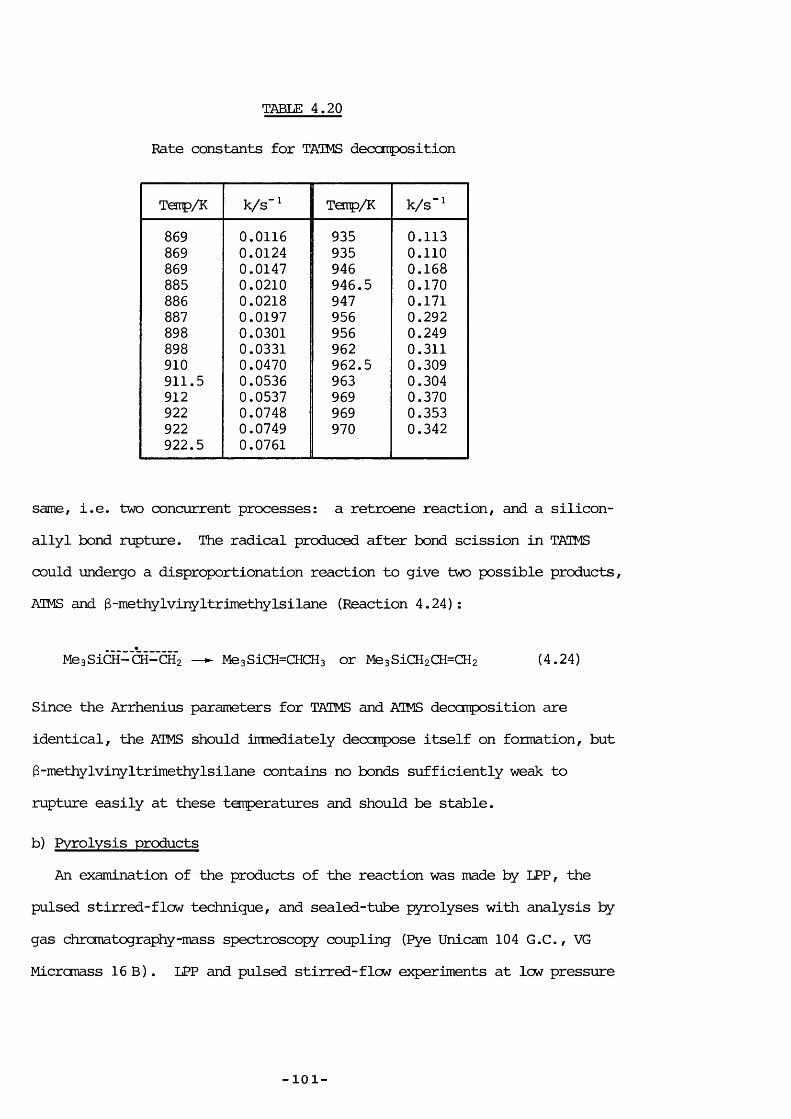
TABLE 4.20
Rate constants for TATMS deccrnposition
Tenp/K k/s"' Tarp/K k/s"'869 0.0116 935 0.113869 0.0124 935 0.110869 0.0147 946 0.168885 0.0210 946.5 0.170886 0.0218 947 0.171887 0.0197 956 0.292898 0.0301 956 0.249898 0.0331 962 0.311910 0.0470 962.5 0.309911.5 0.0536 963 0.304912 0.0537 969 0.370922 0.0748 969 0.353922 0.0749 970 0.342922.5 0.0761
same, i.e. two concurrent processes: a retroene reaction, and a silicon-allyl bond rupture. The radical produced after bond scission in TATMS could undergo a disproportionation reaction to give two possible products, ATMS and 3-methylviny Itrimethy Isilane (Reaction 4.24) :
MesSiCH-CH-CHz Me3SiCH=CHŒ3 or Me3SiŒ2CH=CH2 (4.24)
Since the Arrhenius parameters for TATMS and ATMS deccrnposition are identical, the ATMS should immediately decompose itself on formation, but 3 -methylviny Itrimethy Is ilane contains no bonds sufficiently weak to rupture easily at these temperatures and should be stable.
b) Pyrolysis productsAn examination of the products of the reaction was made by LPP, the
pulsed stirred-flow technique, and sealed-tube pyrolyses with analysis by gas chrcmatography-mass spectroscopy coupling (Pye Unicam 104 G.C., VG Micranass 16 B). LPP and pulsed stirred-flew experiments at low pressure
-101-

gave as products: methane, ethene, propene, trimethylsilane, 1,1-dimethyl-1 -silacyclopent- 3 -ene (m/e = 112'*’, 97"'’: M'*', M' -Me) , DSCB anda product with peaks m/e = 114"*", 99’’’ vhich eluted with the same retention time as authentic ATMS (thought to be 3 -methylviny Itrimethy Isilane, its boiling point, 86.3°C, being similar to that of ATMS, 84.9°C). No significant 116'*' was formed, so propyltrimethylsilane, the conpound vhich would be formed by a hydrogen abstraction by the initially-formed radical, was not a product. At 920 K the propene yield frcm TATMS was between 50 and 90% of the yield frcm the pyrolysis of a similar pressure of ATMS, and the DSCB yield frcm TAIMS was up to double that frcm ATMS because it resulted frcm retroene reactions of both TATMS and ATMS in the TATMS experiments. Minor products were exactly the same as for ATMS pyrolysis, as expected.
A pyrolysis reaction at 855 K in a sealed-tube, achieving about 60% deccrnposition, at higher initial TATMS pressure (ça. 8 Torr) was subjected to gas chromatography - mass spectroscopy coupling, and revealed the following products : methane, ethene, propene, a small amount of dimethylsilane, trimethylsilane, tetramethylsilane, vinyltrimethylsilane, 1,1- dimethyl- 1-silacyclopent-3-ene, DSCB, several minor products, unchanged TATMS (m/e = 186’'', 171" : M'*’, M'"'-Me) and two peaks with approximately thesame retention time as for ATMS. Their mass spectra contained identical mass fragments to those of ATMS but in different ratios. For both, the 99'""/114' ratio was greater, and the 73'"'/ll4' ratio less, than for ATMS. This is consistent with the products being
Me 3 Si (' and
on the basis of ease of fragmentation in the mass spectrometer. Pulsed stirred-flow experiments showed that the yields of tetramethylsilane and
-102-

g
•H
i
"k+ I—Ir~-+vo00
I
i t
/
/ ~ v _•H[/]â
i -i iu+j
+
coI
dü• Hi+Js
m
wI+
• §in
I
œI
g i
+IT)+CM
i
•HWI
W
-103-

vinyltrimethylsilane were approximately equal and were dependent on initial pressure. These two products were a result of trimethylsilyl radical reaction with AIMS; they did not react with TAIMS (Schone 4.15). Whilst the products frcm terminal addition could go unnoticed cwing to their mass spectra being similar to that of the original TAIMS, there was no evidence for m/e 172'*' or 157'*' even at high pressures; also, the tetramethylsilane yield - approximately equal to that of vinyltrimethylsilane - was too low to allcw a further route to its production.
4.9.3 DiscussionThese results indicate that at lew pressures the pyrolysis mechanism
operating was that shewn in Schone 4.16.
Me3SiCH=CH CH2 SiMea
Me2Si=Œ2 + Me3SiCH2CH=CH2
/SiSi Me2Si—CH2 +
CH 3C H K H 2
MeaSi ΠCH CH2 + -SiMeaIMeaSiCH=CHCH2
HSiMea
ta Y r & C H l + -SiMea
Scheme 4.16
At higher pressures bimolecular reactions resulted in the formation of other products. The Arrhenius parameters for TAIMS deccnposition are identical to those for AIMS, the reactions closely resembling each other: TAIMS undergoes a retroene reaction in addition to silicon-allylbond rupture.
-104-

Pyrolysis of allyltxichlorosilane4.10.1 Introduction
The pyrolysis of ally1trichlorosilane was expected to be less carplex: no retroene reaction is possible; the deccrnposition should be daninated by initial silicon-allyl bond rupture.
4.10.2 ExperimentalAllyltrichlorosilane was pyrolysed between 910 and 1020 K at initial
pressures of ça. 0.35 Torr using IPP. The products were: mainly propeneand silicon tetrachloride (m/e = IGS"*": M'*’) , and smaller quantities of methyltrichlorosilane (m/e = 148''’: M”*") , vinyltrichlorosilane (m/e = 160^: M*’) with traces of methane, ethene and possibly trichlorosilane (m/e = 134'*': M'*') , along with several other products in low yield. These findings are consistent with the expected mechanism: initial silicon-allylhemolysis giving trichlorosilyl and allyl radicals (leading to propene formation), followed by reactions of trichlorosilyl radicals : mainlychlorine abstraction frcm allyltrichlorosilane with a small amount of addition (Scheme 4.17).
ClgSi— \ + ClaSi* — ClgSiSiCl:
CI3S1 , ySiCla ClaSiCHz + ^ SiCl^
Scheme 4.17
Preliminary kinetic measurenents showed that decomposition was occurring in the unimolecular fall-off region.
-105-

4.11 DISCUSSION
At low pressures (<0.3 Torr) ATMS pyrolyses between 860 and 960 K via two major decarposition pathways: silicon allyl-bond hemolysis and SEplus propene formation. At higher pressures (>0.3 Torr) the addition of trimethylsilyl radicals to ATMS becomes irrportant, such that the products resulting frcm this reaction, vinyltrimethylsilane and tetramethylsilane, are significant. TATMS was found to adopt similar decomposition behaviour between 869 and 970 K, but allyltrichlorosilane, for which a retroene process is not possible, requires slightly higher torperatures for pyrolysis, 910 to 1020 K, décomposition occurring only by silicon- allyl bond cleavage. Further discussion can be found in Chapter 7.
— 10 6 —

CHAPTER 5PYROLYSIS OF VINYLDIiyETHYLCARBINOXYDIMETIHYI^ILANE
AND 1,1,3,3-TETRAMEIHYL-l-VINYLDISILOXANE

Pyrolysis of vinyldirnethvlcarbinoxvdimethvlsilane and 1,1,3,3- tetramethyl-1-vinyIdisiloxane
5.1.1 IntroductionThe pyrolyses of vinyIdimethyIcarbinoxydimethy Is i lane (VCS) and
1,1,3,3-tetramethyI-1-vinyIdisiloxane (TVS), both gifts frcm Professor C. Frye, USA, were undertaken to study their capabilities of acting as dimethylsilanone (Di) precursors.
5.1.2 Pyrolysis of VCSThe themolytic production of Di frcm VCS would also form 2-methyIbut-
2-ene if a retroene mechanism operated:
Me MeC.. /C
oIISi/
' (5.1)
— Si ) / CHz Me MeMe^ I
(MezSiOa + (Me2SiO)4
5.2 EXPERIMENTAL
A further VCS sample was prepared by the same method as devised by Frye ' (Appendix 5.1), that of base - catalysed alcoholysis of dimethylsilane, easily generated in situ frcm base-catalysed redistribution of the silicon-hydrogen and silicon-oxygen bonds in 1,1,3,3-tetramethyIdisiloxane:
(HMe2Si)20 ^ Me2SiH2 + (Me2SiO)^ (5.2)
Me2SiH2 + HOCMe2CH=CH2 ^ HMe2SiOCMe2CH=CH2 + H2 (5.3)
- 108-

VCS easily reverted back to 1,1,3,3-te trame thy Idisiloxane and 2-methyl-3-buten-2-ol so VCS sairples were of variable purity; consequently some studies of its reactions required that small amounts of these inpurities be taken into consideration.
5.3 LPP PRODUCTS
VCS was pyrolysed using LPP between 710 and 830 K at initial pressures of 0.05 to 0.5 Torr. The products - D3, D^ and 2-methylbut-2-ene, identified by their mass spectra, shewn in Table 5.1 - were consistent with reaction 5.1. The yield of 2-methylbut-2-ene varied from 80 to 100% with the combined D3 and D yield accounting for ip to 60% of the reacted silicon (the rest being lost to the walls of the reaction vessel since no other significant silicon-containing products were detected).The relative yield of D3 and Di+ depended on the initial VCS pressure, but was typically in the region 5:1.
TABLE 5.1
Mass spectral details of VCS and pyrolysis products
Conpound Mass fragments
(^bzSiO)3 222+ (M+) , 207+ (M+-]Vfe) , 193+, 191+, 177+
(MezSiO)^ 296+ (M+) , 281+ (M+-]yfe) ,265+, 249+, 234+, 221+, 207+, 193+, 191+
]yfe2C=C(H)]yfe 70+ (M+) , 55+ (M*--Me) , 42+, 41+, 39+
HMez SiCTJyfe 2 CH=CH2 144+ (M+) , 143+ (M+-H) ,129+ (M+-Me) , 117+ (M+-CH=CH2)
-109-

5.4 KINETIC MEASUREMENTS
The first order rate constants for VCS décomposition, shewn in Table 5.2, vhich were measured between 728 and 819.5 K using a 5:1 mixture of VCS/msthyltrimethylsilyl ether at pressures of ça. 0.3 Torr (see 5.5.2 and Chapter 6) , were obtained by monitoring the decay of the m/e = 129^ fragment and gave (Figure 5.1 ) : -
logio k/s"^ = (12.45 1 0.06) - (205.45 10.84 kJ mDl‘^)/2.303 RI
Repetition of these measuranents between 747.5 to 804 K using neat VCS at initial pressures of 0.2 Torr gave parameters in agreement with these: 18 data points gave:-
logio k/s-i = (12.61 1 0.14) - (208.10 1 2.09 kJ itDl-^)/2.303 RT
These results, and the products obtained, confirm that VCS is an excellent Di source and argue in favour of a molecular mechanism, i.e. retroene (discussed in detail in Chapter 7) and not a radical process : the weakest bond in the molecule towards unimolecular hemolysis should be carbon-oxygen vhose strength (288 kJ mol" ) " is well above the measured activation energy. In addition VCS pyrolysis in excess toluene - a radical trap - gave the same decomposition rates. To investigate the effect of adding radicals to the system, several VCS/ mercury dimethyl co-pyrolyses were carried out; their deconposition rates were approximately equal since mercury dimethyl was within its unimolecular fall-off region. Production of 2-methylbut-2-ene and D 3 was unaffected, further proof that the route to their formation was a molecular one.
-110-
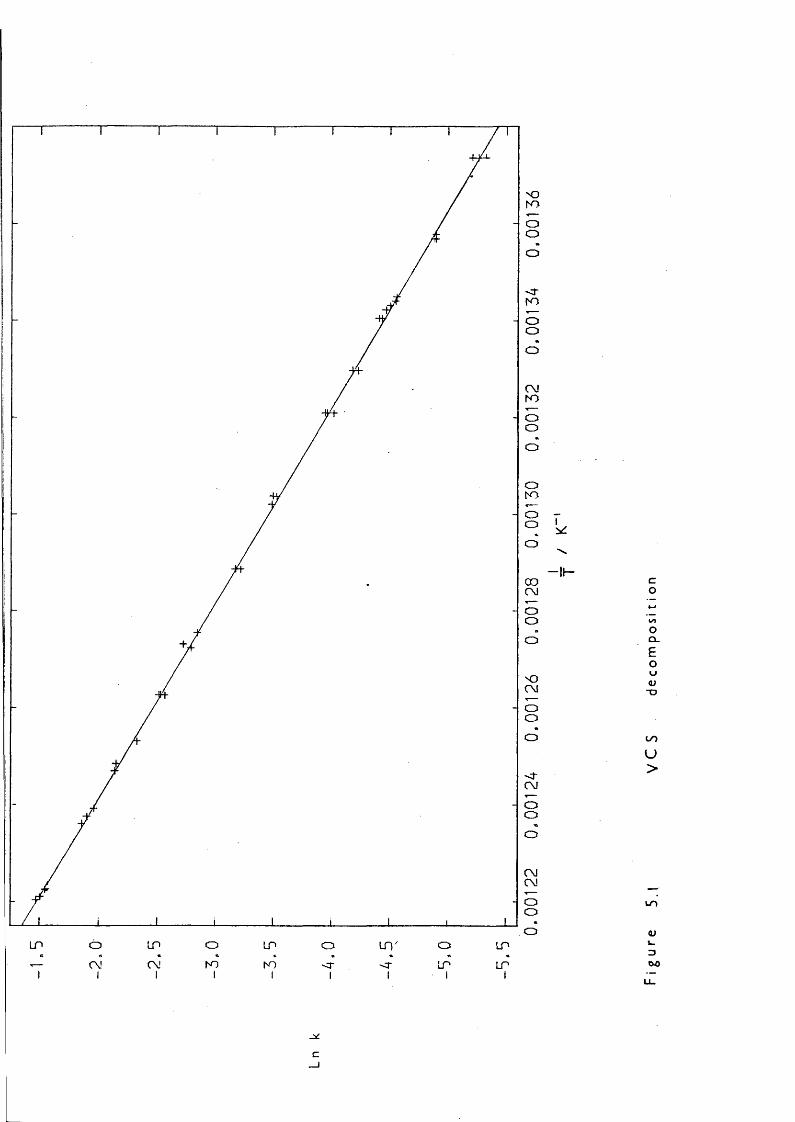
ooo
ooo
ooo
oooo
COC \i
ooo
ooo
CM
ooo
CMCM
ooo
— Ik-
LPI
Oa.£oua-o
LTt
o LP O LP O Ul" o LP 3CM CM hO <r <r LP LP CkO
c

TABLE 5.2
Rate constants for VCS deccnposition
Tenp/K k/s“^ Tertp/K k/s-i
728 0.00487 768 0.0306728 0.00518 776 0.0401728 0.00547 776 0.0418736.5 0.00750 776 0.0420737 0.00750 784 0.0580737 0.00760 785.5 0.0656743.5 0.0105 786 0.0613744 0.0106 792 0.0769744.5 0.0111 792 0.0798745 0.0115 792 0.0808746 0.0119 798 0.0975746 0.0122 801 0.117752 0.0146 802 0.118752 0.0153 807 0.141757 0.0180 808 0.150757 0.0190 809 0.157757 0.0193 818 0.213767 0.0293 819 0.222767 0.0302 819.5 0.230
Reactions of Di
5.5.1 Insertion into oxygen-hvdrocren bondsAlthough no report of a silanone insertion into the oxygen-hydrogen
bond, e.g. in alcohols, exists to the best of our knowledge, this process is to be expected by corparison with the reactions of silaalkenes (Chapter 1) . Under conditions in which there was sane 2-methyl-3-buten- 2-ol impurity in the VCS sample, a Di insertion process occurred, to give mass fragments consistent with the product from reaction 5.4 (m/e = 150^, 149+, 135+, 123+, M" , M' -CH=CH2)
Me2Si=0 + H0CMe2CH=CH2 H0S1MS2 Oayie2 CH=CH2 (5.4)
Pyrolysis of the VCS sample in added excess of the alcohol caused the mass fragments to become more important without altering the VCS
-111-

deccarposition rate. At higher temperatures, e.g. 840 K, the product decarposed v e r y slowly, coinciding with small increases in the m/e 92'*’, 91'"' and 1 1 ' ^ fragments, explained by the production of MezSi(OH)2 (M" ,
M^-Me) , formed following carbon oxygen bond rupture:
HOSiMe20Ciyfe2CH=CH2 —^ H0Siiyfe20' + Me2C"CH'"%
IH0SiJSfe20H (5.5)
A further example of Di insertion into an oxygen-hydrogen bond was found by pyrolysing VCS in the presence of methanol: an efficientreaction ensued, giving MeOSi]\1e20H, identified by m/e = 106'’’, 105‘‘', 91'*’ (M+, M+-H, M'-iyfe) :
]yfe2Si=0 + jyfeOH — ► WfeOSi]Vfe20H (5.6)
That insertion had occurred into the oxygen-hydrogen bond and not carbon - oxygen (which could give the same products) was confirmed by Di reaction with water - to give M02Si(OH)2 - but not diethylèther in the LPP apparatus :
Me2Si=0 + H2O — ^ Me2Si(OH)2 (5.7)
5.5.2 Insertion into silicon-oxygen bondsIn the presence of small quantities of 1,1,3,3-tetramethyldisiloxane,
the other possible impurity in VCS samples, no reaction occurred, but when it was added in five-fold excess, very small peaks with m/e = 207" , 193'*’ formed, corresponding to the production of HMe2Si0SiMe20SiMe2H (M -H, M" -Me) by Di insertion into the silicon-oxygen bond:
1X 2 Si=0 + HMe2SiOSiMS2H — ► HMe2SiOSi]Xk20SiMe2H (5.8)
The same peaks could have been formed by insertion into the silicon-
-112-

hydrogen bond, but no reaction occurred with a ten-fold excess of trimethylsilane in LPP experiments, thereby sipporting Barton's suspicions'^ concerning the reported efficiency of this process^^(Chapter 1).
Further processes involving the facile insertion into silicon-oxygen bonds were established by pyrolysing VCS in the presence of D 3 ( forming
41 42 44 "ÏQDit, reaction 1.21) ’ ’ ’ and methyltrimethylsilyl ether (formingMesSiOSiMezOMe, m/e = 178*, 163*: M*, M*-Me) , the kinetics of which waslater examined (Chapter 6) :
Me2Si=0 + IXfeaSiOiyfe — ► Me3SiOSil^20]Xk (5.9)
If the excess concentration of methyltrimethylsilyl ether over VCS was raised to 8:1 a trace of m/e = 237* (M*-Me) was formed, further Di insertion having occurred:
]yfe2Si=0 + iyte3SiOSi]yfe20iyfe — ► jyte3Si0Si]yte20SiMe20iyfe (5.10)
Under these conditions Dit formation was carp le te ly suppressed whilst D3was still a significant product, although only in low yield. Thissuggested that most of the Dit formed from neat VCS, under LPP oonditions,resulted from Di insertion into D3 and not dimérisation of D2 which was
57considered to be an equally important process by Razuvaev et al.
5.5.3 Other insertion reactionsFurther reactions which were investigated were the fast Di insertion
into hydrogen chloride (forming H0SiMe2Cl: m/e 110*, 109*, 95*: M*,M*-H, M*-Me) :
Me2Si=0 + HCl — ► H0SiMe2Cl (5.11)
and the slower reaction with trimethyIchlorosilane, giving Me3SiOSiMe2Cl (m/e 182*, 167*: M*, M*-Me) by insertion into the silicon-chlorine
-113-

bœd:®®’®®
lSfe2Si=0 + lyfesSiCl — MesSiOSimzCl (5.12)
5.5.4 Further canmentsDuring neat VCS pyrolysis reactions at typical LPP pressures, e.g.
0.2 Torr, no significant mass fragments corresponding to Di reaction with the original material were observed. However, at 0.5 Torr a trace of m/e 203* was formed, probably a result of the product formed by Di insertion into the silicon-oxygen bond (HMe2Si0SiMe20CMe2CH=CH2, M*-Me) as this is a more efficient process than insertion into either silicon- hydrogen or carbon-oxygen bonds (vide supra) . Addition to the carbon- carbon - double bond was also an unlikely process : VCS pyrolyses in aone hundred-fold excess of either 2-methylbut-2-ene or 2,3-dimethylbut- 2-ene produced no significant products. Should an addition reaction occur via a biradical intermediate or an ene process, the products (Chapter 7) would have m/e = 143* fragments, but only a 1% reduction in the observed rate of decay of this peak was found on changing fran neat VCS to the mixtures, indicating insignificant product formation.
5.6 DISCUSSION
VCS has been confirmed to be a good thermal Di source, the mechanism for its production being a molecular process, almost certainly retroene (see Chapter 7). Several Di insertion reactions with a variety of substrates have been studied. It is estimated that their relative reaction rate efficiencies are in the order : HCl > MeOH, H2O > H0CMe2CH=CH2> MeaSiOiyfe, D 3 > Hiyfe2SiOSiMe2H > IXfegSiCl.
— 114 —

Pyrolysis of 1,1,3,3-tetramethvl-l-vinvldisiloxane5.7 INTRODUCTION
The pyrolysis of TVS was of interest because, if a similar retroene reaction occurred, the products would be two corpounds each containing a doubly-bonded silicon: Di and 2-methyl-2-silabut-2-ene:
\/Si
/Si
OSiMe 2
+\/Si
(5.13)
These reactive intermediates would then form the products shown in Table5.3 - which also indicates their predicted mass spectral fragments andthose measured for Dg, Di+ and TVS - i.e. 1,1,2,3,3,4-hexamethy 1-1,3-disilacycldbutane, the dimer of 2-methyl-2-silabut-2-ene, Dg, D.+ and
28two mixed cyclic trimers.
TABLE 5.3
Mass spectral details of TVS and possible pyrolysis products
Corrpound Mass fragments
(Me2SiCHMe)2 172+ (M+) , 157+ (M+-]yfe)(MszSiO) 3 222+ (M+) , 207+ (M+-Me) , 193+, 191+(Me2SiO)4 296+ (M+), 281+ (M+-Me), 265+, 251+, 249+
234+, 221+, 207+, 205+, 193+, 191+(Me2SiO)2]Vfe2SiCHlfe 230+ (M+) , 215+ (M+-Me)(Me2SiCHMe)2 (]Nfe2SiO) 238+ (M+) , 223+ (M+-Me)HMe2SiOSiMe2CH=CH2 160+ (M+) , 159+ (M+-H) , 145+ (M+-]Vfe)
(TVS) 144+, 133+ (M+-CH=CH2) , 132+119+, 118+ (M+-Me,-CH=CH2) , 117+
-115-

Experimental
5.8.1 Pyrolysis productsIn LPP experiments between 860 and 1075 K it was found that the TVS
decomposition rate was so much slower than VCS that a retroene mechanism could be discounted. This was confirmed by a product analysis: of theproducts shewn in Table 5.3 only Dg was formed, in very low yield (<2%). No 1,1,2,3,3,4-hexamethyl-1,3-disilacyclobutane was observed; it was thought that this compound would itself pyrolyse at a slower rate thanTVS by comparison with the rate data obtained in LPP in our laboratoryfor 1,1,3,3-tetramethyl-1,3-disilacyclobutane. Instead, at low pressure (ça. 0.1 Torr) the products were: mainly hydrogen (m/e 2+: M+) ,methane (m/e = 16+, 15+: M+, M+-Me) , ethene (m/e 28+, 27+, 26+: M+,M+-H, M+-2H) in approximate yield 3:3:1, with most of the silicon being lost from the system: several minor peaks formed, in addition to m/e207+ (Dg) , vhich became more important at higher pressures. Above 0.2 Torr these fragments included: m/e 160+, 158+, 145+, 143+ (the largestpeak), and 117+. To aid product identification n.m.r. spectroscopy was used. Because of the lew concentrations of products in LPP experiments, pyrolyses were carried out at 790 K in a sealed tube ofapproximate volume 500 cm^ with initial TVS pressures between 5 and 16Torr. Unfortunately under these different conditions, these experiments gave different products as the most important ones: these were thoughtto be the siloxanes MegSiOSiMeg, MegSiOSiMezH and HMezSiOSiMezH. It was unclear whether they had arisen from homogeneous or heterogeneous processes which had perhaps involved water or oxygen impurities. To circumvent this problem the products from several LPP experiments at 0.4 Torr were combined by freezing the contents of the reaction vessel into a liquid nitrogen-cooled tube containing carbon tetrachloride
— 116“

solvent. Subsequent n.m.r. analysis (Jeol JNM-PS-100 at 100 MHz usingIMS as external reference) allowed two major products to be positivelyidentified (Appendix 5.2) when canbined with the mass spectral data. Tworesonances, both singlets, occurred at 67.0 and 0.7 ppm, consistent withthe reported n.m.r. spectra of 1,1,3,3-tetramethyl-1,3-disila-2-oxa-
184cyclopentene (I) and 1,1,3,3-tetramethyl-1,3-disila-2-oxacyclopentane(II)
/Si Si< /Si Si\\ / \ /C H = C H CHz— CHz
67.0 ppm — 60.7 ppmI II
The resonance at 7.0 ppm provided particularly unambiguous proof:Ienclosing hydrogens of this type (i.e. — Si— CH = CH— ) in a five-menbered
ring moves their resonance position fr( m ~5.8 ppm (e.g. in HMe2SiœiMe2CH=CH2) to the unusually low-field value of ~7.0 ppm (ccmparable to chemical shift in benzene) e.g.:^^^
^ M e
^ Si ^ S i <
H H7.02 ppm
The ratio of I:II in the produch mixture was approximately 6:1. These two compounds accounted for nearly all of the silicon-containing species not lost (ça. 70%). At lower pressure their concentrations were reduced considerably such that they were of ccmparable importance with other silicon-containing products, all being in much Icwer yield than hydrogen.
-117-

methane and ethene. The observed mass fragments are consistent with the formation of the products shown in Table 5.4; the route to their production is shown in Scheme 5.1 in which three major TVS decomposition pathways compete: silicon-hydrogen, silicon-methyl, and silicon-vinylbond rupture, leading to the formation of I, II, III, IV, V and VI. At low pressures the hydrogen, methyl and vinyl radicals abstract hydrogen mainly at the walls of the reaction vessel, but as the pressure is raised hydrogen abstraction from the silicon-hydrogen bond (the most weakly- bound hydrogen in TVS) becomes much more important and I and II are formed in increased yield. Minor peaks (<1%) probably due to products formed following methyl or vinyl radical addition to TVS were also observed. These and most of the other products are unstable in this temperature range and decompose themselves in time.
Further minor products (<1%) probably resulting from Di insertion into the cyclics^^ I to IV were formed (m/e 217+, 219+, 205+, 203^, all M+-Me for I, II, III, IV respectively) e.g.
\/,0 0 ^ ^ 0
]yfe2Si=0 + ^ S i Si :Si Si/V _ J - " w
m/e 219’’’
(5.14)
If a sample of VCS was added to the reaction vessel on top of the TVS pyrolysis product mixture, the 217+, 219+, 205+, 203+ peaks all increased in size as Di was generated.
5.8.2 Trapping ExperimentsThe extent of Di formation was, however, confirmed to be minimal by
pyrolysing TVS in excess methyltrimethylsilyl ether at 970 K:
— 118 —
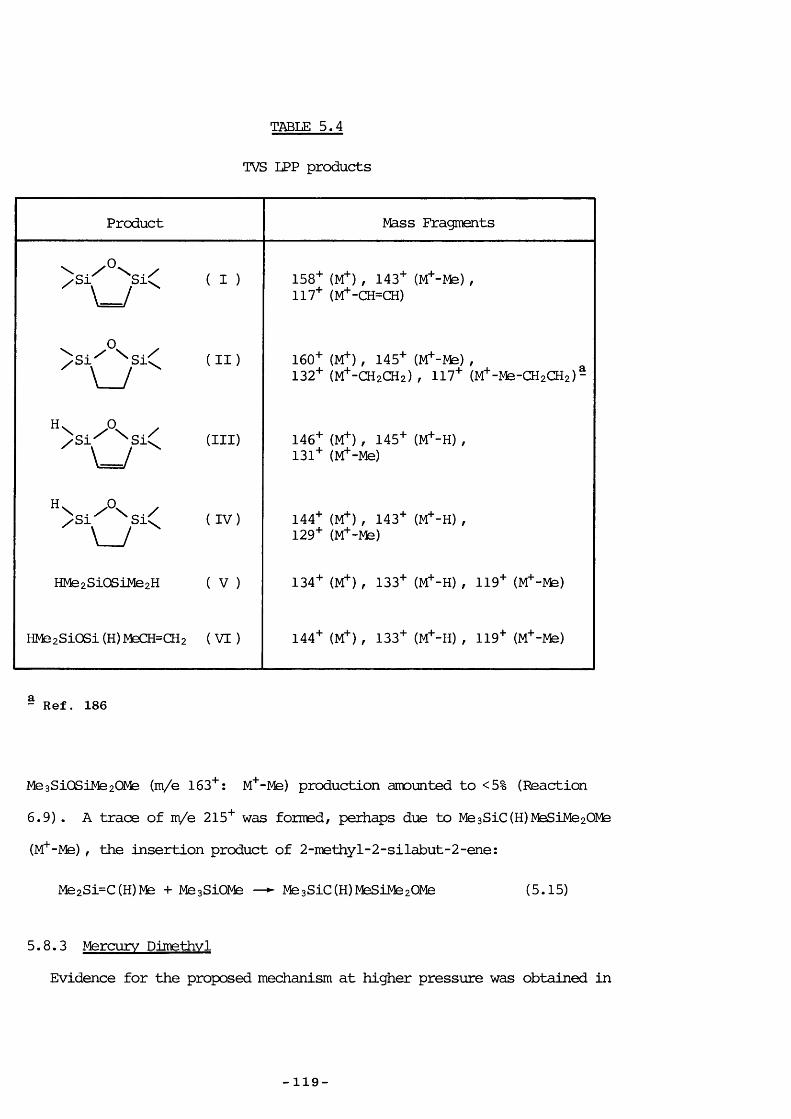
TABLE 5.4
TVS LPP products
Product Mass Fragments
siQs<
w
Si^ ^Siw \
( I )
(II)
(III)
(IV)
HMezSiOSiMezH ( V )
HMezSiOSi (H) MeCH=Œ2 ( VI )
158+117+
160+132+
146+131+
144+129+
134+
144+
M+) , 143+ (M+-Me) , M+-CH=CH)
M+) , 145+ (M+-]Vfe) ,M+-CH2CH2), 117+ (M+-Me-CH2CH2)-
M+) , 145+ (M+-H) , M+-Me)
M+) , 143+ (M+-H) , M+-Me)
M+) , 133+ (M+-H) , 119+ (M+- fe)
M+) , 133+ (M+-H) , 119+ (M+-Me)
- Ref. 186
M03SiOSiJyig2OMe (m/e 163+: M+-Me) production amounted to <5% (Reaction6.9) . A trace of m/e 215+ was formed, perhaps due to Me3SiC(H)MeSiMe20Me (M+-Ms) , the insertion product of 2-methyl-2-silabut-2-ene:
Me2Si=C(H)Me + MegSiOMs — ► MegSiC(H)MeSiMe20Me (5.15)
5.8.3 Mercury DimethylEvidence for the proposed mechanism at higher pressure was obtained in
-119-
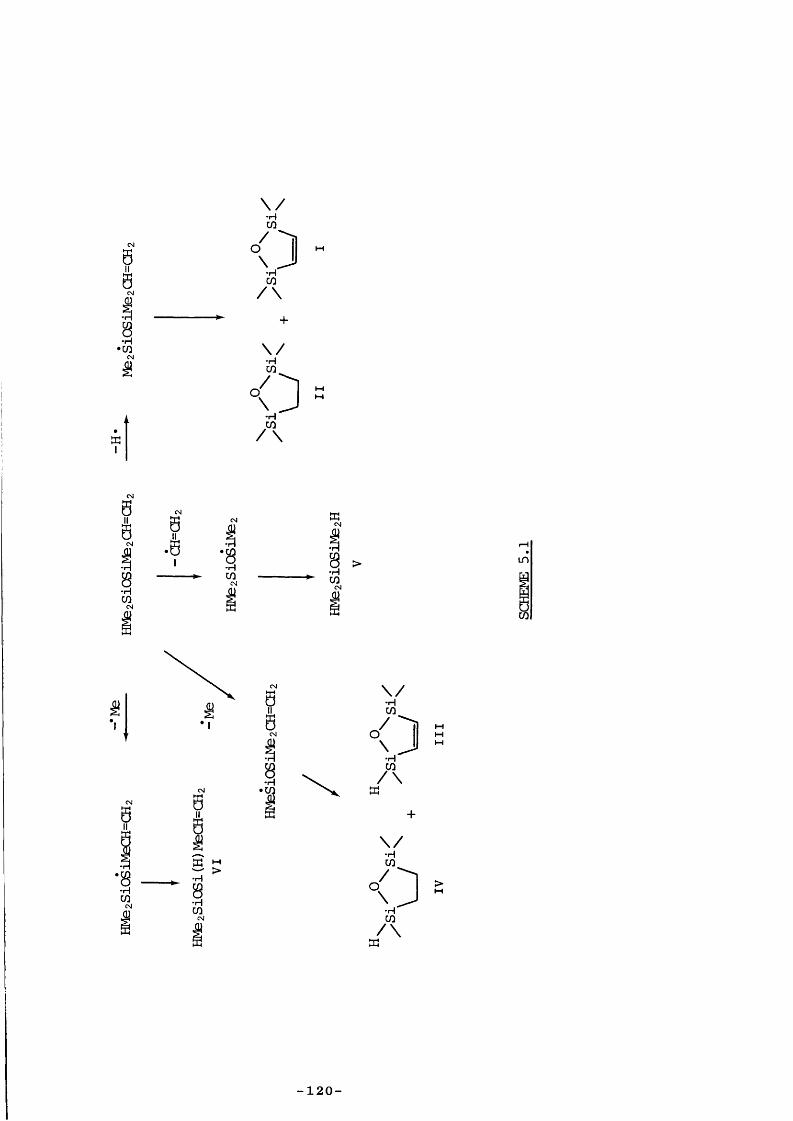
•H•W
mI
\/•HW/O\•HW/\+
\/-HW
<•H
A
S
enw
B
•W
\/•Hœ/\
•H
A3 =
m
3 >
co
\/•Hen
<œ/\
-120-

experiments in which a 1:1 mixture of TVS and mercury dimethyl (see Chapter 4) was pyrolysed at 780 K, a temperature at which TVS did not unimolecularly decaipose. A large quantity of I was formed, resulting from hydrogen abstraction from TVS by methyl radicals. Formation of II was unclear because of its similar mass spectrum to TVS. No Dg was produced, indicating that the Di in the TVS pyrolysis experiments did not result from a radical reaction of this type.
5.8.4 LPP Kinetic MeasurementsAs expected, the rate of deccnposition of TVS increased with
increasing initial TVS pressure which was kept low, at 0.05 Torr, for the kinetic measurements, performed between 945 and 1004 K. Under these conditions the formation of II was minimal and the decay of m/e = 145/ was equated to TVS decomposition which was found to obey first order kinetics. The rate constants, shewn in Table 5.5, gave (Figure 5.2):-
logio k/s‘ = (14.9 ± 0.3) - (305.9 ± 5.3 kJ mol"^)/2.303 RT
Since the activation energy is much lower than the strength of any of_ 22the bonds that rupture - silicon-hydrogen ~ 378 kJ mol" , silicon-
22 -1 methyl, silicon-vinyl ~370 kJ mol" (see Chapter 3) , a chain mechanismprobably operates although the exact nature of it is unclear: TVSdeconposition rate constants were decreased by a factor of approximatelyone-third in excess propene which could have acted as a chain inhibitor.
5.9 DISCUSSION
TVS is not a good thermal source of Di: it is produced in smallquantities only. The retroene mechanism, v^ich operates for VCS pyrolysis, has too high an activation energy; this can be shewn by estimating AH for the two processes :
-121-
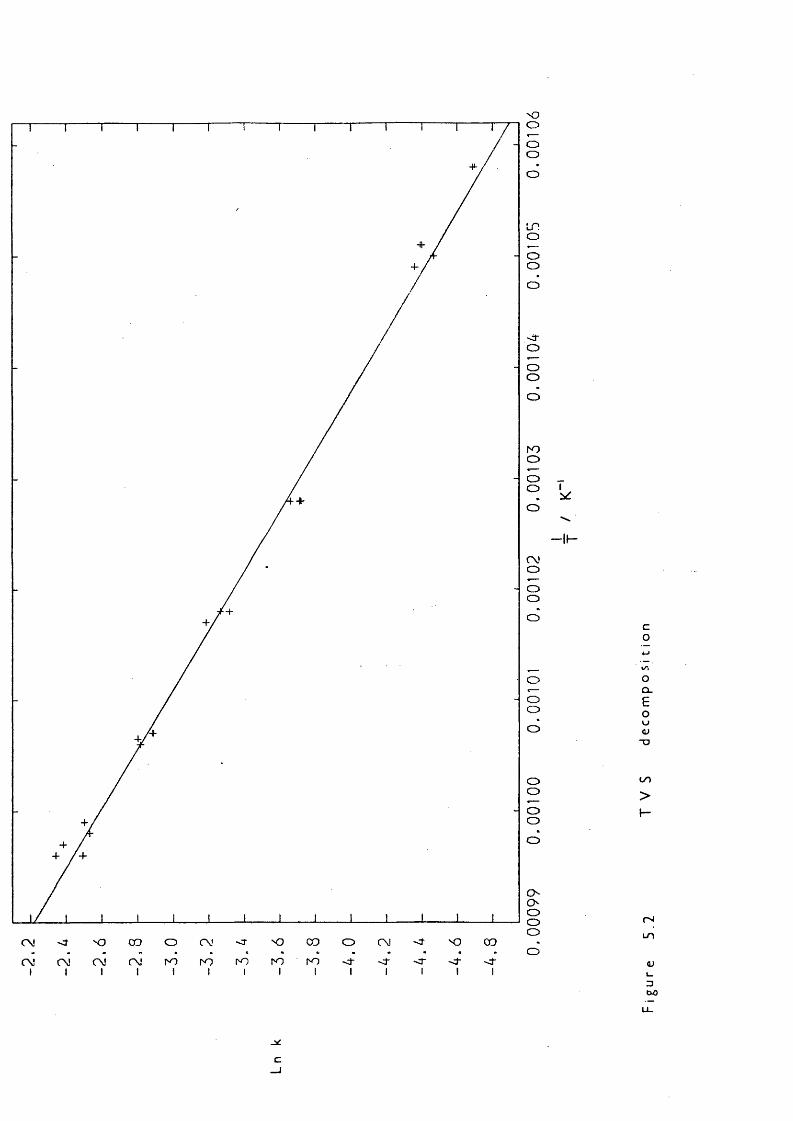
OOOO
OOO
oooo
ooo
ooo
oooo
ooooo
ooooCM ND CO O CM ND oo
OJ CM CM (M lO M r o r o r oI
CMI
<rI
'O
~d-I
OQ
I
-II-
oo
oû_EoVJOJ"O
oo>I-
r~iLT>
3CMD
C

TABLE 5.5
Rate constants for TVS décomposition
Terrp/K k/s"i Tenp/K k/s"i945 0.00910 982.5 0.0382945 0.00920 983 0.0414952 0.0123 993 0.0559952 0.0124 993.5 0.0608952.5 0.0115 994 0.0600953 0.0128 1001 0.0817973 0.0242 1002 0.0794973 0.0244 1003 0.0921973 0.0257 1004 0.0825982.5 0.0363 1004 0.0960
All bond strengths are taken from references 22 and 146 except those for13 22 237T bonds to silicon, all taken to be equal to 175 kJ mol" ’ ’
VCS:HMe2Si(XMe2CH=CH2 —^ M e2Si=0 + Me2C=GHiyfe
AH = D(ESi-H) + D(EC-O) + D7t(-C=C)- Dtt( )si=0) - Dtt( >C=/) - D ( / ^ )
= 378 + 294 + 246 - 175 - 234 - 371= 138 kJ mol"
Measured activation energy = 205 kJ mol"
TVS:HMe2SiOSiMe2CH=CH2 — ► Me2Si=0 + Me2Si=CHMe AH = D(ESi-H) + D(ESi-O) + Dtt(-C=C)
- D tt( >Si=0) - D tt( )Si=/) - D ( y ^ )= 378 + 535 + 246 - 175 - 175 - 371= 438 kJ mol"
The mode of production of the small quantities of dimethyIsilanone (and 2 -methyl - 2 - silabut- 2-ene, if it was formed) is unclear but perhaps
— 12 2 —

resulted from the follcwing unimolecular decomposition process (although there was no evidence for the formation of vinyldimethylsilane) :
Me2Si-0-Sil^2CH=CH2 —► iyfe2Si=0 + JY^2SiCH=CH2
Hiyfe2SiCH=Œ2 + Me2Si=CHiyfe (5.16)
Pyrolysis of TVS proceeds by bond rupture processes : silicon-hydrogen, silicon-methyl and silicon-vinyl, vÆiich form hydrogen, methane and ethene along with compounds I to VI in low yield at lew pressures, e.g. 0.1 Torr. At higher pressures (e.g. 0.3 Torr) the main silicon- containing products are I and II in the ratio of approximately 6:1.
-123-

CHAPTER 6KINETICS OF REACTIONS OF 1,1-DIMETHYI5ILAEIHENE
AND DIMEIHYLSILANONE

Kinetics of reactions of 1,1-dimethylsilaethene and dimet±ivlsilanone6.1 INTRODUCTION
The paucity of kinetic data relating to reactions of 1,1-dimethylsila-ethene (SE) and the ccmplete lack of any measurements for dimethyIsilanone(Di) processes has hampered any detailed carparisons of their reactions.
46In a recent study on the insertion of SE, generated by thermolysis of 1,1-dimethyl-1-silacyclobutane (DMSCB), into methanol, secondary regiospecific insertion of SE into the primary product, methyltrimethylsilyl ether, was observed (Reaction 6.4 in Scheme 6.1).
MezSi— I — Me2Si=CH2 + CH2=CH2 (6.1)
Me2Si=CH2 + MeOH — MegSiOMe (6.2)
ks Me 2 Si-2Me2Si=CH2 Me2bi— I— SiMe2
(6.3)
Me2Si=CH2 + MeaSiCMe — ► MeaSiŒ2SiMe20Me (6.4)
Scheme 6.1
6.2 EXPERIMENTAL
The original intention was to measure the kinetics of reaction (6.2) using the low-pressure pyrolysis (LPP) apparatus as in earlier work in
28our laboratory on the insertions of SE into hydrogen halides and oj^gen.DMSCB/methanol co-pyrolysis experiments were performed; although they
followed the expected course (reactions 6.1 to 6.3), reaction 6.4 was significant and reaction 6.2 was too fast to be measured by our technique in its present form. In addition, there were problems caused by the polar nature of methanol: it tended to adsorb onto the sides of thereaction vessel easily, giving rise to inaccurate rate measurements. However, the LPP technique was suitable for measuring the kinetics of
-125-

reaction 6.4 by pyrolysing EMSCB in the presence of methyltrimethylsilylether itself. n.m.r. analysis of the product, resulting frcm combiningthe reaction mixture of several co-pyrolyses, gave identical results to
46those of John et al. (Appendix 6.1) ; their conclusion as to the regiospecific nature of the insertion process was therefore confirmed. [It is noted that the same product could be obtained by insertion into the silicon-methyl, instead of the silicon-oxygen bond - a process without precedent - but this is considered unlikely.] No evidence was found for any other insertion and no further insertion into the product was observed under IPP conditions.
Difficulties occasionally arose because of the formation of several other products in small quantities, two of which were identified as MeaSiOSiMea (m/e = 172/, 147'*’: M'*’, M'"'-Me) , and Me a SiOSiMezCMe (m/e =178/, 163+: r/, M^-Me) a result of reactions with background water or oxygen on the walls of the reaction vessel, as shown in reactions 6.5 to 6.9'7»28 Scheme 6.2.
MezSi-CHz + H2O — ^ MeaSiOH (6.5)MezSi^CHz + O2 — ► Me2Si=0 + CH2=0 (6.6)
Me2Si=CH2 + MeaSiOH — MeaSiOSiMea (6.7)2MeaSiOH — ^ MeaSiOSiMea + H2O (6.8)
Me2Si=0 + MeaSiCMe — ► Me a SiOSiMe zOMe (6.9)
Scheme 6.2
Although it was not possible to detect formaldehyde or trimethyIsilanol because of their indistinctive mass spectra and very low concentrations, the formation of MeaSiOSiMea and Me a SiOSiMe zOMe and the fact that reaction 6.9 was known to occur under IPP conditions - as was shown in Chapter 5 by the use of HMe2SiOCMe2CH=CH2 as a Di source - provided
— 12 6 —

reasonable evidence for the validity of Scheme 6.2.Cleaning out the reaction vessel by pyrolysing neat EMSCB completely
removed Me3SiOSiMezCMe frcm the product mixture, and MeaSiOSiMea was reduced to a negligible amount. Traces (<0.1%) of the cyclic compounds (MezSiO) a, (MezSiO) zMezSiCHz, MezSiO (MezSiCHz) 2 were also observed (Scheme 4.1) .
6.3 KINETIC MEASUREMENTS
Quantitative measurements were done using a mixture of DMSCB and methyltrimethylsilyl ether in the ratio 1:2, vÆiich was pyrolysed at temperatures between 732 and 789.5 K, initial pressures being ça. 0.3 Torr. The normal kinetic method of measuring ki+ relative to (ka) was used; the formation of DSCB (m/e = 129^: M' -Me) and MeaSiCHzSiMezCMe (m/e = 161*: M*-Me) were taken as a measure of the rate of reaction 6.3and 6.4 respectively:
MSE = MeaSiOMeP = MeaSiCHzSiMezCMeV = initial rate of formation (mol dm"^ s"M concentrations are initial values (mol dm” )
Va = _d [DSCB] = ka [SE] , Va^ = ka^ [SE] dt
V4 = d [P] = U [SE][MSE]dt
V4 = k^ [MSE]
kit “ Yii 1ka^ Va^ [MSE]
Let v' = observed initial slope (s"M [MSE]' = initial peak height MSE
-127-

Sit = sensitivity of mass spectrometer to MesSiCHzSiMezOMeS3 = sensitivity to DSCBF = conversion factor: peak height— ►concentration
(mol dm~^) for MSE[MSE] = [MSE]' F
V = V ' FS
kit — 1 Vit' F I S ;
Aks^ [MSE]'F Sit Vvs'F
kit -___^ / S 3
[MSE]' S» VVa'pj
The (kit/k; ) values shewn in Table 6.1 give:-logio (ki,A3'’)/dm’''‘ mol"’’ s"’’ = (2.01 ±0.22) - (6.3 ±3.2 kJ rrol‘')/2.303 KT(Figure 6.1).
But kit = Ait A 3~* exp [ E 3 - 2Eit | k3 \ 2RT /
If we assume that k3 = 10 ' dm^ mol” s“ with zero activation energy,^®then this gives : -
k^ = Ait A 3 ^ exp /-E^X ka’’ \RT/
and the rate constants (Table 6.1) give:-logio kit/(dm mol"^ s” ) = (5.3 ±0.2) - (6.3 ±3.2 kJ mol’M/2.303 RT.
Further experiments were done in v^ch a series of pyrolyses vere carried out using different mixture carpositions, ranging from 4:1 to 1:6 (DMSCB : methyltrimethylsilyl ether) and extended temperature ranges, 725 to 825 K, confirming the above parameters (see Table 6.2). Results fran different days cannot be combined'because the small day-to-day variation in the sensitivity of the mass spectrometer - which affects calculations of rate constants of formation - gives too much scatter in
— 128“
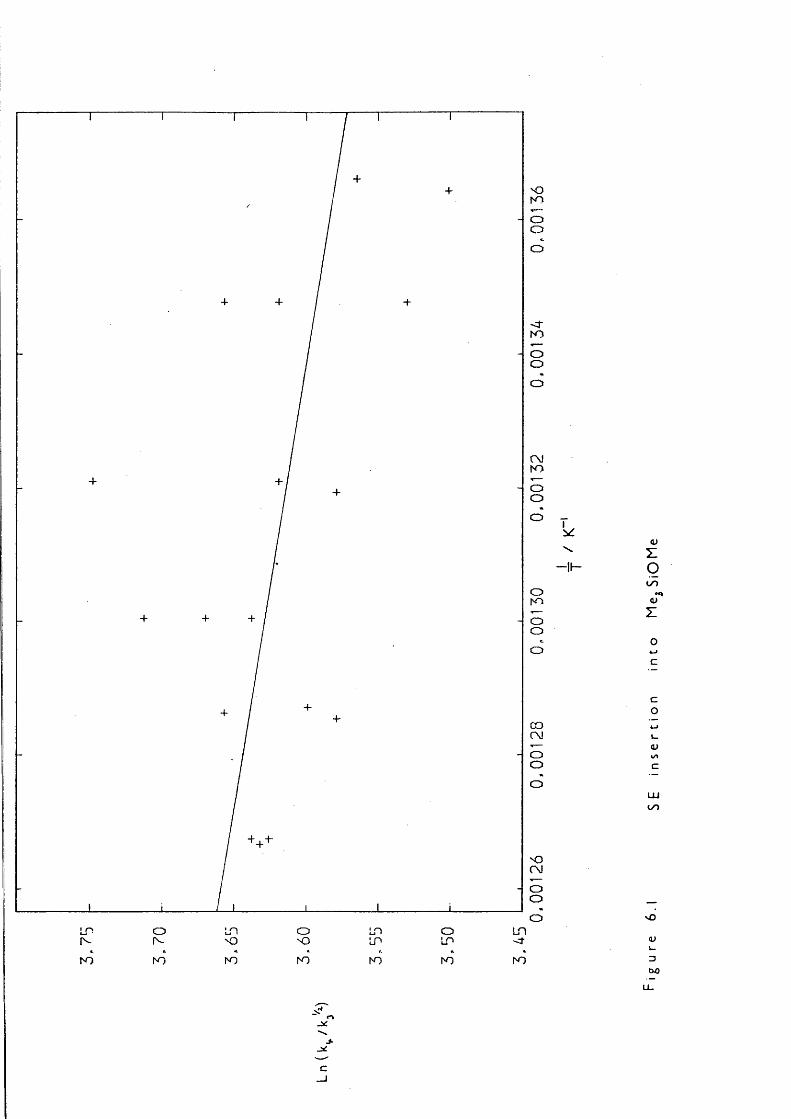
ooo
ooo
ooo
oNO
ooo
coOJooo
ooo
— It~<uzoLO<uzoc
vOLP O LO O CO O LP(V •O nO LP LP (U
c « *-N I N I N I N I N I K ) N 3 3
-M
c
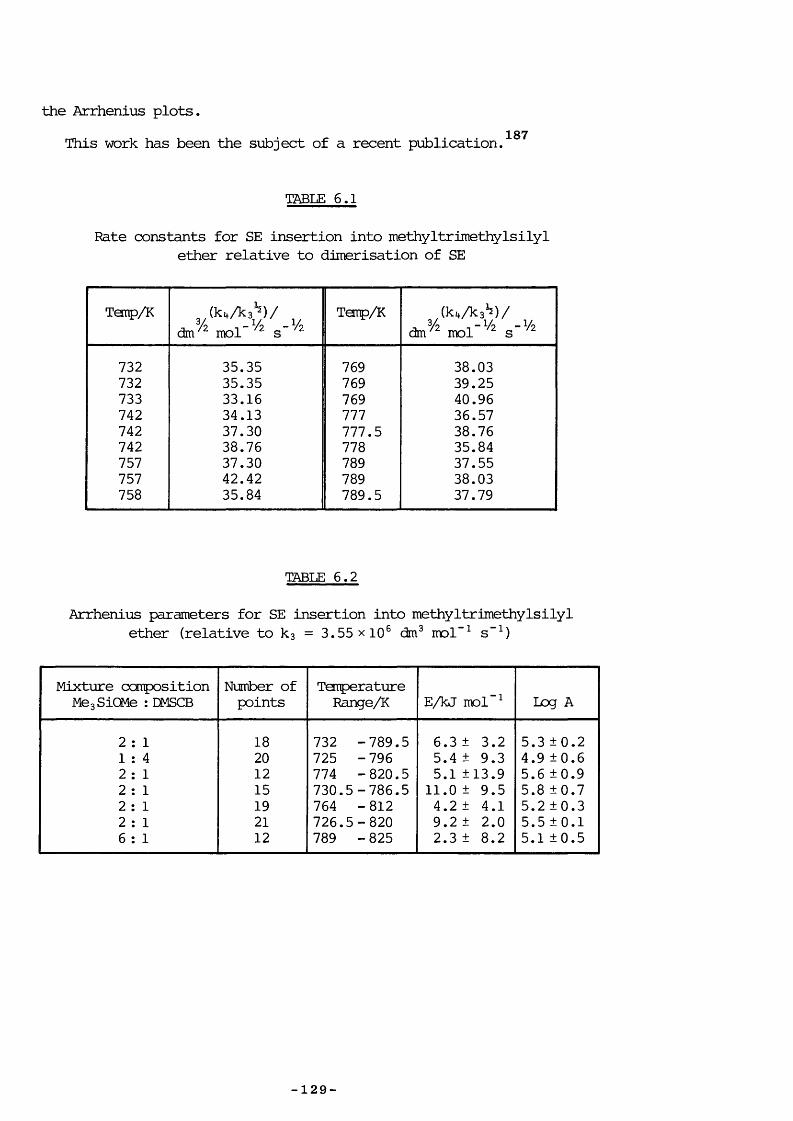
the Arrhenius plots,This work has been the subject of a recent publication. 187
TABLE 6.1
Rate constants for SE insertion into methyltriinethylsilyl ether relative to dimérisation of SE
Tonp/K Torp/K (ki^As^)/ dm% s-''
732 35.35 769 38.03732 35.35 769 39.25733 33.16 769 40.96742 34.13 1 1 1 36.57742 37.30 777.5 38.76742 38.76 778 35.84757 37.30 789 37.55757 42.42 789 38.03758 35.84 789.5 37.79
TABLE 6.2
Arrhenius parameters for SE insertion into methyltrimethylsilylether (relative to kg = 3.55x10'" dm" mol"^ s"M
Mixture ccnposition MegSiQMe : DMSCB
Number of points
TemperatureRange/K E/kJ mol~^ Log A
2 1 18 732 -789.5 6.3 ± 3.2 5.3 ±0.21 4 20 725 -796 5.4 ± 9.3 4.9 ±0.62 1 12 774 -820.5 5.1 ±13.9 5.6 ±0.92 1 15 730.5 -786.5 11.0 ± 9.5 5.8 ±0.72 1 19 764 -812 4.2 ± 4.1 5.2 ±0.32 1 21 726.5-820 9.2 ± 2.0 5.5 ±0.16 1 12 789 - 825 2.3 ± 8.2 5.1 ±0.5
— 129“

A dimethylsilanone reaction6.4 INTRODUCTION
A useful corparison was obviously to study the kinetics of insertion of Di into the same corpound, methyltrimethylsilyl ether, using vinyl- dimethyIcarbinoxydimethylsilane (VCS) as the Di source (Scheme 6.3) .
HMe2SiOCMe2CH=CH2 — ^ Me2Si=0 + Me2C=C(H)Me (6.10)kii
3Me2Si=0 (Me2SiO)g (6.11)kgMe2Si=0 + MegSiCMe — ► MegSiOSiMe2CMe (6.9)
Schane 6.3
6.5 EXPERIMENTAL
Samples of a mixture (VCS : MegSiCMe) at initial pressures of ça. 0.3 Torr were pyrolysed between 728 and 819.5 K using LPP. Under these conditions it was found that: no Di insertion into VCS occurred; only avery small quantity of m/e 135^, 149^ formed, along with a trace of Di+ - (Di preferred to insert into MegSiCMe rather than Dg) ; the HMe2SiOSiMe2H impurity remained unaffected during the course of the reaction; no Di insertion into Me g SiOSiMe 2CMe occurred. All these factors were discussed in more detail in Chapter 5 . kg was measured relative to (kn) ; Dg (m/e = 207^: M'"'-Me) formation was taken as a measure of the rate of reaction 6.11 and Me g SiOSiMe 2CMe (m/e = 163*: M*-Me) of reaction 6.9.The expression for (kgAn ^) can be derived in a similar fashion to that shown before in the SE plus methyltrimethylsilyl ether system:
kg = Ag All exp-/Eg - 3Ei i\kn \ 3RT /
kg = 1 Yi / Si 1 \kn [MSE]' $9 \Vn' F/
-130-

The (kgAl 1 values are shown in Table 6.3 in two sections: sensitivitychanges do not allow the results to be ccmbined; identical Arrhenius parameters are nevertheless obtained by treating each set of data separately: between 728 and 768 K:-logio (kgAll ^^)/(dm^ mol" ^ s"^^) = (3.6 ±0.4) - (28.4 ±6.4 kJ mol"M/2.303 RT (Figure 6.2) and between 714 and 819.5 K:-logio (kgAll ^^)/(dm* mol" ^ s" ^) = (3.8 ±1.0) - (28.0 ±15 kJ mol"^)/2.303 RT.
The implications of these measurements are discussed in Chapter 7.
TABLE 6.3
Rate constants for Di insertion into methyltrimethylsilyl ether relative to the trimérisation of Di
TempA (kgAl/^')/ dm" mol“"/' s-%
TempA (kgAll dm" mol"^ s-%
728 34.34728 35.57728 37.53 784 93.03736.5 36.36 785.5 70.94737 34.76 786 83.97737 35.85 792 70.49743.5 35.83 792 82.71744 39.76 792 88.04744.5 41.72 798 109.5745 44.36 801 82.37746 39.20 802 72.99746 39.87 807 96.17752 45.86 808 87.41752 48.06 809 112.1757 37.41 818 98.20757 39.56 819 94.08757 41.46 819.5 91.50767 42.39767 45.01768 47.83
-131-
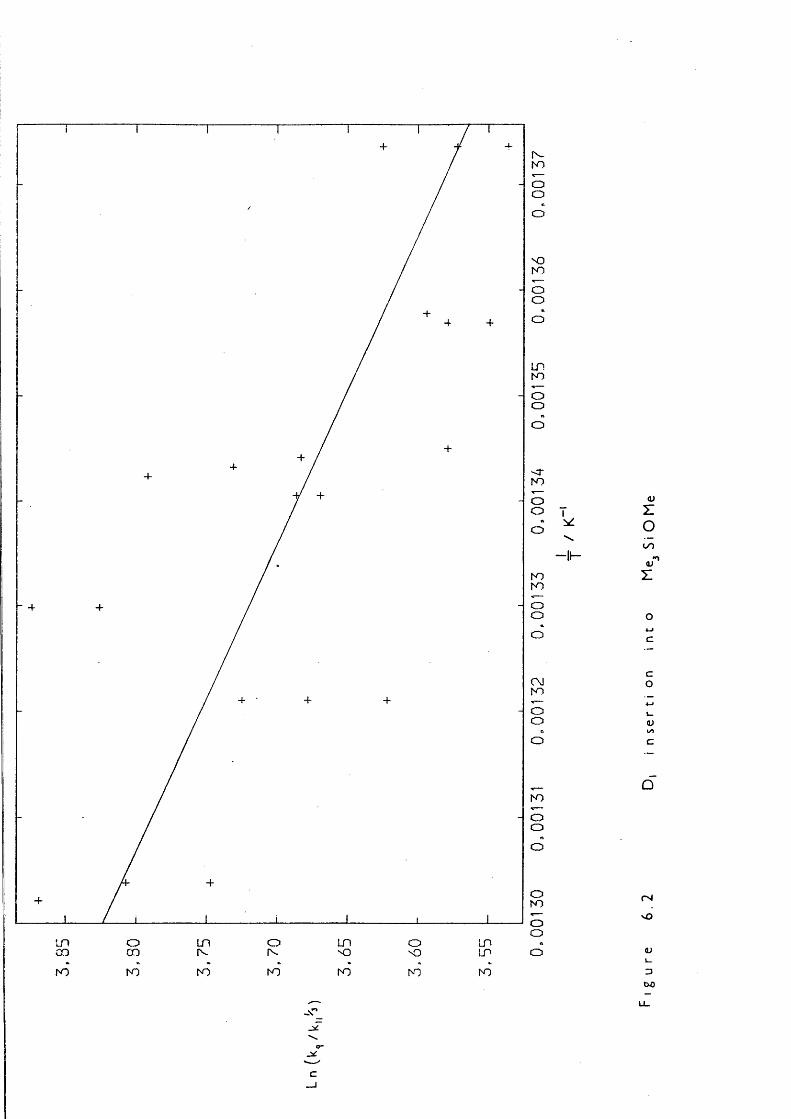
- o oo
ooo
- o oo
ooo
ooo
OJooo
ooo
ooocn LnN o LPLPLPCO
o'O o
-II-
(Uzouoa»"’z
ac
Q
(NvO
3W)

CHAPTER 7DISCUSSION

7.1 INTRODUCTION
The experimental results described in this thesis prarpt several general discussions on the chemistry of organosilicon canpounds, as well as more specific carments. Whilst silylenes and silyl radicals have been encountered, silaethenes and silanones provided the most important part of the research, and in particular 1,1-dimethylsilaethene (SE) and dimethylsilanone (D i ) .
7.2.1 Reaction of SE with unsaturated substratesThese reactions were discussed in part in Chapter 1. SE reacts with
SE (dimérisation) , ethene, and oxygen to give cyclic four-membered ring products (with oxygen the product is unstable) , and with alkenes containing a suitably-positioned hydrogen, such as cis but-2-ene or 2-methyl- propene, the hydrogen-transfer product. With propene both reaction pathways are followed; the latter, giving allyltrimethylsilane (AIMS) , is more important. Conjugated dienes give Diels-Alder adducts in excess over the hydrogen-transfer products; reaction with butadiene gives entirely the Diels-Alder adduct. There are therefore three distinguishable types of reaction, but the mechanisms are not so clearly defined. Measured Arrhenius parameters are shown in Table 7.1.
7.2.2 Diels-Alder reactionsDiels-Alder reactions are fairly well understood, reaction occurring
by a symmetry- allowed 2tt + 4tt cycloaddition process via a six-membered89cyclic transition state :
t ( '
‘Si
(7.1)
-133-
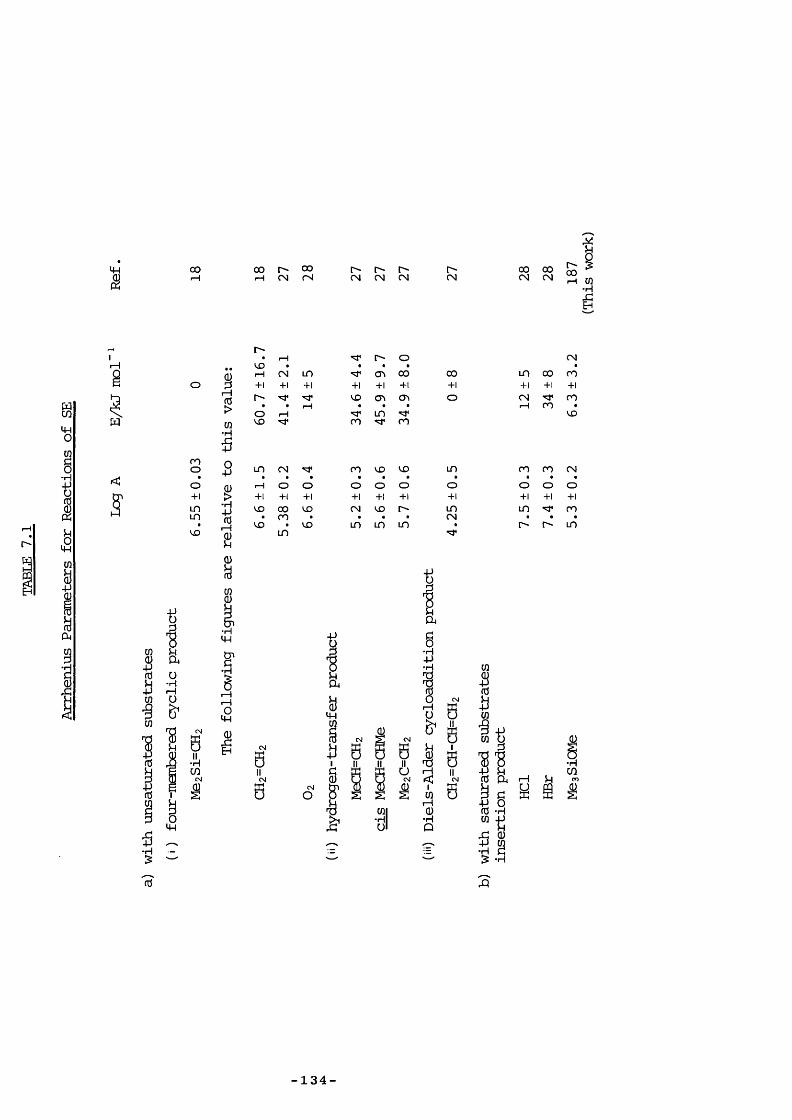
00 CO 00I—I CM rs CM CM CM CM 00 00 CM CM rH C/1
g
r~-i
0s1P4I
I—Ii
H
<COo
o+1LDLT)VX)
5w
CM
a
Icnss
I5!I
•HM-l
gI
. iH O CMCX)rH CM m CT> 00 00 LO 00 cn+1 +1 +1 +1 +1 +1 +1 +1 +1 +1
Tf VO <T» CT» o CM cniH iH cn
o rH LO VOCD
B
cn cn
LO CM cn VO VO LOiH O o o O o o+1 4-1 4-1 4-1 4-1 4-1 4-1VO 00 VO CM VO LOcn CMVO lO VO LO LO LO
BII
cn cn CMo o o4-1 4-1 4-1LO cn
LO
IPI Icn 4-)
I I
u
maI
(tj
-134-

27The low A factor (log A = 4.25 ±0.5) reflects this tightly held transition state.
7 .2 .3 2tt + 2tt reactions
SE reactions giving a four-manbered cyclic product could occur by oneof two separate mechanisms: either via a biradical intermediate or by a
892tt + 2tt pericyclic process.The A factor for the gas-phase dimérisation of C2F4 to Ci+Fa, which
occurs via a biradical species, has been estimated: log A = 8.0^^® andlog A = 7.4.189
Whilst 2tt + 2tt concerted processes are formally syirmetry-disallowed, several reactions are known for certain types of alkene, e.g. cumulenes. The rates of sane of these reactions have been investigated in solutionand found to be almost solvent independent, indicating a concertedprocess with only a small amount of charge build-up in the transition state. Log A factors have been calculated by the author fran the reported entropies of activation, shown in Table 7.2, and range from 3.6 to 5.8,indicating a more restricted transition state.
Although the Woodward-Hoffmann symmetry rules are relaxed for larger atans than carbon, the reaction of SE with SE (dimérisation), ethene, and oxygen (which have approximately equal measured A factors - log A ~ 6 .6, between the two sets discussed above) are thought to proceed via a biradical intermediate. For example, the reaction of SE with ethene is the reverse of 1,1-dimethyl-1-silacyclobutane deccmposition which has Arrhenius parameters consistent with biradical formation (see Chapter 1):
Me2Si-CH2 + CH2-CH2 ^ Me^Si— - Me2Si- (7.2)
-135-
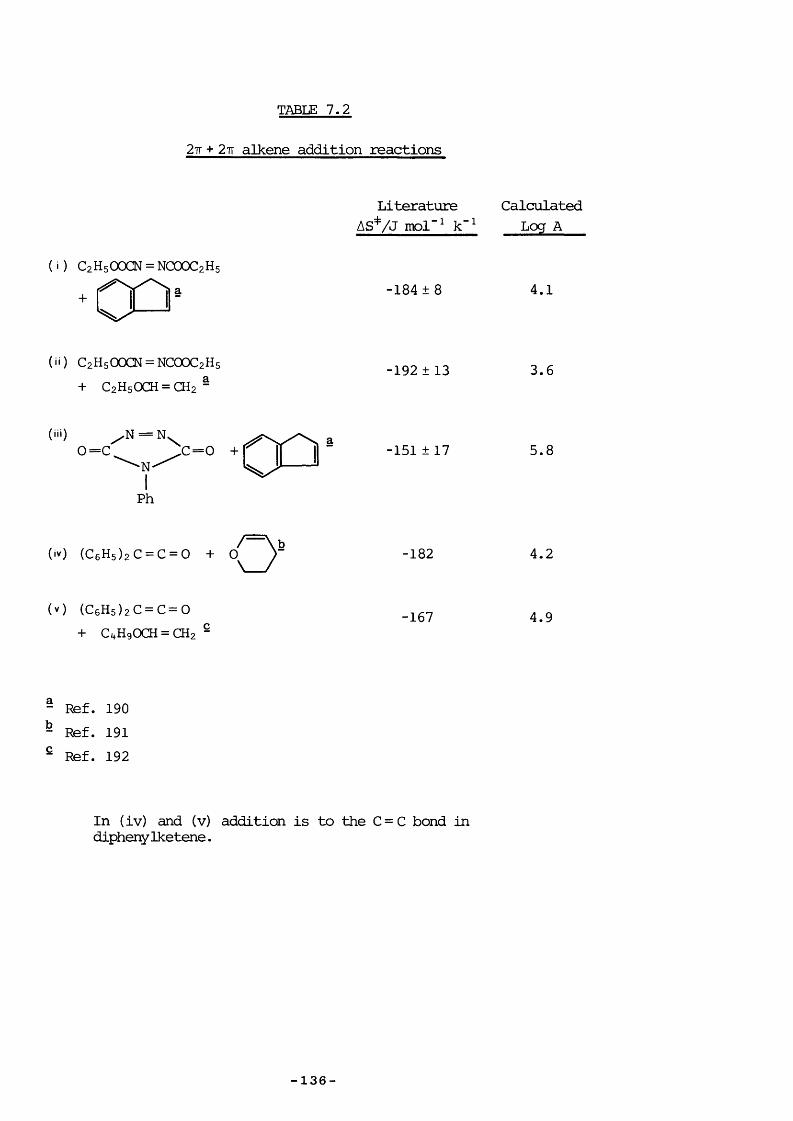
TABLE 7.2
2tt + 2tt alkene addition reactions
(i) C2H500CN = NC00C2H5
Literature Calculated AS*/J mol~^ k~^ Log A
-184 ±8 4.1
(ii) C2H500CN = NC00C2H5 + C2H50CH = CH2-
-192 ±13 3.6
(iii) / N = N. ^0 = 0 . _C= 0 + r IT il"' -151 ±17 5.8
IPh
(iv) (C6H5)2C = C = 0 + 0^ -182 4.2
(V) (C6H5)2C = C = 0 _i67 4.9+ C4HgOCH=CH2 -
- Réf. 190- Réf. 191- Réf. 192
In (iv) and (v) addition is to the C=C bond in diphenyIketene.
-136-

7.2.4 Hydrogen-transfer reactionsThe hydrogen-transfer reaction can also occur either by initial
biradical formation or by a pericyclic process, which in this case isan ene reaction, involving a six-membered transition state (as with
89Diels-Alder reactions; the two processes are often catpared ) . The193 194subject of ene reactions has been reviewed by Hoffmann and Sniekus
et al; these processes are not well understood, particularly as far as biradical (polar) character in the transition state is concerned. For example, if the reaction between SE and propene is a concerted process, the transition state would probably have a character scmevAere between I and II:
\/.Si.GHz'H
I IIIf the amount of biradical character increases the freedom in the transition state, then the A factor for the process will be increased; log A varies from 5.2 to 7.4. [These values were calculated by the
193author from the quoted entropies of activation in Hoffinann's review. ]The A factors for all the hydrogen-transfer reactions of SE (Table 7.1)lie within this range and are therefore consistent with an ene process.Although the reactions for the carbon analogues do not occur by concertedreactions, it is known that silicon has an enhanced ability to
83participate in pericyclic transition states (see Chapter 1).The reaction between SE and propene has been described (shown in
Chapter 1) as occurring via an initially-formed biradical in a four- membered cyclic transition state which could then rearrange to a six-
-137-

membered species to transfer a hydrogen (Reaction 1.5). Chemical intuition argues in favour of the latter preferentially forming since the four-membered requires a much more difficult geometry of approach. The mechanism can be re-written:
— Hmajor route /]yfe2Si=CH2 + C3H6 ^ MezSi^ yCHz — ^ IVfeaSi ^
majorproduct
IVfez Si M92 Si-
Scheme 7.1
The major route, which gives ATMS, could now be an ene reaction, in line36 37 38with proposals for this or similar reactions by Conlin, Wiberg ’
42and Sommer. The reverse reaction, i.e. the decomposition of ATTYB togive SE plus propene, has been described as a retroene process by
39 83Bailey, and Kwart, with reactions resembling this being described in similar terms by Block^^ and Barton.
7.3 RETROENE PROCESSES
The kinetics of two reactions vhich were thought to adopt a retroene mechanism have been measured: the decomposition of ATMS to SE pluspropene (Chuter 4) , and the pyrolysis of vinyldimethylcarbinoxydimethyl- silane (VCS) (Chapter 5):
Log Ajyfe3SiCH2CH=CH2 — ^ Me2Si=CHz + ]yfeCH=CH2 10.55 (7.3)
Hiyfe2SiOCMe2CH=CH2 — ^ f- 2Si=0 + ]Yk2C=C(H)Me 12.46 (7.4)
-138-

These two A factors are consistent with a retroene process since they liewith"the usual range for six-centre pericyclic reactions : log A =11.5± 1.5. The validity of these measured A factors could be assessedby comparisons with the reactions of their carbon analogues; unfortunatelythe appropriate reactions do not appear in the literature to the best ofour knowledge. They can, however, be compared with a series of kinetic
196measurements by Kwart et al. on systems of the type:
R3Ri,C=0 + HR2C=CRiMe (7.5)
From the measured A factors it was concluded that the mechanism adopted was a six-centre pericyclic process. As the R groups were changed from hydrogen to larger groups, e.g. methyl, phenyl, the A factor increased and ranged from:
Log A = 11.09 (Ri,R2,R3/R4 = H) to Log A = 12.21 (Ri,R3,Rit = H; R2 = CgHs)
VCS has four methyl groups attached to the cyclic transition state:
Si
and the measured A factor is towards the upper end of the range.ATl/IS, on the other hand, has no exterior groups on its transition
state, causing it to be less floppy, and giving rise to a lower A factor. Indeed, the measured value suggests that the transition state has little biradical character and I is probably a better approximation than II.
-139-

This is perhaps to be expected since hydrogen transfer occurs by breaking one carbon-hydrogen bond and forming another, vhich is likely to occur simultaneously before a radical centre has chance to develop; this would cause a tighter transition state. For VCS a carbon-hydrogen bond replaces a weaker silicon-hydrogen vhich could be nearly completely broken before the transition state is reached; this would give rise to a freer system and a higher A factor. It is reasonable to conclude, therefore, that both ATMS and VCS deccmpose by a retroene process.
7.4 REVERSIBILITY OF RCTROENE REACTIONS
The possibility that the two retroene reactions studied are reversible(though in LPP the pressure is too low to observe such slow bimolecular processes ; this fact incidentally indicates that 1,1,3-triraethy1-1-silacyclobutane takes no part in our ATMS reaction since it results from SE plus propene, as shown in Scheme 7.1) can be investigated by enthalpy calculations. There are two possible orientations of either SE ordimethylsilanone (Di) to the appropriate alkene:
(i) SE plus propene
\/Si) s i ^
CHz
A B
For both A and B a silicon-carbon double bond and a carbon-hydrogen linkage is broken ; the enthalpy change difference between the two is given by the remaining bonds formed:
— 140“

for A:Si-C ~ 370 kJ irol"
for B:Si— H
146C-H ~ 410 " C-CTotal ~ 780 " TotalAH therefore favours A by 61 kJ mol" .
378 kJ mol"^341719
146
(ii) Di plus 2-methyIbut- 2 -ene
O\/Si
Si
H
O
D
for C:Si— C - 370 kJ mol O — H ~ 380
Total ~ 750
-1146
for D:
AH therefore favours C by 84 kJ mol
Si— H 0-C Total -1
378 kJ mol 288 666
-1146
This means that, whilst Reaction 7.3 (the décomposition of ATMS) is reversible. Reaction 7.4 (the décomposition of VCS) is not likely to be, since orientation C would not re-generate VCS. There are further complications because there are also two possible orientations of the 2-methyIbut-2-ene.
7.5 ENTROPY CALCULATIONS
The plausibility of our measured A factor for ATMS decomposition can27be assessed by using John's figure for the reverse process (log A = 5.2)
along with the value of the entropy change, AS, for the reaction. Since the entropies of the relevant silicon containing species are not known to any degree of accuracy, AS must be calculated using the schane devised by
-141-

O'Neal and Ring,^^^ which is used in a similar way to that for heats offormation, discussed in Chapter 1, and is analogous to the scheme devised
25by Benson. Calculations were based on free-rotor entropies: noallowance was made for hindered rotations because of the large uncertain - ties^^^ and since these contribute only a minor correction compared to
the other approximations involved.
fMe3SiCH2CH=CH2 ^ m2S±=CB2 + ]XfeCH=CH2 (7.3)r
25There are the following symmetry terms:
In Me3SiCH2CH=CH2 :
a xT = 1 (Point group Ci), UiNT = 3* (3 methyl groups, 1 trimethylsilyl)In MeCH=CH2 :
Qext = 1 (Cs), Q|nt = 3^ (1 methyl group)In Me2Si=CH2 :
GEXT = 2 (C2v), cjint = 3^ (2 methyl groups)
AS^ = S^(MS2Si=CH2) + S^(]VfeCH=CH2 ) - S^(]^fe3SiCH2CH=CH2)
AS^ = {[Cd(H)2] + [Sid(C)2 ] +2[C(H)3Si] -Rln2-2Rln3}+ {[C(H)3C] + [Cd(H)(0] + [Cd(H)2 ] - R Li 3}- {3 [C(H)3Si] + [Si(C)U + [C(H)2 CSi] + [Cd(H)(C)J + [Cd(H)2 l -4Rln3}
AS^ = {[Cd(H)2 ] + [Sid(C)2 ] + [C(H)3C]}- {[C(H)3Si] + [Si( O U + [C(H)2 CSi]}- R In 2 + R In 3
Terms [Cd(H)2 ] and [Sid(C)2 l:
There are no entropy terms available for silicon double-bonded species; these can be estimated by comparison with those for M 0 2C=CH2 , i.e.
[Cd(H)2 ] + [Cd(C)2 ] .An approximation for the difference in entropy on "substitution" of
silicon for carbon can be obtained by comparing entropies at 800 K for analogous compounds, e.g. Me3SiH, MS3CH:
-142-

S29s/J niol" k” Cp29e/J mol” k“ Cpeoo/J inol“ k“ Ref.MsaSiH 343.05 106.73 205.18 105lyfeaCH 294.55 94.49 202.92 25
Using the method in reference 25, p.33Estimate: Cpj^ jyfeaSiH ~ 166 J mol"^ k"^
MegCH ~ 159 J mol"^ k"^tfeaSiH sîoo = S?9s + cZ_ In |H
>8 0 0
'TM= 343 + 164 = 507 J mol"^ k"^
lyfeaCH Seoo = 294 + 157= 451 J mol"^ k“^
.*. At 800 K the entropy difference is 507 - 451 = 56 J mol" k"U Thesame value is obtained by using additivity terms rather than theentropies of the molecules directly. There is only a minor correctionin allowing for the lower strength of the silicon-carbon double bondcompared to carbon-carbon. This correction can be calculated from thevibrational frequencies of the two bonds:
C = C 1670 cm"iSi = C 1004 cm“^
Benson (reference 25, p.304) has compiled a table relating entropy andtemperature to the frequency of an oscillator. From the table thecorrection is 0.4 J mol"^ at 298 K and 2.9 J mol"^ at 800 K. This mustbe added to both the terms [Cd(H)2 ] and [Sid(C)2]. The overall correctionis therefore 56 + 5.8 ~ 62 J mol"^ k"U
AS = {[Cd(H)2] + [Cd(C)2] + [C(H)3C]}- {[C(H)3Si] + [Si(C)U + [C(H)2CSi]} - 5.8 + 9.1 + 62 J mol"^ k"^
A correction must be made for AS at elevated temperature: O'Neal andRing^^^ give Cp values at 800 K:
-143-

Cp^gg/J mol"^ k"i Cp8oo/J mol"^ k"[Cd(H)2 ] 21.34 42.13[Cd(C)2 ] 17.15 24.27[C(H)3C] 25.90 54.48[C(H)3Si] 25.90 54.48[Si(C)U 21.63 28.49[C(H)2CSi] 20.42 46.57
ACp298 = (21.34 + 17.15+ 25.9) - (25.9 + 21.63 + 20.42)= -3.56 J mol"^ k"
ACpeoo = (42.13 + 24.27 + 54.48) - (54.48+ 28.49 + 46.57)= -8.66 J mol"^ k"^
Estimate ACp^^ = -6.62 J mol"^ k
I Kr"PtmASaoo — AS298 + ACn,., In 298
1 1-- 1 25
8 0 0
ASsoo = AS298 - 6.54 J mol"^ k"^ASsoo = (115.5- 53.1 + 127.2)
- (127.2- 84.3 + 51.2) - 5.8 + 9.1 + 62 - 6.54 = 154 J mol"^ k"^
The rate constant for the forward reaction is given by:kf = ekT exp(ASf/R) exp(-E/RT)
h ^and the reverse by:
kr = e^kTC^exp(ASr/R) exp(- /RT) h
where is the concentration in the standard state to which the 198entropies refer; this correction factor is necessary to obtain
consistency in units.Af _ 1 exp(ASf/R) exp(-ASr/R)Ar eC^Since AS = ASf"*" - ASr^
è L = 1 - Gxp (AS/R)Ar eC^
-144-

Af = Ar exp (AS/R) eC°
The calculated entropies are for compounds at 1 atmosphere pressure; converting to concentration units at 800 K gives equal to 22.4 x 800/273 = 65.6 dm^ mol”^.
Using John's^^ measured value for Ar of 10 ' dm^ mol” this gives:
Af = 10 ' Xe X 65.6
Af = 10^°’ s"^
This compares favourably with our measured value of 10 °' s"^ and we can conclude that our figure is therefore entirely plausible.
7.6 STRENGTH OF tt BCND IN Me2Si=CH2
The measured Arrhenius parameters for the following reactions v j e r e
(Chapter 4) :ki]yfe3SiCH2CH=CH2 — ► ]Vfe2Si=CH2 + jyfeCH=CH2 (7.3)k2]Vfe3SiCH2CH=CH2 — ^ m s S i - + -C3H5 (7.6)
Log A E/kJ mol-1ki 10.55 ± 0.32 215.5 ±5.5k2 15.60 ± 0.50 306.6 ±8.5ki+k2 12.51 ± 0.27 245.8 ±4.6k_i 5.2 ± 0.3 34.6 ±4.4 (P. John)
The activation energy for Reaction 7.6 can be identified with the silicon-ally 1 bond strength (see below). AH for Reaction 7.3 is given ty Ef-Er = 216-35 kJ mol" . Using appropriate bond strength data in references 22 and 146 we obtain:
-145-

AHi = DCiyfegSi-CsHs) + D(]yte2SiCH2-H) - DTr(]yfe2Si=CH2) -DfCgHs-H)216-35 = 307 + 415 - Dtt (Me2Si=CH2) - 371
D7r0yfe2Si=CH2) = 170 ± 20 kJ mol"^
This value lies between the two most recent estimates : 163 ± 20 kJ mol” (Walsh) and 188 ±20 kJ mol” (Davidson, Potzinger and Peimann).^^
7.7 STRENGTH OF THE SILICON-ALLYL BOND
The strength of the silicon-ally 1 bond can be estimated by catparisons with carbon analogues:
tBu-Me = 342 kJ mol" MegSi - Me = 370 kJ mol" ^Bu-Allyl = 286 kJ mol"^Difference = 56 kJ mol"^
On this basis the silicon-ally 1 bond strength should be 370 - 56 = 314 kJ mol" , which agrees with our measurement within experimental error, whilst being on the upper side of the range covered by our error limits. This is consistent with calculations by Weidner and Schweig^^^'^^^ who demonstrated that in ATIVB there is a significant amount of overlap between the silicon-allyl bond and the tt system, which causes a weakening of the sigma bond (and gives rise to the [1,3] sigmatrcpic shift behaviour at 775 K). This conjugative effect, known as the "silicon 3 effect" is much stronger than in the carbon analogue, ^Bu-allyl, and could account for the small discrepancy. In this light, the measured silicon-allyl bend strength in ATJYG seems a reasonably reliable figure.
7.8 HEAT OF FORMATION OF ATMS
jyfe3SiCH2CH=CH2 — ► Me3Si* + 'C3H5 (7.6)
AH;^ (Me3SiCH2CH=CH2) can be calculated frcm the relationship :
-146-

AH = AHf dyfeaSi*) +AHf^(-C3H5) - AHf (jyfe3SiCH2CH=CH2)AH = Ef-ErIf Er (the activation energy for the corbination of two radicals) is
zero, thenAH = Ef = 307 kJ mol"^ andAHf^(Me3SiCH2CH=CH2) = AHf^(Me3Si') +AHf^(-C3H5) - AH
r\ 22 ^ 25Using Walsh's figure for AHf (]yfe3Si-) and Benson's for AHf (•C3H5),
AHf^(I/k3SiCH2CH=CH2) = -3.0 + 170 - 307-1= -140 kJ mol
O'Neal and Ring's schone^^^ gives:AHf^(Me3SiCH2CH=CH2) = 3[C(H)3Si] + [Si(C)4] + [C(Si)(C)(H)2 ]
+ [Cd (H)(0)] + [Cd(H)2 ]= -3(42.7) - (76.6) - (12.9) + (35.9) + (26.2)= -155.5 kJ mol"^
Therefore our calculated value, based on the measured activation energy for Reaction 7.6 is in reasonable agreement.
7.9 HEAT OF FORMATION OF SE
Me3SiCH2CH=CH2 — iyfe2Si=CH2 + iyfeCH=CH2 (7.3)AHf^(Me2Si=CH2) can be calculated using the relationship:AH = AHf^(Me2Si=CH2) + AHf (]yfeCH=CH2) - AHf^(]^3SiCH2CH=CH2)
Using AHf^(MeCH=CH2) = 20.5 kJ mol"\^^181 = AHf (jyfe2Si=CH2) + 20.5 - AHf^(^3SiCH2CH=CH2)AHf^(Me2Si=CH2) = 160.5 + AHf^(Me3SiCH2CH=CH2)
Walsh^^ has calculated AHf^(Me2Si=CH2) = 29.3 kJ mol" , a figure with which better agreement is obtained with our value for AHf^(Me3SiCH2CH=CH2 )rather than that frcm O'Neal and Ring's scheme: we calculate
= 160.5 - 140 = 20.5 kJ mol”^.
AHf (]yfe2Si=CH2) = 160.5-140
-147-

7.10 1,1,3-TRIMETHYL-1 -SILACYCLOBUTANE FORMATION
The activation energy for the reaction:Me2Si = Œ 2 + Me2Si— (7.7)
can be calculated from the relationship:AH = AHfU]Vk2Si— I AHf (]yfe2Si=Œ2) -(AHf
Q ,
O'Neal and Ring's scheme gives :Atlf / /te2Si-| \ = 2[C(H)3Si] + [Si(C)U +2[C(H)2CSi] + [C(H)(C)3 ]V + [C(H)3 C] + ring strain
= -2(42.7 ) - (76.6) - 2(12.9) - (7.9) - (42.7) + (70.3)
= -168 kJ mol"^AH = Ef — Er
Since Er = 264 kJ mol"^ then AH = Ef - 264.
Using our value for AHf^(Me2Si=CH2) , we obtain Ef - 264 = 1 6 8 - 2 0 . 5 - 20.5
Ef = 55 kJ mol"^
Since this figure compares favourably with those in Table 7.1 for the equivalent SE reaction with ethene, we conclude that our measurements onthe ATI/IS system give a series of thermochemical quantities vbich areinternally consistent and in reasonable agreement with other measurements and estimations. The figures are combined in the energy diagram (Figure 7.1) .
7.11 INSERTION OF Di AND SE INTO METHYLTRIMETHYLSILYL ETHER
The kinetics of the reactions :
Me2Si=CH2 + ]yfe3SiOMe ^ iyfe3SiCH2Si]yfe20]yfe (7.8)
]yfe2Si=o + j/fe3Siojyfe ^ iyfe3Siosijyte20]yfe (7.9)
-148-
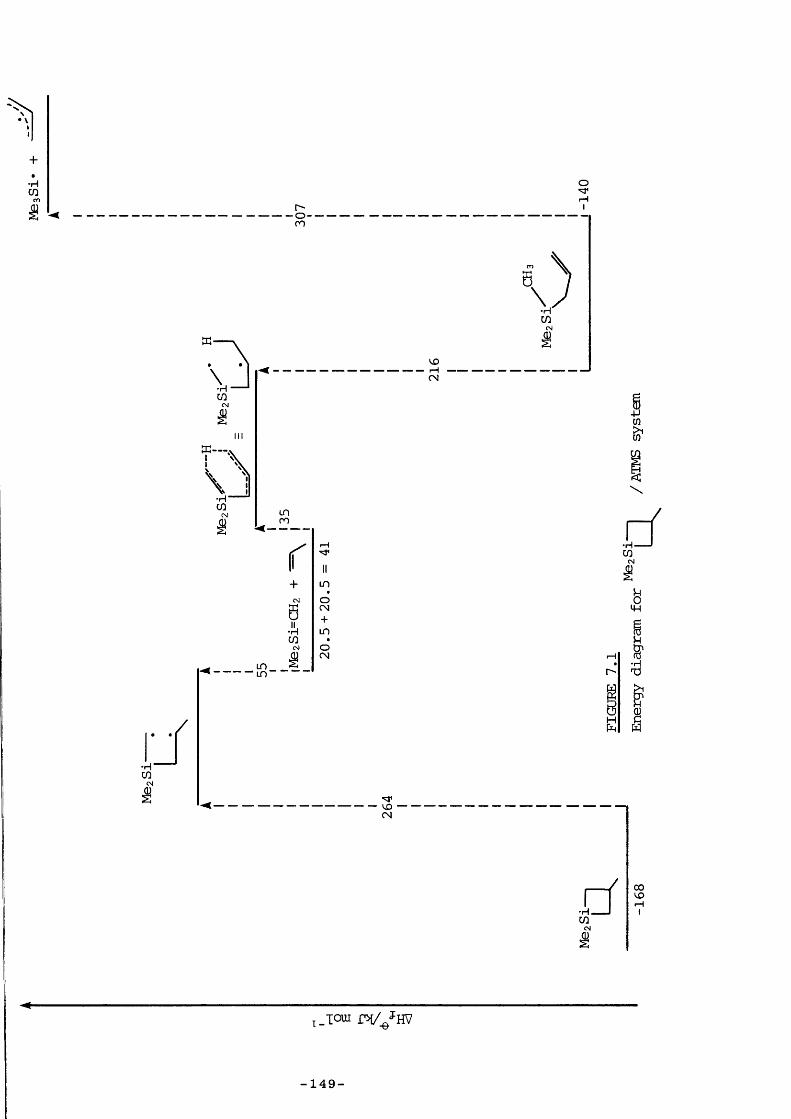
•Hen o
• o -m
vorHCN
LD
r•Hen
min '
LDoCN+moCN
.cfen
I/ I
enI eo
CN
cf•HenCMI
00CD
i.TOüi r>[/Jhv
-149-

have been measured (Chapter 6) relative to the rates of SE dimérisation(for Reaction 7.8) and Di trimérisation (for Reaction 7.9), kg. Using
18Gusel'nikov's parameters for SE dimérisation,logio k3/(dm^ mol"^ s" ) = (5.3 ±0.2) - (6.3 ±3.2 kJ mol”^)/2.303 RT
as shown in Table 7.1, and1/ 2/ 2/
logio (k^As ^)/(dm^ rrol s ) = (3.6 ± 0.4) - (28.4 ± 6.4 kJ mol"^)/2.303 RT.To obtain k^, it is necessary to estimate ks since it has not been measured. The expression for ki+ is not particularly sensitive to changes in ks because its cube root must be taken. If Di trimérisation occurs via Dz, then Di dimérisation is likely to be the rate determining step since D z is unstable, ks can therefore be equated with Di dimérisation. Frcm Table 7.1, the reaction between SE and SE (dimérisation) , ethene, and oxygen, have all measured A factors equal to ~10 ' . If this is taken to be the A factor for Di dimérisation, with the activation energy >0 kJ mol"^, thenlogio ki+/(dm mol"^ s"M = (5.8 ±0.4) - (x± 6.4 kJ mol"^)/2.303 RT vhere x > 28.4.
Both this A factor, and that for SE insertion into methyl trimethylsilyl ether are lower than those for the SE reactions with SE, ethene and oxygen, which is consistent with a tighter transition state: insertionsof this type are thought to occur via a four-centure transition state (of. A factors for 2tt + 2tt systems. Table 7.2). The figure for Reaction 7.9 is slightly higher than that for Reaction 7.8 vhich is to be expected since the transition state for Di insertion into methyItrimethylsilyl ether contains two oxygens and two silicons in its four-centre cyclic structure (III) , a system prcbably more unstable than that in Reaction7.8 (IV); (cf. D z f DSCB, etc.) and hence having greater freedcm. The
-150-
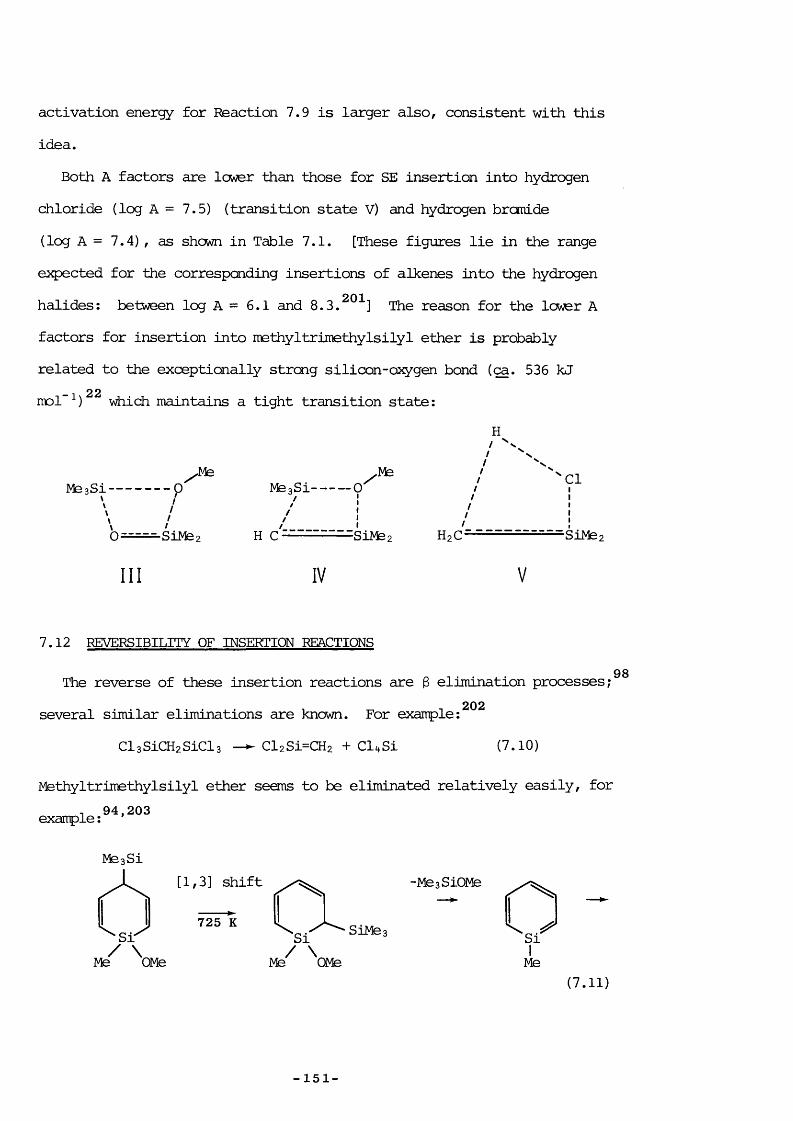
activation energy for Reaction 7.9 is larger also, consistent with this idea.
Both A factors are lower than those for SE insertion into hydrogenchloride (log A = 7.5) (transition state V) and hydrogen bromide(log A = 7.4) , as shown in Table 7.1. [These figures lie in the rangeexpected for the corresponding insertions of alkenes into the hydrogen
201halides: between log A = 6.1 and 8.3. ] The reason for the lower Afactors for insertion into methy Itrimethylsilyl ether is probably related to the exceptionally strong silicon-oxygen bond (ça. 536 kJ mol" 1)22 maintains a tight transition state :
H
MesSi------- O.Me
Me,Si 0Me ^C1
b- -Sim: H C -Sim: HoC- ■sim2
III IV V
7.12 REVERSIBILITY OF INSERTION REftCTICMS
The reverse of these insertion reactions are 3 elimination processes;202several similar eliminations are known. For example:
ClsSiCHzSiCls — ^ Cl2Si=CH2 + Cl^Si (7.10)
98
mthyItrimethylsilyl ether seems to be eliminated relatively easily, for, 94,203example :
MesSi[1,3] shift
725 K
m ' OMe
-MeaSiOm
SiMea - s /IMe
(7.11)
-151-

Me 3 Si SiMezOMe
773 K _\ H 10"^ Torr \ j j (7.12)
45Gusel'nikov et al. have demonstrated that the reverse of Reaction 7.8 occurs at higher tenperatures than those used to study the insertion process (Chapter 6):
MeaSiCHzSiMezOMe — ► MezSi=CHz + MeaSiOMe (7.13)
Our A factor for the insertion process can be canbined with the entropy change for the reaction to calculate the A factor for Reaction (7.13) viiich can then be carpared with A factors for other 3 - elimination reactions. This should give an indication as to the reliability of our measurement. The entropy change for Reaction (7.13) can be estimated using O'Neal and Ring's scheme as was done before for ATMS deccmposition,
7.13 ENTROPY CALCULATIONS
Symmetry terms:MeaSiCHzSiMezOMe
tEXT - 1 (Cl), aiNT = 3 (6 methyl groups, 1 trimethylsilyl group) MeaSiOMe
cEXT = 1 (Cl), tiNT = 3 (4 methyl groups, 1 trimethylsilyl)MezSi=CHz
' EXT = 2 (Point group , tiNj = 3 (2 methyl groups)
MezSi=CHz + S®" MeaSiOMe - S^ MeaSiCHzSiMezOMe
The absence of entropy terms for Si-0 bonds is not a restriction as these terms cancel.
ÛS^= {[Cd(H)z) + [Sid(C)2] +2[C(H)3Si] - R In 2 - 2R In 3}
-152-

+ {3[C(H) 3Si] + [SiO(C)a] + [0(Si)(O] + [C(H)aO] -5Rln3}- {5[C(H)aSi] - [Si(C)4] - [C(H)2(Si) 2] - [SiO(C)a]- [0(Si)(O] - [C(H)aO] + 7Rln3}
AS^ = {[Cd(H)2] + [Sid(C)2]} -{[Si(C)^] + [C(H)2(Si) 2] } ~ Rln 2
We add 56 J mol” k“ for "substitution" of silicon for carbon and 6 kJ mol"^ for the silicon double bond to correct for ASsoo; the remaining corrections are done in the same way:
Cp^gg/J mol"^ k"^ Cpggg/J mol"^ k'^[Si(C)4] 21.63 28.49[C(H)2(Si)2] 19.58 46.61[Cd(H)2l 21.34 42.13[Cd(C)2] 17.15 24.27
ACp298 = (21.34 + 17.15) - (21.63 + 19 . 58)= -2.72 J mol k"^
ACpgoo = (42.13 + 24.27) -(28.49 + 46.61)= -8.70 J mol"^ k"^
Estimate ACp^^ = -6.3 J mol"^ k"^ASsoo = AS298 “ 6.3 In
= AS298 - 6.2ASsoo = (115.5 - 53.1) - (-84.3 + 53.39) + 56 + 6 - 6.20 - 5.76
= 143.3 J mol"^ k-iASsoo ~ 143 J mol"^ k"
From our measured value of 10 ' for the reverse reaction :
Af = X 10 -ex 65.6
Af = 10 °' ^
This is a reasonable figure when carpared with the A factor of the 98reaction:
MeaSiCzH^Cl — ^ MeaSiCl + C2H4 (7.14)
for which A = 10^ ° s"^.
-153-
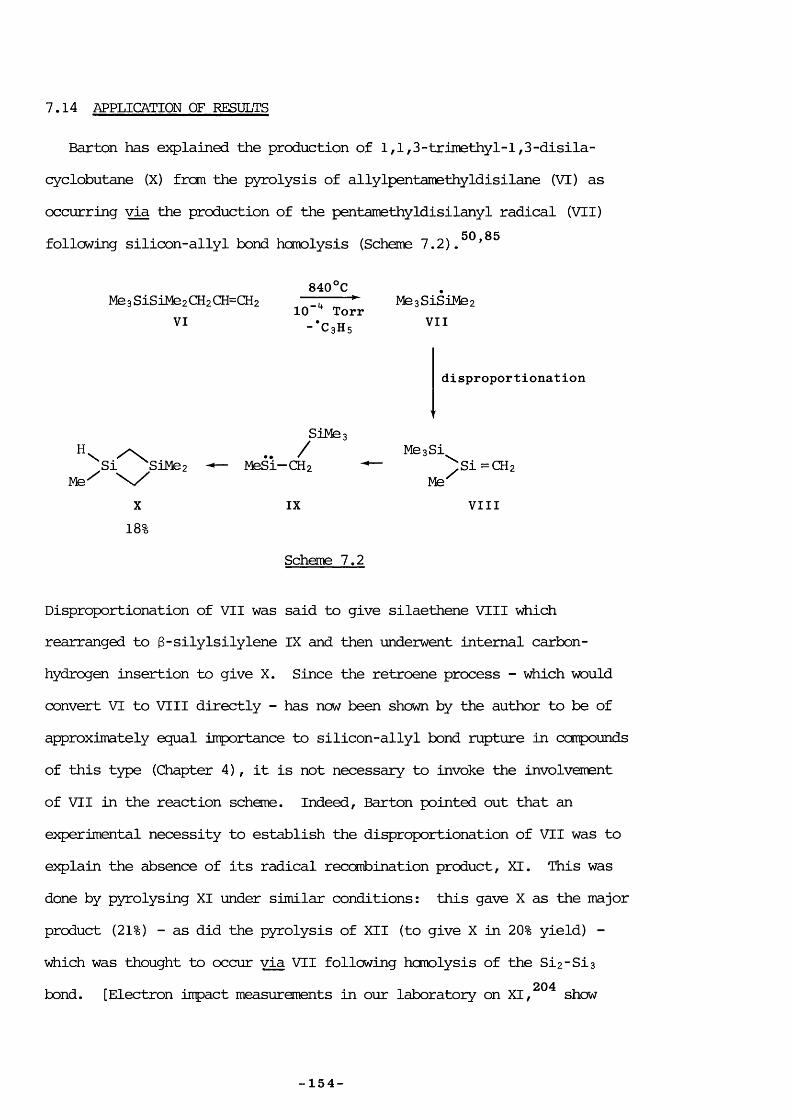
7.14 APPLICATION OF RESULTS
Barton has explained the production of 1,1,3-trimethyl-l,3-disila- cyclobutane (X) from the pyrolysis of allylpentamethyldisilane (VI) as occurring via the production of the pentamethyIdisi lany 1 radical (VII) following silicon-allyl bond hemolysis (Scheme 7.2).^^'^^
Me 3 SiSiMez CHz CH=CH2 VI
840°C10”** Torr -‘C3H 5
MesSiSiMe2VII
disproportionation
SiMeiSi ^SlMe2
Me" ^X18%
.. /MeSi-CH2Me 3 Si
MeIX
Scheme 7.2
Si=CH2
VIII
Disproportionation of VII was said to give silaethene VIII viiich rearranged to 3-silyIsilylene IX and then underwent internal carbon- hydrogen insertion to give X. Since the retroene process - which would convert VI to VIII directly - has now been shown by the author to be of approximately equal importance to silicon-allyl bond rupture in compounds of this type (Chapter 4), it is not necessary to invoke the involvement of VII in the reaction scheme. Indeed, Barton pointed out that an experimental necessity to establish the disproportionation of VII was to explain the absence of its radical recoribination product, XI. This was done by pyrolysing XI under similar conditions: this gave X as the majorproduct (21%) - as did the pyrolysis of XII (to give X in 20% yield) - which was thought to occur via VII following hemolysis of the Si2-Si3 bond. [Electron impact measurements in our laboratory on X I , s h o w
-154-
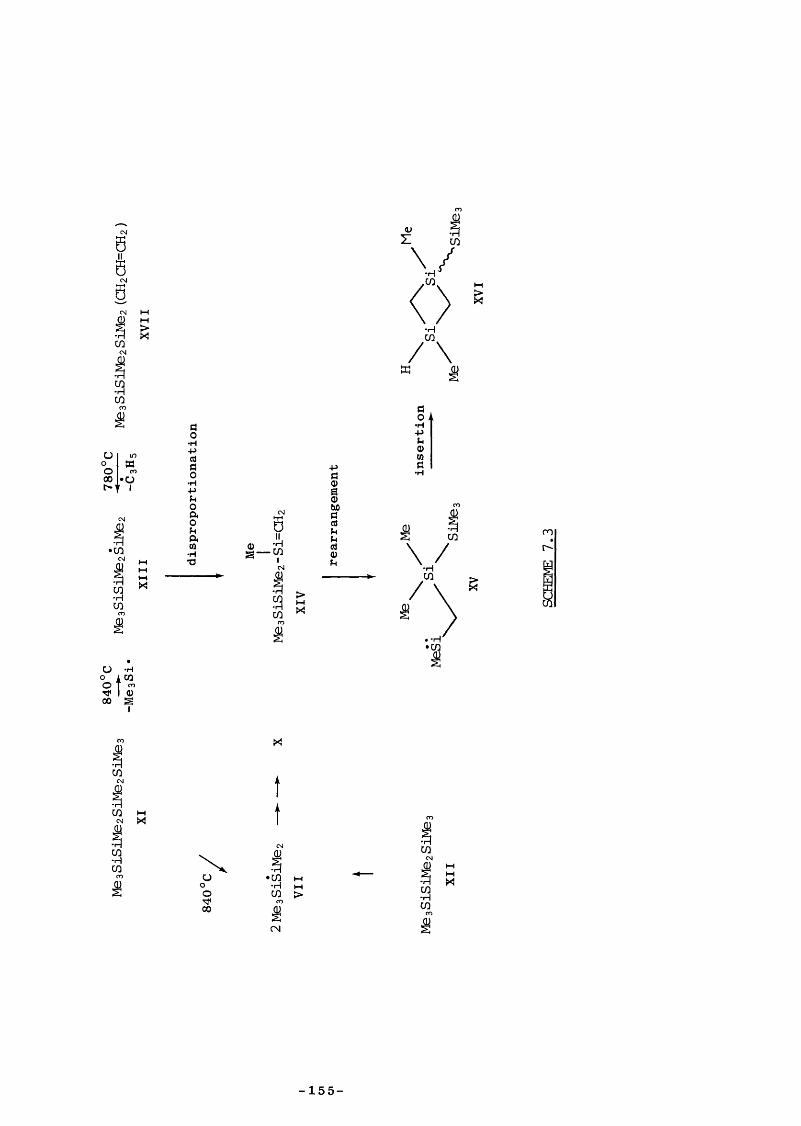
s èS
s
Oo aO00 • ü! > ' ' 1
•en
X
uoo00 t l
ao•H-pco0o•H4Jf-iO(XouAta
SS — CM
m
3en
-p§ibOÜaupcdQ)P
X
ao•H-puotaa•H
enm
en Mcsl X
X
ooo00
I•aenICN
X
-155-

that the weakest silicon-silicon bond is, in fact, the Sii-Si2 linkage, rupture of which would give XIII.] Barton assumed the minor production of XVI resulted frcm disproportionation, rearrangement and insertion of XIII, resembling that for VII. This was said to be substantiated when XVII also gave XVI on pyrolysis, again thought to occur via XIII (Scheme 7.3) . A similar argument as before can be applied : XVII can directly give XIV bya retroene reaction; XIII need not be present in the reaction sequence. Nor need it be invoked in the conversion of XI to XVI. The radical VII is also not a necessary intermediate in forming X frcm either XI or XII: a reasonable alternative is to form the silaethenes VIII and XIV frcm hydrogen abstraction reactions and unimolecular decompositions, as shown in Schane 7.4.
In conclusion, the evidence of the inportance of retroene reactions in the pyrolysis of allyl silanes has thrown doubt on published mechanisms involving initial silicon-allyl bond rupture: products can be equallywell explained by invoking significant retroene reactions.
7.15 SUMMARY
The pyrolysis of several carpounds have been studied by LPP. 1,1- dimethyl-2,3-bis(trimethylsilyl)-1-silirene was found not to be a thermal source of dimethylsilylene: the major (rearrangement) product between440 and 550 K was Me3SiSiMe2C ECSiMeg, probably formed via a biradical intermediate. ATMS pyrolysed between 860 and 960 K via two major path ways, both of approximately equal importance : silicon-allyl bond rupture,and SE plus propene formation by a retroene process. Above ça. 0.3 Torr the secondary bimolecular reaction of trimethylsilyl radical addition to ATMS becomes significant, resulting in the formation of vinyl trimethyl - silane and tetramethylsilane. Pulsed stirred-flow and photochanical
-156-
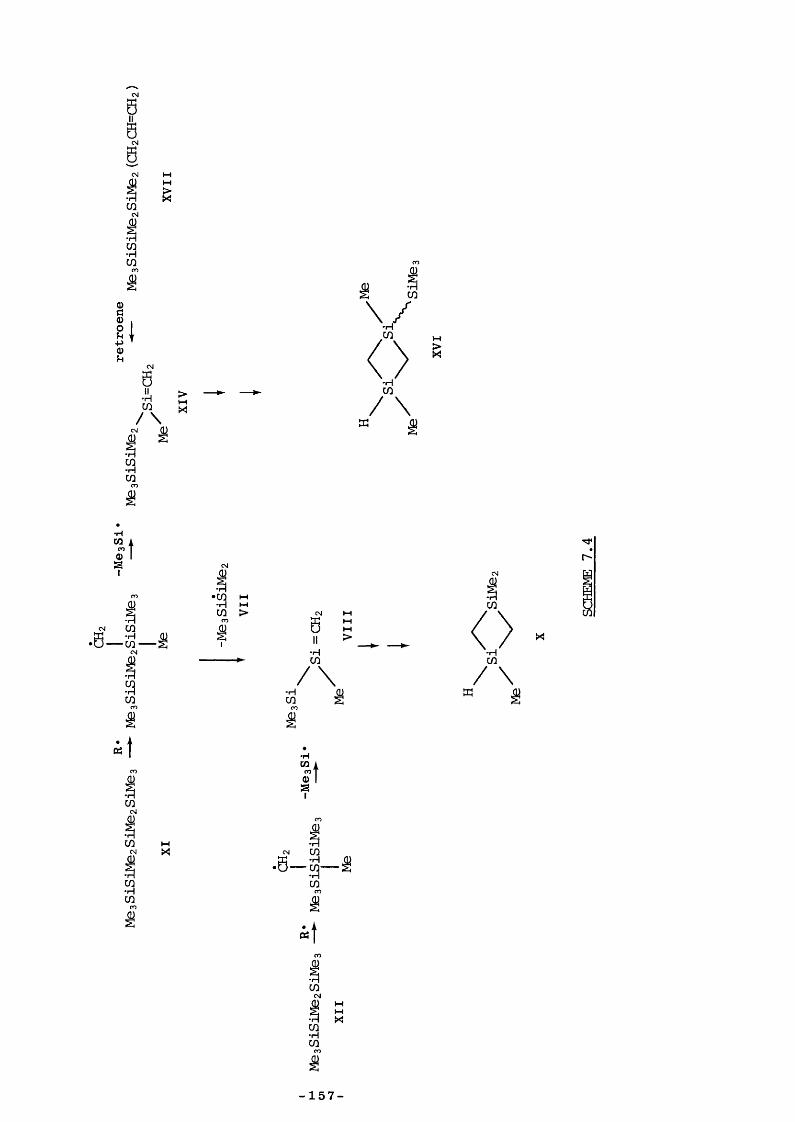
wCM
12-Pd>-Ü4in
/ \a
\/•H•HW/ \
23in
ItsIm -H •u— in —
CMm5[/]
•■'3in0 :•Hin
-HCO/\
CO
X
CO/ \
tf tœ
CM
.#CO
CM
5CO
I
It
.0-1CO
ai t
CO
aCO
X
-157-

experiments confirmed this. The pyrolysis of two related ccmpounds was also studied. A retroene mechanism also operated in the pyrolysis of VCS which gave Di and 2-methyIbut- 2-ene between 710 and 830 K, but not for 1,1,3,3-tetramethy1-1-vinyIdisiloxane: this required much higher teiperatures for décomposition, 860 to 1075 K, and pyrolysed by a radical rather than a molecular mechanism; Di was not a major product. The kinetics of the insertion of both SE and Di into methyItrimethylsilyl ether were measured; the Arrhenius parameters indicated that a similar mechanism, via a four-centre transition state, operated. Several thermochemical quantities can be calculated as a result of kinetic measuranents made on various systens; these agree fairly well with those in the literature.
— 15 8 —

REFERENCES

REFERENCES
1. I. Fleming, "Comprehensive Organic Chemistry" Vol. 3, part 13, p.539.
2. R. West and T. J. Barton, J. Chem. Ed. (1980), 57, 165; 335.3. I. M. T. Davidson, "Reaction Kinetics I" Chem. Soc. Spec. Period.
Report (1975), 212.4. I. M. T. Davidson, Quarterly Reviews (1971), g5. 111.5. P. Cadman, G. M. Tilsley and A. F . Trotman-Dickenson, J. Chem.
Soc. (A), (1969), 1370.6. I. M. T. Davidson and J. I. Matthews, J. Chem. Soc., Farad. Trans.
I (1981), 77, 2277.7. A. C. Baldwin, I. M. T. Davidson, M. D. Reed, J. Chem. Soc.,
Farad. Trans. I (1978), 74, 2171.8. P. Cadman, G. M. Tilsley and A. F . Trotman-Dickenson, J. Chem.
Soc., Faraday I (1972), 68, 1849.9. H. P. Schuchmann, A. Ritter and C. von Sonntag, J. Organomet.
Chem. (1978), 148, 213.10. S. K. Tokach and R. D. Koob, J. Am. Chem. Soc. (1980), 102, 376.11. B. Cornett, K. Choo and P. Caspar, J. Am. Chem. Soc. (1980), 102,
377.12. L. Gammie, I. Safarik, 0. P. Strausz, R. Roberge and C. Sandorfy,
J. Am. Chem. Soc. (1980), 102, 378.13. I. M. T. Davidson, P. Potzinger and B. Reimann, Ber. Bunsenges.
Phys. Chem. (1982), 13.14. R. N. Haszeldine, S. Lythgoe and P. J. Robinson, J. Chem. Soc. (B),
(1970), 1634.15. L. E. Gusel’nikov and N. S. Nametkin, Chem. Rev. (1979), 79, 529.16. N. S. Nametkin, L. E. Gusel'nikov, V. M. Vdovin, P. L. Grinberg,
V. I. Zav’yalov and V. D. Oppengeim, Dokl. Akad. Nauk. SSSR(1966), 171, 630.
17. M. C. Flowers and L. E. Gusel’nikov, J. Chem. Soc. (B), (1968),419; 1396.
18. L. E. Gusel’nikov, K. S. Konobeyevsky, V. M. Vdovin and N. S. Nametkin, Dokl. Akad. Nauk. SSSR (1977), 235. 1086.
19. S. Basu, I. M. T. Davidson, R. Laupert and P. Potzinger, Ber. Bunsenges. Phys. Chem. (1979), 83, 1282.
20. 0. L. Chapman, C. C. Chang, J. Kolc, M. E. Jung, L. A. Lowe,T. J. Barton and M. L. Tumey, J. Am. Chem. Soc. (1976), ^ , 7844.
21. M. R. Chedekel, M. Skoglund, R. L. Kreeger and H. Shechter,J. Am. Chem. Soc. (1976), ^ , 7846.
22. R. Walsh, Accts. Chem. Res. (1981), ^ , 246.23. I. M. T. Davidson, A. M. Fenton, P. Jackson and F . T . Lawrence,
J . Chem. Soc., Chem. Commun. (1982), 806.
-160-

24. R. W. Carr and W. D. Walters, J. Phys. Chem. (1963), 67, 1370.25. S. W. Benson, "Thermochemical Kinetics" 2nd. Ed., Wiley (1976).26. T. J. Barton and E. Kline, J. Organomet. Chem. (1972), ^ , C21.27. P. John, personal communication to I. M. T. Davidson.28. I. M. T. Davidson, C. E. Dean and F. T. Lawrence, J. Chem. Soc.,
Chem. Commun. (1981), 52.29. A. G. Brook and J. W. Harris, J. Am. Chem. Soc. (1976), 98, 3381.30. N. S. Nametkin, L. E. Gusel'nikov, R. L. Ushakova and V. M.
Vdovin, Dokl. Akad. Nauk. SSSR (1971), 201, 1365.31. N. S. Nametkin, L. E. Gusel'nikov, R. L. Ushakova, V. M. Vdovin,
Izv. Akad. Nauk. SSSR Ser. Khim. (1971), 8, 1840.32. N. S. Nametkin, R. L. Ushakova, L. E. Gusel'nikov, E. D. Babich
and V. M. Vdovin, Izv. Akad. Nauk. SSSR Ser. Khim. (1970), 7,1676.
33. L. E. Gusel'nikov, N. S. Nametkin and N. N. Dolgopolov, J. Organomet. Chem. (1979), 169. 165.
34. T. J. Barton, G. Marquardt and J. A. Kilgour, J. Organomet. Chem,(1975), 85, 317.
35. B. G. Gowenlock, paper presented at the 7th. Int. Symposium on Gas Kinetics, Gottingen, (1982).
36. R. T. Conlin, M. P. Bessellieu, P. R. Jones and R. A. Pierce,Organometallics (1982), 396.
37. N. Wiberg, G . Preiner and 0. Schieda, Chem. Ber. (1981), 114. 3518.
38. N. Wiberg, G . Preiner, 0. Schieda and G . Fischer, Chem. Ber.(1981), 11^, 3505.
39. W. J. Bailey and M. S. Kaufmann, Abs. 157th. Nat. Meeting of Am. Chem. Soc., Minneapolis, April (1969), P. Org. 57; Chem. Eng. News (1969), 47, 35.
40. H. M. Frey, A. Kashoulis, L. M. Ling, S. P. Lodge, I. M. Pidgeon and R. Walsh, J. Chem. Soc., Chem. Commun. (1981), 915.
41. D. N. Roark, L. H. Sommer, J. Chem. Soc., Chem. Commun., (1973),167.
42. C. M. Golino, R. D. Bush, D. N. Roark and L. H. Sommer, J. Organomet. Chem. (1974), 66, 29.
43. L. E. Gusel'nikov, N. S. Nametkin and V. M. Vdovin, Accts. Chem. Res. (1975), 8, 18.
44. R. D. Bush, C. M. Golino, G . D . Homer and L. H. Sommer, J. Organomet. Chem. (1974), 80, 37.
45. N. S. Nametkin, L. E. Gusel'nikov, E. A. Volpina and V. M.Vdovin, Dokl. Akad. Nauk. SSSR (1975), 2^, 386.
46. P. John, B. G. Gowenlock and P. Groome, J. Chem. Soc., Chem. Commun., (1981), 806.
47. C. M. Golino, R. D. Bush, P. On and L. H. Sommer, J. Am. Chem.Soc., (1975), 1957.
— 16 1 —

48. R. D. Bush, C. M. Golino and L. H. Sommer, J. Am. Chem. Soc.,(1974), 7105.
49. R. T. Conlin and D. L. Wood, J. Am. Chem. Soc. (1981), 103. 1843.50. T. J. Barton, S. A. Burns and G. T. Burns, Organometallics,
(1982), 1, 210.51. T. J. Barton, G. T. Burns, W. F. Goure and W. D. Wulff, J. Am.
Chem. Soc. (1982), Ij^, 1149.52. I. M. T. Davidson and J. F. Thompson, J. Chem. Soc., Chem. Commun.
(1971), 251.53. I. M. T. Davidson and J. F. Thompson, J. Chem. Soc., Farad. Trans.
I, (1975), 71, 2260.54. T. H. Lane and C. L. Frye, J. Organomet. Chem. (1979), 172. 213.55. T. J. Barton and W. D. Wulff, J. Am. Chem. Soc. (1979), 101. 2735.56. T. J. Barton, S. K. Hoekman and S. A. Burns, Organometallics
(1982), 1, 721.57. A. V. Tomadze, N. V. Yablokova, V. A. Yablokov and G. A. Razuvaev,
J. Organomet. Chem. (1981), 212. 43.58. M. E. Delf, Ph.D. Thesis, Leicester University (1975).59. C. M. Golino, R. D. Bush and L. H. Sommer, J. Am. Chem. Soc.,
(1975), 97, 7371.60. H. Okinoshima and W. P. Weber, J. Organomet. Chem. (1978), 155.
165.61. W. Ando, M. Ikeno and A. Sekiguchi, J. Am. Chem. Soc. (1977).62. W. P. Weber and H. Okinoshima, 5th. Int. Symposium on Organosilicon
Chem. (1978), p.200.63. D. Seyferth, T. F. Lim and D. P. Duncan, J. Am. Chem. Soc. (1978),
100. 1626.64. W. H. Atwell and D. R. Weyenberg, Angew. Chem. Int. Ed., (1969),
8, 469.65. I. M. T. Davidson and J, I. Matthews, J. Chem. Soc., Farad. Trans.
I (1976), 72, 1403.66. I. M. T. Davidson and M. E, Delf, J. Chem. Soc., Farad. Trans. I
(1976), 72, 1912.67. P. P. Caspar, React. Intermed. (1978), 1, 229; P. P. Caspar,
React. Intermed. (1981), 2, 335.68. D. Seyferth and D. Annarelli, J. Organomet. Chem. (1976), 117. C51.69. D. Seyferth, D. Annarelli and S. Vick, J. Am. Chem. Soc., (1976),
M , 6382.70. R. T. Conlin and P. P. Caspar, J. Am. Chem. Soc. (1976), ^ , 3715.71. R. T. Conlin and P. P. Caspar, J. Am. Chem. Soc. (1976), ^ , 868.72. W. H. Atwell and J. G . Uhlmann, J. Organomet. Chem. (1973), 52,
C21.73. D. Seyferth and S. C. Vick, J. Organomet. Chem. (1977), 125. Cll.
-162-

74. I. M. T. Davidson and N, A. Ostah, J. Organomet. Chem. (1981), 206.149.
75. M. Ishlkawa, K. Nakagawa, S. Katayama and M. Kumada, J. Organomet. Chem. (1981), C48.
76. T. y. Gu and W. P. Weber, J. Organomet. Chem. (1980), 184. 7.77. A. Chickl and W. P. Weber, J. Organomet. Chem. (1981), 210. 163.78. A. Chickl and W. P. Weber, Inorg. Chem. (1981), 20, 2822.79. T. J. Barton, W. D. Wulff and W. F. Goure, J. Am. Chem. Soc. (1978),
100. 6236.80. M. E. Childs and W. P. Weber, J. Org. Chem. (1976), 1799.81. T. J. Drahnak, J. Michl and R. West, J. Am. Chem. Soc. (1981), 103.
1843.82. H. Sakurai, A. Hosoml and M. Kumada, J. Chem. Soc., Chem. Commun.
(1970), 767.83. J. Slutsky and H. Kwart, J. Org. Chem. (1973), 38, 3658.84. S. M. Neider, G. R. Chambers and M. Jones Jr., Td. Lett. (1979),
40, 3793.85. T. J. Barton and S. A. Jacobi, J. Am. Chem. Soc. (1980), 102. 7979.86. T. J. Barton, personal communication to I. M. T. Davidson.87. H. Kwart and J. Slutsky, J. Am. Chem. Soc. (1972), 94, 2515.88. J. Slutsky and H. Kwart, J. Am. Chem. Soc. (1973), M , 8678.89. T. L. Gilchrist and R. C. Storr, "Organic Reactions and Orbital
Symmetry", Cambridge University Press, London (1972).90. E. Block and L. K. Revelle, J. Am. Chem. Soc. (1978), 100. 1630.91. T. J. Barton and G. T. Burns, J. Organomet. Chem. (1981), 216.
C5.92. T. J. Barton and G. T. Burns, J. Am. Chem. Soc. (1978), 100. 5246.93. T. J. Barton and G. T. Burns, J. Organomet. Chem. (1979), 179,
C17.94. T. J. Barton, G. T. Burns, E. V. Arnold and J. Clardy, Td. Lett.
(1981), 22, 7.95. V. F. Mironov, G. M. Rad'kova, V. D. Sheludyakov and V. V.
Shcherbinin, Dokl. Akad. Nauk. SSSR (1972), 207. 114.96. D. Seyferth, K. R. Wursthorn, T. F. 0. Lim and D. J. Sepelak,
J. Organomet. Chem. (1981), 205. 301.97. G. Manuel, G. Bertrand, P. Mazerolles and J. Ancelle, J. Organomet.
Chem. (1981), 311.98. I. M. T. Davidson, M. R. Jones and C. Pett, J. Chem. Soc. (B),
(1967), 937.99. P. R. Jones and D. D. White, J. Organomet. Chem. (1978), 154. C33.
100. J. Barthelat, G. Trinquier and G. Bertrand, J. Am. Chem. Soc.(1979), 101, 3785.
— 16 3 —

101. M. Ishikawa, T. Fuchikami and M. Kumada, J. Organomet. Chem.(1977), 142, C45.
102. M. Ishikawa, K. Nakagawa and M. Kumada, J. Organomet. Chem.(1980), 1^, 117.
103. M. Ishikawa, K. Nishimura, H. Sugisawa and M. Kumada, J. OrganometChem. (1980), 1^, 147.
104. C. Haas and M. A. Ring, Inorg. Chem. (1975), 14, 2253.105. H. E. O'Neal and M. A. Ring, J. Organomet. Chem. (1981), 213. 419.106. CATCH Tables, University of Sussex (1972); updated (1977).107. T. N. Bell, K. A. Perkins and P. G. Perkins, J. Chem. Soc., Far.
Trans. I (1981), 77, 1779.108. S. J. Band, I. M. T. Davidson and C. A. Lambert, J. Chem. Soc.
(A), (1968), 2068.109. P. Potzinger and F. W. Lampe, J. Phys. Chem. (1969), 73, 3912.110. R. Ellul, P. Potzinger, B. Reimann and in part P. Camilleri,
Ber. Bunsenges. Phys. Chem. (1981), 85, 407.111. P. Potzinger and F. W. Lampe, J. Phys. Chem. (1970), 74, 719.112. D. Quane, J. Phys. Chem. (1971), 75, 2480.113. P. Potzinger, A. Ritter and J. Krause, Z. Naturforsch. (1975),
30a, 347.114. T. L. Allen, J. Chem. Phys. (1959), 1039.115. S. R. Gunn and L. G. Green, J. Phys. Chem. (1961), 65, 779;
J. Phys. Chem. (1964), 68, 946.116. I. M. T. Davidson, J. Organomet. Chem. (1979), 170. 365.117. S. W. Benson and M. Luria, J. Am. Chem. Soc. (1975), ^ , 704.118. C. E. Dean, Ph.D. Thesis, Leicester University, appearing shortly.119. D. M. Golden and S. W. Benson, Chem. Rev. (1969), 69, 125.120. R. Walsh and J. M. Wells, J. Chem. Soc., Far. Trans. I (1976), 72,
100.121. A. M. Doncaster and R . Walsh, J. Chem. Soc., Far. Trans. I (1979),
75, 1126.122. I. M. T. Davidson and C. A. Lambert, J. Chem. Soc. (A) (1971),
882.123. S. W. Benson and G . N . Spokes, J. Am. Chem. Soc. (1967), 89, 2525.124. D. M. Golden, G. N. Spokes and S. W. Benson, Angew. Chem. Int. Ed.
(1973), 12, 534.125. M. J. Pilling, "Reaction Kinetics" Oxford University Press (1975),
p. 49.126. I. M. T. Davidson and M. A. Ring, J. Chem. Soc., Far. I (1980),
76, 1520.127. I. M. T. Davidson, N. A. Ostah, D. Seyferth and D. P. Duncan,
J. Organomet. Chem. (1980), 187. 297.
-164-

128. J. H. Beynon, "Mass Spectrometry and its Application to Organic Chemistry" Elsevier Publishing Co. (1960), p.26.
129. M. D. Reed, Ph.D. Thesis, Leicester University, (1978).130. V. G. Micromass Q801K, Makerfe Manual, Chapter 5.131. P. J. Robinson and K. A. Holbrook, "Unimolecular Reactions"
Wiley (1972).132. I. M. T. Davidson and A. V. Howard, J. Chem. Soc., Far. Trans. I
(1975), 71, 69.133. A. C. Baldwin, I. M. T. Davidson and A. V. Howard, J. Chem. Soc.,
Far. Trans. I (1975), 71, 972.134. F . T. Lawrence, Ph.D. Thesis, Leicester University, appearing
shortly.135. W. E. Falconer, T. F . Hunter and A. F . Trotman-Dickenson, J. Chem
Soc. (1961), 609.136. E. J. York, W. Dittmar, J. R. Stevenson and R. G . Bergman, J. Am.
Chem. Soc. (1973), 95, 5680.137. V. J. Tortorelli and M. Jones, J. Am. Chem. Soc. (1980), 102.
1425.138. N. P. Neureiter, J. Am. Chem. Soc. (1966), 88, 558.139. D. Seyferth, D. P. Duncan and S. C. Vick, J. Organomet. Chem.
(1977), 1^, C5.140. P. S. Skell and E. J. Goldstein, J. Am. Chem. Soc. (1964), 86.
1442.141. E. A. Chernyshev, N. G . Komalenkova, S. A. Bashkirova and V. V.
Sokolov, Zh. Obshch. Khim. (1978), 48, 830.142. M. Ishikawa, H. Sugisawa, T. Fuchikami, M. Kumada, T . Yamabe,
H. Kawakami, K. Fukui, Y . Ueki and H. Shizuka, J. Am. Chem. Soc.(1982), 104, 2872.
143. H. Gilman, S. G . Cottis and W. H. Atwell, J. Am. Chem. Soc. (1964), 86, 5584.
144. F . Johnson, R. S. Gohlke and W. H. Nasutavicus, J. Organomet. Chem. (1965), 3, 233.
145. M. S. Gordon, J. Am. Chem. Soc. (1980), 102, 7419.146. K. W. Egger and A. T. Cocks, Helv. Chim. Acta (1973), ^ , 1516.147. M. Barber, A. M. Doncaster and R. Walsh, Int. J. Chem. Kinetics
(1982), 14, 669.148. K. Shiina and M. Kumada, J. Org. Chem. (1958), ^ , 139.149. I. M. T. Davidson, F . T . Lawrence, G . Fritz and E. Matern, to be
published shortly in Organometallics.150. E. Bastian, P. Potzinger, A. Ritter, H. P. Schuchmann, C. von
Sonntag and G . Weddle, Ber. Bunsenges. Phys. Chem. (1980), 84, 56151. A. L. J. Beckwith, C. J. Easton and A. K. Serelis, J. Chem. Soc.,
Chem. Commun. (1980), 482.
-165-

152. N. Ya. Chernyak, R. A. Khmel'nitskii, T. V. D'yakova, V. M. Vdovin and T. N. Arkipova, Zh. Obshch. Khim. (1966), 36, 96.
153. I. M. T. Davidson and F. T. Lawrence, unpublished results.154. N. Auner and J. Grobe, J. Organomet. Chem. (1980), 197, 13.155. I. M. T. Davidson and F. T. Lawrence, unpublished results.156. I. M. T. Davidson and I. T. Wood, J. Organomet. Chem. (1980), 202.
C65.157. S. K. Tokach, P. Boudjouk and R. D. Koob, J. Phys. Chem. (1978), 82,
1203.158. S. K. Tokach and R . D . Koob, J. Phys. Chem. (1979), 83, 774.159. P. Cadman, G. M. Tilsley and A. F. Trotman-Dickenson, J. Chem. Soc.,
Far. Trans. I (1973), 69, 914.160. A. V. Topchiev, N. S. Nametkin, T. I. Chernysheva and S. G. Drgaryan,
Dokl. Akad. Nauk. SSSR (1956), 110, 97.161. H. Sakurai, A. Hosomi and M. Kumada, J. Org. Chem. (1969), 34, 1764.162. A. W. P. Jarvie, Organomet. Chem. Rev. A (1970), 6, 153.163. A. W. P. Jarvie and R. J. Rowley, J. Chem. Soc. (B) (1971), 2439.164. M. Kosugi, K. Yano, M. Chiba and T. Migita, Chem. Lett. (1977), 7,
801.165. L. Lunazzi, G. Placucci, L. Grossi and M. Guerva, J. Chem. Soc.,
Perkin Trans. II (1982), 43.166. T. Migita, M. Kosugi, K. Takayama and Y. Nakagawa, Tet. (1973), 29,
51.167. K . W. Egger and A. T. Cocks, Helv. Chim. Acta (1973), 56, 1537.168. Z . G. Szabo, "Advances in the Kinetics of Homogeneous Gas Reactions",
Methuen (1964), p.127.169. J. I. Matthews, Ph.D. Thesis, Leicester University (1975).170. K. Y . Choo and P. P. Gaspar, J. Am. Chem. Soc. (1974), ^ , 1284.171. H. Melville and B. G . Gowenlock, "Experimental Methods in Gas
Reactions", Macmillan (1964), pp.273, 295.172. R. B. Cundall and A. Gilbert, "Photochemistry", Nelson (1970), p.12.173. M. A. Nay, G. N. C. Woodall, 0. P. Strausz and H. E. Gunning, J. Am.
Chem. Soc. (1965), 87, 179.174. D. Mihelcic, R. N. Schindler and P. Potzinger, Ber. Bunsenges. Phys.
Chem. (1971), 15, 1137.175. H. Nicki and G . J . Mains, J. Phys. Chem. (1964), 68, 304.176. L. C. Glasgow, G. Olbrich and P. Potzinger, Chem. Phys. Lett. (1972),
14, 466.177. E. R. Austin and F . W. Lampe, J. Phys. Chem. (1977), ^ , 1134.178. G . Carlo and J. Franck, Z. Phys. (1922), ^ , 161.179. P. Potzinger, B. Reimann and R. S. Roy, Ber. Bunsenges. Phys. Chem.
(1981), 85, 1119.
— 16 6 —

180. H. M. Frey and R. Walsh, Chem. Rev. (1969), 69, 103.181. R. J. Cvetanovic, Âdvan. Photochem. (1963), 1, 115; W. E. Falconer
and R. J. Cvetanovic, unpublished results.182. R. Neat and K. Packer, Leicester University (1982).183. H. K. Spencer and R. K. Hill, J. Org. Chem. (1976), ^ , 2485.184. J. Saam and H. Bank, unpublished studies from Dow Corning Corp.185. T. J. Barton and W. D. Wulff, J. Organomet. Chem. (1979), 168. 23.186. L. E. Gusel'nikov, Y. P. Polyakov and N. S. Nametkin, Dokl. Akad.
Nauk. SSSR (1980), 2^, 1133.187. I. M. T. Davidson and I. T. Wood, J. Chem. Soc., Chem. Commun.
(1982), 550.188. G. A. Drennan and R. A. Matula, J. Phys. Chem. (1968), 72, 3462.189. I. M. Butler, J. Am. Chem. Soc. (1962), 84, 1393.190. E. K. von Gustorf, D. V. White, B. Khim, D. Hess and J. Leitich,
J. Org. Chem. (1970), 1155.191. W. T. Brady and H. R. O'Neal, J. Org. Chem. (1967), 612.192. R. Huisgen, L. A. Feiler and P. Otto, Chem. Ber. (1969), 102, 3444.193. H. M. R. Hoffman, Angew. Chem. Int. Ed. (1969), 8, 556.194. W. Oppolzer and V. Sniekus, Angew. Chem. Int. Ed. (1978), 3^, 476.195. H. E. O'Neal and S. W. Benson, J. Phys. Chem. (1967), 71, 2903.196. H. Kwart, S. F . Sarner and J. Slutsky, J. Am. Chem. Soc. (1973),
95, 5234.197. 0. M. Nefedov, A. K. Maltsev, V. N. Khabashesku and V. A. Korolev,
J. Organomet. Chem. (1980), 201. 123.198. P. J. Robinson, J. Chem. Ed. (1978), 55, 509.199. U. Weidner and A. Schweig, J. Organomet. Chem. (1972), 39, 261.200. U. Weidner and A. Schweig, Angew. Chem. Int. Ed. (1972), 3A, 146.201. S. W. Benson and A. N. Bose, J. Chem. Phys. (1963), 39, 3463.202. G. Fritz and E. Matern, Z. Anorg. Allg. Chem. (1976), 426. 28.203. T. J. Barton and M. Vuper, J. Am. Chem. Soc. (1981), 103, 6788.204. M. Jones, Ph.D. Thesis, Leicester University (1971).
-167-

APPENDICES

APPENDIX 1.1
ABBREVIATIONS FOR CHEMICAL SPECIES
SE 1,1-diinethyl-l-silaetheneDMSCB 1,1- dimethyl-1 - silacyclobutaneDSCB 1,1,3,3-tetramethy 1-1,3-disilacyclQbutaneATMS allyltrlmethylsilaneTATMS trans -a- trimethy Isilylally ItrimethylsilaneDTS 1,1-dime thy 1-2,3-bis (trimethylsilyl) -1-silireneVCS viny IdiraethylcarbinojQ^dimethylsi laneTVS 1,1,3,3- tetramethy 1 -1 - viny Idisiloxane
— 169*”

APPENDIX 2.1
Kinetic runs arc stored on cassette tapes and can be easily accessed and processed rapidly.A typical data file is shown below,the first 4 rows being baseline cycles.
-.E:
- - ; “ . -C" * .t. — . ^ - -» ■ ~
r-
' *- - A-w*- - â. 1 * - ♦ 1 .....
.1 - - - - . - V - .. W V t- : _.L :
— — 1 - : : c• ' •- T - ■ - - . -J
- - » —--r- - : - - T — _ *7,r
* - * - “ -k- ' - ' " • -: ■ - - ■■ : r * r *. u r .
T . •• ** t-u
-170-
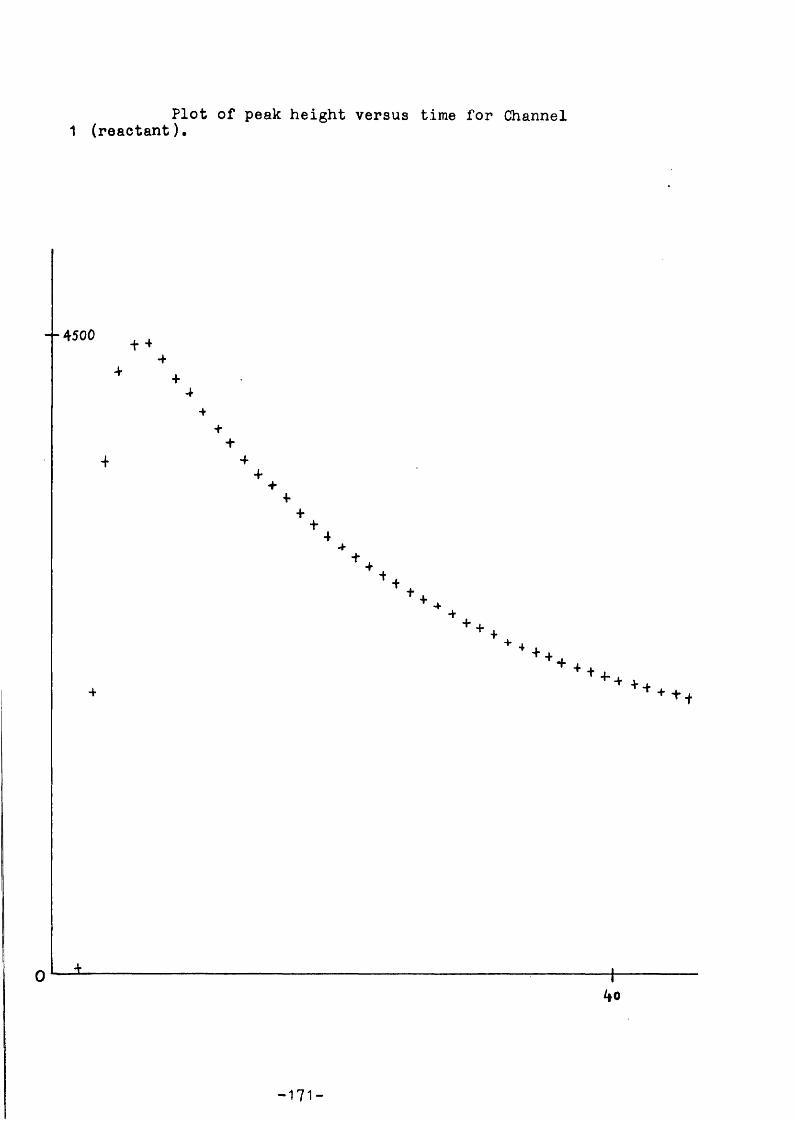
Plot of peak height versus time for Channel1 (reactant).
--4500
40
- 171-
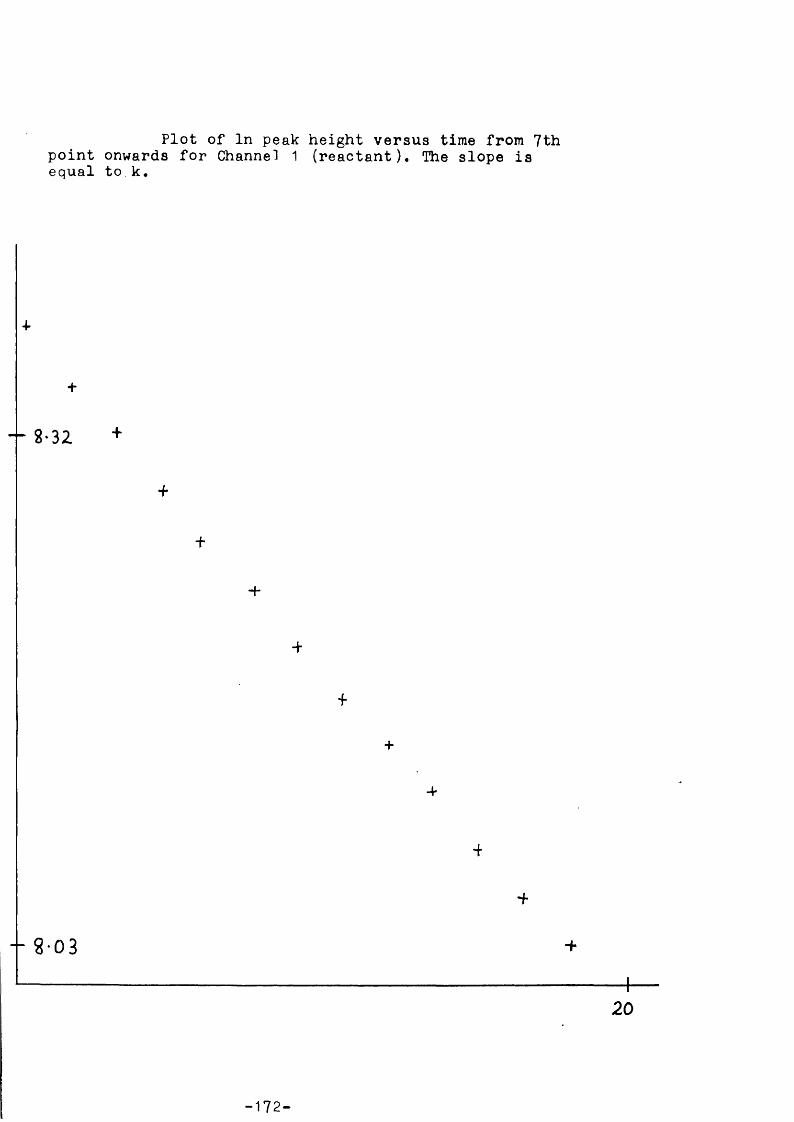
Plot of In peak height versus time from 7th point onwards for Channel 1 (reactant). The slope is equal to k.
— 8 - 3 2
20
-172-
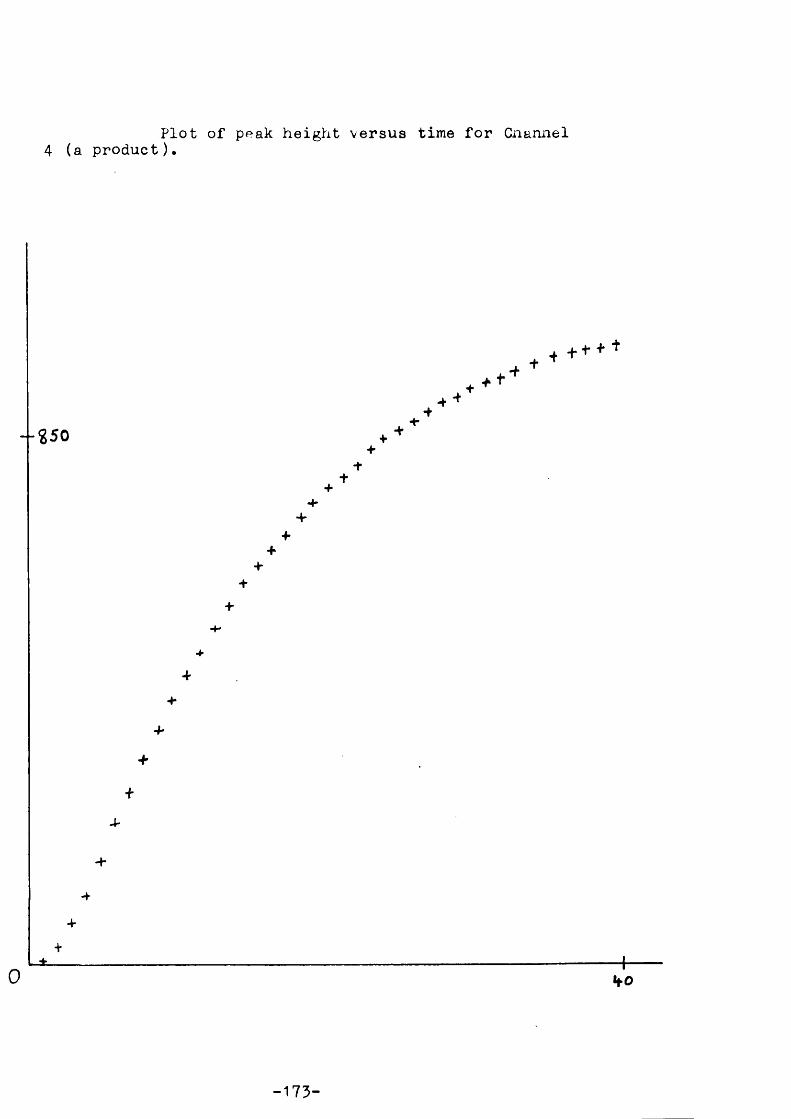
Plot of peak height versus time for Cnannel4 (a product ) •
-175-

APPENDIX 4.1
G.C. Petention Times
B.p./K vhereAverage retention appropriate and
time/min Assigned Coirpomd vhere known4.1 C 1114.1 C = C 1694.1 SiHg 2164.2 CC = C 2254.2 Si Hz 2534.8 SIR 2825.0 Hz Si C = C5.5 Si 3007.2 Hz Si Si Hz7.8 Hz SiCC = C8.8 SiC = C 3289.3 C C C C
669.6 C C C C C
C
338-343
13.8 SiC = C 356IC
14.1 SiCCC
14.3 SiCC = C 35814.4 SiC = CC 359.414.5 SiCCC 36316.0 SiCC = CC 372.617.4 Si Si 386
I-------118.2 Si C Si C 39220.3 Si CCC = C
C20.7 Sic Si23.2 SiCCCC
I ICC
Continued-174-

APPENDIX 4.1 (Continued)
B.p./K vhereAverage retention appropriate and
time/min Assigned Compound vhere known24.9 SiCC SiIC25.1 SiCC Si 435IC25.3 SiC = CCSi (c/t) 44425.7 Si C C C Si 445.628.0 Si C C C C C
Si C30.5 Si C C = CC C Si
CorSi C = C C C C Si (c/t)
C30.8 SiCCCCCSi
C31.7 SiCCC Si
Si
-175-

APPENDIX 4.2
Glossary of Shorthand Notation for Identifying Canpounds
c lyfeHC = C Œ2=CH2SiHa PteSiHaCC = C CHaCH=Œ2Si H2 jyfe2SiH2SiH lyfeaSlHH2 Si C = C H2 (]yfe) SiCH=Œ2Si jyfeitSiH2 Si Si H2 H2 (]yfe) SiSi (Me) H2H2 Si C C = C H2 (jyfe) SiCH2CH=Œ2SiC = C MeaSiCH=Œ2CCCC Me2CHCHiyfe2
66CCCCC 2CHPrCSiC = C MsaSiC=CH2C Me
SiCC MeaSiCmyfeI IC MeSiCC = C MeaSiCH2CH=CHzSi C = C C MeaSiCH=CHMeSiCCC JYkaSiPrSiCC = CC jyfe2Si<r~|Si Si MaSiSiMeaSi C Si C jyfe2Si;^Si]yfe2
Si C Ç C = C jyfeaSiCH2CHCH=CH2C Me
SiCSi MeaSiCH2Si]SkSiCCCC MfeaSiCH2CHCH]yte2
CC m
SiCC Si jyfeaSiCH2CSiJyfeaC Œ
“I76- Continued

APPENDIX 4.2 (Continued)
Si C C Si 6
SiC = CCSiSiCCC SiSi C C C C C
Si CSi C C = C C C Si
ICSiC = CCCCSi
6Si C C C C C Si
ICSi C C C Si
s'i
MeaSiCHzCHSiMeaMe
lyfe aSiCH=CHCH2SiJyfe a
MeaSiCHzCHzCHzSiMea ]yte a SiCHzCHCHzCHiyfe z
MeaSijyfe aSiCHzCH=CHCHCHzSiJ^ a
MeMe3SiCH=CHCHzCHCHzSiMea
MeMe a SiŒzCHzŒzÇHŒzSiJyfe 3
Mea SiCHzCHCHzSi]^ a MeaSi
Radicals
H-C"Hz Si*CCCCCCSi*Si C* SiCCC SiCCC SiCCCC
66 SiCC CC
I IC CSi
H*Me*HziyfeSi*HMezC*EtCHzMeaSi*MeaSiCHzIVfeaSiCHzCHiytejyfeaSiCHzCHz&zlyfeaSiCHzCHCHCHz
I I jyfeMe jyfeaSiCHzCH CHCHz
Me CHzSiMea
-177-

APPENDIX 5.1
PREPARATION OF yiNYIJDIMETHYLCAI^INQXyDIiyEIHYTKqTTANE
25g 1,1,3,3-tetramethyIdisiloxane (0.19 mol), 11.6g 2-methyl-3-buten- 2-ol (0.13 mol) and \ pellet of potassium hydroxide were added to a 100 cm^ flask fitted with a thermometer, magnetic stirring bead and a reflux condenser topped with a dry ice/acetone cold finger condenser and drying tube. This allowed dimethylsilane to be retained and hydrogen to be vented vhilst preventing moisture entering the system. Refluxing at 333K took place over a period of 4 hrs 10 mins. The crude reaction product was distilled away frcm the alkaline catalyst on a vacuum line. Purification was achieved on a Fischer Spaltrohr HMS 500 fractional distillation system vhich gave 9.6g (0.07 mol) of vinyldimethylcarbinoxy- dimethylsilane (51% yield), b.p. 383 K.
n.m.r. (Varian EM 390 80 MHz in CCI4 solvent) :(6, ppm): 0.13, 0.16, d (6H, Si-CHa); 1.30, s (6H, C-CHa) ; 4.66, m (IH,Si-H); 5.00, m (2H, CH=CH2); 5.85, m(IH, CH=CH2).
— 178“

APPENDIX 5.2
n.m.r. spectra frcm TVS pyrolysis experiments.
Original TVS (H]Vb2SiOSiJY 2CH=CH2 )(6, ppm): 0.3 (6H) ; 0.7 (6H) ; 4.6, septet, (IH) ; 5.8, multiplet, (3H)The product spectra contained these, and additional resonances at:(6, ppm) : 0.7, s; 7.0, s.Because their integrations varied, two compounds were present.
-179-

APPENDIX 6.1
n.m.r. of M^aSiCHzSiJY^zOMe.Purification was not possible owing to the low concentrations so
integration gave inaccurate results. However, resonances occurred at46exactly the same chemical shifts as reported by John.
(6 ppm): 3.40 (s, ]yfeO) ; 0.12 (SiMez); 0.04 (s, SiMea) ; -0.14 (s, CHz) .
-180-

CHEMICALS
HMe2 SiOCMez CH=CH2 HMG2 Si06iî4e2CH=CH2 Gift frcm C. Frye, USA
MegSi ySlMesC = CV/ \Me jyfe
Me3SiCH2CH=CH2
Me2Si— I
HMe2SiOSiMe2H Cl3SiCH2CH=CH2 (CH3)2C=C(H)CH3 PhCH2SiJyie3 Me3SiOMe (CH3)2C=C(CH3)2 ]yfe3SiCH=CH2 (Me) 2 Hg TrikloneTreble One Cherniclene
Gift fran D. Seyferth, USA
AldrichCambrian Chemicals
Field Instruments
K & K, ICN Pharmaceuticals, USA
FlukaFlùkaKodak, USA B.D.H.Grant & West
— 181 —

SOME GAS KINETIC STUDIES OF REACTIVE ORGANOSILICON INTERMEDIATES Ian T. Wood
ABSTRACTThe study of organosilicon reactive intermediates is an area of
expanding interest. There are many gaps in our knowledge in this relatively new sphere, in addition to the lack of definitive thermo- chemical data and pyrolysis reaction mechanisms for organosilicon catpounds in general. The research towards this thesis was designed to encompass all these topics, and consisted of the investigation, kinetically vhere possible and vhere appropriate, of processes which thermally generate organosilicon reactive intermediates, and some reactions in vhich they participate, using low-pressure pyrolysis (LPP) . Some supporting work was done using pulsed stirred-flow, sealed tube, and mercury photosensitization techniques.
Siliranes have proved useful as silylene sources, but doubt has existed whether silirenes could exhibit similar thermolytic behaviour. The pyrolysis of 1,l-dimethyl-2,3-bis (trimethylsilyl) -1- silirene was investigated between 420 and 550 K and found to give mainly the rearranged product Ms3SiSiMe2CECSi^3, probably via a biradical intermediate. No significant silylene extrusion occurred.
Considerable controversy has surrounded the décomposition of allyltrimethylsilane. Its pyrolysis was studied between 860 and 960 K and the mechanism found to be strongly pressure dependent. At low pressures (<0.3 Torr) thermolysis proceeded by two concurrent uni- molecular processes: silicon-allyl bond rupture, and formation of1,1-dimethyl-l-silaethene plus propene by a retroene mechanism. At higher pressures the bimolecular reaction of trimethylsilyl radical addition to allyltrimethylsilane giving vinyltrimethylsilane and tetramethyIs ilane became inportant. These results clear up many of the disputes over this ooitpound's pyrolysis mechanism and enabled several thermochemical quantities to be calculated.
Between 710 and 830 K viny Idime thy Icarbinoxydime thy Is ilane pyrolysis gave 1,1 -dimethylsilanone plus 2 -methylbut-2 -ene, the Arrhenius parameters being consistent with a retroene reaction. 1,1,3,3 Tetramethyl- 1 -vinyldisiloxane required higher temperatures, between 860 and 1075 K, for decomposition, which occurred by a radical mechanism. No significant production of 1,1-dimethylsilanone occurred.
Several reactions of 1,1-dimethylsilanone were studied, one kinetically, that of insertion into methyItrimethylsilyl ether, along with the equivalent 1,1-dimethyl-l-silaethene process. The Arrhenius parameters showed that similar four-meirbered cyclic transition states were formed. Seme general comments on reactions involving these species have been made.





























