Embed Size (px)
Citation preview

SÍNDROME ANTIFOSFOLÍPIDO. PRUEBAS DIAGNÓSTICAS Y DE
INVESTIGACIÓN. ACTUALIZACIÓN 2020

Título original: Síndrome antifosfolipido. Pruebas diagnósticas y de investigación. Actualización 2020 Autores: María del Mar Ramírez Morales, María Dolores Gálvez Fuentes,
Samantha María Garrido Garnier, María José Sánchez Pino Especialidad: TSS de Laboratorio de Diagnóstico Clínico y Biomédico Edita e imprime: FEDERACIÓN ANDALUZA DE TÉCNICOS
Avda. de Guerrita, s/n. Centro de negocios “Los Azahares” Planta 1ª, Oficina 21-22 14005 Córdoba Teléfono 957 43 02 37 www.sindicatotecnos.es
ISBN: Diseño y maquetación: Alfonso Cid Edición: julio 2020
Araceli Orizo Valverde

ÍNDICE
UNIDAD TEMÁTICA I PRESENTACIÓN Y METODOLOGÍA DEL CURSO. GUÍA DEL ALUMNO 5 1.1 Objetivos 7 1.2 Pertinencia 7 1.3 Programa Docente 8 1.4 Metodología del Curso 11 1.5 Guía del alumno 11 1.6 Estructura del Curso 15 1.6.4 Duración de los Cursos 20 1.7 Guía de uso de la Web 21 UNIDAD TEMÁTICA II LA SANGRE 27 2.1 Introducción 29 2.2 Composición 29 2.3 Plasma 29 2.4 Células sanguíneas o elementos figurados 32 2.5 Funciones de la sangre 52 UNIDAD TEMÁTICA III SÍNDROME ANTIFOSFOLÍPIDO 67 3.1 Definición 69 3.2 Anticuerpos antifosfolípidos 70 3.3 Manifestaciones clínicas 74 UNIDAD TEMÁTICA IV PRUEBAS DIAGNÓSTICAS 139 4.1 Introducción 141 4.2 Diagnóstico en laboratorio 143 UNIDAD TEMÁTICA V PRUEBAS PARA LA INVESTIGACIÓN DE LAS MUESTRAS 161 5.1 Requisitos diagnósticos para el síndrome antifosfolipídico 163 5.2 Procesamiento y preparación de las muestras 164 5.3 Estudio de investigación a tres tiempos 170 5.4 Perspectivas futuras en la investigación 170 5.5 Conclusiones 191 CUESTIONARIO Cuestionario 195


UNIDAD TEMÁTICA I PRESENTACIÓN Y METODOLOGÍA
DEL CURSO. GUÍA DEL ALUMNO


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
7
1.1 Objetivos
1.1.1 Objetivo general El síndrome antifosfolípido (SAF) es una enfermedad autoinmune
caracterizada por trombosis (venosa y/o arterial), y/o pérdidas fetales recurrentes, asociada con la presencia de anticuerpos antifosfolípidos (aPL).
La prevalencia de la enfermedad es desconocida, esto debido en parte a múltiples cambios en los criterios diagnósticos. En lupus eritematoso sistémico (LES), 30 a 40% de los pacientes presentan aPL, y un tercio de estos (10 a 15% de los pacientes con LES en general) tienen manifestaciones clínicas de SAF
Conocer los Avances en Técnicas de Investigación y Desarrollo en relación con el Síndrome Antifosfolípido y sus patologías primarias o asociadas es de vital importancia en el diagnóstico de las enfermedades autoinmunes.
1.1.2 Objetivos específicos 1. Componer el mecanismo de formación, composición y coagulación de
la sangre.
2. Reconocer y comprender el Síndrome Antifosfolípido y los anticuerpos implicados.
3. Analizar y evaluar el Síndrome Antifosfolípido a través las pruebas analíticas e inmunológicas al efecto.
4. Escoger y comparar los procedimientos, preparaciones y técnicas de las muestras necesarias para la investigación del Síndrome Antifosfolípido.
5. Valorar y concluir las Pruebas Diagnósticas y de Investigación con el fin de obtener resultados que amplíen los conocimientos sobre la enfermedad.
1.2 Pertinencia Ante el avance de las enfermedades autoinmunes es necesario la
formación de los T.S. en Laboratorio de Diagnóstico Clínico en Técnicas de Investigación concretas del Diagnóstico de Enfermedades Inmunitarias que se puedan aplicar, con el fin de reducir el tiempo y proceso de diagnóstico en pacientes con patología que respondan a dicha demanda.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
8
El síndrome antifosfolípido (SAF) o síndrome de Hughes es una enfermedad autoinmune sistémica en la cual el proceso patológico subyacente es la trombosis. El síndrome ha sido definido desde el punto de vista clínico por la presencia de trombosis arteriales, venosas o de pequeño vaso y/o por una historia de problemas obstétricos (abortos repetidos, muertes fetales recurrentes o nacimientos prematuros debidos a preeclampsia, eclampsia o insuficiencia placentaria). Dichas alteraciones se asocian a la positividad de anticuerpos antifosfolípido (AAF), de los cuales, los más conocidos son el anticoagulante lúpico (AL), los anticuerpos anticardiolipinas (AAC) y los anticuerpos anti-ß2glicoproteína I (ß2GPI). Las manifestaciones clínicas, muy variadas, pueden afectar cualquier órgano o tejido y cualquier tipo o tamaño de vaso sanguíneo (1-4). El SAF es, probablemente, la trombofilia adquirida más frecuente.
Los avances tecnológicos de los métodos de diagnóstico requieren una actualización en la formación de estos técnicos, que ha sido incorporada en el Real Decreto 771/2014, de 12 de septiembre, por el que se establece el título de Técnico Superior en Laboratorio Clínico y Biomédico y se fijan sus enseñanzas mínimas, comprendidos en los artículos 4, 5, 6, 7, 8, 9 y 10 del citado Real Decreto. Concretamente, el artículo 10 sobre módulos profesionales, desarrolla en el anexo I del presente real decreto, cumpliendo lo previsto en el artículo 10 del Real Decreto 1147/2011, de 29 de julio, código 1372. Técnicas de inmunodiagnóstico.
Esta actualización de conocimientos, redunda en una mejora de la gestión de los recursos del Área de Laboratorio, y como consecuencia colateral, una mejora en la asistencia sanitaria de calidad hacia los usuarios del SSPA y SNA
1.3 Programa Docente El Curso “SÍNDROME ANTIFOSFOLÍPIDO PRUEBAS DIAGNÓSTICAS Y DE
INVESTIGACIÓN” consta de 33 horas lectivas y se divide en las siguientes Unidades Temáticas:
Encuesta previa.
Antes de comenzar el curso se plantea la realización de una encuesta previa de conocimientos sobre la materia que tratará la actividad docente.
Se trata de un cuestionario compuesto por 10 ítems de respuesta múltiple. Se trata de una evaluación previa que servirá al tutor para poder determinar cuál es el conocimiento que el alumno tiene sobre la materia en

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
9
cuestión, e incidir a lo largo de las diferentes unidades temáticas, en aquellos campos que considere son más desconocidos para el alumno.
Duración estimada: 1 hora.
Unidad Temática I. Presentación y metodología del Curso. Guía del alumno.
Enunciamos los objetivos generales y específicos del Curso, así como la pertinencia y la necesidad de esta formación para los profesionales de la materia.
Se detalla la metodología de enseñanza a distancia que el alumno deberá seguir, así como los procedimientos de estudio y protocolos de evaluación. Igualmente, se adjunta una detallada guía para que el discente pueda realizar el curso de forma correcta.
Unidad Temática II. La sangre
En esta Unidad Temática se hace una incursión por el origen del síndrome antifosfolípido, enfermedad autoinmune poco frecuente, que presenta múltiples manifestaciones clínicas, de graves consecuencias, considerándose por tanto como un importante problema clínico.
Se procede a enumerar y explicar cuáles son los componentes de la sangre, sus funciones y se incide en el concepto de hematopoyeses conjunto de procesos, por el cual se produce la formación de células sanguíneas y su desarrollo a partir de células madre pluripotenciales o STEM- CELL, y el concepto de hemostasia, uno de los principales sistemas de defensa del organismo, que actúa ante la lesión de un vaso sanguíneo para mantener la integridad biológica del sistema vascular.
Duración estimada: 7,5 horas.
Unidad Temática III. Síndrome antifosfolípido
Se analiza de forma exhaustiva el síndrome antifosfolípido, enfermedad autoinmune, caracterizada, por un desorden del estado de hipercoagulidad, que provoca un estado trombofílico. Los anticuerpos antifosfolípidos y las diferentes manifestaciones clínicas serán otros de los puntos destacados que se van a tratar en este apartado.
Duración estimada: 14 horas.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
10
Unidad Temática IV. Pruebas diagnósticas
Se estudian las diferentes pruebas de laboratorio que resultan necesarias para diagnosticar el síndrome antifosfolípido entre las que se encuentran algunas como la homocisteína, el examen del cariotipo, la mutación del gen de la protrombina, el hemograma, o la determinación de los anticuerpos antifosfolípidos.
Duración estimada: 4 horas.
Unidad Temática V. Pruebas para la investigación de muestras
Se detalla cual es el procesamiento adecuado tanto en el proceso como en la preparación de las muestras, partir de especímenes de pacientes que poseen dicha enfermedad y personas sanas, las cuales actuarán como control, para comparar los resultados obtenidos.
Duración estimada: 6,5 horas.
Test Evaluación Con la intención de que los/as Técnicos/as obtengan el máximo
aprovechamiento sobre este Curso, el alumno realizará un test que consta de 30 preguntas tipo a), b), c), para evaluar los conocimientos adquiridos en dicho Curso.
En la evaluación final tendrá un peso del 100 % con la aportación del cuestionario final resuelto. Para aprobar se requiere que se supere con éxito el 80% del cuestionario.
Tutorías
A través de nuestra plataforma de foros (https://docencia.sindicatotecnos.es/foros/) se podrán llevar a cabo tutorías de manera grupal.
En este foro se dará respuesta de todas las dudas que planteen los alumnos de la actividad formativa, además de ofrecer una oportunidad de que se establezcan debates entre los diferentes alumnos enriqueciendo de este modo el aprendizaje y el conocimiento.
Además, en el foro anteriormente citado, los alumnos dispondrán de un sistema de mensajes privados con los que contactar con su correspondiente tutor para la resolución de dudas o cuestiones de forma más directa. El docente responderá de la forma más rápida posible a las dudas surgidas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
11
1.4 Metodología del Curso Nuestra metodología online, con un campus virtual propio (disponible en
https://docencia.sindicatotecnos.es), está basada en la atención personalizada en la que se ofrece un seguimiento constante en el proceso de estudio. Se pone a disposición del alumno un equipo de profesionales que planificarán y guiarán el estudio.
Aprovechando las ventajas de la formación online, se pretende hacer protagonista al alumno de su formación. Con un horario totalmente flexible, conocimientos adaptados a sus objetivos, seguimiento individualizado por tutores y profesores y un conjunto de profesionales para poder compartir dudas, soluciones y experiencias.
1.5 Guía del alumno El curso se estructura de forma que tenga tiempo suficiente para el
estudio y la asimilación de los contenidos de las materias que lo forman. Este material está diseñado específicamente para la formación a distancia, con él, podrá seguir la secuencia de contenidos de una forma clara y sencilla, ya que intentan facilitarle lo más posible el proceso de aprendizaje.
1.5.1 Régimen de Enseñanza La metodología de la enseñanza a distancia, por su estructura y
concepción, le ofrece un ámbito de aprendizaje donde puede acceder, de forma flexible en cuanto a ritmo individual de dedicación, estudio y aprendizaje, a los conocimientos que profesional y personalmente le interesan. Todo curso que quiera ser eficaz, efectivo y eficiente en el cumplimento de su objetivo, debe adaptarse a los conocimientos previos de las personas que lo cursarán.
A continuación, desarrollaremos cuestiones sobre presentación del Curso, procedimientos de estudio, conocimiento de su estructura y el método que se ha de seguir para la ejecución de la actividad formativa.
1.5.2 Introducción. Antes de comenzar el curso, se realiza una encuesta de
conocimientos previos del alumno, relacionada con el tema de la actividad docente. Esta encuesta, tendrá solo valor a efectos meramente informativos, no evaluable para la realización de actividad formativa, y sí para conocimientos de la Organización de la evolución del alumno al finalizar la actividad.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
12
Una vez accedido a la actividad formativa, el alumno tendrá una introducción a la metodología de enseñanza a distancia, procedimientos de estudio y conocimiento de su estructura y el método que se ha de seguir.
Ayuda a asimilar y comprender la enseñanza a distancia y las instrucciones de funcionamiento de la plataforma online. Este es el sentido de la presente introducción.
1.5.3 Presentación. 1. Sistema de Cursos a Distancia
En este apartado al alumno aprenderá una serie de aspectos generales sobre las técnicas de formación que se van a seguir para el estudio.
2. Orientaciones para el estudio.
Si el alumno no conoce la técnica empleada en los cursos a distancia, se le recomienda que lea atentamente los epígrafes siguientes, los cuales le ayudarán a realizar el Curso en las mejores condiciones.
Se dan una serie de recomendaciones generales para el estudio y las fases del proceso de aprendizaje propuesto por el equipo docente.
3. Estructura del curso
Se muestra cómo es el curso, las Unidades Temáticas de las que se compone, el sistema de evaluación y cómo enfrentarse al tipo test.
1.5.3.1 Sistema de Cursos a Distancia
Régimen de Enseñanza
La metodología de enseñanza a distancia, por su estructura y concepción, ofrece un ámbito de aprendizaje donde pueden acceder, de forma flexible en cuanto a ritmo individual de dedicación, estudio y aprendizaje, a los conocimientos que profesional y personalmente le interesen. Tiene la ventaja de estar diseñada para adaptarse a las disponibilidades de tiempo y/o situación geográfica de cada alumno. Además, es participativa y centrada en el desarrollo individual y orientado a la solución de problemas.
La Formación a Distancia facilita el acceso a la enseñanza a todos los Técnicos Sanitarios.
Características del Curso y del alumnado al que va dirigido
Todo curso que pretenda ser eficaz, efectivo y eficiente en alcanzar sus objetivos, debe adaptarse a los conocimientos previos de las personas que lo estudiarán (lo que saben y lo que aún no han aprendido). Por tanto, la dificultad de los temas presentados se ajustará a sus intereses y capacidades.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
13
Un buen curso producirá resultados deficientes si lo estudian personas muy diferentes de las inicialmente previstas.
Los cursos se diseñan ajustándose a las características del alumno al que se dirige.
Orientación de los Tutores
Para cada curso habrá, al menos, un tutor en función del número de alumnos inscritos, siendo 6 los tutores para un máximo de 45 alumnos, estableciendo una ratio de aproximadamente 7 alumnos por tutor. Esta ratio, puede variar en el caso de las diferentes actividades formativas, en función del número de tutores. Los alumnos podrán dirigir todas sus consultas y plantear las dificultades que le surjan a lo largo de toda la actividad docente.
Las tutorías están pensadas partiendo de la base de que el aprendizaje que se realiza en esta formación es totalmente individual y personalizado. En esta línea se habilitará foros o puntos de encuentro para poder resolver las dudas de forma rápida, sencilla y directa (Tutorización en grupo/feedback grupal), desde un foro habilitado para dicho fin en la dirección https://docencia.sindicatotecnos.es/foros/.
Además, también se podrá establecer contacto con el tutor vía email. El tutor responderá en un plazo mínimo a las dudas planteadas a través de correo electrónico: [email protected].
La Organización, es consciente de que los alumnos pueden o no, contactar con el tutor o no asistir a los foros o puntos de encuentro. No obstante, en necesario que el alumno, deba superar un nivel de conocimientos del 80% de los contenidos tratados en la actividad docente, para la obtención de la certificación. Esta acción y requerimiento, será realizado de forma autónoma por la plataforma de teleformación, y volcado los datos a las bases de seguimiento de esta actividad.
1.5.3.2 Orientaciones para el estudio
Los resultados que un alumno obtiene, no están exclusivamente en función de las aptitudes que posee y del interés que pone en práctica, sino también de las técnicas de estudio que utiliza. Aunque resulta difícil establecer unas normas que sean aplicables de forma general, es más conveniente que cada alumno se marque su propio método de trabajo. Les recomendamos las siguientes pautas, que pueden ser de mayor aprovechamiento.
Por tanto, aun dando por supuestas la vocación y preparación de los alumnos y respetando su propia iniciativa y forma de plantear el estudio, parece conveniente exponer algunos patrones con los que se podrá guiar más

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
14
fácilmente el desarrollo académico, aunque va a depender de la situación particular de cada alumno y de los conocimientos de la materia del Curso:
• Decidir una estrategia de trabajo, un calendario de estudio y mantenerlo con regularidad. Es recomendable tener al menos dos sesiones de trabajo por semana.
• Elegir el horario más favorable para cada alumno. Una sesión debe durar mínimo una hora y máximo tres. Menos de una hora es poco, debido al tiempo que se necesita de preparación, mientras que más de tres horas, incluidos los descansos, puede resultar demasiado y descendería el rendimiento.
• Utilizar un sitio tranquilo a horas silenciosas, con iluminación adecuada, espacio suficiente para extender apuntes, etc.
• Estudiar con atención, sin distraerse. Nada de radio, televisión o música de fondo. También es muy práctico subrayar los puntos más interesantes a modo de resumen o esquema.
a) Fase receptiva.
• Observar en primer lugar el esquema general del Curso.
• Hacer una composición de lo que se cree más interesante o importante.
• Leer atentamente todos los conceptos desarrollados. No pasar de uno a otro sin haberlo entendido. Recordar que en los Cursos nunca se incluyen cuestiones no útiles.
• Anotar las palabras o párrafos considerados más relevantes empleando un lápiz o rotulador transparente, en caso de libro de texto o impresión de la actividad formativa. En caso de seguimiento en formato PDF, subrayar. No abusar de las anotaciones para que sean claras y significativas.
• Esquematizar en la medida de lo posible sin mirar el texto el contenido de la Unidad.
• Completar el esquema con el texto.
• Estudiar ajustándose al horario, pero sin imbuirse prisas o impacientarse. Deben aclararse las ideas y fijarse los conceptos.
• Resumir los puntos considerados primordiales de cada tema.
• Marcar los conceptos sobre los que se tengan dudas tras leerlos detenidamente. No insistir de momento más sobre ellos.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
15
b) Fase reflexiva.
• Reflexionar sobre los conocimientos adquiridos y sobre las dudas que hayan podido surgir, una vez finalizado el estudio del texto. Pensar que siempre se puede acudir al tutor y a la bibliografía recomendada y la utilizada en la elaboración del tema que puede ser de gran ayuda.
• Seguir paso a paso el desarrollo de los temas.
• Anotar los puntos que no se comprenden.
• Repasar los conceptos contenidos en el texto según va siguiendo la solución de los casos resueltos.
c) Fase creativa.
En esta fase se aplican los conocimientos adquiridos a la resolución de pruebas de autoevaluación y a los casos concretos de su vivencia profesional.
• Repasar despacio el enunciado y fijarse en lo que se pide antes de empezar a solucionarla.
• Consultar la exposición de conceptos del texto que hagan referencia a cada cuestión de la prueba.
• Solucionar la prueba utilizando el propio cuestionario del manual.
1.6 Estructura del Curso Todo lo relativo a los cursos de Formación Continuada será gestionado
personalmente por el alumno a través de la página web:
https://docencia.sindicatotecnos.es/
En esta Web le proponemos una metodología especial de enseñanza: la formación a distancia, diferente a los métodos tradicionales en los cuales el formador y el alumnado asisten diariamente a un aula donde se desarrollan las clases. Esta nueva metodología le ofrece la ventaja de ser uno mismo quien estructure su tiempo y tareas orientado por el tutor del curso.
1.6.1 Contenidos del Curso • Encuesta Previa de Conocimientos.
• Temario del curso en PDF, con un cuestionario tipo test.
• Test de Evaluación, para contestar las respuestas al cuestionario final.
• Encuesta de Satisfacción (aspectos sobre el curso y la plataforma).

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
16
• Encuesta de Impacto posterior a la realización del curso. El seguimiento de la actividad, no debe de acabar cuando finaliza la convocatoria de esta. Entendemos como vital, hacer una encuesta sobre la aplicación de los conocimientos adquiridos, dentro del campo laboral, si es que se ha tenido la oportunidad de desarrollarlos. En caso de no aplicación en el campo laboral, servirá para tener datos sobre esta materia formativa de cara al futuro de nuevas ediciones.
• Foros en los que se plantean dudas acerca de los cursos que se están cursando, así como temas e información de interés, y donde los alumnos y tutores pueden participar para entre todos establecer un debate y solucionar las cuestiones, aportar nuevos conocimientos, debatir asuntos relativos al curso, etc.
1.6.1.1 Encuesta previa.
Antes de comenzar el curso se plantea la realización de una encuesta previa de conocimientos sobre la materia que tratará la actividad docente.
Se trata de un cuestionario compuesto por 10 ítems, en los que hay tres respuestas, siendo tan solo una de ellas la opción verdadera. En esta evaluación no es necesario que el alumno tenga correctas todas las preguntas, ni siquiera un porcentaje mínimo de ellas. Esto se debe a que, como su propio nombre indica, se trata de una evaluación previa que servirá al tutor para poder determinar cuál es el conocimiento que el alumno tiene sobre la materia en cuestión, e incidir a lo largo de las diferentes unidades temáticas, en aquellos campos que considere son más desconocidos para el alumno.
Esta encuesta previa también le servirá al tutor para, una vez finalizado el curso, poder ver el avance del alumno.
1.6.1.2 Los Cursos
Los cursos se presentan en un archivo PDF cuidadosamente diseñado en Unidades Temáticas, conforme a los requerimientos de la Agencia Acreditadora.
NOTA: Un aspecto muy útil para cuidar el medio ambiente, es utilizar herramientas informáticas que nos ayuden a corregir nuestros trabajos. Las mismas cumplen las funciones de subrayado, destacado, tachadura o revisión ortográfica. Utilizándolas nos ahorraremos las impresiones de todos los documentos que requieran hacer este trabajo. Debemos ser conscientes de que imprimir, es una acción que implica consecuencias, sobre todo para el medio ambiente.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
17
Unidades Temáticas
Son unidades básicas de estos Cursos a distancia. Contienen diferentes tipos de material educativo distinto:
- Texto propiamente dicho, dividido en temas.
- Bibliografía utilizada y recomendada.
- Cuestionario tipo test.
Los temas comienzan con un índice con las materias contenidas en ellos. Continúa con el texto propiamente dicho, donde se desarrollan las cuestiones del programa. En la redacción del mismo se evita todo aquello que no sea de utilidad teórico/práctica.
El apartado de preguntas test serán con los que se trabajen, y con los que posteriormente se rellenará el FORMULARIO de respuestas a remitir. Los ejercicios de tipo test se adjuntan al final del temario.
Cuando están presentes los ejercicios de autoevaluación, la realización de éstos resulta muy útil para el alumno, ya que:
• Tienen una función recapituladora, insistiendo en los conceptos y términos básicos del tema.
• Hacen participar al alumno de una manera más activa en el aprendizaje del tema.
• Sirven para que el alumno valore el estado de su aprendizaje, al comprobar posteriormente el resultado de las respuestas.
• Son garantía de que ha estudiado el tema, cuando el alumno los ha superado positivamente. En caso contrario se recomienda que lo estudie de nuevo.
Dentro de las unidades hay distintos epígrafes, que son conjuntos homogéneos de conceptos que guardan relación entre sí. El tamaño y número de epígrafes dependerá de cada caso.
1.6.1.3 Evaluación Global En este apartado se pretende evaluar los resultados formativos. Se
valora el trabajo de formación y la eficacia del programa de actuación. Se realizará un cuestionario que permita comprobar y verificar los resultados de la actividad planificada y emprendida y en qué medida se han cumplido los objetivos previstos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
18
En la misma página donde se encuentra el enlace de descarga del PDF, se habilitará un botón que posibilitará la realización de la evaluación del curso.
La evaluación consta de 30 ítems, con tres respuestas alternativas. Para aprobar se requiere que se supere con éxito el 80% del cuestionario. La temática de las cuestiones versará sobre el contenido de las distintas unidades de las que se compone la actividad docente, algunas de ellas dirigidas a situaciones prácticas. Los ítems en cuestión que hay que responder, se adjuntan al final del temario.
Posteriormente, el cuestionario lo remite el alumno mediante un formulario web que corrige automáticamente los aciertos.
En el caso de que no se supere el cuestionario con un mínimo del 80% correcto, se tendrá la posibilidad de recuperación. En la pantalla aparecerá un mensaje donde se le indicará al alumno para que lo repita.
1.6.1.4 Evaluación de docentes, recursos y otros aspectos. Se lleva a cabo una evaluación de la satisfacción del alumno mediante
una Encuesta de Satisfacción en la cual se le preguntará sobre: la presentación del material, contenidos, estructura, calidad del profesorado, tutorías, consecución de expectativas del curso, modalidad de formación y aplicación de conocimientos adquiridos en el ámbito profesional.
También se evaluará al tutor / docente: Se valorará su responsabilidad, disciplina, capacidad docente, nivel de interacción con el alumno, con el resto de tutores/docentes y con la propia entidad organizadora, capacidad de respuesta, habilidad para descubrir los intereses y expectativas de los alumnos implicándolos en el proceso de formación…
Esta evaluación es necesaria para poder rectificar posibles errores y mejorar el funcionamiento de nuestra formación. En la misma página donde se encuentra el enlace de descarga del PDF, se habilitará un botón que posibilitará la realización de la encuesta de satisfacción del curso.
Así mismo, cuando se finalice el curso se habilitará un cuestionario de evaluación de la calidad de los cursos virtuales de FATE, y será requisito necesario su realización, para poder recibir el Título del Curso.
1.6.2 Contacto con el tutor. La consulta y resolución de dudas se realizará mediante correo
electrónico en cualquier momento. Además, el feedback también se podrá establecer por medio de los foros que permanecerán abiertos durante toda la duración de la convocatoria del curso: Tutorización en grupo/feedback grupal y Tutorización personalizada/feedback tutor-alumno.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
19
El primer paso que debe realizar el alumno es registrarse en nuestra plataforma de foros (https://docencia.sindicatotecnos.es/foros/), y a partir de ahí podrá entrar en el foro correspondiente al curso o cursos que está realizando y participar en él: respondiendo a temas ya creados, consultando dudas resueltas, creando nuevos temas, etc.
Además, por medio de este foro, se pretende establecer un punto de encuentro con el resto de alumnos que va a suponer un enriquecimiento de conocimientos y una nueva vía de debate sobre las diferentes cuestiones relacionadas con el curso, además de aportar una nueva línea en la que los tutores pueden resolver dudas surgidas, y servir de glosario de consulta a otros usuarios, en caso de que hayan tenido algún problema o cuestión.
Se habilita un correo electrónico al que poder dirigirse en cualquier momento para poder resolver cuestiones que le surjan a lo largo de todo el curso. El alumno deberá dirigir al correo [email protected], indicando en el asunto el nombre y apellidos del alumno y en el cuerpo del mensaje las cuestiones a resolver.
1.6.3 Tutor o Docente. Cada actividad docente contará con un máximo de 45 alumnos y 6
tutores que serán Técnicos de la especialidad correspondiente y que estarán pendientes de las dudas que surjan a los alumnos en todo momento.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
20
Los tutores resolverán las dudas planteadas en el foro por los usuarios en cuanto reciban el aviso mediante email de que se ha generado una nueva consulta. Los alumnos podrán preguntar los problemas que les ha surgido o las dudas que tengan y de esta forma solucionarlas en el foro, de manera que sirva de referencia para otros alumnos que puedan tener esas mismas cuestiones. Además, esta herramienta también permite que se establezcan debates entre los diferentes alumnos enriqueciendo de este modo el aprendizaje y el conocimiento.
Además, en el foro anteriormente citado, los alumnos dispondrán de un sistema de mensajes privados con los que contactar con su correspondiente tutor para la resolución de dudas o cuestiones de forma más directa. El docente debe responder, de la forma más rápida posible a las dudas surgidas.
1.6.4 Duración de los Cursos Los cursos tendrán una duración determinada en función de las horas
estipuladas según criterios de la agencia acreditadora, y una fecha de entrega del cuestionario final, desde el momento de su adquisición en la siguiente plataforma de formación:
https://docencia.sindicatotecnos.es/
El no cumplimiento de la fecha de entrega, llevará consigo el cierre de la convocatoria y pérdida del curso. En el caso que exista alguna causa excepcional y el alumno no pueda terminarlo en la fecha establecida, deberá ponerse en contacto a través del correo “[email protected]” para subsanar esta situación o solicitar un aplazamiento. El equipo docente estudiará las alegaciones recibidas y decidirá cuál es la solución a tomar.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
21
1.7 Guía de uso de la Web
1.7.1 Inscripción En primer lugar, el alumno se tiene que inscribir en la plataforma,
mediante el botón ‘Registro’ que aparece al cargar la página.
Aparece un formulario en el cual hay que rellenar una serie de campos obligatorios. Es muy importante rellenar todos los campos de forma correcta, ya que estos datos son los que se van a utilizar para la emisión de los diplomas y su posterior envío.
Registro
Tras picar en registro se abre un formulario en el cual se solicita los datos personales del alumno (nombre, apellidos, dirección, población, DNI, móvil, correo electrónico...)

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
22
Es importante rellenar los campos con * y en letra mayúscula. Si estos campos no se rellenan, la aplicación bloqueará el paso a la siguiente pestaña.
El alumno debe revisar bien los datos introducidos puesto que son esos datos se emitirán los diplomas. La emisión de un nuevo diploma con error en datos personales correrá por cuenta del alumno.
Finalmente, el usuario tiene que elegir un password o contraseña y pinchar en “registrarse”.
Acceder
Una vez registrados, podremos iniciar sesión en el menú principal antes mencionado con las credenciales que hemos indicado. En caso de que se cierre la sesión, se podrá volver a conectar mediante dicho panel, donde se tiene que introducir el correo electrónico y la contraseña con la que se ha registrado.
1.7.2 Menú principal
Mis Datos
Dicho botón del menú principal, abre un formulario donde podremos ver los datos que registramos para acceder a la plataforma, además de darnos la posibilidad de modificarlos.
Matrícula
Aparecen una serie de iconos con las distintas especialidades disponibles.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
23
Accediendo a ellos, se localizan todos los cursos disponibles de cada categoría.
El usuario tiene que hacer clic en el botón ‘Seleccionar’ de aquel que le interese, y se le abre una página donde hay que aceptar los Términos y Condiciones. Una vez aceptados, podremos elegir la opción de ‘Confirmar’ la matrícula en dicho curso, o seleccionarlo como ‘Curso Gratuito’ del semestre:

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
24
Cuando seleccione dichas opciones, aparecerá un mensaje de confirmación. Solo se podrán incluir un máximo de 4 cursos.
Liquidación
Una vez seleccionados ese máximo de 4 cursos, iremos a la pestaña ‘Liquidación’, desde el propio mensaje de confirmación, o desde la opción disponible en el menú principal de la web.
Ahí se nos indicará el total a pagar (según seamos afiliados o no, y si solo queremos PDF o también libro físico), además de la cuenta bancaria donde hacerlo.
En un plazo máximo de 3 días laborales, el departamento de docencia se encargará de activar el curso para que el alumno lo tenga disponible para su realización.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
25
Historial
Este botón lleva al alumno al listado de cursos en los que se encuentra matriculado.
Desde ahí podremos acceder a la Encuesta Previa, al PDF del curso, al Test de Evaluación, al foro, a la Encuesta de Satisfacción Obligatoria, así como al Pretítulo cuando se considere que el curso está completado.
Guía del alumno
Desde ahí podremos acceder una Guía para el Alumno, donde se le explica el funcionamiento de la web, y además se le indica cómo contactar con nuestro equipo ante cualquier duda.
Para cualquier consulta el alumno se puede dirigir a:
− Sedes provinciales: Málaga 952 61 54 61; Córdoba 957 43 02 37
Evaluación
La evaluación debe ser realizada por el alumno al finalizar el estudio del curso, realizando el Test que se encuentra en la plataforma de formación: https://www.docencia.sindicatotecnos.es/

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
26
La elaboración y posterior corrección de los test ha sido diseñada por el personal docente seleccionado para el Curso con la intención de acercar el contenido de las preguntas al temario asimilado.
Si no se supera el cuestionario con un mínimo del 80% correcto, se tendrá la posibilidad de recuperación.
Al superar el cuestionario automáticamente verá un mensaje de aceptación, y aparecerán las respuestas correctas del cuestionario.
Es IMPRESCINDIBLE haber rellenado y superado el Test de Evaluación y completar la Encuesta de Satisfacción para recibir el certificado o Diploma de aptitud del Curso.
El plazo máximo de entrega de las evaluaciones será de un mes y medio a partir de la recepción del material del curso. Si este plazo caduca, puede solicitar un aplazamiento (2 veces como máximo) a través del correo: [email protected].
1.7.3 Envío de diplomas Una vez estudiado el material docente, realizado y superado el test del
Curso y la Encuesta de Satisfacción Obligatoria, se procederá al envío del Diploma Acreditativo del mismo.
La entrega de los certificados del Curso se realizará, como mínimo, a los quince días de la fecha de finalización de la convocatoria del Curso y NUNCA antes de la fecha de finalización de la convocatoria de dicho Curso. El diploma se enviará con posterioridad a la fecha de finalización por correo.

UNIDAD TEMÁTICA II LA SANGRE


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
29
2.1 Introducción La sangre es un tejido conectivo especializado ligeramente alcalino (pH
7,4), en el que se encuentran en suspensión las células sanguíneas, dentro de un líquido llamado plasma. Esta, constituye aproximadamente el 7% del peso corporal, lo que corresponde a unos 70 ml por Kg.
2.2 Composición La sangre se compone de elementos formes (células sanguíneas) y de
una matriz extracelular a la que denominamos plasma:
2.3 Plasma Es un líquido transparente, de color amarillento, en el que se encuentran
suspendidos todos los componentes de la sangre. Durante la coagulación sanguínea, se pierde el fibrinógeno debido a su transformación en fibrina, formando una red en la que quedan englobadas las células sanguíneas, y también se perderán componentes del plasma, orgánicos e inorgánicos, que pasan a formar parte del coágulo sanguíneo, a partir de este momento el plasma se convierte en suero.
El plasma sanguíneo además de transportar a las células sanguíneas, lleva los nutrientes y sustancias de desecho recogidas por las células. Está formado en su mayoría por agua (90%) en la que se encuentran en disolución, proteínas (9%) y otros componentes (1%). De todos los componentes del plasma sanguíneo, sólo procederemos a explicar las proteínas por su implicación en el proceso de la coagulación.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
30
2.3.1 Concentración de las sustancias que componen el plasma sanguíneo
1.3.1.1 Proteínas del plasma
El plasma está formado por numerosas proteínas, entre las cuales destacamos:
1. Albúmina o seroalbúmina
Es la proteína más abundante, estable, homogénea y soluble de todas las proteínas del plasma sanguíneo, formando aproximadamente un 50% de la concentración total de proteínas plasmáticas, encontrándose entre 3,5 a5,0 gramos por litro de sangre. Se sintetiza en el hígado y sus principales funciones son:
Mantenimiento de la presión coloidosmótica, lo que impide la pérdida de plasma a nivel de los capilares.
Transporte de metabolitos insolubles.
2. Globulinas
Su síntesis suele tener lugar en el hígado, aunque algunas son sintetizadas en el bazo, ganglios linfáticos… Existen tres tipos de globulinas que son:
α y β –globulinas: Los valores normales en sangre son, entre 0,3-0,7 gramos por decilitro para las α-globulinas y de 0,4-0,8 gramos por decilitro para β–globulinas. Ambas se sintetizan en el hígado y sus funciones son: Transportar iones metálicos, líquidos fijos en proteínas y vitaminas liposolubles.
γ-globulinas: Sus valores normales oscilan entre los 0,6-1,1 gramos por decilitro. Se sintetizan en las células plasmáticas, ya que son anticuerpos o inmunoglobulinas que son los encargados de defender nuestro organismo, existiendo 5 tipos de inmunoglobulinas:
IgM: Se forman rápidamente ante un estímulo antigénico y su función es activar el sistema del complemento.
IgE: Se encuentra en concentraciones muy bajas en el suero, aumentando su concentración en procesos alérgicos.
IgG: Son las únicas inmunoglobulinas capaces de atravesar la placenta, siendo por tanto las responsables, de la inmunidad fetal y del recién nacido. Otra de sus funciones es la de unirse macrófagos y neutrófilos con el fin de destruir a los antígenos.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
31
IgA: Actúa protegiendo la superficie de nuestro cuerpo y los conductos secretores, encontrándose por tanto en secreciones serosas y mucosas.
IgD: Se cree que su función está relacionada con la activación de los linfocitos B.
3. Proteínas de la coagulación
Estas proteínas son las responsables de la modificación del estado físico de sangre, mediante la formación de los filamentos de fibrina, que darán lugar a la coagulación sanguínea. Además del fibrinógeno, para que se produzca el proceso de la coagulación, es necesaria la presencia de otras proteínas a las que llamamos, factores de la coagulación, que se denominan con el nombre de “Factor” seguido de un número romano. Todas estas proteínas de la coagulación, son sintetizadas en el hígado. Estos factores de la coagulación son:
- Factor I o Fibrinógeno
- Factor II o Protrombina
- Factor III o Tromboplastina tisular y Factor III plaquetario
- Factor IV o Calcio iónico
- Factor V o Acelerina
- Factor VI
- Factor VII o Proconvertina
- Factor VIII
- Fracción VIII-C o Factor anti hemofílico A
- Fracción VIII V-W o Factor de Von Willebrand
- Factor IX o Factor anti hemofílico B
- Factor X o Factor de Stuart-Prower
- Factor XI o Antecedente plasmático de la tromboplastina
- Factor XII o Factor de Hageman
- Factor XIII o Factor estabilizante de la fibrina

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
32
Otros factores que intervienen en la coagulación son:
- Proteína S
- Proteína C
- Prekalicreína o Factor de Fletcher
- Globulinas aceleradoras…
4. Lipoproteínas plasmáticas
Estas proteínas son las encargadas del transporte de lípidos por todo el organismo a través de la sangre. Y se distinguen varios tipos dentro de estas:
- Quilomicrones: Son ricas en triglicéridos. Se forman a partir de las células epiteliales del intestino y su función es, el transporte de triglicéridos hasta el hígado a través de la sangre.
- VLDL o Lipoproteínas de muy baja densidad: Estas al igual que las anteriores, presentan gran cantidad de triglicéridos. Se sintetizan en el hígado y su función es, el transporte de triglicéridos desde el hígado hasta las células de nuestro organismo.
- LDL o lipoproteínas de baja densidad: También llamado colesterol malo, puesto que es la lipoproteína que se encarga de acumular el exceso de colesterol en las arterias. Estas lipoproteínas están cargadas de colesterol y se forman en el hígado, desde el cual, transportan el colesterol hacia todas las células del cuerpo.
2.3.1.2 Proteínas del complemento
Son un conjunto de unas nueve proteínas (C1, C2, C3…C9), su síntesis tiene lugar en el hígado. Las funciones de estas proteínas del complemento son:
Propiciar una respuesta inmunológica adecuada mediante la destrucción de microorganismos.
Formar parte en el inicio de la inflamación.
2.4 Células sanguíneas o elementos figurados Es la parte sólida de la sangre y constituye aproximadamente el 40% de
la volemia (volumen total de sangre). Todas las células de la sangre, proceden de un tejido altamente especializado, que es el llamado tejido hematopoyético,
Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
33
que se encuentra en la médula ósea roja, donde se originan los tres tipos de células sanguíneas: glóbulos rojos, eritrocitos o hematíes, glóbulos blancos o leucocitos y plaquetas.
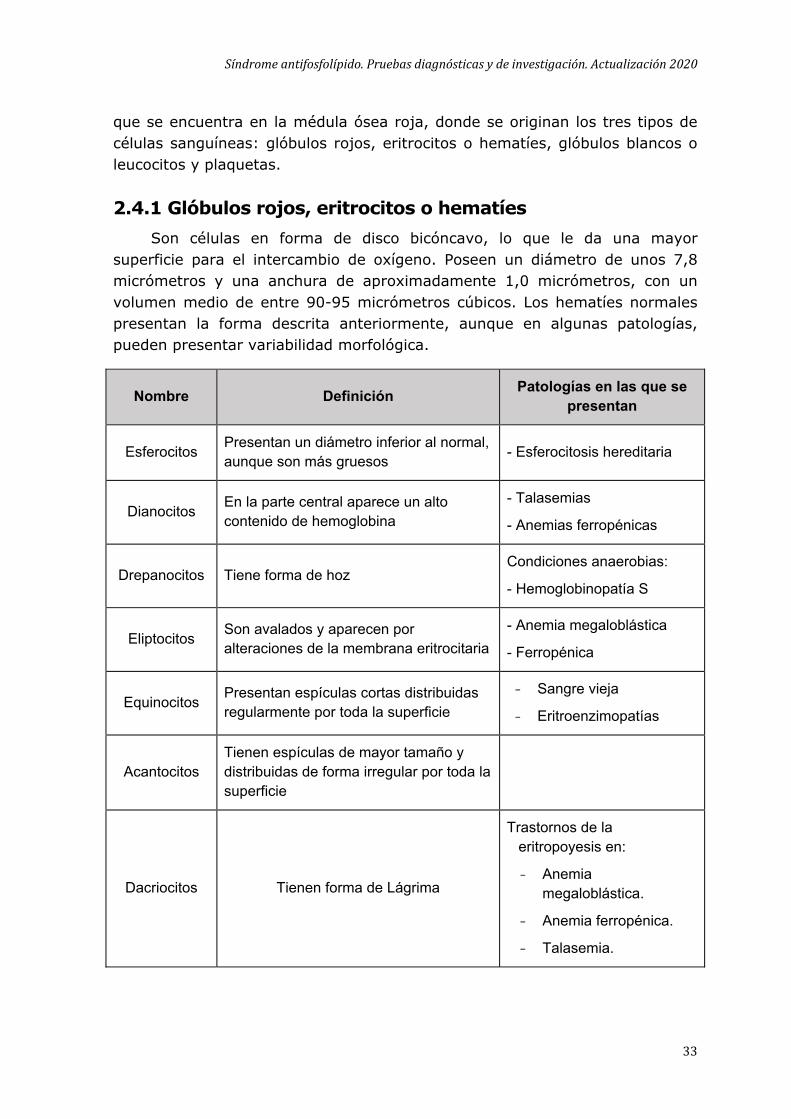
2.4.1 Glóbulos rojos, eritrocitos o hematíes Son células en forma de disco bicóncavo, lo que le da una mayor
superficie para el intercambio de oxígeno. Poseen un diámetro de unos 7,8 micrómetros y una anchura de aproximadamente 1,0 micrómetros, con un volumen medio de entre 90-95 micrómetros cúbicos. Los hematíes normales presentan la forma descrita anteriormente, aunque en algunas patologías, pueden presentar variabilidad morfológica.
Nombre Definición Patologías en las que se presentan
Esferocitos Presentan un diámetro inferior al normal, aunque son más gruesos - Esferocitosis hereditaria
Dianocitos En la parte central aparece un alto contenido de hemoglobina
- Talasemias
- Anemias ferropénicas
Drepanocitos Tiene forma de hoz Condiciones anaerobias:
- Hemoglobinopatía S
Eliptocitos Son avalados y aparecen por alteraciones de la membrana eritrocitaria
- Anemia megaloblástica
- Ferropénica
Equinocitos Presentan espículas cortas distribuidas regularmente por toda la superficie
- Sangre vieja
- Eritroenzimopatías
Acantocitos Tienen espículas de mayor tamaño y distribuidas de forma irregular por toda la superficie
Dacriocitos Tienen forma de Lágrima
Trastornos de la eritropoyesis en:
- Anemia megaloblástica.
- Anemia ferropénica.
- Talasemia.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
34
Nombre Definición Patologías en las que se presentan
Esquistocitos Son hematíes Fragmentados - Anemia hemolítica por ejercicio físico
Estomatocitos Presentan una invaginación central - Estomatocitosis congénita
Keratocitos Poseen dos espículas terminales
Presentan una membrana flexible, que facilita el paso de los hematíes por capilares estrechos. En su estado maduro, carecen de orgánulos citoplasmáticos e incluso de núcleo, presentando un color rojo intenso, debido a la presencia de un pigmento que contiene en su interior moléculas de hierro, las cuales se unen temporalmente a las moléculas de oxígeno presentes en los pulmones, estas moléculas son la hemoglobina a la cual pueden unirse un máximo de cuatro moléculas de oxígeno. La hemoglobina, es la encargada de llevar el oxígeno desde los pulmones hasta los tejidos. Cuando la hemoglobina se une a las moléculas de oxígeno recibe el nombre de oxihemoglobina. Una vez en los tejidos el oxígeno es liberado y la hemoglobina se transforma en desoxihemoglobina o hemoglobina reducida, que captará el dióxido de carbono producido por dichos tejidos, recibiendo el nombre de carbaminohemoglobina, que transportará el dióxido de carbono, hasta los pulmones, donde será expulsado. Los hematíes poseen una enzima, llamada anhidrasa carbónica que facilita la formación de ácido carbónico a partir del dióxido de carbono de los tejidos y agua, disociándose este en ión
Hematíes en suspensión

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
35
bicarbonato y H+. Este ión bicarbonato es transportado al plasma ayudando a la regulación del pH de la sangre y de ahí a los pulmones, donde será eliminado.
CO2 + H2O HCO3- + H+
Los hematíes pueden presentar además de hemoglobina, inclusiones eritrocitarias, que pueden ser: Cuerpo de Howell-Jolly, cuerpos de pappenheimer, punteado basófilo, anillos de Cabot o parásitos.
Nombre Definición Patología
Cuerpo de Howell-Jolly Restos de ARN - Anemias megaloblásticas.
- Anemias hemolíticas.
Cuerpo de Pappenheimer A cúmulos de hierro - Anemias.
Punteado basófilo Gránulos ribosómicos
- Saturnismo
- Intoxicaciones por plomo
Anillos de Cabot Restos de la membrana nuclear - Anemias megaloblásticas.
Parásitos Parásitos - Infecciones producidas frecuentemente por el plasmodium
Los valores de referencia del número de hematíes en la sangre son los siguientes:
Unidades Convencionales
Hombre 4,5 a 6,5 millones/mm3
Mujer 3,8 a 5,8 millones/mm3
Recién nacido 4 a 6 millones/mm3

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
36
La alteración de dichos valores puede producir diversas enfermedades tales como:
Disminución del número de hematíes
Anemia
- Disminución de la concentración de hemoglobina
- Pérdida de hematíes por hemorragia
- Insuficiencia medular
- Modificaciones de la dieta
- Enfermedades sistémicas
Otras alteraciones - Cáncer
- Embarazo…
Aumento del número de hematíes
Poliglobulia - Aumento de Hemoglobina
Policitemia - Aumento de Hemoglobina, leucocitos y plaquetas
Otras alteraciones - Cardiopatías
- Enfermedades pulmonares crónicas…
Para la producción de los hematíes es necesaria la intervención de ciertos elementos tales como:
− Hierro: Interviene en la síntesis de la hemoglobina.
− Vitamina B12: Es un factor esencial para la síntesis y multiplicación de las células sanguíneas.
− Ácido fólico: Se utiliza para la síntesis de glóbulos rojos.
2.4.2 Glóbulos blancos o leucocitos Estas células, al contrario que los hematíes, carecen de pigmentos por lo
que se les califica como glóbulos blancos y miden aproximadamente entre 2.0 a 8 micras de diámetro. En el tejido conectivo, son pleomórficos aunque en la sangre, son esféricos.
Todos los leucocitos poseen un único núcleo, que puede formar diversos lóbulos, y en su citoplasma presentan, mitocondrias y algunos orgánulos

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
37
celulares, así como gránulos inespecíficos, llamados también, gránulos azurófilos, que son lisosomas.
Podemos calificar a los leucocitos como constituyentes del sistema inmune, ya que tienen la capacidad de reaccionar, para paliar el daño producido por los agentes patógenos o sus toxinas, mediante la fagocitosis de sustancias extrañas o la producción de toxinas contra esos antígenos y a través del desarrollo de una respuesta inmunitaria contra los antígenos presentes en nuestro organismo (inmunidad humoral). Estos, no ejercen sus funciones dentro de la sangre, sino que la emplean para transportarse a otras partes de nuestro cuerpo y una vez allí poder salir de los vasos sanguíneos mediante un mecanismo llamado diapedesis, penetrando después en los espacios de tejido conectivo donde ejercen su función.
Los valores normales de leucocitos en sangre son los siguientes:
Unidades Convencionales
Hombre 4,5 a 10 mil/mm3
Mujer 4,5 a 10 mil/mm3
Recién nacido 10 a 26 mil/mm3
Morfología de los distintos tipos de leucocitos

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
38
Célula Recién nacido 1 mes 4 años 6 años Adulto
Neutrófilo 37-57 % 25-35 % 25-45 % 45-50 % 55-65 %
Cayado 0,5 % 0,5 % 0,5 % 0,5 % 0,5 %
Eosinófilo 0,5-4 % 0,5-4 % 0,5-4 % 0,5-4 % 0,5-4 %
Basófilo 0,5 % 0,5 % 0,5 % 0,5 % 0,5 %
Monocitos 0-8 % 0-8 % 0-8 % 0-8 % 0-8 %
Linfocitos 25-35 % 45-65 % 40-60 % 40-45 % 25-40 %
Valores de referencia leucocitarios en distintas edades, en sangre periférica (Laboratorio clínico de hematología)
Según aumenten o disminuyan dichos valores, se distinguen dos tipos de patologías que pueden ser debidas a diversas enfermedades:
Disminución del número de leucocitos en sangre o también llamado, leucopenia, esta disminución puede deberse a:
- Fallo de la médula ósea.
- Enfermedades autoinmunes, como es el caso del síndrome antifosfolípido, lupus eritematoso…
- Enfermedades del hígado o riñón.
- Exposición a radiaciones.
- Presencia de sustancias cito tóxicas.
El aumento de leucocitos o leucocitosis puede deberse a causas fisiológicas como puede ser el ejercicio físico o a enfermedades como:
- Daño de los tejidos por quemaduras.
- Fallos renales.
- Enfermedades infecciosas.
- Enfermedades inflamatorias.
- Estrés.
- Leucemia.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
39
Los leucocitos se pueden clasificar atendiendo a la morfología de su núcleo, denominándose, polimorfo nucleares o mono nucleares o bien como, granulocitos o agranulocitos, por la presencia o ausencia de gránulos específicos en su citoplasma.
2.4.2.1 Granulocitos o polimorfo nucleares
Dentro de esta clasificación, se engloban las siguientes células: neutrófilos, basófilos y eosinófilos, cada uno de los cuales, presenta un núcleo con varias lobulaciones y con numerosos orgánulos y en su citoplasma presentan gránulos, tanto específicos como inespecíficos.
1. Neutrófilos
Tienen un diámetro aproximado de entre 10 a 12 micras de diámetro y una vida media de entre 1 a 4 días. Son los tipos de leucocitos más numerosos constituyendo entre un 55 a 65% de la concentración total en sangre.
Su núcleo está constituido, por numerosos lóbulos (de 3 a 4) unidos entre sí, por puentes de cromatina condensada. El número de lóbulos depende de la edad del neutrófilo, a mayor edad, mayor número de lóbulos y viceversa. Este tipo de neutrófilos recibe el nombre de neutrófilos segmentados, debido a la presencia de varios lóbulos y si el número de lóbulos fuese mayor de lo habitual, se les llamará, neutrófilos hipersegmentados. Otro tipo serían los neutrófilos en banda que si aparecen aumentados, representará una desviación a la izquierda, que es representativa de algún tipo de patología, ya sea infecciosa, inflamatoria o aguda. Este tipo de neutrófilos presenta un núcleo sin lobulaciones y su forma típica es arriñonada.
Los neutrófilos, migran a los sitios del organismo, que están siendo invadidos, gracias a la interacción de estos, con agentes quimiotácticos y una vez allí su función es, fagocitar a las sustancias extrañas y células envejecidas, englobándolas con sus pseudópodos para que pasen a su citoplasma donde liberará las sustancias contenidas en sus gránulos específicos, iniciándose así el proceso inflamatorio. Estos gránulos contienen multitud de enzimas y agentes farmacológicos los cuales intervendrán en la defensa del organismo mediante la digestión de la partícula extraña o célula envejecida. En algunas ocasiones también puede liberarse el contenido de los gránulos inespecíficos o azurófilos, lo que conlleva, a la lesión de los tejidos de nuestro cuerpo. Una vez realizada su función, estos neutrófilos mueren y al conjunto de neutrófilos muertos, bacterias y líquido de los tejidos, es a lo que se le conoce con el nombre de pus. Debido a estos procesos fagocíticos, también se le conoce al neutrófilo con el nombre de macrófagos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
40
La proporción en la que se encuentran los neutrófilos en sangre periférica es de entre un 55 a 65%. Cuando estos valores se encuentran disminuidos, se dice que hay neutropenia y puede ser debido:
- Toma de algunos medicamentos.
- Infecciones víricas o bacterianas.
- Enfermedades reumáticas y autoinmunes.
Si, por el contrario, dichos valores se encontrasen aumentados, se produciría una neutrofília, que puede deberse a causas fisiológicas o patológicas, siendo estas las más importantes:
- Inflamaciones no infecciosas.
- Infecciones bacterianas, como:
- Síndromes mieloproliferativos
- Toma de medicamentos o sustancias químicas
- Enfermedades metabólicas
2. Basófilos
Son células, con un tamaño de entre 8 a 10 micras de diámetro y reciben este nombre, puesto que tienen afinidad por los colorantes básicos. Tienen forma redondeada cuando se encuentran en suspensión, pero al ser observadas al microscopio pueden adoptar diferentes formas, al igual que las anteriores.
Su núcleo se tiñe de color violeta oscuro y presenta una morfología más o menos bilobulada, aunque no se observa bien, debido a la presencia de grandes gránulos específicos en su citoplasma.
El citoplasma de los basófilos, presenta también numerosos orgánulos, entre los cuales se encuentran, los gránulos específicos, que tienen forma esférica y son de mayor tamaño que los gránulos de los otros tipos de granulocitos, midiendo aproximadamente unas 0,5 micras de diámetro. Son de naturaleza ácida, por lo que al teñirlos con colorantes básicos tomarán color azul intenso. Los gránulos azurófilos o inespecíficos, son muy parecidos a los de los neutrófilos tanto en función como en tamaño.
Los gránulos de los basófilos, presentan como parte de su contenido, un tipo de inmunoglobulina, que es la Inmunoglobulina E, por lo que cuando los antígenos se unen a los receptores de los basófilos, estos, para eliminarlos, liberan el contenido de sus gránulos, originando una respuesta alérgica.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
41
Los valores normales de basófilos en sangre son de 0,5% más o menos. Si estos valores se encuentran aumentados se dice que hay una basofília y se debe a:
- Estados de hipersensibilidad.
- Cirrosis hepáticas.
- Infecciones víricas.
- Síndromes mieloproliferativos crónicos.
- Anemias crónicas.
Se dice que hay basofilopenia, cuando el número de basófilos está disminuido, aunque es muy difícil de apreciar debido al escaso número de basófilos en sangre periférica, aunque podría deberse a:
- Trastornos endocrinos.
- Trastornos metabólicos.
3. Eosinófilos
Son de mayor tamaño que los neutrófilos (entre 10 a 14 micras de diámetro). Poseen una vida media inferior a dos semanas.
Su núcleo ocupa gran parte del citoplasma, es bilobulado y se une entre sí, por un denso puente de cromatina más o menos grueso, adquiriendo una forma característica de anteojo.
Presentan numerosos orgánulos citoplasmáticos como son, el aparato de Golgi, algunas mitocondrias, retículo endoplásmico…y gránulos tanto específicos, como inespecíficos, al igual que los demás tipos de leucocitos. Los gránulos específicos, tienen un diámetro de 1,5 micras de diámetro, aproximadamente y en la tinción toma color rosa intenso o anaranjado.
El interior de los M gránulos, presenta una zona densa llamada internum, que está rodeada por una zona llamada externum. El internum contiene proteínas básicas, catiónica y neurotoxinas, que ayudan a la eliminación de las larvas de los gusanos parásitos para que puedan ser digeridas por los neutrófilos o macrófagos.
Otra función de los eosinófilos es la de regular y modular las reacciones alérgicas, apareciendo elevado, el número de eosinófilos en personas alérgicas.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
42
Los eosinófilos suelen encontrarse en sangre periférica en una proporción de entre 0,5 a 4%. Cuando varía la concentración de eosinófilos en sangre, puede ser debido a patologías, como:
- Eosinofília: aumento del número de eosinófilos.
- Presencia de parásitos
- Respuesta alérgica
- Ingesta de medicamentos
- Enfermedad de Hodgkin
- Enfermedades de la piel
- Eosinofilopenia: se debe a una disminución del número de eosinófilos y aparece cuando se dan las mismas patologías que en la basofilopenia.
2.4.2.2 Agranulocitos o mononucleares Como ya hemos dicho anteriormente, estas células, están formadas por
un núcleo unilobulado y carecen de gránulos específicos, aunque si presentan gránulos inespecíficos o azurófilos y este grupo lo forman, los linfocitos y los monolitos.
1. Linfocitos
Son células muy variables en cuanto a tamaño, pudiendo medir desde 7 hasta 18 micras de diámetro. Según la variedad de linfocito de que se trate, su vida media puede variar entre unos pocos días, hasta meses o incluso años. Al igual que las demás células, estas suelen tener forma más o menos redondeada, aunque pueden adoptar diversas morfologías, al migrar a los tejidos conectivos.
Poseen un núcleo redondeado acéntrico, que ocupa casi la totalidad del citoplasma celular y la cromatina es densa y homogénea, por lo que en la tinción se observa de color azul o violeta intenso
El citoplasma es muy escaso, ya que está ocupado casi en su totalidad por el núcleo, aun así, contiene algunos orgánulos como mitocondrias, ribosomas libres, retículo endoplásmico rugoso, aparato de Golgi y gránulos azurófilos. El citoplasma suele teñirse de azul claro.
La proporción de linfocitos en sangre periférica es de entre un 25 a 35%. Al aumento del número de linfocitos en sangre se le denomina, linfocitosis y se debe a la producción de patologías, como:
- Infecciones bacterianas o víricas

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
43
- Leucemia linfoide crónica
- Linfocitosis fisiológica de la infancia: En esta patología aparece un número mayor de linfocitos que de neutrófilos, dando lugar a una fórmula leucocitaria invertida. Ésta alteración de la fórmula se da en niños menores de dos años de forma fisiológica y será patológica cuando se dé a mayores edades.
Cuando el número de linfocitos es menor a los valores normales se llamará linfopenia y se producirá en:
- Enfermedad de Hodgkin.
- Insuficiencia cardiaca.
- Administración de corticosteroides.
- Tratamiento con inmunosupresores.
- SIDA.
- Aplasia del timo.
Dentro de los linfocitos se distinguen dos tipos de células que son, los linfocitos B y los linfocitos T, aunque algunos autores distinguen un tercer tipo celular llamadas, células nulas. Son idénticas en cuanto a morfología, distinguiéndose únicamente por sus marcadores de superficie, además de por sus funciones.
Células nulas:
Están compuestas por dos poblaciones celulares que son, las células madre y las células“natural killer”:
Células madre o Stem cell: Son células circulantes, muy indiferenciadas, que, al dividirse por mitosis, producen una célula idéntica así misma, (que se mantiene como célula madre, lo que hace que la población de células madre permanezca intacta) y otra célula especializada (en cualquier tipo de tejido). Por tanto la función de estas células madre, es la de producir todos los elementos formes de la sangre.
Células asesinas naturales o natural killer: Presentan una citotoxicidad natural, ya que son capaces de destruir a células extrañas, además contribuyen a la defensa frente a células infectadas por bacterias, hongos, parásitos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
44
Linfocitos B:
Estos linfocitos B, suelen tener mayor tamaño que los demás tipos de linfocitos, aunque son indistinguibles. Presentan numerosos orgánulos citoplasmáticos, siendo ricas en, retículo endoplasmático, mitocondrias, gránulos inespecíficos o azurófilos…
La mayor diferencia radica en la presencia de inmunoglobulinas, que son los receptores específicos de los linfocitos B a los que se unirán los antígenos, siendo su función principal la producción y secreción de grandes cantidades de estos anticuerpos.
Su síntesis tiene lugar en la médula ósea desarrollándose a partir de células madre. Cuando maduran, pueden seguir en la médula ósea o también pueden pasar a los nódulos linfáticos, Bazo, algunas partes del intestino y una pequeñísima proporción, libres por el fluido sanguíneo.
Cuando la célula se une a los antígenos se produce su activación, experimentando una mitosis que dará lugar a dos tipos celulares que serán:
- Células de memoria: Estas células, no participan en la reacción inmunógena, pero permanecen como clones de las células anteriores, que poseen una memoria inmunológica, por lo cual si una vez eliminado el antígeno o sustancia extraña causante de la enfermedad, volviese a penetrar en nuestro organismo, estas células de memoria actuarían más rápidamente, desencadenando una nueva reacción para eliminar al antígeno o sustancia extraña.
- Células plasmáticas: Su función, es la de producir los anticuerpos, estos anticuerpos producidos por las células plasmáticas, son liberados a la sangre o líquido linfático, por lo que se dice que estos linfocitos B, son los encargados de producir la reacción inmunitaria mediada de manera humoral. Al ponerse en contacto con el antígeno se formará una reacción primaria, en la que se producirá Inmunoglobulina M y una vez que el antígeno a estimulado a la célula, se producirán las inmunoglobulinas G, A y E, la única inmunoglobulina que no presentan es la inmunoglobulina D. Estos anticuerpos llegarán hasta el fluido sanguíneo, secreciones respiratorias e intestinales y lágrimas, para defendernos frente al ataque de cualquier sustancia extraña. A todo este proceso se le denomina cambio de fase.
Hay antígenos que no requieren la participación de linfocitos B antes de producir una reacción inmunológica humoral, estos antígenos son conocidos como, antígenos independientes del timo, los cuales sólo pueden producir Inmunoglobulinas M y no se podrán formar células de memoria.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
45
Linfocitos T:
Son otro tipo de células inmunológicas, al igual que las células anteriores, estas también presentan numerosos orgánulos, aunque no poseen la capacidad de síntesis de anticuerpos moleculares (inmunoglobulinas).
Son una población, formada por tres tipos celulares y todos ellos poseen receptores específicos para los antígenos, conocidos con el nombre de TCR o receptores de linfocitos T, que se encuentra asociado a la superficie celular del complejo CD3, cuya función es activar al linfocito T. En general, las funciones de los linfocitos T, son las de atacar a los antígenos, tales como virus, hongos, bacterias… y actuar como reguladoras del sistema inmunológico.
La síntesis de este tipo celular tiene lugar (al igual que la anterior) en la médula ósea, generándose a partir de células madre, aunque su maduración se lleva a cabo en el timo. Una vez maduros, dejarán este órgano para ocupar otros, como el bazo, nódulos linfáticos, médula ósea y sangre.
Los linfocitos T poseen en su membrana diversos marcadores, siendo los más importantes los linfocitos T CD4 o T4 y los CD8 o T8:
- T4: Presentan una proteína denominada CD4 que se encuentra en la superficie de uno de los tipos de linfocitos T que son los linfocitos T colaboradores. La función de estos marcadores es la de fijar el epítopo del antígeno, al receptor TCR y la activación de la célula T cooperadora, lo que hace posible la eliminación de bacterias y otros parásitos que viven en el interior celular.
- T8: son otro tipo de proteína, la CD8, que se encuentran en la superficie de las células supresoras y citotóxicas, que como veremos a continuación son otro tipo de linfocitos T. Al igual que la anterior también ayuda a la fijación del epítopo al receptor TCR e interviene en la activación de la célula T citotóxica que producirá la eliminación de virus más específicos.
Los tipos de linfocitos T son muy distintos entre sí en cuanto a funcionalidad se refiere, distinguiéndose los siguientes tipos:
- Linfocitos T colaboradores: Son un tipo de linfocitos T, que como hemos dicho anteriormente, poseen en su membrana unos marcadores llamados CD4, que secretan sustancias que intervienen en casi todos los procesos de inmunidad, por ello también se conocen a estas células con el nombre de ayudantes. Estos marcadores colaboran con los linfocitos B y producen una sustancia que estimula la producción de estos y de anticuerpos específicos libres en sangre.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
46
- Linfocitos T citotóxicos: Son los responsables de la destrucción del microorganismo invasor. Una vez activados, son los linfocitos más agresivos, ya que pueden destruir células blanco o diana, infectadas por virus o bacterias, o incluso células cancerosas, fijándose al antígeno y secretando sustancias como, citosinas, perforinas… que degradan la membrana de la célula infectada, para que al llegar los macrófagos y otros fagotitos, estos puedan destruir a las células dañadas, siendo por tanto los responsables de los fenómenos de citotoxicidad, de la respuesta inmune celular.
Estos linfocitos también son los responsables, de los rechazos de transplantes, ya que responden frente a tejidos u órganos extraños en nuestro cuerpo. Como hemos visto antes, estas células presentan el marcador CD8, cuyas funciones, han sido explicadas con anterioridad.
- Linfocitos T supresores o reguladores: Su función, es regular la respuesta inmune, que es de gran importancia en el desarrollo de la tolerancia frente a los componentes propios del organismo, suprimiendo a los linfocitos T colaboradores, lo que impide que el organismo siga trabajando después de la infección.
Los linfocitos T colaboradores y supresores actúan conjuntamente, regulando todo el conjunto de linfocitos, dejándolos actuar el tiempo necesario para paliar el ataque de los antígenos.
Por último, decir, que al igual que los linfocitos T citotóxicos, presentan en su membrana, moléculas del tipo CD8.
Reconocimiento del antígeno

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
47
Los antígenos que circulan por nuestro organismo no pueden ser reconocidos directamente por los linfocitos T, para ello una célula procesadora de antígenos, rodea e ingiere al antígeno. Por otro lado, las enzimas de la célula procesadora de antígenos, lo rompe, fragmentándolo y algunos de estos fragmentos se unen a las moléculas del complejo mayor de histocompatibilidad y son enlazados a la superficie de la membrana celular. Los receptores de superficie del linfocito T, reconocen el fragmento del antígeno unido a la molécula del complejo mayor de histocompatibilidad y se adhiere a él.
2. Monocitos
Son las células más grandes de la sangre, su tamaño oscila entre 14 a 20 micras de diámetro. Presentan un gran núcleo de forma arriñonada, más o menos centrado, que ocupa gran parte del citoplasma celular. Su cromatina suele ser laxa. El núcleo puede presentar dos nucléolos aunque no suelen ser visibles al microscopio.
El citoplasma presenta gránulos azurófilos o inespecíficos, aparato de Golgi, diversas vacuolas, gránulos de glucógeno, retículo endoplasmático rugoso, algunos ribosomas libres y mitocondrias.
Los monocitos se originan, en la médula ósea y pasan a la circulación sanguínea donde permanecen algunos días y después migrarán hacia los tejidos, donde se diferencian en macrófagos, constituyendo las células defensivas del sistema inmunitario. Estos se localizan por todo el organismo, pero alcanzan grandes concentraciones en pulmones, hígado, bazo, médula ósea y algunas membranas, donde sobreviven algunos meses.
Al llegar a los tejidos y transformarse en macrófagos, su función será la de fagocitar y destruir a células muertas, antígenos y sustancias extrañas, englobándolas en su interior y digiriéndolas mediante enzimas y otras sustancias (como son el superóxido, peróxido de hidrógeno y ácido hipocloroso). También producen, citoquinas que activan la reacción inflamatoria, proliferación y maduración de otras células. Para fagocitar sustancias extrañas de gran tamaño, se produce la fusión de los macrófagos formando un macrófago lo suficientemente grande como para fagocitar y digerir a la partícula.
Los monocitos, presentan diversos receptores de membrana:
- CD11b y CD16, actúan como receptores de la inmunoglobulina G.
- CD33, su función se desconoce.
- CD 14, que es un receptor de alta afinidad para los lipopolisacáridos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
48
Los valores normales son de entre un 4 a 8%.
El aumento de monocitos en sangre, corresponden a una monocitosis y se debe a diversas enfermedades como:
- Infecciones bacterianas.
- Enfermedad de Hodgkin y otras neoplasias.
- Leucemias.
- Enfermedades reumáticas.
Además, puede deberse a causas fisiológicas como ocurre en el recién nacido.
La disminución del número de monocitos se denomina, monocitopenia aunque no tiene ninguna significación clínica.
Este gráfico, nos indica las concentraciones máximas y mínimas (en porcentaje) de los distintos tipos de leucocitos que podemos encontrar en sangre periférica:

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
49
2.4.3 Plaquetas Son los elementos formes más pequeños de la sangre, su tamaño oscila
entre 2 a 4 micras de diámetro, y va disminuyendo a medida que van madurando. Son conocidas también, con otros nombres como trombocitos o tromboplástidos. Son muy frágiles, no presentan núcleo y proceden de fragmentos citoplasmáticos no nucleados de células gigantes y en forma de disco que derivan de los megacariocitos originados en la médula ósea. Las plaquetas tienen una vida media de unos catorce días.
Micrografía de un conjunto de plaquetas activadas.
Se pueden distinguir dos zonas dentro de las plaquetas, que son:
Hialómero: Es la parte de la periferia, presenta una coloración clara, pueden emitir pseudópodos. En el interior de las plaquetas se localizan entre 10 a 15 microtúbulos paralelos entre sí, que se disponen formando un túbulo, cuya función es la de proporcionar a la plaqueta su morfología discoidal.
Granulómero: Es la región central, que tiene una coloración más oscura. En este, se encuentran algunas mitocondrias, glucógeno, peroxisomas, un sistema de enzimas (que permite a las plaquetas catalizar el glucógeno, consumir oxígeno y generar ATP) y tres tipos gránulos: gránulos α, gránulos δ y gránulos γ.
- Gránulos α: están compuestos por, factores de la coagulación, factores del crecimiento, fibrinógeno, tromboplastina plaquetaria y trombospondina, que facilitan la reparación vascular, la agregación plaquetaria y la coagulación de la sangre.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
50
- Gránulos δ: presentan en su interior, calcio, ADP, ATP, serotonina, histamina y tirofosfatasa, entre otras, favoreciendo la agregación, adhesión plaquetaria y la vasoconstricción.
- Gránulos γ: Son básicamente enzimas hidrolíticas, que ayudan a la reabsorción del coágulo.
Las plaquetas, tienen gran capacidad de adhesión, a células o tejidos y se aglutinan entre ellas formando coágulos, gracias a la presencia de anticoagulantes, que actúan fijando el ión calcio de la sangre.
Se forman en la médula ósea (aunque nunca podemos encontrarlas en ella, sino en sangre periférica) a partir de células madre, activándose o inhibiéndose su producción según las necesidades plaquetarias del organismo.
De la concentración total de plaquetas, encontramos una tercera parte de estas en el bazo y el resto en sangre periférica, habiendo un intercambio entre estos, aunque siempre respetando esas concentraciones, en ambos espacios.
Las plaquetas son destruidas en el hígado y en el bazo, donde son fagocitadas por los macrófagos del sistema mononuclear fagocítico. En algunas ocasiones, las plaquetas pueden adherirse a la superficie de los neutrófilos, llamándose satelismo aunque no se conoce cuales la causa ni la función.
La función de las plaquetas es, la de detener la hemorragia, mediante la formación del trombo blanco plaquetario y factores plaquetarios, interviniendo por tanto en la hemostasia primaria y a nivel de la coagulación. En las heridas, las plaquetas aceleran la coagulación, obstruyendo y contrayendo algunos vasos sanguíneos para cortar la hemorragia.
Las funciones que desempeñan las plaquetas son las siguientes:
• Funciones dinámicas:
- Adhesividad.
- Agregación.
- Metamorfosis.
- Función trombodinámica.
- Función retráctil.
• Funciones plasmáticas:
- Liberación de los factores II, III y IV plaquetarios.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
51
Los niveles de plaquetas en sangre, oscilan entre 150.000 a 400.000 células por mm3, cuando el número de plaquetas se encuentra disminuido, se denomina trombocitopenia o trombopenia y puede deberse a:
- Trombopoyesis ineficaz de origen hereditario o bien debido a falta de vitamina B12 o ácido fólico.
- Púrpura trombocitopenia.
- Lupus eritematoso.
- Coagulación intravascular diseminada.
- Anemia hemolítica microangiopática.
- Aumento de la función del bazo (Hiperesplenismo).
- Hipersensibilidad a ciertos medicamentos.
- Enfermedad de Gaucher (desplazamiento de plaquetas hacia el bazo).
- Invasión de la médula ósea.
- Quimioterapia.
- Leucemias.
- Transfusión sanguínea…
Cuando el número de plaquetas está aumentado se denomina trombocitosis y puede ser por:
- Síndromes mieloproliferativos.
- Anemia ferropénica.
- Enfermedad de Kawasaki.
- Colitis ulcerosa.
- Esplenectomía (Extirpación del bazo).
- Traumatismos.
- Trombocitosis primarias…

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
52
2.5 Funciones de la sangre La sangre realiza múltiples funciones, llevadas a cabo por los distintos
elementos que componen la sangre, destacando como más importantes:
- Respiratoria: Esta función la llevan a cabo los hematíes, que transportan el oxígeno de los pulmones hacia los tejidos, para oxigenarlos, retirando el dióxido de carbono, para ser llevado a los pulmones y eliminado, mediante la expiración.
- Nutritiva: La sangre permite el aporte de sustancias nutritivas para las células, a partir de la digestión.
- Inmunitaria o defensiva: Protege al organismo frente a antígenos, gracias a la presencia de glóbulos blancos o leucocitos.
- Excretora: Recogen los residuos y desechos del organismo, para su posterior eliminación.
- Transportadora: La sangre transporta a las distintas partes de nuestro organismo las secreciones y hormonas producidas por las glándulas.
- Reguladora: Interviene manteniendo la regulación de la temperatura corporal, el volumen del líquido intersticial, el pH…
- Hemostática: Limita y paraliza la pérdida de sangre a nivel de los vasos dañados, mediante la hemostasia, en la que intervienen las plaquetas y los factores de la coagulación, entre los que se encuentran, el fibrinógeno, trombina, protrombina…
2.5.1 Hematopoyesis
1.5.1.1 Definición
Es un conjunto de procesos, por el cual se produce la formación de células sanguíneas y su desarrollo a partir de células madre pluripotenciales o STEM- CELL, pasando a sangre periférica como células maduras, para ejercer su función, eliminándose las células envejecidas, para que el equilibrio entre la producción y destrucción no varíe.
2.5.1.2 Tejidos y órganos hematopoyéticos
La hematopoyesis, tiene lugar a partir de las primeras semanas de vida del embrión y finaliza con la muerte del individuo. Durante toda la vida del individuo las células sanguíneas, se reemplazan continuamente por el proceso de la hematopoyesis, a partir de las células madre pluripotentes,

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
53
experimentando numerosas divisiones, siendo regulado por factores de crecimiento, que controlan la concentración y tipo de células que se van a formar, para mantener un equilibrio.
El sistema hematopoyético está formado por numerosos órganos y tejidos que varían según la edad del individuo, interviniendo en todos los procesos de formación de células.
Estos son:
- Saco vitelino. La hematopoyesis en el embrión, tiene lugar a las pocas semanas de vida y ocurre en el saco vitelino, en el que se producen agregaciones de células llamadas Islotes sanguíneos que en su periferia formará la pared vascular y las células centrales darán lugar a eritroblastos que por maduración se convertirán en eritrocitos. A esta fase se le denomina fase mesoblástica.
- Hígado. Una vez concluida la fase anterior tendrá lugar la fase hepática, que se produce también en la etapa embrionaria, entre la 5ª o 6ª semana de gestación. En esta fase los eritrocitos siguen siendo nucleados (igual que en la fase anterior) y sobre la 8ª semana de gestación aparecerán los leucocitos.
- Bazo. Este órgano interviene en la hematopoyesis teniendo una duración más corta y menos intensa que la anterior iniciándose durante el tercer mes y desapareciendo aproximadamente al séptimo mes de gestación. Esta es la fase esplénica.
- Médula ósea. Esta es la fase mieloide, que tiene lugar alrededor de la 6ª semana de gestación alcanzando valores máximos tras el nacimiento, produciéndose hematopoyesis en casi toda la médula ósea de nuestro organismo y durante toda la vida del individuo.
En ésta, tendrá lugar la formación de linfocitos B y T. La médula alberga en su interior arterias, venas y sinusoides, que forman el compartimento vascular, entre los cuales se encuentran islotes de células hematopoyeticas, que se fusionan formando, el compartimento hematopoyético, que es el lugar donde se encuentran las células sanguíneas en las distintas etapas de maduración y los distintos macrófagos,cuya función será, destruir los núcleos libres de las células precursoras de los eritrocitos, células anormales y citoplasma excesivo.
- Timo. Es el órgano en el que se produce la maduración y distribución (a otros tejidos linfoides) de los linfocitos T.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
54
- Ganglios Linfáticos. Son glándulas productoras de linfa, en la que se almacenan los linfocitos y células encargadas de la defensa del organismo, formando parte del sistema inmune y es donde maduran los linfocitos B, pudiéndose transformar en células plasmáticas.
- Sistema mononuclear fagocítico (SMF). Son células encargadas de la defensa del organismo frente a sustancias extrañas, procesando y presentando el antígeno al linfocito y además eliminan células anormales envejecidas, mediante acciones inmunológicas y fagocíticas. Dentro de estos tipos celulares, se encuentran los monocitos, histiocitos y macrófagos.
2.5.1.3 Células que intervienen en el proceso hematopoyético
Como ya sabemos, todas las células sanguíneas, derivan de un precursor común e indiferenciado, denominado célula madre pluripotente o Stem-cell, que posee gran capacidad de maduración y es conocida también como unidad formadora de colonias (CFU) la cual puede madurar convirtiéndose en células multipotenciales originando dos poblaciones:
- CFU-S (unidades esplénicas formadoras de colonias) o línea mieloide: dará lugar a eritrocitos, monocitos, leucocitos granulados y megacariocitos.
- CFU-Ly (unidades formadoras de colonias de linfocitos) o línea linfoide: es la encargada de producir los distintos tipos de linfocitos (T y B).
Cada una de las líneas madurará, perdiéndose la capacidad proliferativa (excepto la de los linfocitos, que puede proliferar mediante la adecuada estimulación) dando lugar a los distintos tipos celulares.
2.5.1.4 Eritropoyesis
Son los procesos de maduración que tienen lugar, hasta la formación de los hematíes. Se originan a partir del precursor mieloide CFU-S, que se diferenciará en BFU-E, seguido de CFU-E, que madurará gracias a una hormona eritropoyética dando lugar al proeritroblasto, que es la forma más inmadura que puede ser reconocida mediante observación microscópica. Estas células, son de gran tamaño y presenta un gran núcleo, que cubre casi la totalidad del citoplasma, el cual se tiñe de azul intenso, debido a la ausencia de hemoglobina.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
55
El siguiente nivel de diferenciación, será el que transforma los proeritroblastos en eritroblastos. El núcleo, pierde los nucléolos y el tamaño de la célula es un poco menor, a medida que va madurando va perdiendo el núcleo hasta transformarse en un reticulocito, el cual presenta capacidad de síntesis de hemoglobina, proteínas y ARN. Una vez saturados de hemoglobina abandona la médula ósea y pasa la sangre como hematíe maduro y debido a la presencia de hemoglobina se tiñe de rojo.
2.5.1.5 Granulopoyesis
Es el proceso de formación de todas las células granulocíticas. Los neutrófilos proceden de la línea CFU-NM (Neutrófilo-Monocito), mientras que los eosinófilos y basófilos proceden cada uno de una línea celular propia que es la CFU-Eosinófila y CFU-Basófila, todas estas líneas maduran dando lugar al mieloblasto, el cual se irá transformando en promielocito, a partir de este momento madurará produciendo células características de cada tipo de granulocito, que serán, el mielocito y metamielocito, diferenciándose en neutrófilos, basófilos y eosinófilos en banda, que finalmente cambiarán su morfología, madurando a eosinófilos, neutrófilos y basófilos.
2.5.1.6 Monocitopoyesis
Proceden de la línea CFU-NM, que es la misma que la de los neutrófilos, pero tras la mitosis se originan dos líneas celulares, CFU-G que originará los neutrófilos y la CFU-M o monoblastos, que por maduración, darán lugar a los promonocitos, que son células grandes de núcleo arriñonado. El citoplasma presenta gránulos azurófilos y muestra (por tinción) coloración azulada. Al madurar originará al monocito, el cual circulará por sangre periférica unos 2 o 3 días, pasando después a órganos o tejidos, denominándose histiocito, donde adquiere actividad macrofágica convirtiéndose en macrófagos.
2.5.1.7 Trombopoyesis
Procede de la línea mieloide, del precursor CFU-S, que al madurar formará la CFU-Megacariocítica, que se transformará en megacarioblasto, que es una célula de gran tamaño (a pesar de ello, es la más pequeña de estas células que forman el proceso de la trombopoyesis), con un núcleo formado por varios nucléolos y lobulaciones. Estas células, aumentan de tamaño por un proceso denominado endocitosis, volviéndose cada vez más grandes, hasta convertirse en un megacariocito, cuyo núcleo carece de lobulaciones. El citoplasma emite numerosos pseudópodos, que al fragmentarse van acumulándose formando proplaquetas que al dispersarse producirán las plaquetas.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
56
2.5.1.8 Linfopoyesis
Procede de la línea linfoide, de la célula madre pluripotencial CFU-Ly, que al dividirse formará dos líneas:
- CFU-LyB: Se producen en la médula ósea de las células, como precursores B (Pre B) que madurarán en el folículo linfoide del ganglio linfático donde podrá continuar con el proceso de transformación, hasta convertirse en una célula plasmática.
- CFU-LyT: También se producen en la médula ósea, donde tendrá lugar la mitosis de estas células y migrarán de la médula ósea como células T inmunocompetentes (Pre T), llegando hasta el timo, donde proliferan y maduran pasando a ser linfocitos T inmunocompetentes.
Hematopoyesis. Origen de las células sanguíneas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
57
2.5.2 Hemostasia
2.5.2.1 Definición
La hemostasia, es uno de los principales sistemas de defensa del organismo, que se pone en marcha ante la lesión de un vaso sanguíneo, produciéndose el proceso hemostático, a través del cual, se detiene la hemorragia y se mantiene la fluidez de la sangre, para mantener la integridad biológica de nuestro sistema vascular.
2.5.2.2 Etapas de la hemostasia
Durante la hemostasia, se producen diversas etapas hasta la completa detención de la hemorragia y reparación del vaso dañado, como son la hemostasia primaria, la coagulación y la fibrinólisis, que son tres etapas que están muy estrechamente relacionadas entre sí:
1ª Etapa: Hemostasia primaria
1.Vasoconstricción
Cuando se produce la lesión del vaso sanguíneo, tiene lugar la salida de las primeras plaquetas del vaso dañado produciéndose una vasoconstricción

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
58
primaria debido a la estimulación de las terminaciones nerviosas. Esta vasoconstricción primaria, se continuará con una vasoconstricción secundaria de mayor duración, debido a la liberación de sustancias contenidas en los gránulos de las plaquetas liberadas.
Además, el endotelio vascular dañado, interviene en los procesos de, agregación plaquetaria, coagulación, tromborresistencia de la pared vascular y fibrinólisis, mediante la liberación de múltiples sustancias producidas por el vaso lesionado. La prostaciclina, glucosaminoglucanos, activadores del plasminógeno… son algunas de las sustancias liberadas por las células endoteliales del vaso dañado, las cuales poseen acción antitrombótica. La liberación de estas sustancias, es imprescindible para el proceso de formación del trombo plaquetario y para su posterior inhibición, una vez reparado el vaso.
2. Adhesión/activación
Como consecuencia de la vasoconstricción se produce la adhesión de las plaquetas a la parte dañada formando una sola capa de plaquetas. La unión al vaso dañado, tiene lugar, gracias a la pérdida de las propiedades tromborresistentes de este y al contacto establecido entre el colágeno y las plaquetas. Como consecuencia, las plaquetas se activan variando su forma, pasando a ser esféricas y emitiendo pseudópodos para una mejor adhesión a la superficie lesionada, lo que es posible, por unos complejos glucoproteicos (GPIa, GPIb, GPIIb, GPIIIa…, GPIX y factor de Von Willebrand) que se encuentran tanto en la superficie de la plaqueta como en el endotelio del vaso lesionado.
La pared subendotelial del vaso dañado presenta unos receptores específicos para las plaquetas que son, la glicoproteína Ia/IIa que junto con otros receptores situados en la membrana de las plaquetas que son la glicoproteína Ib/IX, se unen a la fibra de colágeno del subendotelio y esta unión será estabilizada por la presencia del factor de Von Willebrand.
3. Agregación
A continuación, y como consecuencia de la unión de las plaquetas, se producirá la unión del fibrinógeno a éstas, a través de la glicoproteína IIb/IIIa, en presencia de Calcio. Cuando la plaqueta se pone en contacto con el subendotelio, le induce a la liberación de sustancias contenidas en sus gránulos, como son, ADP, Adrenalina y Serotonina, permitiendo la agregación de más plaquetas, formando un trombo plaquetario, que es inestable ya que si cesa la liberación de estas sustancias, las plaquetas retomarían su forma original y se desharía el trombo plaquetario, pero, si por el contrario, las

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
59
sustancias siguiesen secretándose se produciría la formación de fibrina haciendo que el trombo plaquetario sea estable causando el sangrado de la parte dañada.
2ª Etapa: Coagulación.
Juegan un importante papel, en el sistema hemostático desarrollándose en numerosas etapas en las que se produce el cambio físico del estado de la sangre, debido a la transformación de una proteína soluble como es el fibrinógeno, en insoluble como la fibrina, que constituye el soporte principal de todo este proceso.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
60
Los factores que intervienen en la cascada de la coagulación son los siguientes:
Factor Nombre Síntesis Vida media Características
Ι Fibrinógeno Hepática 4 a 6 Días Es coagulable por la Trombina
II Protrombina Hepática 4 a 6 Días Precursor plasmático de la Trombina
III Tromboplastina tisular Lipoprotéica Activación del factor X
IV Calcio Activación de varias etapas de la coagulación
V Acelerina Hepática 12 a 20 Horas Se consume en el proceso de la coagulación
VI No asignado
VII Proconvertina Hepática 4 a 6 Horas Actúa sobre el F.X con ayuda de F.III y F.IV
VIII Antihemofílico A Células endoteliales 10 a 12 Horas Sensible a la Trombina
Von Willebrand Proporciona adhesividad y agregabilidad a las plaquetas
IX Christmas
Antihemofílico B Hepática 15 a 30 Horas Tiene actividad enzimática
X Stuart Prower Hepática 45 a 75 Horas Activa la Protrombina
XI Antecedente tromboplastina Hepática 45 a 100
Horas Se activa en presencia de F.III sin necesidad de calcio
XII Hageman 60 Horas Se activa por enzimas o por contacto con superficies cargadas negativamente
XIII Estabilizador de la fibrina 3 a 5 Días Estabiliza a la fibrina
Fletcher Hepática Posee actividad enzimática
Proteína S Hepática Actúa como cofactor de la Proteína C y es antifibrinolítico
Proteína C Hepática Inhibe a F.V y F.VIII, activa el plasminógeno y es antifibrinolítico
En condiciones normales, la coagulación se produce solamente, en las zonas vasculares que se encuentran lesionadas, debido a dos mecanismos que son, la capacidad diluidora del flujo normal de la sangre, lo que hace que las

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
61
formas activas de los factores de la circulación lleguen hasta el hígado donde serán destruidos y junto con todo ello, también interviene la presencia en plasma de sustancias que neutralizan estas formas activadas y que reciben el nombre de factores inhibidores de la coagulación, destacándose algunos como:
- Anticardiolipina: Son anticuerpos de tipo Inmunoglobulina G y M, que actúan neutralizando a los fosfolípidos, interviniendo en la iniciación del proceso de la coagulación. Estos anticuerpos anticardiolipina están presentes, en el síndrome antifosfolípido.
- Anticoagulante lúpico: aparece en enfermos de lupus eritematoso, además de ser otro de los anticuerpos del síndrome antifosfolípido.
- Proteínas C y S: actúan desactivando los factores V y VIII.
- Cofactor de la heparina o Antitrombina III: actúa inactivando a los factores IX, X, XI y XII, inhibiendo la acción de la trombina y acelerando el proceso anticoagulante de la heparina.
- Β2-Glicoproteína I: Actúa como anticoagulante natural por su capacidad de unirse a fosfolípidos e inhibir la formación de la trombina.
En la cascada de la coagulación intervienen tres vías, que son:
1. Vía Extrínseca
Se desencadena mediante sustancias liberadas tras las lesiones de la pared vascular, que son sustancias procoagulantes, procedentes del espacio intersticial. Esta vía requiere para su activación, la presencia de un factor no plasmático que es el factor III o tromboplastina tisular (Factor tisular), que estimula la coagulación al formar un complejo con el factor VII en presencia de calcio, lo que conlleva a la activación del factor X.
2. Vía Intrínseca
Esta vía tiene lugar cuando la sangre entra en contacto con superficies cargadas negativamente, es decir con las fibras de colágeno de la capa subendotelial de los vasos dañados. En esta vía todos los factores necesarios se encuentran en sangre periférica. Cuando el factor XII entra en contacto con una superficie cargada negativamente o mediante intervención enzimática se producirá la activación de este factor, lo que dará lugar a la consecuente activación del factor X. Esta es una vía larga y lenta.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
62
3. Vía Común
Es común tanto a la vía intrínseca como extrínseca. En esta vía se produce la activación del factor X, siendo necesario, que esta esté unida a la superficie fosfolipídica plaquetaria, por medio de puentes de calcio. Una vez activado, este formará un complejo enzimático con el factor V (como cofactor), fosfolípidos y calcio, llamado protrombinasa, que producirá la activación de la protrombina (Factor II) y esta es la responsable del paso de protrombina a trombina, que activará a los factores V, VIII y XIII y estimulará la adhesión y agregación plaquetaria, además de actuar sobre el fibrinógeno,

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
63
haciendo que este se transforme en fibrina. A partir de este momento tendría lugar la formación del trombo plaquetario soluble o inestable que para convertirlo en insoluble se necesitará la presencia del factor XIII cuya función es la de polimerizar la fibrina para estabilizarla, creando el coágulo estable. Además, facilita la adherencia del coágulo a la superficie dañada.
La última etapa de este proceso será la retracción del coágulo, que está ligada a al número y funcionamiento de las plaquetas.
Nuevo modelo celular de coagulación
Recientemente ha surgido un modelo nuevo de la teoría de la coagulación, el modelo celular, que deja atrás el modelo clásico anteriormente explicado que se basaba en dos vías separadas: la vía intrínseca y la vía extrínseca.
Este nuevo modelo celular ha surgido en el tiempo y es que se ha visto que ciertas enfermedades deficientes en ciertos factores no producían los resultados esperados, es decir, nos basábamos en el hecho de que al haber una deficiencia en algún factor concreto en algún punto de la vía, toda ella se veía afectada y por ende el sangrado sería igual si faltaba un factor u otro. Esto fue debatido porque, por ejemplo, enfermedades deficientes en el factor VII conllevan una hemorragia mayor que las deficientes en el factor V. Además, si realmente existieran dos vías separadas esto no explicaría que la formación del factor Xa de la vía intrínseca no compense la falta del factor VIII en los pacientes con hemofilia. Otro punto a tener en cuenta que cuestiona el modelo clásico de coagulación es que la activación de la cascada de la coagulación por la vía intrínseca es bastante cuestionable si tenemos en cuenta que la deficiencia del factor XII o precalicreína no producen sangrado, aunque incluso el TPTA sea altísimo. Por otro lado, el déficit de factor X, V y VII puede producir un gran sangrado por el contrario al déficit del factor XI que es menos predecible. Por tanto el modelo clásico no podemos decir que explique la hemostasia in vivo.
El nuevo modelo celular explica el inicio de la coagulación basándose en el complejo formado por el factor tisular (FT) y el factor VII activado (FT-FVIIa) que se activan donde hay daño tisular. Este complejo actúa sobre la superficie de las células que portan factor tisular activando distintos factores como el factor V y el IX lo que inicia el proceso de coagulación. Este nuevo modelo por tanto explica la regulación de la coagulación por la interacción del complejo FVIIa-FT y por las propiedades de la superficie celular y no por tanto a través de dos vías humorales distintas como era el modelo clásico. El modelo celular hace especial hincapié en los receptores específicos para factores de

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
64
coagulación, fundamentalmente en las plaquetas y células endoteliales que tienen un contenido parecido en fosfatidilserina.
Según el modelo celular la coagulación se explica en tres fases superpuestas:
• Iniciación: al producirse daño tisular se expone el factor tisular (FT) que en condiciones normales se encuentra en el interior de las células del endotelio a salvo del flujo sanguíneo y al exponerse activa al factor VII. Cuando se produce daño en el endotelio el factor tisular sale a la sangre donde rápidamente se une al factor VII creando el complejo FT-FVIIa. Este complejo inicia la formación de la trombina tras la activación del factor X. La trombina inicia la siguiente fase de la coagulación que es la amplificación activando los factores V, VIII y XI.
• Amplificación: tras formarse la trombina se activan las plaquetas expresando fosfolípidos de carga negativa en su superficie como la fosfatidilserina que sirven para la activación del factor X y para que se produzca más trombina. Las plaquetas son moduladoras y amplificadoras de la coagulación y como bloqueantes de esta fase están los inhibidores de la proteasa que impiden el paso del factor Xa de la fase fluida a la superficie de las plaquetas, que es clave para la siguiente fase que es la propagación.
• Propagación: los factores activados y sus cofactores en la superficie plaquetaria amplifican exponencialmente la formación de trombina que activa al fibrinógeno y por tanto resulta en la

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
65
polimerización de la fibrina. En esta fase a parte de los factores de coagulación son necesarios los inhibidores de fibrinólisis y estabilizadores de fibrina como el factor XIII que forma un coágulo duradero y resistente. El inhibidor de la fibrinólisis dependiente de trombina necesita para activarse altas concentraciones de trombina. De este modo se produce una autorregulación con la interacción FVIIa-FT, plaquetas, trombina, fibrina e inhibidores fibrinolíticos dependientes de trombina.
3ª Etapa: Regulación del sistema coagulativo. Fibrinólisis
Es la etapa final de la coagulación, cuya función es actuar como regulador coagulativo, eliminando la fibrina que no sea necesaria para el proceso de la hemostasia, lo que hace precisar a su vez de un sistema de control que evite su activación excesiva y con ella la hemorragia. Para que esto no tenga lugar, ocurre la lisis del coágulo y la reparación del vaso dañado.
La fibrinólisis puede ocurrir de forma natural, es decir eliminando un coágulo producido tras la reparación del vaso, llamándose fibrinólisis primaria o bien puede ocurrir antes de tiempo, es decir antes de que el vaso llegue a repararse y esto puede deberse a diversas enfermedades o medicamentos.
Como ya sabemos los coágulos se forman a partir de una proteína llamada fibrina, cuya producción puede verse incrementada por ejercicio físico intenso, oxigenación inadecuada de los tejidos, azúcar bajo en sangre o infecciones bacterianas. Para inhibir el crecimiento del coágulo y su posterior eliminación, habrá que limitar la formación de fibrina, para ello intervienen proteínas como la antitrombina III que es la más importante, ya que es el principal inhibidor fisiológico de la trombina y de los factores activados X, IX, XI, XII y plasmina.
La fibrina es eliminada de la circulación por acción del sistema fibrinolítico, que está regulado por:
- Los inhibidores de la plasmina, que transformarán el plasminógeno en plasmina, que es una enzima proteolítica capaz de degradar la fibrina.
- Los inhibidores de los activadores del plasminógeno.
La transformación de plasminógeno a plasmina es un proceso enzimático irreversible. Estos activadores del plasminógeno pueden encontrarse en sangre, tejidos u orina.
La plasmina interviene en la degradación de la fibrina, Factores V, VII, VIII y fibrinógeno. Mediante la degradación de la fibrina, por parte de la

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
66
plasmina, se forman tres tipos de fragmentos, que inhiben la polimerización de la fibrina y son los fragmentos X, D, Y y un cuarto fragmento que inhibe la agregación plaquetaria y puede actuar como antitrombina. Los productos de degradación inhiben también el mecanismo de la coagulación y así como los de la agregación plaquetaria.

UNIDAD TEMÁTICA III SÍNDROME ANTIFOSFOLÍPIDO


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
69
3.1 Definición El síndrome antifosfolípido, también conocido como “Síndrome de la
sangre pegajosa” o Síndrome de Hughes, es una enfermedad autoinmune, rara y sistémica, que puede aparecer en cualquier momento de la vida de una persona y dicha enfermedad está caracterizada, por un desorden del estado de hipercoagubilidad, lo que provoca un estado trombofílico debido a la presencia en sangre de anticuerpos dirigidos contra el complejo protrombina-fosfolípidos o contra factor de coagulación-fosfolípidos que afectan la conversión del complejo protrombina-fosfolípidos, en trombina y la activación del factor de coagulación fosfolípidos.
Estos fosfolípidos, son un tipo de grasa especial que contiene el fosfato, el cual constituye las paredes externas de las células de nuestro cuerpo. La misión de estos anticuerpos antifosfolípidos, es la de atacar a los fosfolípidos de las paredes celulares, lo que ocasiona diversos problemas, como por ejemplo el estado de hipercoagubilidad de la sangre. Por tanto como la cardiolipina es un tipo de fosfolípido, podrán desarrollarse anticuerpos anticardiolipinas específicos.
También se le puede definir como una alteración del sistema inmune, caracterizándose como una enfermedad autoinmune, en la que nuestro cuerpo produce grandes concentraciones de anticuerpos antifosfolípidos. Siendo por tanto el responsable de pérdidas fetales de repetición, trombosis arteriales, venosas o ambas al mismo tiempo (los coágulos en los vasos sanguíneos del sistema nervioso central como son el cerebro y médula espinal, lo que puede provocar accidentes cerebrovasculares) y trombocitopenia (que se caracteriza por una disminución del número de plaquetas, siendo un factor muy perjudicial, en el mecanismo de la coagulación de la sangre). Además de las patologías anteriores pueden darse otras manifestaciones patológicas de menor importancia diagnóstica tales como, lesiones cardiacas, neurológicas, cutáneas, oculares… y es una enfermedad que puede estar asociada en algunas ocasiones a otras enfermedades autoinmunes como puede ser el lupus eritematoso sistémico (LES).
El síndrome antifosfolípido presenta tres tipos de anticuerpos que son utilizados clínicamente para la identificación de la enfermedad, que son:
- Anticuerpos anticoagulantes lúpico.
- Anticuerpo anticoagulante anticardiolipina.
- Anticuerpo anti β2-Glicoproteína I.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
70
El síndrome antifosfolípido suele manifestarse sobre todo en mujeres (80 %) de entre 20 a 40 años, aunque la posibilidad de padecer esta enfermedad, aumenta con la edad, sobre todo si la persona padece algún tipo de enfermedad crónica.
3.2 Anticuerpos antifosfolípidos Los anticuerpos antifosfolípidos, son una familia de inmunoglobulinas,
IgA, IgG o IgM, que reaccionan contra los complejos formados por fosfolípidos aniónicos de las membranas celulares y proteínas plasmáticas, que actúan como cofactores.
Estos cofactores son la β2-glucoproteìna I, la protrombina, la proteína C, la proteína S y la anexina V, que reaccionan contra complejos proteína-fosfolípido y contra proteínas ligadoras de fosfolípidos, sobre todo reacciona contra la β2-glicoproteínaI, que es una proteína sérica que actúa como anticoagulante natural por su capacidad de unirse a los fosfolípidos e inhibir la formación de la trombina., la unión de esta a los fosfolípidos, induce un cambio conformacional que expone el sitio antigénico reconocido por los anticuerpos, aunque también existen otros anticuerpos, que reconocen a la proteína directamente, por ello se dice que reaccionan contra los complejos fosfolípido-proteínas.
Estos anticuerpos antifosfolípidos se han detectado principalmente en pacientes con lupus eritematoso sistémico, aunque también puede aparecer en otras enfermedades autoinmunes como la artritis reumatoide, esclerodermia, síndrome de Sjögren primario, enfermedad de Crohn, vasculitis y púrpura trombocitopenia autoinmune, entre otras.
La especificidad de los anticuerpos será mayor cuanto mayor sea su título, siendo aún mayor para las inmunoglobulinas del tipo Ig M e Ig G.
Los anticuerpos antifosfolípidos se clasifican actualmente en una familia heterogénea de anticuerpos dirigidos contra los fosfolípidos aniónicos (cardiolipina y fosfatidilserina) o neutros (fosfatidiletanolamina) y/o las proteínas plasmáticas ligadas a los fosfolípidos como la β2-glicoproteína I y la protrombina.
Los anticuerpos antifosfolípidos, que se detectan comúnmente en esta enfermedad son estos tres tipos:
- Anticoagulante lúpico.
- Anticuerpos anticardiolipinas.
- β2-glicoproteína I.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
71
3.2.1 Anticoagulante lúpico Este anticoagulante está asociado a fenómenos tromboembólicos. Las
inmunoglobulinas son las responsables de la actividad del anticoagulante lúpico, siendo la inmunoglobulina G la principal responsable y con menor frecuencia las inmunoglobulinas M o A.
“In vitro” estos anticuerpos, actúan por formación de un complejo con los fosfolípidos exógenos o endógenos y los cofactores β2-GP-I o protrombina.
Estos anticoagulantes son identificados mediante el empleo de un test de coagulación, llamado test de screening que mide el tiempo parcial de tromboplastina activada (TPTA) o tiempo de cefalina-caolín. Este tiempo aparecerá prolongado si existe este anticoagulante en el suero del paciente, aún así la sensibilidad y especificidad de esta prueba varía dependiendo de la composición, cantidad e incluso del estado del fosfolípido que contienen los reactivos, además puede ser debida a un déficit de algún factor de la coagulación, por lo tanto para confirmar la presencia del anticoagulante lúpico será necesaria la realización de otros estudios para que se pueda concretar la presencia o ausencia de este, por tanto como este tiempo puede variar, algunos expertos recomiendan usar otros test, basados en diferentes principios de la coagulación.
Para confirmar que la alteración en alguna de las pruebas de coagulación es debida a la presencia de un inhibidor, puede añadirse plasma normal al del paciente y si al realizar nuevamente la prueba, sigue dando el tiempo alterado se confirmará la presencia de un inhibidor, es decir que al añadir el plasma normal al plasma del paciente el tiempo de tromboplastina tendría que corregirse de lo contrario demostraría la presencia del anticoagulante lúpico.
Otra prueba para la identificación del anticoagulante lúpico es la prueba de neutralización con plaquetas lavadas, que se usa para demostrar que el inhibidor es dependiente del fosfolípido, pudiendo dar falsos positivos en presencia de heparina o inhibidor del factor V.
3.2.2 Anticuerpos anticardiolipina Son otro tipo de anticuerpos presentes en personas con síndrome
antifosfolípido, en definitiva son fosfolípidos que ayudan a formar las membranas celulares. Los anticuerpos anticardiolipinas se asocian con la formación de coágulos de sangre (trombosis) dentro de condiciones autoinmunes.
La detección de los anticuerpos anticardiolipina, es el test de mayor sensibilidad para el síndrome antifosfolipídico, mientras que el anticoagulante

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
72
lúpico es el más específico. En estos pacientes se puede observar mediante técnicas reagínicas una serología luteica falsamente positiva, lo que nos indica la presencia de anticuerpos dirigidos contra la cardiolipina, lecitina y colesterol.
Para que se produzca la unión de estos anticuerpos a su antígeno, es necesaria la presencia del cofactor enzimático β2-Glicoproteína I, a consecuencia de ello existen muchos anticuerpos anticardiolipina dependientes de β2-Glicoproteína I, asociados a procesos trombóticos y otros anticuerpos anticardiolipina no dependientes de β2-Glicoproteína I que se identifican principalmente en el curso de diferentes infecciones y no tienen relación con las manifestaciones clínicas del síndrome.
Para la identificación de estos anticuerpos se emplean técnicas de inmunoensayo como son las técnicas de ELISA, en la que los anticuerpos anticardiolipinas teóricamente se unen a la cardiolipina. Las inmunoglobulinas M y G, son los dos tipos que se identifican habitualmente mediante esta técnica, aunque si los resultados dan negativo para ambos tipos de inmunoglobulinas se procedería a la determinación de la inmunoglobulina A u otros fosfolípidos aniónicos. Los títulos altos y persistentes en el tiempo de inmunoglobulina G, suelen asociarse a fenómenos trombóticos y por tanto al síndrome antifosfolípido.
3.2.3 Anticuerpos anti B2-glicoproteína I La β2-Glicoproteína I (β2 GPI) es un anticoagulante natural que inhibe la
vía intrínseca de la coagulación y la actividad protrombinasa de las plaquetas. La unión y alteración del cofactor (β2 GPI) puede producir estados protrombóticos.
La β2-glicoproteína I tiene una estructura dimérica, que se reproduce seguidamente y explica su afinidad por los fosfolípidos, pudiendo ser activada por la plasmina. Los anticuerpos patógenos reconocen una secuencia del dominio 1 e inducen una dimerización de la molécula que aumenta la afinidad de la β2-glicoproteína por los fosfolípidos aniónicos procoagulantes.
Los anticuerpos β2-Glicoproteína I o el complejo β2-Glicoproteína I/cardiolipina son comunes en pacientes con síndrome antifosfolípido, aunque puede ser que sólo uno de los dos anticuerpos de positivo, por tanto para que se dé la enfermedad no es necesaria la presencia de ambos anticuerpos en el organismo de la persona enferma.
Los anticuerpos anti β2-Glicoproteína I son detectados por inmunoensayos que miden la unión del anticuerpo al complejo fosfolípido -β2-

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
73
Glicoproteína, respectivamente. Estos anticuerpos están fuertemente asociados con las trombosis y otras manifestaciones del síndrome antifosfolipídico, por lo que se recomienda su detección en pacientes cuyos test para anticoagulante lúpico y para los anticuerpos anticardiolipina fueron negativos.
Alrededor de un 60 % de los pacientes que presentan anticuerpos anticardiolipina y trombosis, presentan también anticuerpos anti β2-Glicoproteína I, siendo estos mejores marcadores de trombosis que los anticuerpos anticardiolipinas.
CLASIFICACIÓN RESUMEN DE LOS ANTICUERPOS ANTIFOSFOLÍPIDOS

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
74
Anticuerpos anticoagulantes lúpicos.
1º Prolongación de la coagulación en alguna de las pruebas “in vitro” de coagulación dependientes de al menos un fosfolípido usando plasma pobre en plaquetas. Estas pruebas pueden ser:
- Tiempo de protrombina (afecta a la vía extrínseca de la coagulación).
- Tiempo de tromboplastina parcial actividad (u otro test que afecta a la vía intrínseca de la coagulación)
Pruebas que afectan la vía común de coagulación (tiempo del veneno de víbora, y otros) 2º Fracaso en prolongar el tiempo de coagulación al mezclar plasma del paciente con plasma normal. 3º Confirmación de la presencia de anticuerpos anticoagulantes lúpicos que acorten el tiempo de coagulación prolongado después de añadir fosfolípidos o plaquetas. 4º Descartar otras coagulopatías mediante ensayos específicos si la prueba es negativa y se sospecha la presencia de otro inhibidor de un factor específico.
Anticuerpos anticardiolipina.
Inmunoensayo en fase sólida (ELISA) en presencia de β2-glicoproteína I, en suero bovino.
- Los anticuerpos anticardiolipinas de pacientes con el síndrome antifosfolípido son dependientes de la glicoproteína.
- Los anticuerpos de pacientes con enfermedades infecciosas son independientes de la glicoproteína.
Anticuerpos antiglicoproteína β2. Inmunoensayo en fase sólida (ELISA) en placas de poliestireno recubiertas de la β2-glicoproteína I, humana.
3.3 Manifestaciones clínicas El síndrome antifosfolípido, se define como un estado de
hipercoagubilidad en el que tiene lugar la asociación entre los anticuerpos antifosfolípido-anticoagulante-lúpico y los anticuerpos anticardiolipinas, con manifestaciones clínicas peculiares como: trombosis venosa y arterial, abortos a repetición o pérdidas fetales y trombocitopenia, como síntomas principales.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
75
Las causas de la prevalencia de esta enfermedad, son aún desconocidas, aunque se están realizando estudios con la finalidad de determinar la importancia de los anticuerpos antifosfolípidos, en diversas enfermedades asociadas a la enfermedad, como accidentes cerebrovasculares, infartos de miocardio, abortos recurrentes…
El factor genético puede jugar un importante papel en la manifestación de esta enfermedad, ya que existen familias en las que varios de sus miembros presentan anticuerpos antifosfolípidos positivos, describiéndose asociaciones entre la presencia de anticuerpos antifosfolípidos y la de antígenos de clase II del complejo mayor de histo-compatibilidad.
La presencia de anticuerpos antifosfolípidos en el suero de los enfermos se cree que es debida a que las células están indemnes, por lo que sus fosfolípidos de membrana no están expuestos al sistema inmunológico, por tanto no habrá ninguna activación de anticuerpos antifosfolípidos, si por el contrario, estos fosfolípidos de membrana, son agredidos o entran en apoptosis, pierden la asimetría en la estructura de la membrana, por lo que los fosfolípidos o las proteínas antigénicas quedan expuestas en su superficie, siendo reconocidas por los macrófagos, que transfieren la información y activan a los linfocitos B, que se multiplican y segregan inmunoglobulinas. Esto puede ocurrir a consecuencia de:
• Alteración del sistema inmune
En el curso de una enfermedad autoinmunitaria, (la situación más frecuente es el lupus eritematoso diseminado, en cuyo caso los niveles de anticuerpos son mantenidos y predomina el isotipo Inmunoglobulina G).
Cuando hay destrucción celular (como en enfermedades infecciosas o malignas).
Cuando se modifica la inmunidad por cambios hormonales drásticos, tal como sucede durante el embarazo (situaciones en las que los APL desaparecen una vez interrumpida la causa determinante, son predominantemente Ig M).
• Sin un hecho desencadenante manifiesto
Los anticuerpos antifosfolípidos/ proteínas son los causantes de estados protrombóticos debido a que los fosfolípidos intervienen en numerosas reacciones de la hemostasia, como son, la β2-Glicoproteina I, la protrombina, la proteína C, la proteína S, los kininógenos, la anexina V, la trombomodulína, el factor X y el factor XI, están implicados en la aparición y desarrollo de esta enfermedad.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
76
Se pueden diferenciar dos tipos de síndrome antifosfolípido:
• Primario: no acompaña a alguna enfermedad autoinmune y en este sólo se producen embolias anormales.
• Secundario: cuando se presenta en el contexto de un lupus eritematoso sistémico, en el cual se presentan manifestaciones asociadas a un lupus eritematoso sistémico.
En el síndrome antifosfolípido primario y secundario, se produce la oclusión de grandes vasos sanguíneos. También puede darse otro tipo de síndrome antifosfolípido que es el llamado síndrome antifosfolípido catastrófico, que produce una obstrucción de pequeños vasos, produciéndose en pacientes totalmente anticoagulados, con embolias muy rápidas, que afectan a varios órganos y tiene un elevado índice de mortalidad. Aunque algunos SAF primarios pueden evolucionar a un LES, la mayoría no demuestran tal progresión.
El síndrome antifosfolípido secundario, está ligado a otro tipo de enfermedad autoinmune, definida como una enfermedad crónica multisistémica, llamada lupus eritematoso sistémico.
El Lupus Eritematoso Sistémico es, una enfermedad incurable del sistema inmunitario, una condición por la cual el mecanismo de defensa de nuestro organismo comienza a atacarse a sí mismo creando un exceso de anticuerpos en el torrente sanguíneo que causan inflamación y dañan las articulaciones, los músculos y otros órganos.
Se cree que un 1% de la población puede tener Lupus en este país, de los cuales el 90% son mujeres, principalmente entre 15 y 55 años de edad y entre un 30 y un 35 % de los pacientes con lupus eritematoso sistémico, generan anticuerpos antifosfolipídos y, de estos, aproximadamente un tercio desarrollan el Síndrome antifosfolípido.
El Lupus no tiene causa conocida, aunque la investigación evidencia la implicación de trastornos de la inmunorregulación, factores genéticos, hormonales y ambientales:
Trastornos de la inmunorregulación: se produce una asociación entre una susceptibilidad genética y determinados factores ambientales desencadenando una respuesta inmunológica anormal, con hiperactividad de los linfocitos T y B, y producción excesiva de autoanticuerpos. Existe además una regulación anómala de dicha hiperactividad por hipofunción de la población supresora CD8 y de las células citotóxicas natural killer, menor eficiencia del sistema mononuclear fagocítico y deficiencia en los receptores del complemento. La

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
77
producción de los autoanticuerpos no es indiscriminada, sino que existe selectividad ante las moléculas de proteínas y ácidos nucleicos. Estos autoanticuerpos son los responsables de la respuesta inflamatoria local y la lesión tisular en los órganos afectados, bien directamente o bien uniéndose a su antígeno específico y formando inmunocomplejos.
Predisposición genética: existe una concordancia en el 25-60 % de gemelos monocigóticos, lo que demuestra la naturaleza poligénica de esta predisposición genética, así como la contribución de factores ambientales.
Factores hormonales (hiperactividad estrogénica): se sabe que los estrógenos inhiben la actividad de los linfocitos T supresores con aumento de la síntesis de auto-anticuerpos por las células B.
Factores ambientales: el lupus eritematoso sistémico se relaciona con la recepción de radiaciones ultravioleta que producen la desnaturalización del DNA modificando su antigenicidad, con determinados fármacos como pueden ser, la hidralazina, procainamida, isoniazida, interferón alfa y estrógenos y con algunos agentes infecciosos como el retrovirus, aunque no se ha demostrado una asociación definitiva.
Las posibles manifestaciones del Lupus son muy variadas, rara vez dos personas tienen exactamente los mismos síntomas, a pesar de ello los más comunes son:
− Dolor en músculos y articulaciones.
− Eritemas permanentes en las mejillas.
− Problemas de riñón.
− Síntomas gripales y/o sudoraciones nocturnas.
− Inflamación de los tejidos que recubren órganos internos con dolor abdominal o pectoral.
− Mala circulación sanguínea.
− Problemas hematológicos como la anemia.
− Ataques, enfermedad mental u otros problemas cerebrales.
− Dolores de cabeza, migrañas.
− Fatiga extrema y debilidad.
− Ulceras bucales o nasales.
− Riesgo de abortos espontáneos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
78
− Pérdida de pelo.
− Eritemas solares.
− Depresión.
Se pueden desarrollar 4 ó 5 de estos síntomas, que pueden desaparecer y/o desarrollar otros. Además de estos síntomas generales, aparecen otras patologías que afectan al corazón, pulmones, piel…
Otro tipo de afectaciones que se producen en esta enfermedad, son las hematológicas en las cuales se pueden encontrar alteraciones en las tres series. Es común la leucopenia, menor de 4.000 células por mililitro, con linfopenia absoluta. La mayoría tienen anemia de enfermedad crónica. La anemia hemolítica aguda coombs positiva, es característica del lupus eritematoso sistémico, pero afortunadamente es poco frecuente. En la serie plaquetaria es frecuente la trombopenia leve, que puede ser grave en el 5 % de los casos con hemorragia y púrpura, en estos casos es muy importante descartar la presencia de anticuerpos anticardiolipina o anticoagulante lúpico. La púrpura trombocitopénica autoinmune puede ser la forma de manifiesto de la enfermedad o bien puede aparecer en el curso evolutivo de dicha enfermedad.
El Lupus parece ser un tipo de auto-alergia y al manifestarse de manera tan variada hace que el perfil de los pacientes sea completamente diferente, por ello para llegar a su diagnóstico existen una serie de condiciones aceptadas internacionalmente, de las cuales el paciente debe presentar al menos cuatro, para poder diagnosticar esta enfermedad, teniendo además en cuenta los resultados analíticos y los antecedentes familiares y estas condiciones son:
− Piel
− Sangre
− Fatiga
− Articulaciones
− Corazón
− Fiebre
− Riñones
− Pulmones
− Pérdida de peso

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
79
− Glándulas inflamadas
− Sistema nervioso
− Sobrepeso
Para el diagnóstico del lupus eritematoso sistémico deben realizarse una serie de pruebas siempre que se sospeche la existencia de esta enfermedad, para ello se realizarán pruebas analíticas como hemograma, VSG, análisis de orina, bioquímica general, coagulación y anticuerpos.
Las alteraciones que se suelen producir, incluyen anemia de enfermedad crónica, anemia hemolítica, leucopenia, linfopenia o trombocitopenia. La presencia de anticoagulante lúpico se pone en evidencia mediante la demostración de un tiempo parcial de tromboplastina prolongado. En los brotes de actividad se detectan reactantes de fase aguda como la VSG. La proteína C reactiva raramente presenta títulos altos durante las fases de actividad, pero sí aumenta en casos de infección intercurrente. La hipoalbuminemia, hiperlipemia, alteración de la función renal y del sedimento urinario sugieren glomerulonefritis.
Se desarrollan pruebas inmunológicas consistentes en la demostración en el suero de leucocitos polimorfonucleares que han fagocitado núcleos de linfocitos aunque esta técnica no suele usarse en la actualidad.
1) Los anticuerpos que se estudian para la detención de la enfermedad son los siguientes:
2) Los anticuerpos órgano-específicos: están dirigidos contra elementos celulares del sistema hematopoyético o contra determinados tejidos como el tiroides, la pared gástrica, el músculo o las glándulas adrenales.
Los anticuerpos antinucleares (ANA), suelen ser positivos en el 95 % de los pacientes y se distinguen 3 grandes grupos en función del antígeno diana:
− Los anticuerpos anti-DNA nativo o bicatenario son muy específicos de lupus eritematoso sistémico. Se detectan en el 40-60 % y su título varía con el grado de actividad.
− Las anti-histonas (proteínas básicas presentes en el núcleo) son proteínas características del lupus inducido por fármacos.
− La anti-ENAs (proteínas no histonas presentes en el núcleo y citoplasma) se han catalogado más de 20 tipos específicos, pero

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
80
los que tienen más interés práctico son los anti-SSA/Ro, anti-SSB/La, anti-RNP y anti-Sm (este último es el más específico del lupus eritematoso sistémico).
3) Los anticuerpos antifosfolípidos se detectan por el método de ELISA, en el que se puede presentar una serología luética falsamente positiva que es un marcador indirecto de estos anticuerpos. Se asocian con fenómenos trombóticos, especialmente los del isotipo inmunoglobulina G y con títulos altos o moderados.
Además de las pruebas anteriores se realizan otro tipo de pruebas inmunológicas como son:
− La concentración baja de C3 y C4 se utiliza como marcador de actividad lúpica.
− La disminución de CH50 (complemento hemolítico total) con valores normales de C3 sugiere la presencia de defectos hereditarios, y es frecuente en casos de lupus eritematoso sistémico con anticuerpos antinucleares negativos.
− La presencia del factor reumatoide indica menor probabilidad de afectación renal.
Las enfermedades renales que presentan más frecuentemente este tipo de pacientes son, la presencia en orina de cilindros celulares o hemáticos y las proteinurias, en las cuales se producen en orina una excreción de proteínas, mayor de 0,5 gramos al día.
Los trastornos hematológicos producidos en esta enfermedad son la presencia de anemia hemolítica, leucopenia con valores menores a 4.000 por mm3 en dos o más ocasiones, linfopenia menor a 1.500 por mm3 también en dos o más ocasiones y trombopenia menor a 100.000 por mm3.
Se producirá además trastornos inmunitarios de los anticuerpos antinucleares, anticuerpos anti –ADN o anticuerpos anti-VDRL falsamente positivos y anticuerpos antifosfolípidos positivos.
Además del síndrome antifosfolípido primario o secundario, los anticuerpos antifosfolípidos, han sido descritos en una amplia variedad de desórdenes, sean estos reumáticos, inducidos por drogas o infecciones.
A continuación, explicaremos las enfermedades características del síndrome, así como las diferentes patologías que se puedan producir dentro de cada enfermedad.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
81
3.3.1 Manifestaciones mayores
3.3.1.1 Trombosis
Los anticuerpos antifosfolípidos se asocian con trombosis a cualquier nivel del territorio vascular y/o venoso, produciendo por tanto dos tipos de trombosis que son la trombosis arterial y la trombosis venosa:
1. Trombosis venosa
Afecta a las venas, así como a vasos de distinto calibre y dimensiones. Las trombosis suelen ser recurrentes y suelen acompañarse de embolismo pulmonar y las más frecuentes son las que tienen lugar en las extremidades inferiores, que pueden producirse tanto en venas superficiales como profundas. Las trombosis venosas pueden producir múltiples enfermedades que pueden afectar a diversas partes de nuestro cuerpo, así pues procederemos a clasificar las enfermedades según la parte del organismo en las que se produzcan:
TROMBOSIS VENOSA
Parte del cuerpo afectada Enfermedad producida
Extremidades Tromboflebitis
Enfermedades hepáticas Grandes vasos: Síndrome de Budd- Chiari
Pequeños vasos: Hepatomegalia
Enfermedades de las Glándulas suprarrenales Enfermedad de Addison
Enfermedades pulmonares Tromboembolismo pulmonar
Manifestaciones cutáneas
Livedo reticularis
Úlceras en miembros inferiores
Lesiones purpúricas
Lesiones oculares Trombosis de las arterias retinianas

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
82
a) Extremidades
- Tromboflebitis
La tromboflebitis, es la obstrucción de una vena, originado por la presencia de trombos dentro de estas, lo que da lugar a su inflamación, debido a que impide el paso de sangre por la vena obstruida.
El 90 % de las trombosis suelen originarse en miembros inferiores y pueden no manifestarse clínicamente. Estos trombos son originados como consecuencia del estancamiento de sangre, la lesión de la pared de la vena y la presencia de sustancias que favorecen la coagulación sanguínea, produciéndose en las paredes de las venas un cúmulo de todos estos componentes, que darán lugar a la obstrucción progresiva de la vena.
El origen de las tromboflebitis se da en personas que permanecen
sentadas o acostadas durante un período de tiempo prolongado, en alteraciones de la coagulación, insuficiencia cardiaca y administración prolongada de anticonceptivos orales.
Los síntomas que presentan tromboflebitis presentan unos síntomas comunes, que son:
− Calor y sensibilidad sobre la vena.
− Dolor en la parte afectada del cuerpo.
− Enrojecimiento de la piel (no siempre presente).
− Inflamación (hinchazón) en la parte del cuerpo afectada.
Para la detección de esta enfermedad, se realizan (entre otras) pruebas de la coagulación.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
83
El tratamiento al que se le somete al paciente, consiste en analgésicos, anticoagulantes, y trombolíticos. La tromboflebitis y otras formas de flebitis usualmente responden a un tratamiento médico rápido.
Los dos tipos de esta enfermedad son: tromboflebitis superficial (afecta venas cercanas a la superficie de la piel) y trombosis venosa profunda (afecta venas más profundas y más grandes).
- Tromboflebitis superficial
Se produce por la presencia de un coágulo, que produce la inflamación de la vena que se encuentra justo por debajo de la piel, por ello se le denomina tromboflebitis superficial. Puede aparecer tras una extracción sanguínea o en personas con riesgo de sufrir tromboflebitis.
Esta tromboflebitis, puede asociarse a enfermedades como, el cáncer abdominal y las trombosis venosas profundas, entre otras.
Los síntomas que suelen producirse en estos enfermos son:
− Enrojecimiento o inflamación de la piel a lo largo de una vena superficial.
− Enrojecimiento del tejido alrededor de una vena superficial.
− Calor en el tejido alrededor de una vena superficial.
− Sensibilidad o dolor a lo largo de una vena superficial, lo cual empeora cuando se aplica presión sobre la vena.
− Endurecimiento de una vena superficial (induración): la vena se siente como un cordón.
− Dolor en una extremidad.
Los riesgos para una tromboflebitis superficial incluyen los siguientes:
− Trastornos que implican aumento de la probabilidad de coagulación.
− Infección.
− Venas varicosas.
− Irritación química del área.
− Permanecer sentado o de pie por mucho tiempo.
− Mujeres en período de gestación.
− Uso de anticonceptivos orales.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
84
La tromboflebitis superficial puede estar asociada a niveles sanguíneos disminuidos, en cuanto a la presencia de antitrombina III, proteína C o proteína S, por lo tanto, habrá que realizar las correspondientes pruebas diagnósticas, además si se sospecha infección en la zona, habrá que realizar un hemocultivo.
- Tromboflebitis venosa profunda
Es originada por la presencia de un coágulo sanguíneo o trombo en una vena profunda (una vena que acompaña a una arteria), afectando principalmente a las venas de las extremidades inferiores, especialmente la parte inferior de la pierna y del muslo, produciéndose la formación de trombos en las venas más grandes, que puede desprenderse, pasando a formar un émbolo, el cual viajará por el torrente sanguíneo, pudiendo alojarse en diversos órganos como, pulmones, cerebro, corazón afectando gravemente al órgano.
Algunos de los riesgos para la producción de esta tromboflebitis son:
− Permanecer sentado, reposar en cama o estar inmovilizado por mucho tiempo.
− Tener antecedentes de policitemia vera, tumores malignos o hipercoagulabilidad.
− Cirugía o trauma reciente.
− Tener más de 60 años (aunque en realidad se puede producir a cualquier edad).
− Fracturas.
− Haber dado a luz en los últimos 6 meses.
− Uso de medicamentos tales como estrógenos y píldoras anticonceptivas.
La sintomatología que presenta este tipo de personas, se da sólo en una de las extremidades inferiores y son:
− Sensibilidad.
− Aumento de la temperatura.
− Cambios en el color de la piel o enrojecimiento.
− Dolor.
− Inflamación.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
85
Muchas de las causas hereditarias y adquiridas de la hipercoagulabilidad (tendencia a la coagulación) se pueden detectar por medio de exámenes de sangre, la antitrombina III, proteína C, proteína S, el factor V Leyden, anticoagulante lúpico y anticuerpos para anticardiolipina.
b) Enfermedades hepáticas
Las enfermedades hepáticas producidas como consecuencia de la presencia de anticuerpos antifosfolípidos, pueden afectar a pequeños o grandes vasos según donde tenga lugar la formación del trombo sanguíneo:
- Afectación de grandes vasos
Síndrome de Budd-Chiari
El síndrome de Budd-Chiari, es otra de las enfermedades que puede provocar el SAF y que está ligada a la trombosis venosa. Este síndrome fue descrito por primera vez en 1885 por Budd y en 1889 por Chiari. Este síndrome es un trastorno raro, generalmente causado por coágulos de sangre que obstruyen, parcial o completamente, el árbol venoso hepático en cualquier localización comprendida entre la vena eferente del lóbulo hepático y la entrada de la vena cava inferior en la aurícula derecha, por lo que se considera una condición grave que tiene múltiples etiologías, manifestaciones clínicas y patologías.
En general, la causa de la aparición del síndrome de Budd-Chiari no se conoce y suele ocurrir espontáneamente con mayor frecuencia, en mujeres jóvenes.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
86
Los síntomas del síndrome de Budd-Chiari pueden comenzar repentinamente y ser devastadores, pero, en general, se presentan gradualmente. A pesar de ello, están particularmente asociados a la administración de estrógenos, pero también después de la administración de quimioterapia para el tratamiento del cáncer y en enfermedades asociadas a un estado de hipercoagulabilidad de la sangre (síndrome antifosfolípido). Este síndrome está asociado también a malformaciones congénitas del hígado y al curso de algunas enfermedades hepáticas. En un 50% de los casos, la etiología del síndrome de Budd-Chiari permanece desconocida.
Clínicamente, se distinguen una forma aguda y otra crónica, que se distinguen por la gravedad y manifestaciones clínicas que presentan:
La forma aguda o fulminante, se distingue por tener un curso breve y relativamente grave, además de presentar un comienzo brusco. Es la forma más frecuente que se suele dar en este tipo de enfermedad. Se inicia con dolor abdominal, vómitos, hepatoesplenomegalia dolorosa (aumento del hígado y bazo), ictericia (coloración amarillenta de la piel, por liberación de bilirrubina) y ascitis (líquido en la cavidad abdominal). Si la obstrucción es completa, se observan rápidamente síntomas de insuficiencia hepática aguda grave con encefalopatía y muerte. Esta forma, es mucho más frecuente, observándose en el 90% de los casos positivos.
En la forma crónica, tiene un curso prolongado por mucho tiempo y el cuadro clínico se desarrolla de un modo progresivo, apareciendo un síndrome de hipertensión portal (aumento de la presión venosa en la circulación del sistema portal, que lleva la sangre de los distintos tejidos al hígado, por compresión u obstrucción de los sistemas vasculares portal o hepático) con esplenomegalia, ascitis, varices esofágicas y hepatomegalia dolorosa sin reflujo hepatoyugular que puede ser difícil de diferenciar de una cirrosis hepática.
En este síndrome, el hígado se llena de sangre y se vuelve doloroso. El líquido se filtra desde la superficie del hígado aumentado de volumen hacia la cavidad abdominal. Puede producirse dolor abdominal y una leve ictericia. La acumulación de sangre en el hígado aumenta la presión en la vena porta, aunque las consecuencias de esto, tales como las hemorragias de las varices en el esófago, pueden tardar semanas o meses en aparecer. Al cabo de varios meses, suelen presentarse ictericia, fiebre y otros síntomas de insuficiencia hepática. A veces los coágulos se agrandan tanto que obstruyen la parte inferior de la vena cava inferior, esta oclusión causa un aumento considerable del volumen de las piernas y del abdomen.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
87
Los síntomas característicos son las claves principales para el diagnóstico, junto con resonancias magnéticas, radiografías de las venas, biopsia de hígado y ecografías, ya que las pruebas de rutina realizadas mediante análisis hematológico como son determinaciones de rutina de las transaminasas séricas, la bilirrubina, las fosfatasas alcalinas y la protrombina suelen ser (en la mayoría de los casos) normales o mínimamente alterados, por lo que no tienen ningún significado para el establecimiento del diagnóstico. Además, la albúmina sérica, que se observa usualmente por debajo de lo normal, debido a la ascitis no puede ser interpretada como un signo de un fallo hepático.
Si el diagnóstico es temprano, el tratamiento de esta enfermedad se podrá realizar con fármacos capaces de disolver los trombos: si la vena se ha estrechado más bien que obstruido, se pueden usar fármacos anticoagulantes (que evitan los coágulos) o trombolíticos (que disuelven los coágulos) aunque en la mayoría de los casos, el tratamiento de elección es el quirúrgico, que se basa en la redistribución del flujo sanguíneo. El trasplante hepático sólo está indicado si existe insuficiencia hepática grave. Si no se trata, evoluciona hasta lograr la muerte del enfermo.
Este tratamiento se ha estructurado en cuatro fases distintas:
1. Descompresión del territorio obstruido. El objetivo de estas técnicas es controlar el síndrome de hipertensión portal y disminuir la progresión de las lesiones hepáticas, mediante tratamientos fibrinolíticos, técnicas de repermeabilización y técnicas de descompresión.
2. Trasplante hepático. El trasplante hepático debe reservarse para pacientes cuya presentación sea en forma de fracaso hepático fulminante, en los cuales la posibilidad de tratamiento médico o cirugía derivativa presentan escasas posibilidades de éxito.
3. Tratamiento de la enfermedad subyacente. El diagnóstico de síndrome de Budd-Chiari requiere una cuidadosa evaluación hematológica destinada a cumplir dos fines fundamentales:
• El primer fin que deben cumplir es la evaluación de la existencia de trastornos hematológicos como enfermedad de base, evaluándose concretamente la presencia de hemoglobinuria paroxística nocturna, policitemia vera, la detección de anticoagulante lúpico y trastornos como la presencia de Factor V Leiden.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
88
• Además, también se evaluará e instaurará el tratamiento anticoagulante y el tratamiento específico de la enfermedad hematológica si la hubiera. El tratamiento agresivo de cualquier enfermedad identificada es esencial para evitar la aparición de enfermedad recurrente, así como manifestaciones trombóticas a otros niveles.
4. Tratamiento de base. La eficacia del tratamiento médico incluyendo el tratamiento de la ascitis y el de la enfermedad de base, ha sido recientemente evaluada, sugiriendo un papel relevante en pacientes respondedores a diuréticos y sin hemorragia por varices.
- Afectación de pequeños vasos
Hepatomegalia
La hepatomegalia, es un aumento del tamaño normal del hígado, que puede palparse por debajo del reborde costal derecho debido a este aumento de tamaño, ya que, en un hígado normal, el borde inferior del hígado llega hasta el borde inferior de las costillas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
89
La hepatomegalia puede ser producto de:
MECANISMO CONDICIÓN
INFLAMACIÓN
- Hepatitis viral
- Síndrome de Torch
- Hepatitis neonatal
- Absceso Hepático
- Colestasia intrahepática
- Colestasia extrahepática
- Drogas (Ioniazida, metildopa, sulfonamidas, propiltiouracilo, etc.)
CONGESTIÓN
- Falla cardíaca congestiva
- Taponamiento pericárdico
- Síndrome de Budd-Chiari
- Obstrucción de la vena cava
- Enfermedad Veno-oclusiva
ENFERMEDADES DE DEPÓSITO
- Glicogenosis (Pompe, von Gierke, etc)
- Lipoidosis (Gaucher, Neimann-Pick, etc)
- Mucopolisacaridosis
- Gangliosidosis
- Amiloidosis
- Deficiencia de alfa-1-antitripsina
- Enfermedad de Wilson
- Depósito de hierro
- Hígado graso secundario a desnutrición, obesidad, fibrosis quística, diabetes mellitus, galactosemia, síndrome de Reye, drogas (etanol, esteroides, tetraciclina, ácido valproico, antineoplásicos, etc.)

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
90
MECANISMO CONDICIÓN
INFILTRACIÓN
- Leucemias
- Linfomas
- Histiocitosis
- Metástasis
- Eritroblastosis fetal
HIPERPLASIA DEL SISTEMA RETÍCULO ENDOTELIAL
- Infección extrínseca del hígado
- Septicemia
- Hepatitis granulomatosas (sarcoidosis, tuberculosis, etc)
- Tumores extrínsecos
- Hipervitaminosis A
TUMORES INTRÍNSECOS
- Hemangioblastoma
- Hamartomas
- Hepatoblastoma
FIBROSIS
- Cirrosis (infecciosa, drogas, idiopática)
- Enfermedad hepática poliquística
- Fibrosis hepática congénita
Los principales síntomas observados en pacientes con hepatomegalia son:
− Dolor e inflamación de la parte superior derecha del abdomen.
− Ictericia (piel y ojos amarillentos).
− Orina de color más oscuro que lo normal.
− Materia fecal de color claro.
Las pruebas de laboratorio que se deben realizar a este tipo de pacientes, son las pruebas de la función hepática, que son pruebas bioquímicas que entregan información sobre aspectos específicos de la función hepática, pero sólo el conjunto de ellos permite tener una visión global del proceso

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
91
fisiopatológico. La mayoría de estas pruebas son una combinación de algunas pruebas específicas que corresponden a diferentes funciones que desempeña el hígado:
Ninguna de estas pruebas da información sobre la etiología de la hepatomegalia, más bien aportan información sobre funciones específicas del hígado. Sin embargo, la combinación de estos test puede sugerir algunos patrones fisiopatológicos.
La presencia de fosfatasas alcalinas elevadas por encima 3 veces el valor normal, con transaminasas sobre las 500 μl/dl, GGT y colesterol muy elevados, con mejoría del tiempo de protrombina después de la administración de vitamina K parenteral, sugiere un proceso obstructivo. Por otra parte, la presencia de elevación moderada de las fosfatasas alcalinas (< 3 veces el
PRUEBAS DE LA FUNCIÓN HEPÁTICA
FUNCIÓN PRUEBA
Marcadores de necrosis celular
- Aspartato aminotransferasa o "transaminasa glutámica oxaloacética" (SGOT)
- Alanino aminotransferasa o "transaminasa glutámico pirúvica" (SGPT)
- Deshidrogenasa láctica (LDH)
FUNCIÓN PRUEBA
Marcadores de transporte de iones orgánicos
- Bilirrubina
- Ácidos biliares
Marcadores de síntesis proteica
- Albúmina sérica
- Lipoproteínas
Factores de coagulación
Marcadores de otras funciones metabólicas
- Glucosa
- Amonio
- Colesterol

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
92
valor normal), pero con transaminasas sobre las 1000 μl /dl, con GGT normal o moderadamente elevada, colesterol bajo, y tiempo de protrombina persistentemente prolongado a pesar de terapia con vitamina K, sugiere la presencia de un proceso hepatocelular.
c) Glándulas suprarrenales
- Enfermedad de Addison.
La definición original de esta enfermedad, fue definida por Addison, que la describió como: "languidez y debilidad general, actividad hipocinética del corazón, irritabilidad gástrica y un cambio peculiar de la coloración de la piel".
La enfermedad de Addison o insuficiencia suprarrenal primaria, se produce como consecuencia de:
− El uso de corticosteroides como tratamiento (por ejemplo prednisona) que causa una disminución en la producción de los corticosteroides naturales por parte de las glándulas adrenales.
− Ciertos medicamentos usados para tratar infecciones por hongos pueden bloquear la producción de corticosteroides en las glándulas adrenales.
Las glándulas suprarrenales, son pequeños órganos que secretan hormonas y que se encuentran localizados en la parte superior de cada riñón. Estas glándulas están formadas por una parte externa (corteza) y una parte interna (médula).
La corteza produce tres tipos de hormonas: hormonas sexuales, hormonas glucocorticoides y hormonas mineralocorticoides.
− Las hormonas sexuales: andrógenos (hombres) y estrógenos (mujeres) afectan el desarrollo sexual y la reproducción.
− Las hormonas glucocorticoides (como el cortisol): mantienen la regulación de la glucosa (azúcar), suprimen la respuesta inmune y ayudan al cuerpo a responder al estrés.
− Las hormonas mineralocorticoides (como la aldosterona): regulan el equilibrio entre el sodio y el potasio.
La atrofia de las glándulas suprarrenales es de causa desconocida y representa, aproximadamente entre el 60 al 70% de los casos de Enfermedad de Addison. Esta es una enfermedad muy rara y se puede presentar a cualquier edad y con igual prevalencia en ambos sexos, aunque es entre los 30 y los 40 años cuando lo hace con mayor frecuencia.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
93
Para que aparezca la Enfermedad de Addison debe destruirse más del 90% de las glándulas suprarrenales de forma bilateral. Entre las causas de la destrucción de las suprarrenales se destaca:
− Infecciones: suelen ser infecciones causadas por virus, como el SIDA o bacterias, como las productoras de la meningitis meningocócica o la tuberculosis.
− Atrofia idiopática: Se piensa que son mecanismos autoinmunitarios, en los que se produce una elevación de la concentración de ácidos grasos de cadenas largas que son las causantes de provocar la insuficiencia suprarrenal
− Hemorragias bilaterales de las glándulas suprarrenales: Ocurren sobre todo en pacientes tratados con anticoagulantes, en los que tienen anticoagulantes circulantes y en los estados de hipercoagulabilidad, como ocurre en el síndrome antifosfolípido.
− Invasión de las glándulas suprarrenales: Ocurre en las metástasis tumorales, sobre todo en la amiloidosis y sarcoidosis.
− Intervención quirúrgica en la que se produce la extirpación de las glándulas suprarrenales.
− Fallo metabólico de la producción hormonal: que provoca una hiperplasia suprarrenal congénita, activación de inhibidores enzimáticos (pueden causar o potenciar una insuficiencia suprarrenal) y producción de agentes citotóxicos, como el mitotano, que es un principio activo adrenal citotóxico.
Para el diagnóstico de la enfermedad se requieren numerosas pruebas bioquímicas, para observar los niveles de los distintos componentes sanguíneos:
− Hiponatremia: Los niveles séricos bajos de sodio se debe a su pérdida por la orina por déficit de aldosterona y al desplazamiento del sodio hacia el compartimento intracelular. Esta pérdida de sodio extravascular reduce el volumen plasmático y acentúa la hipotensión.
− Hiperpotasemia: Aumento de los niveles séricos de potasio. Se debe a los efectos combinados del déficit de aldosterona, la reducción del filtrado glomerular y la acidosis.
− Hipocortisolemia: Los niveles de cortisol y aldosterona son bajos y no aumentan con la administración de ACTH.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
94
− Hipercalcemia: Aumento de los niveles séricos de calcio. Ocurre en un 10-20% de los pacientes de causa desconocida.
− Hemograma: Puede haber anemia normocítica, linfocitosis relativa y eosinofilia moderada.
− Prueba de estimulación de ACTH: Es la prueba principal que nos confirma el diagnóstico de insuficiencia suprarrenal, al evaluar la capacidad de las suprarrenales para producir esteroides, que suelen estar ausentes o disminuidos tanto en sangre como en orina tras la estimulación de ACTH.
− Determinación de la ACTH: En la insuficiencia suprarrenal primaria o Enfermedad de Addison, la ACTH y sus péptidos afines, están elevados en plasma ante la pérdida del mecanismo de retroalimentación del eje hipotálamo-hipofisario-suprarrenal.
d) Pulmones
- Tromboembolismo pulmonar
El tromboembolismo pulmonar, es el resultado de la obstrucción de la circulación de un vaso sanguíneo que desemboca en el pulmón, por un émbolo procedente, en un 95% de los casos del sistema venoso profundo de las extremidades inferiores (grandes venas proximales) y en menor frecuencia de las venas de las extremidades superiores, renales o uterinas, puede desprenderse formando un émbolo que podrá llegar hasta los pulmones, constituyendo una importante causa de muerte. Un 30% son mortales en ausencia de diagnóstico y tratamiento.
La enfermedad tromboembólica venosa, engloba a la trombosis venosa profunda y su consecuencia más grave la del tromboembolismo pulmonar. El tromboembolismo pulmonar es un problema de salud muy importante ya que:
− Es la tercera causa de muerte en hospitales.
− Tiene una incidencia de 10 casos por cada 100.000 habitantes y una prevalencia en hospitalizados del 1%.
− Sin tratamiento tiene una mortalidad del 30%.
− Es de difícil diagnóstico, sólo el 30% de los tromboembolismos pulmonares con un mal desenlace se diagnostican en vida.
− A largo plazo produce hipertensión arterial.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
95
Las personas propensas a padecer un tromboembolismo pulmonar son:
− Personas mayores de 40 años.
− Enfermos de cáncer.
− Infarto de miocardio, insuficiencia cardiaca congestiva e insuficiencia venosa crónica.
− Personas con problemas genéticos de la coagulación.
− Sobrepeso.
− Permanecer en reposo durante un período de tiempo más o menos largo (más de cuatro días).
− Embarazo.
− Accidentes cerebrovasculares.
− Padecer estados de hipercoagulabilidad primaria.
− Consumo de anticonceptivos y terapia estrogénica.
Fracturas pelvianas y de los miembros inferiores, debido a la inmovilización a la que obligan.
Los síntomas son inespecíficos, su intensidad depende del grado de oclusión del lecho vascular pulmonar y de la reserva cardiorrespiratoria previa del paciente, es decir dependen del tamaño del trombo, mediante el cual cada enfermo presentará unos u otros síntomas:
Si el trombo es pequeño:
- Dificultad respiratoria.
- Dolor torácico súbito.
- Tos y esputo con algo de sangre.
- Sensación de malestar general.
- Fiebre y malestar, con sensación de ahogo.
Si el trombo es de mayor tamaño puede ser letal para el enfermo presentando:
- Síntomas que aparecen de manera súbita.
- Palidez y sudor frío.
- Pérdida del conocimiento.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
96
Las pruebas de laboratorio realizadas para el diagnóstico de la enfermedad son pruebas secundarias, en las que se produce una anomalía de los valores normales:
- Leucocitosis moderada con desviación a la izquierda.
- Aumento de productos de degradación del fibrinógeno.
- Aumento del dímero-D en plasma: el dímero-D, se presenta en sangre tras la existencia de fibrinólisis, sirve como marcador de la presencia de trombos endovasculares.
- Aumento de la LDH.
- Gasometría arterial: hipoxemia con hipocapnia y alcalosis respiratoria (30% de los casos la PO2 es normal). Aumento de la diferencia alveolo-arterial de O2.
e) Manifestaciones cutáneas
Las manifestaciones cutáneas asociadas al síndrome antifosfolípido son:
- Lívedo reticularis
El Livedo reticularis es una enfermedad o desorden, en el cual los vasos sanguíneos se contraen, produciendo un estancamiento de sangre en los capilares superficiales dilatados y vénulas, lo que implica cambios patológicos en los grandes vasos profundos, inflamación generalmente de pies y brazos y la aparición de un punteado cianótico (amoratado) persistente en la piel, en grandes áreas de pies y brazos (especialmente) con un aspecto reticular típico que puede afectar a cualquier región de las extremidades y del tronco, pudiéndose acentuar con la exposición al frío.
No es una entidad diagnóstica por sí misma sino un patrón reaccional, en el cual se ha reportado su asociación con diversas enfermedades del tejido conectivo, tales como dermatomiositis, esclerodermia, artritis reumatoidea, fiebre reumática y fundamentalmente con el lupus eritematoso sistémico, el cual, por ser una enfermedad multisistémica, es capaz de producir una gran
Lívedo reticularis en glúteos y pie.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
97
variedad de manifestaciones cutáneas tanto específicas como inespecíficas, también los pacientes con el síndrome de anticuerpos anticardiolipina presentan Livedo reticularis extensa y trombosis arteriales y venosas recidivantes que afectan a varios sistemas orgánicos.
No se conocen sus causas exactas, aunque se cree, que es debido a espasmos de los vasos sanguíneos, producidos con mayor frecuencia en mujeres y sucede después de los treinta años.
- Úlceras en miembros inferiores
Las úlceras de miembros inferiores, son una ruptura de la piel consistente en la pérdida de toda la epidermis y toda o parte de la dermis, comprometiendo también el tejido celular subcutáneo y músculo con diversos grados de profundidad.
Aproximadamente el 70% de las úlceras de la pierna son venosas, siendo su ubicación en la cara interna de la misma, en el tercio inferior y/o medio, de bordes irregulares y fondo sucio. No son dolorosas, al no ser que esta, esté infectada.
Las úlceras venosas ocurren como un estado final del proceso de hipoxia y malnutrición cutánea.
Úlcera venosa trófica en paciente con insuficiencia venosa.
Las paredes de las venas son más delgadas que las de las arterias y la capa media muscular es mucho más débil. Las venas a diferencia de las arterias poseen válvulas semilunares que impiden que el reflujo sanguíneo, retorne de nuevo al corazón, este retorno de la sangre hacia el corazón, sólo se produce cuando la sangre es dirigida desde el sistema venoso superficial, al profundo a través del sistema de comunicantes, favorecido por el vaciamiento por presión y la contracción de los músculos de la pantorrilla, de este modo es impulsada hacia los muslos y posteriormente al abdomen y al corazón por lo que cualquier anomalía de estos mecanismos produciría un incremento en la presión venosa capilar desarrollando una serie de procesos que dará lugar a la

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
98
fibrosis del tejido circundante y en consecuencia una alta probabilidad de desarrollar ulceras venosas. Esto puede ser debido a:
− El incremento persistente de la presión venosa capilar, tiene como consecuencia la liberación de fibrinógeno, proteínas, glóbulos rojos y suero desde el espacio intravascular hacia la dermis circundante, originando un edema e inflamación, que con el tiempo este proceso lleva a la formación de un anillo fibroso pericapilar que impide el intercambio de oxígeno y nutrientes en la dermis, disminuyendo además la llegada de factores de crecimiento al tejido afectado, produciéndose así, hipoxia tisular que lleva a fibrosis y posteriormente a una ulceración.
− El incremento de la presión venosa capilar produce daños en el endotelio vascular, produciéndose una activación de leucocitos, que a su vez estimulan la liberación de enzimas proteolíticas y radicales libres, los cuales se depositan en la dermis circundante a través de los poros endoteliales dilatados los cuales producirán daños en el tejido produciéndose la fibrosis y como consecuencia la ulceración.
Los síntomas comunes que presentan los pacientes con insuficiencia venosa, son:
− Ardor.
− Prurito.
− Dolor pulsátil.
− Calambres.
− Dolor sordo.
− Pesadez.
− Agitación.
− Cansancio en los miembros inferiores.
Estos síntomas suelen ser más molestos al principio de la enfermedad que en los estados intermedios, empeorando en las fases avanzadas.
La insuficiencia venosa de miembros inferiores es más frecuente en mujeres que en hombres, con una relación mujeres: hombres de 4:1, dándose

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
99
sobre todo en mujeres mayores de 50 años y especialmente en aquellas con sobrepeso.
Las úlceras venosas pueden volverse crónicas, pudiendo generar gran morbilidad e incluso mortalidad, si no se tratan en forma adecuada, lo que conllevaría a la aparición de osteomielitis, en pacientes que presentan úlceras profundas, carcinoma escamocelular en úlceras venosas de muchos años de evolución, estas úlceras se denominan úlceras de Majorlin, en cuyo caso son de mal pronóstico pues el carcinoma escamocelular allí generado, es más agresivo y con mayor potencial metastásico.
En los tejidos afectados por el aumento de la presión venosa capilar se producen una serie de signos clínicos debido a la extravasación de sustancias desde el espacio intravascular al tejido subcutáneo y al pobre intercambio de oxígeno y nutrientes, originando:
− Edemas: Se produce como consecuencia de la acumulación de líquido en el espacio intersticial, debido al aumento sostenido en la presión venosa capilar. Pero el edema también puede ser producido por el exceso de proteínas en el intersticio, debido al mal funcionamiento de los vasos linfáticos.
− Eczema gravitacional: Se define como, una placa eritematosa y descamativa, que se desarrolla en las piernas y es secundario a una hipertensión venosa, que lleva a hipoxia tisular por formación de barreras fibrosas pericapilares.
− Atrofia blanca o vasculitis livedoide: No se sabe cuáles son las causas exactas que lo producen, aunque se piensa que puede ser debido a alteraciones en la cascada de la coagulación, de manera que se presentan trombos y depósitos de fibrina alrededor de las vénulas (vasos pequeños).
− Dilataciones venosas o venas varicosas: Son inflamaciones de distinto grado, producidas en las venas, que se acompañan de otros cambios. Su presencia se debe a factores hormonales, por lo que es más frecuente en mujeres de entre 30 a 40 años.
Su etiología puede ser primaria por defectos heredados de la pared de los vasos y/o del sistema valvular, o secundaria al síndrome postflebítico.
− Lipodermatoesclerosis: Es una enfermedad producida en las piernas, produciendo durezas, inflamación y pigmentación de la parte afectada, llevando a una malnutrición e hipoxia de las células afectadas.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
100
Aproximadamente el 10% de las úlceras son de tipo arterial o isquémico, debido a la falta de irrigación y suele ubicarse en el lado externo de la pierna, de bordes netos y su causa pueden ser la aterosclerosis de los vasos arteriales, o la hipertensión arterial. La úlcera arterial suele ser dolorosa, incrementándose durante el reposo o con la pierna elevada.
El consumo de cigarrillos, de alcohol y la diabetes son los principales desencadenantes de estas patologías, por tanto la prevención y la educación sanitaria son imprescindibles.
Muchas úlceras pueden ser de origen arterial y venosa, llamándose úlceras mixtas.
- Lesiones purpúricas
Las púrpuras son una extravasación de hematíes en la piel, originada por la rotura de pequeños vasos, dando lugar a lesiones, que se caracterizan porque al ejercer una presión sobre la zona, esta no se clarea completamente, sino que se observan, puntos o manchas. Si las manchas de púrpura, tienen un tamaño menor a dos milímetros se llamarán petequias y si miden más de un centímetro se les llama equimosis y pueden ser palpables o no, pudiéndose presentar en las membranas mucosas (como en el revestimiento de la boca) y en los órganos internos.
Las púrpuras pueden ser de dos tipos:
1. Púrpuras no trombocitopénicas: cuando el número de plaquetas está dentro de los valores normales y pueden ser debidos a:
− Cambios de la presión vaginal, durante el parto.
− Vasculitis, como la púrpura de Henoch-Schonlein (púrpura anafilactoide.).
− Citomegalovirus congénito.
− Síndrome de rubéola congénita (cambios en el bebé que pueden ocurrir cuando una mujer embarazada tiene rubéola).
− Disfunción plaquetaria inducida por medicamentos (algunos medicamentos pueden afectar la acción de las plaquetas).
− Púrpura senil (los vasos sanguíneos se vuelven más frágiles a medida que el individuo envejece).

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
101
2. Púrpuras trombocitopénicas: si el número de plaquetas se encuentra por debajo de los valores de referencia. Como ocurre en:
− Púrpura trombocitopénica idiopática.
− Trombocitopenia neonatal inmune (una enfermedad que se puede presentar en niños cuyas madres sufren de púrpura trombocitopénica idiopática).
− Consumo de plaquetas en hemangioma.
− Trombocitopenia inducida por medicamentos (algunos medicamentos pueden impedir la formación de plaquetas).
− Meningococemia (una infección causada por la bacteria meningococo).
− Valores de referencia plaquetarios: de 150.000 a 400.000 células por mm3.
f) Oculares
Las patologías oculares, asociadas al síndrome antifosfolípido son la oclusión de las venas retinianas por trombosis en las que pueden aparecer otras patologías secundarias asociadas a esta, como son la isquemia o el infarto de retina, lo cual está relacionado con la presencia de anticuerpos antifosfolípidos.
- Trombosis de las venas retinianas
La trombosis de las venas retinianas, se debe a la obstrucción de la arteria central de la retina (la principal arteria de la retina) por un trombo, que provoca una severa pérdida de visión más o menos grave, que en muchos casos es irreversible.
Las oclusiones venosas retinianas son la segunda patología vascular de la retina tras la diabetes mellitus y aparece fundamentalmente en personas de más de 60 años. En muchos casos se origina una pérdida significativa de visión en el ojo afectado.
En el fondo del ojo podemos distinguir los signos propios de estasis venosa, con aparición de edema de papila, venas dilatadas y tortuosas con profusión de hemorragias en la retina.
Se presentan abundantes hemorragias que rodean un disco óptico desde el que parten venas ingurgitadas y muy tortuosas. Esta trombosis puede complicarse, produciéndose la aparición tardía de un glaucoma neovascular.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
102
Normalmente este tipo de glaucoma tardaba unos 3 meses y medio en aparecer que es el tiempo que tardaba en desarrollarse las neovascularizaciones secundarias a la isquemia retiniana, por eso se le llamaba glaucoma de los 100 días.
El tratamiento de este proceso trombótico se basa en la terapia anticoagulante y en el tratamiento del proceso sistémico causante del mismo.
La aparición de una Trombosis Venenosa Profunda se debe a una oclusión venenosa espontánea, sin ningún determinante patológico, médico o quirúrgico aparente, existe una alta probabilidad de que la persona afectada pudiera albergar algún sustrato trombofílico.
2. Trombosis arterial
Afectan a las arterias produciéndose, oclusiones arteriales que constituyen otro hallazgo prominente del síndrome antifosfolípido, principalmente de los vasos intracraneales, originando accidentes isqúemicos transitorios o accidentes cerebrovasculares, que van desde lesiones isquémicas únicas, hasta infartos múltiples diseminados, traduciéndose esto por manifestaciones demenciales o psiquiátricas. Las afecciones arteriales coronarias con producción de angina e infartos han sido descritas, puede existir afectación de las válvulas cardiacas especialmente la mitral, pudiendo ocasionar disfunciones valvulares, o eventos embólicos.
TROMBOSIS ARTERIAL
Parte del cuerpo afectada Enfermedad producida
Extremidades Gangrena
Lesiones cerebrales Isquemia cerebral
Enfermedades cardiovasculares
Grandes vasos: Trombosis de las arterias coronarias
Angina de pecho
Pequeños vasos: Miocardiopatía
Arritmias
Enfermedades renales Grandes vasos: Trombosis arterial renal
Pequeños vasos: Microangiopatía arterial
Enfermedades hepáticas Grandes vasos: Infarto hepático
Pequeños vasos: Hiperplasia nodular regenerativa

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
103
a) Extremidades
- Gangrena
La gangrena es la muerte de un órgano o tejido de una parte del cuerpo, producida mediante la evolución de una necrosis, dando color negruzco a la parte de piel afectada debido al insuficiente aporte de sangre. Cuando este suplemento sanguíneo es suprimido, los tejidos no adquieren suficiente oxígeno, entonces comienzan a morir. Si la gangrena se extiende ampliamente, puede causar un shock generalizado.
Gangrena por oclusión vascular arterial, en paciente con síndrome antifosfolípido
Se distinguen dos tipos de gangrenas:
1. Gangrena isquémica.
Se produce en la piel y tejidos blandos subyacentes, con mayor frecuencia en las extremidades inferiores debido a obstrucción arterioesclerótica. La necrosis se produce por la isquemia y sobre el tejido necrótico actúan secundariamente los gérmenes saprófitos de la piel. Según cuáles sean las condiciones del tejido comprometido, se produce una gangrena isquémica seca o húmeda:
1.1. Gangrena seca. Tiene lugar una evaporación del agua lo que produce rápidamente una desecación de la piel comprometida, que se transforma en una lámina acartonada, pardo negruzca y seca. El territorio comprometido queda bien demarcado, los gérmenes no penetran en la profundidad, no se produce intoxicación del organismo.
1.2. Gangrena húmeda. Especialmente cuando hay edema o la piel está húmeda, los gérmenes penetran en los tejidos subyacentes, donde proliferan y dan origen a un estado tóxico. El territorio comprometido es de color pardo verduzco y no queda bien delimitado.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
104
2. Gangrena infecciosa.
La gangrena es producida por gérmenes anaeróbicos, que actúan sobre tejidos ya desvitalizados generalmente por una inflamación. Esta forma de gangrena se observa en las vísceras, en que el territorio comprometido aparece reblandecido y a veces con burbujas de gas producido por los gérmenes. La gangrena infecciosa es altamente tóxica, se la encuentra como complicación de bronconeumonías o neumonías, apendicitis, colecistitis, metritis y otras inflamaciones.
La gangrena puede ocurrir cuando una parte del cuerpo pierde su suministro sanguíneo, lo cual puede suceder por ejemplo a raíz de una lesión o una infección. Una persona tiene un mayor riesgo de presentar gangrena si tiene:
− Diabetes.
− Enfermedades vasculares (como arterioesclerosis en los brazos o las piernas), ya que impiden el flujo sanguíneo a los tejidos.
− Infección tras una lesión o cirugía.
− Inmunosupresión (por ejemplo, por VIH o quimioterapia).
Los factores de riesgo que incrementan la posibilidad de adquirir una enfermedad o afección, son:
− Fumar.
− Consumo de alcohol.
− Lesión traumática, especialmente lesiones por aplastamiento.
− Heridas infecciosas después de una cirugía.
− Congelación.
− Quemaduras.
− Aterosclerosis.
− Diabetes.
− Enfermedad de Raynaud.
− Coágulos sanguíneos.
− Ruptura del apéndice.
− Hernia.
− Uso de drogas intravenosas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
105
La sintomatología que presentan este tipo de personas son:
− Decoloración azul o negra si la piel está afectada o roja o bronce si el área afectada está por debajo de la piel.
− Gases en los tejidos, bajo la piel.
− Sensación de malestar general.
− Pérdida de la sensibilidad que puede ocurrir después de dolor severo en el área.
− Secreción maloliente.
− Fiebre.
− Shock séptico.
Los exámenes llevados a cabo en el laboratorio para el posterior diagnóstico de la gangrena, son:
− Exámenes de sangre: un conteo sanguíneo completo puede mostrar un conteo alto de glóbulos blancos (GB).
− Examen microscópico de tejido para buscar células muertas.
− Cultivo de tejido o fluidos de las heridas para identificar la infección bacteriana.
El pronóstico depende de la parte del cuerpo afectada, la magnitud de la gangrena, su causa y la condición del paciente. El paciente puede morir si el tratamiento se demora, si el área afectada es extensa o si el paciente tiene otras condiciones médicas significativas.
b) Cerebro
- Isquemia cerebral
Esta enfermedad ocupa el tercer lugar en la morbilidad y mortalidad del adulto en países desarrollados, presentándose en el 80 % del total de las enfermedades cerebro vascular.
La isquemia cerebral, es una enfermedad cerebro-vascular que tienen lugar en los vasos sanguíneos, en los cuales se produce una llegada deficitaria de sangre y por lo tanto de oxígeno, a una determinada área del cerebro, produciéndose una lesión más o menos importante según la localización y el tamaño de la zona afectada y del tiempo durante el cual el paciente permanece sin el tratamiento adecuado.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
106
La isquemia cerebral puede producirse por varios factores:
− Por oclusión total "in situ" de una arteria (trombosis) en general debida a una lesión arteriosclerótica.
− Por estenosis (estrechamientos) arteriales de origen hereditario o debidas a causas diversas.
− Por embolia, generalmente de origen cardíaco, más frecuente en pacientes con valvulopatías o arritmias.
Como consecuencia de la falta de oxígeno, por el escaso aporte circulatorio, se podría producir un infarto cerebral, lo que originaría un déficit neurológico de más de 24 h de duración que es expresión de una necrosis tisular.
El mecanismo patofisiológico más frecuente es la oclusión de las arterias
cerebrales, producidas como consecuencia de una trombosis o embolia. La arterioesclerosis puede provocar la oclusión de las arterias cerebrales, impidiendo la correcta circulación de la sangre hasta el cerebro, debido a la formación de la placa de ateroma (depósito de lípidos y material fibroso), las cuales pueden crecer obstruyendo aún más la luz de la arteria, que si se produjese su desprendimiento daría lugar a una embolia o servir de iniciador para la formación de una agregación plaquetaria. Además de la arterioesclerosis hay otras enfermedades que pueden provocar una isquemia aunque son menos frecuentes, afectando sobre todo a personas jóvenes
Las enfermedades cardíacas pueden también ocasionar isquemia cerebral bien por la incapacidad del corazón de bombear suficiente sangre (fallo cardíaco, hipotensión, hipovolemia) o bien por la formación de trombos que pueden viajar hasta las arterias del cerebro causando la obstrucción de dichas arterias (valvulopatías, arritmias).
Oclusión de arteria cerebral, lo que impide el paso de sangre hasta las diferentes zonas del cerebro

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
107
La hipertensión arterial, la diabetes mellitus, el alcoholismo, el hábito de fumar, el sedentarismo, el colesterol alto, las tensiones emocionales mantenidas (estrés), la edad, cuanto mayor sea la persona mayor será la incidencia a padecer esta enfermedad y la etnia, son los factores de riesgo fundamentales para que pueda darse esta enfermedad, además de todo ello también repercuten los factores genéticos, ya que en personas con antecedentes familiares de infartos de corazón o afecciones cerebrovasculares son las que tienen mayor riesgo a padecer una isquemia cerebral.
Los principales síntomas clínicos que se producen ante la presencia de una isquemia cerebral son, trastornos del lenguaje, se origina también una pérdida súbita de la fuerza muscular en una u otra parte del cuerpo y se produce una desviación de la boca, fundamentalmente.
c) Enfermedades cardiovasculares
En el síndrome antifosfolípido pueden darse lesiones cardiovasculares, siendo las más frecuentes, las trombosis de las arterias coronarias.
- Afectación de grandes vasos
Trombosis de las arterias coronarias
La trombosis de las arterias coronarias se produce como consecuencia de la formación de un trombo o coágulo sanguíneo, en una de las tres arterias coronarias, encargadas de proporcionar la sangre y el oxígeno necesario al corazón, lo que conlleva a la interrupción del flujo sanguíneo hasta el músculo cardiaco, lo que provocaría infarto de miocardio y la de la válvula mitral, seguida de la arteria aorta, lo que suele ser asintomático, aunque en algunos pacientes puede producir síntomas de insuficiencia valvular.
Se suele originar mediante la ruptura de una placa arterioesclerótica, situada en las arterias coronarias, produciéndose:
- Dolor repentino detrás del esternón (el hueso del pecho), o en la parte delantera izquierda del pecho.
- Una posible extensión del dolor hacia el brazo izquierdo.
- El dolor también puede extenderse hacia las manos, la mandíbula, el oído, el estómago o el brazo derecho.
- Una sensación de opresión en, o alrededor, de la garganta.
- Pueden producirse dificultades respiratorias graves y esporádicas con, o sin dolor.
- Desfallecimiento repentino o mareo fuerte, a menudo, acompañado de dolor.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
108
Si una determinada zona del corazón no recibe el suficiente aporte de oxígeno, este dejará de funcionar adecuadamente, hasta que se produzca la disolución del coágulo.
Angina de pecho
La angina se caracteriza por una sensación de dolor, malestar, constricción o presión incómoda en el centro del pecho. Estos síntomas generalmente duran unos pocos minutos. La sensación asociada con la angina puede también extenderse a los brazos, espalda, cuello, mandíbula o estómago. La angina es originada por un cúmulo de la placa aterómica en las arterias que van hacia el corazón.
Los síntomas de angina son temporales y muy similares a los de un ataque de corazón y se presentan cuando el suministro de sangre y oxígeno al corazón es insuficiente y puede deberse a la realización de actividades que requieren de un esfuerzo físico, como subir escaleras o correr, situaciones de alto estrés emocional o exposición a temperaturas muy cálidas o frías.
En estas situaciones, la frecuencia cardíaca y la presión arterial aumentan, y el corazón requiere de un mayor suministro de sangre y oxígeno. Pero en alguien que padece de angina, las arterias coronarias no pueden satisfacer completamente el incremento en la demanda por parte del corazón.
Hay dos tipos de anginas, angina estable y angina inestable:
1. Angina estable.
En las personas con angina estable, los síntomas generalmente se presentan de manera previsible, porque se presentan al hacer actividad física o durante momentos de estrés o ansiedad.
2. Angina inestable.
Los síntomas de una angina inestable, se presentan sin aviso, y pueden ocurrir incluso en momentos de reposo. Durante un episodio de angina inestable, la sensación de malestar puede ser más aguda y prolongada que en alguien con angina estable.
- Afectación de pequeños vasos
Miocardiopatía
La miocardiopatía es una enfermedad del músculo cardíaco que se caracteriza por inflamación del músculo cardíaco y una reducción en la capacidad de bombeo del corazón. En algunos casos, se produce una alteración del ritmo cardíaco, lo que lleva a latidos irregulares o arritmias.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
109
Esta enfermedad suele presentarse en personas jóvenes y constituye una de las principales causas de trasplante de corazón ya que es una enfermedad progresiva y que suele producirse un rápido empeoramiento, pudiendo estar asociado a otras enfermedades.
Su origen puede ser debido a infecciones virales que infectan el corazón, estas son una de las principales causas de la miocardiopatía. En algunos casos, la miocardiopatía es el resultado de otra enfermedad o de su tratamiento, como la cardiopatía congénita (de nacimiento) compleja, las deficiencias de nutrición, los ritmos cardíacos rápidos e incontrolables o ciertos tipos de quimioterapia para cánceres infantiles. A veces, la miocardiopatía se puede asociar a una anomalía genética, en cambio, otras veces, se desconoce la causa.
Hay tres tipos de miocardiopatía, que son, la miocardiopatía dilatada o congestiva, miocardiopatía hipertrófica y miocardiopatía restrictiva (esta es menos común que las demás):
1. Miocardiopatía dilatada o congestiva.
Esta, es la forma más común de miocardiopatía, en ella hay un agrandamiento y dilatación de la cavidad del corazón (dilatación cardíaca) lo que produce gran debilidad del corazón y una incapacidad para bombear sangre suficientemente. La sangre fluye con menos velocidad a través de un corazón dilatado, de modo que coágulos de sangre se pueden formar fácilmente.
En la mayoría de los casos, el enfermo desarrolla una insuficiencia cardiaca, anormalidades del ritmo cardiaco o trastornos de la conducción eléctrica del corazón, además de algunas infecciones que pueden causar este tipo de miocardiopatía. También se sabe que el contacto con toxinas o con drogas terapéuticas muy poderosas (como ciertos tipos de quimioterapia contra el cáncer) causan la miocardiopatía dilatada.
Debido a que el músculo cardíaco es débil e incapaz de bombear suficiente sangre para suplir las demandas del organismo, éste intenta preservar el flujo sanguíneo a los órganos esenciales como el cerebro y los riñones reduciendo el flujo sanguíneo a las restantes zonas del cuerpo, como la piel y los músculos.
Entre los síntomas más frecuentes de la miocardiopatía dilatada, se pueden incluir:
− Piel pálida.
− Piel fría y sudorosa.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
110
− Frecuencia cardíaca rápida.
− Frecuencia respiratoria rápida.
− Dificultad al respirar.
− Fatiga.
− Irritabilidad.
− Dolor en el pecho.
− Poco apetito.
− Crecimiento lento.
2. Miocardiopatía hipertrófica.
Esta afección se caracteriza por el agrandamiento o hipertrofia de la musculatura del ventrículo izquierdo del corazón, es decir, la pared (septo) entre los dos ventrículos (cámaras de bombeo) se agranda y obstruye el flujo de sangre proveniente del ventrículo izquierdo.
El engrosamiento del músculo o del tabique también puede afectar una de las valvas de la válvula mitral, que separa la aurícula y el ventrículo izquierdo. La valva de la válvula permite que la sangre se escape y retroceda desde el ventrículo izquierdo a la aurícula izquierda en vez de avanzar hacia el cuerpo.
En más de la mitad de los casos, la enfermedad es hereditaria y esta afección se presenta más comúnmente en adultos jóvenes.
Los síntomas incluyen:
− Dificultad para respirar con el esfuerzo físico.
− Mareos.
− Desmayos.
− Angina de pecho.
− Arritmias cardiacas, que pueden llevar a la muerte súbita.
3. Miocardiopatía restrictiva.
Esta es la forma menos común de miocardiopatía, caracterizada por la rigidez del miocardio (la musculatura del corazón) de los ventrículos, la cual resulta en la incapacidad de los ventrículos para llenarse adecuadamente de

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
111
sangre entre los latidos del corazón. Por lo general, este tipo de miocardiopatía se debe a alguna otra enfermedad.
Los síntomas incluyen:
− Fatiga.
− Hinchazón de las manos y los pies.
− Dificultad para respirar con esfuerzo físico.
Arritmias
Las arritmias son alteraciones del ritmo cardíaco. Cada latido es impulsado por una señal eléctrica generada desde el nódulo sinusal, situado en el lado derecho del corazón. Si existe alguna anormalidad en la secuencia de señales eléctricas que llegan al corazón, se produce una arritmia.
Las causas que provocan las arritmias, son en muchos casos desconocidas, aunque hay numerosos factores contribuyentes, incluyendo la enfermedad coronaria, la presión arterial alta, la diabetes, el fumar, el consumo excesivo de alcohol, el abuso del consumo de drogas, y el estrés. Algunas sustancias, como los medicamentos de receta, suplementos dietéticos, y remedios herbales pueden causar las arritmias en algunas personas.
Algunas arritmias se caracterizan por un ritmo cardíaco anormalmente rápido, y otras por un ritmo anormalmente lento:
1. Braquicardia.
La braquicardia es una frecuencia cardiaca de menos de 60 latidos por minuto. Ésta puede causar fatiga, mareo, o desmayos. Los síntomas se pueden tratar, y en ciertos casos se pueden corregir implantando un marcapasos electrónico por debajo de la piel para acelerar la frecuencia cardiaca.
2. Taquicardia.
La taquicardia es una frecuencia cardiaca de más de 100 latidos por minuto. Ésta puede producir síntomas como palpitaciones, latido rápido del corazón, dolor en el pecho, mareo, o desmayos. Si la taquicardia ocurre dentro de los ventrículos del corazón, puede ser muy peligrosa, porque impide el bombeo de sangre desde el corazón hacia el resto del cuerpo. La taquicardia que ocurre en los ventrículos también puede causar un trastorno altamente peligroso, la fibrilación ventricular, la cual se caracteriza por la palpitación rápida de las cámaras inferiores del corazón. Cuando se presenta la fibrilación

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
112
ventricular, el corazón no puede bombear sangre, situación que puede resultar en muerte si no se presta atención médica inmediatamente.
La taquicardia se puede tratar con medicamentos, o en algunos casos, con una descarga eléctrica mediante un desfibrilador cardioversor implantable.
d) Renales
Pueden ocurrir complicaciones renales ligadas a la presencia de anticuerpos antifosfolípidos, originadas por trombosis arteriales, trombosis de la arteria renal, infarto renal y microangiopatía trombótica (una lesión estructural de la pared vascular (principalmente arteriolas y capilares), con engrosamiento de la misma, trombosis plaquetaria intraluminal y obstrucción parcial o completa de la luz vascular, la presencia de trombocitopenia y anemia hemolítica son características constantes de la microangiopatía trombótica y reflejan el consumo y la disrupción de plaquetas y hematíes en la microvasculatura).
- Grandes vasos
Trombosis de arteria renal
La trombosis de arteria renal, es la formación de un coágulo en una arteria renal. La trombosis de una arteria renal, puede causar insuficiencia renal, debido a una obstrucción del flujo sanguíneo al riñón.
La obstrucción de la arteria renal, se debe a cualquier reducción del flujo sanguíneo en la arteria renal, lo que produciría el deterioro de su función. La obstrucción total del flujo sanguíneo, si es prolongado, ocasionará una insuficiencia permanente del riñón.
La falta de funcionamiento de uno de los riñones, puede que no cause síntomas, ya que el segundo riñón, filtra adecuadamente la sangre, aunque el individuo podría presentar hipertensión. Si por el contrario ninguno de los dos riñones tuviese una correcta funcionalidad, la obstrucción de la arteria renal pude provocar síntomas de insuficiencia renal aguda.
La oclusión arterial aguda del riñón, puede ocurrir después de una lesión o trauma en el abdomen, el costado u ocasionalmente la espalda, pudiéndose alojar en la arteria renal los émbolos (coágulos de sangre que viajan a través del torrente sanguíneo), que se presentarán con mayor frecuencia si hay antecedentes de ciertos trastornos cardíacos como, estenosis mitral o fibrilación auricular. Los individuos con trastornos de hipercoagulación, pueden ser particularmente vulnerables a las oclusiones agudas de la arteria renal.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
113
Ocasionalmente, la estenosis de la arteria renal, puede incrementar el riesgo de una oclusión súbita, debido a la formación de coágulos.
- La sintomatología que el paciente suele presentar, es:
- Dolor en el costado
- Dolor abdominal
- Dolor de espalda
- Hematuria
- Pequeños vasos:
Microangiopatía trombótica
La microangiopatía trombótica es una lesión de la pared vascular presentando una tumefacción y descamación del endotelio y acúmulo de un material mucoide, translucido, en el espacio subendotelial. Estas lesiones producen una oclusión parcial o total de la luz de los pequeños vasos, (principalmente en arteriolas y capilares) con engrosamiento de la misma, edema y desprendimiento de las células endoteliales de la membrana basal, acumulación de material algodonoso en el espacio subendotelial, trombosis plaquetaria intraluminal y obstrucción parcial o completa del volumen vascular.
La presencia de trombocitopenia y anemia hemolítica, son características constantes de la microangiopatía trombótica y reflejan el consumo y la disrupción de plaquetas y hematíes en la microvasculatura.
Pueden presentar diversos signos clínicos dependiendo de la distribución de las lesiones microvasculares y de la prevalencia de lesiones renales o cerebrales.
El síndrome urémico hemolítico y la púrpura trombocitopénica trombótica son dos entidades patológicamente indistinguibles pero clínicamente diferentes:
Se piensa que las células endoteliales, son el principal factor que interviene en los sucesos producidos por esta enfermedad. La toxina-shiga y otras endotoxinas bacterianas, como también anticuerpos, inmunocomplejos, ciertas drogas y virus (en particular el VIH), son tóxicos endoteliales “in vitro” y pueden causar síndrome urémico hemolítico o la púrpura trombocitopénica trombótica “in vivo”.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
114
En algunos casos de la forma esporádica del síndrome urémico hemolítico y de la púrpura trombocitopénica trombótica, se han reportado anormalidades plaquetarias y del factor Von Willebrand, junto con el aumento de los niveles de marcadores tempranos de la activación de la coagulación y la fibrinólisis.
La pérdida de la tromborresistencia fisiológica, la adhesión leucocitaria al endotelio dañado, el consumo del complemento, la anormal liberación y fragmentación del factor de Von Willebrand, pueden sostener e incluso amplificar el proceso microangiopático.
El síndrome urémico hemolítico puede aparecer esporádicamente a cualquier edad, mientras que la forma epidémica predomina en niños expuestos a infección por Escherichia coli y Shigella disenteriae, caracterizándose por la presencia de anemia hemolítica, trombocitopenia e insuficiencia renal aguda. Las exotoxinas producidas por estas bacterias al ser absorbidas en el tracto gastrointestinal provocaran diarreas con hemorragia e insuficiencia renal, desencadenando la liberación de citoquinas, deficiencia en la síntesis de prostaciclina, activación plaquetaria con biosíntesis aumentada de tromboxano, así como activación de leucocitos y de los sistemas de coagulación y fibrinólisis. Los niños tienen un pronóstico bueno y no suele tener repercusiones en la salud de paciente una vez curado, sin embargo no ocurre lo mismo en adultos, estos tienen un pronóstico malo y un cuadro clínico grave, además los agentes desencadenantes de la enfermedad pueden ser, además de los citados anteriormente, infecciones virales, trasplante de órganos, embarazo, uso de ciertas drogas, cáncer o enfermedades inmunológicas.
La púrpura trombótica trombocitopénica es una enfermedad esporádica y aguda, que afecta a los adultos, con predominio de las mujeres (relación mujer-hombre de 3:2) los únicos síntomas clínicos que los distinguen del síndrome urémico hemolítico (además de enfermedades neurológicas) son, la anemia pleiocrómica, fiebre, petequias, parálisis y coma, con frecuencia se acompaña de un síndrome constitucional inespecífico, con malestar, náuseas y vómitos.
Se piensa que los niveles de citotoxinas, aumentados en pacientes con microangiopatía trombótica intervienen en la patotoxicidad de la enfermedad, ya que algunas citoquinas (IL-6) inducen la activación de la coagulación, mientras que otras (como la TNF) parecen estar involucradas en la activación de la fibrinólisis, por lo que se cree que las anormalidades hemostáticas producidas durante el desarrollo de esta enfermedad podrían estar ligadas a la interacción entre estas citoquinas y células endoteliales, monocitos y neutrófilos.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
115
Los elementos claves del laboratorio del síndrome urémico hemolítico y púrpura trombótica trombocitopénica, son la presencia de anemia hemolítica microangiopática y trombocitopenia:
La trombocitopenia por lo general es severa, con recuentos de plaquetas inferiores a 60.000/mm3 en la mayoría de los casos. Hay presencia de plaquetas gigantes en la observación de frotis sanguíneos.
La anemia, es generalmente severa, los niveles de hemoglobina en el 99% de los casos son inferiores a 10 mg/dl y menor a 6,5 mg/dl en el 49 % de los casos. Los niveles de LDH están incrementados, reflejando además de la hemólisis una isquemia tisular difusa. La hiperbilirrubinemia (principalmente no conjugada), reticulocitosis, hemoglobina libre circulante y niveles bajos o indetectables de haptoglobina, todos son indicadores específicos de la acelerada disrupción y producción de hematíes. La detección de fragmentos de hematíes (esquistocitos) juntos a un test de Coombs negativo (con la excepción del síndrome urémico hemolítico asociado a neumonía por Streptococcus pneumoniae) son necesarios, para confirmar la naturaleza microangiopática de la hemólisis.
La anemia hemolítica, es probablemente la consecuencia de la fragmentación mecánica de los hematíes durante su paso por las estrechas arteriolas o capilares. Este proceso puede estar favorecido por la liberación de radicales libres de los neutrófilos que median la peroxidación lipídica de la membrana de los hematíes. Como consecuencia de ello, las membranas se vuelven más rígidas y susceptibles al daño inducido por la exotoxina, cuando ellos pasan a través de la microcirculación estenosada. Pueden estar involucrados mecanismos adicionales, los receptores de las glicoproteínas Ib y glicoproteínas IIb-IIIa (que pueden unirse al factor de Von Willebrand proveniente de las células endoteliales) y a los receptores de la trombospondina que pueden estar presentes en las superficies de las membranas celulares de los hematíes jóvenes. Las GPIb-IIIa están presentes en las membranas de las células endoteliales por lo que se observa que durante la fase aguda del síndrome urémico hemolítico/púrpura trombótica trombocitopénica, los reticulocitos y los hematíes jóvenes se adhieren a las células endoteliales microvasculares a través de los puentes moleculares del factor de Von Willebrand y trombospondina.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
116
e) Hígado
- Grandes vasos
Infarto hepático
El infarto hepático, es la necrosis de parte o todo el parénquima del hígado, producido por una isquemia tisular, normalmente debido a una oclusión del flujo de la arteria hepática o una de sus ramas. Entre las causas más frecuentes están la embolización terapéutica de alguna rama de la arteria hepática, la ligadura accidental de la arteria hepática y la trombosis de la anastomosis arterial tras el trasplante hepático.
Debido a la doble circulación que presenta el hígado (procedente de la arteria hepática y del sistema porta), sumado a la abundancia de vasos accesorios del órgano, hacen que la aparición de infartos sea algo excepcional.
- Pequeños vasos
Hiperplasia nodular regenerativa
La hiperplasia nodular regenerativa es definida como, la nodularidad difusa del hígado, producida por nódulos regenerativos que no están asociados con la fibrosis. Se caracteriza por la presencia de nódulos en la superficie externa del hígado que al corte son discretos, redondos, y planos, que se asemejan a un carcinoma metastásico.
Este padecimiento es raro y se asocia a veces a síndromes mieloproliferativos, linfoproliferativos, alteraciones vasculares y de la colágena, así como al uso de esteroides y medicamentos antineoplásicos.
- Pérdidas fetales recurrentes
La pérdida fetal recurrente es la interrupción reiterada de la evolución del embarazo produciéndose abortos espontáneos sucesivos de dos, tres o más embarazos por debajo de las 20 semanas, ya sea en etapa preembrionaria (no se visualizan latidos cardiacos por medio de la ecografía) embrionaria (latidos cardiacos presentes, hasta las 10 semanas) o fetal (latidos cardiacos presentes, entre las 10 y 34 semanas), lo que representa un problema frecuente y de compleja resolución en mujeres que padecen el síndrome antifosfolípido.
Este concepto, es poco aplicable del punto de vista clínico ya que habría que esperar a que se produjesen 3 abortos para empezar a tratar a una paciente con el fin de diagnosticar si la causa de estos abortos es debida a la presencia de anticuerpos antifosfolípidos, por lo tanto se propone considerar como síndrome antifosfolípido del embarazo a la presencia de uno o más tipos

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
117
de anticuerpos antifosfolípidos asociados a una o más de las siguientes complicaciones del embarazo: DPP, Preeclampsia, óbito fetal...
− DPP: es la separación parcial o total de la placenta de la pared uterina, en un embarazo de veinte o más semanas de edad gestacional, producido a consecuencia de un hematoma retroplacentario, evidente sólo por ecografía o con manifestaciones clínicas (hipertonía uterina, genitorragia, sufrimiento fetal agudo, etc.).
− Preeclampsia (PE): es una patología propia del embarazo en las primeras 72 horas del puerperio caracterizada, en su forma completa, por hipertensión arterial, proteinuria, edemas, convulsiones epileptoides o coma.
La Preeclampsia es una de las tres principales causas de mortalidad materna en el mundo. La preeclampsia presenta además complicaciones, tanto para la madre: falla renal, coagulopatía, convulsiones, síndrome Hellp, como para el feto: retardo del crecimiento intrauterino, sufrimiento fetal agudo, prematurez y muerte fetal.
Debido a la variedad de alteraciones que pueden intervenir en su etiología se ha definido su origen multifactorial. Los factores de riesgo para la preeclampsia, han sido agrupados en alteraciones placentarias y/o enfermedades maternas subyacentes se hayan o no presentado expresión clínica antes del embarazo, con disfunción endotelial como compromiso final común.
La preeclampsia involucra a la falla de la invasión trofoblástica (por mal adaptación inmune, genética o metabólicamente determinada) a las arterias espirales, hipoxia, presentes desde el primer trimestre de la gestación pero solo detectables en la clínica por la ausencia de la hipotensión fisiológica del segundo trimestre y/o la positividad del test de la rodada (test de Gant) a partir de la semana 26.
El curso natural de la enfermedad se manifestará tardíamente con afección multiorgánica, casi siempre de predominio renal, pero a veces con cierta sobreexpresión placentaria, hepática, cerebral y raramente miocárdica. En todo caso siempre habrá activación endotelial, microangiopatía trombótica, anemia hemolítica, consumo de plaquetas y depósito de fibrina a nivel sistémico. Se producirá un incremento del “estrés oxidativo” por la alta disponibilidad y baja capacidad de limpieza del anión superóxido y los radicales libres. La caída de la presión oncótica por la hipoalbuminemia secundaria a la proteinuria, predispone a hipovolemia, hemoconcentración y desequilibrio hidroelectrolítico.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
118
El 10% de las pacientes preeclámpticas, se presentan temprano en la gestación y casi siempre severas y complicadas (semanas 20 a 28).
La presencia de anticuerpos antifosfolípidos, está relacionado con la presentación temprana y con graves complicaciones de la enfermedad y se distinguen dos tipos:
Las inmunoglobulinas G (IgG) y M (IgM) intervienen en el desarrollo de esta enfermedad, produciéndose la unión a la β2-glicoproteína I, formando complejos donde participan fosfolípidos aniónicos de disposición bilaminar, que pueden o no prolongar los tiempos de coagulación dependientes de fosfolípidos y se denominan anticuerpos anticardiolipina.
La segunda clase de anticuerpos antifosfolípidos se identifican por su interferencia con ensayos de coagulación dependientes de fosfolípidos (anticoagulante lúpico, tiempos de: caolina, de veneno de víbora Russel y de tromboplastina activado) los cuales se unen a fosfolípidos de fase hexagonal o formando complejos con protrombina o β2-glicoproteína I.
- Óbito fetal: consiste en la muerte de un feto cuyo peso es mayor de 500 g (20 semanas de edad gestacional), que ocurre durante el embarazo.
Hoy en día un 50% las causas de estos abortos pueden atribuirse a alteraciones genéticas, diabetes y otras endocrinopatías, infecciones materno-fetales, alteraciones anatómicas del aparato genital femenino e inmunopatías.
El organismo materno, tolera un tejido que le es inmunológicamente ajeno, cuanto más difieren madre y padre en sus antígenos de histocompatíbilidad, mayor tolerancia inmunológica al embrión existe. Estos antígenos determinan la aparición de anticuerpos supresores, que los bloquean e impiden el desarrollo de anticuerpos citotóxicos. Por otra parte, la secreción de progesterona desde el inicio de la gestación tiene acción inmunosupresora. También puede influir la modificación de los corticoides maternos y fetales.
En algunos casos, de la modulación hormonal modificada o de la respuesta del sistema inmune, se inician respuestas inmunitarias, por lo que se explica la aparición de anticuerpos antifosfolípidos durante el período de gestación y su desaparición una vez finalizado este proceso.
En el embarazo, desde el momento de la implantación hay un incremento de la vascularización en la placenta. A ese nivel el flujo sanguíneo es alto y la sangre no coagula. El principal mecanismo anticoagulante es la presencia sobre la superficie del trofoblasto de una proteína llamada Anexina V, que es un potente anticoagulante. Al mismo tiempo el trofoblasto rompe y penetra en

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
119
la pared de las arteriolas que alimentan la placenta, colonizan y destruyen la capa músculo elástica, por esta razón, en condiciones normales, estos vasos se comportan como de mínima resistencia y no son capaces de reaccionar a los estímulos vasoconstrictores. También por esta causa, el flujo por el espacio intervelloso depende normalmente de la presión de perfusión del sistema arterial.
Cuando los anticuerpos antifosfolípidos, están presentes, parecen inhibir la anexina V, con lo que aumenta la coagulabilidad de la sangre, constituyéndose depósitos de fibrina, trombos e infartos, que engrasan el espesor y disminuyen la superficie de la membrana de intercambio materno fetal. Por sus otras propiedades protrombóticas los anticuerpos antifosfolípidos, determinan la desfuncionalización progresiva o aguda de la unidad feto placentaria. La consecuencia de ello es un fallo en la implantación o bien la muerte del embrión o el feto en diferentes momentos de la gestación, según la época de aparición de los anticuerpos antifosfolípidos y su actividad biológica. Al mismo tiempo parece ser que los anticuerpos antifosfolípidos alteran la invasión arteriolar, con lo que la perfusión placentaria se compromete y se reduce frente a estímulos vasoconstrictores, lo que altera la nutrición fetal y lleva a retardo del crecimiento fetal.
La mayoría de las mujeres con síndrome antifosfolípido primario no progresan a lupus eritematoso sistémico y pueden presentar períodos de remisión (clínica y de laboratorio) con escaso riesgo de manifestaciones trombóticas. Sin embargo, la mayoría de las pacientes con síndrome antifosfolípido no presentan un embarazo normal, produciéndose la pérdida recurrente del embarazo.
Muchas mujeres con síndrome antifosfolípido, presentan una trombofilia durante el período de gestación, presentando un embarazo sano, aun así, las trombofilias contribuyen a una serie de complicaciones, incluyendo la pérdida del feto al final del primer trimestre de la gestación o durante el segundo o tercer trimestres (nacimientos sin vida), desprendimiento de la placenta (cuando la placenta se separa de la pared uterina, en forma parcial o total, antes del parto) y crecimiento insuficiente del feto. Las trombofilias también pueden causar una forma severa de preeclampsia. Se cree que la mayoría de estos problemas se producen como resultado de coágulos en los vasos sanguíneos placentarios que causan cambios en la placenta y reducen el suministro de sangre al feto. Las mujeres embarazadas con una trombofilia también tienen un riesgo mayor de desarrollar una tromboembolia venosa profunda. Sin embargo, incluso las mujeres embarazadas sin trombofilias tienen más probabilidades de desarrollarla que las mujeres no embarazadas. Esto se debe a cambios normales en la coagulación sanguínea relacionados con el embarazo que limitan la pérdida de sangre durante el parto.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
120
Cuando evoluciona espontáneamente, la gestación complicada por el síndrome antifosfolípido es de pobre pronóstico, pero este es mejorado al instaurar un tratamiento preventivo, como las combinaciones de aspirina a bajas dosis y corticosteroides, aspirina y heparina, y otras modalidades inmunosupresivas (tales como inmunoglobulinas) a pesar de ello no hay una terapia de elección para paliar estas pérdidas fetales.
Trombocitopenia
Las manifestaciones hematológicas más frecuentes son la trombocitopenia y la anemia hemolítica autoinmune, que si se manifiestan conjuntamente reciben el nombre de síndrome de Evans.
La trombocitopenia es relativamente frecuente en pacientes con anticuerpos antifosfolípidos, aunque no suele ser tan severa como para causar hemorragia. El número de plaquetas suele permanecer estable durante años y sin razones aparentes, producirse en un momento dado, un descenso del número de plaquetas, que suele ser muy severo.
El número de plaquetas en una persona sana suele oscilar entre 150.000 a 400.000 células por mm3, mientras que en el caso de pacientes con síndrome antifosfolípido se aprecia una gran disminución del número de plaquetas, siendo este inferior a 50.000 plaquetas por mm3.
Los anticuerpos antifosfolípidos, actúan sobre las plaquetas mediante la unión de estos anticuerpos a los fosfolípidos presentes en la superficie externa de la membrana plaquetaria, causando daño a las plaquetas, lo que provoca el descenso de estas en sangre, también se piensa que al unirse los anticuerpos a las plaquetas se produce un aumento de la captación y destrucción por el sistema reticuloendotelial con acortamiento de la supervivencia de la plaqueta.
Algunos pacientes pueden presentar sólo trombocitopenia y conforme se va desarrollando la enfermedad se pueden ir desarrollando las pérdidas fetales o las trombosis. Algunos pacientes con anticuerpos antifosfolípido y trombocitopenia, también desarrollan anemia hemolítica con test de Coombs positivo.
3.3.2 Manifestaciones menores En el síndrome antifosfolípido existen otro tipo manifestaciones clínicas,
clasificadas como manifestaciones menores, estas aparecen con menos frecuencia y las más destacadas son las neurológicas, (epilepsia, migraña, corea, mielitis transversa y síndrome de Guillain-Barré), las cutáneas, y la anemia hemolítica.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
121
3.3.2.1 Manifestaciones neurológicas
1. Epilepsia.
La epilepsia, también llamada, crisis convulsiva o “el gran mal”, es un trastorno cerebral que involucra convulsiones recurrentes, producidas debido a cambios breves y repentinos del funcionamiento del cerebro, dando lugar a una descarga desordenada de las células de nuestro cerebro. Por esta razón, se trata de una afección neurológica, la cual no es contagiosa ni está causada por ninguna enfermedad o retraso mental (aunque algunas personas con retraso mental pueden experimentar ataques epilépticos).
Las crisis epilépticas, también conocidas como el gran mal, son una serie de movimientos de las cuatro extremidades tipo sacudidas, que produce pérdida de la conciencia y del control de los esfínteres, con una duración de segundos o algunos minutos, y cuando persiste se llama estado epiléptico. Son más frecuentes de lo que comúnmente se piensa y afectan al 1 % de la población. Pueden comenzar a cualquier edad, si bien se inician más frecuentemente durante la niñez y la adolescencia.
Algunas epilepsias aparecen sólo en determinada edad, otras se acompañan de otros problemas neurológicos, otras se producen sólo frente a un estímulo definido, como descargas de luz, otras más son hereditarias, o se acompañan de problemas en otras funciones cerebrales o en otros órganos.
Los síntomas que presente una persona durante una crisis epiléptica dependerán entonces de la o las zonas del cerebro que estén siendo afectadas por la descarga. Estas crisis epilépticas, producen una alteración momentánea del funcionamiento cerebral, debida a la descarga súbita y desproporcionada de los impulsos eléctricos que habitualmente utilizan las células del cerebro.
Se distinguen distintos tipos de crisis, que son:
− Crisis generalizadas: pueden afectar a todo el cerebro, manifestándose con pérdida brusca de conocimiento, con caída al suelo, contractura de los músculos de las extremidades y de la cara seguidas de sacudidas rítmicas. En otras ocasiones, especialmente en niños y adolescentes, las crisis se presentan con una pérdida del conocimiento, sin caída al suelo ni convulsiones, de segundos de duración, con rápida recuperación.
− Crisis parciales: afectan a una parte del cerebro y pueden presentarse con sensaciones subjetivas extrañas o difíciles de describir o con fenómenos auditivos, visuales, sensación de hormigueo, etc. Estos síntomas pueden aparecer en forma aislada o dar paso a una pérdida de conocimiento con movimientos

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
122
automáticos de la boca, de las manos o de otra parte del cuerpo. En otras oportunidades las crisis parciales pueden presentarse con sacudidas de una extremidad o de la mitad de la cara, sin pérdida del conocimiento.
− Crisis de ausencias: En este tipo de crisis no hay ninguna manifestación de movimientos anormales, ya que depende del sitio del cerebro en el que se origine. Estos pacientes pueden tener sensaciones extrañas, escuchar, ver u oler algo determinado o en los niños es frecuente que haya periodos en blanco, donde el niño se desconecta brevemente del medio.
Las convulsiones (ataques) son episodios de alteración de la función cerebral que producen cambios en la atención o el comportamiento y se producen por una excitación eléctrica anómala del cerebro. Este trastorno convulsivo afecta a cerca del 0,5% de la población, del 1,5 al 5,0% de la población puede presentar una convulsión en su vida, pudiéndose producir varios tipos de convulsiones:
− Idiopática: Suele ser debido a herencia familiar, comenzando antes de los 20 años, sin encontrarse problemas cerebrales.
− Secundarios a problemas de desarrollo o genéticos producidos en el nacimiento: En este caso las convulsiones se originan en la infancia.
− Alteraciones metabólicas: Pueden producirse a cualquier edad y algunas de ellas pueden ser producidas por, diabetes mellitas, síndrome de abstinencia de alcohol o drogas, fenilcetonuria, deficiencias nutricionales, alteraciones del equilibrio electrolítico…
− Lesión cerebral: Se da especialmente en adulto de corta edad, (aunque también pueden ocurrir a cualquier otra edad) que hayan sufrido lesiones cerebrales y tras un período de latencia de 1 a 3 años comienza a convulsionar.
− Tumores y otras lesiones cerebrales: Se da en personas mayores de 30 años, apareciendo en ellos convulsiones parciales o focales, apareciendo posteriormente una convulsión tónico-crónica generalizada.
− Enfermedades de los vasos sanguíneos: es la causa más común de las convulsiones después de los 60 años de edad.
− Enfermedades degenerativas: afecta a personas de muy avanzada edad, estas son enfermedades de tipo senil.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
123
− Infecciones: Suelen ser reversibles, afectando por igual a todas las edades. Este tipo de infecciones son, meningitis, complicaciones del SIDA, encefalitis…
Algunos factores como, el embarazo, la falta de sueño, no administrar un tratamiento para la epilepsia, el consumo de alcohol u otras drogas…pueden presentar el riesgo de empeorar las convulsiones en una persona con un trastorno convulsivo bien controlado con anterioridad.
Una vez que se identifica el tipo de crisis que presenta el paciente y si no existe tratamiento específico para la causa detectada o ésta no se pudo establecer porque todos los estudios resultaron normales, la epilepsia debe ser tratada con medicamentos que eviten la presentación repetida de las crisis convulsivas.
2. Migraña
La migraña, también llamada jaqueca, es un tipo de dolor de cabeza primario que afecta a algunas personas de manera repetitiva con el tiempo. Se caracteriza por dolores fuertes y palpitantes que normalmente afectan a un solo lado de la cabeza, además de producir, náuseas y vómitos, distorsión de la visión, vértigo e hipersensibilidad a la luz.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
124
Una migraña es ocasionada por una actividad anormal del cerebro, la cual es desencadenada por el estrés, por alimentos o por algún otro factor. Se desconoce la cadena exacta de eventos, sin embargo parece involucrar diversos procesos químicos y vías nerviosas del cerebro. Los cambios afectan el flujo sanguíneo del cerebro y de las membranas circundantes.
Tiene un carácter pulsátil y se acompaña de un malestar generalizado, este dolor empeora normalmente con la actividad física y mejora con el reposo. Afecta a 17 de cada 100 mujeres y a un 5 por ciento de los hombres.
Las migrañas o jaquecas normalmente no representan una amenaza significativa para la salud general, sin embargo, pueden volverse crónicas, recurrentes, frustrantes y pueden interferir en la vida diaria de la persona.
El accidente cerebro vascular, es una complicación extremadamente poco común de la migraña intensa, este riesgo puede presentarse a causa del estrechamiento de los vasos sanguíneos que limita el flujo de sangre a algunas partes del cerebro por un largo período de tiempo.
La mayoría de los dolores de cabeza no son serios y se curan solos, sin embargo, frecuentes migrañas pueden reducir la calidad de vida, aunque se desconoce el motivo, estudios recientes indican que quienes las sufren tienen más riesgo de padecer un infarto y se cree que el dolor puede ser originado por:
− Inflamación o irritación de los vasos sanguíneos del cuero cabelludo: Esta inflamación, produce su contracción y posteriormente su dilatación, lo cual produce inflamación y dolor palpitante.
− Hormonal: Puesto que el 70 % de las personas que padecen esta enfermedad son mujeres, se cree, que los cambios hormonales durante la menstruación y ovulación provocan, a menudo, ataques.
− Factores ambientales: Algunos factores ambientales provocan migrañas en personas propensas, entre ellos los más comunes son, el alcohol, especialmente el vino tinto o burdeos, ciertos alimentos o aditivos, quesos crudos, hígado de aves, chocolate, vino tinto, glutamato monosódico y conservadores que se encuentran en los productos ahumados y carnes en conserva.
− Otros estímulos: Pueden afectarle estímulos como son, los cambios en la presión atmosférica, cambios en la altitud, el resplandor solar...

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
125
− Herencia: Aunque la forma de herencia no está totalmente establecida, en algunas formas especiales de migraña ya se ha identificado el gen que la transmite situado en el cromosoma 9.
− Edad: En la infancia la migraña se presenta por igual en niños y niñas. A partir de la pubertad y debido a los cambios hormonales, se dispara la incidencia de migraña en las mujeres.
− Estrés y ansiedad: Es necesario aprender a relajarse, buscar alguna distracción en momentos estresantes.
− Falta o exceso de sueño: También puede ser un desencadenante de la migraña.
Los síntomas más frecuentes son:
− Dolor de cabeza fuerte y palpitante, normalmente limitado a un solo lado.
− Distorsiones visuales y otros síntomas precediendo al dolor.
− Náuseas, vómitos, vértigo, manos frías, temblores o sensibilidad a la luz y al sonido.
Las migrañas se clasifican como:
• Migraña sin aura:
No produce síntomas de alerta. Entre algunos de los síntomas comunes se incluyen:
Dolor de cabeza con palpitación, pulsátil, usualmente peor a los lados de la frente, que también puede localizarse a un solo lado de la cabeza, con una duración de entre 6 a 48 horas.
− Mareos.
− Vértigo.
− Pérdida del apetito.
− Náuseas.
− Vómito.
− Fatiga.
Una vez que el ataque ha terminado se produce en la persona otros síntomas como pueden ser, pesadez, dolor de cuello, necesidad de dormir más…

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
126
• Migraña con aura o migraña clásica:
La mayoría de las personas que sufren de migrañas no tienen ningún signo de advertencia antes de que se presenten, sin embargo, en algunas personas el dolor de cabeza está precedido de una perturbación visual denominada, aura, que son un grupo de síntomas neurológicos, en la cual la persona ve luces intermitentes, formas y colores distorsionados y otras ilusiones ópticas.
La migraña clásica comienza con un aura que dura de 10 a 30 minutos antes de empezar el dolor. Durante el ataque, el aura desaparece y el dolor se hace más intenso y palpitante, este dolor ataca a un solo lado de la cabeza y a veces el vomitar puede aliviar el ataque. Una migraña puede durar desde unos 30 minutos a 5 días o más.
Los cambios visuales son comunes en uno o ambos ojos y es común que ocurran en cualquier combinación:
− Visión de líneas en zigzag.
− Visión de luces intermitentes.
− Otras alucinaciones visuales.
− Puntos ciegos temporales.
− Sensibilidad a la luz intensa.
− Visión borrosa.
− Dolor ocular.
Algunos de los síntomas que pueden preceder o acompañar el dolor de cabeza son:
− Pérdida del apetito.
− Náuseas.
− Vómitos.
− Escalofríos.
− Aumento de la micción.
− Aumento de la sudoración.
− Inflamación facial.
− Irritabilidad.
− Fatiga.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
127
• Migraña mixta sensorial:
Es un dolor de cabeza con rasgos, tanto de dolor de cabeza por tensión, como de jaqueca.
Los síntomas asociados con la migraña mixta sensorial pueden incluir:
− Dolor de cabeza en uno o en ambos lados.
− Sensación pulsátil.
− Sensación de tensión como si una banda estuviera apretando.
− Dolor cuya intensidad varía de molesto a incapacitante.
− Puede empeorar con las actividades físicas de rutina.
− Náuseas o vómitos.
− Sensibilidad a la luz o al sonido.
− Centelleos visuales.
Puede durar de 4 a 72 horas, sin embargo, puede evolucionar a un dolor diario crónico, provocando, irritabilidad, depresión, pereza, entumecimiento, hormigueo, debilidad, dolor en el cuello…
Aunque todas las migrañas están asociadas con el dolor, difieren en su severidad y su frecuencia. Por ello es necesario crear un tratamiento a medida que atienda las necesidades individuales de cada enfermo. Los especialistas en dolor de cabeza han descubierto que una migraña puede desaparecer si se actúa con rapidez durante la etapa de aura.
Una estrategia común es tomar aspirina con café u otra fuente de cafeína. La aspirina inhibe la producción de prostaglandina y la cafeína combate los vasculares.
Otra manera es recostarse a oscuras con una compresa fría en la frente. En casos leves esto puede prevenir la dilatación de los vasos del cuero cabelludo y parar o minimizar el ataque.
3. Corea
En 1872, el médico americano, George Huntington, describió la enfermedad. Uno de los primeros nombres de esta enfermedad fue corea que proviene del griego y significa “danza” con lo que nos describe los movimientos incontrolados que sufren las personas afectadas por la enfermedad, más tarde otros nombres usados fueron "corea hereditaria" que de alguna forma recalcaba como la enfermedad se transmitía de padres a hijos y más tarde se le llamó "Corea crónica progresiva" lo que indicaba como la

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
128
enfermedad progresa y empeoraba al transcurrir el tiempo, aunque hoy en día se usa normalmente el término de enfermedad de Huntington, para describir este trastorno tan complejo.
La enfermedad de Huntington es el resultado de una degeneración genéticamente programada de las células del cerebro (neuronas) en unas áreas concretas de este, específicamente las células afectadas son las del ganglio basal, (estructura profunda del cerebro que tiene importantes funciones, incluyendo la coordinación de los movimientos), en la que la enfermedad de Huntington ataca a las neuronas. También está afectada otra parte del cerebro que es el córtex, que controla el pensamiento, la percepción y la memoria. Esta enfermedad se hereda por la alteración de un único gen en el cromosoma cuatro.
Los principales síntomas de la enfermedad de Huntington son los siguientes:
- Cambios del comportamiento como la irritabilidad.
- Cambios en el estado de ánimo, inquietud, agitación, conducta antisocial, psicosis, paranoia, alucinaciones.
- Movimientos faciales, muecas.
- Necesidad de girar la cabeza para desviar la mirada.
- Demencia progresiva con pérdida de memoria, falta de juicio, alteraciones del lenguaje, pérdida de funciones cognitivas como el cálculo, cambios de la personalidad, desorientación, confusión.
- Marcha inestable.
- Desarrollo progresivo de movimientos coreiformes del tipo de movimientos bruscos, súbitos, rápidos y repetitivos de brazos, piernas, cara, cuerpo o movimientos lentos incontrolables del cuerpo y las extremidades.
- Deterioro de la capacidad del lenguaje.
- Ansiedad, estrés y tensión.
- Dificultad para tragar.
En los niños además pueden aparecer movimientos lentos, temblor y rigidez.
Los primeros síntomas de la enfermedad de Huntington varían mucho de una persona a otra, volviéndose irritables, apáticos, pasivos, depresivos o

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
129
enfadados, afectando a la razón, memoria y otras funciones, lo que les conlleva a tener gran dificultad para la conducción, aprendizaje, toma de decisiones En algunos enfermos la enfermedad puede comenzar con torpeza o problemas con el equilibrio, movimientos incontrolados, en los dedos, pies, cara o tronco, que se intensifican cuando la persona está ansiosa. Estos síntomas pueden desaparecer con el avance de la enfermedad, o en algunos individuos pueden continuar e incluir además explosiones de hostilidad o profundos ataques de depresión.
Lo que sí es común, es que cuanto antes aparecen los síntomas más rápidamente progresa la enfermedad, dificultando el desarrollo de funciones intelectuales.
Con el progreso de la enfermedad el lenguaje puede convertirse en incomprensible, y funciones vitales como, tragar, comer, hablar y especialmente andar pueden empeorar grandemente y algunas pueden llegar a no recordar a personas de su entorno aunque otras si pueden recordar y seguir expresando sus sentimientos.
En general el curso de la enfermedad abarca desde 10 a 30 años. La causa más común de muerte es debido a las infecciones (sobre todo neumonía), heridas debidas a caídas, u otras complicaciones. En la actualidad no existe cura para la enfermedad de Huntington y no existe terapia conocida que sea capaz de detener la progresión de la enfermedad que suele ser fatal para el enfermo, en un período de tiempo de entre 15 a 20 años.
4. Mielitis transversa
La mielitis transversa es un trastorno neurológico, causado por la inflamación en ambos lados de un nivel, o segmento, de la médula espinal. El término mielitis se refiere a la inflamación de la médula espinal y transversa describe simplemente la posición de la inflamación, es decir, que abarca el ancho de la médula espinal. Los ataques de inflamación pueden dañar o destruir la mielina, la sustancia grasa aisladora que recubre las fibras de las células nerviosas. Estos daños causan cicatrices en el sistema nervioso que interrumpen la comunicación entre los nervios de la médula espinal y el resto del cuerpo.
Los síntomas de la mielitis transversa incluyen la pérdida de función de la médula espinal durante varias horas o varias semanas. Lo que comienza generalmente por un dolor repentino en la espalda, debilidad muscular o sensaciones anormales en los pies y los dedos de los pies, puede progresar rápidamente a síntomas más severos, incluyendo parálisis, retención urinaria y la pérdida de control del intestino. Aunque algunos pacientes se recuperan

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
130
de la mielitis transversa con pocos o ningún problema residual, otros sufren daños permanentes que afectan a su capacidad de realizar tareas normales de la vida diaria.
El segmento de la médula espinal en el cual ocurre el daño determina qué partes del cuerpo se ven afectadas. Los nervios de la región cervical (cuello) controlan las señales que viajan hacia el cuello, los brazos, las manos y los músculos respiratorios (el diafragma). Los nervios de la región torácica (parte superior de la espalda) envían señales al torso y a algunas partes de los brazos. Los nervios de la región lumbar (parte media de la espalda) controlan las señales a las caderas y las piernas. Finalmente, los nervios sacros, situados dentro del segmento más bajo de la médula espinal, retransmiten señales a la ingle, a los dedos de los pies y a algunas partes de las piernas. Los daños que ocurren en un segmento afectan las funciones de ese segmento y los segmentos inferiores. En pacientes que padecen de mielitis transversa, la desmielinización ocurre generalmente a nivel torácico, causando problemas de movimiento en las piernas y el control del intestino y de la vejiga, los cuales requieren señales de los segmentos inferiores de la médula espinal.
No existe evidencia de predisposición genética. Esta enfermedad se suele dar sobre todo en pacientes de entre los 10 y 19 años y entre los 30 y 39 años de edad.
Se desconocen las causas exactas que producen esta enfermedad aunque se le relaciona a infecciones virales, trastornos autoinmunes (como el Síndrome antifosfolípido) reacciones inmunes anormales, escasez de la sangre que atraviesa los vasos sanguíneos situados en la médula espinal, sífilis, sarampión, la enfermedad de Lyme y algunas vacunas, incluyendo las de la varicela y la rabia.
En enfermedades autoinmunes, como el síndrome antifosfolípido, el sistema inmunológico, (que normalmente protege el cuerpo contra organismos extraños), ataca por error a los propios tejidos del cuerpo causando inflamación y, en algunos casos, daños a la mielina de la médula espinal.
La mielitis transversa puede ser:
− Aguda: la mielitis transversa aguda es una enfermedad que se desarrolla en cuestión de horas o varios días, considerándose como una enfermedad intrínseca de la médula espinal, poco frecuente y potencialmente devastadora.
− Sub-aguda: la mielitis transversa sub-aguda es una enfermedad que se desarrolla en un corto período de tiempo que suele oscilar entre una y dos semanas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
131
Los síntomas iniciales incluyen generalmente dolores en la parte inferior de la espalda, parestesias repentinas en las piernas (sensaciones anormales tales como ardor, cosquilleo, pinchazos u hormigueo), pérdida sensorial y la parálisis parcial de las piernas, que a menudo se convierte en parálisis completa de las piernas y de la parte inferior del tronco, con disfunción de la vejiga urinaria y del intestino, espasmos musculares, sensación general de malestar, dolores de cabeza, fiebre y pérdida del apetito y dependiendo de en qué segmento de la médula espinal esté involucrado, algunos pacientes también pueden padecer problemas respiratorios.
Todos estos síntomas se pueden englobarse en:
− Debilidad de las piernas y de los brazos.
La mayoría de los pacientes padecen diversos grados de debilidad en las piernas, algunos también en los brazos. Inicialmente, los pacientes con mielitis transversa pueden notar que tropiezan, arrastran un pie o que sus piernas parecen más pesadas de lo normal. La coordinación de los movimientos de la mano y del brazo, así como también la fuerza de ambos, pueden verse afectadas. La progresión de la enfermedad durante varias semanas conduce a menudo a la parálisis completa de las piernas.
− Dolores.
El dolor es el síntoma principal de la mielitis transversa entre aproximadamente un tercio y la mitad de todos los pacientes. El dolor se puede localizar en la parte inferior de la espalda o puede consistir en breves sensaciones punzantes que se desplazan hacia las piernas, los brazos o alrededor del torso.
− Alteración sensorial.
Los pacientes que padecen de problemas sensoriales utilizan a menudo términos tales como entumecimiento, hormigueo, sensación de frío o ardor para describir sus síntomas. Hasta 80 % de los pacientes que padecen de mielitis transversa señalan áreas con aumento en la sensibilidad al tacto, por lo que la ropa o el tacto ligero pueden causar malestar o dolores significativos (una condición denominada alodinia). Muchos también padecen un aumento en la sensibilidad a los cambios en la temperatura, o al calor o al frío extremos.
− Disfunción del intestino y de la vejiga.
Los problemas de la vejiga y del intestino pueden involucrar un aumento en la frecuencia de las ganas de orinar o evacuar, incontinencia, evacuación

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
132
dificultosa, la sensación de evacuación incompleta y estreñimiento. Durante el curso de la enfermedad, la mayoría de las personas con mielitis transversa padece uno o varios de estos síntomas.
Se diagnostica realizando un examen neurológico completo, resonancia magnética y mielografía, además se pueden realizar análisis de sangre para descartar varios trastornos tales como el lupus eritematoso sistémico, la infección por VIH y una deficiencia en la vitamina B12. En algunos pacientes con mielitis transversa, el líquido cerebroespinal que recubre la médula espinal y el cerebro contiene más proteínas de lo normal y un número creciente de leucocitos (glóbulos blancos), indicando una posible infección. Se puede realizar una punción espinal para obtener el líquido y estudiar estos factores.
Si ninguna de estas pruebas sugiere una causa específica, se presume que el paciente padece de mielitis transversa idiopática (casos en los que no se pueden determinar las causas de la mielitis transversa).
No existe actualmente ninguna cura eficaz para los pacientes con mielitis transversa. Los tratamientos se establecen para manejar y aliviar los síntomas y dependen en gran parte de la severidad de la situación neurológica. En las primeras semanas para disminuir la inflamación, reduciendo la actividad del sistema inmunológico (esto se debe a que se sospecha que los mecanismos autoinmunes influyen en el trastorno), tomarán corticosteroides aunque no se ha demostrado que alteren el curso de la mielitis transversa.
Después de terapia inicial, se tiene que mantener funcionando el organismo de los pacientes mientras se espera que ocurra una recuperación espontánea completa o parcial del sistema nervioso. A veces, esto puede requerir que el paciente sea conectado a un respirador.
5. Síndrome de Guillain-Barré
El síndrome de Guillain-Barré, recibe el nombre de síndrome en lugar del de enfermedad debido a que no está claro si existe un agente específico causante de la enfermedad. Un síndrome es una condición médica que se caracteriza por un conjunto de síntomas (lo que siente el paciente) y señales (lo que el médico puede observar o medir). Las señales y síntomas del síndrome de Guillain-Barré son bastante variados por lo que, en raras ocasiones, se puede realizar el diagnóstico en las etapas iniciales.
El síndrome de Guillain-Barré es un trastorno neurológico en el que el sistema inmunológico de nuestro cuerpo, ataca a una parte del sistema nervioso periférico, produciéndose la inflamación aguda de un nervio, dañando porciones de las células nerviosas (pérdida de la vaina de mielina que recubre al axón de la neurona, lo cual disminuye la velocidad de conducción de

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
133
impulsos a través del nervio, pudiendo producir además una denerviación, que conlleva a la destrucción del axón de la neurona), lo que provoca en el enfermo debilidad muscular o parálisis y pérdida sensorial.
Puede aparecer de forma muy brusca e inesperada y la causa exacta de este trastorno no se conoce. El paciente está más débil en las dos primeras semanas después de la aparición de los síntomas.
Aunque es poco frecuente, ya que lo sufren aproximadamente entre una y dos personas de cada 100.000, el síndrome de Guillain-Barré, puede afectar a personas de cualquier edad, aunque es más común en personas de entre 30 a 50 años, produciéndose por igual, tanto en hombres como en mujeres. El trastorno suele aparecer, unos días o unas semanas, después de que la persona haya tenido síntomas de infección viral (como la mononucleosis y el herpes simple) o después de infecciones bacterianas (como micoplasma y algunos tipos de diarrea), aunque estos síntomas suelen desaparecer antes de que comiencen los signos del síndrome de Guillain-Barré. En algunas ocasiones, el embarazo, la cirugía, un traumatismo o las vacunas (como vacunas para la rabia o la influenza) pueden desencadenar el síndrome.
En la actualidad no se sabe por qué el síndrome de Guillain-Barré afecta a algunas personas, lo que sí se sabe es que el sistema inmunológico del cuerpo comienza a atacar al propio organismo.
Normalmente, las células del sistema inmunológico atacan sólo a la materia extraña o a los organismos invasores, pero en el síndrome de Guillain-Barré el sistema inmunológico comienza a destruir las vainas de mielina que rodean los axones de muchas células nerviosas y, a veces, a los propios axones. Cuando esto sucede, los nervios no pueden enviar las señales de forma eficaz, los músculos pierden su capacidad de responder a las órdenes del encéfalo y el encéfalo recibe menos señales sensoriales del resto del cuerpo. El resultado es la incapacidad de sentir calor, dolor y otras sensaciones.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
134
Los síntomas iniciales de la enfermedad, se dan en las tres primeras semanas y se desarrollan rápidamente. Incluyen diversos grados de debilidad o de sensaciones de hormigueo en las piernas que, en ocasiones, se extienden a los brazos y a la parte superior del cuerpo, presentándose normalmente, debilidad y parálisis de manera simétrica desde los pies y las piernas hacia arriba por el cuerpo y se le llama parálisis ascendente. A medida que la parálisis alcanza el nivel del tórax, el paciente puede necesitar ventilación asistida para respirar y los cambios en el movimiento y la sensibilidad ocurren de manera simultánea.
La enfermedad progresa muy rápidamente y cada individuo puede experimentar síntomas distintos, aunque hay unos síntomas que suelen ser los más comunes del síndrome de Guillain-Barré, que son:
La debilidad puede comenzar:
− En los pies y las piernas y puede progresar hacia arriba hasta los brazos y a los nervios craneanos (cabeza).
− Comenzar por los brazos y progresar hacia las extremidades inferiores.
− En los brazos y las piernas al mismo tiempo.
− En los casos leves puede ocurrir en los nervios craneanos.
Cambios en la sensibilidad, entumecimiento, disminución de la sensibilidad o dolor muscular (puede ser similar al dolor por calambres) que usualmente acompaña o precede la debilidad muscular a pesar de ello es posible que no ocurra.
Algunos síntomas adicionales que pueden estar asociados con esta enfermedad son:
− Visión borrosa.
− Dificultad para mover los músculos de la cara.
− Mareo.
− Palpitaciones (sensación táctil de los latidos del corazón).
− Dificultad para empezar a orinar.
− Sensación de vaciamiento incompleto de la vejiga.
− Incontinencia (fuga) urinaria.
− Estreñimiento.
− Contracciones musculares.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
135
Los siguientes síntomas indican que se presenta una emergencia:
− Dificultad para tragar, babeo.
− Dificultad respiratoria.
− Sensación de "incapacidad de respirar profundamente".
− Desmayo.
Tras los síntomas iniciales le sigue una fase estable, en donde no se presentan cambios y posteriormente, una fase de recuperación, que puede durar entre cuatro a seis meses o más, cuando hay mejoría de los síntomas. Aunque estos síntomas pueden o no irse agravando.
Los síntomas pueden llegar a poner en peligro la vida del enfermo, pero a pesar de ello es posible una recuperación parcial incluso en los casos más graves del síndrome de Guillain-Barré. Sin embargo, puede persistir cierto grado de debilidad.
Los procedimientos de diagnóstico son los siguientes:
1. Punción raquídea (punción lumbar): Con esta prueba se pretende extraer una pequeña cantidad de líquido cefalorraquídeo para posteriormente ser enviada al laboratorio para comprobar si existe una infección o algún otro tipo de problema. Para la extracción (será realizada por el médico especialista) se coloca una aguja especial en la parte baja de la espalda, en el interior del conducto raquídeo, que es la zona que rodea la médula espinal, pudiéndose medir la presión que existe en la médula espinal y en el encéfalo, extrayendo una pequeña cantidad de líquido para realizar su correspondiente análisis.
2. Exámenes de electrodiagnóstico (electromiografía y velocidad de conducción nerviosa): Son unas pruebas que sirven para evaluar y diagnosticar los trastornos de los músculos y de las neuronas motoras. Para ello el médico introduce unos electrodos en el músculo o se sitúan sobre la piel que recubre un músculo o un grupo de músculos y se registra la actividad eléctrica y la respuesta del músculo.
En la actualidad no existe una cura conocida para el síndrome de Guillain-Barré, pero sí que hay un tratamiento cuyo objetivo de este tratamiento consiste en reducir la gravedad de la enfermedad y contribuir a la recuperación.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
136
El tratamiento puede incluir:
− Terapia con altas dosis de inmunoglobulina: Consiste en la administración intravenosa de proteínas para atacar a los organismos invasores.
− Terapia hormonal.
− Fisioterapia: Para aumentar la flexibilidad y la fuerza de los músculos.
− Plasmaféresis: Es un proceso por el cual se extrae sangre del cuerpo del paciente y el plasma líquido y las células se separan por centrifugación. El plasma, junto con linfocitos, se descarta y se reemplaza por plasma normal o albúmina humana para evitar la pérdida de proteínas y fluido. La sangre “reconstituida” se devuelve entonces al paciente. Este proceso se puede repetir un cierto número de veces. Se cree que sustancias que pueden dañar la mielina y/o deteriorar la conducción nerviosa se extraen con este método.
La mayor parte de las personas que padecen el síndrome de Guillain-Barré, sobreviven y se recuperan completamente, que son aproximadamente un 95 % de los enfermos.
3.3.2.2 Manifestaciones cutáneas: Dentro de estas se incluyen (entre otras) Livedo reticularis, úlceras en
miembros inferiores, lesiones purpúricas, siendo éstas las manifestaciones cutáneas más características de la enfermedad. (Estas manifestaciones han sido explicadas en las trombosis venosas).
3.3.2.3 Anemia hemolítica: Se caracteriza por la presencia de un número inadecuado de glóbulos
rojos en sangre y es ocasionada por la destrucción prematura de los mismos.
La anemia hemolítica se presenta cuando la médula ósea es incapaz de compensar la destrucción prematura de los glóbulos rojos por medio del aumento en su producción.
Existen varios tipos de anemia hemolítica que se clasifican según el sitio en que se ubica el defecto, el cual puede estar dentro del glóbulo rojo sanguíneo (anemia hemolítica intrínseca) o fuera de éste (anemia hemolítica extrínseca):
− Anemia hemolítica intrínseca: se produce una destrucción de los glóbulos rojos, debido a un defecto en los glóbulos rojos, lo que

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
137
además acorta la vida media del hematíe. Las anemias hemolíticas intrínsecas son a menudo hereditarias, como la anemia drepanocítica y la talasemia.
− Anemia hemolítica extrínseca o autoinmune: los glóbulos rojos se producen sanos pero más tarde son destruidos al quedar atrapados en el bazo o al ser destruidos por una infección o por fármacos que pueden afectar a los glóbulos rojos. La anemia hemolítica extrínseca puede ser provocada por: infecciones (hepatitis, el citomegalovirus, el virus Epstein-Barr...), medicamentos,(como la penicilina las sulfamidas o el acetaminofén...), la leucemia o el linfoma, los trastornos autoinmunes (como el lupus sistémico eritematoso, la artritis reumatoidea, el síndrome de Wiskott-Aldrich o la colitis ulcerosa), trastornos hereditarios o por diversos tumores. Algunos tipos de anemia hemolítica extrínseca son temporales y se curan tras un período de varios meses, en cambio otros tipos de anemias extrínsecas, pueden volverse crónicas con períodos de remisiones y recidivas.
La anemia hemolítica se caracteriza por una serie de síntomas clínicos, palidez, fatiga, dificultad al respirar, astenia, disnea de esfuerzo, hipoxia cardiaca, lo que da lugar a palpitaciones, amenorrea, hipoxia cerebral…
Para el diagnóstico adecuado de la anemia hemolítica, son necesarios los siguientes análisis de laboratorio:
− Análisis de sangre: la finalidad de este análisis es la de medir la cifra de hemoglobina y reticulocitos, indicando la cantidad de glóbulos rojos nuevos que se producen.
− Análisis de sangre adicionales: se utilizan para verificar el funcionamiento del hígado así como la presencia de ciertos anticuerpos.
− Análisis de orina: para observar la presencia de hematuria.
− Biopsia por aspiración y por punción de la médula ósea: es un examen empleado para estudiar la cantidad, tamaño y madurez de los glóbulos y, o de las células anormales. Este procedimiento comprende la extracción de una pequeña cantidad de líquido de la médula ósea (aspiración) y/o de tejido sólido de la médula ósea (biopsia core o por punción), generalmente de los huesos de la cadera.


UNIDAD TEMÁTICA IV PRUEBAS DIAGNÓSTICAS


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
141
4.1 Introducción
De acuerdo al criterio de clasificación más reciente (Sydney 2006), un diagnóstico de SAF requiere una manifestación clínica y una prueba de laboratorio (anticuerpos antifosfolípidos).
Criterios de clasificación del SAFC `
1. Afectación de al menos 3 órganos, sistemas y/o tejidos.
2. Desarrollo de los síntomas simultáneamente o en menos de una semana.
3. Confirmación anatomopatológica de una oclusión microvascular en al menos un órgano o tejido.
4. Confirmación serológica de la presencia de anticuerpos antifosfolípidos (presencia de un anticoagulante circulante de tipo lúpico y/o de anticuerpos anticardiolipinas) ƒ
SAFC definitivo: Presencia de 4 criterios
SAFC probable:
− Presencia de los criterios 2, 3 y 4 pero afectación solamente de 2 órganos, sistemas o tejidos.
− Presencia de los criterios 1, 2 y 3 pero ausencia de confirmación serológica con al menos 6 semanas de intervalo, debido al fallecimiento precoz de un paciente al que nunca se le hicieron pruebas para determinar la presencia de anticuerpos antifosfolípidos antes de la aparición del SAFC
− Presencia de los criterios 1, 2 y 4
− Presencia de los criterios 1, 3 y 4 con desarrollo de un tercer episodio clínico en un período de más de una semana pero menos de un mes, a pesar del tratamiento anticoagulante.
4.1.1 Criterios o Manifestaciones Clínicas
• Trombosis vascular
− Trombosis arterial, venosa o de pequeño vaso en cualquier órgano o tejido confirmada con métodos objetivos. En caso de histiología, trombosis sin presencia de inflamación.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
142
• Complicaciones en el embarazo
− Una o más muertes fetales inexplicadas después de la décima semana de gestación con feto normal.
− Una o más muertes prematuras de un feto normal antes de la 34 semana de gestación debida a preeclampsia severa, eclampsia o insuficiencia placentaria.
− Tres o más abortos espontáneos consecutivos antes de la décima semana de gestación tras excluir causas anatómicas o genéticas en la madre.
4.1.2 Criterios o Pruebas de Laboratorio (anticuerpos antifosfolípidos)
• Anticoagulante lúpico en plasma, confirmado en dos determinaciones separadas 12 semanas, detectado según las guías de la Sociedad Internacional de Trombosis y Hemostasia.
• Anticuerpos anticardiolipina (ACA) isotipo IgG o IgM por ELISA con título moderado o alto confirmado en dos ocasiones separadas 12 semanas.
• Anticuerpos anti B2-GP1, isotipo IgG o IgM por ELISA confirmado en dos ocasiones separadas 12 semanas.
Asimismo en la reunión de Sidney de 2006 se acordó clasificar a los pacientes en subtipos:
− Subtipo I: si presentan más de un anticuerpo antifosfolípido
− Subtipo IIa: si sólo presentan anticoagulante lúpico
− Subtipo IIb: si sólo presentan anticuerpos anticardiolipina
− Subtipo IIc: si sólo presentan anticuerpos anti-beta-2-glicoproteína I
Las pruebas de laboratorio que demuestran la presencia de un anticuerpo antifosfolípido pueden dividirse en dos categorías:
1. Inmunológicas (ELISA): detectan autoanticuerpos dirigidos principalmente contra las cardiolipinas y Beta 2 glicoproteínas (B2-GP1).
2. Ensayos de coagulación: demuestran la presencia del anticoagulante Lúpico (AL) que son inmunoglobulinas que inhiben la coagulación in vitro pero están asociadas a trombosis.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
143
4.2 Diagnóstico en laboratorio Como hemos visto anteriormente según el consenso de la reunión de
Sidney de 2006 para el diagnóstico de este síndrome es principal presentar una manifestación clínica y una prueba de laboratorio refiriéndose en esta reunión como prueba de laboratorio a la presencia de al menos uno de los anticuerpos antifosfolípido que ya sabemos que son el anticoagulante lúpico, el anticuerpos anti-beta-2-glicoproteína I y el anticuerpo anticardiolipina. Sin embargo en este trabajo trataremos también otros anticuerpos que también nos ayudan y orientan en el estudio de esta enfermedad así como otras pruebas de laboratorio, obligatorias que se le piden a cualquier paciente siempre que se le realiza una analítica básica.
En cuanto a las manifestaciones clínicas, la anamnesis en primer lugar debe ir orientada especialmente en la búsqueda de posibles antecedentes de trombosis, obstétricos y otro tipo de manifestaciones de SAF u otra enfermedad autoinmune sistémica junto con una exploración física del paciente completa.
Determinación de anticuerpos antifosfolipidos (AAF)
• Anticuerpos antifosfolípido (AAF)
− Anticuerpos anticardiolipina (AAC), isotipos IgG e IgM por ELISA
− Anticoagulante lúpico
− Anticuerpos antibeta-2-glicoproteína I (cofactor de anticuerpos anticardiolipina)
Otros anticuerpos
• Antifosfolípidos
− Anticuerpos antifosfatidilserina
− Antiácido fosfatídico
− Antifosfatidilinositol
− Antifosfatidilglicerol
− Anticuerpos antifosfatidiletanolamina.
• Antiprotrombina
• Antianexina V
• Anticomponentes del sistema de la proteína C

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
144
• AntiLDL oxidada
• Anticélula endotelial
Pruebas de Laboratorio obligatorias
• Estudio analítico básico. Hemograma completo, coagulación básica con tiempo de protrombina y cefalina, VSG, glucemia, función renal con sedimento de orina y proteinuria en orina de 24 horas, ionograma básico, perfil lipídico y hepático, etc.
• Estudio inmunológico básico. Anticuerpos antinucleares, factor reumatoide, cuantificación de inmunoglobulinas IgG, IgM e IgA junto con fracciones de C3, C4 y CH50 del complemento. Si existe sospecha de lupus eritematoso sistémico (LES), se determinarán también los anticuerpos anti-DNA y anti-ENA (Sm, RNP, Ro, La), etc.
4.2.1 Determinación de anticuerpos antifosfolipidos (AAF) Como ya sabemos, el síndrome antifosfolípido (SAF) es una trombofilia
autoinmune adquirida caracterizada por la presencia de anticuerpos antifosfolípido, procesos trombóticos y/o pérdidas fetales recurrentes, por ello es clave su determinación.
4.2.1.1 Anticuerpos anticardiolipina (AAC) isotipos IgG e IgM por ELISA.
En el ELISA la cardiolipina se usa como antígeno. Para ello se fija previamente al ensayo a los pocillos de la placa. Esta es la etapa de adhesión o coating en la que es necesaria la presencia de beta-2-glicoproteína I en el medio al igual que lo será en la etapa de adhesión del anticuerpo a la cardiolipina..
La beta-2-glicoproteina I se obtiene de proteína humana purificada o también puede obtenerse a partir de suero bovino adulto o fetal. Cuando se añade el suero o plasma del paciente, si existen anticuerpos anticardiolipina presentes en él, se fijarán a la cardiolipina presente en la placa y los detectaremos con un anticuerpo antiinmunoglobulina humana marcado con un cromógeno que se revelará al final del ELISA con un sustrato.
Se han desarrollado también inmunoensayos de quimioluminiscencia para determinar anticuerpos anticardiolipinas mediante la fijación de partículas magnéticas y detectando este complejo con anticuerpos antiinmunoglobulina humana con isoluminol. También existen en el mercado ensayos por revelado fluoroenzimático.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
145
Un requisito indispensable para detectar anticuerpo anticardiolipina es el uso de calibradores valorados frente a un estándar internacional. Esto permite expresar los resultados en unidades GPL (G phospholipid unit) para los IgG y MPL para los IgM. Recientemente se está investigando la posibilidad de sustituir el estándar actual obtenido de mezclas de plasmas humanos por uno basado en anticuerpos monoclonales.
También pueden detectarse anticuerpos anticardiolipinas por citometría de flujo. En este ensayo se utilizan partículas de poliestireno recubiertas de fosfolípidos. Esto se incuba con el plasma del paciente y los anticuerpos que se hayan fijado son revelados con un anticuerpo antiinmunoglobulina humana marcado con un fluorocromo que será leído en el citómetro de flujo.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
146
Un paso más es emplear partículas de dos tamaños distintos para detectar simultáneamente anticuerpos anticardiolpina y antifosfatidilserina.
Las técnicas de citometría de flujo actualmente presentan la ventaja de ser más rápidas que el ELISA convencional.
En cuanto a los resultados, los anticuerpos anticardiolipinas suelen expresarse cuantitativamente en GPL ó MPL, sin embargo, entre distintos laboratorios la concordancia dado el empleo de productos diferentes debería orientarse a ofrecer resultados semicuantitativos.
Para que estos anticuerpos sean aceptados como criterio diagnóstico es imprescindible que su título sea medio o alto, es decir, normalmente más de 40 GPL o MPL por encima del percentil 99 de la población general y que además se haya probado su positividad al menos en dos ocasiones con una separación de doce semanas entre un análisis y otro. También es requisito indispensable que en el ensayo utilizado sea beta-2-glicoproteína I dependiente.
Como hemos mencionado ya, los isotipos relevantes para el diagnóstico son los IgG e IgM, siendo incluso la subclase IgG2 la que predominante se asocia con las manifestaciones clínicas. En cuanto al isotipo IgA, según el consenso de Sidney parece ser que podría ser útil para el pronóstico de determinadas manifestaciones clínicas en afroamericanos pero no como valor diagnóstico.
4.2.1.2 Anticoagulante lúpico
La determinación del anticoagulante lúpico se realiza en tres etapas:
1. Cribado
• Tiempo tromboplastina parcial activada
• Tiempo de tromboplastina parcial activada diluida
• Test de inhibición de la tromboplastina tisular diluida
• Tiempo de coagulación con caolín
• Test de veneno de víbora de Russell diluido
• Tiempo de textarina
• Tiempo de veneno de Taipán
Lo importante de estas pruebas de cribado es obtener una alta sensibilidad.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
147
2. Identificación del inhibidor
Si alguna de estas pruebas está alargada se debe probar la presencia de un inhibidor en experimentos de mezcla. Para esto debe repetirse la prueba que aparece alargada pero esta vez mezclando el plasma del paciente con plasma normal a partes iguales. Si el resultado sigue igual es que hay algún inhibidor plasmático.
3. Confirmación del inhibidor
Finalmente para terminar de confirmar la presencia del anticoagulante lúpico se repite la prueba coagulométrica añadiendo un exceso de fosfolípidos a la muestra procedentes de un lisado plaquetario o bien sintéticos. Esto debería corregir la alteración observada en la prueba. Como alternativa al uso de fosfolípidos se puede usar el cociente entre el tiempo de textarina y el de ecarina.
4. Descartar la presencia de un inhibidor específico de un factor de la coagulación
En algunos casos es necesario este paso.
A parte de los ensayos basados en el cribado, identificación, confirmación y descarte de un inhibidor específico de algún factor de la coagulación, existen otros ensayos para determinar el anticoagulante lupico como es el ASLA (Activated Seven Lupus Anticoagulant) que activa el anticoagulante con una dilución de fosfolípidos de cerebro y lo neutraliza con plaquetas. Esta prueba se basa en la exploración de la vía extrínseca de la coagulación permitiendo una alta identificación de positivos (87%) de los cuales un 9% es detectable solo en ensayos de la vía intrínseca. También han surgido modificaciones del test de veneno de víbora de Russell diluido reduciéndole la concentración de calcio.
Es muy importante saber que las pruebas coagulométricas son especialmente sensibles a las condiciones pre analíticas y analíticas ya que el plasma se deteriora rápidamente y hay que tener especial cuidado en la separación de las plaquetas para evitar que sus fosfolípidos interfieran en los resultados. La separación de las plaquetas suele hacerse mediante una doble centrifugación o también por microfiltración para separar el plasma siempre que la muestra vaya a congelarse antes de ser procesada.
Otro punto a tener en cuenta es que no es recomendable determinar el anticoagulante lúpico en la fase aguda de las trombosis ya que ciertos reactantes de la fase aguda pueden interferir en las pruebas de cribado. Del mismo modo, la presencia de heparina no fraccionada, heparina de bajo peso

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
148
molecular o anticoagulantes orales interfieren o incluso imposibilitan la interpretación de los resultados.
En cuanto a los resultados el anticoagulante lupico se valora como positivo o negativo. Con el objetivo de reducir la variabilidad de las distintas pruebas se recomienda el uso de razones normalizadas pero aunque esto aumenta la precisión y la especificidad puede llegar a disminuir la sensibilidad. A veces los resultados se expresan únicamente en razón del tiempo de víbora de Russell diluido con y sin fosfolípidos lo cual no es del todo correcto ya que debería informarse como positivo o negativo siguiendo todos los pasos vistos anteriormente y utilizando varias pruebas de cribado, ya que ninguna de ellas tiene una sensibilidad del 100% de forma aislada.
En cualquier caso la concordancia interlaboratorios no es del todo buena por lo que es conveniente seguir un programa de evaluación externa de calidad.
El hallazgo clínico del anticoagulante lúpico es importante ya que es un criterio de laboratorio para el diagnóstico del síndrome antifosfolípido. Para que sea considerado como criterio analítico debe identificarse al menos dos veces con una separación mínima entre una y otra de doce semanas.
En lo referente a la titulación o cuantificación del anticoagulante se hace imprescindible disponer de material de referencia adecuado y valorado. El uso de estándares empleando anticuerpos monoclonales podría utilizarse en la cuantificación siempre y cuando tenga una buena linealidad.
4.2.1.3 Anticuerpos antibeta-2-glicoproteína I (cofactor de anticuerpos anticardiolipina)
Su determinación se hace por técnicas de ELISA fijando beta-2-glicoproteína I generalmente humana a los pocillos de la placa en ausencia de fosfolípidos. Primeramente deben irradiarse las placas con rayos gamma para oxidar la superficie de la placa generando así cargas negativas a las que se unirán las moléculas de beta-2-glicoproteína I, lo cual genera cambios conformacionales en ellas que hacen que aparezcan neoepítopes que serán reconocidos por los anticuerpos antibeta-2-glucoproteína I si están presentes en la muestra.
Hasta hoy no existen materiales de referencia debidamente estandarizados para la determinación de los anticuerpos anti-beta-2-glicoproteína I.
En cuanto a los resultados al no existir un material de referencia validado se considera un resultado positivo si supera el percentil 99 de la población

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
149
general. Cada fabricante de reactivos tiene sus propias unidades arbitrarias aplicando un nivel propio de positividad. Debido a esto la comparación entre laboratorios no es muy útil.
Estos anticuerpos en un paciente con el síndrome aparecen son en torno al 50% del total. Si el paciente presenta estos anticuerpos tienen un riesgo aumentado de sufrir trombosis. Son por sí mismos considerados como un criterio de laboratorio para el diagnóstico del síndrome siempre y cuando aparezcan al menos dos veces con una separación mínima entre una y otra de doce semanas.
4.2.2 Otros anticuerpos
4.2.2.1 Anticuerpos antifosfolípidos
Los principales son los anticuerpos antifosfatidilserina, los antiácido fosfatídico, los antifosfatidilinositol, los antifosfatidilglicerol y los anticuerpos antifosfatidiletanolamina.
Se detectan individualmente por técnicas de ELISA que emplean suero bovino que aporta beta-2-glicoproteína I.
Recientemente se ha desarrollado un ensayo llamado “anticuerpos aPhL” que se hace con ELISA para determinar conjuntamente algunos de estos anticuerpos. En esta prueba se emplea una mezcla compleja de fosfolípidos aniónicos para la unión de los anticuerpos del paciente. Actualmente no se utiliza en la práctica clínica habitual pero se piensa que podría emplearse ya que podría considerarse como una evolución de la determinación de los anticuerpos anticardiolipina diferenciando más específicamente los anticuerpos patogénicos frente a los no patogénicos.
Actualmente no existen materiales de referencia internacionales para ninguna de estas pruebas ni cuentan con una metodología estandarizada. No existe por tanto una buena reproducibilidad entre laboratorios.
Representan una prevalencia del 30% en paciente con lupus eritematoso sistémico pero no está muy definido el papel que juegan en la enfermedad.
La determinación de estos anticuerpos no se realiza de manera habitual en la práctica clínica porque resulta caro y no están muy definidos todavía.
4.2.2.2 Anticuerpos antiprotrombina
Se determinan por ELISA empleando protrombina humana purificada como antígeno fijada en los pocillos de la placa. Igual que ocurría con la beta-2-glicoproteína I, la placa debe ser irradiada con rayos gamma para aumentar

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
150
las cargas negativas de la protrombina humana y que se fije mejor a la placa. A parte de la irradiación también pueden emplearse complejos de protrombina con fosfatidilserina para la adhesión a la placa. Al unirse a la placa la protrombina cambia su conformación haciendo que aparezcan neoepítopes a los que se unirán los anticuerpos antiprotrombina en caso de haberlos.
Presentan una alta prevalencia en los pacientes y hasta el momento se han descrito asociados a casos de trombosis venosa profunda y cardiopatía isquémica sin presentar enfermedad inmunológica alguna ni presencia de otros anticuerpos antifosfolípido.
Ni hay materiales de referencia ni metodología estándar establecida. Al faltar calibradores y debido a la gran diferencia entre laboratorios en cuanto a la determinación de estos anticuerpos la comparación entre ellos es difícil. Su utilidad clínica hoy por hoy está por determinar.
4.2.2.3 Anticuerpos antianexina V Estos anticuerpos se determinan por ELISA utilizando placas irradiadas
que aumentan las cargas negativas a las que se unirán las anexinas humanas purificadas que serán los antígenos de nuestro ensayo. Esto generará cambios conformacionales en los antígenos que aumentarán la probabilidad de unión de los anticuerpos antianexina V que estamos buscando.
Aparecen en el 19% de los pacientes diagnosticados de lupus eritematoso sistémico y parecen asociados a una mayor prevalencia de trombosis arteriales y venosas siendo asociados en su mayoría a las manifestaciones clínicas obstétricas del síndrome antifosfolípido.
Tampoco existen materiales de referencia ni tienen una metodología estandarizada.
4.2.2.4 Anticuerpos frente a componentes del sistema de la proteína C Habitualmente se determinan por ELISA pero ni los materiales están
estandarizados ni hay metodología de referencia por tanto no hay una buena comparabilidad entre laboratorios. Se determinan únicamente en el ámbito de la investigación ya que hasta el momento se desconoce su implicación en el síndrome.
4.2.2.5 Anticuerpos anti-LDL-oxidada Se determinan por ELISA principalmente aunque también puede hacerse
por quimioluminiscencia.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
151
La ELISA en este caso es más complicada porque es necesario separar primero y purificar las LDL y oxidarlas in vitro. Esto se hace para que expresen los neoepítopes, luego se fijan a los pocillos y finalmente se sigue con la adhesión de los posibles anticuerpos en el ELISA. No hay muchos laboratorios que realicen esta prueba por la complejidad que representa. Tampoco están estandarizados los métodos ni existen materiales de referencia, por ello la comparación entre laboratorios no es buena.
Estos anticuerpos suelen estar presentes en pacientes con lupus eritematoso sistémico y en un 25% en pacientes con síndrome antifosfolípido. La presencia de estos anticuerpos parece demostrar una tendencia a padecer trombosis arteriales.
4.2.2.6 Anticuerpos anticélula endotelial
Se determinan con ELISA modificada. En primer lugar deben crearse cultivos de células endoteliales en los micropocillos de la placa y sobre ellas se incuba el plasma diluido de los pacientes. Luego se revelan los anticuerpos que se hayan unido a las células con anticuerpos antiinmunoglobulina humana marcados con cromógenos. Las células endoteliales pueden proceder de cordón umbilical o de grasa abdominal.
El problema de este método son las uniones inespecíficas y la poca reproducibilidad entre pocillos precisamente por tratarse de células vivas. Esto obliga a tener que utilizar varios controles negativos y también a tener que hacer réplicas de una misma muestra. Parece ser que si se usan células de cordón fijadas a la placa en lugar de en cultivo mejora la reproducibilidad entre los pocillos.
Actualmente no existen materiales estandarizados ni métodos de referencia.
No está muy clara la importancia de estos anticuerpos en la enfermedad, aunque prevalecen en un 67% de los pacientes con trombosis. Parece ser que la influencia de estos anticuerpos es distinta si proceden de cordón o de grasa abdominal. Se estudian únicamente en investigación clínica.
4.2.3 Pruebas de Laboratorio obligatorias
4.2.3.1 Factores del complemento
Las deficiencias de complemento se producen debido a la presencia de algunas enfermedades reumáticas como es el caso del lupus eritematoso sistémico que suele estar asociado al síndrome antifosfolípido.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
152
El estudio de los factores del Complemento es la determinación de los componentes de uno de los factores que influyen directamente en la inflamación llamada cascada del Complemento, entre estos factores se suelen determinar principalmente los factores C3, C4, y la actividad del complemento total ó CH50.
La cascada puede ser iniciada por diversos factores, principalmente la unión de complejos antígeno-anticuerpo, siendo el producto final de la cascada la unidad de ataque de la membrana celular, que genera agujeros en las bacterias pero también en ocasiones en células del propio cuerpo ocasionando enfermedades auto-inmunes.
La actividad del complemento CH 50 mide la actividad que le queda a la cascada por activarse, si esta actividad es baja es que el complemento ya está siendo activado por otros factores y se encuentra agotada su capacidad.
La actividad de complemento (CH50 o las proteínas individuales de complemento) se miden para determinar si el complemento está involucrado en el origen o en el proceso de diferentes enfermedades. La actividad de complemento se mide también para controlar la gravedad o evolución de una enfermedad, tanto como diagnóstico como para comprobar la eficacia del tratamiento.
Por ejemplo, los pacientes con Lupus Eritematoso Sistémico activos pueden tener niveles bajos de C3 y C4, y estos niveles del Complemento pueden seguirse como un índice de la actividad de la enfermedad. Los valores normales del complemento son:
− CH 50 de entre 75 a 160 unidades por mililitro.
− C1 Inhibidor entre 16 a 33 miligramos por decilitro.
− C3 Hombres 88 a 252 miligramos por decilitro.
− C3 Mujeres 88 a 206 miligramos por decilitro.
− C4 Hombres 12 a 72 miligramos por decilitro.
− C4 Mujeres 13 a 75 miligramos por decilitro.
La actividad disminuida de complemento aparece el caso de pacientes con enfermedades autoinmunes, como es el caso del síndrome antifosfolípido.
4.2.3.2 Análisis del factor IX Es un examen para medir la actividad del factor IX: una de las sustancias
utilizadas para la coagulación en la sangre.
Esta prueba se utiliza para detectar la causa específica del sangrado excesivo (coagulación sanguínea disminuida).

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
153
Los valores normales son del 50 al 200% del valor de referencia o de la muestra control del laboratorio. La disminución de la actividad del factor IX puede ser producto de una deficiencia congénita del factor IX (hemofilia B), malabsorción de grasa, enfermedades hepáticas (como la cirrosis), deficiencia de vitamina K, o administración de warfarina.
4.2.3.3 Recuento de plaquetas
Las plaquetas también intervienen en la formación del coágulo sanguíneo, es decir la hemostasia, produciéndose su adhesión a la zona dañada, activando al factor VIII y liberando los fosfolípidos necesarios para el proceso de la coagulación.
Con este examen se pretende hacer un recuento del número de plaquetas presentes en el organismo del paciente, para ver si este, presenta trombocitopenia lo que estaría asociado al síndrome antifosfolípido.
Los valores de plaquetas normales que deben existir en el organismo del paciente oscilan entre 150.000 a 400.000 células por milímetro cúbico.
4.2.3.4 Hemograma
Se le realizará al paciente un hemograma básico para ver si los demás parámetros están o no alterados.
− El recuento bajo glóbulos rojos en este caso sería indicativo de una hemorragia.
− El conteo bajo de glóbulos blancos se denomina leucopenia y pueden ser indicio de enfermedades vasculares del colágeno como es el caso del lupus eritematoso.
− El hematocrito suele verse aumentado debido a la presencia de trombosis.
4.2.3.5 Tiempo parcial de tromboplastina
La tromboplastina es también otro de los factores que intervienen en el proceso de la coagulación sanguínea.
Con este análisis medimos el tiempo de coagulación en el plasma para evaluar una amplia variedad de trastornos de la coagulación sanguínea y sirve para la evaluación de los trastornos de coagulación y sangrado excesivos.
Los valores normales son de entre 25 a 35 segundos en el caso del síndrome antifosfolípido debido a la disminución del número de plaquetas el tiempo parcial de tromboplastina se verá aumentado.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
154
4.2.3.6 Fibrinógeno
Otro factor de la coagulación es el fibrinógeno que es una proteína producida por el hígado. Este examen sirve para medir la cantidad de fibrinógeno en la sangre realizándose cuando la coagulación del individuo se encuentra anormal, relacionándose además con el desprendimiento prematuro de la placenta lo que daría lugar a los abortos recurrentes y también se realiza para la prueba de la coagulación intravascular diseminada, explicada con anterioridad.
Los valores normales de fibrinógeno en el organismo del paciente son de entre 200 a 400 miligramos por decilitro y los valores anormales pueden ser debidos a una insuficiencia en la producción de fibrinógeno (adquirida o congénita), al uso excesivo de fibrinógeno (como en la coagulación intravascular diseminada), a la fibrinólisis o descomposición anormal del fibrinógeno (ya sea primaria o secundaria) o a una hemorragia con transfusión de sangre deficiente en fibrinógeno.
4.2.3.7 Tiempo de protrombina Es un examen que mide el tiempo de coagulación del plasma (la porción
líquida de la sangre), que se utiliza para evaluar trastornos de la coagulación sanguínea, usualmente sangrado.
El rango normal es de 11 a 13,5 segundos viéndose aumentado en pacientes con síndrome antifosfolípido, debido a deficiencia de vitamina K, terapia con cumadina (warfarina), deficiencia del factor VII, deficiencia del factor X, deficiencia del factor II (protrombina), deficiencia del factor V, deficiencia del factor I (fibrinógeno).
4.2.3.8 Antitrombina
Es un examen de sangre que mide la cantidad de antitrombina III que es una proteína que ayuda a prevenir y regular la coagulación sanguínea, realizándose este examen cuando hay episodios repetidos de coágulos sanguíneos, entre otros.
El rango normal se ubica entre 0,20 y 0,45 miligramos por mililitro.
4.2.3.9 Tiempo de euglobulina o fibrinólisis
Es un examen que mide el tiempo de fibrinólisis en la sangre que es una medida de la actividad del sistema fibrinolítico (disolución de coágulo).
Este es uno de los mejores exámenes para diferenciar la fibrinólisis primaria de la coagulación intravascular diseminada.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
155
Los valores normales están entre 90 minutos a 6 horas. La lisis del coágulo normalmente se completa entre 2 y 4 horas.
El aumento de la fibrinólisis puede deberse a:
− Realización de un ejercicio muy intenso.
− Envejecimiento.
− Administración de estreptocinasa o urocinasa.
− Corticosteroides y la HACT (hormona adrenocorticotrópica).
La disminución de la fibrinólisis (aumento del tiempo de lisis) pude ser originada por:
− La prematurez.
− Deficiencia hereditaria de fibrinógeno.
− Complicaciones obstétricas (por ejemplo, hemorragia preparto, mola hidatidiforme, embolia de líquido amniótico).
4.2.3.10 Análisis del factor VIII
Usualmente el factor sérico VIII se mide para diagnosticar o controlar el tratamiento para la hemofilia. Este examen se puede utilizar para detectar la causa específica del sangrado excesivo (disminución en la capacidad de coagulación de la sangre).
4.2.3.11 Vitamina K
La vitamina K es una vitamina liposoluble conocida como vitamina de la coagulación, que juega un papel importante en la coagulación de la sangre, ya que en ausencia de esta, la sangre no coagularía.
4.2.3.12 Fragilidad capilar
Este examen se utiliza para ver la fragilidad de los capilares evaluando la fragilidad de las paredes capilares o la deficiencia de plaquetas sanguíneas (trombocitopenia), para ello se coloca un torniquete utilizado para medir la presión sanguínea en el brazo, ejerciendo suficiente presión para evitar el retorno venoso de la sangre. El incremento en la presión en los vasos capilares resultante puede ocasionar su ruptura capilar, la cual se manifiesta en forma de petequias (áreas pequeñas de color púrpura sobre la piel).
Después de cinco minutos, se retira la presión, se cuenta la cantidad de petequias que se encuentren en un área de cinco centímetros y se clasifican de acuerdo con un sistema de puntaje.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
156
4.2.3.13 Dímero D
El Dímero-D es uno de los productos de degradación de la fibrina, es utilizado en otras patologías ya que no es normal tener altas concentraciones del Dímero-D en la circulación puesto que las concentraciones normales son de 0 a 200 miligramos decilitro, excepto en pacientes que han sufrido de episodios trombóticos y que han sido anticoagulados, en estos casos se utiliza la medición del Dímero-D en el seguimiento del paciente, demostrando que la medicina le está funcionando, en la degradación del trombo.
En la TVP el Dímero-D determinado mediante la técnica de ELISA, tiene un valor predictivo positivo del 44% y un valor predictivo negativo del 98%, por lo tanto, un resultado negativo del Dímero-D en pacientes con Bajo Riesgo de TVP según el modelo de Wells permite descartar la trombosis sin necesidad de recurrir a otras técnicas.
El Dímero-D aumenta significativamente durante el embarazo normal y parto. Un estudio muestra que el dímero-D aumenta en forma significativa durante el embarazo. Además dichos incrementos pueden no correlacionarse con un aumento de la actividad trombogénica.
4.2.3.14 Factor Anti-XA
El factor Anti-Xa permite realizar detectar los niveles de heparina en sangre. Los valores deben ser entre 0,3 y 0,6 en el embarazo tratado con heparina (porque cambia si el tratamiento con heparina se aplica a otros trastornos...). Debe realizarse a las cuatro horas siguientes tras la aplicación, donde llega a su máximo valor.
4.2.3.15 Niveles de activador de plasminógeno tisular (T-PA)
El plasminógeno tisular es una enzima circulante en la sangre relativamente inactiva que cuando se incorpora al coágulo de fibrina convierte activamente el plasminógeno unido a la fibrina en plasmina, la cual degrada el coágulo de fibrina.
4.2.3.16 Actividad del activador inhibidor del plasminógeno I
Es una glucoproteína que está presente en el plasma y se concentra principalmente en el hígado. Es un precursor inactivo de la plasmina.
La ruptura del enlace Arg560-Val561 convierte el plasminógeno en plasmina. Entre los activadores fisiológicos del plasminógeno están, el tiempo de plastina activada y la urokinasa.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
157
4.2.3.17 Pruebas analíticas de los anticuerpos antinucleares Es una determinación analítica sanguínea que mide las inmunoglobulinas
Ig G que son específicas contra antígenos del núcleo de células del propio cuerpo. Esta medición de las inmunoglobulinas será explicada en las pruebas de investigación, pudiéndose medir mediante el método de ELISA o por inmunofluorescencia directa.
Se utiliza para comprobar la presencia de dichos anticuerpos antinucleares, en enfermedades autoinmunes y también se puede realizar este examen cuando un paciente tiene síntomas inexplicables tales como artritis, erupciones o dolor en el pecho.
Para la realización del examen sanguíneo se extrae la sangre del paciente por punción venosa usualmente de la parte interior del codo o del dorso de la mano, limpiando la zona para desinfectarla con algún antiséptico y se coloca una banda elástica alrededor del antebrazo, con el fin de ejercer presión y restringir el flujo sanguíneo a través de la vena, lo cual hace que las venas bajo la banda se dilaten y llenen de sangre. Y posteriormente recogemos la sangre con una aguja y la colocamos en un tubo con anticoagulante
Los valores normales son negativos, es decir ausencia de anticuerpos antinucleares detectables en sangre, aunque también se pueden considerar como valores negativos, los resultados positivos a una dilución mayor de 1:20, en algunas ocasiones en personas sin ninguna enfermedad específica puede tener bajos niveles de estos anticuerpos sin una razón evidente.
Cuando los valores son anormales los anticuerpos antinucleares pueden encontrarse en estos enfermos siendo producidos por el sistema inmunológico y atacando a los propios tejidos del cuerpo en vez de toxinas extrañas. Frecuentemente se encuentran en personas con lupus eritematoso sistémico y, con menos frecuencia, en caso de otras enfermedades.
4.2.3.18 Homocisteína La hiperhomocisteinemia es un factor de riesgo establecido de la
arteriosclerosis y otras enfermedades vasculares. En la homocistinuria clásica, la mitad de las complicaciones vasculares son de origen venoso.
El diagnóstico de la homocisteína se emplea para la investigación de factores de riesgo de arteriosclerosis precoz y trombosis.
Este factor de riesgo es mayor entre las mujeres en particular después de la menopausia, aumenta con la edad y es significativo después de excluir otros factores de riesgo. Los niveles elevados de homocisteína puede ser el resultado de una deficiencia de ácido fólico, de vitamina B6 o B12 o presentarse en caso de algunas enzimopatías hereditarias.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
158
4.2.3.19 Examen del cariotipo
Es un análisis cromosómico que se puede realizar a partir de una muestra de sangre, de médula ósea, de líquido amniótico o de tejido placentario.
Este examen se realiza generalmente para evaluar una apariencia anormal o un desarrollo que sugiere una anomalía genética, para evaluar una pareja con antecedentes de abortos. El examen de líquido amniótico se hace para evaluar anomalías cromosómicas en un feto en desarrollo.
La muestra se deja crecer en un cultivo de tejido en el laboratorio y luego se toman las células, se tiñen y se observan bajo el microscopio. Las células se fotografían para obtener un cariotipo que muestra la organización de los cromosomas. Las anomalías se pueden detectar a través de la organización de los cromosomas.
Para examinar el líquido amniótico se realiza una amniocentesis y para examinar un espécimen de médula ósea se requiere de una biopsia de médula ósea. El examen del tejido placentario se hace después de un aborto.
4.2.3.20 Mutación del factor V de Leiden
El factor V Leiden es el factor de riesgo genético más frecuente para la trombosis venosa. El factor V es uno de los factores normales de coagulación sanguínea. El factor V Leiden es una forma cambiada o "mutada" del factor V que se inactiva diez veces más lentamente que el factor V normal. Esto provoca que permanezca más tiempo en la circulación, produciendo un estado de hipercoagulación, con lo que la sangre sigue coagulándose, produciéndose una posible obstrucción de los vasos sanguíneos.
Una copia del gen del factor V Leiden aumenta el riesgo de trombosis venosa de 4 a 8 veces, mientras que dos copias del gen aumentan el riesgo 80 veces.
Otros defectos coexistentes de la coagulación pueden producirse con el factor V Leiden y, en general, el riesgo de trombosis aumenta en los pacientes que tienen más de un defecto genético.
La mutación del factor V Leiden está implicada en el 20 al 40 % de los casos de trombosis venosa, y se sospecha en individuos que tienen antecedentes médicos de trombosis venosa o en familias en las que existe una alta incidencia de trombosis venosa.
La medición de este factor se realiza mediante un estudio funcional que se basa en la comparación del tiempo de coagulación de la tromboplastina parcial activada en el plasma del paciente.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
159
Este test tiene una sensibilidad y especificidad para el factor V Leiden del 85 al 90% aunque una desventaja importante que presenta este tipo de test es que no puede ser utilizado para el análisis de pacientes en tratamiento con anticoagulantes.
El test más directo para la identificación del factor V Leiden es el análisis genético que incluye la utilización de ADN genómico como molde para la amplificación de fragmento del gen del factor V que contiene la mutación. Se puede detectar la presencia o ausencia de la mutación mediante restricción enzimática, uso de sondas alelo-específicas o secuenciación directa del fragmento de PCR amplificado.
4.2.3.21 Mutación gen de la protrombina (G20210A)
La variante alélica protrombina 20210A es una mutación en el gen de la protrombina asociada a un aumento de la concentración plasmática de protrombina y que confiere un mayor riesgo de trombosis venosa a los individuos portadores de esta mutación. Se ha observado una incidencia de 18% en pacientes con historia familiar de trombosis, mientras que en sujetos sanos es de aproximadamente 2%. Debido a que este defecto es causado por una mutación puntual, el análisis molecular es considerado un pilar diagnóstico.
Esta mutación ha sido encontrada en 1%-2% de individuos sanos, 6,2% de pacientes con un primer episodio de trombosis venosa profunda y 18% de los pacientes con trombofilia familiar no explicada.
El diagnóstico de laboratorio para demostrar la presencia de protrombina 20210A está basado únicamente en el análisis del ADN. La detección de esta variante alélica se realiza mediante técnicas de genética molecular, las cuales se basan en la amplificación del ADN por PCR seguida de digestión enzimática.
4.2.3.22 Factor reumatoide
El factor reumatoide es una prueba que mide la presencia y nivel de Inmunoglobulina M específicas contra las inmunoglobulinas G anormales, producidas por los linfocitos de las articulaciones de las personas afectadas de artritis reumatoide, produciéndose la unión de estas a las inmunoglobulinas M formando inmunocomplejos Ig G-Ig M, que producen la activación del complemento, así como de otros factores inflamatorios, siendo por tanto una enfermedad autoinmune. La medición del factor reumatoide puede realizarse por el método de nefelometría o por el método de aglutinación.
La toma de muestras se realizará por punción venosa, extrayendo la sangre e introduciéndola en un tubo con anticoagulante para bioquímica.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
160
Los resultados suelen aparecer positivos en un 80 % de las personas con artritis reumatoide y en enfermedades autoinmunes no asociadas con la enfermedad. Los valores normales que deben darse para descartar la presencia del factor reumatoide, serán menores a 60 U/ mililitro (si la determinación se hace por nefelometría) o un título menor a 1:80 (si se efectúa por el método de aglutinación).

UNIDAD TEMÁTICA V PRUEBAS PARA LA INVESTIGACIÓN
DE LAS MUESTRAS


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
163
5.1 Requisitos diagnósticos para el síndrome antifosfolipídico
5.1.1 Requisitos clínicos Mayores (Sapporo)
A.1. Trombosis vascular Uno o más episodios clínicos de trombosis arterial, venosa o de pequeños vasos en cualquier tejido u órgano. Debe confirmarse por técnicas de imagen, estudios Doppler y/o histopatología. Se excluye la trombosis superficial venosa. Histopatológicamente la trombosis debe estar presente sin evidencia de inflamación en la pared de los vasos
A.2. Morbilidad gestacional
• A.2.1. Una o más muertes fetales inexplicadas de fetos morfológicamente normales (documentados por ecografía o examen directo del feto) en la semana 10 o posterior de gestación
• A.2.2. Uno o más nacimientos prematuros de neonatos normales en la semana 34 de gestación o anterior debidos a preeclampsia, eclampsia o insuficiencia placental grave
• A.2.3. Tres o más abortos consecutivos espontáneos inexplicados antes de la semana 10 de gestación. Se excluyen anormalidades anatómicas u hormonales maternas, o bien cromosómicas tanto maternas como paternas
Menores (Tours)
A.3. Dos pérdidas fetales espontáneas consecutivas antes de la semana gestacional 10. Accidente isquémico transitorio. Trombocitopenia. Anemia hemolítica autoinmune. Corea. Mielopatía transversa. Anormalidades valvulares no reumáticas (por engrosamiento o vegetaciones). Livedo. Úlceras en pierna. Hemorragia adrenal bilateral. Historia familiar de LES o SAF. ¿ACV? ¿Hipertensión pulmonar primaria? ¿Nefropatía con aFL?
5.1.2 Requisitos analíticos Mayores (Sapporo)
B.1) IgG y/o IgM aCL presentes a títulos medios o altos (> 10 GPL/MPL U/ml) en dos o más ocasiones (separadas por lo menos 6 semanas), determinadas por un método ELISA con β2GPI como cofactor

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
164
B.2) AL presente por lo menos en dos o más ocasiones (separadas por lo menos 6 semanas) determinado según las normas de la Sociedad Internacional de Trombosis y Hemostasis (Subcomité de Anticoagulante Lúpico/Anticuerpos Dependientes de Fosfolípidos), siguiendo los pasos:
− Tiempo de coagulación dependiente de fosfolípidos (TTPA/TVVR/TPD/tiempo de textarina/tiempo de Taipan) prolongado
− Fallo en la corrección del tiempo de coagulación prolongado mediante la adición de plasma pobre en plaquetas
− Corrección por adición de fosfolípidos en exceso
− Exclusión de otras coagulopatías
Menores (Tours)
B.3. Presencia de aβ2GPI. Presencia de IgA aCL. Anticuerpos antimitocondriales (M5). ¿Anticuerpos antifosfatidiletanolamina? ¿Antifosfatidilserina? ¿aPT? ¿Anti-LDL-oxidada?
− SAF «cierto» (Sapporo): ≥ 1 requisito clínico mayor (A.1 y/o A.2.) + ≥ 1 analítico mayor (B.1 y/o B.2)
− SAF «probable» (Tours): ≥ 1 requisito clínico mayor (A.1 y/o A.2) + ≥ 1 requisito analítico menor (B.3)
• ≥ 2 requisitos clínicos menores (A.3) + ≥ 1 requisito analítico mayor (B.1
− y/o B.2.)
− SAF «posible» (Tours): ≥ 1 requisito clínico menor (A.3) + ≥ 1 requisito analítico mayor (B.1 y/o B.2.)
5.2 Procesamiento y preparación de las muestras Las siguientes técnicas, son las realizadas en la unidad de investigación
del Hospital Universitario Reina Sofía de Córdoba, algunas de estas técnicas han sufrido algunas modificaciones, ya que estos cambios han sido requeridos para una mejor adaptación a la hora de realizar las investigaciones sobre el síndrome antifosfolípido.
Las investigaciones sobre el síndrome antifosfolípido son realizadas a partir de muestras de pacientes con dicha enfermedad. Estas muestras son la sangre de los pacientes, además de muestras de personas sanas, que son las que actuarán como control, para comparar los resultados obtenidos del paciente.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
165
5.2.1 Separación de las células mononucleadas (FICOLL-HYPAQUE)
Se utiliza para la separación de células sanguíneas, con el fin de poder realizar la separación de los distintos componentes sanguíneos, obteniendo finalmente la separación de las células mononucleadas, ya que se ven alteradas en pacientes con síndrome antifosfolípido.
Para ello se usa un compuesto llamado ficoll, que es muy espeso, lo que hace que las células más grandes como los monocitos queden retenidas en la superficie filtrándose a través de este, sólo las células más pequeñas como los hematíes, que por acción de la gravedad quedarán retenidas en el fondo del tubo.
Para poder llevar a cabo esta técnica son necesarios los siguientes reactivos: ficoll, PBS y la sangre del paciente.
Realización de la técnica.
La realización de la técnica consta de varios pasos:
− Primero se echa la sangre del paciente, junto con el PBS y el ficoll, en proporción 1/3.
− Se toma la misma cantidad de sangre que de PBS, se echa en un tubo universal y se mezcla por inversión.
− Añadir el ficoll en otro tubo y echar la mezcla de PBS + Sangre muy lentamente deslizándolo por las paredes y teniendo la precaución de no mezclarlos. Una vez echado, se coloca el tubo verticalmente con mucho cuidado observándose las dos distintas fases.
− Se centrifuga durante 20 minutos, a 2500 revoluciones por minuto, a temperatura ambiente y se le quita el freno a la centrífuga.
− Una vez terminada la centrifugación, se coge con cuidado la fracción de células mononucleadas, con una pipeta pasteur, muy lentamente para no mezclarlo con el ficoll y se echa en otro tubo con PBS, aproximadamente el triple del volumen recogido para lavar las células, y volvemos a centrifugar 5 minutos, 4ºC a 1800 revoluciones por minuto. Una vez lavado tendremos todas las células sanguíneas a excepción de los hematíes, todas estas células deberán separarse de los monocitos ya que es lo que realmente queremos obtener.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
166
Una vez centrifugado obtendremos el pellet por decantación del sobrenadante , para posteriormente proceder a la separación de los monocitos, mediante la adición de FcR Blocking Reagent, cuya función es bloquear el receptor CD14 de los monocitos para que al añadir el Biotin-antibody cocktail, se una a todas las células menos a los monocitos, puesto que tienen bloqueados todos sus receptores.
El antibiotin-antibody son unas bolitas magnéticas que hacen que al pasar la mezcla por la columna metálica, todas las células que llevan adheridas el antibioting microbeads quedarán unidas a la columna metálica, cayendo sólo las que no tienen el antibiotin-antibody, es decir los monocitos.
Para la realización de esta técnica son necesarios los siguientes reactivos: PBS estéril, buffer, biotin-antibody, antibioting microbeads y azul tripán, (para realizar el recuento).
Realización de la técnica
La realización de esta técnica se lleva a cabo en oscuridad, ya que los reactivos son cromóforos.
− Una vez realizado el Ficoll-hypaque y recogidas las células blancas o monocitos, se procede a su aislamiento, para ello en primer lugar habrá que realizar un recuento:
Pellet diluido en PBS (2 ó 3 mililitros) _ 15 μl
Azul Tripán ______________________ 15 μl
− Se anotan los resultados, se lava el pellet en 1 mililitro de PBS y se centrifuga durante 5 minutos a 4ºC y a 1800 revoluciones por minuto.
− Tras la centrifugación habrá que eliminar completamente el sobrenadante con una micropipeta y resuspender el pellet en PBS, añadiendo 30 μl de PBS por cada 10.000.000 de células contadas, es decir :
− Si contamos 20 millones de células habrá que añadir 30 μl x 2 = 60 μl
− Después se echa 10 μl de FcR blocking reagent por cada 10 millones de células contadas y 10 μl de biotin-antibody cocktail, también por cada 10 millones de células contadas. Se mezcla y se incuba en hielo durante 20 minutos.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
167
− Mientras esperamos prepararemos la columna, a la que tendremos que echar 3 mililitros de buffer:
BSA _______________________________ 0,125 gramos
PBS estéril __________________________ 25 mililitros
EDTA ______________________________ 100 microlitros
Plasma antólogo (del propio paciente) ____ 2,5 mililitros
Se mezcla todo y se centrifuga para quitar los gases que desprende el suero, ya que, si entran burbujas a la columna, las células no podrán pasar a través de esta. Estos 3 mililitros se recogen al final de la columna y se vuelve a echar varias veces, para que la columna no se seque.
Pasado el tiempo de incubación se añade 30 μl de PBS y 20 μl de antibiotin-microbead por cada 10 millones de células contadas. Se mezcla bien y se incuba 15 minutos en hielo.
A continuación hay que lavar las células en PBS añadiendo aproximadamente 1 mililitro y se centrifuga 5 minutos a 4ºC a 1800 revoluciones por minuto. Después habrá que eliminar el sobrenadante y resuspender el pellet en 500 μl de PBS, procediendo posteriormente a la separación de los monocitos por la columna metálica.
Una vez recogidos los monolitos en el extremo de la columna, se lavan con 2 mililitros de PBS, se centrifugan a 1800 revoluciones por minuto, 5 minutos a 4ºC obteniendo el pellet al que habrá que añadir 1 mililitro de PBS y realizar un recuento de estos monocitos.
5.2.2 Extracción de proteínas plasmáticas y citosólicas Esta técnica se utiliza para extraer proteínas citoplasmáticas y nucleares,
conjuntamente o por separado, mediante la adición de detergentes que lisan la pared celular y nuclear, con el objetivo de separar dichas proteínas para realizar los posteriores estudios y ver en pacientes con síndrome antifosfolípido que proteínas permanecen normales y cuáles no, mediante el empleo de técnicas como la proteómica.
Los reactivos necesarios para llevar a cabo este proceso son: proteínas del paciente, PBS, PMSF, DTT, Buffer A, aprotinina, igepal 10% y buffer C.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
168
Realización de la técnica
Una vez extraída la sangre del paciente se procederá a la extracción de las distintas proteínas:
• Extracción de proteínas citoplasmáticas:
Partimos de un pellet, lavado con PBS y preparamos (la cantidad de buffer a preparar dependerá del número de muestras ya que para cada una necesitamos 500 μl)
Buffer A:
Buffer A _______________________ 1 mililitro
PMSF _________________________ 10 μl
DTT __________________________ 10 μl
Aprotinina _____________________ 15 μl
Buffer C:
Buffer C _______________________ 500 μl
PMSF _________________________ 2,5 μl
DTT __________________________ 5 μl
Aprotinina _____________________ 10 μl
Se le echa a cada muestra 400 μl de la mezcla de buffer A y se incuba en hielo durante 15 minutos, pasado este tiempo de incubación, suponiendo que se tuviesen dos muestras habríamos echado antes del tiempo de incubación 400 μl de mezcla buffer A, a cada una por tanto se habrían gastado 800 μl de mezcla, por tanto, como se ha preparado 1000 μl nos quedarán 200 μl a los que se le añadirán Igepal al 10% y de lo cual se echará 50 μl a cada muestra, se voltea, y se centrífuga 5 minutos a 12000 revoluciones por minuto y a 4º C y se alícuota el sobrenadante, que es lo que nos servirá para realizar las distintas investigaciones.
El Igepal, actúa como detergente, lisando las células, haciendo que las proteínas del citosol queden libres y se puedan recoger junto con el sobrenadante.
• Extracción de proteínas nucleares:
Una vez centrifugado y quitado el sobrenadante, se le añade al pellet 50 μl de la mezcla de buffer C a cada muestra e incubamos 15 minutos en hielo y volteamos cada 5 minutos.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
169
Se centrifuga 5 minutos a 12000 revoluciones por minuto a 4º C y se extrae el sobrenadante, que serán las proteínas nucleares, se alícuota y se conserva, para realizar los posteriores estudios.
5.2.3 Extracción de ARN El ARN de las células se extrae para realizar técnicas como la RT-PCR,
partiendo siempre de una población celular, la cual será obtenida a partir de la realización de la técnica del ficoll-hypaque, para ello necesitamos los siguientes reactivos: TRI Reagent, cloroformo, isopropanol, etanol (75%), buffer 10x, DNAsa y stop Solution.
Realización de la técnica.
− Se añade 1 mililitro de TRI Reagent al pellet y se esperan 5 minutos a temperatura ambiente. Tras la espera de los 5 minutos se le añaden 200 μl de cloroformo, mezclar con el vortex, esperar 15 minutos a temperatura ambiente y centrifugar a 12000 revoluciones por minuto durante 15 minutos a 4ºC.
− Una vez hecho lo anterior se transfiere el sobrenadante a otro vial sin coger nada del halo blanco (donde están las proteínas) y se le añaden 500 μl de isopropanol, volvemos a mezclar en el vortex y esperamos 10 minutos a temperatura ambiente. Pasado este tiempo centrifugamos a 12000 revoluciones por minuto durante 15 minutos a 4ºC.
− Se elimina el sobrenadante y se le añade al pellet 1 mililitro de etanol al 75% (no resuspender) y centrifugar a 7500 revoluciones por minuto durante 5 minutos a 4ºC.
− Seguidamente se elimina el etanol y se deja secar el pellet durante 5 ó 10 minutos, dejando el vial abierto. Se disuelve el pellet en agua libre de ARN-asas (de 50-200 μl según el tamaño del pellet) y se incuba el ARN extraído, durante 10 minutos a 65 ºC.
− A continuación hay que tratar el ARN con Desoxirribonucleasa para eliminar cualquier contaminación con ADN añadiendo por cada μg de ARN:
Buffer 10X ____________________ 5 μl
ADN-asa _____________________ 5 μl
Y se incuba a 30ºC durante 30 minutos.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
170
Después se le añaden 5 μl de “Stop Solution” para inactivar el efecto de la DNA-asa, se incuba a 70ºC durante 10 minutos para desnaturalizar a la enzima, se pone en hielo y se congela a –80ºC.
Una vez procesadas y preparadas las muestras, tienen que ser conservadas a -80ºC,
5.3 Estudio de investigación a tres tiempos Las pruebas de laboratorio para investigación se realizan a cada una de
las muestras de todos los pacientes con síndrome antifosfolípido y se hacen varios estudios a tres tiempos:
− T=0 Antes de la administración, por parte del paciente, del fármaco con el que se va a realizar la investigación.
− T=3 Tres meses tras la administración del medicamento.
− T=6 Seis meses tras la administración del medicamento.
Al realizar este estudio en tres tiempos distintos, lo que se pretende es analizar cada uno de los componentes sanguíneos que se ven afectados durante el desarrollo de dicha enfermedad y ver como actuaría el paciente tras la administración de dicho fármaco, viendo si se restablecen o no los valores que el enfermo de síndrome antifosfolípido tiene alterados.
Por ejemplo:
Algunas investigaciones están demostrando que las estatinas (fármaco sintético) intervienen en la regulación del factor tisular, que está estrechamente ligado al desarrollo de la enfermedad.
Otras investigaciones, han demostrado que la aspirina (ácido acetil salicílico) además de combatir el dolor y la fiebre, también ejerce un efecto anticoagulante y antitrombótico, por lo que es una buena terapia para los enfermos con síndrome antifosfolípido.
Otro tratamiento esencial para las personas con síndrome antifosfolípido es la administración de anticoagulantes, como la heparina o la warfarina, que actúan previniendo la producción de trombos.
5.4 Perspectivas futuras en la investigación Las técnicas empleadas, para llevar acabo futuras investigaciones sobre
la enfermedad, son las siguientes:

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
171
5.4.1 Western-Blot Esta técnica se utiliza para separar proteínas según su peso molecular,
con el fin de ver qué función ejercen determinadas proteínas en pacientes con síndrome antifosfolípido, observando si determinadas proteínas (MAP kinasas) están o no activas en estos pacientes. Una de las funciones finales de estas proteínas es la de activar al factor tisular, por ello es necesario ver si se encuentran o no presentes.
Para separar las proteínas necesitamos lisar las células, esto podemos hacerlo, lisándolas por completo obteniendo conjuntamente tanto las proteínas del núcleo como las del citosol o también podemos lisar 1º el citosol y recoger las proteínas, y después las del núcleo y recogerlas aparte (éste es el método que emplearemos).
Para llevar a cabo esta técnica necesitamos: geles de poliacrilamida que son los geles usados para la investigación, este gel de poliacrilamida se utiliza para electroforesis, formando una especie de entramado, consiguiendo la separación de los componentes de la muestra según densidad, peso molecular… la concentración del gel dependerá del tamaño de la partícula a separar, a mayor tamaño de la partícula menor concentración del gel y viceversa. Este gel lleva poliacrilamida que es cancerígena por lo que hay que tener especial cuidado en su manipulación ya que éste es capaz de penetrar por vía parenteral produciendo a largo plazo o tras un contacto prolongado sin usar protección, mutación en nuestras células.
Los reactivos empleados para la elaboración de los geles de poliacrilamida son: separating 2x, stacking 2x, poliacrilamida 40%, agua bidestilada, TEMED y APS al 10%.
Realización de la técnica
Para la realización de la técnica, primero habrá que montar los cristales uniendo la parte lisa del cristal con otro cristal que tiene unos rebordes laterales, de modo que quede entre ellos un espacio donde se solidificará el gel. Se unen ambos laterales con unas pinzas y se colocan los cristales con las pinzas en un soporte que presiona dichos cristales hacia abajo para impedir que salga el gel.
Preparamos el stacking y separating 1x:
− Separating 1x
Separating 2x ----------------------------- 10 mililitros.
Solución al 40% de poliacrilamida ------- 5 mililitros.
H2O destilada ----------------------------- 5 mililitros.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
172
− Stacking 1x
Stacking 2x ------------------------------ 5 mililitros.
Solución al 40% de poliacrilamida ------ 1 mililitro.
H2O destilada --------------------------- 4 mililitros.
A partir del separating 1x se preparará el sello del fondo del gel:
Separating 1x ------------------- 4 mililitros.
TEMED --------------------------- 10 µl.
APS 10% ------------------------ 60 µl.
El APS 10% es lo último que se echa ya que su función es, la de polimerizar el gel.
Se echa la mezcla entre los cristales con una pipeta rápidamente, puesto que solidifica muy rápido, hasta que alcance una altura aproximadamente de 1 centímetro.
Con el separating 1x que ha sobrado se preparará la parte media que es donde se separarán los componentes de la muestra.
Resto de separating de la etapa anterior.
TEMED ----------------------- 10 µl.
APS 10% -------------------- 200 µl.
Una vez solidificado el sello del fondo se le echa la mezcla con una pipeta dejando el espacio justo para colocar el peine, es decir, se echará mezcla hasta que queden unos 2 centímetros de espacio y seguidamente se le añade 1 mililitro de agua bidestilada (con cuidado de no mezclarla con el gel). Esto se hace para que el gel solidifique en anoxia.
Una vez solidificado se retira el agua introduciendo tiras de papel de filtro pero sin que toque el gel para evitar que éste se rompa. Y el siguiente y último paso es preparar la parte superior en la que se colocará el peine y se formarán los pocillos donde se depositarán las muestras.
Al stacking preparado echamos:
TEMED ----------------------- 5 µl.
APS 10% -------------------- 100 µl.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
173
Se echa con una pipeta hasta rebosar por los cristales y se le coloca el peine que tiene que tener la misma medida que los cristales.
Una vez solidificado, se retira el peine con cuidado de no romper los pocillos y se le echa H2O destilada en estos para lavarlos.
Si en lugar de preparar el gel al 10% lo queremos a otra concentración, tendríamos que aplicarle esas muestras sólo a la poliacrilamida y al agua.
Ejemplo:
Gel poliacrilamida 8% Gel poliacrilamida 10%
Separating 2x -- 5 mililitros. Separating 2x -- 5 mililitros.
Poliacrilamida --- 2 mililitros. Poliacrilamida -- 2,5 mililitros.
H2O destilada -- 3 mililitros. H2O destilada -- 2,5 mililitros.
(El volumen preparado sería para 2 geles).
El loading buffer es un tampón que sirve para aumentar el peso molecular de las proteínas y además las tiñe de azul intenso para hacer más fácil la visualización de las bandas, añadiéndolo junto con la muestra para que ésta tenga una mejor separación de sus componentes.
Los reactivos empleados son: TRIS a pH 6,8 DTT, SDS, glicerol puro, bromoferol blue.
Se añade en el siguiente orden:
TRIS 1 M pH 6,8 ------------- 1,5 ml.
DTT --------------------------- 0,07 g.
SDS --------------------------- 0,5 g
Se disuelve bien para que no queden brumos, se mete en estufa a 37 ºC unos 2 o 3 minutos y se agita con fuerza hasta que quede completamente disuelto.
Glicerol puro ----------------- 2,5 ml.
Se vuelve a agitar ya que el glicerol es muy denso.
Bromoferol blue -------------- 0,5 ml.
Agitar de modo que tome una coloración azul oscuro.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
174
Para la preparación de las muestras es necesario un tubo ependorf pequeño para cada paciente, que se rotulará con el tiempo y nº de paciente. Se le echa a cada tubo 5 µl de loading buffer y la cantidad exacta de proteína que se haya obtenido tras la cuantificación de proteínas, resuspender y en otro tubo echar 5 µl de marcador (el volumen depende de la concentración a la que esté el producto).
Preparación de Running buffer 1x.
Running buffer 5x ................... 200 ml
H2O destilada ........................ 800 ml
Se monta la cubeta y se le añade un poco de running buffer 1x a la cubeta (hasta la mitad aprox.) y añadir running buffer 1x entre la zona de geles hasta que casi llegué al primer cristal, (hay que tener cuidado de que el running de la zona de geles no se mezcle con el del resto de la cubeta).
Se echan las muestras depositando el contenido de cada tubo ependorf en su correspondiente pocillo, sin que se formen burbujas.
Una vez cargado el gel se cierra la cubeta, teniendo la precaución de que coincidan los colores de los electrodos de la cubeta con las de la tapa y conectar a la fuente de alimentación a un voltaje máximo de 100 V. (la intensidad no debe superar los 80 mA).
La transferencia a papel de nitrocelulosa se hace para que el manejo y visualización de las proteínas sea más fácil ya que el gel se puede romper con facilidad.
Mientras corre el gel se preparan dos trozos de filtro para transferencia de 8 x 5 cada uno y una membrana de 8,5 x 5 cm. La membrana debe ser de mayor tamaño que los filtros (ya que si ambos filtros se tocan entre sí, no habría una transferencia correcta de la proteína), y se deja todo el tiempo que tarda en correr el gel metidos en el líquido de transferencia.
Cuando acabe la electroforesis, se sacan los cristales y se abren con cuidado haciendo palanca entre ellos con una espátula y le cortamos y tiramos el stacking (que es la parte donde se formaron pocillos), y pasamos el gel al buffer de transferencia donde se montará el "sándwich" colocando primero el filtro, después la membrana, seguidamente el gel y por último el otro filtro y se pasa rápidamente al aparato de transferencia (trans-blot) electrónico y se conecta poniendo 230 mA y 15 V (cuidando que no llegue a 50 V), manteniéndolo durante una hora.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
175
Una vez transferido del gel a la membrana se tiñe con rojo ponceau para ver las bandas y homogeneidad de proteína puesta en los lisados, se lava con H2O destilada varias veces y después con TTBS durante unos 15 minutos, se pone en BSA y se incuba 2 horas en agitación, pasado este tiempo lavar 3 veces con TTBS, 15 minutos cada vez, desechar el TTBS y añadir 10 mililitros de anticuerpo primario diluido al 1/1000 en TTBS + BSA 3%.
Preparación del BSA 3%:
BSA ------------- 3 g
TTBS ------------ csp 100
Preparación del Anticuerpo primario:
Ac primario ----- 10 µl
BSA 33% ------- 10 mililitros
Incubamos en agitación a 4 ºC durante toda la noche.
Reciclamos el Ac 1rio y echamos TTBS haciendo 2 lavados de 15 minutos cada uno.
Tiramos el TTBS y añadimos el anticuerpo secundario marcado con peroxidasa diluido al 1/5000 - 1/10000 en TTBS + BSA 3%.
Preparación del Anticuerpo secundario:
Ac 2rio ---------- 1 µl
BSA 3% -------- 10 ml
E incubamos durante 1 hora en agitación a temperatura ambiente.
Tiramos el Ac 2rio y lavamos 3 veces con TTBS 5 minutos cada vez.
Cuando analizamos proteínas fosforiladas, el protocolo de bloqueo e incubación con Ac 1rio y 2rio se hace con BSA 3% (en TTBS). Y para el resto de proteínas empleamos leche (5%).
Preparación de leche:
Leche ----------- 5 g.
TTBS ------------ csp 100 ml.
La última etapa del western blot, es el marcaje y revelado mediante autoradiografía, empleando el luminol, que es un reactivo que se une al anticuerpo primario emitiendo fluorescencia para que pueda ser posible su visualización mediante autorradiografía.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
176
Los reactivos empleados son: luminol reagent, líquido revelador y líquido fijador.
Tras la incubación con el anticuerpo primario, se lava 3 veces con TTBS, y se escurre en posición vertical el gel y se pone sobre un cristal (preferiblemente de color oscuro para que pueda distinguirse bien, el papel de nitrocelulosa ya transferido).
Se prepara el luminol, que hace que las proteínas emitan fluorescencia y lo vamos echando sobre el papel de nitrocelulosa con una micropipeta, de forma que lo cubra totalmente y que no quede ninguna burbuja de aire (ya que si ocurriese así, en esa parte no se marcarían las proteínas, por lo que no emitirían fluorescencia y no sería posible su visualización tras el revelado) e incubamos durante 5 minutos.
Escurrimos el papel de nitrocelulosa en posición vertical y lo pasamos al papel glad para cubrir las dos caras, esto se hace para que el papel de nitrocelulosa no esté en contacto directo con la placa fotográfica. Al cubrir hay que tener la precaución de no formar burbujas y una vez cubierto pasamos un trozo de papel sobre este, presionando ligeramente para eliminar burbujas que hayan podido quedar y después cortamos el papel glad que sobra.
A partir de aquí todo el proceso se hará en el cuarto oscuro, con la bombilla roja de revelado.
Preparamos 3 recipientes de izquierda a derecha: líquido revelador, agua y líquido fijador. Sacamos de la caja la placa fotográfica y cortamos un trozo para cada papel de nitrocelulosa que tiene que ser de un tamaño un poco superior a la membrana de nitrocelulosa y lo colocamos sobre esta. Y volvemos a guardar las placas fotográficas para que estén fuera el menor tiempo posible (no le puede dar la luz).
Metemos la membrana con la placa en el casete (la placa se pone encima de la membrana) y lo cerramos y esperamos aproximadamente 1 minuto.
Sacamos la membrana del casete de revelado e incubamos en el líquido revelador hasta que las bandas se pongan oscuras (pero no mucho ya que al fijarse se oscurecen aún más y hace que no se vea bien), mientras incubamos vamos balanceándolo. Después lo sacamos y lo metemos en el recipiente con H2O unos 30 segundos para lavarlo y después en el fijador hasta que la membrana quede transparente y sólo se vean las bandas. Todo este proceso se hace balanceando los recipientes manualmente. Una vez fijado se vuelve a lavar y se deja secar al aire.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
177
Si no sale bien se pueden variar los tiempos, es decir, si sale muy oscuro lo dejaremos menos tiempo en el casete y en el revelador, y si sale muy claro lo dejaremos más tiempo, según vayamos viendo.
Este sería el último paso del Western Blot, ya que hemos obtenido impreso en papel fotográfico cada una de las proteínas que queremos estudiar.
Si no sale bien porque las bandas salgan muy oscuras o muy claras, podemos volver a marcar la membrana mediante un proceso por el cual eliminamos los anticuerpos primarios y secundarios adheridos a las proteínas que es el Streeping para nuevo mareaje, para ello usaremos glicina al 2%:
Glicina ---------------- 2 g.
H2O destilada -------- csp 100 ml.
Disolvemos bien y lo calentamos en estufa hasta alcanzar los 95 ºC y echamos un poco en un recipiente sobre la membrana, poniéndolo en agitación durante 5 min. y realizamos la misma operación tantas veces como podamos (más de 3 veces).
Una vez pasado el tiempo lavamos con TTBS 3 veces 5 minutos cada lavado. Después ya estaría lista la membrana para realizar de nuevo la inmunotinción.
5.4.2 EMSA Es un ensayo de cambio de movilidad electroforética (del inglés,
electrophoretic mobility shift assay).
Se utiliza para detectar factores de trascripción, que para activarse deben unirse al ADN.
Se necesitan varios reactivos y la realización de dicha técnica es muy parecida a la de Western Blot.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
178
Realización de la técnica
Primero se preparan los cristales para los geles y en un tubo universal se echa:
Acrilamida __________ 8 ml.
TBE 10X ___________ 2 ml.
Agua destilada ______ 30 ml.
TEMED _____________ 20 μl.
APS 10% ___________ 400 μl.
Una vez echados todos los reactivos anteriores, se va rellenando el interior de los cristales hasta que rebose, se coloca el peine y se espera a que polimerice.
Se llena la cubeta con TBE 0,5X y se conecta a la fuente de alimentación, dejándola unos 10 minutos corriendo (sin poner las muestras), sin poner las muestras. Con esto se consigue eliminar cualquier resto que quede en los pocillos para que no haya interferencias con las muestras.
Se carga el gel con 25 μg de proteínas por tanto se tendrán que realizar los cálculos correspondientes y echar cada muestra en un tubo ependorf pequeño y llevarla hasta 10 μl con agua destilada.
En un tubo ependorf preparar la mezcla compuesta por: buffer de unión, poly [ d ( I – C )], poly L- Lysina y sonda DYG, unida a la secuencia de ADN, (todo esto se va añadiendo por orden). El volumen de cada componente con el que se preparará la mezcla dependiendo del número de muestras que se tengan y está protocolizado, por ejemplo:
Si tenemos 6 muestras, tendremos que preparar mezcla para 7, para evitar posibles errores por mal pipeteo y echaremos:
Buffer de unión ______ 28 μl.
Poly [d ( I – C )] _____ 7 μl.
Poly L- Lysina _______ 7 μl.
Sonda DYG _________ 28 μl.
De esta mezcla echamos a cada muestra (+ agua) 10 μl, resuspendemos e incubamos 15 minutos a temperatura ambiente.
Tras la incubación, echamos 5 μl de loading buffer (para EMSA) y cargamos el gel, conectándolo a la fuente de alimentación a 120 V.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
179
El gel se transfiere igual que en la técnica de Western Blot, sólo que una vez transferido tenemos que secarlo en estufa a 120ºC durante 5 minutos. Transcurrido este tiempo, echamos 1 μl de NFHB sobre la membrana y la colocamos en un transluminador, poniendo hacia abajo la parte donde se encuentran las proteínas. Esto sirve para que las cadenas de ADN queden bien fijadas a la membrana.
A continuación, colocamos la membrana en un recipiente y le añadimos 5 mililitros de buffer de lavado (tween).
Después incubamos 30 minutos con el bloqueo.
Preparación del bloqueo:
Reactivo _______________ 5 ml
Buffer de lavado tween ____ 45 ml
Disolvemos en agitación continua a 65ºC
Seguidamente, centrifugamos 5 minutos a 10.000 revoluciones por minuto, desechamos el sobrenadante y le ponemos el anticuerpo e incubamos durante 30 minutos.
Volvemos a lavar 3 veces, durante 15 minutos cada vez, con el buffer de lavado, tras los tres lavados, añadimos buffer de detección y lo dejamos actuar durante 5 minutos.
Preparación de buffer de detección:
Tris __________________ 50 ml
NaCl _________________ 10 ml
Agua destilada__c.s.p ____ 500 ml
Colocamos la membrana en una hoja de papel glad y le añadimos el sustrato CSPD y cubrimos la otra cara de la membrana, cortamos los bordes y eliminamos el exceso de líquido con papel a la vez que se distribuye uniformemente la solución.
Preparación del CSPD:
− Diluir en proporción 1:100 en detection buffer
− Todo el proceso que queda es igual que la técnica de Western blot

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
180
5.4.3 Citometría de flujo Sirve para detectar la presencia de proteínas implicadas en la activación
del factor tisular, utilizando los reactivos: PBS, FBS 5%, suero humano y anticuerpos que queremos medir.
Realización de la técnica
Una vez descongeladas las células y realizado el recuento de estas, se preparan dos tubos para cada uno de los anticuerpos que se quieran medir: uno para la muestra y otro actuará como control.
Se coge un volumen (dependerá del número de células contadas, ya que queremos tener 150.000 células para realizar la citometría) del tubo de partida para contar (pellet + PBS), haciendo para ello una regla de tres, echando este volumen + 1 mililitros de PBS a cada tubo (el PBS servirá para hacer los lavados) y centrifugamos 5 minutos a 4ºC y a 1800 revoluciones por minuto.
Una vez centrifugado, invertimos el tubo tirando el sobrenadante quedando sólo el pellet y preparamos un tubo ependorf para cada muestra, echando:
Suero humano __________ 200 μl
PBS __________________ 800 μl
De esta mezcla se echan 100 μl al pellet y resuspender e incubar 15 minutos en hielo, para bloquear las uniones inespecíficas.
Mientras se incuba prepararemos 3 mililitros de PBS + FBS 5%:
3 mililitros __________ 100%
X mililitros __________ 5%
X = 5x3 = 0,150 μl de FBS al 5%
100 mililitros – 0,150 μl = 2,850 mililitros de PBS.
Tras la incubación en hielo, se le añade 1 mililitro de PBS + FBS y se centrifuga.
A continuación todo el proceso se hará en oscuridad.
Se quita el sobrenadante con la pipeta pasteur y se le añaden 10 μl del anticuerpo que queremos. Esperamos 30 minutos en hielo poniéndole papel de aluminio para impedir que entre luz.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
181
Tras la incubación añadimos 1 mililitro de PBS + FBS y centrifugamos.
Después eliminamos el sobrenadante y resuspendemos el pellet en 500 μl de PBS + FBS e incubamos en hielo y finalmente medimos en el citómetro.
Tras la lectura en el citómetro obtendremos una serie de gráficas, que nos indicarán la presencia normal o anormal del factor tisular, en el organismo del paciente con síndrome antifosfolípido.
En la gráfica obtendremos dos curvas, la primera, (sombreada) corresponde a la medición de las proteínas de una persona sana que actuará como control, para poder compararla con la gráfica del enfermo.
La siguiente curva (no sombreada) corresponde a la del enfermo, si esta está desplazada hacia la derecha con respecto a la curva de la persona sana, significará la presencia anormal del factor tisular en el organismo del paciente y si la gráfica del enfermo se encuentra sobrepuesta con la de la persona sana, significará la presencia normal del factor tisular.
En este caso, se observa la presencia anormal del factor tisular ya que la curva del enfermo se encuentra desplazada hacia la derecha con respecto a la del paciente.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
182
Las gráficas que obtendremos tras cualquier medición por citometría serán las siguientes (para una misma muestra):
5.4.4 ELISA Esta técnica la hemos visto en el tema anterior aplicada a numerosas
pruebas de laboratorio que se le piden al paciente con síndrome antifosfolípido.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
183
5.4.5 Cuantificación de proteínas por el método de Brad Ford Es una técnica de cuantificación de proteínas, mediante medición del
color (absorbancia) obteniéndose unos resultados en concentración, comparándolos con una curva estándar.
Los reactivos necesarios son: NaCH, proteína estándar, dye reagent concentrado y las proteínas del paciente.
Realización de la técnica
La primera fase consiste en la realización de la curva estándar, que es necesaria para extrapolar los resultados.
Los pasos son:
Realizar la mezcla: En un tubo preparamos la mezcla de concentración de 25 µg/ml y echamos:
H2O _______________ 4,660 ml.
NaOH (1N) _________ 250 µg/l.
Proteína estándar ____ 90 µg/l.
A continuación mezclamos con el voltex.
A partir de la mezcla preparamos los siguientes tubos de concentración decreciente:
− Tubo 20 µg/ml 0,3 ml H2O +1,2 ml de solución 25.
− Tubo 15 µg/ml 0,6 ml H2O +0,9 ml de solución 25.
− Tubo 10 µg/ml 0,9 ml H2O +0,6 ml de solución 25.
− Tubo 5 µg/ml 1,2 ml H2O +0,3 ml de solución 25.
− Tubo 2,5 µg/ml 1,35 ml H2O +0,15 ml de solución 25.
− Tubo 1,5 µg/ml 1,44 ml H2O +60 ml de solución 25.
Volumen final de cada tubo: 1,5 ml.
Volteamos cada tubo y preparamos viales, uno para cada tubo con la concentración de ésta y echamos en cada vial:
Mezcla _________________ 800 µg/l.
Dye Reagent concentrado __ 200 µg/l.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
184
La segunda fase consiste en la preparación del blanco, con el que eliminaremos las posibles interferencias.
H2O destilada ___________ 800 µg/l.
Dye Reagent concentrado __ 200 µg/l.
Y por último la preparación de las muestras, preparando tantos viales como muestras tengamos (uno para cada muestra) y echamos por orden:
NaOH __________________ 50 µg/l.
Proteína ________________ 1-5 µg/l. Hemos echado 2 µg/l.
H2O destilada - la cantidad de proteína que hayamos echado
Colorante: Dye Reagent concentrado ___________200 µg/l.
Volteamos cada vial y esperamos de 10 a 20 minutos. Volteamos pasado el tiempo y cargamos por triplicado las muestras y la curva estándar en una placa de 96 pocillos, 200 µg/l en cada pocillo.
Medimos en el lector de placas, poniéndolo a modo absorbancia.
5.4.6 Rt-PCR Se parte de un ARN procedente de las células de pacientes enfermos con
síndrome antifosfolípido, de las cuales a partir de la enzima retrotranscriptasa, obtendremos copias de un fragmento de ADN (ADNc).
Los reactivos empleados son :ARN de cada paciente, kit comercial para PCR, genes y primers o cebadores.
En primer lugar necesitaremos un kit comercial para PCR, que consta de:
− SYBR Green: Es una sustancia que hace que, al unirse a las cadenas de doble hélice del ADN, estas emitan fluorescencia para permitirnos ver cuántas cadenas de ADNc se forman en cada momento, por ello se llama PCR a tiempo real.
− RT: Enzima retrotranscriptasa.
− H2O destilada y desionizada.
Genes:
− Gen que queremos amplificar que en este caso es el gen PAR – 3.
− Gen GAP que nos servirá como referencia para comparar los resultados obtenidos, ya que se expresa en todos los genes.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
185
Primers o cebadores de cada gen:
− Gen PAR – 3: necesitamos dos primers:
- FOR 3´5´
- REV 5´3´
− Gen GAP:
- FOR 3´5´
- REV 5´3´
Por tanto tendremos que preparar un vial para cada mezcla.
Los primers vienen liofilizados y para reconstituirlos tenemos que añadir una determinada cantidad de agua destilada, estéril y libre de ADN-asa que es una enzima presente en pelo, piel…que degrada el ADN, por lo cual, debemos tener mucho cuidado en su manipulación.
Dejamos incubar 30 minutos en hielo volteándolo cada 7 minutos (aproximadamente).
El volumen que tenemos que añadir de cada compuesto depende del número de muestras. Estos volúmenes están estandarizados, como tenemos 16 muestras, prepararemos mezcla para 17, por si hubiese algún error en el pipeteo.
Para la mezcla del PAR – 3, añadiremos en el siguiente orden:
− SYBR – Green
− RT
− H2O destilada y desionizada
− Primer del PAR – 3: FOR y REV
Para la mezcla del gen GAP añadiremos exactamente lo mismo menos los primers que serán los correspondientes a este gen.
Una vez preparada la mezcla, cogemos el ARN de los pacientes y preparamos las muestras en capilares para ARN, (tenemos que preparar las mismas muestras para el PAR – 3 que para el GAP) echando en cada uno de ellos:
Mezcla_______________19 μl.
Muestra______________1,5 μl.
También tenemos que preparar un control de cada gen a partir de una muestra de una persona sana.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
186
A continuación hacemos un sprint (centrifugación rápida) de unos 10 segundos y después introducimos los capilares en el termociclador y ponemos los datos necesarios para que se lleve a cabo el proceso.
5.4.7 Proteómica Con la proteómica se pretende ver cuáles son las proteínas anormales
que presentan los distintos enfermos de síndrome antifosfolípido para su posterior estudio y tratamiento.
Realización de la técnica
1. Realización de lisados celulares.
Primero prepararemos la solución Urea + CHAPS:
Urea (9,8) M ________ 29,4 g.
H2O destilada _______ 25 ml.
Agitar en agitado a no más de 35 ºC.
CHAPS (4%) ________ 2 g.
H2O destilada _______ csp 50 ml.
Alicuotar en ependorf de 1 mililitro y congelar (si no se va a usar) a -20 ºC.
Una vez preparada la solución, lavamos el pellet de las células con PBS unas 2 o 3 veces dependiendo de la transparencia del sobrenadante.
Centrifugamos a 1800 revoluciones por minuto durante 5 minutos a 4 ºC.
A cada ependorf de 1 ml. de urea + CHAPS añadimos:
DTT (0,5 µ) _________ 100 µl.
Anfolitos (0,02%) ____ 5 µl (entre 3-10 µl).
Quitamos el sobrenadante de las células y dejamos el pellet añadiéndole 400 µl de la solución anterior y volteamos.
Centrifugar a 14000 revoluciones por minuto a 4 ºC durante 5 minutos.
Alicuotamos el sobrenadante en 50 µl pasándolo antes a un ependorf para mezclarlo bien.
2. Cuantificación de proteínas por el método de Bradfor. (Método explicado anteriçormente)

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
187
Con los resultados obtenidos tras la realización de la curva y muestras mediante un programa informatizado, averiguaremos las absorbancias de cada muestra y con ello la concentración y el volumen de muestra en µl que tenemos que poner para tener 500 µl de proteínas, sabiendo así el volumen de proteína necesaria, teniendo en cuenta que el volumen final que queremos obtener es 150 µl, por tanto si la cantidad de proteínas es menor se le añade H2O bidestilada hasta 150 µl y no debe pasar este volumen ya que la cantidad máxima de proteínas que es capaz de absorber la tira es de unos 170 µl.
Ejemplo: Tabla de absorbancias tras la cuantificación de proteínas.
1 2 3 4 5 6
0.278 0.291 0.291 0.374 0.389 0.390
0.679 0.679 0.711 0.362 0.371 0.371
0.502 0.512 0.520 0.358 0.373 0.382
0.441 0.445 0.447 0.370 0.389 0.388
0.417 0.420 0.428 0.392 0.405 0.408
0.370 0.365 0.369 0.366 0.383 0.382
0.320 0.322 0.328 0.365 0.373 0.370
0.306 0.321 0.313 0.367 0.379 0.385
y’ es la abs de cada muestra y tenemos que despejar 'x' (concentración). Una vez obtenido el % 500 entre cada concentración obtenida (x) dando la µl de muestra no tenemos que parar en cada tira para tener 500 µg de proteína.
- 1º Dimensión.
Preparamos 1 tubo ependorf para cada paciente y en cada uno echamos:
• µl de la muestra del paciente (proteínas) según cálculos.
• µl de H2O destilada hasta completar los 150 µl que es el volumen final.
• 2 µl de colorante azul de bromofenol.
Preparamos la caja con los 12 carriles y en cada carril ponemos la mezcla de cada paciente, extendiéndolo a lo largo del carril.
Cogemos una tira de gel de pH 3-10, le despegamos la tira que lo cubre y colocamos una tira en cada carril con el gel hacia debajo de modo que quede totalmente cubierta.

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
188
Dejamos actuar durante 1 hora y después echamos 1 mililitro de aceite en cada carril y tapamos la caja con los carriles.
Pasadas 24 horas, preparamos la placa de enfoque, para ello depositaremos 1 wicks (papel absorbente de 0,5 x 0,5) sobre cada electrodo y sobre este wicks echamos 8 µl de H2O destilada.
Escurrimos las tiras en papel absorbente con el gel hacia arriba sin tocarlo y una vez escurrido colocamos el gel hacia abajo en cada carril de la placa de enfoque y echamos 1 mililitro de aceite a cada carril sin que sobrepase.
Colocamos la placa de enfoque en el aparato de electroenfoque de manera que coincidan los colores del ánodo y cátodo de la placa con el del aparato y lo programamos.
- 2ª Dimensión.
Se basa en separar las proteínas según su peso molecular.
Primero prepararemos el tampón de equilibrado base
Urea _________________ 36 gr.
SDS (20%) ____________ 10 ml.
Tris-HCl (1,5m pH=8,8) __ 3,3 ml.
Glicerol (50%) __________ 40 ml.
Agua destilada _________ 46,7 ml.
Volumen final = 100 mililitros.
Preparamos 12 tubos y en cada uno echaremos 8 mililitros tampón de equilibrado base, y si no se va a utilizar se guarda a -20ºC.
- Preparación de DTT y yodoacetamida
Cogemos un tubo de tampón de equilibrado base de 8 mililitros y le añadimos 160 miligramos de DTT.
En otro tubo de tampón equilibrado base echaremos 200 miligramos de yodoacetamida y preparamos dos placas de 12 carriles (según la cantidad de muestras). Una servirá para el DTT y la otra para la yodoacetamida.
Ponemos en cada carril 2 mililitros. de tampón equilibrado base + DTT y metemos cada tira en su carril con el gel hacia arriba y lo dejamos durante 10 min. en agitación. Pasado este tiempo escurrimos las tiras.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
189
En la otra caja ponemos 2 mililitros de tampón equilibrado base + yodoacetamida en tantos carriles como tiras tengamos e introducimos las tiras ya escurridas con el gel hacia arriba y lo dejamos otros 10 minutos en agitación.
- Preparación del gel
Mientras esperamos vamos haciendo el gel. Este gel no lleva sellado el fondo, por lo tanto, tendremos que apretar muy bien los cristales para que no se pierda el separativo. Tampoco lleva Stacking.
Gel al 10%.
Separating 2x ________ 5 ml.
Archilamida (40%) ____ 2,5 ml.
H2O destilada ________ 2,5 ml.
TEMED ______________ 5 µl.
APS ________________ 50 µl.
Volumen final 10 mililitros.
Llenamos los cristales hasta la 2ª marca verde y echamos 1 mililitros de H2O destilada para que solidifique en anoxia.
Una vez pasados los 10 min. Y terminado el gel: calentamos la agarosa en el microondas (más 10 g.) hasta que se licue, introducimos las tiras con el gel mirando hacia fuera en el gel de poliacrilamida preparado anteriormente, y en el hueco que queda sin gel, ponemos un wicks cortado por la mitad y le echamos 3 µl de marcador de peso molecular y echamos 1 mililitros de agarosa sobre la tira y lo dejamos solidificar.
Después montamos la cubeta y la llenamos de buffer 1x y lo conectamos a la fuente de alimentación a 200 V. y esperamos a que bajen las bandas con las proteínas (más de 20 min.).
Una vez corrido el gel lo sacamos y los colocamos en un recipiente y le añadimos 50 mililitros de metanol-acético (40%-10%) y lo dejamos 1 hora en agitación.
Pasado el tiempo hacemos 3 lavados de 5 minutos con H2O destilada.
Echamos 50 mililitros de Cómase y lo dejamos 1 hora en agitación.
Lavamos con H2O destilada cada 15-20 min. hasta que quede transparente (unas 2 o 3 veces).

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
190
Escaneamos los geles en un programa para proteómica y conservamos los geles en ácido acético al 1%.
Después podemos teñir el gel con plata si no se ven bien las proteínas ya que la resolución de ésta es 100 veces mayor que la tinción con Coomassie.
Sacamos las tiras del ácido acético 1% y lavamos con metanol acético y se tiñen con Ag y cuando esté teñido (15-20 min.) pero no muy oscuro, se le añade ácido acético al 5% para detener la tinción con antígeno. Luego se lava con H2O unos 15 minutos por cada lavado haciendo 3 lavados en total.
Y el último paso sería comparar las proteínas del gel del paciente con síndrome antifosfolípido con un control, que serán proteínas de un paciente sano y picar aquellas proteínas que sean anormales y analizarlas por técnicas densitométricas.
Se comparan ambos geles y se tican las proteínas anormales, para poder ser analizadas.

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
191
5.4.8 Pronóstico del SAF Los estudios prospectivos de cohortes de pacientes con SAF indican que
la supervivencia a largo plazo es comprometida. La presentación clínica del primer episodio trombótico parece condicionar la probabilidad de desarrollar otro; así, los pacientes que presentan aß2GPI y hemólisis autoinmune parecen ser los de mayor riesgo para desarrollar un segundo episodio trombótico; por el contrario, los que carecen de aß2GPI y presentan abortos recurrentes son los de menor riesgo.
Los daños irreversibles causados por los fenómenos trombóticos ocasionan disfunción orgánica y morbilidad. En cualquier caso, la prognosis a largo plazo del SAF, ya sea primario o secundario, es mala. En un seguimiento a 10 años de pacientes con SAF primario, un tercio desarrolló daño orgánico y la quinta parte se vio afectada en su funcionalidad. La supervivencia de los pacientes afectos de LES y SAF secundaria está más comprometida que la de los enfermos de LES. La mortalidad de los pacientes con SAF ingresados en unidades de cuidados intensivos es de un 53%, y la afectación renal preingreso un factor de mal pronóstico.
5.5 Conclusiones Desde que fue definido por Hughes, el SAF se ha investigado
exhaustivamente. Sin embargo, todavía hoy no han podido elucidarse con claridad su etiología ni sus mecanismos patogénicos. La multiplicidad de hipótesis propuestas pone de manifiesto un conocimiento sólo parcial de su origen y de su mecanismo de acción, que probablemente sean multifactoriales. Ni siquiera se puede afirmar con certeza que los aFL sean patogénicos en el ser humano, y por tanto la causa de los fenómenos trombóticos y de los abortos de repetición. Por consiguiente, la definición del SAF que ha servido y sirve para estudiar, diagnosticar y tratar esta enfermedad todavía no se apoya en una evidencia sólida. La debilidad de esta base afecta a los criterios diagnósticos de SAF y al resto de clasificaciones propuestas que intentan acercarse a la realidad clínica cotidiana.
Con todo ello, no es de extrañar que existan contradicciones en los resultados de los estudios, ya que al no estar los pacientes perfectamente ajustados, en muchas ocasiones, a los criterios diagnósticos, pueden surgir discrepancias en sus conclusiones.
Se han efectuado indudables avances analíticos, tratando de incrementar la especificidad y la sensibilidad analítica y clínica mediante la estandarización de la determinación de los aFL. Los aß2GPI aportan mayor especificidad,

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
192
aunque no mayor sensibilidad diagnóstica, y no están incluidos, de momento, entre los criterios diagnósticos para el SAF «cierto». También se están evaluando otros muchos aFL, aunque ninguno de ellos parece aportar una mejora diagnóstica sustancial a los ya conocidos.
La diversa presentación clínica del SAF, fruto de su afectación multiorgánica, se enriquece día a día con una gran variedad y cantidad de casos clínicos que se publican continuamente en la bibliografía médica.
En cuanto al tratamiento, hay acuerdo en que los pacientes con SAF y algún episodio trombótico deben ser anticoagulados, que las pacientes afectadas de SAF con abortos de repetición embarazadas se deben tratar con AASBD y HBPM (o sólo ácido AASBD según otros autores), que los pacientes con aFL y LES pero sin criterio de SAF deben ser antiagregados, y que no deben ser tratados los pacientes con aFL sin LES ni criterios de SAF. Diversos autores reclaman la instauración de tratamientos individualizados ajustados al riesgo trombótico del paciente en concreto. Mención aparte merece el SAFC, en el que se aconseja un tratamiento combinado y agresivo.
En definitiva, pese a lo avanzado y al gran interés internacional sobre el tema, estamos lejos de conocer en profundidad esta enfermedad, que ofrece, pese al tratamiento y a los avances para obtener un diagnóstico temprano, un pronóstico comprometido.

CUESTIONARIO


Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
195
Cuestionario
1. Durante la coagulación sanguínea se pierde a) Fibrinógeno b) Componentes del plasma orgánicos e inorgánicos c) Ambas son correctas
2. Para la producción de los hematíes son necesarios: a) Ácido fólico b) Vitamina D c) Calcio
3. ¿Cuál de éstas es función de los eosinófilos? a) Regular y modular las reacciones alérgicas b) Paliar el daño producido por agentes patógenos c) Transportan el oxígeno a la sangre
4. Las células nulas están compuestas por: a) Linfocitos B b) Células natural killer c) Neutrófilos
5. Los monocitos se encuentran en grandes concentraciones en: a) Pulmones, hígado, bazo b) En el aparato óseo c) Ni ninguna de las 2 es correcta
6. ¿A qué puede deberse la monocitosis? a) A un aumento de temperatura b) A una reacción alérgica c) A causas fisiológicas
7. Los gránulos β favorecen: a) La agregación b) La adhesión plaquetaria y la vasoconstricción c) Las dos son correctas

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
196
8. ¿Qué es la hematopoyesis? a) Es el conjunto de procesos por el cual se produce la formación de
plaquetas b) Es el conjunto de procesos por el cual se produce la formación de
las células sanguíneas c) Es el conjunto de procesos por el cual se produce la maduración y
formación de hematíes
9. La hemostasia: a) Se pone en marcha tras la lesión de un vaso sanguíneo b) Es uno de los principales sistemas de defensa del organismo c) Ambas son correctas
10. El trombo plaquetario tiene lugar: a) Durante la etapa de agregación b) Durante la etapa de adhesión c) Durante la etapa de activación
11. El factor X puede activarse: a) Sólo por la vía intrínseca b) tanto por la vía intrínseca como extrínseca c) Sólo por la vía extrínseca
12. La antitrombina III: a) Inhibe la acción de la trombina b) No inhibe el crecimiento del coágulo c) Desacelera el proceso anticoagulante
13. En cuanto al nuevo modelo celular de la coagulación: a) Existen la vía intrínseca y la vía extrínseca b) Existen tres fases superpuestas: iniciación, amplificación y
propagación c) Ninguna de las 2 es correcta
14. El síndrome antifosfolípido es una enfermedad: a) Muy común en asiáticos menores de 15 años b) Autoinmune c) Ninguna de las 2 es correcta
15. El síndrome antifosfolípido: a) También es conocido como “el síndrome de la sangre pegajosa” b) Es una enfermedad autoinmune, rara y sistémica c) Las dos son correctas

Síndrome antifosfolípido. Pruebas diagnósticas y de investigación. Actualización 2020
197
16. Con la técnica de ELISA suele detectarse: a) Solamente las inmunoglobulinas M y A b) Inmunoglobulina A c) Inmunoglobulinas M y G e inmunoglobulinas A
17. La obstrucción de la vena cava inferior produce: a) Aumento del tamaño normal del hígado b) Un aumento del volumen de piernas y abdomen c) Sensación de malestar general y sudor frío
18. La enfermedad de Addison es debida a: a) El uso de corticosteroides como tratamiento b) una trombosis arterial c) un aumento de presión arterial
19. El tromboembolismo pulmonar: a) Es una enfermedad tromboembólica venosa b) Es el resultado de la obstrucción de la circulación de un vaso
sanguíneo c) Las dos son correctas
20. La angina estable puede presentarse: a) En momentos de reposo b) En momentos de estrés o ansiedad c) Los síntomas se presentan sin aviso
21. ¿Qué es el síndrome de Evans? a) La manifestación de trombocitopenia b) La manifestación de la anemia hemolítica c) La manifestación conjunta de trombocitopenia y anemia hemolítica
autoinmune
22. El síndrome de Guillain-Barré se debe: a) Es difícil de diagnosticar en las etapas iniciales b) Los pacientes no presentan problemas sensoriales c) A un trastorno hormonal
23. El síndrome de Guillain-Barré: a) Es una enfermedad extinta b) Lo padecen sólo afroamericanos c) Lo sufren 1 o 2 personas de cada 100.000

Técnico Superior Laboratorio de Diagnóstico Clínico y Biomédico
198
24. Una cirrosis puede: a) Disminuir la activación del factor IX b) Aumentar la activación del factor IX c) No tiene relación con el factor IX
25. La lisis del coágulo de euglobulina se completa entre: a) 2 y 4 horas b) 2 y 4 minutos c) 2 y 4 días
26. El examen del cariotipo se realiza para: a) Realizar análisis tumorales b) Evaluar cualquier anomalía genética c) Ninguna es correcta
27. El tratamiento de pacientes con síndrome antifosfolípido con aspirina produce un efecto: a) Analgésico y antipirético b) Anticoagulante y antitrombótico c) No es recomendable este tratamiento para éste síndrome
28. El Western-Blot se utiliza para: a) Separar células unidas b) Unir proteínas según su peso molecular c) Separar las proteínas según su peso molecular
29. El EMSA: a) Se utiliza para detectar los factores de trascripción que deben unirse
al ADN b) Es una técnica similar a la de Western Blot c) Las dos son correctas
30. El cálculo de la concentración de proteínas por el método de Brad-Ford se hace mediante: a) Necesita como reactivos NaCH, proteína estándar, dye reagent
concentrado y las proteínas del paciente b) Es una técnica de cuantificación de la concentración de los reactivos c) Como reactivo se utiliza el ARN de los pacientes





























