Embed Size (px)
Citation preview

SPECIAL ISSUE REVIEWS–A PEER REVIEWED FORUM
Simple Vertebrate Models for ChemicalGenetics and Drug Discovery Screens: LessonsFrom Zebrafish and XenopusGrant N. Wheeler1* and Andre W. Brandli2*
Chemical genetics uses small molecules to modulate protein function and, in principle, has the potential toperturb any biochemical event in a complex cellular context. The application of chemical genetics to dissectbiological processes has become an attractive alternative to mutagenesis screens due to its technicalsimplicity, inexpensive reagents, and low-startup costs. In vertebrates, only fish and amphibians areamenable to chemical genetic screens. Xenopus frogs share a long evolutionary history with mammals andso represent an excellent model to predict human biology. In this review, we discuss the lessons learnedfrom chemical genetic studies carried out in zebrafish and Xenopus. We highlight how Xenopus can beemployed as a convenient first-line animal model at various stages of the drug discovery and developmentprocess and comment on how they represent much-needed tools to bridge the gap between traditional invitro and preclinical mammalian assays in biomedical research and drug development. DevelopmentalDynamics 238:1287–1308, 2009. © 2009 Wiley-Liss, Inc.
Key words: Xenopus; zebrafish; chemical genetics; chemical library; chemical screening; drug discovery; drugdevelopment; embryo; tadpole; embryogenesis; organogenesis
Accepted 25 March 2009
INTRODUCTION
Large-scale genetic screens in modelorganisms have been used very suc-cessfully for many years to identifygenes involved in diverse developmen-tal and physiological processes. Theyhave been particularly powerful inidentifying key genes and geneticpathways underlying axis specifica-tion and pattern formation during em-bryogenesis in Drosophila (Nusslein-Volhard and Wieschaus, 1980), andprogrammed cell-death in Caenorhab-ditis elegans (Metzstein et al., 1998).However, genetic screens are expen-
sive and time-consuming, particularlyif performed in vertebrate model sys-tems. In addition, these screens are oflimited use with respect to the geneticdissection of late development and or-ganogenesis. Embryonic developmentis controlled by a surprisingly smallnumber of genes and signaling path-ways, which are redeployed at multi-ple times and places as developmentproceeds (Davidson et al., 2002; VanRaay and Vetter, 2004; Reya andClevers, 2005). Most mutations recov-ered from genetic screens are not con-ditional, i.e., they cannot be reversed
at will. Therefore, the mutations willonly define the first biological processand time point, where a particulargene is required during embryogene-sis. Frequently, the mutation may ar-rest development and the mutant or-ganism is no longer viable. Hence, itbecomes difficult to study late genefunctions in organogenesis, physiol-ogy, and behavior using a classical ge-netic approach.
Chemical genetics provides a com-plementary approach to loss-of-func-tion mutations in the study of complexbiological processes (Stockwell, 2000;
1School of Biological Sciences, University of East Anglia, Norwich, United Kingdom2Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zurich, Zurich, SwitzerlandGrant sponsor: MRC; Grant number: G0100722; Grant sponsor: Swiss National Science Foundation; Grant number: 3100A0-114102; Grantsponsor: European Community; Grant number: EuReGene LSHG-CT-2004-005085; Grant sponsor: ETH Zurich.*Correspondence to: Grant N. Wheeler, School of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK.E-mail: [email protected]; or Andre W. Brandli, Institute of Pharmaceutical Sciences, Department of Chemistry andApplied Biosciences, ETH Zurich, Wolfgang-Pauli-Strasse 10, Zurich, CH-8093, Switzerland. E-mail: [email protected]
DOI 10.1002/dvdy.21967Published online 15 May 2009 in Wiley InterScience (www.interscience.wiley.com).
DEVELOPMENTAL DYNAMICS 238:1287–1308, 2009
© 2009 Wiley-Liss, Inc.

Min et al., 2007). It makes use of smallorganic molecules with a molecularweight of typically less than 2,000Dalton (Da) to alter the function of agene product within a complex cellu-lar context. Chemical genetic strate-gies are conditional and readily appli-cable to cells derived from complexorganisms such as vertebrates. Com-pounds can be added or removed atany time, enabling the kinetic analy-sis of protein functions in vivo. In mul-ticellular organisms, as alreadystated, chemical genetics offers a com-plementary approach to loss-of-func-tion mutations or knockdowns withsiRNA or morpholino oligonucleotidesin the analysis of complex biologicalprocesses, such as organogenesis. Inaddition, chemical genetic screens canalso be employed to study the functionof maternal gene products, which arenot targeted by conventional geneticscreens. One approach of chemical ge-netics, forward chemical genetics,uses the screening of annotated librar-ies of small organic compounds withexperimentally verified biologicalmechanisms and activities to study bi-ological systems (Stockwell, 2000).This approach circumvents the well-known problems of target identifica-tion and lack of mechanistic under-standing associated with activecompounds recovered from screens us-ing conventional chemical libraries(Root et al., 2003). Forward chemicalgenetics has, therefore, become in-creasingly used in cell cultures toidentify signaling pathways involvedin cellular functions in vitro (Root etal., 2003; Rickardson et al., 2006; Dia-mandis et al., 2007), and more re-cently, whole organisms such as Dro-sophila, C. elegans, and zebrafishhave been used for compound discov-ery (Min et al., 2007; Tran et al., 2007;Chang et al., 2008). Furthermore, bi-ologically active small molecules re-covered from chemical geneticsscreens also provide important struc-tural information for the developmentof novel therapeutic agents.
Beyond applications in chemical ge-netics, small multicellular model or-ganisms offer new opportunities in thedrug discovery and drug developmentpipeline. They can be treated withsmall molecules in a multi-well formatfor high-throughput phenotypic anal-ysis to identify novel drug candidates
that are bioactive in a whole organ-ism, where complex cell–cell and cell–matrix interactions remain intact incontrast to in vitro cell-based screen-ing approaches. Furthermore, smallmodel organisms are also beginning tobe used at various other stages of thedrug discovery process, where theycan be useful and cost-effective alter-natives to mammalian models. Theseuses include drug target identificationand validation, lead compound char-acterization and optimization, and theassessment of drug toxicity. Simpleand cost-effective maintenance to-gether with abundant experimentaltechniques and molecular tools havemade zebrafish the only vertebratemodel used to date for chemical genet-ics and large-scale in vivo drugscreens (Zon and Peterson, 2005). Am-phibians offer many of the same ex-perimental advantages that have fa-vored the use of zebrafish in the past,such as rapid extrauterine develop-ment, the transparency of developingtadpoles, and the permeability of theskin for small molecules, but theyhave until recently not been employedfor large-scale chemical screening. Inthis review, we will introduce Xenopusembryos and tadpoles as alternativevertebrate model organisms for chem-ical genetics and whole-organism-based drug discovery screens. Fur-thermore, we show that Xenopusrepresents a cheap and efficient invivo bioassay tool that can contributeto several aspects of the drug develop-ment process, including drug targetidentification, lead compound discov-ery, and estimation of drug toxicity.
ANIMAL MODELS FORORGANISM-BASEDCHEMICAL SCREENING
Organism-based chemical screeningtests the ability of each compound of alibrary to induce a specific phenotypein an organism. Such screens are,therefore, analogous to classic for-ward-genetic screens in model organ-isms. There are two purposes for car-rying out chemical screens in modelorganisms. One is to promote researchin a given area by obtaining smallmolecules, such as antagonists, thatcan be used as conditional researchtools to investigate fundamental ques-tions in development, physiology, and
behavior. The second and increasinglymore important application of thesescreens is to identify drug candidatesthat can be potentially used for ther-apeutic purposes. An important ad-vantage of organism-based chemicalscreening is the fact that compoundsare tested in the context of an intactorganism rather than under artificialin vitro conditions. Many diseases af-fect organs as a whole, and most or-gans cannot be reconstituted in vitro.Small molecules discovered by virtueof their ability to induce a specific,desirable phenotype in a whole organ-ism are likely to fulfill efficacy andspecificity requirements that ulti-mately need to be met by promisingdrug candidates entering clinical de-velopment. They have to be cell-per-meable, devoid of obvious toxicities,effective, and possess favorable, phar-macodynamic and pharmacokineticprofiles. Drug discovery in the wholeorganism, therefore, combines screen-ing and animal testing in one step.
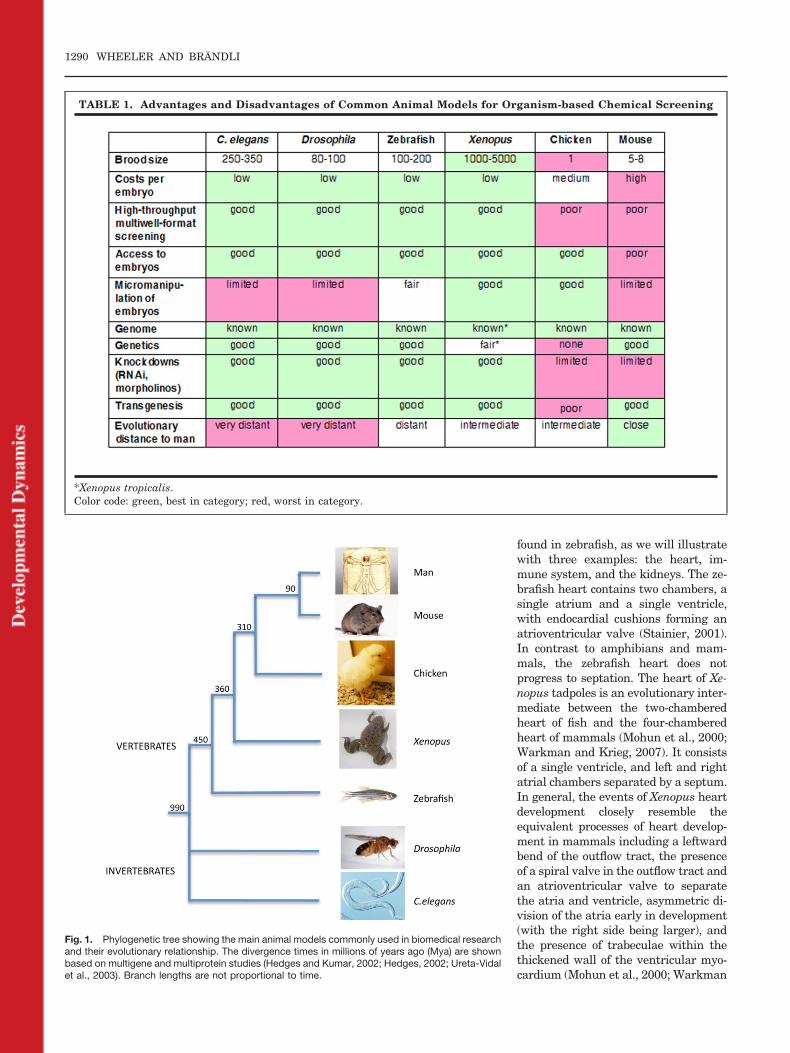
Biomedical research exploits arange of model organisms to gain in-sights into fundamental biologicalprocesses and disease mechanisms.The nematode C. elegans and the fruitfly Drosophila melanogaster are popu-lar genetic model organisms and theyhave recently also been used asscreening tools in drug discovery (Se-galat, 2007), An important limitationof invertebrate models is, however,the fact that they lack many directlyextrapolatable complex organs, suchas a cardiovascular system, an im-mune system, and kidneys, relevantto human biology and physiology. Ver-tebrate models, by contrast, usuallyhave all the tissues afflicted by com-mon human disease. However, not allvertebrate animal models are equallysuitable for organism-based drug dis-covery screens. Table 1 compiles someof the general advantages and disad-vantages of the commonly used multi-cellular model organisms. Animalmodels to be employed for organism-based chemical screens have to besmall, low-cost, and compatible withsimple culture conditions to be suit-able for the technologies of modernhigh-throughput screening. There-fore, organisms producing large num-bers of embryos are essential, whichmeans that chicken and mouse are notsuitable for chemical screens. The or-
1288 WHEELER AND BRANDLI

ganisms also have to allow rapid pen-etration of small molecules. Drosoph-ila and C. elegans are surrounded by athick cuticle that is a physical barrierto the penetration of small moleculeslimiting the access to tissues and or-gans in these organisms.
Among the vertebrate models onlyzebrafish and Xenopus fulfill theabove-mentioned criteria for high-throughput organism-based chemicalscreening. The animals have simplehusbandry requirements, are fecund,and generate large clutches of eggs.Fertilization and embryonic develop-ment are external. The resulting em-bryos and larvae are small enough togrow in microformat screening plates.Xenopus scores well in most of the cat-egories. Their eggs can be easily ob-tained in numbers of several thou-sands at any time during the year bysimple hormone injection and theycan then be synchronously fertilizedin vitro. This facilitates biochemical,pharmacological, and statistical anal-yses. They develop in simple salt solu-tions at room temperature. Com-pounds can be added to the bathingmedia and the vitelline membranearound the embryo is highly porousand thus accessibility of compounds tothe embryo is assumed to be good.Later, drugs and small molecules canbe rapidly absorbed through the skinand gills, though this has not beenstudied in detail. Xenopus laevis em-bryos are bigger than zebrafish, butthey can still be screened in 48- or96-well plates (Brandli, 2004; Tomlin-son et al., 2005). Embryos of Xenopustropicalis, the diploid sister species ofXenopus laevis, are half the size ofXenopus laevis embryos and are,therefore, more suitable for assayingin 96-well plates. Overall, zebrafishand Xenopus share similar experi-mental advantages with regard totheir utility for high-throughputchemical screening.
ADVANTAGES ANDDISADVANTAGES OFXENOPUS AS AVERTEBRATE MODELORGANISM FOR CHEMICALSCREENING
Zebrafish and Xenopus are both un-doubtedly well suited to whole-organ-
ism based small-molecule screens be-cause large numbers of embryos canbe arrayed in multiwell plates, alongwith compounds from a chemical li-brary. The first large-scale chemicallibrary screen using a vertebratemodel was reported by Peterson et al.(2000) who screened 1,100 compoundsagainst zebrafish embryos arrayed in96-well plates to identify moleculesthat modulate embryogenesis. Theuse of Xenopus as an alternative ver-tebrate model for chemical screenshas been proposed in the past(Brandli, 2004; Tomlinson et al.,2005), but until recently zebrafish hasremained the only vertebrate modelused for large-scale chemical screens(Zon and Peterson, 2005). This hasbeen in part due to the availability ofchemically induced zebrafish mutantsthat model human disease (Shin andFishman, 2002; Rubinstein, 2003).The versatility of Xenopus must,therefore, be compared to the well-es-tablished properties of the zebrafishsystem. The recent report of geneticscreens performed in Xenopus tropica-lis (Goda et al., 2006) indicate thatXenopus mutants that phenocopy hu-man disorders may be recovered in thefuture. The X. tropicalis genome isnow nearly complete (JGI X. tropicalisgenome assembly 4.1; http://genome-.jgi-psf.org/Xentr4/), public databasesharbor the nucleotide sequences of al-most 2 million expressed sequence tag(EST) from X. tropicalis and X. laevis(dbEST; http://www.ncbi.nlm.nih.gov/dbEST/), and DNA microarrays areavailable for both Xenopus species.Thus, Xenopus is rapidly becoming amodel organism armed with an im-pressive collection of genomic andtranscriptomic tools.
The closer a model organism is inevolutionary terms to the target or-ganism, i.e., humans, the more reli-ably results from studies in the modelorganism translate to humans. Figure1 shows a phylogenetic tree highlight-ing the evolutionary distances be-tween various animal model organ-isms used in biomedical research.Xenopus has a common evolutionaryhistory with mammals that is an esti-mated 90–100 million years longerthan between zebrafish and mam-mals. Since both are tetrapods, Xeno-pus and mammals share extensivesynteny at the level of the genomes
and have many similarities in organdevelopment, anatomy, and physiol-ogy (Christensen et al., 2008; Raciti etal., 2008). A high degree of sequenceconservation and structure betweenthe proteins of model species and hu-mans is desirable, since this will in-crease the likelihood that bioactivecompounds recovered in chemicalscreens in model organisms will retainactivity and specificity for preclinicalstudies in mice and eventually forclinical studies in humans. As stated,Xenopus as a tetrapod is evolutionar-ily closer to humans than zebrafish.There are also significant differencesat the genomic level. Genome and ESTsequence data support the notion thatthe X. tropicalis genome is diploid innature (Hellsten et al., 2007). In con-trast, the teleost lineage of fish under-went a genome duplication event afterdiverging from tetrapods about 450million years ago (Postlethwait et al.,1998; Postlethwait, 2007). Zebrafish,therefore, possess two copies of manymammalian genes. Such redundancycan confound extrapolation of the ef-fect of single gene knockdown or mu-tation from fish to mammals and com-plicate the generation of zebrafishmodels of congenital human disease.In addition, comparative genomicshas identified entire gene families,such as the KRAB domain containingC2H2 zinc finger (KRAB-ZF) tran-scription factors (Emerson andThomas, 2009) that first arose in tet-rapod vertebrates and are absent inthe zebrafish genomes.
There are drawbacks that apply toboth animal models. Most advantagesof zebrafish and Xenopus as a model forchemical screens are limited to embry-onic and larval stages and do not applyto adult organisms, since they are typi-cally too large to be employed in whole-organism-based screens. Despite theselimitations, zebrafish larvae and Xeno-pus tadpoles contain most organs andtissues affected by common human dis-eases, including a cardiovascular sys-tem, a digestive tract, excretory organs,sensory organs, a hematopoietic sys-tem, and a central nervous system. Be-ing tetrapods, Xenopus tadpoles will de-velop lungs and limbs, which are absentfrom zebrafish. Importantly, the organsof Xenopus tadpoles are morphologi-cally and functionally more similar totheir human counterpart than those
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1289
found in zebrafish, as we will illustratewith three examples: the heart, im-mune system, and the kidneys. The ze-brafish heart contains two chambers, asingle atrium and a single ventricle,with endocardial cushions forming anatrioventricular valve (Stainier, 2001).In contrast to amphibians and mam-mals, the zebrafish heart does notprogress to septation. The heart of Xe-nopus tadpoles is an evolutionary inter-mediate between the two-chamberedheart of fish and the four-chamberedheart of mammals (Mohun et al., 2000;Warkman and Krieg, 2007). It consistsof a single ventricle, and left and rightatrial chambers separated by a septum.In general, the events of Xenopus heartdevelopment closely resemble theequivalent processes of heart develop-ment in mammals including a leftwardbend of the outflow tract, the presenceof a spiral valve in the outflow tract andan atrioventricular valve to separatethe atria and ventricle, asymmetric di-vision of the atria early in development(with the right side being larger), andthe presence of trabeculae within thethickened wall of the ventricular myo-cardium (Mohun et al., 2000; Warkman
Fig. 1. Phylogenetic tree showing the main animal models commonly used in biomedical researchand their evolutionary relationship. The divergence times in millions of years ago (Mya) are shownbased on multigene and multiprotein studies (Hedges and Kumar, 2002; Hedges, 2002; Ureta-Vidalet al., 2003). Branch lengths are not proportional to time.
TABLE 1. Advantages and Disadvantages of Common Animal Models for Organism-based Chemical Screening
*Xenopus tropicalis.Color code: green, best in category; red, worst in category.
1290 WHEELER AND BRANDLI

and Krieg, 2007). Differences are alsoevident with the immune system. Whilethe antibody-based, adaptive immunesystem is present in all jawed verte-brates (gnathostomes) (Klein and Niko-laidis, 2005), zebrafish and Xenopus dif-fer considerably with regard to thepresence of major lymphoid tissues, im-munoglobulin gene organization, andthe ability to perform immunoglobulinclass switching (Du Pasquier et al.,1989; Traver et al., 2003). Bone mar-row, gut-associated lymphoid nodules,and primitive lymph nodes are absentfrom zebrafish but present in Xenopus.There are also significant differences inthe gene organization, gene usage, andgene number of immunoglobulin genes,whereby Xenopus shares many featureswith mammals (Du Pasquier, 2001). Ze-brafish express a new immunoglobulinheavy chain subtype, immunoglobulinZ, which does not have a counterpart inmammals (Danilova et al., 2005). Fur-thermore, isotype class switching is aprocess that has its earliest evolution-ary roots in amphibians and does notoccur in fishes (Traver et al., 2003). Insummary, Xenopus display an efficientimmune system that is very similar tomammals and includes rearranging T-cell receptors (TCR) and immunoglobu-lin genes, as well as major histocom-patibilitiy complex (MHC) class I- andclass II-restricted T cell recognition.With regard to the excretory system,the pronephric kidneys of zebrafish andXenopus kidneys differ in their globalstructure from the adult mammaliankidneys, but fulfill the same essentialphysiological functions in solute reab-sorption, water and ion homeostasis,pH regulation, and waste product ex-cretion. This is underscored at the mo-lecular level by a recent large-scale geneexpression study that revealed a re-markable conservation between thenephron organization of Xenopus tad-pole kidneys and adult mammalian kid-ney (Raciti et al., 2008). Overall, thesetraits favor the use of amphibians forlarge-scale in vivo drug screens andsuggest that they may unveil mecha-nisms and pathways relevant to humandisease and therapy. There is no doubtthat mammals, such as mice and rats,clearly reflect human physiology better.However, there are no other vertebratesof the tetrapod superclass that offer thesame advantages of Xenopus. Theirfree-living embryos are amenable to
large-scale, high-throughput chemicalscreens. Therefore, Xenopus providesthe optimal trade-off between experi-mental use, cost-efficient performance,and biological relevance to humans.
LESSONS FROMPHENOTYPE-BASEDCHEMICAL LIBRARYSCREENS IN ZEBRAFISH
Assessing the Feasibility ofPhenotypic ChemicalLibrary Screens in Zebrafish
Compounds of chemical libraries areusually stored as stock solutions indimethyl sulfoxide (DMSO), whichalso serves as a vehicle to improvesolubility of the compounds in theaqueous solutions used to culture em-bryos and facilitates compound per-meation into cells. Both zebrafish andXenopus embryos are surprisingly tol-erant to a range of DMSO concentra-tions (Brandli, 2004; Chan and Ser-luca, 2004). A final concentration of1% DMSO is compatible with normaldevelopment in both species, which in-dicates that compounds can bescreened at a wide range of differentcompound concentrations in larvaeand tadpoles. Embryos from both spe-cies are small enough (about 1 mmdiameter) to be arrayed in 48- or 96-well plates and the analysis for re-sponses to chemical treatment can beperformed by microscopy, in situ hy-bridization, or reporter readout. De-spite these shared advantages, whole-organism chemical screens had untilrecently not been carried out in Xeno-pus and zebrafish have led the way(Table 2).
The potential of zebrafish to be usedas a vertebrate model for chemicalscreens was first assessed by Petersonand colleagues (Peterson et al., 2000).They used wild-type zebrafish em-bryos to screen for chemical modifiersof development using a strategy anal-ogous to genetic screens, where muta-tions are identified that affect devel-opment of different organ systems. Intheir screen, 1,100 synthetic smallmolecules were randomly chosen fromthe DIVERSet library (ChembridgeCorp.) and added to the embryos. Theeffects of the individual compounds onthe embryo’s morphology and physiol-ogy were examined visually under the
dissection microscope. From thisscreen, 2% of the compounds cause le-thality, and 1% caused specific pheno-types leading to the identification ofcompounds that affect development ofthe central nervous system, the car-diovascular system, pigmentation,and the ear. Several of the identifiedcompounds were highly potent andspecific by acting in the nanomolarrange and frequently affecting onlyone organ. For example, the molecule32P6, subsequently renamed concen-tramide, affects heart chamber pat-terning by causing the ventricle toform in the atrium (Peterson et al.,2000, 2001). Concentramide acts withan EC50 of 2 nM and does not appearto cause additional side effects (Peter-son et al., 2001). Peterson and col-leagues also took advantage of the factthat active compounds can be admin-istered at different times during ze-brafish development, which allowedthem to determine the exact timepoint when a target protein’s activitywas required during development.This provides temporal insights intodevelopment, which frequently cannotbe obtained using conventional ge-netic mutants, as the mutant proteinsmay not permit survival beyond earlyembryogenesis. While the precise tar-gets of the active molecules recoveredin the in vivo chemical screen remainunknown to date, the work by Peter-son and colleagues demonstrated thatthe zebrafish embryo is permeable tomany small molecules underscoringthe utility of zebrafish as a high-throughput in vivo bioassay system.Importantly, it was subsequentlydemonstrated that most of the com-pounds identified by Peterson et al.(2001) gave comparable results whenapplied to Xenopus embryos (Tomlin-son et al., 2005).
The Spectrum of ChemicalScreens Performed inZebrafish
Over the last few years, phenotype-based developmental screens usingwild-type zebrafish embryos haveidentified multiple compounds thatcause general defects in embryogene-sis and now await further character-ization (Sternson et al., 2001; Moon etal., 2002; Spring et al., 2002; Wong etal., 2004). Chemical screens were also
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1291
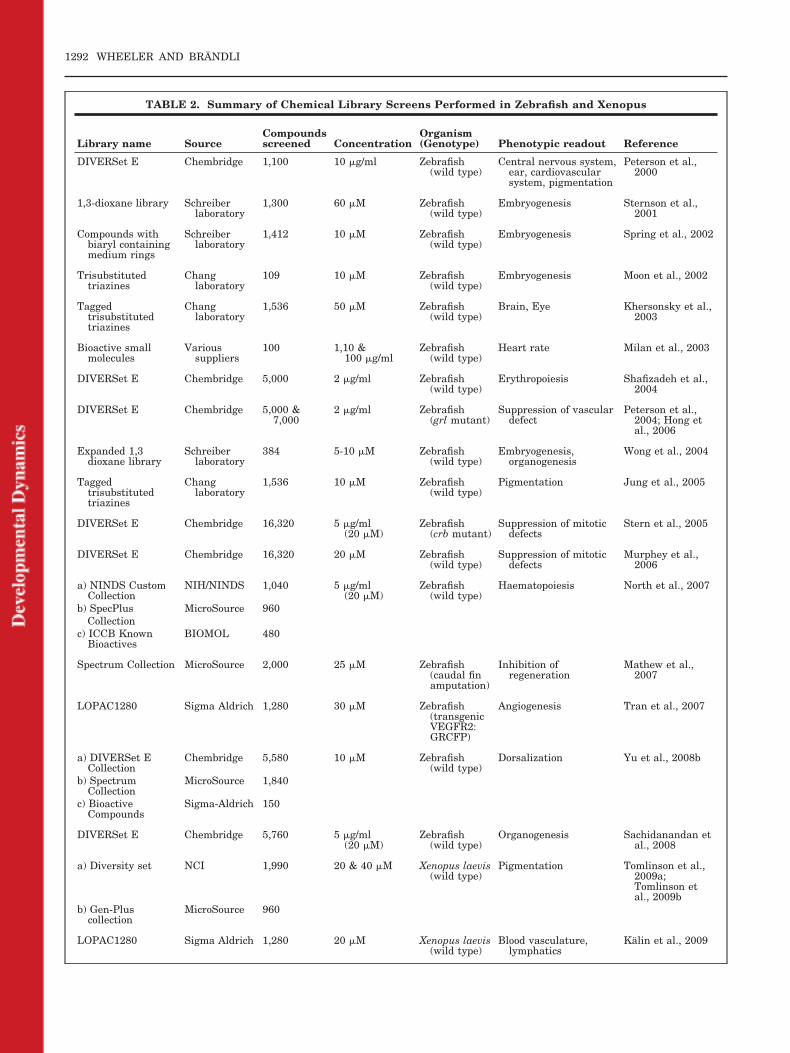
TABLE 2. Summary of Chemical Library Screens Performed in Zebrafish and Xenopus
Library name SourceCompoundsscreened Concentration
Organism(Genotype) Phenotypic readout Reference
DIVERSet E Chembridge 1,100 10 �g/ml Zebrafish(wild type)
Central nervous system,ear, cardiovascularsystem, pigmentation
Peterson et al.,2000
1,3-dioxane library Schreiberlaboratory
1,300 60 �M Zebrafish(wild type)
Embryogenesis Sternson et al.,2001
Compounds withbiaryl containingmedium rings
Schreiberlaboratory
1,412 10 �M Zebrafish(wild type)
Embryogenesis Spring et al., 2002
Trisubstitutedtriazines
Changlaboratory
109 10 �M Zebrafish(wild type)
Embryogenesis Moon et al., 2002
Taggedtrisubstitutedtriazines
Changlaboratory
1,536 50 �M Zebrafish(wild type)
Brain, Eye Khersonsky et al.,2003
Bioactive smallmolecules
Varioussuppliers
100 1,10 &100 �g/ml
Zebrafish(wild type)
Heart rate Milan et al., 2003
DIVERSet E Chembridge 5,000 2 �g/ml Zebrafish(wild type)
Erythropoiesis Shafizadeh et al.,2004
DIVERSet E Chembridge 5,000 &7,000
2 �g/ml Zebrafish(grl mutant)
Suppression of vasculardefect
Peterson et al.,2004; Hong etal., 2006
Expanded 1,3dioxane library
Schreiberlaboratory
384 5-10 �M Zebrafish(wild type)
Embryogenesis,organogenesis
Wong et al., 2004
Taggedtrisubstitutedtriazines
Changlaboratory
1,536 10 �M Zebrafish(wild type)
Pigmentation Jung et al., 2005
DIVERSet E Chembridge 16,320 5 �g/ml(20 �M)
Zebrafish(crb mutant)
Suppression of mitoticdefects
Stern et al., 2005
DIVERSet E Chembridge 16,320 20 �M Zebrafish(wild type)
Suppression of mitoticdefects
Murphey et al.,2006
a) NINDS CustomCollection
NIH/NINDS 1,040 5 �g/ml(20 �M)
Zebrafish(wild type)
Haematopoiesis North et al., 2007
b) SpecPlus MicroSource 960Collection
c) ICCB KnownBioactives
BIOMOL 480
Spectrum Collection MicroSource 2,000 25 �M Zebrafish(caudal finamputation)
Inhibition ofregeneration
Mathew et al.,2007
LOPAC1280 Sigma Aldrich 1,280 30 �M Zebrafish(transgenicVEGFR2:GRCFP)
Angiogenesis Tran et al., 2007
a) DIVERSet ECollection
Chembridge 5,580 10 �M Zebrafish(wild type)
Dorsalization Yu et al., 2008b
b) SpectrumCollection
MicroSource 1,840
c) BioactiveCompounds
Sigma-Aldrich 150
DIVERSet E Chembridge 5,760 5 �g/ml(20 �M)
Zebrafish(wild type)
Organogenesis Sachidanandan etal., 2008
a) Diversity set NCI 1,990 20 & 40 �M Xenopus laevis(wild type)
Pigmentation Tomlinson et al.,2009a;Tomlinson etal., 2009b
b) Gen-Pluscollection
MicroSource 960
LOPAC1280 Sigma Aldrich 1,280 20 �M Xenopus laevis(wild type)
Blood vasculature,lymphatics
Kalin et al., 2009
1292 WHEELER AND BRANDLI

designed to identify novel bioactivecompounds that affect the develop-ment of specific organs, physiologicalactivities, or cellular processes in ze-brafish (Table 2). These includescreens to identify compounds affect-ing dorsoventral patterning (Yu et al.,2008b), hematopoiesis (North et al.,2007), erythropoiesis (Shafizadeh etal., 2004), vasculature (Tran et al.,2007), pigmentation (Jung et al.,2005; North et al., 2007), fin regener-ation (Mathew et al., 2007), and mor-phogenesis of the brain, eyes, andother tissues and organs easily identi-fied by visual inspection of compound-treated embryos (Khersonsky et al.,2003; Sachidanandan et al., 2008). Fi-nally, screens to identify modifiers orsuppressors of zebrafish mutant phe-notypes have been developed (Peter-son et al., 2004; Stern et al., 2005;Hong et al., 2006). We will now reviewthe specifics of these screens ingreater detail to highlight key lessonsthat can be learned from the carefulanalysis of the published literature.Technical aspects of performing chem-ical screens in zebrafish have been re-viewed elsewhere in detail (Chan andSerluca, 2004; Peterson et al., 2004;Murphey and Zon, 2006; Berger andCurrie, 2007).
Exploring the Advantages ofIn Vivo Screens Over Cell-Based Assays
One of the great hopes of phenotypicwhole-organism-based drug screens isthat they may lead to the identi-fication of novel bioactive compoundsthat could not be recovered usingstandard cell culture–based pheno-typic screens. In contrast to cell cul-tures, cells in whole organisms arenon-transformed and found in theirnormal context of three-dimensionalcell–cell and cell–matrix interactions,ingested compounds may become ac-tive as metabolites, and compoundsexerting specific biological effects indi-rectly by acting on adjacent tissuemay be recovered.
As an alternative approach to stan-dard cell culture–based drug screens,a chemical screen in zebrafish em-bryos was devised to identify leadcompounds that have effects on thecell cycle and hence could act as novelchemotherapeutics for cancer treat-
ment (Murphey et al., 2006). The ze-brafish-based screen was performedwith a chemical library that was pre-viously screened for mitotic inhibitorswith cell lines (Haggarty et al., 2000).Of the 29 identified compounds affect-ing cell cycle activity in vivo, 14 com-pounds were novel. Seven compoundswere also active in mammalian and/orzebrafish cell cultures. The remainingseven compounds were only active inthe context of the intact organism.Whether these compounds also elicitin vivo cell cycle activity in mamma-lian animal models has not beentested to date. Interestingly, threecompounds were inactive in vitro dueto the presence of serum. The serum-free culture conditions for zebrafishembryos, therefore, enabled the iden-tification of biologically active com-pounds that would absorb to serumproteins and score as inactive in thecell culture screens. Taken together,the study demonstrates that whole or-ganism–based screens can indeedidentify compounds with novel activi-ties, which had been missed in cellculture–based phenotypic. Cell-basedand organism-based chemical screens,therefore, complement each other inidentifying novel lead compounds.
Overcoming the Limits ofVisual Screens in Wild-TypeZebrafish Embryos
Despite the high degree of transpar-ency during embryogenesis, not all tis-sues of the zebrafish embryo are easilyidentified and monitored by light mi-croscopy. Furthermore, as develop-ment proceeds, pigmentation willhamper phenotype detection. Thisproblem can be overcome by supple-menting the growth medium with thetyrosinase inhibitor 1-phenyl-2-thio-urea (PTU), which is routinely used toinhibit pigmentation production andis usually well tolerated by zebrafishembryos and larvae (Peterson et al.,2000; Peterson and Fishman, 2004).The recent generation of casper(White et al., 2008), a robust, fertile,and easily maintained transparent ze-brafish mutant line that lacks all me-lanocytes and iridophores in embryos,larvae, and adulthood, may in futurereplace the use of PTU in chemicalscreens. In addition, the developmentof transgenic zebrafish expressing re-
porter genes under the control of tis-sue-specific promoters significantlyexpands the possibilities of phenotypedetection in wild-type zebrafish em-bryos subjected to chemical screening.Burns and colleagues reported a firstassay for small molecules that modu-late the heart rate in transgenic ze-brafish embryos (Burns et al., 2005).They developed an automated 96-mul-tiwell plate assay for heart rate usingautomated fluorescence microscopy oftransgenic embryos expressing greenfluorescent protein (GFP) in the myo-cardium. The assay can be used torapidly assess the differing pharmaco-kinetic profiles for small moleculesthat influence the heart rate, and issuitable for high-throughput chemicalscreening. As a note of caution, itshould be mentioned that the practi-cal use of zebrafish as a model to as-sess cardioactive drug candidates wasrecently questioned because of thehigh concentrations that must bereached to see the pharmacological ef-fects (Mittelstadt et al., 2008). Trans-genic zebrafish expressing GFP in theembryonic vasculature were used forthe first time to screen chemical li-braries for compounds eliciting anti-angiogenic effects in vivo (Tran et al.,2007). Automated image analysis wasused to identify active compounds,which were subsequently shown to in-hibit human endothelial cell tube for-mation in vitro. Many additionalscreens can be envisioned as moretransgenic models expressing fluores-cent markers of organ morphogenesisand physiology are being developed inzebrafish. Crossing these fluorescentreporter lines to the transparentcasper line will extend the range ofdetecting fluorescently tagged organsinto the adult fish.
Suppression Screens inZebrafish Mutants
Over the last few years, several ze-brafish mutants have been isolatedthat mimic human disease such aspolycystic kidney disease, heart dis-ease, and anaemias (Shin and Fish-man, 2002; Zon and Peterson, 2005;Lieschke and Currie, 2007). This of-fers the possibility to screen chemicallibraries for small molecules that con-fer therapeutic effects in zebrafishmutants. Such chemical suppressors
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1293

of a disease process in a whole, verte-brate organism would represent po-tential lead compounds for drug devel-opment. The feasibility of thisapproach was first demonstrated bythe identification of a novel class ofcompounds capable of suppressing thegridlock mutation (Peterson et al.,2004). Gridlock mutants have a hypo-morphic mutation in the hey2 gene, ahairy/enhancer of split-related basichelix-loop-helix transcription factor,leading to the malformation of the lat-eral dorsal aorta preventing blood cir-culation to the trunk and tail (Zhonget al., 2000). Coarctation of the aortais a common human congenital cardio-vascular malformation that is mor-phologically similar to the gridlockmutant (Towbin and McQuinn, 1995).After screening a diverse library of5,000 drug-like compounds, two struc-turally related compounds were iden-tified that completely rescued gridlockmutants in a dose-dependent mannerto develop a normal vasculature with-out causing additional developmentaldefects (Peterson et al., 2004). Themechanism of action is unknown, butthe more potent compound, GS4012,induced expression of vascular endo-thelial growth factor (VEGF). In sup-port of this notion, overexpression ofVEGF is sufficient to rescue the grid-lock mutation and, similar to VEGF,GS4012 promotes human endothelialtube formation in vitro (Peterson etal., 2004). GS4898, which is structur-ally distinct from GS4012, was identi-fied as a second compound capable ofsuppressing the gridlock phenotype ina second unbiased screen of 7,000 un-characteristic compounds (Hong et al.,2006). The molecular target ofGS4898 is unknown, but pharmaco-logical studies suggest that GS4898acts by partially inhibiting the phos-phatidylinositol-3-kinase (PI3K)/Aktkinase branch of VEGF signaling,which is a negative regulator of VEGFsignaling (Hong et al., 2006).
In another example of a chemicalsuppressor screen in zebrafish, Sternand colleagues screened a chemical li-brary using the recessive cell cyclemutant crash&burn (crb), a homozy-gous viable mutation of the bmybgene, that has an increased number ofmitotic cells as detected by pH3 anti-body staining (Stern et al., 2005).Screening of a 16,320-compound li-
brary resulted in the identification ofone compound, named persynth-amide, which suppressed the pheno-type of the crb mutant. Because of itsability to suppress a specific cell cycledefect in crb mutants without affect-ing wild-type embryos, persynth-amide might be a useful anticanceragent. An interesting feature of thisspecific suppressor screen was that8 –10 compounds were pooled for em-bryo treatment to enable faster pro-cessing of the chemical library. Amatrix pooling strategy was devisedto ensure that each compound wasrepresented in two independentpools, which enabled rapid deconvo-lution of putative suppressors bycross-referencing (Stern et al., 2005;Murphey et al., 2006). Matrix pool-ing can greatly increase the through-put, but can complicate identifica-tion of the active compound andincrease the frequency of toxicity inthe screen.
Finally, chemical suppressor screensin zebrafish mutants can also be car-ried out in transgenic mutant animalsexpressing a fluorescent reporter genethat permits the in vivo monitoring oforgan morphology and physiology. Forexample, the application of this tech-nology to zebrafish mutants with car-diac rhythm disturbances may lead tothe discovery of novel lead compoundsfor the treatment of heart disease inhumans as was recently suggested(Burns et al., 2005). It is worth noting,however, that few zebrafish mutantswill be suitable for suppressor screensas one typically needs to identify therare homozygous mutants that escapeembryonic and larval lethality (Mar-golis and Plowman, 2004). Further-more, there is the issue of obtaininglarge numbers of zebrafish with a mu-tant genotype to screen large, chemi-cally diverse libraries. Taken to-gether, chemical suppressor screenspromise to provide a rapid, alternativeapproach to identify lead compoundsfor diseases whose underlying biologyand pathological mechanisms are notwell understood. Further therapeuticdevelopment to optimize for specificitywill, however, benefit from identifica-tion of the molecular targets and un-derstanding of the mode of action ofthe recovered lead compounds.
Considerations RegardingCompound Concentration,Penetration, and Metabolism
Three key variables in chemicalscreens that need to be taken into con-sideration when screening chemical li-braries on whole vertebrate organ-isms are compound concentration,penetration, and metabolism. Theconcentration of a compound forwaterborne treatment of embryos hastypically to be increased by an order ofmagnitude greater than the effectiveconcentrations required for cell cul-ture experiments, where the com-pounds are directly accessible to allcells. Ideally, each compound of achemical library would be screened ata full range of concentrations. How-ever, this is impossible in the contextof large-scale screens and, therefore,primary screens are usually per-formed at a single compound concen-tration. Regarding the choice of an op-timal compound concentration, dose-response studies of tyrosine kinaseinhibitors provide some guidance. Ex-periments using the VEGF inhibitorsSU5416 and PTK787/ZK222584 dem-onstrated a complete block of angio-genesis in zebrafish at concentrationsof 2 and 5 �M, respectively (Ser-bedzija et al., 1999; Chan et al., 2002).Furthermore, the FGF receptor inhib-itor SU5402 has been extensivelyused to block FGF signaling in ze-brafish and Xenopus embryos withdoses ranging from 20–160 �M(Raible and Brand, 2001; Shinya etal., 2001; Maroon et al., 2002; Delauneet al., 2005). A survey of the concen-trations used to date for chemicalscreening of zebrafish and Xenopus re-veals that they ranged between 1 and100 �g/ml or 5 and 60 �M (Table 2).Overall, an average screening concen-tration of 10 �g/ml corresponding to20 �M for small molecules with molec-ular weights of 500 Da seems to con-stitute a good compromise in balanc-ing low toxicity yet ensuring thatmany small molecules reach an activedose in vivo. This notion is also sup-ported by several studies using ze-brafish and Xenopus, where toxicitiesassociated with higher compound con-centrations were reported (Serbedzijaet al., 1999; Tomlinson et al., 2009b).
Waterborne exposure of zebrafish orXenopus embryos to small molecules
1294 WHEELER AND BRANDLI

typically leads to compound uptakeinitially via the skin and gills, and atlater stages by oral adsorption in thegastrointestinal tract through wateringestion. The absorption and bio-availability of a compound in theselower vertebrate animal models can-not be predicted due to the absence ofspecific data, but will be dependent onthe compound’s specific physicochem-ical properties. As in mammalian sys-tems, the compound’s molecularweight, hydrophobicity, and numberof hydrogen bond donors and accep-tors are considered to be critical fac-tors (Lipinski et al., 2001). One pa-rameter, the logarithm of thepartition ratio between octanol andwater (logP), can be measured empir-ically or calculated on the basis of thecompound’s chemical structure. ThelogP values correlate well with a com-pound’s membrane permeability. Acomparison of the logP values of 23drugs, which were previously testedfor bioactivity in zebrafish embryos(Milan et al., 2003), demonstratedthat compounds with logP valueshigher than �1 were absorbed in thezebrafish (Peterson and Fishman,2004). These findings were confirmedin a subsequent study of 175 bioactivecompounds where the logP values ofthe majority of compounds clusteredbetween �1 and �15 with a markedabsence of hydrophilic compounds(Sachidanandan et al., 2008). In termsof the size distribution, the activecompounds exhibited no bias and in-cluded the full-size range of the li-brary from molecular weights of 200to 700 Da. The penetration problem ofhydrophilic compounds with logP val-ues below 1 can be largely eliminatedby microinjection of the compoundinto the cytoplasm of fertilized eggs orthe blood circulation of larvae (Milanet al., 2003). However, these ap-proaches are not suited for large-scalechemical library screening. The drugpenetration problem associated withwaterborne drug administration canalso be seen as an advantage of thescreening approach as it allows for theselection of bioactive compounds withfavorable permeability or uptakeproperties in whole organisms.
Drug metabolism is an importantfactor in the conservation of drug ac-tivity across species. The cytochromeP450 (CYP) gene family is a well-rec-
ognized participant in detoxificationand drug metabolism. These enzymesnot only metabolize a large number oftoxic and endogenous compounds, butalso participate in biotransformationof ingested drugs. The cytochromeP450 gene family is, therefore, partic-ularly relevant to clinical and phar-macological studies in humans(Danielson, 2002). Metabolism of ex-ogenous compounds in zebrafish andXenopus by the cytochrome P450(CYP) family enzymes in the liver willundoubtedly affect the compound’sstability and may produce a number ofbioactive metabolites. The sequenc-ing of mammalian genomes has re-vealed that the cytochrome P450gene family is subject to rapid evolu-tion in vertebrate genomes (Gibbs etal., 2004). Compared with humangenes, there are clear expansions ofseveral rodent P450 subfamilies, butthere are also significant differencesbetween rat and mouse subfamilies.A comprehensive survey of the cyto-chrome P450 gene content in the ze-brafish and Xenopus genomes andtheir phylogenetic relationship withthe human cytochrome P450 genes isstill lacking and, therefore, it cannotbe predicted how exogenous com-pounds would be metabolized in ze-brafish or Xenopus. Furthermore,the metabolic activities of the devel-oping livers in zebrafish larvae andXenopus tadpoles may differ signifi-cantly from those in adult organ-isms. Overall, this suggests that spe-cies-specific and developmentaldifferences in drug metabolism haveto be taken into consideration, whenextrapolating data from animal mod-els, even from rodents, to humans.
Despite these limitations, in vivochemical screens in whole organismscan identify lead compounds thatcould not have been recovered in invitro cell-based screens. For example,a screen for compounds affectingerythropoiesis in zebrafish resulted inthe identification of a compound thatneeds to be metabolized in vivo to be-come effective (Shafizadeh et al.,2004). While the question whetherdrugs found in zebrafish screens willhave similar effects in humans cannotbe answered in a general sense, it hasbeen demonstrated that drugs withknown effects in humans can causeanalogous effects in zebrafish. Milan
et al. (2003) tested 23 compounds inzebrafish known to alter heart rates inhumans, which can lead to fatal ar-rhythmia and constitutes an undesir-able drug side effect. Of the 23 drugstested, 22 caused an analogous prolon-gation of the cardiac cycle (bradycar-dia) in zebrafish. In a separate study,the examination of 17 known cell cycleinhibitors in zebrafish embryos re-vealed that the majority of the testeddrugs (9 out 17) exhibited the pre-dicted cell cycle effects in vivo (Mur-phey et al., 2006). A further six drugswere shown to be active in a zebrafishcell line but not in embryos, suggest-ing that they are poorly absorbed intoembryos by waterborne treatment.Only two drugs in zebrafish were nei-ther active in vivo nor in vitro indicat-ing their targets are not conserved be-tween zebrafish and mammals. Otherdrugs that have similar effects inmammals and fish include compoundsdisrupting angiogenesis by blockingVEGF receptor signaling (Chan et al.,2002). Collectively, these studies sug-gest that drug targets are generallywell conserved between zebrafish andhumans, and, therefore, lead com-pounds identified in zebrafish-basedchemical screens are likely to havesimilar activities in humans.
Approaches to ElucidateMolecular Targets ofBioactive Compounds
While phenotypic whole-organism-based screens have the ability to ex-plore the chemical space for novelcompounds with unique biological ac-tivities in vivo, the identification ofthe molecular targets of compoundscausing specific phenotypes is notstraightforward and offers a majorchallenge. The identification of themolecular target and mechanism ofaction is, however, a prerequisite toconvert the phenotype-inducing activ-ity of a compound into a molecularunderstanding of the affected biologi-cal process or disease pathway. Oneapproach is to use affinity purificationto identify biochemically the specificprotein targets of a biologically activesmall molecule. These experimentsare unbiased and do not require as-sumptions about a molecule’s mecha-nism of action. Strategies on how toperform affinity purifications have
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1295

been published elsewhere in detail(Peterson and Fishman, 2004). Thereare, however, two drawbacks to affin-ity purification. First, hits recoveredfrom chemical library screens are of-ten moderately potent with a low mi-cromolar affinity (Burdine andKodadek, 2004). This can lead to non-specific interactions rendering theidentification of the primary bindingpartners difficult. The use of high-af-finity compounds would, therefore, in-crease the success rate of affinity-pu-rification approaches. Secondly, theconjugation of a bioactive compound toa solid resin to enable affinity purifi-cation of a target protein may result inthe loss of the compound’s binding ac-tivity. It is, therefore, not surprisingthat this time-consuming and labori-ous approach has lead to failures intarget identification. One alternativeapproach to overcome the problems isthe construction of tagged chemical li-braries, where each compound of thelibrary possesses the same functional-ized linker that can be used for effi-cient coupling to solid supports (Ahnand Chang, 2007). Khersonsky andcolleagues generated a tagged triazinecombinatorial library consisting of1,536 compounds allowing affinity pu-rification of the target protein andidentification by mass spectroscopy(Khersonsky et al., 2003). The taggedtriazine library was screened in vivoto recover compounds causing brain,eye, or pigmentation defects in ze-brafish embryos (Khersonsky et al.,2003; Jung et al., 2005). One pheno-typic screen led to the isolation of acompound that suppressed develop-ment of eyes and brain. The tag-freecompound named encephalazine waseven more potent, suggesting that thetag was not necessary for its activity.Affinity purification led to the identi-fication of four ribosomal subunit pro-teins (S5, S13, S18, and L28), whichwere previously reported to be in-volved in brain and eye developmentby a genetic mutation (Khersonsky etal., 2003). Other screens using tagged-triazine libraries focused on identify-ing novel pigmentation modulators incultured melanocytes or zebrafish em-bryos and have been recently re-viewed (Ni-Komatsu and Orlow,2007). It should be noted that evenhigh-affinity compounds could bind toproteins that are not responsible for
producing the in vivo phenotype of in-terest. Therefore, it is important toconfirm the identity of candidate pro-teins by performing gene knockdownsusing antisense morpholino oligonu-cleotides. The example given abovedemonstrates the power of tagged li-braries in identifying drug targetswithout the need for cumbersomestructure-activity-relationship stud-ies. It is hoped that the use of taggedlibraries will facilitate the process ofmoving from compound discovery totarget identification.
The Screening of AnnotatedChemical Libraries ProvidesNovel Mechanistic Insights
The selection of an adequate chemicallibrary is a crucial step in the processof performing chemical screens. Acompilation of the chemical librariesused to date for screens in zebrafishand Xenopus is given in Table 2. Twomain types of chemical libraries, syn-thetic combinatorial libraries and col-lected libraries, can be distinguished.Synthetic combinatorial libraries con-sist of compounds that are generallyall synthesized in parallel through aseries of synthetic reactions that joinsmall numbers of building blocks invarious combinations to generatelarge collections of distinct molecules(Schreiber, 2000). Examples of combi-natorial libraries used for chemicalscreens in zebrafish include trisubsti-tuted triazine libraries (Moon et al.,2002; Khersonsky et al., 2003), andlibraries containing 1,3-dioxanes(Sternson et al., 2001; Wong et al.,2004) and biaryl-containing mediumrings (Spring et al., 2002). Despite thelarge numbers of distinct compoundscontained in these libraries, many ofthe compounds are structurally re-lated and share common core struc-tures. Hence, they cover only a rela-tively small area of the chemical spaceeven if they contain thousands of dis-tinct molecules. In contrast, collectedlibraries are assembled from naturalproducts and/or synthetic compoundsthat were selected on the basis ofchemical diversity, proven biologicalactivity, lack of nonspecific toxicity,and physicochemical properties thatare compatible with efficient absorp-tion and bioavailability (Ghose et al.,1999; Lipinski, 2000; Lipinski et al.,
2001; Stockwell, 2004). Generally, thecollected chemical libraries frequentlypossess a larger structural diversitythan combinatorial libraries. Consis-tent with this notion, the hit rates inbroad chemical screens for develop-mental defects are low and compara-ble for both types of libraries, butfewer distinct phenotypes are ob-served with combinatorial libraries(Peterson and Fishman, 2004). A fur-ther advantage of collected chemicallibraries is that they can be obtainedfrom various commercial and non-profit sources as shown in Table 2 andlisted in greater detail elsewhere(Peterson and Fishman, 2004). Asmentioned above, matrix poolinggreatly increases the throughput ofchemical library screens (Stern et al.,2005; Murphey et al., 2006). The strat-egy is, however, primarily suitable forsynthetic, combinatorial libraries asmany compounds will have no biolog-ical activity. In contrast, pooling ofcompounds from collected bioactive li-braries is undesirable as this leads toa very high rate of toxicity (Murpheyand Zon, 2006). The choice to poolcompounds will, therefore, have to bedetermined empirically in pilot stud-ies.
Irrespective of the type of librarychosen for chemical screening, signif-icant efforts are required to illuminatethe mechanistic basis of each com-pound’s activity. To address this prob-lem, Stockwell and colleagues devel-oped a powerful strategy to facilitatethe screening step and the study of thelead compound’s mode of action (Rootet al., 2003). They set out to assemblea chemical library composed of 2,036well-studied organic compounds withdiverse, experimentally confirmed bi-ological mechanisms and activities.Each compound was assigned to one of169 broad biological descriptors andall published information on its activ-ity was compiled. The library includedapproved drugs, failed drug candi-dates, and chemicals with someknown biological activity such as an-tibiotic, anticancer, antidiabetic, andneurological activities among others.The resulting annotated chemical li-brary was structurally more diverseand more enriched in active com-pounds than conventional, commer-cially available libraries (Root et al.,2003). Annotated chemical libraries
1296 WHEELER AND BRANDLI

are a central resource for forwardchemical genetics approaches to studybiological systems in vitro and in vivoas was postulated by Stockwell (2000).In recent years, annotated chemicallibraries have become a valuable re-source for both chemists and biologistsinterested in carrying out chemical ge-netic screens. As a consequence, non-profit organizations and companieshave developed different annotatedchemical libraries. They include theNINDS Custom Collection (NationalInstitute of Health/National InstituteNeurological Disease and Stroke), theLOPAC1280 library (Sigma-Aldrich),the Spectrum Collection (Micro-Source), and the ICCB Known Bioac-tives library (Biomol). These librarieshave also been recently used for chem-ical screens using zebrafish embryos(Table 2) and the outcome of thesestudies will now be reviewed ingreater detail.
The first forward chemical geneticsscreen was published by North andcolleagues in 2007 and was aimed atidentifying new pathways modulatingdefinitive hematopoietic stem cell for-mation during zebrafish embryogene-sis (North et al., 2007). They used insitu hybridization to monitor changesin hematopoietic gene expression toidentify compounds inducing alter-ations in zebrafish embryos. Most ofthe non-toxic 2,357 compoundsscreened were inactive, whereas 35(1.4%) and 47 (1.9%) increased or re-duced hematopoietic stem cell geneexpression. Among these substances,10 affected the prostaglandin path-way. Compounds that increased pros-taglandin synthesis promoted hema-topoietic stem cell numbers, whereasthose that block prostaglandin syn-thesis reduced the stem cell numbers.The fact that the chemical screen re-covered several different agonists andantagonists of prostaglandin synthe-sis corroborates the findings. Further-more, the effects of prostaglandins onhematopoietic stem cells were ex-tended into mouse models.
A chemical genetics approach todefine the pathways governing ver-tebrate regeneration was developedusing a zebrafish early life stage finregeneration model (Mathew et al.,2007). The authors screened an an-notated chemical library of 2,000 bi-ologically active small molecules in
wild-type zebrafish embryos thathad been subjected to caudal fin am-putation. They identified 17 com-pounds including five glucocorti-coids, which specifically inhibitedregeneration. All functional studiesdemonstrating a role of glucocorti-coids in limiting the regenerative ca-pacity of the fin were performed us-ing a prototype glucocorticoidreceptor agonist. Since the identityof the biologically active compoundswas not disclosed, the study is of lim-ited value to researchers active inthe field of tissue regeneration. Bycontrast, all hits were documented ina chemical genetics screen for anti-angiogenic activities in transgeniczebrafish (Tran et al., 2007). Screen-ing of the LOPAC1280 library re-sulted in the identification of twoknown antiangiogenic compoundsand one compound, indirubin-3�-monoxime (IRO), not previouslyknown to possess antiangiogenic ac-tivity. IRO was subsequently shownto inhibit human endothelial tubeformation in vitro. IRO is a cell-permeable derivative of indirubinand was shown to inhibit a numberof kinases including Lck, cyclin-dependent kinases, and AMP-acti-vated protein kinases. Therefore, theantiangiogenic activity of IRO maybe multimodal resulting from the in-hibition of multiple kinases (Tran etal., 2007).
Another example demonstrating thepower of screening annotated chemicallibraries in whole organisms led to theidentification of dorsomorphin, the firstsmall molecule inhibitor of the bonemorphogenetic protein (BMP) signalingpathway (Yu et al., 2008b). The authorsdecided to use an in vivo screening ap-proach that would identify compoundsthat inhibit BMP signaling while select-ing against those with non-specific bio-logical effects. Since gene mutationsthat disrupt BMP signaling affect dor-soventral patterning resulting in dor-salization, the authors screened severalsmall molecule libraries for compoundsthat would phenocopy these mutants.The chemical libraries screened totaledover 7,500 compounds and containedamong others known bioactive mole-cules and approved drugs. However,only dorsomorphin produced substan-tial and reproducible dorsalization inzebrafish embryos. In a series of in vitro
studies, the authors show that dorso-morphin inhibits phosphorylation of thereceptor SMAD proteins, probably byblocking type I BMP receptors. Interest-ingly, dorsomorphin was previouslyshown to antagonize AMP-activated ki-nase (AMPK) in vitro (Zhou et al.,2001), an enzyme that has no role indorsoventral patterning in zebrafish asdemonstrated by Yu and colleagues (Yuet al., 2008a). Dorsomorphin was laterused as a valuable novel tool to probethe requirement for BMP signaling inosteoblast differentiation in vitro andbone mineralization in vivo, and to ex-plore the physiological role of hepaticBMP signaling in iron metabolism (Yuet al., 2008a). On the basis of the latterfindings, dorsomorphin has been sug-gested as a promising lead compoundfor the treatment of anemia in chronicdisease. Furthermore, preclinical stud-ies of dorsomorphin and its subse-quently developed more potent and spe-cific derivatives (Cuny et al., 2008) havealso suggested a rational therapy totreat fibrodysplasia ossificans progres-siva (FOP), a congenital disorder of pro-gressive and widespread postnatal ossi-fication of soft tissues, and otherheterotopic ossification syndromes as-sociated with excessive BMP signaling(Yu et al., 2008a).
Finally, a phenotype-based chemi-cal library screen of 5,760 compoundsfor novel regulators of zebrafish em-bryogenesis resulted in the identifica-tion of DTAB, a novel retinoid thatselectively activates the retinoic acidreceptor � (RAR�) and disrupts pat-terning along the anterioposteriorbody axis in vivo (Sachidanandan etal., 2008). Overall, these studies dem-onstrate that the screening of anno-tated chemical libraries in whole or-ganisms can yield novel regulators ofbiological processes such as mitosis,regeneration, stem cell proliferation,or organogenesis, and can identifynovel, previously elusive inhibitors ofimportant signaling pathways. Fur-thermore, they highlight the great ad-vantages that the novel compoundsemerging from these in vivo screensprovide with their exquisite temporalcontrol in the dissection of signalingpathways and their promising poten-tial as novel lead compounds for drugdevelopment.
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1297

XENOPUS AS A NEWMODEL FOR LARGE-SCALECHEMICAL SCREENS
A General Outline forChemical Screens inXenopus
Despite the fact that Xenopus makean ideal tetrapod model for in vivochemical screens due to the small sizeof the embryos, accessibility duringdevelopment ex utero, optical trans-parency of tadpoles, permeability tosmall molecules, and favorable evolu-tionary position relative to humans,their utility has remained untesteduntil recently. The feasibility of per-forming large-scale phenotype-based
chemical screens in Xenopus is illus-trated by two recent examples inwhich novel compounds affecting ei-ther pigmentation or angiogenesisand lymphangiogenesis were identi-fied (Tomlinson et al., 2009a,b; Kalinet al., 2009).
The general procedure of perform-ing large-scale chemical screens withXenopus embryos is outlined in theflow diagram shown in Figure 2. Typ-ically, compound treatment is not ini-tiated before the completion of gastru-lation, unless the screen is targetedfor biological processes occurring dur-ing blastula stages or gastrulation. Pi-lot studies have shown the suggestedtiming of chemical exposure is benefi-
cial as the toxicity of compounds ismuch less pronounced in older em-bryos (Tomlinson et al., 2005) (Kalinand Brandli, unpublished observa-tions). Unfertilized eggs and mal-formed embryos can be removed,whose death would otherwise contrib-ute to poor health, developmental de-lays, and often death of the remainingembryos in the well. In addition, it iseasier to select healthy, developmen-tally synchronized embryos for chem-ical treatments at later stages. Em-bryos or tadpole, usually five per well,are arrayed in 96- or 48-well microti-ter plates, respectively, containing theculture medium supplemented withthe test compounds at the desired con-
Fig. 2. Flow diagram of a Xenopus chemical library screen. Compounds are arrayed in multi-well plates and five embryos at stage 15 are added perwell. These are incubated for 3 days and then scored for phenotypic changes in comparison to control tadpoles. Examples of some of the possiblepigmentation phenotypes observed are shown. Positive hits are independently verified. Reproduced by permission of Royal Society of Chemistry(Tomlinson et al., 2009b).
1298 WHEELER AND BRANDLI

centration. Typically, a low-magnifi-cation dissection microscope is used tomonitor the embryos for phenotypicchanges during cultivation in pres-ence of the compounds. The examplesof embryos displaying differences inpigment patterns, which are easily de-tected by visual inspection, are shownin Figure 2. Other rapidly detectedphenotypes occurring in response tocompound treatment include defectsin brain and eye morphogenesis (e.g.,small eyes), and effects on heart func-tion and the shape of the trunk, tail,and fins.
A Chemical Screen forCompounds AffectingPigment Cell Development
Pigment cells (melanophores or mela-nocytes as they are more commonlyknown in higher vertebrates) are aparticularly useful cell type to focuson for high-throughput chemicalscreening. This is due to the ease ofvisual scoring and because melano-phores and retinal pigment epithelial(RPE) cells are capable of acting as anin vivo model for many aspects of cel-lular biology, including cell migration,morphology, and biochemical path-ways (such as the production of pig-ment). In addition, the development ofpigmented tissue is related to a num-ber of disease states and so a screenfocusing on melanocytes could alsoplay a role at the early stages of drugdiscovery. Examples of such diseasesinclude albinism, piebaldism, an auto-somal disorder leading to localized hy-popigmentation (Thomas et al., 2004),vitiligo, an autoimmune disease lead-ing to the destruction of melanocytesand pigment-free patches of skin inpatients (Kelsh et al., 2000), hyperpig-mentation, and finally skin cancer ormelanoma (Kelsh et al., 2000; vanKempen and Coussens, 2002; Whiteand Zon, 2008; Sturm, 2009).
We have carried out a developmentalchemical genetic screen of 2,950 com-pounds to identify phenotypes associ-ated with pigment cell development inXenopus (Tomlinson et al., 2009b). Werecovered 41 compounds producing var-ious phenotypes (Tomlinson et al.,2009b). Two compounds showed a re-duction in the size of the eye and sev-eral compounds caused general devel-opmental defects and edema in the
heart and kidneys. The largest numberof compounds discovered in one pheno-typic category were those causing apartial or total loss of pigment in themelanophores and retinal pigment epi-thelium (17 compounds). Five com-pounds were identified as affecting mel-anophore morphology in the developingembryo. Seven compounds were identi-fied that affected the normal migrationof melanophores. Of these, we have fur-ther characterized compound NSC84093, which gives a striking verticalbanding pattern on the dorsal side ofthe embryo and a decrease in the mela-nophores on the ventral side of the tail.We identified this compound as an8-quinolol derivative and further dem-onstrated that it functions as a potentmatrix metalloproteinase (MMP) inhib-itor. MMPs are a family of proteinsknown to have important roles in cellmigration, inflammation, angiogenesis,and cancer (Page-McCaw et al., 2007).Potential targets for NSC 84093 in-cluded MMP-14 and MMP-2, which areexpressed in or close to migrating neu-ral crest cells and will give rise to mela-nophores. Interestingly, knockdown ofMMPs using morpholinos partially phe-nocopied the effect of NSC 84093 (Tom-linson et al., 2009a). NSC 84093, there-fore, represents a novel and uniquemolecular tool to investigate neuralcrest cell migration and subsequentmelanophore development in vivo. Fur-thermore, NSC 84093 may contributetowards a better understanding of theroles of MMPs in cell migration pro-cesses under normal and pathologicalconditions, such as in cancer.
A Chemical Screen forCompounds AffectingAngiogenesis andLymphangiogenesis
Angiogenesis and lymphangiogenesisare essential for embryonic develop-ment and organogenesis, but also playimportant roles in tissue regeneration,chronic inflammation, and tumor pro-gression (Carmeliet, 2003; Alitalo et al.,2005; Cueni and Detmar, 2006). De-spite the significant advances in devel-oping antiangiogenic therapies of recentyears, the identification of novel drugstargeting the blood and lymph vascula-ture is of high priority. This is particu-larly true for lymphangiogenesis, wherethe lack of an appropriate, simple ani-
mal model has hampered progress inelucidating the key molecular playersand signaling pathways. In addition,large-scale in vivo screens to identifydrug-like small molecule modulators oflymphatic vessel formation are missingto date. The recent imaging of an elab-orate lymph vessel system and its mo-lecular characterization in Xenopus tad-poles (Ny et al., 2005) has created newopportunities for studying vascular de-velopment and chemical screens.
We have applied an unbiased for-ward chemical genetics approach incombination with a simple pheno-typic readout to uncover signalingpathways and small molecule regu-lators of lymphatic and blood vascu-lar system development in Xenopustadpoles (Kalin et al., 2009). In asecondary screen, Xenopus embryoswere treated with the hits recoveredfrom the primary screen and ana-lyzed by in situ hybridization fordefects in angiogenesis and/or lym-phangiogenesis. An annotated chem-ical library (LOPAC1280, Sigma-Al-drich) representing small organicmolecules with diverse well-definedpathway specificities was analyzedusing this novel two-step chemicalscreening strategy. After screening1,280 small molecules, 65 com-pounds scored positive in the pri-mary screen and were subsequentlysubjected to the secondary screen.This resulted in the identification of32 compounds modulating distinctaspects of vascular development invivo. Specifically, we found that 18compounds selectively blocked bloodvessel development, six compoundsinterfered with both blood and lym-phatic vessel development, and eightcompounds affected lymphatic devel-opment only The bioactive com-pounds fell into 15 distinct pharma-cological classes with modulators ofphosphorylation representing thelargest activity class (13 out of 32).Positive hits included inhibitors of theVEGF signaling pathway, which is es-sential for vascular development (Ols-son et al., 2006), and thus validates thein vivo chemical screening approach.This point is further underscored by thefact that the chemical screen in Xeno-pus was able to recover five of the eightknown antiangiogenic compounds rep-resented in the LOPAC1280 library. Bycontrast, a screen of the LOPAC1280
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1299

library for antiangiogenic activity intransgenic zebrafish resulted in the re-covery of only three out of the eight an-tiangiogenic compounds (Tran et al.,2007). This indicates that the chemicallibrary screening strategy in Xenopus ismore sensitive in recovering compoundswith vascular activity than the one em-ploying transgenic zebrafish.
The chemical screen in Xenopus alsoresulted in the identification of small-molecule modulators of pathways notpreviously known to mediate vascularand/or lymphatic development. Theseincluded, for example, an adenosineA1 receptor antagonist that inhibitedboth lymphatic and blood vessel for-mation in Xenopus tadpoles. In aproof-of-principle study, we subse-quently validated this compound in amammalian animal model by demon-strating that it blocked VEGFA-in-duced neovascularization in adultmice. Taken together, the study estab-lished a rapid and sensitive in vivotwo-step method for large-scale chem-ical screens using Xenopus tadpoles toidentify novel pathways and lead com-pounds with selectivity for lymphaticand blood vessel formation in a time-and cost-saving manner. The recov-ered compounds represent a rich re-source for in-depth analysis in the fu-ture, and their drug-like features willfacilitate further evaluation in pre-clinical models of inflammation andcancer metastasis.
Inverse Drug Screening
Whereas large-scale chemical libraryscreens have until recently not beencarried out in Xenopus, the use ofpharmacological agents to study de-velopmental processes has a longertradition. Defined small-molecule an-tagonists are valuable tools in phar-macological loss-of-function studies invivo. They can be used at any time ofdevelopment and are ideal to studythe role of maternal proteins, which isotherwise difficult to achieve using ge-netic or knockdown approaches. Theuse of individual chemical inhibitorsas tools for functional analysis of Xe-nopus development has, for example,been demonstrated using cyclopam-ine, an inhibitor of hedgehog signal-ing, SU5402, a potent fibroblastgrowth factor receptor (FGFR) inhib-itor, and the retinoic acid inhibitor
BMS453 (Koebernick et al., 2003;Chen et al., 2004; Chung et al.,2004). On a larger scale, the effectsof treating Xenopus embryos with anarray of 13 protein kinase inhibitorsdemonstrated that several com-pounds caused specific developmen-tal defects (Tomlinson et al., 2005).In general, these approaches are fre-quently limited by the lack of suit-able, highly specific drugs thateither inhibit or activate a given pro-tein function or signaling pathway invivo. For example, to date there areno reliable small-molecule inhibitorsof the Wnt signaling pathway known(Barker and Clevers, 2006). By con-trast, rich collections of pharmaco-logic agents are available for certainprotein families, such as ion chan-nels, neurotransmitter receptors,and pumps, which could be tapped toimplicate specific protein families ina chosen biological process.
Levin and co-workers took advan-tage of this resource and have devel-oped a chemical genetic screeningstrategy coupled with a simple pheno-typic read-out to rapidly gain molecu-lar insight into endogenous ion flowsthat control the establishment of left-right asymmetries of the body plan inXenopus embryos (Levin et al., 2002).In their screening strategy, fertilizedeggs were treated with drugs of in-creasing specificity in successive reit-erations of the screen to narrow downthe lead candidate protein(s). Using14 broad-range inhibitors, most pro-tein families tested, such as sodiumand chloride channels, were ruled outto play a role in left-right asymmetry.However, the broad-range screen im-plicated that blocking H� and K� ionflux induced heterotaxia, an abnormalarrangement of organs or parts of thebody in relation to one another. Theapplication of more specific inhibitorsidentified the H�/K�-ATPase as theion pump responsible for establishingdifferential hydrogen/potassium fluxesas an early step in the determination ofleft-right asymmetry in Xenopus em-bryos. This conclusion was then sup-ported by genetic disruption of endoge-nous H�/K�-ATPase activity, and bypharmacological studies in chicken em-bryos (Levin et al., 2002).
Adams and Levin subsequently for-malized their iterative screeningstrategy, which they termed inverse
drug screening (Adams and Levin,2006). The pharmacological “loss-of-function” strategy uses known chemi-cal agents to rapidly implicate specificcandidates for roles in any chosen bi-ological process. The hierarchical test-ing procedure allows the assessmentof large numbers of pharmacologicalagents in a manner that is more effi-cient than performing exhaustivescreens of entire compound families.No more than 20–30 compounds needto be tested before implicating a shortlist of targets that are ultimately val-idated. Hence, the strategy quickly re-veals a manageable number of specificmolecular candidates that can then betargeted and validated in detail usinghighly specific pharmacological agentsand genetic approaches, respectively.To date, inverse drug screening inXenopus embryos has been used to im-plicate H�-V-ATPase-dependent pro-ton fluxes in early left-right pattern-ing and tail regeneration (Adams etal., 2006, 2007) and to uncover novelpre-nervous roles for the neurotrans-mitter serotonin in left-right pattern-ing (Fukumoto et al., 2005a,b). In-verse drug screening, therefore,represents an interesting, alternativechemical genetic approach to dissectbiological process using small-mole-cule effectors.
Using Cell-Free XenopusExtracts for ChemicalScreens
While the focus of this review is onwhole-organism-based chemical screens,Xenopus has also been instrumental inthe development of chemical screen-ing methods using cell-free extracts.Unique to Xenopus among model organ-isms is the use of oocyte and egg ex-tracts as cell-free systems for the studyof various cellular and biochemicalmechanisms (Liu, 2006). Xenopus oo-cytes and eggs contain the complete bio-chemical machineries necessary for cellcycle progression and DNA replication.Cell-free Xenopus extracts can be ob-tained in large quantities, are easily ac-tivated in vitro, and can recapitulateimportant cellular processes such ascell cycle progression, DNA replication,and centrosome duplication in vitro.Given these properties, it is not surpris-ing that cell-free extracts from Xenopus
1300 WHEELER AND BRANDLI

have been used successfully in chemicalscreens to identify small-molecule mod-ulators of actin assembly, microtubulestability, ubiquitin-dependent proteindegradation, cell cycle machinery, andDNA damage response (Verma et al.,2004; Wignall et al., 2004; Peterson etal., 2006; Dupre et al., 2008; Landais etal., 2009). Chemical screening in cell-free Xenopus egg extracts takes place ina fully soluble in vitro context. There-fore, compound delivery is simple andproblems associated with compoundtoxicity and bioavailability are mini-mized. The utility of the compoundsemerging from these screens for thetreatment of cells or organisms needs tobe established in each case. Despite thislimitation, chemical screens in Xenopusextracts have the potential to revealnovel drugable components in impor-tant biochemical pathways (Hathawayand King, 2005).
XENOPUS AS ASCREENING TOOL IN THEDRUG DISCOVERY ANDDEVELOPMENT PROCESS
Basic Strategies in DrugDiscovery and Development
There are two broad strategies fordrug development, which converge atthe level of the lead validation stepand are followed by preclinical andclinical studies (Fig. 3). The target-based strategy has been applied indrug discovery and molecular phar-macology over the past decade. It re-quires a functional understanding ofthe disease pathway to determine spe-cific target proteins or genes directlyassociated with the disease. Sus-pected targets are subjected to valida-tion to determine whether they arecritical to disease pathogenesis. Con-
firmed targets will serve as a basis forlarge-scale high-throughput in vitroscreens to identify active compounds.The cell-free assays typically monitorthe ability of a compound to bind tothe target and/or inhibit its function.The in vitro approach has the advan-tage that target– compound interac-tions are identified in the absence ofconfounding variables. Active com-pounds recovered from the screeningof chemical libraries will be used asleads that will have to undergo vali-dation and optimization before theyor their optimized chemical deriva-tives can enter preclinical and clini-cal studies. In the activity-basedstrategy, the molecular basis of adisease is unknown and active com-pounds cannot be identified on thebasis of their inhibition of a specificprotein target. Therefore, large-scalechemical screening is performedagainst an intact biological systemto identify lead compounds thatinduce interesting phenotypicchanges. Activity-based approachesare also known as phenotype-baseddrug discovery approaches. The bio-logical systems suitable for pheno-typic chemical screening can consistof prokaryotic or eukaryotic single-cell organisms, normal or pathologi-cal mammalian cells, or even wholemulticellular organisms. Oncebiologically active compounds thatinduce a desired phenotype are iden-tified, they can enter the lead opti-mization and development process.In addition, efforts are directed atstudying the mechanistic basis of thephenotype by identifying the target.The structural and biological in-formation of the target can be usedin target-based approaches to iden-tify and develop more potent com-pounds that disrupt the function ofthe target. The end point of any drugdiscovery and development processis a drug, a registered and approvedchemical substance that is used in adose-dependent manner in treat-ment of a medical condition.
Identifying Drug TargetsIn Vivo
Regardless of the strategy chosen, Xe-nopus embryos and tadpoles can serveas cheap and efficient bioassay tools atseveral stages of the drug discovery
Fig. 3. The utility of Xenopus in drug discovery and development. The two broad strategies ofdrug discovery and development are shown. The target-based strategy requires a functionalunderstanding of the disease pathway to determine target genes or proteins for drug discovery anddevelopment. The activity-based strategy selects for drug candidates on the basis of biologicalactivities implicated with a therapeutic benefit. The Xenopus embryos and tadpoles can serve ascheap and efficient bioassay tools at several stages of the drug-discovery process regardless of thestrategy chosen. SAR, structure-activity-relationship.
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1301

process (Fig. 3). At present, the utilityof Xenopus for therapeutic targetidentification is unproven, but prom-ising examples are emerging (Kalin etal., 2007; Tomlinson et al., 2009a).Ample genomic and transcriptomic in-formation and the similarity to mam-mals make Xenopus amenable to func-tional analysis relevant to uncoveringhuman disease pathways and theidentification of targets suitable fordrug development. For example, alarge-scale in situ hybridization anal-ysis of solute carrier (SLC) gene ex-pression has provided compelling evi-dence that the Xenopus orthologues ofeleven SLC genes causing inheritedrenal diseases in humans are ex-pressed in the equivalent segments ofthe Xenopus pronephric kidney (Racitiet al., 2008). The Xenopus pronephroscan, therefore, serve as a novel modelfor the study of human renal diseasemechanisms. Similarly, the molecularpathways regulating angiogenesisand lymphangiogenesis, two processesthat play central roles in wound heal-ing processes and in the pathology ofinflammatory diseases and cancer, areshared between Xenopus and mam-mals (Cleaver and Krieg, 1998; Hel-bling et al., 2000; Ny et al., 2005; Coxet al., 2006; Kalin et al., 2007). Molec-ular dissection of these pathways canbe done efficiently and rapidly by tar-geted gene knockdowns using anti-sense morpholino oligonucleotides asdemonstrated first for the canonical,�-catenin-dependent Wnt signalingpathway in Xenopus (Heasman et al.,2000). Alternatively, the screening ofannotated chemical libraries can iden-tify novel targets as demonstrated formatrix metalloproteinases, whichwere recently shown to play an essen-tial role in pigment cell migration(Tomlinson et al., 2009a). Finally,large-scale morpholino knockdownscreens (Kenwrick et al., 2004; Ranaet al., 2006), genetic screens by ran-dom mutagenesis in Xenopus tropica-lis (Goda et al., 2006), and novel tar-geted mutagenesis approaches (seeFuture Directions section) will makein vivo functional analysis of diseasepathways and therapeutic targetidentification in this vertebrate modelorganism an even more attractive al-ternative to expensive and time-con-suming mouse models.
Validating Drug Targets InVivo
Target validation encompasses thedemonstration that a target gene orprotein is critical to a signaling path-way underlying a medical condition orto the pathogenesis of a disease. A rig-orous evaluation must occur to demon-strate that modulation of the target willhave the desired therapeutic effect.This involves intensive in vitro, as wellas in vivo studies that provide gain- andloss-of-function information on the pos-sible effects of therapeutic intervention.The result of these efforts is to establishsufficient knowledge so that physiologi-cally relevant model systems can be de-veloped into assays for downstreamchemical screening. Current validationmethods require time-consuming andoften expensive studies, particularly iftransgenic mouse models have to be de-veloped to validate a target. Target val-idation has, therefore, become a bottle-neck in drug development. On the otherhand, the failure to correctly determinethe biological relevance of a target canresult in large accumulated costs due tohigh attrition rates on the later stagesof drug development. Thus, alternativeapproaches that streamline and speedup the target prioritization efforts are ofimmense value to the drug developmentcommunity. Lower vertebrate animalmodels such as Xenopus offer a cheapalternative to demonstrate the causal-ity of target gene candidates in vivo.Gene overexpression and morpholinoknockdown experiments in Xenopusembryos can be used to validate targetsand to identify epistatic relationshipsbetween individual targets and otherpathway members. For example, the Gprotein–coupled receptor APJ and itsligand have been implicated in regulat-ing tumor angiogenesis (Kalin et al.,2007). Using gain- and loss-of-functionexperiments in Xenopus, the role of APJand apelin in regulating embryonic an-giogenesis was determined and the ep-istatic relationship between VEGF andapelin-APJ signaling was established(Cox et al., 2006; Kalin et al., 2007).
Validating Lead CompoundsIn Vivo
Once a target has been validated,large-scale high-throughput chemicalscreens are carried out to identify lead
compounds. In vitro chemical screen-ing against a defined molecular targetexploits a protein’s function, i.e., enzy-matic activity, signaling activity, orits interaction with its partners, toidentify small-molecule binders. Al-ternatively, activity-based approachesuse large-scale in vivo chemicalscreening in whole organisms to iden-tify leads on the basis of phenotypicchanges. The utility of Xenopus as anovel vertebrate model for in vivochemical screening has recently beenestablished (Tomlinson et al., 2009a,b;Kalin et al., 2009) and is extensivelydiscussed above. Regardless of the pri-mary screening strategies, the identi-fied small-molecule hits are typicallyconfirmed using a series of secondaryassays. In this confirmative processknown as lead validation, Xenopusembryos and tadpoles can be em-ployed as powerful in vivo models.This is best documented for drug dis-covery screens aimed at interferingwith the Wnt signaling pathway,where the �-catenin-dependent trans-activation of Tcf-dependent genes rep-resents the key molecular lesion caus-ing colorectal cancer (Barker andClevers, 2006). Various cell-basedchemical libraries screens have iden-tified small molecules that modulate�-catenin-dependent Wnt signaling invitro, including the agonists 6-bro-moindirubin-3�-oxime (BIO) (Meijer etal., 2003) and a pyrimidine derivate(Liu et al., 2005); the co-activatorQS11 (Zhang et al., 2007); the antag-onists NSC668036 (Shan et al., 2005),a natural product compound (Lep-ourcelet et al., 2004); and other syn-thetic compounds identified using avirtual compound screening strategy(Heligon and Brandli, unpublished ob-servations). In all these cases, Xeno-pus embryos served as effective andrapid in vivo validation systems thatprovide a basis for rational design ofhigh-affinity compounds modulatingWnt signaling in vivo. Other promis-ing areas in pharmaceutical research,where the use of Xenopus as an in vivobioassay tool for lead validation is ex-pected to accelerate the drug discov-ery process, include the developmentof small-molecule inhibitors of (lym-ph)angiogenesis and skin pigmenta-tion.
1302 WHEELER AND BRANDLI

Optimizing Lead CompoundsIn Vivo
Before validated lead compounds en-ter preclinical studies, they are opti-mized through structure-activity-rela-tionship (SAR) studies to improvetheir properties, such as increasingtheir potency, specificity, and enhanc-ing their pharmacokinetic properties.SAR studies require the synthesis ofanalogous synthetic compounds withrefined properties. Besides increasedpotency and target selectivity, drug-like characteristics that are compati-ble with testing in biological modelsystems are improved. The syntheticanalogues are subjected to assay-based screening to compare theirphysicochemical and biological prop-erties with those of the initial leadcompounds. Repeated rounds of SARstudies give rise to progressive leadoptimization. SAR studies are typi-cally performed in vitro either by as-sessing binding to the purified targetprotein or using cell culture–based as-says. Apart from the intrinsic biologi-cal activity on the target, the absorp-tion, distribution, metabolism, andexcretion (ADME) are fundamentalpharmacokinetic properties that de-termine the in vivo efficacy of a drugcandidate. Structural changes thatimprove the potency of a compoundmay have detrimental effects onADME, which go undetected in tradi-tional in vitro SAR studies. By per-forming SAR studies in whole organ-isms, structures can be identified thatimprove potency without loss of invivo efficacy or increased toxicity. Xe-nopus embryos and tadpoles arehighly amenable to in vivo SAR stud-ies as recently demonstrated for NCS84093, an inhibitor of pigment cell mi-gration, and other structurally relatedmolecules (Tomlinson et al., 2009a).One danger associated with perform-ing in vivo SAR studies in Xenopus isthat the screening may select for com-pounds that are species-specific andmay no longer recognize the humanprotein target. Validation of optimizedleads in human in vitro assays is,therefore, imperative. However, giventhe ease of performing the SAR exper-iments in Xenopus embryos and tad-poles, it is likely that in vivo SARstudies will become increasingly pop-ular. Another attractive aspect of per-
forming in vivo SAR studies is thatthey couple the characterization ofbinding affinities with ADME analy-sis. Compound testing in Xenopus canfacilitate the identification of lead de-rivatives with improved absorptionproperties or favorable metabolic con-version rates resulting in enhancedbioavailability and drug efficacy.Given the differences in physiologyand metabolism, it is evident that theADME properties of small moleculesin Xenopus embryos and tadpoles willnot reflect all aspects of ADME inadult mammalian animal models orhumans. Nevertheless, structural op-timization of leads in Xenopus repre-sents a simple, rapid, and cost-effec-tive experimental approach that canassist in the prioritization process ofcompounds for subsequent testing inmore sophisticated mammalian ani-mal models.
Assessing Toxicity andTeratogenicity
Toxicology and teratology testing areother important areas of drug devel-opment, where Xenopus serves asconvenient vertebrate animal model.In fact, Xenopus have a long historyin the fields of teratology and toxi-cology. The Frog Embryo Teratogen-esis Assay–Xenopus (FETAX) is a96-hr whole-embryo developmentaltoxicity test that uses late blastula-stage Xenopus embryos to measurethe effects of chemicals on mortality,malformation, and growth inhibition(Courchesne and Bantle, 1985). FETAXhas been used for many years as ascreening assay to identify potential hu-man teratogens, toxic compounds, andecological hazards as demonstrated bytwo recent examples, where humanpharmaceuticals and an antimalarialdrug were assessed (Richards and Cole,2006; Longo et al., 2008). The advan-tages of FETAX include its speedand low cost. It is, however, not clearhow predictive FETAX will be of humantoxicity, since Xenopus have limited xe-nobiotic metabolism through the first96 hr of embryogenesis. These limita-tions can be partially overcome using anin vitro metabolic activation system,where test compounds are pretreatedwith activated rat liver microsomes(Fort et al., 1988). Using this pretreat-ment procedure, the teratogenic risk
of proteratogenic compounds, such asthalidomide and aflatoxins, can be ac-curately assessed (Fort et al., 2000; Vis-mara and Caloni, 2007). Taken to-gether, Xenopus represents a promisingbioassay system in the drug discoveryand development process that will com-plement cell culture assays, and in vivoexperiments in mice. Ideally, Xenopus-based assays are positioned at an earlystage in the lead validation and optimi-zation pipeline.
FUTURE DIRECTIONS
Phenotype-based chemical libraryscreens in zebrafish and Xenopus em-bryos have emerged as powerful ap-proaches to recover bioactive smallmolecules, identify novel therapeuticlead compounds, and characterize or-gan-specific toxicities of drug candi-dates in preclinical development.Analogous approaches are not possi-ble in rodents because their develop-ment occurs in utero and embryos arelimited in number. With the increas-ing costs of drug development, theidentification of cheaper ways to eval-uate lead compounds in an in vivo sit-uation will be of interest to pharma-ceutical companies. Xenopus andzebrafish also offer complementarymodels to the mouse as they can pro-vide an initial assessment of theADME properties of a compound un-der development, which tests in cellcultures are unable to do.
As shown in Table 2, the number oflarge-scale chemical screens carriedout to date in Xenopus is fairly lim-ited. There is, therefore, plenty ofroom for further chemical screens tar-geting different tissue and organ sys-tems. By varying the time point andlength of compound exposure, differ-ent aspects of organogenesis, such asspecification, patterning, and termi-nal differentiation, can be targeted,enabling the chemical dissection ofthe complex biological processes un-derlying organogenesis. In future,chemical genetic screens will also takeadvantage of Xenopus as a model tet-rapod to study organs and appendagesnot found in fish, such as lungs, lymphhearts, and limbs. This asset and, incomparison to zebrafish larvae, the in-creased similarities of the tissues andorgans of Xenopus tadpoles to theirmammalian counterparts will trans-
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1303

late into a higher predictive power offindings made in Xenopus for our un-derstanding of human biology. Al-though highly informative and power-ful, the large-scale chemical geneticapproaches used to date remain labo-rious, especially if the desired pheno-types cannot be scored by simple vi-sual inspections of intact embryos andtadpoles and, therefore, require in situhybridization or immunohistochemi-cal methods to detect chemically in-duced changes. Promising automationof the screening process and intelli-gent compound pooling strategies toincrease the through-put of chemicalscreens have been reported (Burns etal., 2005; Stern et al., 2005; Tran etal., 2007) and recent developments,such the use as transgenic GFP re-porter lines (Tran et al., 2007), willenable more sophisticated screens tobe carried out in the future. These willinclude screens targeting internal or-gans, such as the gut, immune system,and vasculature inaccessible to visualinspection of intact embryos. Fur-thermore, chemical screens may bedevised that use GFP reporters tomonitor changes in physiological ac-tivities.
A further focus of intense researchin the future will be on the develop-ment of lower vertebrate models of hu-man disease for whole-organism-based chemical library screening.First examples of chemical screens toidentify small molecules that sup-press disease phenotypes in mutantzebrafish and thus provide therapeu-tic benefit to the affected animals havebeen reported (Peterson et al., 2004;Stern et al., 2005; Hong et al., 2006).The identification of genetic mutantsin X. tropicalis (Goda et al., 2006) willcomplement the many mutants al-ready available in zebrafish. Pres-ently, chemical suppressor screens arelimited to the pool of mutants thathave emerged from various randommutagenesis screens carried out in ze-brafish and Xenopus. Many of thesemutants are not suitable for chemicalsuppressor screens as they do nothave robust or reproducible pheno-types and there is frequently the issueof obtaining sufficient amounts of mu-tant individuals (Margolis and Plow-man, 2004). The development of ze-brafish and Xenopus models thataccurately phenocopy human congen-
ital diseases requires the generationof tailor-made mutations in the ortho-logues of human disease genes. Tar-geted knockout approaches and theengineering of genes carrying prede-termined mutations has been verysuccessful in mice, but are not feasiblein zebrafish and Xenopus due to theinability to grow embryonic stem cells.A step forward towards recoveringmutations in a gene of interest hasbeen the introduction of TILLING(Targeting Induced Local Lesions INGenomes) methodology (Wienholds etal., 2003; Stemple, 2004; Moens et al.,2008). This reverse genetics strategyutilizes the screening of chemicallymutagenized genomes by resequenc-ing and TILLING to identify muta-tions in specific genes. In addition tozebrafish, TILLING has recently alsobeen successfully applied to recoverrecessive mutations in X. tropicalis(D.L. Stemple, personal communica-tion). Although specific regions withingenes can be analyzed for disruptions,the mutations obtained by TILLINGare random. Moreover, the requiredresources and technical knowhow arebeyond the scope of most laboratories.Recently, zinc-finger nuclease technol-ogy has been developed that offers thepossibility to engineer mutations inspecific zebrafish genes (Doyon et al.,2008; Meng et al., 2008).
The technique employs zinc-fingernucleases, chimeric molecules con-sisting of a DNA binding zinc-fingerdomain and the FokI restrictionendonuclease, to induce targetedmutations in any gene of interest.While not proven to date, the tech-nique offers a rational and straight-forward strategy to engineer precisemodels of human inherited diseasesin zebrafish and X. tropicalis. Giventhe diploid nature of the X. tropicalisgenome and the extensive syntenyshared with the human genome, weanticipate that the engineering ofanimal models carrying mutations inhuman disease genes will be moresuccessful in X. tropicalis thanzebrafish, whose genome has under-gone an additional genome duplica-tion relative to tetrapods. The mod-eling of human diseases in Xenopuscarries the great hope that thou-sands of mutant embryos can be ob-tained from a single mating, whichwill subsequently be employed for
large-scale chemical suppressorscreens to identify novel compoundsconferring therapeutic activity.While this approach is presently al-ready possible using transgenicmouse models, costs and limitationsin obtaining sufficient amounts ofmutant animals have prohibited thepractical execution of such screens.In our opinion, the modeling of hu-man inherited diseases affectingskin pigmentation, the heart, thekidney, the blood vasculature andlymphatics will be of particular in-terest as defects in these tissues andorgan systems will not cause earlyembryonic lethality and first becomeevident once tadpole stages havebeen reached. Affected tadpoles willdisplay phenotypes that are easilyidentified by visual inspection, suchas loss of pigmentation, edema for-mation, and irregular heart beating,which offers the advantage thatsmall molecules suppressing the dis-ease pathology can be easily scoredand recovered. The coming years willshow whether these exciting newpossibilities can be exploited. Over-all, the development and use of mul-ticellular vertebrate organisms, suchas zebrafish and Xenopus embryos,as in vivo drug discovery tools hasexpanded our possibilities to investi-gate the chemical universe for novelsmall bioactive molecules that couldserve as lead compounds for drug de-velopment and therapeutic interven-tion.
ACKNOWLEDGMENTSWe thank Derek Stemple for sharingunpublished results. Research in thelaboratories of G.N.W. and A.W.B.was supported by grants from theMedical Research Council to G.N.W.,and the Swiss National Science Foun-dation, the European Community,and the ETH Zurich to A.W.B.
REFERENCES
Adams DS, Levin M. 2006. Inverse drugscreens: a rapid and inexpensive methodfor implicating molecular targets. Gene-sis 44:530–540.
Adams DS, Robinson KR, Fukumoto T,Yuan S, Albertson RC, Yelick P, Kuo L,McSweeney M, Levin M. 2006. Early,H�-V-ATPase-dependent proton flux isnecessary for consistent left-right pat-terning of non-mammalian vertebrates.Development 133:1657–1671.
1304 WHEELER AND BRANDLI

Adams DS, Masi A, Levin M. 2007. H�pump-dependent changes in membranevoltage are an early mechanism neces-sary and sufficient to induce Xenopustail regeneration. Development 134:1323–1335.
Ahn YH, Chang YT. 2007. Tagged smallmolecule library approach for facilitatedchemical genetics. Acc Chem Res40:1025–1033.
Alitalo K, Tammela T, Petrova TV. 2005.Lymphangiogenesis in development andhuman disease. Nature 438:946–953.
Barker N, Clevers H. 2006. Mining theWnt pathway for cancer therapeutics.Nat Rev Drug Discov 5:997–1014.
Berger J, Currie P. 2007. The role of ze-brafish in chemical genetics. Curr MedChem 14:2413–2420.
Brandli AW. 2004. Prospects for the Xeno-pus embryo model in therapeutics tech-nologies. Chimia 58:694–702.
Burdine L, Kodadek T. 2004. Target iden-tification in chemical genetics: the (of-ten) missing link. Chem Biol 11:593–597.
Burns CG, Milan DJ, Grande EJ, Rott-bauer W, MacRae CA, Fishman MC.2005. High-throughput assay for smallmolecules that modulate zebrafish em-bryonic heart rate. Nat Chem Biol1:263–264.
Carmeliet P. 2003. Angiogenesis in healthand disease. Nat Med 9:653–660.
Chan J, Serluca FC. 2004. Chemical ap-proaches to angiogenesis. Methods CellBiol 76:475–487.
Chan J, Bayliss PE, Wood JM, Roberts TM.2002. Dissection of angiogenic signalingin zebrafish using a chemical genetic ap-proach. Cancer Cell 1:257–267.
Chang S, Bray SM, Li Z, Zarnescu DC, HeC, Jin P, Warren ST. 2008. Identificationof small molecules rescuing fragile Xsyndrome phenotypes in Drosophila. NatChem Biol 4:256–263.
Chen Y, Pan FC, Brandes N, Afelik S,Solter M, Pieler T. 2004. Retinoic acidsignaling is essential for pancreas devel-opment and promotes endocrine at theexpense of exocrine cell differentiation inXenopus. Dev Biol 271:144–160.
Christensen EI, Raciti D, Reggiani L, Ver-roust PJ, Brandli AW. 2008. Gene ex-pression analysis defines the proximaltubule as the compartment for endocyticreceptor-mediated uptake in the Xeno-pus pronephric kidney. Pflugers Arch456:1163–1176.
Chung HA, Hyodo-Miura J, Kitayama A,Terasaka C, Nagamune T, Ueno N.2004. Screening of FGF target genes inXenopus by microarray: temporal dis-section of the signalling pathway usinga chemical inhibitor. Genes Cells9:749 –761.
Cleaver O, Krieg PA. 1998. VEGF medi-ates angioblast migration during devel-opment of the dorsal aorta in Xenopus.Development 125:3905–3914.
Courchesne CL, Bantle JA. 1985. Analysisof the activity of DNA, RNA, and proteinsynthesis inhibitors on Xenopus embryo
development. Teratog Carcinog Mutagen5:177–193.
Cox CM, D’Agostino SL, Miller MK, He-imark RL, Krieg PA. 2006. Apelin, theligand for the endothelial G-protein-cou-pled receptor, APJ, is a potent angio-genic factor required for normal vasculardevelopment of the frog embryo. Dev Biol296:177–189.
Cueni LN, Detmar M. 2006. New insightsinto the molecular control of the lym-phatic vascular system and its role indisease. J Invest Dermatol 126:2167–2177.
Cuny GD, Yu PB, Laha JK, Xing X, Liu JF,Lai CS, Deng DY, Sachidanandan C,Bloch KD, Peterson RT. 2008. Structure-activity relationship study of bone mor-phogenetic protein (BMP) signaling in-hibitors. Bioorg Med Chem Lett18:4388–4392.
Danielson PB. 2002. The cytochrome P450superfamily: biochemistry, evolution anddrug metabolism in humans. Curr DrugMetab 3:561–597.
Danilova N, Bussmann J, Jekosch K,Steiner LA. 2005. The immunoglobulinheavy-chain locus in zebrafish: identifi-cation and expression of a previously un-known isotype, immunoglobulin Z. NatImmunol 6:295–302.
Davidson EH, Rast JP, Oliveri P, RansickA, Calestani C, Yuh CH, Minokawa T,Amore G, Hinman V, Arenas-Mena C,Otim O, Brown CT, Livi CB, Lee PY,Revilla R, Rust AG, Pan Z, Schilstra MJ,Clarke PJ, Arnone MI, Rowen L, Cam-eron RA, McClay DR, Hood L, Bolouri H.2002. A genomic regulatory network fordevelopment. Science 295:1669–1678.
Delaune E, Lemaire P, Kodjabachian L.2005. Neural induction in Xenopus re-quires early FGF signalling in additionto BMP inhibition. Development132:299–310.
Diamandis P, Wildenhain J, Clarke ID, Sa-cher AG, Graham J, Bellows DS, LingEK, Ward RJ, Jamieson LG, Tyers M,Dirks PB. 2007. Chemical genetics re-veals a complex functional ground stateof neural stem cells. Nat Chem Biol3:268–273.
Doyon Y, McCammon JM, Miller JC,Faraji F, Ngo C, Katibah GE, Amora R,Hocking TD, Zhang L, Rebar EJ, Greg-ory PD, Urnov FD, Amacher SL. 2008.Heritable targeted gene disruption in ze-brafish using designed zinc-finger nucle-ases. Nat Biotechnol 26:702–708.
Du Pasquier L. 2001. The immune systemof invertebrates and vertebrates. CompBiochem Physiol B Biochem Mol Biol129:1–15.
Du Pasquier L, Schwager J, Flajnik MF.1989. The immune system of Xenopus.Annu Rev Immunol 7:251–275.
Dupre A, Boyer-Chatenet L, Sattler RM,Modi AP, Lee JH, Nicolette ML, Kopelov-ich L, Jasin M, Baer R, Paull TT, GautierJ. 2008. A forward chemical geneticscreen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol4:119–125.
Emerson RO, Thomas JH. 2009. Adaptiveevolution in zinc finger transcription fac-tors. PLoS Genet 5:e1000325.
Fort DJ, Dawson DA, Bantle JA. 1988. De-velopment of a metabolic activation sys-tem for the frog embryo teratogenesisassay: Xenopus (FETAX). Teratog Car-cinog Mutagen 8:251–263.
Fort DJ, Stover EL, Bantle JA, Finch RA.2000. Evaluation of the developmentaltoxicity of thalidomide using frog embryoteratogenesis assay-xenopus (FETAX):biotransformation and detoxification.Teratog Carcinog Mutagen 20:35–47.
Fukumoto T, Blakely R, Levin M. 2005a.Serotonin transporter function is anearly step in left-right patterning inchick and frog embryos. Dev Neurosci27:349–363.
Fukumoto T, Kema IP, Levin M. 2005b.Serotonin signaling is a very early stepin patterning of the left-right axis inchick and frog embryos. Curr Biol 15:794–803.
Ghose AK, Viswanadhan VN, WendoloskiJJ. 1999. A knowledge-based approachin designing combinatorial or medicinalchemistry libraries for drug discovery. 1.A qualitative and quantitative charac-terization of known drug databases.J Comb Chem 1:55–68.
Gibbs RA, Weinstock GM, Metzker ML,Muzny DM, Sodergren EJ, Scherer S,Scott G, Steffen D, Worley KC, BurchPE, Okwuonu G, Hines S, Lewis L, De-Ramo C, Delgado O, Dugan-Rocha S,Miner G, Morgan M, Hawes A, Gill R,Celera, Holt RA, Adams MD, Amanati-des PG, Baden-Tillson H, Barnstead M,Chin S, Evans CA, Ferriera S, Fosler C,Glodek A, Gu Z, Jennings D, Kraft CL,Nguyen T, Pfannkoch CM, Sitter C, Sut-ton GG, Venter JC, Woodage T, Smith D,Lee HM, Gustafson E, Cahill P, Kana A,Doucette-Stamm L, Weinstock K, Fech-tel K, Weiss RB, Dunn DM, Green ED,Blakesley RW, Bouffard GG, De Jong PJ,Osoegawa K, Zhu B, Marra M, Schein J,Bosdet I, Fjell C, Jones S, Krzywinski M,Mathewson C, Siddiqui A, Wye N,McPherson J, Zhao S, Fraser CM, ShettyJ, Shatsman S, Geer K, Chen Y, Abram-zon S, Nierman WC, Havlak PH, ChenR, Durbin KJ, Egan A, Ren Y, Song XZ,Li B, Liu Y, Qin X, Cawley S, Cooney AJ,D’Souza LM, Martin K, Wu JQ, Gonza-lez-Garay ML, Jackson AR, Kalafus KJ,McLeod MP, Milosavljevic A, Virk D,Volkov A, Wheeler DA, Zhang Z, BaileyJA, Eichler EE, Tuzun E, Birney E, Mon-gin E, Ureta-Vidal A, Woodwark C,Zdobnov E, Bork P, Suyama M, TorrentsD, Alexandersson M, Trask BJ, YoungJM, Huang H, Wang H, Xing H, DanielsS, Gietzen D, Schmidt J, Stevens K, VittU, Wingrove J, Camara F, Mar Alba M,Abril JF, Guigo R, Smit A, Dubchak I,Rubin EM, Couronne O, Poliakov A,Hubner N, Ganten D, Goesele C, Hum-mel O, Kreitler T, Lee YA, Monti J,Schulz H, Zimdahl H, Himmelbauer H,Lehrach H, Jacob HJ, Bromberg S, Gull-ings-Handley J, Jensen-Seaman MI,Kwitek AE, Lazar J, Pasko D, Tonellato
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1305

PJ, Twigger S, Ponting CP, Duarte JM,Rice S, Goodstadt L, Beatson SA, EmesRD, Winter EE, Webber C, Brandt P,Nyakatura G, Adetobi M, ChiaromonteF, Elnitski L, Eswara P, Hardison RC,Hou M, Kolbe D, Makova K, Miller W,Nekrutenko A, Riemer C, Schwartz S,Taylor J, Yang S, Zhang Y, LindpaintnerK, Andrews TD, Caccamo M, Clamp M,Clarke L, Curwen V, Durbin R, Eyras E,Searle SM, Cooper GM, Batzoglou S,Brudno M, Sidow A, Stone EA, PayseurBA, Bourque G, Lopez-Otin C, PuenteXS, Chakrabarti K, Chatterji S, DeweyC, Pachter L, Bray N, Yap VB, Caspi A,Tesler G, Pevzner PA, Haussler D,Roskin KM, Baertsch R, Clawson H,Furey TS, Hinrichs AS, Karolchik D,Kent WJ, Rosenbloom KR, TrumbowerH, Weirauch M, Cooper DN, Stenson PD,Ma B, Brent M, Arumugam M, Shteyn-berg D, Copley RR, Taylor MS, RiethmanH, Mudunuri U, Peterson J, Guyer M,Felsenfeld A, Old S, Mockrin S, CollinsF. 2004. Genome sequence of the BrownNorway rat yields insights into mamma-lian evolution. Nature 428:493–521.
Goda T, Abu-Daya A, Carruthers S, ClarkMD, Stemple DL, Zimmerman LB. 2006.Genetic screens for mutations affectingdevelopment of Xenopus tropicalis. PLoSGenet 2:e91.
Haggarty SJ, Mayer TU, Miyamoto DT,Fathi R, King RW, Mitchison TJ, Schre-iber SL. 2000. Dissecting cellular pro-cesses using small molecules: identifica-tion of colchicine-like, taxol-like andother small molecules that perturb mito-sis. Chem Biol 7:275–286.
Hathaway NA, King RW. 2005. Dissectingcell biology with chemical scalpels. CurrOpin Cell Biol 17:12–19.
Heasman J, Kofron M, Wylie C. 2000. Be-ta-catenin signaling activity dissected inthe early Xenopus embryo: a novel anti-sense approach. Dev Biol 222:124–134.
Hedges SB. 2002. The origin and evolutionof model organisms. Nat Rev Genet3:838–849.
Hedges SB, Kumar S. 2002. Genomics.Vertebrate genomes compared. Science297:1283–1285.
Helbling PM, Saulnier DM, Brandli AW.2000. The receptor tyrosine kinaseEphB4 and ephrin-B ligands restrict an-giogenic growth of embryonic veins inXenopus laevis. Development 127:269–278.
Hellsten U, Khokha MK, Grammer TC,Harland RM, Richardson P, Rokhsar DS.2007. Accelerated gene evolution andsubfunctionalization in the pseudotet-raploid frog Xenopus laevis. BMC Biol5:31.
Hong CC, Peterson QP, Hong JY, PetersonRT. 2006. Artery/vein specification isgoverned by opposing phosphatidylinosi-tol-3 kinase and MAP kinase/ERK sig-naling. Curr Biol 16:1366–1372.
Jung DW, Williams D, Khersonsky SM,Kang TW, Heidary N, Chang YT, OrlowSJ. 2005. Identification of the F1F0 mi-tochondrial ATPase as a target for mod-ulating skin pigmentation by screening a
tagged triazine library in zebrafish. MolBiosyst 1:85–92.
Kalin RE, Kretz MP, Meyer AM, Kispert A,Heppner FL, Brandli AW. 2007. Para-crine and autocrine mechanisms of ape-lin signaling govern embryonic and tu-mor angiogenesis. Dev Biol 305:599–614.
Kalin RE, Banziger-Tobler NE, Detmar M,Brandli AW. 2009. An in vivo chemicalscreen in Xenopus tadpoles reveals novelpathways involved in angiogenesis andlymphangiogenesis. Blood (in press).
Kelsh RN, Schmid B, Eisen JS. 2000. Ge-netic analysis of melanophore develop-ment in zebrafish embryos. Dev Biol 225:277–293.
Kenwrick S, Amaya E, Papalopulu N.2004. Pilot morpholino screen in Xeno-pus tropicalis identifies a novel gene in-volved in head development. Dev Dyn229:289–299.
Khersonsky SM, Jung DW, Kang TW,Walsh DP, Moon HS, Jo H, JacobsonEM, Shetty V, Neubert TA, Chang YT.2003. Facilitated forward chemical ge-netics using a tagged triazine library andzebrafish embryo screening. J Am ChemSoc 125:11804–11805.
Klein J, Nikolaidis N. 2005. The descent ofthe antibody-based immune system bygradual evolution. Proc Natl Acad SciUSA 102:169–174.
Koebernick K, Hollemann T, Pieler T.2003. A restrictive role for Hedgehog sig-nalling during otic specification in Xeno-pus. Dev Biol 260:325–338.
Landais I, Sobeck A, Stone S, LaChapelleA, Hoatlin ME. 2009. A novel cell-freescreen identifies a potent inhibitor of theFanconi anemia pathway. Int J Cancer124:783–792.
Lepourcelet M, Chen YN, France DS,Wang H, Crews P, Petersen F, Bruseo C,Wood AW, Shivdasani RA. 2004. Small-molecule antagonists of the oncogenicTcf/beta-catenin protein complex. Can-cer Cell 5:91–102.
Levin M, Thorlin T, Robinson KR, Nogi T,Mercola M. 2002. Asymmetries in H�/K�-ATPase and cell membrane poten-tials comprise a very early step in left-right patterning. Cell 111:77–89.
Lieschke GJ, Currie PD. 2007. Animalmodels of human disease: zebrafishswim into view. Nat Rev Genet 8:353–367.
Lipinski CA. 2000. Drug-like propertiesand the causes of poor solubility and poorpermeability. J Pharmacol Toxicol Meth-ods 44:235–249.
Lipinski CA, Lombardo F, Dominy BW,Feeney PJ. 2001. Experimental and com-putational approaches to estimate solu-bility and permeability in drug discoveryand development settings. Adv Drug De-liv Rev 46:3–26.
Liu J, Wu X, Mitchell B, Kintner C, Ding S,Schultz PG. 2005. A small-molecule ago-nist of the Wnt signaling pathway. An-gew Chem Int Ed Engl 44:1987–1990.
Liu XJ. 2006. Xenopus protocols: cell biol-ogy and signal transduction. Springer.512 p.
Longo M, Zanoncelli S, Della Torre P, RosaF, Giusti A, Colombo P, Brughera M,Mazue G, Olliaro P. 2008. Investigationsof the effects of the antimalarial drugdihydroartemisinin (DHA) using theFrog Embryo Teratogenesis Assay-Xeno-pus (FETAX). Reprod Toxicol 25:433–441.
Margolis J, Plowman GD. 2004. Overcom-ing the gridlock in discovery research.Nat Biotechnol 22:522–524.
Maroon H, Walshe J, Mahmood R, KieferP, Dickson C, Mason I. 2002. Fgf3 andFgf8 are required together for formationof the otic placode and vesicle. Develop-ment 129:2099–2108.
Mathew LK, Sengupta S, Kawakami A,Andreasen EA, Lohr CV, Loynes CA,Renshaw SA, Peterson RT, Tanguay RL.2007. Unraveling tissue regenerationpathways using chemical genetics. J BiolChem 282:35202–35210.
Meijer L, Skaltsounis AL, Magiatis P, Poly-chronopoulos P, Knockaert M, Leost M,Ryan XP, Vonica CA, Brivanlou A, Da-jani R, Crovace C, Tarricone C, Musac-chio A, Roe SM, Pearl L, Greengard P.2003. GSK-3-selective inhibitors derivedfrom Tyrian purple indirubins. ChemBiol 10:1255–1266.
Meng X, Noyes MB, Zhu LJ, Lawson ND,Wolfe SA. 2008. Targeted gene inactiva-tion in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26:695–701.
Metzstein MM, Stanfield GM, Horvitz HR.1998. Genetics of programmed cell deathin C. elegans: past, present and future.Trends Genet 14:410–416.
Milan DJ, Peterson TA, Ruskin JN, Peter-son RT, MacRae CA. 2003. Drugs thatinduce repolarization abnormalitiescause bradycardia in zebrafish. Circula-tion 107:1355–1358.
Min J, Kyung Kim Y, Cipriani PG, KangM, Khersonsky SM, Walsh DP, Lee JY,Niessen S, Yates JR, 3rd, Gunsalus K,Piano F, Chang YT. 2007. Forwardchemical genetic approach identifies newrole for GAPDH in insulin signaling. NatChem Biol 3:55–59.
Mittelstadt SW, Hemenway CL, Craig MP,Hove JR. 2008. Evaluation of zebrafishembryos as a model for assessing inhibi-tion of hERG. J Pharmacol Toxicol Meth-ods 57:100–105.
Moens CB, Donn TM, Wolf-Saxon ER, MaTP. 2008. Reverse genetics in zebrafishby TILLING. Brief Funct Genomic Pro-teomic 7:454–459.
Mohun TJ, Leong LM, Weninger WJ, Spar-row DB. 2000. The morphology of heartdevelopment in Xenopus laevis. Dev Biol218:74–88.
Moon HS, Jacobson EM, Khersonsky SM,Luzung MR, Walsh DP, Xiong W, LeeJW, Parikh PB, Lam JC, Kang TW, Ro-sania GR, Schier AF, Chang YT. 2002. Anovel microtubule destabilizing entityfrom orthogonal synthesis of triazine li-brary and zebrafish embryo screening.J Am Chem Soc 124:11608–11609.
1306 WHEELER AND BRANDLI

Murphey RD, Zon LI. 2006. Small moleculescreening in the zebrafish. Methods 39:255–261.
Murphey RD, Stern HM, Straub CT, ZonLI. 2006. A chemical genetic screen forcell cycle inhibitors in zebrafish embryos.Chem Biol Drug Des 68:213–219.
Ni-Komatsu L, Orlow SJ. 2007. Identifica-tion of novel pigmentation modulators bychemical genetic screening. J Invest Der-matol 127:1585–1592.
North TE, Goessling W, Walkley CR, Leng-erke C, Kopani KR, Lord AM, Weber GJ,Bowman TV, Jang IH, Grosser T,Fitzgerald GA, Daley GQ, Orkin SH, ZonLI. 2007. Prostaglandin E2 regulatesvertebrate haematopoietic stem cell ho-meostasis. Nature 447:1007–1011.
Nusslein-Volhard C, Wieschaus E. 1980.Mutations affecting segment numberand polarity in Drosophila. Nature 287:795–801.
Ny A, Koch M, Schneider M, Neven E,Tong RT, Maity S, Fischer C, PlaisanceS, Lambrechts D, Heligon C, TerclaversS, Ciesiolka M, Kalin R, Man WY, SennI, Wyns S, Lupu F, Brandli A, VleminckxK, Collen D, Dewerchin M, Conway EM,Moons L, Jain RK, Carmeliet P. 2005. Agenetic Xenopus laevis tadpole model tostudy lymphangiogenesis. Nat Med 11:998–1004.
Olsson AK, Dimberg A, Kreuger J, Claes-son-Welsh L. 2006. VEGF receptor sig-nalling: in control of vascular function.Nat Rev Mol Cell Biol 7:359–371.
Page-McCaw A, Ewald AJ, Werb Z. 2007.Matrix metalloproteinases and the regu-lation of tissue remodelling. Nat Rev MolCell Biol 8:221–233.
Peterson JR, Lebensohn AM, Pelish HE,Kirschner MW. 2006. Biochemical sup-pression of small-molecule inhibitors: astrategy to identify inhibitor targets andsignaling pathway components. ChemBiol 13:443–452.
Peterson RT, Fishman MC. 2004. Discov-ery and use of small molecules for prob-ing biological processes in zebrafish.Methods Cell Biol 76:569–591.
Peterson RT, Link BA, Dowling JE, Schre-iber SL. 2000. Small molecule develop-mental screens reveal the logic and tim-ing of vertebrate development. Proc NatlAcad Sci USA 97:12965–12969.
Peterson RT, Mably JD, Chen JN, Fish-man MC. 2001. Convergence of distinctpathways to heart patterning revealedby the small molecule concentramideand the mutation heart-and-soul. CurrBiol 11:1481–1491.
Peterson RT, Shaw SY, Peterson TA, MilanDJ, Zhong TP, Schreiber SL, MacRaeCA, Fishman MC. 2004. Chemical sup-pression of a genetic mutation in a ze-brafish model of aortic coarctation. NatBiotechnol 22:595–599.
Postlethwait JH. 2007. The zebrafish ge-nome in context: ohnologs gone miss-ing. J Exp Zool B Mol Dev Evol 308:563–577.
Postlethwait JH, Yan YL, Gates MA,Horne S, Amores A, Brownlie A, Dono-van A, Egan ES, Force A, Gong Z, Goutel
C, Fritz A, Kelsh R, Knapik E, Liao E,Paw B, Ransom D, Singer A, ThomsonM, Abduljabbar TS, Yelick P, Beier D,Joly JS, Larhammar D, Rosa F, Wester-field M, Zon LI, Johnson SL, Talbot WS.1998. Vertebrate genome evolution andthe zebrafish gene map. Nat Genet 18:345–349.
Raciti D, Reggiani L, Geffers L, Jiang Q,Bacchion F, Subrizi AE, Clements D,Tindal C, Davidson DR, Kaissling B,Brandli AW. 2008. Organization of thepronephric kidney revealed by large-scale gene expression mapping. GenomeBiol 9:R84.
Raible F, Brand M. 2001. Tight transcrip-tional control of the ETS domain factorsErm and Pea3 by Fgf signaling duringearly zebrafish development. Mech Dev107:105–117.
Rana AA, Collart C, Gilchrist MJ, SmithJC. 2006. Defining synphenotype groupsin Xenopus tropicalis by use of antisensemorpholino oligonucleotides. PLoSGenet 2:e193.
Reya T, Clevers H. 2005. Wnt signalling instem cells and cancer. Nature 434:843–850.
Richards SM, Cole SE. 2006. A toxicity andhazard assessment of fourteen pharma-ceuticals to Xenopus laevis larvae. Eco-toxicology 15:647–656.
Rickardson L, Fryknas M, Haglund C,Lovborg H, Nygren P, Gustafsson MG,Isaksson A, Larsson R. 2006. Screeningof an annotated compound library fordrug activity in a resistant myeloma cellline. Cancer Chemother Pharmacol 58:749–758.
Root DE, Flaherty SP, Kelley BP, Stock-well BR. 2003. Biological mechanismprofiling using an annotated compoundlibrary. Chem Biol 10:881–892.
Rubinstein AL. 2003. Zebrafish: from dis-ease modeling to drug discovery. CurrOpin Drug Discov Dev 6:218–223.
Sachidanandan C, Yeh JR, Peterson QP,Peterson RT. 2008. Identification of anovel retinoid by small molecule screen-ing with zebrafish embryos. PLoS ONE3:e1947.
Schreiber SL. 2000. Target-oriented anddiversity-oriented organic synthesis indrug discovery. Science 287:1964 –1969.
Segalat L. 2007. Invertebrate animal mod-els of diseases as screening tools in drugdiscovery. ACS Chem Biol 2:231–236.
Serbedzija GN, Flynn E, Willett CE.1999. Zebrafish angiogenesis: a newmodel for drug screening. Angiogenesis3:353–359.
Shafizadeh E, Peterson RT, Lin S. 2004.Induction of reversible hemolytic anemiain living zebrafish using a novel smallmolecule. Comp Biochem Physiol C Toxi-col Pharmacol 138:245–249.
Shan J, Shi DL, Wang J, Zheng J. 2005.Identification of a specific inhibitor of thedishevelled PDZ domain. Biochemistry44:15495–15503.
Shin JT, Fishman MC. 2002. From Ze-brafish to human: modular medical mod-
els. Annu Rev Genomics Hum Genet3:311–340.
Shinya M, Koshida S, Sawada A, KuroiwaA, Takeda H. 2001. Fgf signallingthrough MAPK cascade is required fordevelopment of the subpallial telenceph-alon in zebrafish embryos. Development128:4153–4164.
Spring DR, Krishnan S, Blackwell HE,Schreiber SL. 2002. Diversity-orientedsynthesis of biaryl-containing mediumrings using a one bead/one stock solu-tion platform. J Am Chem Soc 124:1354–1363.
Stainier DY. 2001. Zebrafish genetics andvertebrate heart formation. Nat RevGenet 2:39–48.
Stemple DL. 2004. TILLING: a high-throughput harvest for functionalgenomics. Nat Rev Genet 5:145–150.
Stern HM, Murphey RD, Shepard JL,Amatruda JF, Straub CT, Pfaff KL,Weber G, Tallarico JA, King RW, ZonLI. 2005. Small molecules that delay Sphase suppress a zebrafish bmyb mu-tant. Nat Chem Biol 1:366 –370.
Sternson SM, Louca JB, Wong JC, Schre-iber SL. 2001. Split-pool synthesis of1,3-dioxanes leading to arrayed stocksolutions of single compounds suffi-cient for multiple phenotypic and pro-tein-binding assays. J Am Chem Soc123:1740 –1747.
Stockwell BR. 2000. Chemical genetics: li-gand-based discovery of gene function.Nat Rev Genet 1:116–125.
Stockwell BR. 2004. Exploring biology withsmall organic molecules. Nature432:846–854.
Sturm RA. 2009. Molecular genetics of hu-man pigmentation diversity. Hum MolGenet 18:R9–17.
Thomas I, Kihiczak GG, Fox MD, Janni-ger CK, Schwartz RA. 2004. Piebald-ism: an update. Int J Dermatol 43:716 –719.
Tomlinson M, Field RA, Wheeler GN. 2005.Xenopus as a model organism in devel-opmental chemical genetic screens. Mo-lecular BioSystems 1:223–228.
Tomlinson ML, Guan P, Morris RJ, Fi-dock MD, Rejzek M, Garcia-Morales C,Field RA, Wheeler GN. 2009a. A chem-ical genomic approach identifies matrixmetalloproteinases as playing an es-sential and specific role in Xenopusmelanophore migration. Chem Biol 16:93–104.
Tomlinson ML, Rejzek M, Fidock M, FieldRA, Wheeler GN. 2009b. Chemicalgenomics identifies compounds affectingXenopus laevis pigment cell developmentMolecular BioSystems 5:376–384.
Towbin JA, McQuinn TC. 1995. Gridlock: amodel for coarctation of the aorta? NatMed 1:1141–1142.
Tran TC, Sneed B, Haider J, Blavo D,White A, Aiyejorun T, Baranowski TC,Rubinstein AL, Doan TN, Dingledine R,Sandberg EM. 2007. Automated, quanti-tative screening assay for antiangiogeniccompounds using transgenic zebrafish.Cancer Res 67:11386–11392.
CHEMICAL SCREENS IN ZEBRAFISH AND XENOPUS 1307

Traver D, Herbomel P, Patton EE, Mur-phey RD, Yoder JA, Litman GW, Catic A,Amemiya CT, Zon LI, Trede NS. 2003.The zebrafish as a model organism tostudy development of the immune sys-tem. Adv Immunol 81:253–330.
Ureta-Vidal A, Ettwiller L, Birney E. 2003.Comparative genomics: genome-wideanalysis in metazoan eukaryotes. NatRev Genet 4:251–262.
van Kempen LC, Coussens LM. 2002.MMP9 potentiates pulmonary metasta-sis formation. Cancer Cell 2:251–252.
Van Raay TJ, Vetter ML. 2004. Wnt/friz-zled signaling during vertebrate reti-nal development. Dev Neurosci 26:352–358.
Verma R, Peters NR, D’Onofrio M,Tochtrop GP, Sakamoto KM, Varadan R,Zhang M, Coffino P, Fushman D, De-shaies RJ, King RW. 2004. Ubistatinsinhibit proteasome-dependent degrada-tion by binding the ubiquitin chain. Sci-ence 306:117–120.
Vismara C, Caloni F. 2007. Evaluation ofaflatoxin B1 embryotoxicity using thefrog embryo teratogenesis assay-Xeno-pus and bio-activation with microsomeactivation systems. Birth Defects Res BDev Reprod Toxicol 80:183–187.
Warkman AS, Krieg PA. 2007. Xenopus asa model system for vertebrate heart de-
velopment. Semin Cell Dev Biol 18:46–53.
White RM, Zon LI. 2008. Melanocytes indevelopment, regeneration, and cancer.Cell Stem Cell 3:242–252.
White RM, Sessa A, Burke C, Bowman T,LeBlanc J, Ceol C, Bourque C, Dovey M,Goessling W, Burns CE, Zon LI. 2008.Transparent adult zebrafish as a tool forin vivo transplantation analysis. CellStem Cell 2:183–189.
Wienholds E, van Eeden F, Kosters M,Mudde J, Plasterk RH, Cuppen E.2003. Efficient target-selected mu-tagenesis in zebrafish. Genome Res 13:2700 –2707.
Wignall SM, Gray NS, Chang YT, JuarezL, Jacob R, Burlingame A, Schultz PG,Heald R. 2004. Identification of a novelprotein regulating microtubule stabilitythrough a chemical approach. Chem Biol11:135–146.
Wong JC, Sternson SM, Louca JB, Hong R,Schreiber SL. 2004. Modular synthesisand preliminary biological evaluation ofstereochemically diverse 1,3-dioxanes.Chem Biol 11:1279–1291.
Yu PB, Deng DY, Lai CS, Hong CC, CunyGD, Bouxsein ML, Hong DW, Mc-ManusPM, Katagiri T, SachidanandanC, Kamiya N, Fukuda T, Mishina Y,Peterson RT, Bloch KD. 2008a. BMP
type I receptor inhibition reduces het-erotopic ossification. Nat Med 14:1363–1369.
Yu PB, Hong CC, Sachidanandan C, BabittJL, Deng DY, Hoyng SA, Lin HY, BlochKD, Peterson RT. 2008b. Dorsomorphininhibits BMP signals required for embry-ogenesis and iron metabolism. Nat ChemBiol 4:33–41.
Zhang Q, Major MB, Takanashi S, CampND, Nishiya N, Peters EC, GinsbergMH, Jian X, Randazzo PA, Schultz PG,Moon RT, Ding S. 2007. Small-moleculesynergist of the Wnt/beta-catenin signal-ing pathway. Proc Natl Acad Sci USA104:7444–7448.
Zhong TP, Rosenberg M, Mohideen MA,Weinstein B, Fishman MC. 2000. grid-lock, an HLH gene required for assem-bly of the aorta in zebrafish. Science287:1820 –1824.
Zhou G, Myers R, Li Y, Chen Y, Shen X,Fenyk-Melody J, Wu M, Ventre J, Doeb-ber T, Fujii N, Musi N, Hirshman MF,Goodyear LJ, Moller DE. 2001. Role ofAMP-activated protein kinase in mecha-nism of metformin action. J Clin Invest108:1167–1174.
Zon LI, Peterson RT. 2005. In vivo drugdiscovery in the zebrafish. Nat Rev DrugDiscov 4:35–44.
1308 WHEELER AND BRANDLI




























