Embed Size (px)
Citation preview

Short-Lived, Intense and Narrow Bluish-Green Emitting Gold ZincSulfide Semiconducting NanocrystalsRiya Bose,†,‡ Umamahesh Thupakula,‡ J. K. Bal,‡ and Narayan Pradhan*,†,‡
†Department of Materials Science and ‡Centre for Advanced Materials, Indian Association for the Cultivation of Science, Jadavpur,Kolkata 700032, India
*S Supporting Information
ABSTRACT: In nanoscale, gold is one of the widely studiedmetals. It is well-known for its size dependent surface plasmonicabsorbance. It has also been reported that clusters of a few atomsof gold can show fluorescence. However, these optical propertiesof gold are mostly associated with Au(0), and little has beenexplored for the compounds of gold in nanoscale. Herein, wereport a new semiconducting nanocrystalline material involvingAu(I), which shows intense, narrow, and stable emission at itsbandedge absorption. These are composed of Au, Zn, and S andsynthesized by introducing Au to zinc sulfide or Zn to gold(I)sulfide nanocrystals in their aqueous dispersion and underambient condition. The obtained emission is short-lived and tunable in a short spectral window. These new semiconductingfluorescent gold based nanomaterials are characterized with UV−visible, photoluminescence spectroscopy, TCSPC, HRTEM,STM, and STS experiments. Further, the electrical and optical sensing properties of these nanocrystals have also been measured.
■ INTRODUCTION
Gold is one of the most important and widely studied materialsin nanoscale. It is widely known for its size-dependentplasmonic absorption properties.1−4 The chemical synthesis,dimension-dependent optical properties and possible imple-mentations comprise the multidisciplinary fields that have beenenormously studied since Faraday’s report of Au colloid.4−11
Typically, when the size of a gold particle reduces to ∼20 nm,the conduction electrons form size-dependent plasmonbands.5−8 But, on further reduction of the size of thesematerials (<3 nm) close to de-Broglie wavelength of theconduction electrons, quasi-continuous electronic bands appearthat make them behave molecule-like, with properties such assize-dependent fluorescence characteristics.12−16 Depending onthe size of the Au core and nature of the stabilizing agent, thesegold nanoparticles can emit in different energies from UV toNIR spectral window.14,16−21 These photophysical properties ofthe tiny gold clusters not only add to the study of thefundamental aspects of nanocrystals/clusters but also makethem suitable for use as biocompatible biolabels and light-emitting sources in nanoscale and, hence, can be applied inimaging, detection, and so on, in conjunction with differentbiomolecules.22−24
But, with progress of science, new optical materials with sizetunable absorption and emission energy variation propertieshave also been developed.25−29 Among these, quantumconfined binary, multinary and their different alloyed semi-conductor nanocrystals are the leading and most studiednanoscale materials.28,30−35 These remained as the work horsein the dispersed colloidal nanomaterials with bandgap depend-
ent wide window absorption and efficient, stable, and colortunable emission. Several reports on fabrication of lightemitting,27,36,37 photovoltaic,38,39 and light sensing40,41 devicesinvolving these materials are already reported and search fornew materials, fabricating more effective devices, and enhance-ment of the device efficiencies is continuously going on to meetthe current demands in the community. In comparison, theemission from noble metal gold remains less focused due to lessintensity and confusion regarding the origin of the emission. Tothe best of our knowledge, there is no report of anysemiconductor nanocrystal composed of gold with other ionsso far which can provide the band edge tunable emission or hasthe visible light absorption, whereas larger size gold particles(>3 nm) have been widely reported as fluorescence quencherand can quench the semiconductor emissions.42
Searching for new nanomaterials for multipurpose applica-tions and with the urge to know more about gold, we reporthere a new multinary gold zinc sulfide semiconductingnanocrystalline material, which can absorb visible light andemit tunable bandedge emission in visible window.These nanocrystals are synthesized simply by introducing
Zn(II) solution to an aqueous mixture of Au(III) and thiol(mercaptopropionic acid, MPA). Alternatively, these are alsodesigned by introducing Au(III) to ZnS nanocrystals in theaqueous dispersion. The obtained emission is intense, stable,narrow, and short-range tunable (blue-green window), and the
Received: May 22, 2012Revised: July 7, 2012Published: July 10, 2012
Article
pubs.acs.org/JPCC
© 2012 American Chemical Society 16680 dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−16686

particle size remains within 2−2.5 nm (diameter). Suchultrasmall nanocrystals, which are synthesized at roomtemperature and in presence of air, have an emission intensity(Quantum Yield, 22%) comparable to several leading semi-conductor nanocrystals reported so far. Details of the synthesis,sequential optical absorption and emission spectra, analysis ofthe products, definition of the emission origin, and so on areprovided in this communication. Further, the electrical andphotoresponse properties of these nanocrystals have beenmeasured and reported here, which support their semi-conducting behavior.
■ RESULTS AND DISCUSSIONAlkyl thiols that have high affinity to gold produce lightemitting nanoclusters/nanocrystals when treated with Au(III)solution.16,43−46 This has been reported since decades withseveral speculation of the origin of this emission. Proceedingone step further, we introduced aqueous solution of Zn(II) tothis Au(III)−thiol mixture that quenched the existing emissionand generated a new but spectacularly intense emission withcompletely different characteristics. Details of the synthesishave been provided in the Experimental section. In a typicalexperiment, aqueous solution of mercaptopropionic acid(MPA) with desired concentration was taken in a test tubealong with required amount of PVA solution that acted as astabilizing agent. Stock solution of NaAuCl4 was addeddropwise to the same test tube under vigorous stirringcondition at room temperature. Within seconds the solutionshowed a faint yellow emission centered at 570 nm (excitationat ∼320 nm) whose intensity enhanced to some extent(quantum yield <1%) with more NaAuCl4 addition. Furtheraddition of Zn(OAc)2 stock solution (see ExperimentalSection) to the same test tube quenched this yellow emissionand instead a new narrow and intense emission in bluish-greenwindow appeared and tuned with the progress of the reaction.A schematic presentation of the reaction has been shown inFigure 1.
Figure 2 shows the successive evolution of UV−visible andphotoluminescence (PL) spectra in a typical reaction. Theabsorption spectra during the reaction of NaAuCl4 with MPAdid not show the band-edge shifting (yellow line) with time andalso its yellow emission did not tune. But, on addition of zinc(stock solution), its absorption band-edge red-shifted con-tinuously with time (blue line), whereas the yellow emissionquenched with appearance of the narrow emission in bluish-green window. The quantum yield of the yellow emissionremained <1% and that of the bluish-green emission wasmeasured to be ∼22%. Photoluminescence excitation (PLE)spectra of both solutions have been provided in SupportingInformation (SI) (see Figure S1).
To understand the origin of both emissions, several opticaland microscopic studies have been carried out for the samplescollected before and after zinc addition. The yellow emission(excitation up to 350 nm) obtained from the mixture of Au(III)and MPA, which is broad (fwhm = ∼60 nm) and less intense,shows large Stokes shift (∼150 nm), and has excited statelifetime in μs (Figure 3a). Emission from the mixture of Au(III)and thiol with these characteristics has already been reported inthe literature.16,44,47 But the new emission that appears at ∼470nm (excitation up to ∼450 nm) is narrow (fwhm, ∼25 nm),intense, appears at the band-edge, and has excited state lifetimein nanoseconds (Figure 3b).Comparing TEM images of both samples, it is observed that
the average particle size increases up to ∼0.3−0.5 nm afterZn(II) addition from the initial ∼1.9 ± 0.2 nm particles inAu(III) and MPA mixture only (Figure 3c,d; also seehistograms and more TEM images in SI, Figure S2). Elementalanalysis from EDAX and ICP suggests the presence of Au and Sfor the yellow emitting and Zn, Au, and S for the final bluish-green emitting sample.Successive XRD of samples during the reaction has been
presented in Figure 3e. The blue line plot obtained from thesample before zinc addition provides direct evidence of thepresence of Au(I) sulfide and its peaks match with cubic Au2S(ICSD #78718). Absence of any peak of fcc Au(0) in spite ofseveral careful experiments suggests absence of gold cluster inthe sample. Hence, we can conclude here that the particlesfrom this yellow emitting sample as observed in the TEMimages are Au(I) sulfide nanocrystals and the emission isexpected from the surface bound Au(I)−thiolate complex, assuggested in the literature.47
On the other hand, the sharp emission obtained after Znaddition which tunes along with the tuning of the absorptionband and has lifetime in nanoseconds resembles the excitonicemission of quantum confined semiconductor nanocrystals.48
To further confirm the semiconductor origin, we have carriedout the STM and STS measurements. Figure 4a shows theSTM image of a single particle (more images in SI, Figure S3)from the sample after Zn addition, and its corresponding STSmeasurement (Figure 4b) shows the bandgap of 2.6 eV, whichmatches with the absorption band edge of the same and isdifferent from ZnS and Au2S of similar dimensions. STSmeasurements on several other particles also provide the sameresult. As this new material is obtained after addition of Zn salt
Figure 1. Schematic presentation of the synthesis of bluish-greenemitting gold zinc sulfide nanocrystals from yellow emitting Au(III)−MPA mixture.
Figure 2. Successive evolution of UV−visible and PL spectra duringthe formation of bluish-green emitting gold zinc sulfide nanocrystals.Excitation wavelength is 320 nm.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616681

to Au2S, it may seem that Zn(II) is forming an alloy with Au2S.To check this, successive XRD evolution process has beenanalyzed. XRD obtained from the final sample (Figure 3e, redline spectra) suggests formation of new nanostructure and thepeak positions are different from Au2S as well as ZnS (FiguresS4 and S5, respectively) whereas the XRD of the intermediatesample shows the presence of mixture of peaks correspondingto both Au2S and the new structure. Emergence of new peaksrather than shifting of existing Au2S peaks rules out the alloyformation. From XPS analysis of the sample (Figure 4c), thebinding energy values of 1022.4 and 1045.5 eV for Zn, 84.5 and88.2 eV for Au, and 161.7 eV for S suggest that Zn is present in+2, Au in +1, and S in −2 oxidation state. Hence, we designatethese nanocrystals as (AuxZnySx/2+y). An average of EDAX (10sets of measurement) and ICP analysis from samples atdifferent stages after Zn(II) addition shows that Zn remains40−60% of the total cations with an average of ∼50%,
suggesting x = y = ∼1. Samples having emission at differentpositions do not show any significant difference in composition,suggesting the tunability is due to change of size rather than thecomposition, which is typical for ternary nanocrystals.Unavailability of any reference JCPDS data made it difficult
for us to determine the exact crystal structure of the finalsample. But from the ratio of d-values corresponding to theXRD peak positions in final pattern (Figure 3e), it appears tobe a cubic structure with most probable lattice parameter a =0.424 nm. Details of the structural analysis have been providedin Supporting Information (Figure S6 and subsequentdiscussion).Apart from mixing of the Zn(II) solution to the aqueous
solution of Au(III) and MPA, these nanocrystals can also besynthesized by adding solution of Au(III) to aqueous dispersionof MPA-capped ZnS nanocrystals. For this, ZnS nanocrystalswere synthesized first following literature reported method andthey were dispersed in water following surface ligand (MPA)exchange. To this solution, required amount of Au(III) stocksolution was introduced under vigorous stirring condition.Soon after the addition, the yellow emission was obtained,which then quenched with the appearance of the bluish-greenemission. Moreover, here the progress of the reaction is notedto be much faster (5 min) to obtain the bluish-green emission.Figure 5a shows the schematic synthetic approach and Figure5b represents the time-dependent photoluminescence spectraof the solution - fwhm, tunability, position, and lifetime ofwhich are the same with the emission spectra presented inFigure 2. TEM of the final sample also shows similar-sizedparticles with prominent d-spacing of 0.21 nm, which matcheswith the d-spacing obtained from XRD. Intermediate samplewith yellow emission shows coupled dots with ZnS whichsuggests that Au ions approach to ZnS surface to nucleate thesenew ternary nanocrystals. But, at the end, we have observedthat all the ZnS nanocrystals are consumed and only the 2−2.5nm ternary nanocrystals remain as the ultimate product.As discussed earlier, sequential XRD pattern of the samples
during the reaction does not support alloy formation. Instead,
Figure 3. (a, b) Excited state lifetime decay of yellow and bluish-greenemitting nanocrystals. Y axes of both plots are in log scale. Insets arethe PL spectra of both nanocrystals. (c, d) TEM images of the yellowand bluish-green emitting samples obtained before and after zincaddition, respectively. Inset of (d) shows the HRTEM of one singlenanocrystal obtained after zinc addition. (e) XRD pattern of goldsulfide (blue line) and the final sample (red line) after excess zincaddition. XRD of the intermediate sample (black line) shows thepresence of peaks corresponding to both gold sulfide (blue line) andfinal product gold zinc sulfide (red line). Blue and red arrows aremarked for disappearance and appearance of respective peaks duringthe progress of reaction.
Figure 4. (a) STM image of a single gold zinc sulfide particle. (b)Current vs voltage plot measured on the particle shown in (a), and dI/dV vs V plot, obtained by numerical differentiation of the I−V curve,arrows indicate the energy separation between valence band andconduction band. (c) XPS of the final sample showing the presence ofZn(II), Au(I), and S(VI).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616682
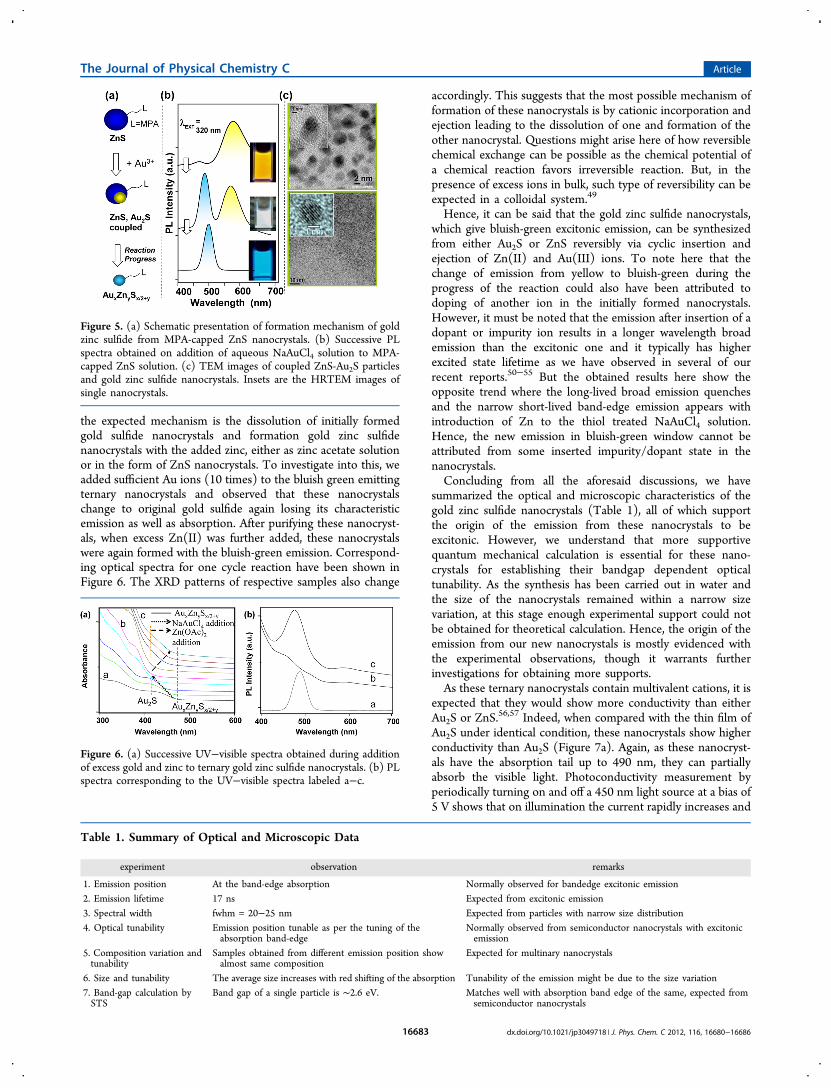
the expected mechanism is the dissolution of initially formedgold sulfide nanocrystals and formation gold zinc sulfidenanocrystals with the added zinc, either as zinc acetate solutionor in the form of ZnS nanocrystals. To investigate into this, weadded sufficient Au ions (10 times) to the bluish green emittingternary nanocrystals and observed that these nanocrystalschange to original gold sulfide again losing its characteristicemission as well as absorption. After purifying these nanocryst-als, when excess Zn(II) was further added, these nanocrystalswere again formed with the bluish-green emission. Correspond-ing optical spectra for one cycle reaction have been shown inFigure 6. The XRD patterns of respective samples also change
accordingly. This suggests that the most possible mechanism offormation of these nanocrystals is by cationic incorporation andejection leading to the dissolution of one and formation of theother nanocrystal. Questions might arise here of how reversiblechemical exchange can be possible as the chemical potential ofa chemical reaction favors irreversible reaction. But, in thepresence of excess ions in bulk, such type of reversibility can beexpected in a colloidal system.49
Hence, it can be said that the gold zinc sulfide nanocrystals,which give bluish-green excitonic emission, can be synthesizedfrom either Au2S or ZnS reversibly via cyclic insertion andejection of Zn(II) and Au(III) ions. To note here that thechange of emission from yellow to bluish-green during theprogress of the reaction could also have been attributed todoping of another ion in the initially formed nanocrystals.However, it must be noted that the emission after insertion of adopant or impurity ion results in a longer wavelength broademission than the excitonic one and it typically has higherexcited state lifetime as we have observed in several of ourrecent reports.50−55 But the obtained results here show theopposite trend where the long-lived broad emission quenchesand the narrow short-lived band-edge emission appears withintroduction of Zn to the thiol treated NaAuCl4 solution.Hence, the new emission in bluish-green window cannot beattributed from some inserted impurity/dopant state in thenanocrystals.Concluding from all the aforesaid discussions, we have
summarized the optical and microscopic characteristics of thegold zinc sulfide nanocrystals (Table 1), all of which supportthe origin of the emission from these nanocrystals to beexcitonic. However, we understand that more supportivequantum mechanical calculation is essential for these nano-crystals for establishing their bandgap dependent opticaltunability. As the synthesis has been carried out in water andthe size of the nanocrystals remained within a narrow sizevariation, at this stage enough experimental support could notbe obtained for theoretical calculation. Hence, the origin of theemission from our new nanocrystals is mostly evidenced withthe experimental observations, though it warrants furtherinvestigations for obtaining more supports.As these ternary nanocrystals contain multivalent cations, it is
expected that they would show more conductivity than eitherAu2S or ZnS.56,57 Indeed, when compared with the thin film ofAu2S under identical condition, these nanocrystals show higherconductivity than Au2S (Figure 7a). Again, as these nanocryst-als have the absorption tail up to 490 nm, they can partiallyabsorb the visible light. Photoconductivity measurement byperiodically turning on and off a 450 nm light source at a bias of5 V shows that on illumination the current rapidly increases and
Figure 5. (a) Schematic presentation of formation mechanism of goldzinc sulfide from MPA-capped ZnS nanocrystals. (b) Successive PLspectra obtained on addition of aqueous NaAuCl4 solution to MPA-capped ZnS solution. (c) TEM images of coupled ZnS-Au2S particlesand gold zinc sulfide nanocrystals. Insets are the HRTEM images ofsingle nanocrystals.
Figure 6. (a) Successive UV−visible spectra obtained during additionof excess gold and zinc to ternary gold zinc sulfide nanocrystals. (b) PLspectra corresponding to the UV−visible spectra labeled a−c.
Table 1. Summary of Optical and Microscopic Data
experiment observation remarks
1. Emission position At the band-edge absorption Normally observed for bandedge excitonic emission2. Emission lifetime 17 ns Expected from excitonic emission3. Spectral width fwhm = 20−25 nm Expected from particles with narrow size distribution4. Optical tunability Emission position tunable as per the tuning of the
absorption band-edgeNormally observed from semiconductor nanocrystals with excitonicemission
5. Composition variation andtunability
Samples obtained from different emission position showalmost same composition
Expected for multinary nanocrystals
6. Size and tunability The average size increases with red shifting of the absorption Tunability of the emission might be due to the size variation7. Band-gap calculation bySTS
Band gap of a single particle is ∼2.6 eV. Matches well with absorption band edge of the same, expected fromsemiconductor nanocrystals
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616683

then sharply returns to the initial value when light is turned off(Figure 7b). The rise and decay times are below our detectionlimit which is 1 ms. The photodetector also shows highly stableand reproducible characteristics. This suggests that thesenanocrystals can be used as efficient light sensor. This alsosupports that the origin of absorption is from the new ternarynanocrystals rather than ZnS or Au2S (2−2.5 nm), whichcannot be excited with 450 nm light source.
■ CONCLUSIONIn summary, we report here intense (QY = ∼22%) bluish-greenemitting ultrasmall AuxZnySx/2+y semiconductor nanocrystals.These nanocrystals are synthesized using all greener chemicalsand just by mixing their aqueous solutions, either zinc to goldsulfide or gold to zinc sulfide, in a test tube at roomtemperature and at ambient condition. Though there areseveral reports on aqueous synthesis of various semiconductornanocrystals;58,59 this is the first of its kind where gold has beeninvolved and shows intense emission at the band-edge. Thecharacterization of these new nanocrystals are carried out withthe support of UV−visible, PL, XPS, TCSPC, XRD, TEM, andSTM/STS, all of which lead to the conclusion that this is a newfamily of ternary semiconductor nanocrystals and shows band-gap dependent emission tunability. We believe that this newmaterial with remarkable emission intensity obtained in theroom temperature aqueous synthesis would be a milestone inthe family of semiconductor nanocrystals not only for providinghighly emittive environment friendly nanomaterials, but also tobring into focus some unknown aspects of gold. Finally, theelectrical (I−V) and photoresponse properties of thesenanocrystals are studied, which suggest that these ternarynanocrystals can have multiple functional applications for bothgenerating as well as harvesting visible light.
■ EXPERIMENTAL SECTIONMaterials. Zinc acetate (Zn(CH3COO)2·2H2O, ≥98%),
zinc stearate (ZnSt2, tech), octadecylamine (ODA, 97%),octadecene (ODE, tech), stearic acid (SA, 95%), S powder(99.9%), mercaptopropionic acid (MPA), tetramethylammo-nium hydroxide (TMAH, 25% in methanol), sodiumtetrachloroaurate (NaAuCl4·2H2O, 99%), and poly(vinylalcohol) (PVA; avg mol wt 130000, 99+% hydrolyzed) werepurchased from Aldrich. All the chemicals were used withoutfurther purification.Preparation of Stock Solutions. Preparation of Zn-
(CH3COO)2 Solution. Stock solution of zinc acetate wasprepared by dissolving 0.022 g of solid Zn(CH3COO)2 in 2mL of water.
Preparation of NaAuCl4 Solution. Stock solution wasprepared by dissolving 0.012 g of solid NaAuCl4 in 5 mL ofwater.
Preparation of PVA Solution. 0.5 g PVA was dissolved in 50mL of water by heating it at 80 °C for ∼2 h. Then the mixturewas kept at room temperature for 1 day to get a clear solution.
Preparation of S Solution. Sulfur stock was prepared bydissolving 0.16 g S powder in 10 mL of ODE with gentleheating under argon.
Synthesis of Bluish-Green Emitting AuxZnySx/2+y Nano-crystals. 1 mL aliquot of 0.5 M aqueous solution of MPA alongwith 1 mL of PVA stock solution were taken in a test tube and150 μL of the NaAuCl4 stock solution was added to it dropwiseand stirred vigorously at room temperature. After 30 min, 100μL of the Zn(CH3COO)2 stock solution was again added to itdropwise and stirred vigorously. A greenish-yellow color wasslowly developed in the solution with the progress of thereaction and intensified. Without PVA, the solution remainedstable for a few minutes (<5 min) and then precipitated. Butwith PVA, the solution can be stored for weeks.
ZnS-Assisted Synthesis of Bluish-Green EmittingAuxZnySx/2+y Nanocrystals. Synthesis of ZnS Nanocrystals.ZnS nanocrystals were synthesized according to our previouslyreported method.51
Water Transfer of ZnS Nanocrystals. Purified nanocrystalswere taken in minimum volume of chloroform and an excessamount of MPA and very little amount of tetramethylammo-nium hydroxide solution was added in it until the solutionbecame cloudy. The mixture was then shaken for 20 min. TheMPA-capped nanocrystals were flocculated, and then thesewere centrifuged. The precipitate was washed 2−3 times withchloroform and ethanol to remove excess MPA. Finally, desiredamount of water was added to the precipitated nanocrystalsdropwise and stirred until all the nanocrystals were transferredto water.
Synthesis of AuxZnySx/2+y Nanocrystals Using MPA-CappedZnS Nanocrystals. 2 mL aliquot of ZnS solution in water (OD= 0.05 at 320 nm) was taken in a vial and 1 mL of PVA solutionwas added to it and stirred vigorously to get a homogeneoussolution. 50 μL of NaAuCl4 stock solution was added to it all ata time and stirred vigorously at room temperature. A greenish-yellow color developed slowly in the solution with time.
■ ASSOCIATED CONTENT
*S Supporting InformationExperimental details, PLE, additional TEM and STM images,and detailed structural characterization from XRD. This
Figure 7. (a) Plot of current vs voltage of a film made of bluish-green emitting gold zinc sulfide and yellow emitting gold sulfide nanocrystals. (b)Photocurrent growth and decay of a film made of gold zinc sulfide nanocrystals under periodic illumination of light (1 min) under 5 V fixed bias.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616684

material is available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSDST and CSIR of India are acknowledged for funding. Weacknowledge Dr. Somobrata Acharya for measurement andanalysis of STM and STS data, Sanjay Mahatha and Dr. K. S. R.Menon for XPS measurement, and Dr. Alok K. Mukherjee foranalysis of crystal structure. Authors U.T. and J.K.B. haveperformed the STM/STS measurement and helped in its dataanalysis.
■ REFERENCES(1) Averitt, R. D.; Sarkar, D.; Halas, N. J. Phys. Rev. Lett. 1997, 78,4217−4220.(2) Link, S.; El-Sayed, M. A. J. Phys. Chem. B 1999, 103, 4212−4217.(3) Link, S.; El-Sayed, M. A. Annu. Rev. Phys. Chem. 2003, 54, 331−366.(4) Orendorff, C. J.; Sau, T. K.; Murphy, C. J. Small 2006, 2, 636−639.(5) Faraday, M. Philos. Trans. 1857, 147, 145.(6) Mie, G. Ann. Phys. 1908, 25, 377−445.(7) Daniel, M.-C.; Astruc, D. Chem. Rev. 2004, 104, 293−346.(8) Eustis, S.; El-Sayed, M. A. Chem. Soc. Rev. 2006, 35, 209−217.(9) Sau, T. K.; Murphy, C. J. J. Am. Chem. Soc. 2004, 126, 8648−8649.(10) Murphy, C. J.; Sau, T. K.; Gole, A. M.; Orendorff, C. J.; Gao, J.;Gou, L.; Hunyadi, S. E.; Li, T. J. Phys. Chem. B 2005, 109, 13857−13870.(11) Huang, X.; El-Sayed, I. H.; Qian, W.; El-Sayed, M. A. J. Am.Chem. Soc. 2006, 128, 2115−2120.(12) Lee, S. T.; Apai, G.; Mason, M. G.; Benbow, R.; Hurych, Z. Phys.Rev. B: Condens. Matter 1981, 23, 505−8.(13) Whetten, R. L.; Khoury, J. T.; Alvarez, M. M.; Murthy, S.;Vezmar, I.; Wang, Z. L.; Stephens, P. W.; Cleveland, C. L.; Luedtke,W. D.; Landman, U. Adv. Mater. 1996, 8, 428−33.(14) Wilcoxon, J. P.; Martin, J. E.; Parsapour, F.; Wiedenman, B.;Kelley, D. F. J. Chem. Phys. 1998, 108, 9137−9143.(15) Zheng, J.; Nicovich, P. R.; Dickson, R. M. Annu. Rev. Phys.Chem. 2007, 58, 409−431.(16) Jin, R. Nanoscale 2010, 2, 343−362.(17) Schaaff, T. G.; Shafigullin, M. N.; Khoury, J. T.; Vezmar, I.;Whetten, R. L.; Cullen, W. G.; First, P. N.; Wing, C.; Ascensio, J.;Yacaman, M. J. J. Phys. Chem. B 1997, 101, 7885−7891.(18) Zheng, J.; Petty, J. T.; Dickson, R. M. J. Am. Chem. Soc. 2003,125, 7780−7781.(19) Link, S.; Beeby, A.; FitzGerald, S.; El-Sayed, M. A.; Schaaff, T.G.; Whetten, R. L. J. Phys. Chem. B 2002, 106, 3410−3415.(20) Bigioni, T. P.; Whetten, R. L.; Dag, O. J. Phys. Chem. B 2000,104, 6983−6986.(21) Huang, T.; Murray, R. W. J. Phys. Chem. B 2001, 105, 12498−12502.(22) Liu, C.-L.; Ho, M.-L.; Chen, Y.-C.; Hsieh, C.-C.; Lin, Y.-C.;Wang, Y.-H.; Yang, M.-J.; Duan, H.-S.; Chen, B.-S.; Lee, J.-F.; Hsiao, J.-K.; Chou, P.-T. J. Phys. Chem. C 2009, 113, 21082−21089.(23) Lin, C.-A. J.; Yang, T.-Y.; Lee, C.-H.; Huang, S. H.; Sperling, R.A.; Zanella, M.; Li, J. K.; Shen, J.-L.; Wang, H.-H.; Yeh, H.-I.; Parak, W.J.; Chang, W. H. ACS Nano 2009, 3, 395−401.(24) Muhammed, M. A. H.; Verma, P. K.; Pal, S. K.; Kumar, R. C. A.;Paul, S.; Omkumar, R. V.; Pradeep, T. Chem.–Eur. J. 2009, 15, 10110−10120, S10110/1−S10110/9.
(25) Peng, Z. A.; Peng, X. J. Am. Chem. Soc. 2002, 124, 3343−3353.(26) Medintz, I. L.; Uyeda, H. T.; Goldman, E. R.; Mattoussi, H. Nat.Mater. 2005, 4, 435−446.(27) Anikeeva, P. O.; Halpert, J. E.; Bawendi, M. G.; Bulovic, V. NanoLett. 2009, 9, 2532−2536.(28) Talapin, D. V.; Rogach, A. L.; Kornowski, A.; Haase, M.; Weller,H. Nano Lett. 2001, 1, 207−211.(29) Murray, C. B.; Norris, D. J.; Bawendi, M. G. J. Am. Chem. Soc.1993, 115, 8706−15.(30) Battaglia, D.; Peng, X. Nano Lett. 2002, 2, 1027−1030.(31) Qu, L.; Peng, X. J. Am. Chem. Soc. 2002, 124, 2049−2055.(32) Xie, R.; Battaglia, D.; Peng, X. J. Am. Chem. Soc. 2007, 129,15432−15433.(33) Torimoto, T.; Adachi, T.; Okazaki, K.-I.; Sakuraoka, M.;Shibayama, T.; Ohtani, B.; Kudo, A.; Kuwabata, S. J. Am. Chem. Soc.2007, 129, 12388−12389.(34) Li, L.; Pandey, A.; Werder, D. J.; Khanal, B. P.; Pietryga, J. M.;Klimov, V. I. J. Am. Chem. Soc. 2011, 133, 1176−1179.(35) Xie, R.; Rutherford, M.; Peng, X. J. Am. Chem. Soc. 2009, 131,5691−5697.(36) Wood, V.; Panzer, M. J.; Caruge, J.-M.; Halpert, J. E.; Bawendi,M. G.; Bulovic, V. Nano Lett. 2010, 10, 24−29.(37) Caruge, J. M.; Halpert, J. E.; Wood, V.; Bulovic, V.; Bawendi, M.G. Nat. Photon. 2008, 2, 247−250.(38) Pattantyus-Abraham, A. G.; Kramer, I. J.; Barkhouse, A. R.;Wang, X.; Konstantatos, G.; Debnath, R.; Levina, L.; Raabe, I.;Nazeeruddin, M. K.; Gratzel, M.; Sargent, E. H. ACS Nano 2010, 4,3374−3380.(39) Bang, J. H.; Kamat, P. V. ACS Nano 2009, 3, 1467−1476.(40) Li, L.; Lee, P. S.; Yan, C.; Zhai, T.; Fang, X.; Liao, M.; Koide, Y.;Bando, Y.; Golberg, D. Adv. Mater. 2010, 22, 5145−5149.(41) Clifford, J. P.; Konstantatos, G.; Johnston, K. W.; Hoogland, S.;Levina, L.; Sargent, E. H. Nat. Nanotechnol. 2009, 4, 40−44.(42) Huang, C.-C.; Chiang, C.-K.; Lin, Z.-H.; Lee, K.-H.; Chang, H.-T. Anal. Chem. 2008, 80, 1497−1504.(43) Wang, Z.; Cai, W.; Sui, J. ChemPhysChem 2009, 10, 2012−2015.(44) Zhou, C.; Sun, C.; Yu, M.; Qin, Y.; Wang, J.; Kim, M.; Zheng, J.J. Phys. Chem. C 2010, 114, 7727−7732.(45) Wu, Z.; Gayathri, C.; Gil, R. R.; Jin, R. J. Am. Chem. Soc. 2009,131, 6535−6542.(46) Forward, J. M.; Bohmann, D.; Fackler, J. P., Jr.; Staples, R. J.Inorg. Chem. 1995, 34, 6330−6336.(47) Susha, A. S.; Ringler, M.; Ohlinger, A.; Paderi, M.; Li Pira, N.;Carotenuto, G.; Rogach, A. L.; Feldmann, J. Chem. Mater. 2008, 20,6169−6175.(48) Nirmal, M.; Brus, L. Acc. Chem. Res. 1999, 32, 407−414.(49) Sarkar, S.; Karan, N. S.; Pradhan, N. Angew. Chem., Int. Ed. 2011,50, 6065−6069, S6065/1−S6065/6.(50) Srivastava, B. B.; Jana, S.; Pradhan, N. J. Am. Chem. Soc. 2011,133, 1007−1015.(51) Srivastava, B. B.; Jana, S.; Karan, N. S.; Paria, S.; Jana, N. R.;Sarma, D. D.; Pradhan, N. J. Phys. Chem. Lett. 2010, 1, 1454−1458.(52) Sarkar, S.; Bose, R.; Jana, S.; Jana, N. R.; Pradhan, N. J. Phys.Chem. Lett. 2010, 1, 636−640.(53) Pradhan, N.; Sarma, D. D. J. Phys. Chem. Lett. 2011, 2, 2818−2826.(54) Karan, N. S.; Sarma, D. D.; Kadam, R. M.; Pradhan, N. J. Phys.Chem. Lett. 2010, 1, 2863−2866.(55) Acharya, S.; Sarma, D. D.; Jana, N. R.; Pradhan, N. J. Phys. Chem.Lett. 2010, 1, 485−488.(56) Tang, J.; Konstantatos, G.; Hinds, S.; Myrskog, S.; Pattantyus-Abraham, A. G.; Clifford, J.; Sargent, E. H. ACS Nano 2009, 3, 331−338.(57) Panthani, M. G.; Akhavan, V.; Goodfellow, B.; Schmidtke, J. P.;Dunn, L.; Dodabalapur, A.; Barbara, P. F.; Korgel, B. A. J. Am. Chem.Soc. 2008, 130, 16770−16777.(58) Rogach, A. L.; Franzl, T.; Klar, T. A.; Feldmann, J.; Gaponik, N.;Lesnyak, V.; Shavel, A.; Eychmueller, A.; Rakovich, Y. P.; Donegan, J.F. J. Phys. Chem. C 2007, 111, 14628−14637.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616685

(59) Rogach, A. L.; Kornowski, A.; Gao, M.; Eychmueller, A.; Weller,H. J. Phys. Chem. B 1999, 103, 3065−3069.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp3049718 | J. Phys. Chem. C 2012, 116, 16680−1668616686





























