Embed Size (px)
Citation preview

Sequence Assembly: Concepts
BMI/CS 576www.biostat.wisc.edu/bmi576/
Sushmita [email protected]
September 2012
BMI/CS 576

Goals for this unit
• Shortest superstring problem• Greedy and non-polynomial time algorithms• Graph-theoretic concepts– Euler path– Hamiltonian path
• k-mer spectrum– Debruijn graphs
• Popular algorithms for assembly

Sequencing problem
• What is the base composition of– A single molecule of DNA– A genome– Many genomes
• We cannot read the entire genomic sequence but can do this in pieces

Finding the genomic sequence
• Fragment the genome into short strings• Sequence/read smaller strings• Assemble these strings into the larger genome

Sequencing as a fragment assembly
Multiple copies of sample DNA
Randomly fragment DNA
Sequence sample of fragments
Assemble reads

Sequence assembly is like solving a big jigsaw puzzle

Different types of sequencing methods
• Sanger sequencing: 800bp-1000 bp– Chain termination method
• 454 sequencing: 300-400 bp• Illumina Genome Analyzer: 35-150 bp• Helicos: 30bp

First and second generation
• First generation– Automated Sanger
sequencing – Amplification through
bacterial colonies (in vivo)
– 800-1000bp– Low error– Slow and expensive
(limited parallelization: in 96 or 384 capillary-based electrophoresis)
• Second generation– 454 sequencing,
Illumina, Helicos, SOLiD
– Amplification on arrays (in vitro)
– 30-150bp– High error– Fast and cheap
(parallel acquisition of many DNA sequences)

De novo assembly: The fragment assembly problem
• Given: A set of reads (strings) {s1, s2, … , sn }• Do: Determine a superstring s that “best explains”
the reads
• What do we mean by “best explains”?

Shortest superstring problem (SSP)
• Objective: Find a string s such that– all reads s1, s2, … , sn are substrings of s– s is as short as possible
• Assumptions:– Reads are 100% accurate– Identical reads must come from the same location on the
genome– “best” = “simplest”

Shortest superstring example
• Given the reads: {ACG, CGA, CGC, CGT, GAC, GCG, GTA, TCG}
• What is the shortest superstring you can come up with?
• This problem is a computationally difficult problem– NP-complete
TCGACGCGTA (length 10)

NP complete problems
• NP (non-deterministic polynomial) problems:– A problem for which we don’t know a polynomial
algorithm– But we don’t whether a polynomial algorithm exists or not– You can check a solution in polynomial time
• NP complete– A problem that is NP– Is at least as hard as other problems in NP

SSP as the traveling salesman problem
• Traveling salesman problem– Given a set of cities and distances between them– Find the shortest path covering all cities, but no city more
than once
• SSP->TSP– replace edge overlap by negative edge overlap– minimize over path length

Algorithms for shortest superstring problem
• Greedy• Overlap Layout Consensus• Eulerian path– Debruijn graphs

Greedy algorithms
• Definition: An algorithm that takes the best immediate, or local steps while finding an answer.
• May find less than-optimal solutions for some ‐ instances of problems.

Greedy algorithm for SSP
• Simple greedy strategy:while # strings > 1 do
merge two strings with maximum overlaploop
• Overlap: size of substring common between two strings
• Conjectured to give string with length ≤ 2 × minimum length

Algorithms for shortest superstring problem
• Greedy• Overlap Layout consensus (OLC)• Eulerian path– Debruijn graphs
The rest require graph theory.

Graph theory basics
• a graph (G) consists of vertices (V) and edges (E)G = (V,E)
• edges can either be directed (directed graphs)
• or undirected (undirected graphs)
1
2
43
1
2
43

Graph theory basics
• degree of a vertex: number of neighbors of a vertex– In degree: number of incoming edges– Out degree: number of outgoing edges
• path from u to v: number of connected edges starting from u and ending at v
• cycle is a path that begins and ends at the same vertex.
1
2
4

Overlap Layout consensus methods
• Overlap graph creation• Layout• Consensus

Overlap graph
• For a set of sequence reads S, construct a directed weighted graph G = (V,E,w)– One vertex per read (vi corresponds to si)– edges between all vertices (a complete graph)– w(vi,vj) = overlap(si ,sj )
• length of longest suffix of si that is a prefix of sj
AGAT
prefix
suffix

Overlap graph example
• Let S = {AGA, GAT, TCG, GAG}
AGA
GAT TCG
GAG
2
2
0
0
10
0
1
1
2
0
1

Overlap graph example
• Let S = {AGA, GAT, TCG, GAG}
AGA
GAT TCG
GAG
2
21
1
1
2
1

Layout: Find the assembly
• Identify paths corresponding to segments of the genome
• The general case is finding the Hamiltonian path

Hamiltonian path
• Hamiltonian path: path through graph that visits each vertex exactly once
AGA
GAT TCG
GAG
2
2
0
0
10
0
1
1
2
0
1
Path: AGAGATCGAre there more Hamiltonian paths?

Hamiltonian path
• A path that visits every vertex only once• Checking whether a graph has a Hamiltonian path or
not is very difficult– NP complete

Assembly as a Hamiltonian path
• finding Hamiltonian path is an NP-complete problem – Similar to the Traveling salesman problem
• nevertheless overlap graphs are often used for sequence assembly

Consensus
• Once we have a path, determine the DNA sequence• Implied by the arrangement of reads along the
chosen path
GTATCGTAGCTGACTGCGCTGCATCGTCTCGTAGCTGACTGCGC
TGCATCGTATCGAAGCGGAC TGACTGCGCTGCATCGTATCGTAGC
Consensus: TGACTGCGCTGCATCGTATCGTAGCGGACTGCGCTGC

Summary
• OLC: Overlap layout consensus• Overlap: compute the maximal substring common
between two strings• Layout: arrange the reads on a graph based on the
overlap– Exact solution requires the Hamiltonian path->NP
complete!– Greedy solution is used in practice

Euler Path and cycles
• A path in a graph that includes all edges• An Euler cycle is an Euler path that begins and ends
at the same vertex• Finding an Euler path is easy– Linear in the number of edges of a graph

Seven Bridges of Königsberg
Euler answered the question: “Is there a walk through the city that traverses each bridge exactly once?”

Properties of Eulerian graphs• cycle: a path in a graph that starts/ends on the same
vertex
• Eulerian cycle: a path that visits every edge of the graph exactly once
• Theorem: A connected graph has an Eulerian cycle if and only if each of its vertices are balanced
• A vertex v is balanced if indegree(v) = outdegree(v)
• There is a linear-time algorithm for finding Eulerian cycles!

Eulerian cycle algorithm
• start at any vertex v, traverse unused edges until returning to v
• while the cycle is not Eulerian– pick a vertex w along the cycle for which there are
untraversed outgoing edges– traverse unused edges until ending up back at w– join two cycles into one cycle

Finding cycles
1) start at arbitrary vertex 2) start at vertex along cycle with untraversed edges

Finding cycles
3) join cycles 4) start at vertex along cycle with untraversed edges

Finding cycles
5) join cycles

Eulerian paths
1 4
6
3
7
5
2
• a vertex v is semibalanced if |indegree(v) – outdegree(v)| = 1
• a connected graph has an Eulerian path if and only if it contains at most two semibalanced vertices

Eulerian path Eulerian cycle
• If a graph has an Eulerian Path starting at w and ending at x then– All vertices must be balanced, except for w and x which are
semibalanced– If and w and x are not balanced, add an edge between
them to balance• Graph now has an Eulerian cycle which can be converted to an
Eulerian path by removal of the added edge

Eulerian path Eulerian cycle
1 4
6
3
7
5
2

Spectrum of a sequence
• k-mer spectrum of s is the set of all k-mers that are present in the sequence
• ATTACAG– 3-spectrum: {ATT, TTA, TAC, ACA, CAG}– 4-spectrum: {ATTA,TTAC,TACA,ACAG}

deBruijn graph
{ATG, TGG, TGC, GTG, GGC, GCA, GCG, CGT}
AT CA
CG
GC
GG
GT
TGATG TGC
TGG GGC
GCA
GCG
CGT
GTG
• in a deBruijn graph
– edges represent k-mers that occur in s– vertices correspond to (k-1)-mers– Directionality goes from the k-1 prefix to the k-1 suffix

deBruijn graphs/Eulerian approach for assembly
• Find a DNA sequence containing all k-mers?
• In a deBruijn graph, can we find a path that visits every edge of the graph exactly once?

Assembly as finding Eulerian paths• Eulerian path: path that visits every edge exactly once• we can frame the assembly problem as finding Eulerian paths in a deBruijn graph• resulting sequences contain all k-mers
AT CA
CG
GC
GG
GT
TGATG TGC
TGG GGC
GCA
GCG
CGT
GTG
• assembly: ATGGCGTGCA

Eulerian path-> Assembly
• But there are exponentially many cycles for a given graph!– Most assemblers pick one
• Better for next-generation sequencing data– Breaking reads into k-mers loses connectivity

Assembly is complicated because
• Of repeats– Hard to know where repeats begin or end– Reads are not long enough– Human genome has many different repeats
• SINEs (Short interspersed elements, 300bp)• LINEs (Long interspersed elements, 1000 bps)• Gene duplications
• Sequencing errors– Distinguishing true from error-based overlaps.

Sequence assembly in practice
• approaches are based on these ideas, but include a lot of heuristics
• “best” approach varies depending on length of reads, amount of repeats in the genome, availability of paired-end reads
• Assemblies are done iteratively– Contigs: contiguous piece of sequence from overlapping
reads– Scaffolds: ordered contigs based on paired end reads

Sequence assembly in practice
• Most sequencing platforms produce a fastq file– Sequence quality scores: Phred scores
• Q=-10log10P, P is the probability of a base being wrong
– Estimated from known sequences
• N50: a statistic used to assess the quality of assembly– The length l of a contig such that all contigs of length l’<l
cover 50% of the assembly– Other NX also defined.– Higher the N50 the better.
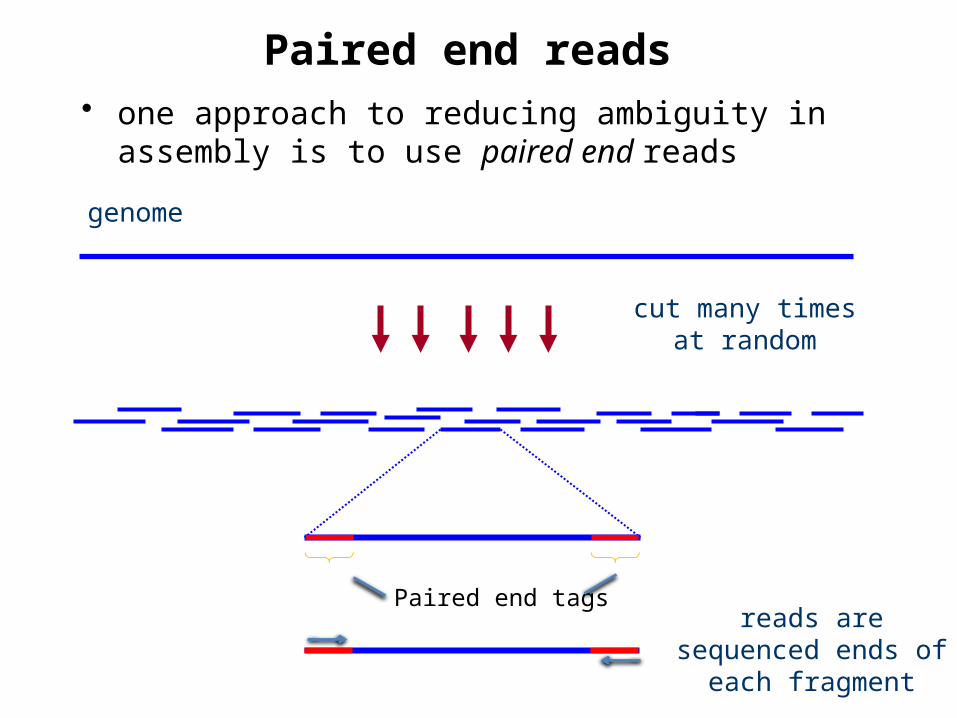
Paired end reads
cut many times at random
genome
reads are sequenced ends of each fragment
• one approach to reducing ambiguity in assembly is to use paired end reads
Paired end tags

Implemented Algorithms for assembly
• Overlap layout consensus– Celera:
• was developed for human genome• Finds uniquely assembled contigs• Throws out regions with very high reads• Resolve forks using mate pairs.
– Arachne• Uses mate pairs from the beginning
– Use efficient k-mer based indexing to estimate read overlap
• Eulerian path/deBruijn path– Velvet

Velvet for assembling genomes from 32-50bp reads
• Debruijn graphs• Generate k-mers per read• Simplify
– Merge linear chains
• Error correction– Remove tips: disconnected ends <2k length– Remove bubbles: Bus tour algorithm
• Do a breadth-first search. • Each time you hit a node already seen consider merging
• Handle repeats– Breadcrumb

From reads to deBruijn graphs
deBruijn Graph
CTCCGA
GGA CTG
TCCCCG
GGG TGG
AAG AGA GAC ACT CTT TTT
4-mer spectrum
AAGAACTCACTGACTTAGACCCGACGACCTCCCTGGCTTTGACTGGACGGGATCCGTGGG
• human genome: ~ 3B nodes, ~10B edges
AAGACAGGACTT
…
Reads

Simplifying the graph in Velvet
CTCCGA
CTGGGA
AAGA
GAC ACT CTTT
CTCCGA
GGA CTG
TCCCCG
GGG TGG
AAG AGA GAC ACT CTT TTT
collapse linear subgraphs

Bus tour algorithm
A B C D E
B1 C1
Z
Trace shortest path from A
A B C D E
B1 C1
Z
D is reached through two paths

Bus tour algorithm
A B C D E
B1 C1
Z
1
2
A B C D E
Z
A B C D E
Z
Align and merge to shorter path
Path 1 < Path 2
Simplify

Breadcrumb for handling repeats
• It all boils down to connecting two long segments/contigs
• Use paired end reads to connect long segments• Connect intermediate smaller segments using other
paired end tags

Genome projects
• 1000 genomes to study human genetic variation• 1001 Arabidopsis genomes• i5K: 5000 insect genomes• 16,000 vertebrate genomes• Human microbiome project

Summary
• Sequence assembly: Shortest superstring problem• Graph theoretic approaches to address this
– Greedy algorithm– Eulerian paths– Debruijn path
• In practice much harder because– Repeats– Errors
• Algorithms use non-trivial post-processing to produce final assembly





























