Embed Size (px)
Citation preview

Applied Catalysis A: General, 107 (1993) 73-81
Elsevier Science Publishers B.V., Amsterdam
73
APCAT A2646
Selective synthesis of light alkenes from carbon monoxide and hydrogen on silicalite supported iron-manganese catalysts
Debasish Das, G. Elavichandran and D.K. Chakrabarty
Solid State Laboratory, Chemistry Department, Indian Institute of Technology, Bombay 400076
(India)
and
S.N. Piramanayagiam and S.N. Shringi
Physics Department, Zndian Institute of Technology, Bombay 400076 (India)
(Received 29 January 1993, revised manuscript received 30 August 1993)
Abstract
Iron-manganese catalyst supported on silicalite 1 was prepared by impregnation. The catalyst prep- aration was followed by X-ray diffraction, temperature-programmed reduction and Mtissbauer spec- troscopy. Hydrogenation of CO was studied in a high pressure reactor by varying CO:H, ratio, temper- ature, and pressure. The silicalite supported catalyst shows very high selectivity for C,-C, alkenes. When acidic ZSM-5 having the same structure as silicalite 1 was used as the support, the product had very little alkenes. Addition of 0.2% potassium improves the alkene selectivity of the Fe-Mn on the silicalite catalyst.
Key words: carbon monoxide hydrogenation; Fe-Mn/silicalite; Fe-Mn/ZSM-5; light alkenes
INTRODUCTION
To meet the ever growing demand for chemical feedstocks, considerable at- tention is being paid to the design of Fischer- Tropsch catalysts with high selectivity for the lower alkenes. It has been shown that iron catalysts pro- moted with manganese oxides show unusually high selectivity for lower al- kenes and suppress methane formation [l-7]. The precise role of manganese is still not clear and the procedure to be used for catalyst preparation has not been unequivocally established. Most of the iron-manganese catalysts re- ported so far are either precipitated, or are catalysts supported on oxides [ 81.
Correspondence to: Dr. D.K. Chakrabarty, Solid State Laboratory, Chemistry Department, In-
dian Institute of Technology, Bombay 400076 (India), fax. (+091-22)5783480, e-mail
0926-860X/93/$06.00 0 1993 Elsevier Science Publishers B.V. All rights reserved.

74 D. Das et al./Appl. Catal. A 107 (1993) 73-81
Efforts are now being directed towards the development of CO hydrogena- tion catalysts supported on zeolites as, because of their high-surface area, shape selective charactier, acidity and well-defined structure, they have some added advantage over conventional oxide supports. While the high-surface area of these supports allows a high degree of metal dispersion, acidity and shape and size constraints can significantly change the selectivity of the catalysts. Thus, carbonyl derived iron catalysts supported on Y and ZSM-5 zeolites showed high selectivity for alkenes [ 9,101, whereas in the case of Fe/mordenite cata- lysts, alkene selectivity was found to be strongly dependent on the exchanged cation in the zeolite [ 111. Similarly, high selectivity for C!,-C, alkenes of the silicalite supported iron [ 121 and cobalt [ 131 catalysts was reported. Further, addition of a small amount of potassium (0.9% ) to iron catalyst (7.8% ) sup- ported on silicalite dramatically improved the selectivity for lower alkenes [ 121. It is well known that addition of small amounts of potassium to iron catalysts causes an increase in CO dissociation, alkene to alkane ratio and chain growth, and suppresses methane formation [ 14,151.
Although high selectivity for lower alkenes was reported for silicalite sup- ported iron catalysts as well as for precipitated iron-manganese catalysts, no work has been do:ne on iron-manganese supported on silicalite. The aim of this work is to invest.igate the behaviour of silicalite supported iron-manganese catalysts. The effect of adding potassium to the silicalite supported catalyst and that of the acidity of the support on alkene selectivity has also been inves- tigated using ZSM-5 zeolite as the support.
EXPERIMENTAL
Catalyst preparation
Silicalite 1 and ZSM-5 supports were synthesized according to a method described in ref. 16. Impregnated catalysts, designated as Fe-Mn/Sil, Fe-Mn/ ZSM-5 and K-Fe-Mn/Sil (x), where Sil stands for silicalite 1 and (x) indi- cates the weight percentage of potassium, were prepared by the incipient wet- ness technique. The supports were calcined at 500’ C overnight before addition of the metal nitrate solutions. Impregnation was carried out by adding requi- site amounts of 0.45 M metal nitrate solutions to 10 g of the support with continuous stirring under vacuum to ensure entry of the metal nitrates into the zeolite pores. The total metal loading was maintained at 20% with an Fe to Mn ratio 1: 1. Potassium was incorporated in the promoted catalysts as po- tassium carbonate in a similar manner before treating with the iron or man- ganese nitrate solutions. The impregnated material was initially dried at 120’ C for 12 h and then finally calcined at 450°C in air for 8 h.

D. Das et al./Appl. Catul. A 107 (1993) 73-81 75
Characterization of catalysts
X-ray diffraction patterns of the supports and the catalysts were recorded in a Philips X-ray diffractometer PW 1820 with nickel filtered Cu Ka radia- tion at a scanning rate of 2 ’ per minute.
Temperature-programmed reduction (TPR) was studied in a conventional flow apparatus. The catalysts were heated in air at 400°C overnight before TPR studies. A high purity premixed gas containing 94% argon and 6% hydro- gen was used for TPR. Traces of oxygen and water were removed by passing through heated copper turnings at 400’ C and activated molecular sieve. About 50 mg of the sample was loaded in a quartz reactor for the TPR studies. The sample was heated in argon at 400’ C for 6 h and cooled down to room temper- ature in an argon flow. It was then reduced in 6% hydrogen in argon (flow rate 55 ml min-’ ) at a heating rate of 10” C min-‘.
Miissbauer spectra of the catalysts after the calcination stage and those of the used catalysts were obtained using a PC based multichannel analyser in the transmission mode. A 5 mCi 57Co(Rh) source was used in the constant acceleration mode. The spectrometer was calibrated with a standard a-Fe ab- sorber. The spectra were fitted using a computer program. Baseline counts and intensities were evaluated by using a non-iterative optimization and iterative grid search procedure was used to determine the linewidths and peak positions of the Lorentzians. x2 was close to one and linewidths were restricted to 0.25 mm/s.
Reaction studies
Carbon monoxide conversion was studied in a a inch I.D. stainless-steel tixed- bed reactor with a:n effective length 30 cm (type BTRS-Jr. of Autoclave En- gineers, USA). About 1 g of catalyst (particle size: 180-300 mesh) was packed between quartz wool plugs in the reactor and was reduced in-situ at 500’ C for 12 h in hydrogen (CHSV = 800 h-l). The feed gas (premixed H2 and CO (99.7% pure) was preheated before entering the reactor. The flow rate was controlled by precision needle valves and the desired pressure was achieved by a back pressure regulator. The reactor inside a tubular furnace and the sampling valves were maintained inside a chamber heated to 200” C. A bypass valve permitted the sampling of either the reactant gas or the product that was carried to a gas chromatograph through a heated transfer line. Hydrocarbons were separated on a 6 ft. x l/8 in. stainless-steel column packed with Porapak Q and analysed using flame ionisation detector. CO, COZ, H2 and Ar were separated on a 6 ft. x $ in. CTR-1 column and analysed using TC detector. The signals from the detector were inte!grated by a Milton Roy CI-1OB integrator. The results of analysis were reproducible within 1%. CO conversion was calculated using ar- gon as the internal standard and expressed as

76 D. Das et al./Appl. Catal. A 107(1993) 73-81
CO conversion ( 7% ) = WIJ [Ad - Wl,/kW, x 1oo
[COlr/ [Arlr where the subscr:ipts r and p stand for reactant and product respectively. The activity and selectivity data reported here are after 3 h of reaction when the catalysts attained steady activity that remained nearly unchanged for 18 h after which the reaction was not studied.
RESULTS AND DISCUSSION
The XRD pattern of the silicalite sample showed that the support was highly crystalline in nature and free from any other crystalline impurities. Unlike precipitated catalysts [ 171, the calcined zeolite supported iron-manganese catalysts did not show any XRD lines due to iron or manganese oxides, indi- cating the highly dispersed nature of the metal oxides. A small decrease in the crystallinity of the support was also observed with metal loading which is how- ever not unexpected in the case of impregnated catalysts. XRD patterns of the used catalysts showed a further decrease in peak intensities, indicating consid- erable loss in crystallinity, but no XRD lines other than those of the support were observed in the used catalysts.
Temperature-programmed reduction patterns of the catalysts indicate a two step reduction process with the first peak at about 500°C and the other at approximately 675’ C. Precipitated iron-manganese catalysts too showed sim- ilar two-peak pattern [ 171 with a small shift in the peak positions (450 and 680” C). Under similar conditions, a-Fe,O, showed only one TPR peak at 610’ C and MnOa obtained by the decomposition of manganese nitrate did not undergo reduction upto 800” C. However, TPR experiments with Fe/silicalite and Mn/ silicalite showed reduction of iron and manganese species at 525 and 450’ C, respectively. Since the calcined Mn/silicalite sample did not show any XRD lines other than lthose due to silicalite, we are not in a position to ascertain whether the calcined Mn/silicalite contained MnO, or some other oxide of manganese. Going by the TPR results alone, it appears that it was not MnO, that is difficult to reduce. It has been reported earlier that in precipitated iron- manganese hydrolxides, Mnz03 and ar-Fe,O, are formed on calcination where Fe and Mn substi.tute each other [ 181. TPR of such samples also show a two peak patterns [ 171. The amount of hydrogen consumed in each step is given in Table 1. Addition of potassium to the iron-manganese silicalite catalyst does not seem to affect the nature and positions of the TPR peaks but the catalyst having 0.5 wt.-% potassium is reduced to a greater extent. However, the hy- drogen:iron mol ratio for each step of reduction does not match with that ex- pected for any simple iron oxide reduction process, and this may be due to the fact that the speci.es involved in the reduction include solid solution of the two metal oxides.

D. Das et al.fAppl. Catd. A 107 (1993) 73-81
TABLE 1
Hydrogen consumed in TPR of the silicalite supported catalysts
77
Catalyst Peaktemp. (“C) Hydrogen/iron ratio
Fe-Mn/Sil 500 0.25 670 0.45
K-Fe-Mn/Sil(0.2) 500 0.17 679 0.57
K-Fe-Mn/Sil(0.5) 500 0.36 680 0.58
Mijssbauer data of the fresh catalysts showed a well resolved central doublet which is characteristic of fine particles of superparamagnetic (SPO) Fe203 [ 19,201. Absence of a six-line pattern, as expected for large particles of (Y- Fe203, indicates that most of the iron oxides are present in a finely dispersed state which was also indicated by the XRD results. If calcination was carried out at 5OO”C, XRD lines due to a-Fe,O, was indeed seen and the Mossbauer showed the usual six-line pattern of a-Fe,O, spectrum. This indicates that at 450’ C, the above-mentioned oxide was present as very small particles.
The size of the superparamagnetic oxide particles can be estimated by using the correlation between particle size and quadrupole splitting (Q.S.) as pro- posed by Kundig et. al. [ 211. The quadrupole splitting is nearly the same for all the calcined samples and this corresponds to about 3.0 nm particle size. The reduced catalysts could not be studied as they are pyrophoric.
Mijssbauer data of the used catalysts are given in Table 2. The spectra showed superposition of patterns for iron carbides and high-spin Fe3+, as has been observed in the case of oxide- supported iron Fischer-Tropsch catalysts [ 221. The computer fitted spectra showed three carbide sextets with hyperfine fields of 178,104 and 218 kG, which corresponds to Hagg carbide, x-Fe5C2 in which iron atoms are present in three different sites. Presence of a doublet with IS. = 0.3 mm/s, and Q.S. = 0.7 mm/s indicate oxidic Fe3+. The results suggest that all the used catalysts contain about 80% of iron as Hagg carbide in addition to trivalent iron. The latter could have been formed from the incompletely car- bided lower oxides of iron during sample transfer. The presence of iron carbide, however, was not shown by the X-ray diffraction patterns of the used catalysts. This is possibly due to the presence of very poorly crystallized carbide particles in which case the diffraction lines for carbides will be masked by strong lines of the silicalite support.

78
TABLE 2
D. Das et al./Appl. Catal. A 107 (1993) 73-81
Mossbauer spectra of the used catalysts
Catalyst
Fe-Mn/Sil
Species
x-Fe&
Fe3+
I.S.(mm/s) Q.S.(mm/s) HF field (kG) Area (%)
0.22 0.04 177 0.44 0.00 104 84 0.27 0.04 218 0.44 0.80 16
0.22 0.04 178 K-Fe-Mn/Sil(0.2) X-Fe,& 0.44 0.00 104 80
0.27 0.04 218 Fe3+ 0.33 0.62 20
0.27 0.04 179 K-Fe-Mn/Sil(O.S) x-FesC2 0.44 0.00 104 79
0.31 0.04 218 Fe3+ 0.24 0.53 21
TABLE 3
Product pattern (wt.-% ) at different CO:H, ratio for Fe-Mn/Sil catalyst at steady conversion Pressure=300psi, temp.=250”C, GHSV= 1000 h-’
CO:H, CO converted C, Cz C,= c3 C3= C, Cd C: G/P
(%i)) CM”)
1:l 16.3 x.1 19.3 4.4 20.3 4.6 27.0 3.2 13.8 7.4 5.0 1:2 20.0 1:2.4 23.5 4.7 16.3 5.0 22.4 4.8 14.5 8.6 3.7 1:3 26.8 112.5 22.7 4.9 13.6 5.8 20.8 4.3 15.6 12.3 3.3
“M= pmoles of CO reacted ‘--l g-1 Fe.
CO hydrogenation
Hydrogenation of CO has been studied at different CO:H, ratios on the Fe- Mn/silicalite catalyst. The results are presented in Table 3. It has been ob- served that methane selectivity is lowest when the ratio is 1: 1. An increase in reaction temperature, on the other hand, increases CO conversion without af- fecting alkene/alkane (O/P) ratio.
A comparison Iof the products formed on Fe-Mn/Sil and Fe-Mn/ZSM-5 catalysts as studied in this work and along with the results reported for Fe/Sil, Fe/ZSM-5 and precipitated Fe-Mn catalysts are given in Table 4. At 275°C the conversion of CO to hydrocarbons was 25% on Fe-Mn/Sil and 13.5% on Fe-Mn/ZSM-5. For the other catalysts shown in Table 4 (taken from pub-
D. Das et al./Appl. Catai!. A 107 (1993) 73-81 79
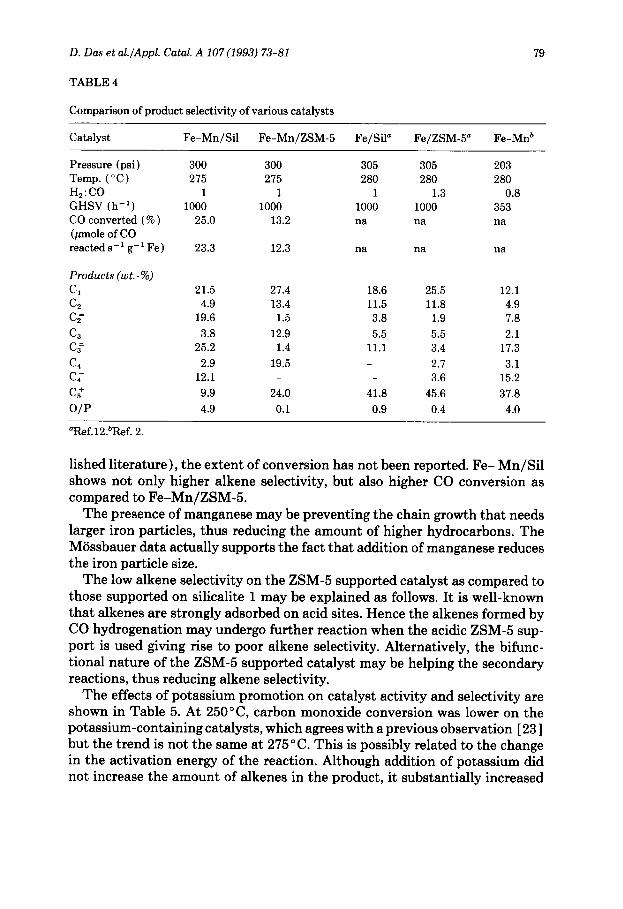
TABLE 4
Comparison of product selectivity of various catalysts
Catalyst Fe-Mn/Sil Fe-Mn/ZSM-5 Fe/Sil” Fe/ZSM-5” Fe-Mnb
Pressure (psi) Temp. (“C) H,:CO GHSV (h-r) CO converted ( % ) (pmole of CO reacted s-l g-r Fe)
Products (wt.-%)
C, C, C;
C3 CT
Cd C;
C:
o/p
300 300 305 305 203 275 275 280 280 280
1 1 1 1.3 0.8 1000 1000 1000 1000 353
25.0 13.2 na na na
23.3 12.3 na na na
21.5 4.9
19.6
3.8 25.2
2.9 12.1
9.9
4.9
27.4 13.4
1.5
12.9 1.4
19.5
18.6 11.5 3.8
5.5 11.1
24.0 41.8
0.1 0.9
25.5 12.1 11.8 4.9
1.9 7.8
5.5 2.1 3.4 17.3
2.1 3.1 3.6 15.2
45.6 37.8
0.4 4.0
“Ref.lZ.“Ref. 2.
lished literature ), tlhe extent of conversion has not been reported. Fe- Mn/Sil shows not only higher alkene selectivity, but also higher CO conversion as compared to Fe-Mn/ZSM-5.
The presence of manganese may be preventing the chain growth that needs larger iron particles, thus reducing the amount of higher hydrocarbons. The Miissbauer data actually supports the fact that addition of manganese reduces the iron particle size.
The low alkene selectivity on the ZSM-5 supported catalyst as compared to those supported on silicalite 1 may be explained as follows. It is well-known that alkenes are strongly adsorbed on acid sites. Hence the alkenes formed by CO hydrogenation may undergo further reaction when the acidic ZSM-5 sup- port is used giving rise to poor alkene selectivity. Alternatively, the bifunc- tional nature of the! ZSM-5 supported catalyst may be helping the secondary reactions, thus reducing alkene selectivity.
The effects of potassium promotion on catalyst activity and selectivity are shown in Table 5. At 25O”C, carbon monoxide conversion was lower on the potassium-containing catalysts, which agrees with a previous observation [ 231 but the trend is not the same at 275°C. This is possibly related to the change in the activation energy of the reaction. Although addition of potassium did not increase the amount of alkenes in the product, it substantially increased
80 D. Dm et al./Appl. Catal. A 107 (1993) 73-81
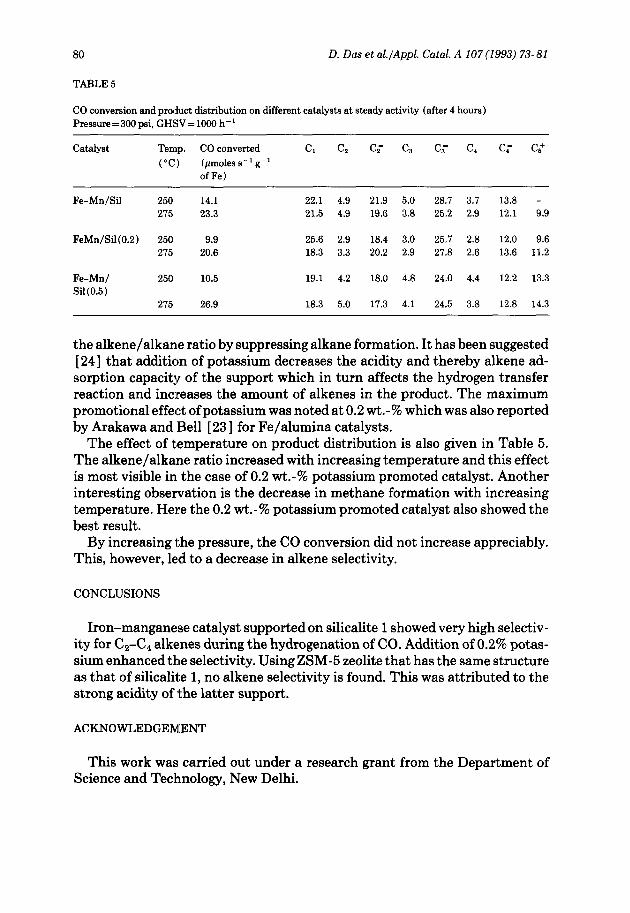
TABLE 5
CO conversion and product distribution on different catalysts at steady activity (after 4 hours) Pressure=300 psi, GHSV= 1000 h-i
Catalyst Temp. CO converted C, C2 CT C, Cb C1 Cp CS+
(“Cl (~moles s-i g-i of Fe)
Fe-Mn/Sil 250 14.1 22.1 4.9 21.9 5.0 28.7 3.1 13.8 - 275 23.3 21.5 4.9 19.6 3.8 25.2 2.9 12.1 9.9
FeMn/Sil(O.Z) 250 9.9 25.6 2.9 18.4 3.0 25.7 2.8 12.0 9.6 215 20.6 18.3 3.3 20.2 2.9 21.8 2.6 13.6 11.2
Fe-Mn/ 250 10.5 19.1 4.2 18.0 4.8 24.0 4.4 12.2 13.3 Sil(O.5)
275 26.9 18.3 5.0 17.3 4.1 24.5 3.8 12.8 14.3
the alkene/alkane ratio by suppressing alkane formation. It has been suggested [24] that addition of potassium decreases the acidity and thereby alkene ad- sorption capacity of the support which in turn affects the hydrogen transfer reaction and increases the amount of alkenes in the product. The maximum promotional effect of potassium was noted at 0.2 wt.-% which was also reported by Arakawa and Bell [ 231 for Fe/alumina catalysts.
The effect of temperature on product distribution is also given in Table 5. The alkene/alka:ne ratio increased with increasing temperature and this effect is most visible in the case of 0.2 wt.-% potassium promoted catalyst. Another interesting observation is the decrease in methane formation with increasing temperature. Here the 0.2 wt.-% potassium promoted catalyst also showed the best result.
By increasing the pressure, the CO conversion did not increase appreciably. This, however, led to a decrease in alkene selectivity.
CONCLUSIONS
Iron-manganese catalyst supported on silicalite 1 showed very high selectiv- ity for C,-C, alkenes during the hydrogenation of CO. Addition of 0.2% potas- sium enhanced the selectivity. Using ZSM-5 zeolite that has the same structure as that of silicalite 1, no alkene selectivity is found. This was attributed to the strong acidity of the latter support.
ACKNOWLEDGEMENT
This work was carried out under a research grant from the Department of Science and Technology, New Delhi.

D. Das et al./Appl. Catal. A 107(1993) 73-81
REFERENCES
81
1 2
3 4 5 6 7 8 9
10 11 12 13
14
15
16 17
B. Bussemeier, C.D. Frohning and B. Cornils, Hydrocarbon Proc., 55 (11) (1976) 105. H. Kolbel, M. RaIek and K.O. Tillmetz, in Proc. of the 13th Intersoc. Energy Conv. Eng. Conf., Society of Automotive Eng., San Diego (1978) p. 482. J. Barrault, C. Forquy and V. Perrichon, Appl. Catal., 5 (1983) 119. H.W. Pennline, M.F. Zarochak, R.E. Tischer and R.R. Schehl, Appl. Catal., 21 (1986) 313. I.S.C. Hughes, J.O.H. Newman and G.C. Bond, Appl. Catal., 30 ( 1987) 303. C.H. Yang, Ph.D. Thesis, Univ. of Utah, Salt Lake City, UT (1979). Y.S. Tsai, MS. Thesis, Univ. of Utah, Salt Lake City, UT (1980). M. Janardana Rao, Ind. Eng. Chem. Res., 29 (1990) 1735. T. Mitsudo, H. Boku, S. Murachi, A. Ishihara and Y. Watanabe, Chem. Lett., (1985) 1463. J. Zwart and J. Vink, Appl. Catal., 33 (198’7) 383. H.G. Yun, S.I. Woo and J.S. Chung, Appl. Catal., 68 ( 1991) 97. V.U.S. Rao and R.J. Gormley, Hydro. Proc., 59 (11) (1980) 139. M. Peuckert and G. Linden, in Proc. of the 8th Int. Congress on Catalysis, Berlin, 1984, Verlag Chemie, Weinheim, 1984, Vol. II, p. 135. M.E. Dry, in J.R. Anderson and M. Boudart (Editors), Catalysis - Science and Technology Vol. 1, Springer-Verlag, Berlin, 1981, p. 159. G. van der Lee, G.T.M. Bastein, I. van Boogert, B. Schuller, H.Y. Luo and V. Ponec, J. Chem. Sot., Faraday Trans., 83 (1987) 2013. C.V.V. Satyanaryana and D.K. Chakrabarty, Appl. Catal., 66 (1990) 1. C.K. Das, N.S. IDas, D.P. Choudhary, G. Ravichandran and D.K. Chakrabarty (communication).
18 G.C. Maiti, R. Malessa and M. Baerns, Appl. Catal., 5 (1983) 151. 19 E.E. Unmuth, L.H. Schwartz and J.B. Butt, J. Catal., 61 (1980) 242. 20 W. Kundig, H. Bommel, G. Constabaris and R.H. Lindquist, Phys. Rev., 142 (1966) 327. 21 W. Kundig, K.J. Ando, R.H. Lindquist and G. Constabaris, Czech. J. Phys., 17 (1967) 467. 22 J. Galuszka, T. Sano and J.A. Sawicki, J. Cat& 136 (1992) 96. 23 H. Arakawa and A.T. Bell, Ind. Eng. Chem. Process Des. Dev., 22 (1983) 97. 24 J.A. Rabo, R.D. Benzman and M.L. Poutsma, Acta. Phys. Chem., 24 (1978) 39.





























