Embed Size (px)
Citation preview

Section-II: Experimental
Section-II: Experimental 46
Section-II
Present Work
In view of encouraging reports about technical applications of some of the
novel reactive dyes, it was thought interesting to undertake synthesis and study of the
dyeing properties of the dyes based on,
Series-1 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichlorophenyl}-6-
bromo-2-phenylquinazolin-4(3H)-one
Series-2 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-
phenylquinazolin-4(3H)-one
Series-3 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-
carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one
Series-4 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluorophenyl}-6-nitro-
2-phenylquinazolin-4(3H)-one
The work is divided in four series.
In Series-1, the hot brand reactive dyes based on the following structure were
prepared.
Where R = various m-nitro anilino cyanurated coupling components.
One mole of diazotized 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-
dichlorophenyl}-6-bromo-2-phenylquinazolin-4(3H)-one was condensing with one
mole of various m-nitro anilino cyanurated coupling components to obtain different
reactive dyes.

Section-II: Experimental
Section-II: Experimental 47
In Series-2, the hot brand reactive dyes based on the following structure were
prepared.
Where R = various p-chloro anilino cyanurated coupling components.
One mole of diazotized 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-
chloro-2-phenylquinazolin-4(3H)-one was condense with one mole of various
p-chloro anilino cyanurated coupling components to obtain different reactive dyes.
In Series-3, the hot brand reactive dyes based on the following structure were
prepared.
Where R = various p-nitro anilino cyanurated coupling components.
One mole of diazotised 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-
2-carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one was condensing with one
mole of various p-nitro anilino cyanurated coupling components to obtain different
reactive dyes.
In Series-4, the hot brand reactive dyes based on the following structure were
prepared.

Section-II: Experimental
Section-II: Experimental 48
Where R = various o-chloro-p-nitro anilino cyanurated coupling components.
One mole of diazotised 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-
difluorophenyl}-6-nitro-2-phenylquinazolin-4(3H)-one was condensing with one
mole of various o-chloro-p-nitro anilino cyanurated coupling components to obtain
different reactive dyes.
The procedure followed in connection with each reactive dye is as follows:
1. The reactive dyes are prepared under proper experimental conditions and
purified. The thin layer chromatography was employed to ascertain purity of
the dyes.
2. Each dye is characterized by IR, 1H NMR Spectral Characteristics and C, H, N
elemental analysis.
3. Absorbance maxima (λmax) of each dye in UV Spectrum were recorded.
4. Application of these dyes on silk, wool and cotton fabrics has been
investigated.
5. Colorimetric data (CIELAB values L*, a*, b*, c*, H* and K/S value) have
been determined.
6. Light fastness has been measured by the standard methods of testing
(BS: 1006-1978). The wash fastness has been measured in accordance with
IS: 765-1979. The rubbing fastness test was carried out with a crockmeter
(Atlas) in accordance with AATCC-1961.
7. Antimicrobial activity data of all the dyes were evaluated.
8. Thermogravimetric analysis (TGA) data were also evaluated.

Section-II: Experimental
Section-II: Experimental 49
Experimental
Series-1: Hot brand reactive dyes
Step-1: Synthesis of 4,4'-methylene bis(2,5-dichloro aniline)[1,2]
(1)
2,5-Dichloro aniline (16.2 g, 0.1 mol) was dissolved in water (125 ml) and
36.5% hydrochloric acid (25 ml) at 50°C. The reaction mixture was then reacted with
3% aqueous formaldehyde (35 ml) solution at 60°C with stirring for 1 hour and
neutralized with 10% sodium hydroxide solution. The white precipitates obtained
were filtered, washed with hot water, dried and recrystallized from acetic acid, yield
75%, m.p. 115-118°C. Rf = 0.75 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3395, 3315
(N-H), 2995 (C-H), 1595 (N-H bend.), 785 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 2.92
(2H, s, CH2), 6.25 (4H, s, NH2), 6.63-7.12 (4H, m, Ar-H). Anal. Calcd. for
C13H10Cl4N2 (336.04): C, 46.46%; H, 3.00%; N, 8.34%. Found: C, 46.41%; H, 2.92%;
N, 8.26%.
Step-2: Synthesis of 6-bromo-2-phenyl-4H-benzo[1,3]oxazine-4-one[3]
(2)
To a stirred solution of 5-bromo anthranilic acid (2.16 g, 0.01 mol) in pyridine
(60 ml), Benzoyl chloride (1.16 ml, 0.01 mol) was added dropwise, maintaining the
temperature near 0-5°C for 1 hour. The reaction mixture was stirred for another
2 hours at room temperature until a solid product was formed. The reaction mixture
was neutralized with saturated sodium bicarbonate solution. Yellow solid which
separated was filtered, washed with water and recrystallized from ethanol. Yield 78%,
m.p. 175-178°C. Rf = 0.65 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3066 (C-H), 1615
(C=N), 1755 (C=O), 1058 (C-O-C), 1182 (C-O), 536 (C-Br). 1H NMR (DMSO-d6) δ
ppm: 7.55-8.14 (8H, m, Ar-H). Anal. Calcd. for C14H8O2NBr (302.12): C, 55.66%;
H, 2.67%; N, 4.64%. Found: C, 55.60%; H, 2.61%; N, 4.56%.
Step-3: Synthesis of 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichlorophenyl}-6-
bromo-2-phenylquinazolin-4(3H)-one[4]
(3)
4,4'-Methylene bis(2,5-dichloro aniline) (1.68 g, 0.005 mol) and 6-bromo-2-
phenyl-4H-benzo[1,3]oxazine-4-one (1.51 g, 0.005 mol) were dissolved in pyridine
(40 ml) and heated under reflux for 6 hours under anhydrous reaction conditions and
then allowed to cool at room temperature. The reaction mixture was then treated with
ice cooled dilute hydrochloric acid and stirred. A solid separated out which was

Section-II: Experimental
Section-II: Experimental 50
filtered off and washed with water to remove any adhered pyridine. The crude
quinazolinone thus obtained was dried under vacuum and recrystallized from ethanol.
Yield 70%, m.p. 125-128°C. Rf = 0.60 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3404,
3365 (N-H) 3028 (C-H), 1596 (C=N), 1666 (C=O), 1507 (C-N), 786 (C-Cl), 550
(C-Br). 1H NMR (DMSO-d6) δ ppm: 2.65 (2H, s, CH2), 6.18 (2H, s, NH2), 7.45-8.05
(12H, m, Ar-H). Anal. Calcd. for C27H16ON3Cl4Br (620.15): C, 52.29%; H, 2.60%;
N, 6.78%. Found: C, 52.22%; H, 2.54%; N, 6.71%.
Step-4: Diazotization of 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichlorophenyl}-
6-bromo-2-phenylquinazolin-4(3H)-one (4)
Concentrated hydrochloric acid (1.88 ml, 0.015 mol) was added to a well
stirred suspension of 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichlorophenyl}-6-
bromo-2-phenylquinazolin-4(3H)-one (3.10 g, 0.005 mol) in water (25 ml) and the
mixture was heated up to 70°C and maintained at that temperature till a clear solution
obtained. After cooling the solution to 0-5°C in an ice bath, a solution of sodium
nitrite (0.35 g, 0.005 mol) in water (10 ml) was added dropwise over a period of 10
minutes with stirring. The reaction was stirred at a temperature below 5°C for an hour.
The excess of nitrous acid (tested for using starch iodide paper) was removed by
adding required amount of sulphamic acid solution (10% w/v). The clear diazonium
salt solution (4) thus obtained was used immediately in the coupling reaction.
Step-5: Cyanuration of H-acid (5)
Cyanuric chloride (1.85 gm, 0.01 mol) was stirred in acetone (25 ml) at a
temperature below 5ºC for a period of an hour. A neutral solution of H acid (3.19 g,
0.01 mol) in aqueous sodium carbonate solution (10% w/v) was then added in small
amount in about an hour. The pH was maintained neutral by simultaneous addition of
sodium carbonate solution (1% w/v). The reaction mass was stirred at 0-5°C for
further 4 hours then clear solution was obtained. The resultant solution was used for
condensation reaction with m-nitro aniline.
Step-6: Condensation with m-nitro aniline (Synthesis of m-nitro anilino
cyanurated H-acid) (6)
The temperature of ice-cooled well stirred solution of cyanurated H-acid (4.67
g, 0.01 mol) was gradually raised to 45°C for half an hour. To this cyanurated H-acid

Section-II: Experimental
Section-II: Experimental 51
the m-nitro aniline (1.38 g, 0.01 mol) was added dropwise at same temperature,
during a period of 30 minutes, maintaining the pH neutral by simultaneous addition of
sodium bicarbonate (1% w/v). After the addition was completed, stirring was
continued for further 3 hours. The m-nitro anilino cyanurated H-acid solution thus
obtained was subsequently used for further coupling reaction.
Step-7: Synthesis of reactive dyes D1
To an icecold and stirred solution of m-nitro anilino cyanurated H-acid (2.83
g, 0.005 mol), a freshly prepared diazo solution (4) (3.34 g, 0.005 mol) was added
dropwise over a period of 10-15 minutes. The pH was maintained at 7.5 to 8.5 by
simultaneous addition of sodium carbonate solution (10% w/v). During coupling the
purple solution was formed. The stirring was continued for 3-4 hours to maintain the
temperature below 5°C. The reaction mixture was heated up to 60oC and sodium
chloride (12 g) added until the coloring material was precipitated. Further stirred for
an hour, filtered and washed with a small amount of sodium chloride solution (5%
w/v). The solid was dried at 80-90oC and extracted with DMF. The dye was
precipitated by diluting the DMF-extract with excess of chloroform. A violet dye was
then filtered, washed with chloroform and dried at 60oC.
Same procedure was followed for other reactive dyes D2 to D10 were
synthesized using m-nitro anilino cyanurated coupling components such as J-acid,
N-methyl-J-acid, N-phenyl-J-acid, Bronner acid, Gamma acid, Tobias acid, Sulpho
Tobias acid, Peri acid and Koch acid.
Characterizations of all the dyes are recorded in Table-1.
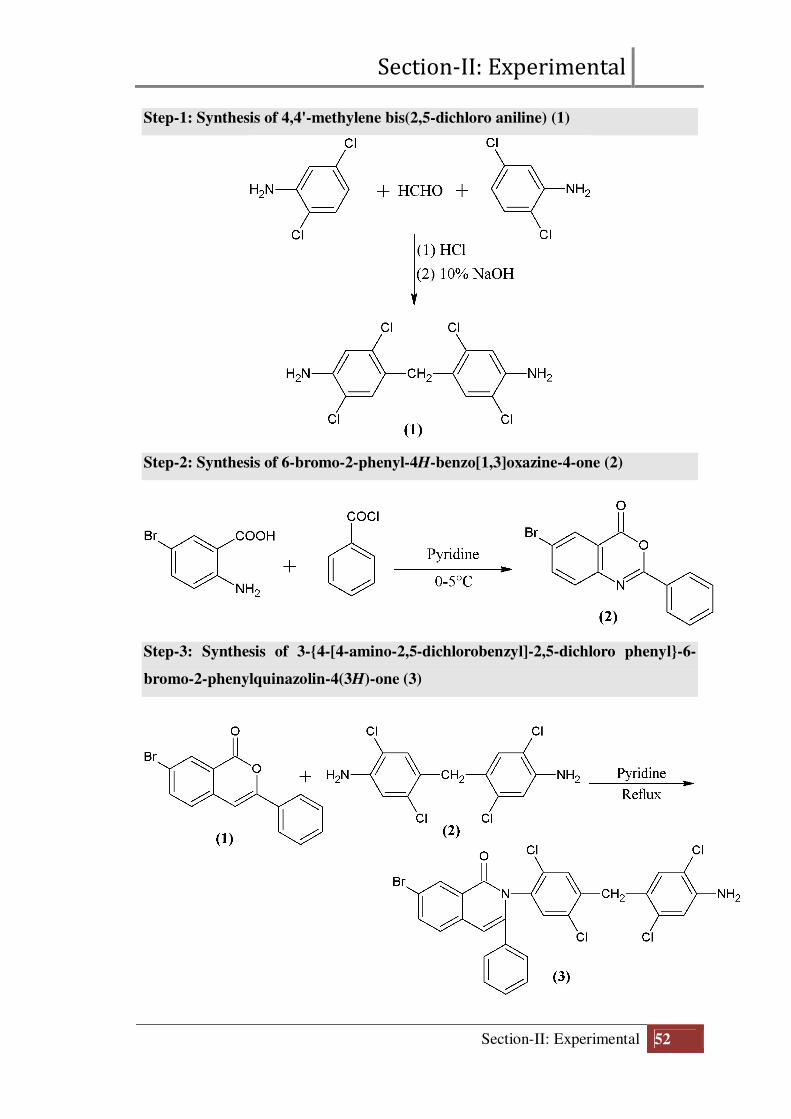
Section-II: Experimental
Section-II: Experimental 52
Step-1: Synthesis of 4,4'-methylene bis(2,5-dichloro aniline) (1)
Step-2: Synthesis of 6-bromo-2-phenyl-4H-benzo[1,3]oxazine-4-one (2)
Step-3: Synthesis of 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichloro phenyl}-6-
bromo-2-phenylquinazolin-4(3H)-one (3)

Section-II: Experimental
Section-II: Experimental 53
Step-4: Diazotization of 3-{4-[4-amino-2,5-dichlorobenzyl]-2,5-dichloro phenyl}-
6-bromo-2-phenylquinazolin-4(3H)-one (4)
Step-5: Cyanuration of H-acid (5)
Step-6: Condensation with m-nitro aniline
(Preparation of m-nitro anilino cyanurated H-acid) (6)

Section-II: Experimental
Section-II: Experimental 54
Step-7: Synthesis of reactive dyes D1

Section-II: Experimental
Section-II: Experimental 55
Chart-1: Coupling components used in the preparation of reactive dyes
(Arrow indicates the coupling position)
Final structure of reactive dyes (D1 to D10)

Section-II: Experimental
Section-II: Experimental 56

Section-II: Experimental
Section-II: Experimental 57

Section-II: Experimental
Section-II: Experimental 58

Section-II: Experimental
Section-II: Experimental 59
Series-2: Hot brand reactive dyes
Step-1: Synthesis of 4,4'-methylene bis(m-nitro aniline)[1,2]
(7)
m-Nitro aniline (13.8 g, 0.1 mol) was dissolved in water (125 ml) and 36.5%
hydrochloric acid (25 ml) at 50°C. The reaction mixture was then reacted with 3%
aqueous formaldehyde (35 ml) solution at 60°C with stirring for 1 hour and
neutralized with 10% sodium hydroxide solution. The reddish precipitates obtained
were filtered, washed with hot water, dried and recrystallized from acetic acid, yield
78%, m.p. 65-68°C. Rf = 0.74 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3430, 3385
(N-H), 3097 (C-H), 1625 (N-H bend.), 1523, 1349 (N=O). 1H NMR (DMSO-d6) δ
ppm: 2.38 (2H, s, CH2), 6.28 (4H, s, NH2), 6.95-7.42 (6H, m, Ar-H). Anal. Calcd. for
C13H12O4N4 (288.26): C, 54.17%; H, 4.20%; N, 19.44%. Found: C, 54.11%; H,
4.12%; N, 19.38%.
Step-2: Synthesis of 7-chloro-2-phenyl-4H-benzo[1,3]oxazine-4-one[3]
(8)
To a stirred solution of 4-chloro anthranilic acid (1.72 g, 0.01 mol) in pyridine
(60 ml), Benzoyl chloride (1.16 ml, 0.01 mol) was added dropwise, maintaining the
temperature near 0-5°C for 1 hour. The reaction mixture was stirred for another 2
hours at room temperature until a solid product was formed. The reaction mixture was
neutralized with saturated sodium bicarbonate solution. A white solid which separated
was filtered, washed with water and recrystallized from ethanol. Yield 70%, m.p. 192-
195°C. Rf = 0.68 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3075 (C-H), 1613 (C=N),
1756 (C=O), 1062 (C-O-C), 1177 (C-O), 779 (C-Cl). 1H NMR (DMSO-d6) δ ppm:
7.58-8.10 (8H, m, Ar-H). Anal. Calcd. for C14H8O2NCl (257.67): C, 65.26%; H,
3.13%; N, 5.44%. Found: C, 65.20%; H, 3.05%; N, 5.38%.
Step-3: Synthesis of 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-
phenylquinazolin-4(3H)-one[4]
(9)
4,4'-Methylene bis(m-nitro aniline) (1.44 g, 0.005 mol) and 7-chloro-2-phenyl-
4H-benzo[1,3]oxazine-4-one (1.29 g, 0.005 mol) were dissolved in pyridine (40 ml)
and heated under reflux for 6 hours under anhydrous reaction conditions and then
allowed to cool at room temperature. The reaction mixture was then treated with ice
cooled dilute hydrochloric acid and stirred. A solid separated out which was filtered
off and washed with water to remove any adhered pyridine. The crude quinazolinone
thus obtained was dried under vacuum and recrystallized from ethanol. Yield 72%,

Section-II: Experimental
Section-II: Experimental 60
m.p. 157-160°C. Rf = 0.55 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3425, 3380 (N-H)
3045 (C-H), 1613 (C=N), 1674 (C=O), 1494 (C-N), 1525, 1350 (N=O), 778 (C-Cl).
1H NMR (DMSO-d6) δ ppm: 2.28 (2H, s, CH2), 6.22 (2H, s, NH2), 7.52-8.10 (14H, m,
Ar-H). Anal. Calcd. for C27H18O5N5Cl (527.92): C, 61.43%; H, 3.44%; N, 13.27%.
Found: C, 61.35%; H, 3.38%; N, 13.20%.
Step-4: Diazotization of 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-
phenylquinazolin-4(3H)-one (10)
Concentrated hydrochloric acid (1.88 ml, 0.015 mol) was added to a well
stirred suspension of 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-phenyl
quinazolin-4(3H)-one (2.64 g, 0.005 mol) in water (25 ml) and the mixture was heated
up to 70°C and maintained at that temperature till a clear solution obtained. After
cooling the solution to 0-5°C in an ice bath, a solution of sodium nitrite (0.35 g, 0.005
mol) in water (10 ml) was added dropwise over a period of 10 minutes with stirring.
The reaction was stirred at a temperature below 5°C for an hour. The excess of nitrous
acid (tested for using starch iodide paper) was removed by adding required amount of
sulphamic acid solution (10% w/v). The clear diazonium salt solution (10) thus
obtained was used immediately in the coupling reaction.
Step-5: Cyanuration of H-acid (11)
Cyanuric chloride (1.85 g, 0.01 mol) was stirred in acetone (25 ml) at a
temperature below 5°C for a period of an hour. A neutral solution of H acid (3.19 g,
0.01 mol) in aqueous sodium carbonate solution (10% w/v) was then added in small
amount in about an hour. The pH was maintained neutral by simultaneous addition of
sodium carbonate solution (1% w/v). The reaction mass was stirred at 0-5°C for
further 4 hours then clear solution was obtained. The resultant solution was used for
condensation reaction with p-chloro aniline.
Step-6: Condensation with p-chloro aniline (Synthesis of p-chloro anilino
cyanurated H-acid) (12)
The temperature of ice-cooled well stirred solution of cyanurated H-acid (4.67
g, 0.01 mol) was gradually raised to 45°C for half an hour. To this cyanurated H-acid
the p-chloro aniline (1.28 g, 0.01 mol) was added dropwise at same temperature,
during a period of 30 minutes, maintaining the pH neutral by simultaneous addition of

Section-II: Experimental
Section-II: Experimental 61
sodium bicarbonate (1% w/v). After the addition was completed, stirring was
continued for further 3 hours. The p-chloro anilino cyanurated H-acid solution thus
obtained was subsequently used for further coupling reaction.
Step-7: Synthesis of reactive dyes D11
To an ice cold and stirred solution of p-chloro anilino cyanurated H-acid (2.79
g, 0.005 mol), a freshly prepared diazo solution (10) (2.88 g, 0.005 mol) was added
dropwise over a period of 10-15 minutes. The pH was maintained at 7.5 to 8.5 by
simultaneous addition of sodium carbonate solution (10% w/v). During coupling the
purple solution was formed. The stirring was continued for 3-4 hours to maintain the
temperature below 5°C. The reaction mixture was heated up to 60oC and sodium
chloride (12 g) added until the coloring material was precipitated. Further stirred for
an hour, filtered and washed with a small amount of sodium chloride solution (5%
w/v). The solid was dried at 80-90oC and extracted with DMF. The dye was
precipitated by diluting the DMF-extract with excess of chloroform. A violet dye was
then filtered, washed with chloroform and dried at 60oC.
Same procedure was followed for other reactive dyes D12 to D20 were
synthesized using p-chloro anilino cyanurated coupling components such as J-acid,
N-methyl-J-acid, N-phenyl-J-acid, Bronner acid, Gamma acid, Tobias acid, Sulpho
Tobias acid, Peri acid and Koch acid.
Characterizations of all the dyes are recorded in Table-2.

Section-II: Experimental
Section-II: Experimental 62
Step-1: Synthesis of 4,4'-methylene bis(m-nitro aniline) (7)
Step-2: Synthesis of 7-chloro-2-phenyl-4H-benzo[1,3]oxazine-4-one (8)
Step-3: Synthesis of 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-
phenylquinazolin-4(3H)-one (9)

Section-II: Experimental
Section-II: Experimental 63
Step-4: Diazotization of 3-{4-[4-amino-2-nitrobenzyl]-3-nitrophenyl}-7-chloro-2-
phenylquinazolin-4(3H)-one (10)
Step-5: Cyanuration of H-acid (11)
Step-6: Condensation with p-chloro aniline
(Preparation of p-chloro anilino cyanurated H-acid) (12)

Section-II: Experimental
Section-II: Experimental 64
Step-7: Synthesis of reactive dyes D11
Final structure of reactive dyes (D11 to D20)
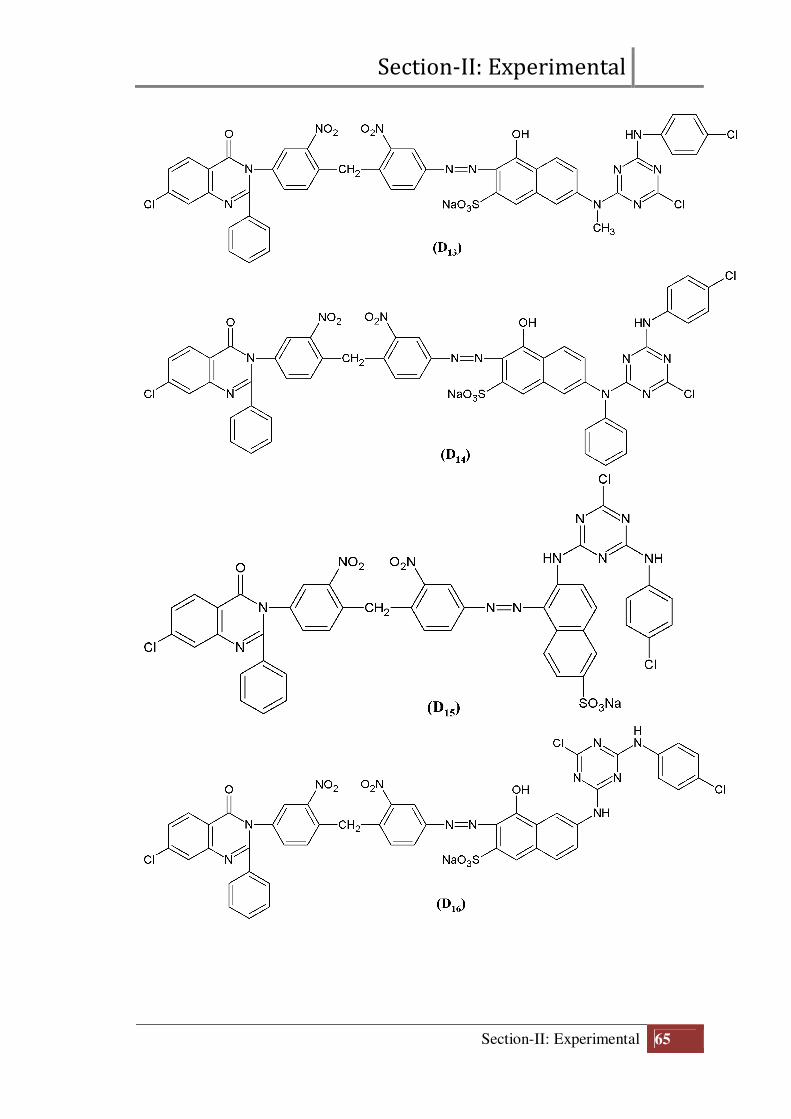
Section-II: Experimental
Section-II: Experimental 65

Section-II: Experimental
Section-II: Experimental 66

Section-II: Experimental
Section-II: Experimental 67

Section-II: Experimental
Section-II: Experimental 68
Series-3: Hot brand reactive dyes
Step-1: Synthesis of 5,5'-methylene bis(2-amino-4-chlorobenzoic acid)[1,2]
(13)
2-Amino-4-chloro benzoic acid (17.2 g, 0.1 mol) was dissolved in water
(125 ml) and 36.5% hydrochloric acid (25 ml) at 50°C. The reaction mixture was then
reacted with 3% aqueous formaldehyde (35 ml) solution at 60°C with stirring for
1 hour and neutralized with 10% sodium hydroxide solution. The reddish precipitates
obtained were filtered, washed with hot water, dried and recrystallized from acetic
acid, yield 72%, m.p. 215-218°C. Rf = 0.76 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
:
3478, 3362 (N-H), 3006 (C-H), 1754 (C=O), 1614 (N-H bend.), 777 (C-Cl). 1H NMR
(DMSO-d6) δ ppm: 2.65 (2H, s, CH2), 6.28 (4H, s, NH2), 11.06 (2H, s, COOH),
6.95-7.32 (4H, m, Ar-H). Anal. Calcd. for C15H12O4N2Cl2 (355.17): C, 50.72%;
H, 3.41%; N, 7.89%. Found: C, 50.65%; H, 3.35%; N, 7.80%.
Step-2: Synthesis of 6-Iodo-2-phenyl-4H-benzo[1,3]oxazine-4-one[3]
(14)
To a stirred solution of 5-Iodo anthranilic acid (2.63 g, 0.01 mol) in pyridine
(60 ml), Benzoyl chloride (1.16 ml, 0.01 mol) was added dropwise, maintaining the
temperature near 0-5°C for 1 hour. The reaction mixture was stirred for another
2 hours at room temperature until a solid product was formed. The reaction mixture
was neutralized with saturated sodium bicarbonate solution. A yellow solid which
separated was filtered, washed with water and recrystallized from ethanol. Yield 75%,
m.p. 175-178°C. Rf = 0.65 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3056 (C-H), 1608
(C=N), 1757 (C=O), 1062 (C-O-C), 1182 (C-O), 539 (C-I). 1H NMR (DMSO-d6)
δ ppm: 7.55-7.95 (8H, m, Ar-H). Anal. Calcd. for C14H8O2NI (349.12): C, 48.16%;
H, 2.31%; N, 4.01%. Found: C, 48.09%; H, 2.24%; N, 3.92%.
Step-3: Synthesis of 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-
carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one[4]
(15)
5,5'-Methylene bis(2-amino-4-chlorobenzoic acid) (1.78 g, 0.005 mol) and
6-Iodo-2-phenyl-4H-benzo[1,3]oxazine-4-one (1.75 g, 0.005 mol) were dissolved in
pyridine (40 ml) and heated under reflux for 6 hours under anhydrous reaction
conditions and then allowed to cool at room temperature. The reaction mixture was
then treated with ice cooled dilute hydrochloric acid and stirred. A solid separated out
which was filtered off and washed with water to remove any adhered pyridine. The
crude quinazolinone thus obtained was dried under vacuum and recrystallized from

Section-II: Experimental
Section-II: Experimental 69
ethanol. Yield 76%, m.p. 102-105°C. Rf = 0.56 (PhMe:EtOAc, 3:1 v/v). IR (KBr)
cm-1
: 3425, 3374 (N-H), 3067 (C-H), 1758 (C=O of COOH), 1668 (C=O of
quinazoline), 1578 (C=N), 1511 (C-N), 778 (C-Cl), 531 (C-I). 1H NMR (DMSO-d6) δ
ppm: 2.72 (2H, s, CH2), 6.32 (2H, s, NH2), 11.12 (2H, s, COOH), 7.52-7.95 (12H, m,
Ar-H). Anal. Calcd. for C29H18O5N3Cl2I (686.28): C, 50.75%; H, 2.64%; N, 6.12%.
Found: C, 50.68%; H, 2.56%; N, 6.05%.
Step-4: Diazotization of 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-
carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one (16)
Concentrated hydrochloric acid (1.88 ml, 0.015 mol) was added to a well
stirred suspension of 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-carboxy
phenyl}-6-iodo-2-phenylquinazolin-4(3H)-one (3.43 g, 0.005 mol) in water (25 ml)
and the mixture was heated up to 70°C and maintained at that temperature till a clear
solution obtained. After cooling the solution to 0-5°C in an ice bath, a solution of
sodium nitrite (0.35 g, 0.005 mol) in water (10 ml) was added dropwise over a period
of 10 minutes with stirring. The reaction was stirred at a temperature below 5°C for an
hour. The excess of nitrous acid (tested for using starch iodide paper) was removed by
adding required amount of sulphamic acid solution (10% w/v). The clear diazonium
salt solution (16) thus obtained was used immediately in the coupling reaction.
Step-5: Cyanuration of H-acid (17)
Cyanuric chloride (1.85 g, 0.01 mol) was stirred in acetone (25 ml) at a
temperature below 5°C for a period of an hour. A neutral solution of H acid (3.19 g,
0.01 mol) in aqueous sodium carbonate solution (10% w/v) was then added in small
amount in about an hour. The pH was maintained neutral by simultaneous addition of
sodium carbonate solution (1% w/v). The reaction mass was stirred at 0-5°C for
further 4 hours then clear solution was obtained. The resultant solution was used for
condensation reaction with p-nitro aniline.
Step-6: Condensation with p-nitro aniline
(Synthesis of p-niro anilino cyanurated H-acid) (18)
The temperature of ice-cooled well stirred solution of cyanurated H-acid (4.67
g, 0.01 mol) was gradually raised to 45°C for half an hour. To this cyanurated H-acid
the p-nitro aniline (1.38 g, 0.01 mol) was added dropwise at same temperature, during

Section-II: Experimental
Section-II: Experimental 70
a period of 30 minutes, maintaining the pH neutral by simultaneous addition of
sodium bicarbonate (1% w/v). After the addition was completed, stirring was
continued for further 3 hours. The p-nitro anilino cyanurated H-acid solution thus
obtained was subsequently used for further coupling reaction.
Step-7: Synthesis of reactive dyes D21
To an icecold and stirred solution of p-nitro anilino cyanurated H-acid (2.85 g,
0.005 mol), a freshly prepared diazo solution (16) (3.67 g, 0.005 mol) was added
dropwise over a period of 10-15 minutes. The pH was maintained at 7.5 to 8.5 by
simultaneous addition of sodium carbonate solution (10% w/v). During coupling the
purple solution was formed. The stirring was continued for 3-4 hours to maintain the
temperature below 5ºC. The reaction mixture was heated up to 60oC and sodium
chloride (12 g) added until the coloring material was precipitated. Further stirred for
an hour, filtered and washed with a small amount of sodium chloride solution (5%
w/v). The solid was dried at 80-90oC and extracted with DMF. The dye was
precipitated by diluting the DMF-extract with excess of chloroform. A violet dye was
then filtered, washed with chloroform and dried at 60oC.
Same procedure was followed for other reactive dyes D22 to D30 were
synthesized using p-nitro anilino cyanurated coupling components such as J-acid,
N-methyl-J-acid, N-phenyl-J-acid, Bronner acid, Gamma acid, Tobias acid, Sulpho
Tobias acid, Peri acid and Koch acid.
Characterizations of all the dyes are recorded in Table-3.

Section-II: Experimental
Section-II: Experimental 71
Step-1: Synthesis of 5,5'-methylene bis(2-amino-4-chlorobenzoic acid) (13)
Step-2: Synthesis of 6-Iodo-2-phenyl-4H-benzo[1,3]oxazine-4-one (14)
Step-3: Synthesis of 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-
carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one (15)

Section-II: Experimental
Section-II: Experimental 72
Step-4: Diazotization of 3-{4-[2-chloro-4-amino-5-carboxybenzyl]-5-chloro-2-
carboxyphenyl}-6-iodo-2-phenylquinazolin-4(3H)-one (16)
Step-5: Cyanuration of H-acid (17)
Step-6: Condensation with p-nitro aniline
(Synthesis of p-niro anilino cyanurated H-acid) (18)

Section-II: Experimental
Section-II: Experimental 73
Step-7: Synthesis of reactive dyes D21
Final structure of reactive dyes (D21 to D30)
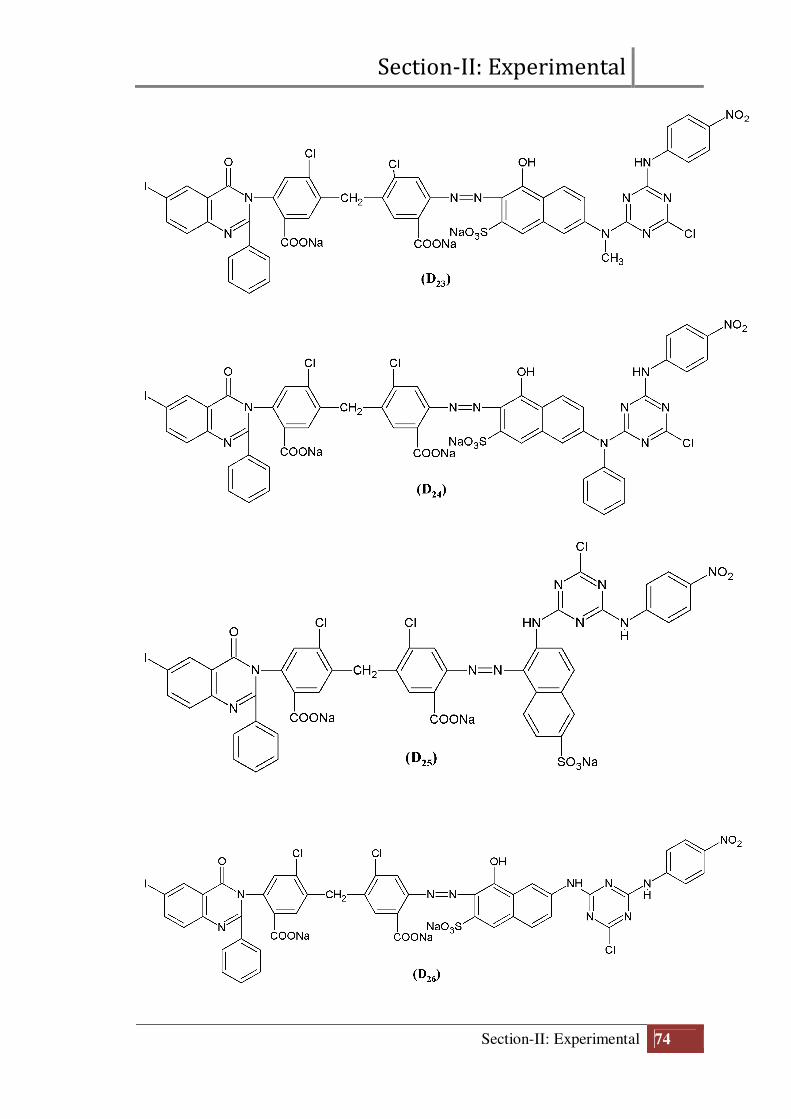
Section-II: Experimental
Section-II: Experimental 74

Section-II: Experimental
Section-II: Experimental 75

Section-II: Experimental
Section-II: Experimental 76

Section-II: Experimental
Section-II: Experimental 77
Series-4: Hot brand reactive dyes
Step-1: Synthesis of 4,4'-methylene bis(3,5-difluoro aniline)[1,2]
(19)
3,5-Difluoro aniline (12.9 g, 0.1 mol) was dissolved in water (125 ml) and
36.5% hydrochloric acid (25 ml) at 50°C. The reaction mixture was then reacted with
3% aqueous formaldehyde (35 ml) solution at 60°C with stirring for 1 hour and
neutralized with 10% sodium hydroxide solution. The orange precipitates obtained
were filtered, washed with hot water, dried and recrystallized from acetic acid. Yield
75%, m.p. 140-142°C. Rf = 0.73 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3435, 3355
(N-H), 2956 (C-H), 1642 (N-H bend.), 1051 (C-F). 1H NMR (DMSO-d6) δ ppm: 2.72
(2H, s, CH2), 6.32 (4H, s, NH2), 6.22-6.28 (4H, m, Ar-H). Anal. Calcd. for
C13H10N2F4 (270.23): C, 57.78%; H, 3.73%; N, 10.37%. Found: C, 57.70%; H,
3.64%; N, 10.30%.
Step-2: Synthesis of 6-nitro-2-phenyl-4H-benzo[1,3]oxazine-4-one[3]
(20)
To a stirred solution of 5-nitro anthranilic acid (1.82 g, 0.01 mol) in pyridine
(60 ml), Benzoyl chloride (1.16 ml, 0.01 mol) was added dropwise, maintaining the
temperature near 0-5°C for 1 hour. The reaction mixture was stirred for another
2 hours at room temperature until a solid product was formed. The reaction mixture
was neutralized with saturated sodium bicarbonate solution. A yellow solid which
separated was filtered, washed with water and recrystallized from ethanol. Yield 78%,
m.p. 130-132°C. Rf = 0.66 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3087 (C-H), 1611
(C=N), 1761 (C=O), 1060 (C-O-C), 1178 (C-O), 1532, 1343 (N=O). 1H NMR
(DMSO-d6) δ ppm: 7.50-7.98 (8H, m, Ar-H). Anal. Calcd. for C14H8O4N2 (268.22):
C, 62.69%; H, 3.01%; N, 10.44%. Found: C, 62.58%; H, 2.94%; N, 10.35%.
Step-3: Synthesis of 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluorophenyl} -6-
nitro-2-phenylquinazolin-4(3H)-one[4]
(21)
4,4'-Methylene bis(3,5-difluoro aniline) (1.35 g, 0.005 mol) and 6-nitro-2-
phenyl-4H-benzo[1,3]oxazine-4-one (1.34 g, 0.005 mol) were dissolved in pyridine
(40 ml) and heated under reflux for 6 hours under anhydrous reaction conditions and
then allowed to cool at room temperature. The reaction mixture was then treated with
ice cooled dilute hydrochloric acid and stirred. A solid separated out which was
filtered off and washed with water to remove any adhered pyridine. The crude

Section-II: Experimental
Section-II: Experimental 78
quinazolinone thus obtained was dried under vacuum and recrystallized from ethanol.
Yield 70%, m.p. 112-115°C. Rf = 0.58 (PhMe:EtOAc, 3:1 v/v). IR (KBr) cm-1
: 3434,
3365 (N-H) 3095 (C-H), 1618 (C=N), 1687 (C=O), 1495 (C-N), 1585 (asym.), 1343
(sym.) (N=O), 1052 (C-F). 1H NMR (DMSO-d6) δ ppm: 2.78 (2H, s, CH2), 6.28 (2H,
s, NH2), 7.40-7.96 (12H, m, Ar-H). Anal. Calcd. for C27H16O3N4F4 (520.43): C,
62.31%; H, 3.10%; N, 10.77%. Found: C, 62.24%; H, 3.02%; N, 10.70%.
Step-4: Diazotization of 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluorophenyl}-6-
nitro-2-phenylquinazolin-4(3H)-one (22)
Concentrated hydrochloric acid (1.88 ml, 0.015 mol) was added to a well
stirred suspension of 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluorophenyl}-6-nitro-
2-phenylquinazolin-4(3H)-one (2.60 g, 0.005 mol) in water (25 ml) and the mixture
was heated up to 70°C and maintained at that temperature till a clear solution
obtained. After cooling the solution to 0-5°C in an ice bath, a solution of sodium
nitrite (0.35 g, 0.005 mol) in water (10 ml) was added dropwise over a period of 10
minutes with stirring. The reaction was stirred at a temperature below 5°C for an hour.
The excess of nitrous acid (tested for using starch iodide paper) was removed by
adding required amount of sulphamic acid solution (10% w/v). The clear diazonium
salt solution (22) thus obtained was used immediately in the coupling reaction.
Step-5: Cyanuration of H-acid (23)
Cyanuric chloride (1.85 g, 0.01 mol) was stirred in acetone (25 ml) at a
temperature below 5°C for a period of an hour. A neutral solution of H acid (3.19 g,
0.01 mol) in aqueous sodium carbonate solution (10% w/v) was then added in small
amount in about an hour. The pH was maintained neutral by simultaneous addition of
sodium carbonate solution (1% w/v). The reaction mass was stirred at 0-5°C for
further 4 hours then clear solution was obtained. The resultant solution was used for
condensation reaction with o-chloro-p-nitro aniline.
Step-6: Condensation with o-chloro-p-nitro aniline
(Synthesis of o-chloro-p-niro anilino cyanurated H-acid) (24)
The temperature of ice-cooled well stirred solution of cyanurated H-acid (4.67
g, 0.01 mol) was gradually raised to 45°C for half an hour. To this cyanurated H-acid
the o-chloro-p-nitro aniline (1.73 g, 0.01 mol) was added dropwise at same

Section-II: Experimental
Section-II: Experimental 79
temperature, during a period of 30 minutes, maintaining the pH neutral by
simultaneous addition of sodium bicarbonate (1% w/v). After the addition was
completed, stirring was continued for further 3 hours. The o-chloro-p-nitro anilino
cyanurated H-acid solution thus obtained was subsequently used for further coupling
reaction.
Step-7: Synthesis of reactive dyes D31
To an icecold and stirred solution of o-chloro-p-nitro anilino cyanurated
H-acid (3.02 g, 0.005 mol), a freshly prepared diazo solution (22) (2.84 g, 0.005 mol)
was added dropwise over a period of 10-15 minutes. The pH was maintained at 7.5 to
8.5 by simultaneous addition of sodium carbonate solution (10% w/v). During
coupling the purple solution was formed. The stirring was continued for 3-4 hours to
maintain the temperature below 5°C. The reaction mixture was heated up to 60oC and
sodium chloride (12 g) added until the coloring material was precipitated. Further
stirred for an hour, filtered and washed with a small amount of sodium chloride
solution (5% w/v). The solid was dried at 80-90oC and extracted with DMF. The dye
was precipitated by diluting the DMF-extract with excess of chloroform. A violet dye
was then filtered, washed with chloroform and dried at 60oC.
Same procedure was followed for other reactive dyes D32 to D40 were
synthesized using o-chloro-p-nitro anilino cyanurated coupling components such as
J-acid, N-methyl-J-acid, N-phenyl-J-acid, Bronner acid, Gamma acid, Tobias acid,
Sulpho Tobias acid, Peri acid and Koch acid.
Characterizations of all the dyes are recorded in Table-4.

Section-II: Experimental
Section-II: Experimental 80
Step-1: Synthesis of 4,4'-methylene bis(3,5-difluoro aniline) (19)
Step-2: Synthesis of 6-nitro-2-phenyl-4H-benzo[1,3]oxazine-4-one (20)
Step-3: Synthesis of 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluorophenyl} -6-
nitro-2-phenylquinazolin-4(3H)-one (21)

Section-II: Experimental
Section-II: Experimental 81
Step-4: Diazotization of 3-{4-[4-amino-2,6-difluorobenzyl]-3,5-difluoro phenyl}-
6-nitro-2-phenylquinazolin-4(3H)-one (22)
Step-5: Cyanuration of H-acid (23)
Step-6: Condensation with o-chloro-p-nitro aniline
(Synthesis of o-chloro-p-niro anilino cyanurated H-acid) (24)

Section-II: Experimental
Section-II: Experimental 82
Step-7: Synthesis of reactive dyes D31
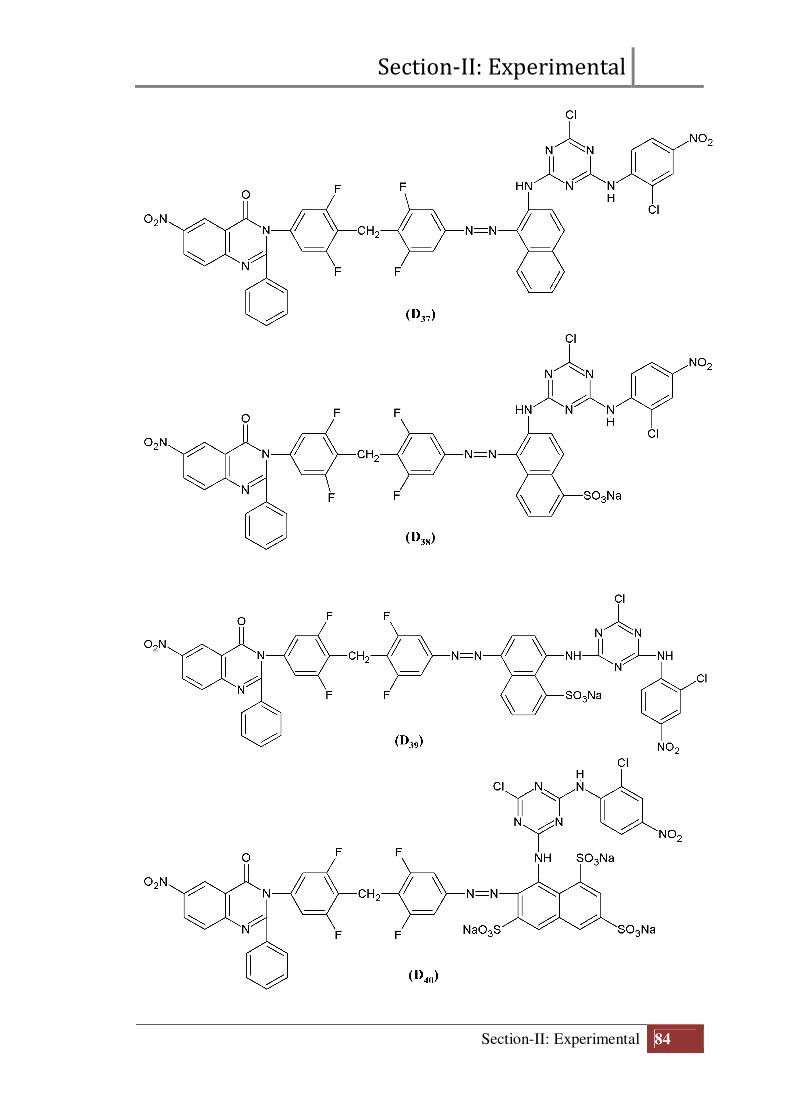
Final structure of reactive dyes (D31 to D40)
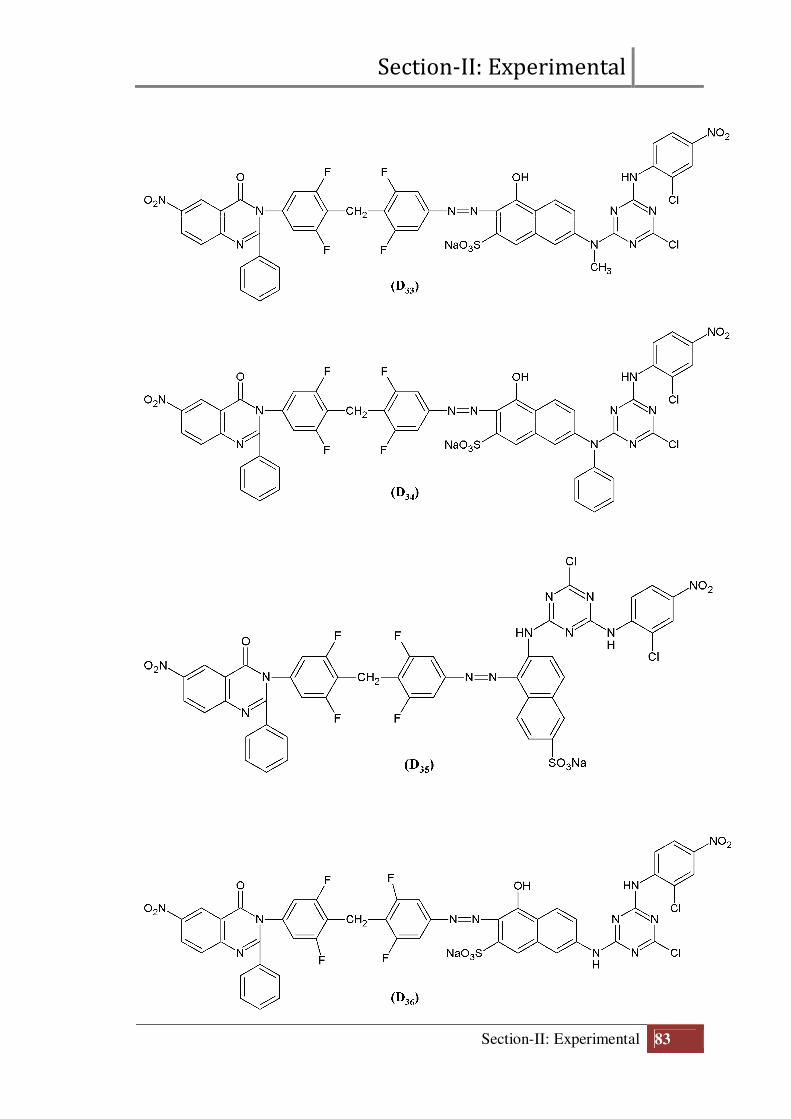
Section-II: Experimental
Section-II: Experimental 83
Section-II: Experimental
Section-II: Experimental 84

Section-II: Experimental
Section-II: Experimental 85

Section-II: Experimental
Section-II: Experimental 86
References
1. D. R. Patel, J. A. Patel, K. C. Patel, Proc. Nat. Acad. Sci. India Sect. A, 80-II
(2010) 109.
2. D. R. Patel, A. L. Patel, K. C. Patel, L. A. Patel, Colorage, 57(4) (2010) 72.
3. X. Gao, X. Cai, K. Yan, B. Song, L. Gao, Z. Chen, Molecules, 12 (2007)
2621.
4. V. K. Pandey, J. Kumar, S. K. Saxena, Mukesh, M. N. Joshi, S. K. Bajpai, J.
Indian Chem. Soc., 84 (2007) 593.





























