Embed Size (px)
Citation preview

Research Collection
Master Thesis
Pharmakologische Eigenschaften von Neurotensin-DerivatenAbhängigkeit von den Markierungsbedingungen
Author(s): Feurer, Fabienne
Publication Date: 2000
Permanent Link: https://doi.org/10.3929/ethz-a-004261168
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Departement Angewandte Biowissenschaften Institut für Pharmazeutische Wissenschaften
Pharmakologische Eigenschaften von Neurotensin-Derivaten: Abhängigkeit von
den Markierungsbedingungen
Diplomarbeit
von
Fabienne Feurer Bürgerin von Alt St. Johann/SG
Referent: Prof. Dr. P.A. Schubiger
Korreferent: Dr. P. Bläuenstein
Zentrum für Radiopharmazie, PSI Villigen/AG 06. März 2000 bis 28. Juli 2000

Mein erster Dank gilt Prof. Dr. P.A. Schubiger für die Ermöglichung der Diplomarbeit am Zentrum für Radiopharmazie, PSI Villigen/AG. Er hat mit freundlichem Interesse meine Diplomarbeit begleitet. Für seine äusserst kompetente fachliche Betreuung und wertvolle Unterstützung mit Rat und Tat möchte ich Dr. Peter Bläuenstein meinen besten Dank aussprechen. Dr. Elisa Garcia danke ich ebenfalls herzlich für die zahlreichen nützlichen Tipps zur Methodik. Für die Unterstützung bei den Markierungen einen grossen Dank an Alain Blanc und an René Bugmann für seine Hilfe bei den Zellkulturen. Frick, den 28. Juli 2000

Diplomarbeit Fabienne Feurer Inhaltsverzeichnis
INHALTSVERZEICHNIS 1. Einleitung .....................................................................................2 2. Aufgabenstellung ........................................................................6 2.1. Analyse der Bindung an lebende Zellen nach Lindmo (Bindungsassay)........ 6 2.2. Untersuchung der Markierungsbedingungen mit NT-VIII und NT-XI .............. 6 3. Zusammenfassung......................................................................7 4. Material und Methoden ...............................................................8 4.1. Apparaturen und Geräte ................................................................................ 8 4.2. Methoden ....................................................................................................... 9 5. Resultate ....................................................................................11 5.1. Binding Assay nach Lindmo......................................................................... 11 5.2. Einfrieren von Zellen für den Binding Assay................................................. 15 5.3. Markierungsbedingungen............................................................................. 16 5.4. Produktepeaks bei NT-VIII und NT-XI.......................................................... 18 5.5. Tabellen zur Resultatezusammenfassung ................................................... 20 5.6. HPLC-Optimierung....................................................................................... 21 6. Diskussion .................................................................................22 6.1. Binding Assay nach Lindmo......................................................................... 22 6.2. Einfrieren von Zellen für den Binding Assay................................................. 25 6.3. Markierungsbedingungen............................................................................. 25 6.4. Zwei Produktepeaks bei NT-VIII und NT-XI ................................................. 26 6.5. Schlussfolgerungen...................................................................................... 26 7. Experimenteller Teil ..................................................................27 7.1. Chemikalien ................................................................................................. 27 7.2. Pufferlösungen ............................................................................................. 27 7.3. HPLC-Gradienten......................................................................................... 28 7.4. Markierung von Neurotensin-Derivaten........................................................ 29 7.5. Reinigung und Sammeln von Fraktionen ..................................................... 29 7.6. Kultivierung von HT-29 Zellen ...................................................................... 30 7.7. Lindmo-Anleitung ......................................................................................... 30 7.8. Messung im Gamma-Counter ...................................................................... 31 7.9. Auswertung der Resultate zum Lindmo ....................................................... 31 7.10. Problembehandlung..................................................................................... 32 8. Literatur......................................................................................33

Diplomarbeit Fabienne Feurer Einleitung
2
1. Einleitung Peptide als Radiopharmazeutika in der Nuklearmedizin Viele Peptide werden sowohl in neuronalem und lymphatischem Gewebe als auch im endokrinen System (das sind Organe, die als steuernde bzw. regulative Zentren funktionieren wie z.B. die Hypophyse, die Schilddrüse, die Nebennieren bzw. Pankreas, aber auch Ovar und Testis) synthetisiert. Ein wesentlicher Unterschied zwischen Peptiden und Proteinen ist ihre Grösse. Peptide bestehen aus bis zu 50 Aminosäuren und einer Molekularen Masse von <10'000 Da. Ebenso wichtig sind dabei die "ex-vivo"-Produktionsmöglichkeiten. Kleine Peptide können leicht chemisch synthetisiert werden, wogegen Proteine oft aus einer biologischen Quelle (z.B. durch DNA-rekombinante Techniken) gewonnen werden müssen. Relativ kurze Aminosäureseqenzen, also Peptide, sind zu hoch selektiver Bindung an Rezeptorbindungsstellen fähig. [1] Biologie Mehrere menschliche Tumorgewebe exprimieren im Überfluss bestimmte Typen von Rezeptoren auf ihrer Zelloberfläche, z.B. Neuropeptid-Rezeptoren, welche als Targets für das in vivo Imaging und Therapie verwendet werden können. Schon lange sind Antikörper gegen verschiedene Rezeptoren, Antigene genannt, auf Tumorzellen bekannt. Für die in vivo Anwendung typisch ist die langsame Anreicherung im Tumorgewebe und langsame Ausscheidung nicht gebundener Antikörper. Demgegenüber bietet der Gebrauch von kleinen Peptiden wie Neurotensin mehrere Vorteile: Einfache Synthese und Modifikation, hohe Affinität zu ihren Rezeptoren, schnelle Plasma-Clearance und oft hohe Konzentration im Zielorgan. Doch der Abbau der Peptide im Plasma durch endogene Peptidasen und Proteasen geschieht ebenfalls schnell und deshalb drängte es sich auf, neue stabilisierte Derivate zu entwickeln, die immer noch eine hohe Bindungsaffinität und eine verbesserte Bioverteilung (v.a. niedrige Leber-und Nierenakkumulation und hohe Tumoranreicherung) besitzen. [1] [2] Neurotensin als Radiopharmazeutikum pGlu1-Leu2-Tyr3-Gly4-Asn5-Lys6-Pro7-Arg8-Arg9-Pro10-Tyr11-Ile12-Leu13-OH [2] Neurotensin (NT) gehört zu der Gruppe der Neuropeptide und bindet an Rezeptoren (NTS1-Rezeptoren [3]) von Zellen verschiedener Gewebe, darunter auch von Tumoren, z.B. kleinzelliges Lungenkarzinom, Pankreaskarzinom. Der am Zentrum für Radiopharmazie, PSI Villigen/AG entwickelte Komplex 99mTc(CO)3(H2O)3
+ reagiert mit verschiedenen Liganden zu stabilen Verbindungen. Das Histidinacetat hat sich als sehr geeignet erwiesen, weil es die drei Wassermoleküle ersetzen kann und eine Carboxylatgruppe frei hat für die Bindung an das Aminoende des Peptids.
Diplomarbeit Fabienne Feurer Einleitung
3
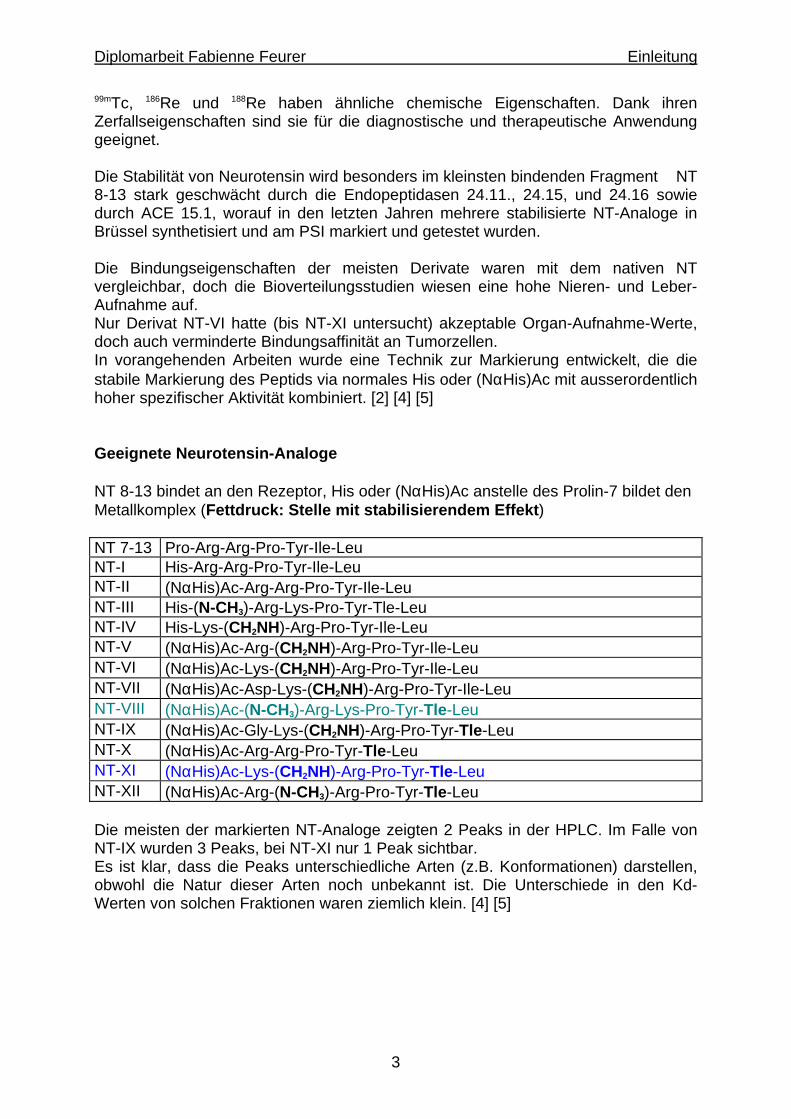
99mTc, 186Re und 188Re haben ähnliche chemische Eigenschaften. Dank ihren Zerfallseigenschaften sind sie für die diagnostische und therapeutische Anwendung geeignet. Die Stabilität von Neurotensin wird besonders im kleinsten bindenden Fragment NT 8-13 stark geschwächt durch die Endopeptidasen 24.11., 24.15, und 24.16 sowie durch ACE 15.1, worauf in den letzten Jahren mehrere stabilisierte NT-Analoge in Brüssel synthetisiert und am PSI markiert und getestet wurden. Die Bindungseigenschaften der meisten Derivate waren mit dem nativen NT vergleichbar, doch die Bioverteilungsstudien wiesen eine hohe Nieren- und Leber-Aufnahme auf. Nur Derivat NT-VI hatte (bis NT-XI untersucht) akzeptable Organ-Aufnahme-Werte, doch auch verminderte Bindungsaffinität an Tumorzellen. In vorangehenden Arbeiten wurde eine Technik zur Markierung entwickelt, die die stabile Markierung des Peptids via normales His oder (NαHis)Ac mit ausserordentlich hoher spezifischer Aktivität kombiniert. [2] [4] [5] Geeignete Neurotensin-Analoge NT 8-13 bindet an den Rezeptor, His oder (NαHis)Ac anstelle des Prolin-7 bildet den Metallkomplex (Fettdruck: Stelle mit stabilisierendem Effekt) NT 7-13 Pro-Arg-Arg-Pro-Tyr-Ile-Leu NT-I His-Arg-Arg-Pro-Tyr-Ile-Leu NT-II (NαHis)Ac-Arg-Arg-Pro-Tyr-Ile-Leu NT-III His-(N-CH3)-Arg-Lys-Pro-Tyr-Tle-Leu NT-IV His-Lys-(CH2NH)-Arg-Pro-Tyr-Ile-Leu NT-V (NαHis)Ac-Arg-(CH2NH)-Arg-Pro-Tyr-Ile-Leu NT-VI (NαHis)Ac-Lys-(CH2NH)-Arg-Pro-Tyr-Ile-Leu NT-VII (NαHis)Ac-Asp-Lys-(CH2NH)-Arg-Pro-Tyr-Ile-Leu NT-VIII (NαHis)Ac-(N-CH3)-Arg-Lys-Pro-Tyr-Tle-Leu NT-IX (NαHis)Ac-Gly-Lys-(CH2NH)-Arg-Pro-Tyr-Tle-Leu NT-X (NαHis)Ac-Arg-Arg-Pro-Tyr-Tle-Leu NT-XI (NαHis)Ac-Lys-(CH2NH)-Arg-Pro-Tyr-Tle-Leu NT-XII (NαHis)Ac-Arg-(N-CH3)-Arg-Pro-Tyr-Tle-Leu Die meisten der markierten NT-Analoge zeigten 2 Peaks in der HPLC. Im Falle von NT-IX wurden 3 Peaks, bei NT-XI nur 1 Peak sichtbar. Es ist klar, dass die Peaks unterschiedliche Arten (z.B. Konformationen) darstellen, obwohl die Natur dieser Arten noch unbekannt ist. Die Unterschiede in den Kd-Werten von solchen Fraktionen waren ziemlich klein. [4] [5]

Diplomarbeit Fabienne Feurer Einleitung
4
Metabolische Stabilität Alle Derivate wurden stabilisiert an Position 12 durch Ersatz von Ile durch Tle. NT-VIII, -IX und -XI wurden ausserdem noch an Position 8-9 entweder durch Methylierung oder durch Reduktion eines Amids zu einem Amin stabilisiert. Arg-8 (in NT-IX und NT-XI) oder Arg-9 (in NT-VIII) wurden ersetzt durch Lys, um die Peptidsynthese zu vereinfachen. Schliesslich wurde in NT-XI das Amid zwischen Position 8 und 9 ebenso zu einem Amin reduziert. Diese Änderungen in der Struktur führten zu einer Verbesserung der Stabilität in allen Fällen. Die Plasma-Halbwertszeit war durch die Stabilisierung angestiegen. NT-X, welches nur an Position 12 stabilisiert worden war, zeigte eine Plasma-Halbwertszeit von 1.5 h, wogegen die anderen drei Derivate (bei Pos. 8-9 und 12 stabilisiert) noch 24 h nach Inkubation intakt waren. [4] [5] Binding Assay und Internalisation Assay Beide Assays wurden in vitro mit der HT-29 Zelllinie (Human adenocarcinoma tumorcells), welche den NTS1-Rezeptor exprimiert, durchgeführt. Alle der 99mTc-markierten Peptide zeigten eine hohe Affinität an diese Rezeptoren mit Kd-Werten im nanomolaren Bereich (zw. 0.2 und 3 nM für alle Peptide) ähnlich dem Wert für iodiniertes Neurotensin (1.6 nM). Nach der Interaktion von Neurotensin mit seinem Rezeptor wird der Peptid-Rezeptor-Komplex rasch internalisiert. Die Internalisierung steigt mit der Zeit bei allen Derivaten in ähnlicher Weise an. In allen Fällen wurde ca. 80% innerhalb der ersten 30 Minuten internalisiert und verblieb in der Zelle für mindestens 2 h. [4] [5]
Diplomarbeit Fabienne Feurer Einleitung
5
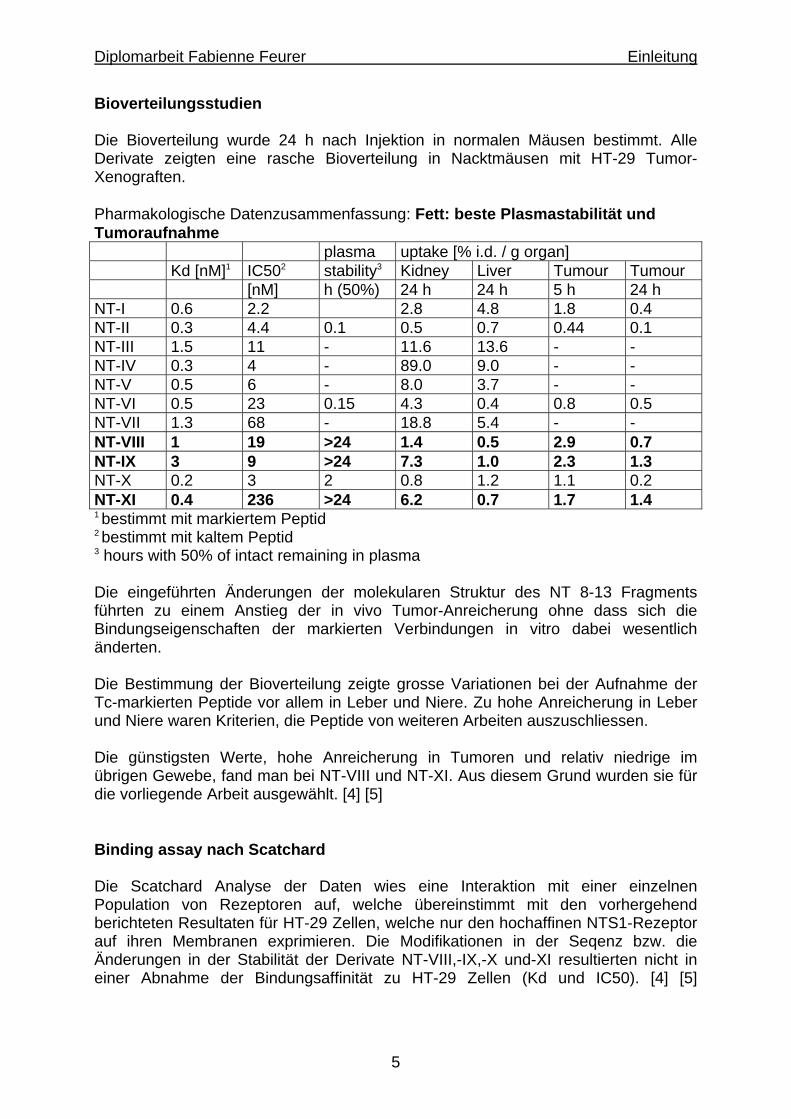
Bioverteilungsstudien Die Bioverteilung wurde 24 h nach Injektion in normalen Mäusen bestimmt. Alle Derivate zeigten eine rasche Bioverteilung in Nacktmäusen mit HT-29 Tumor-Xenograften. Pharmakologische Datenzusammenfassung: Fett: beste Plasmastabilität und Tumoraufnahme plasma uptake [% i.d. / g organ] Kd [nM]1 IC502 stability3 Kidney Liver Tumour Tumour [nM] h (50%) 24 h 24 h 5 h 24 h NT-I 0.6 2.2 2.8 4.8 1.8 0.4 NT-II 0.3 4.4 0.1 0.5 0.7 0.44 0.1 NT-III 1.5 11 - 11.6 13.6 - - NT-IV 0.3 4 - 89.0 9.0 - - NT-V 0.5 6 - 8.0 3.7 - - NT-VI 0.5 23 0.15 4.3 0.4 0.8 0.5 NT-VII 1.3 68 - 18.8 5.4 - - NT-VIII 1 19 >24 1.4 0.5 2.9 0.7 NT-IX 3 9 >24 7.3 1.0 2.3 1.3 NT-X 0.2 3 2 0.8 1.2 1.1 0.2 NT-XI 0.4 236 >24 6.2 0.7 1.7 1.4 1 bestimmt mit markiertem Peptid 2 bestimmt mit kaltem Peptid 3 hours with 50% of intact remaining in plasma Die eingeführten Änderungen der molekularen Struktur des NT 8-13 Fragments führten zu einem Anstieg der in vivo Tumor-Anreicherung ohne dass sich die Bindungseigenschaften der markierten Verbindungen in vitro dabei wesentlich änderten. Die Bestimmung der Bioverteilung zeigte grosse Variationen bei der Aufnahme der Tc-markierten Peptide vor allem in Leber und Niere. Zu hohe Anreicherung in Leber und Niere waren Kriterien, die Peptide von weiteren Arbeiten auszuschliessen. Die günstigsten Werte, hohe Anreicherung in Tumoren und relativ niedrige im übrigen Gewebe, fand man bei NT-VIII und NT-XI. Aus diesem Grund wurden sie für die vorliegende Arbeit ausgewählt. [4] [5] Binding assay nach Scatchard Die Scatchard Analyse der Daten wies eine Interaktion mit einer einzelnen Population von Rezeptoren auf, welche übereinstimmt mit den vorhergehend berichteten Resultaten für HT-29 Zellen, welche nur den hochaffinen NTS1-Rezeptor auf ihren Membranen exprimieren. Die Modifikationen in der Seqenz bzw. die Änderungen in der Stabilität der Derivate NT-VIII,-IX,-X und-XI resultierten nicht in einer Abnahme der Bindungsaffinität zu HT-29 Zellen (Kd und IC50). [4] [5]

Diplomarbeit Fabienne Feurer Aufgabenstellung
6
2. Aufgabenstellung 2.1. Analyse der Bindung an lebende Zellen nach Lindmo
(Bindungsassay) Bei den zu untersuchenden Neurotensin-Derivaten wurden bisher zwei Produktepeaks in der HPLC beobachtet. Ist die Bindung an die NTS1-Rezeptoren der HT-29 Zelllinie signifikant verschieden bei den beiden Produkten ? Hat die Puffersubstanz einen Einfluss ? Ist die Methode nach Lindmo geeignet, um eine regelmässige Endkontrolle der markierten Peptide durchzuführen ? 2.2. Untersuchung der Markierungsbedingungen mit NT-VIII und
NT-XI Vergleich von MES- und Phosphatpuffer. Welche Unterschiede findet man ? Vergleich der beiden NT's, der Puffersubstanzen, mehrerer pH-Werte und verschiedener Markierungszeitpunkte.

Diplomarbeit Fabienne Feurer Zusammenfassung
7
3. Zusammenfassung In dieser Arbeit wurden zwei 99mTc-Carbonyl-markierte Neurotensin-Derivate auf pharmakologische Eigenschaften, insbesondere auf ihre Bindungseigenschaften an HT-29 Zellen mit NTS1-Rezeptoren untersucht. Es galt, den Binding Assay nach Lindmo zu etablieren und damit unter anderem direkt verschiedene Markierungs-bedingungen zu testen. Die zu untersuchenden Markierungsbedingungen waren die Puffersubstanzen MES-Puffer/HCl und Phosphatpuffer/HCl, mehrere pH-Werte und verschiedene Markierungszeitpunkte. Als Analysemethoden standen dazu eine HPLC-Apparatur mit Aktivitätsdetektor und der Binding Assay nach Lindmo zur Verfügung, welcher wie der Binding Assay nach Scatchard zur Kd-Berechnung diente und sich zusätzlich zur Qualitätsüberprüfung eignete. Der Binding Assay nach Lindmo liess sich dabei unter folgenden Bedingungen durchführen: Man benötigte eine 4x6 Well-Platte mit mindestens vier identischen fixierten HT-29-Zellverdünnungsreihen. Diese wurden mit einer konstanten Menge des 99mTc-markierten Neurotensin-Derivats (ca. 4 fmol auf 20 kBq / well in 0.2 ml) für 2 h bei 37°C inkubiert. Zur Bestimmung der Nicht Spezifischen Bindung wurde in zwei der Reihen eine konstante Menge (0.2 ml von 2 µM) an nicht-markiertem Neurotensin 8-13 zugegeben, welches zur Blockierung der spezifischen Bindungsstellen diente. Aus diesen Parametern wurde die Spezifische Bindung (SB/Standard) bei jeder Zellkonzentration bestimmt, worauf durch Invertierung der Daten zum Lindmo-Plot aus der Steigung der Kd-Wert sowie der bindende Anteil r aus dem y-Achsenabschnitt (x=0 --> y=1/r) bestimmt werden konnten. Die verschiedenen Markierungsbedingungen wurden jeweils mit HPLC kontrolliert und Kd nach Lindmo ausgewertet. Für die Markierung, pH-Einstellung und für die darauffolgenden pharmakologischen Untersuchungen eignete sich die Phosphat-puffer/HCl-Neutralisationslösung gleichgut bis besser als die früher verwendete MES-Puffer/HCl-Lösung. Unterschiedliche pH-Werte zeigten keinen Effekt auf die Kd-Werte, doch der optimale pH-Bereich wurde zwischen pH 6-7.5 gewählt. Bei den beiden untersuchten Peptiden NT-VIII und NT-XI erschienen jeweils nach der Markierung bei 75°C zwei Produktepeaks in der HPLC, wobei die Peakverhältnisse bei den Markierungszeiten 30', 60' und 120' verschieden waren. Diese beiden Substanzen zeigten bezüglich Bindungseigenschaften (Kd) nur kleine, statistisch nicht signifikante Unterschiede, jedoch immer mit der gleichen Tendenz: Dabei korrelierte der erste und kleinere Peak von NT-VIII mit einer besseren Bindung an die HT-29 Zellen als der zweite doppelt so grosse Peak und bei NT-XI konnte diese Tendenz bei anderen Peakverhältnissen in umgekehrter Richtung beobachtet werden (2. Peak>1. Peak). Die beiden Konfigurationen der markierten Peptide waren bei >75°C nicht stabil und erreichten nach 2 h ein Gleichgewicht. Der Binding Assay nach Lindmo ist wegen seiner geringen Aktivität und kleinen Strahlenbelastung für eine regelmässige Endkontrolle besser geeignet als der Binding Assay nach Scatchard. Eine Abschätzung der Qualität der Markierung ist dabei möglich. Beide Methoden ergaben gleiche Kd-Werte im Bereiche von 0.1-1 nM.

Diplomarbeit Fabienne Feurer Material und Methoden
8
4. Material und Methoden Details sind unter Kapitel 7 "Experimenteller Teil" beschrieben. 4.1. Apparaturen und Geräte 4.1.1. HPLC Die HPLC (High performance liquid chromatography) bestand aus zwei Waters 510 Pumpen, einem Waters Gradient Controller 610, einem UV-Detektor (Kontron 710), einem NaI Scintillation Detektor (Picon Model P14 well counter, verbunden mit einem Verstärker und einem Zähler) und eine Reversed Phase Säule (Nucleosil 100-5, C18, 5 µm, 250 mm Länge, 4.6 mm Durchmesser). Laufmittel A war 0.1% TFA in H2O, Laufmittel B enthielt Ethanol/Wasser im Verhältnis 70/30 und ebenfalls 0.1% TFA. Der optimierte Gradient lief unter folgenden Parametern:
Gradient-Nr. Time [min] A [%] B[%] Flow [ml/min]6 Initial 100 0 0.00
3.00 100 0 1.005.00 60 40 1.008.00 60 40 1.0025.00 50 50 1.0032.00 50 50 1.0034.00 0 100 0.2036.00 0 100 0.2040.00 100 0 0.20
100.00 100 0 0.20101.00 100 0 0.00
Das Pertechnetat wurde dabei nach 3 Minuten eluiert, der freie Carbonyl-Tc-Komplex nach 9-11 Minuten und die Peptide je nach Struktur nach 19-25 Minuten. Das inaktive Peptid war nur sichtbar im UV bei 215 nm während der Reinigung (Injektion grösserer Mengen Aktivität, Detektionslimit bei ca. 5 nmol) und wurde etwa 2-4 Minuten vor dem markierten Peptid eluiert. Nach 32 Minuten Laufzeit folgten die Parameter für die Reinigung und Konditionierung der Säule. 4.1.2. Gamma counter Die Aktivität wurde mit einem Packard Canberra Cobra II Auto-Gamma-Counter gemessen. Die Resultate wurden in counts per minute (cpm) herausgegeben, wobei die obere Grenze einer linearen Messung bis etwa 2 Millionen cpm reicht. Jede Probenserie wurde von 3 Referenzröhrchen mit den Standardwerten (z.B. total zugegebene Aktivität pro well) begleitet. Somit konnte die Zerfallskorrektur experimentell bestimmt werden und alle Werte befanden sich im Bezug auf die Standardwerte bereits innerhalb dieser Zerfallskorrektur.

Diplomarbeit Fabienne Feurer Material und Methoden
9
4.2. Methoden 4.2.1. Synthese von Neurotensin-Derivaten Die Neurotensin-Derivate wurden in Brüssel von Prof. D. Tourwé durch Festphasen-Synthese auf einem "Merrifield Harz" unter Verwendung eines halbautomatischen Labortec Peptide Synthesizer SP640B hergestellt. Die Strukturen von NT-VIII, NT-XI, NT-XII und NT 8-13 sind in untenstehender Tabelle ersichtlich: NT 7-13 Pro-Arg-Arg-Pro-Tyr-Ile-Leu NT-VIII (NαHis)Ac-(N-CH3)-Arg-Lys-Pro-Tyr-Tle-Leu NT-XI (NαHis)Ac-Lys-(CH2NH)-Arg-Pro-Tyr-Tle-Leu NT-XII (NαHis)Ac-Arg-(N-CH3)-Arg-Pro-Tyr-Tle-Leu 4.2.2. Radiomarkierung von Neurotensin-Derivaten 1. Schritt: Herstellung des 99mTc-Tricarbonylkomplexes (Carbonylierung) 0.1-40 GBq Pertechnetat in 2-4 ml Generatoreluat (siehe Kapitel 7.1.) wurden zu einer Mischung von 4 mg Na2CO3, 5.5 mg NaBH4 und 15 mg NaKTartrat in einem Penicillin Vial, das zuvor mit CO 1-2 ml/sec während 30 Min. gespült wurde, gegeben und anschliessend mindestens 30 Min. bei 75°C erhitzt. Die Lösung wurde abgekühlt und auf verschiedene pH-Werte (pH 6-8.5) neutralisiert unter Verwendung von einer MES-Puffer/HCl- oder Phosphatpuffer/HCl-Lösung. 2. Schritt: Markierung des NT-Analogs (Reaktion mit Neurotensin) Je nach benötigter Menge wurden 0.2 bis 0.6 ml (max. 1 ml) der neutralisierten Tc-Carbonyllösung mit 10-15 µl 1 mM N2-gespülten Lösung des entsprechenden Neurotensin-Analogs (= minimale Peptidendkonzentration: 0.05 mM) gemischt und erneut bei 75°C für 30-60 (max.120) Minuten erhitzt. Das Produkt wurde mit HPLC analysiert und konnte bei Bedarf gereinigt werden. Unvollständige CO-Spülung oder Anwesenheit von O2 führte anschliessend zu mehr Pertechnetat (1.Schritt). Ein pH-Wert weit unter pH 6 (pH 2-3) führte zu mehr freiem Carbonyl-Tc-Komplex. Ein sehr hoher pH-Wert > 8.5 führte hingegen sowohl zu mehr Pertechnetat als auch zu einer späteren Denaturierung der Peptide (2. Schritt). Längere Markierung als nur 30 Min. (2. Schritt) führte zu einer besseren Ausbeute des markierten Peptids im Verhältnis zum Tc-Carbonyl. Während der Reinigung wurden Fraktionen von 1 ml je Minute entnommen und einzelne mit genügend hoher Aktivität oder jene von Interesse (1.-2. Peak) konnten für weitere Untersuchungen (z.B. Kd-Bestimmung nach Lindmo oder Scatchard) verwendet werden. Die gewählte Fraktion wurde mit 1N NaOH-Lösung und PBS 5x (konzentrierter Phosphatpuffer mit physiologischer Salzkonzentration) neutralisiert auf pH 6.5-7. Die relativ hohe Ethanolkonzentration konnte für den Versuch auch durch Abdampfen reduziert werden. Für den Lindmo war dies allerdings nicht nötig,

Diplomarbeit Fabienne Feurer Material und Methoden
10
da mit sehr kleinen Konzentrationen und einer hohen Verdünnung im Versuch selber gearbeitet wurde. 4.2.3. Zellkultur Die humane Zelllinie HT-29 wurde von der "European Collection of Cell Culture" (ECACC, Salisbury, England) erhalten. Die Zellen wurden in McCoy's 5A-GLUTAMAX ITM , ergänzt mit 10% FBS, 100 I.U./ml Penicillin, 100 µg/ml Streptomycin und 0,25 µg/ml Amphotericin B bei 37°C in einem feuchten Inkubator unter einer Atmosphäre mit 7.5% CO2 kultiviert und wöchentlich umgesetzt. [9] 4.2.4. Vorbereiten von gefrorenen Zellen in der 4x6 Well-Platte Die Zellen wurden wie für den Lindmo-Assay ausplatiert und über Nacht im Inkubator fixiert. Dann wurde das Medium entfernt, die Zellen während drei Tagen im Laminar Flow getrocknet und schliesslich bei –20°C gelagert. Bei einer zweiten Methode wurden die Zellen nach der Fixierung in Binding Buffer bei 4°C aufbewahrt. 4.2.5. Binding Assay nach Lindmo Details siehe unter Kapitel 7 "Experimenteller Teil" Der Binding Assay nach Lindmo wurde in lebenden fixierten HT-29 Zellen durchgeführt. Man benötigte dabei eine Zellverdünnungsreihe 1:2 in sechs wells, in welchen die höchste Zellkonzentration 1-1.75 mio Zellen / well enthalten sollte. Während des Binding Assays wurden alle Zellen mit einer konstanten Menge des 99mTc-NT-Derivats während 2 h bei 37°C inkubiert. Die zugegebene Aktivität betrug 20 kBq / well. Zur Bestimmung der nichtspezifischen Bindung wurde in zwei Reihen kaltes NT 8-13 (2 µM) hinzugefügt. [7] [8] 4.3.6. Kd-Berechnung nach Lindmo Aus der Regressionsgerade des Lindmo-Plots (Standard/SB vs. 1/mio cells* ml-1) von Punkten der Spezifischen Bindung (SB) wurde die Steigung [mio cells* ml-1] mit dem Faktor 7.5*10-11 [mol Bindungsstellen*103/mio cells] multipliziert. Der erhaltene Wert stellte den Kd-Wert in mol/l [M] dar. Alle Kd-Werte lagen im nanomolaren Bereich. Der Achsenabschnitt ist gleich 1/r, wobei r den reaktiven (extrapoliert auf unendlich viele Mole Bindungsstellen) Anteil des markierten Peptids darstellt, d.h. der Anteil, welcher maximal zur Bindung fähig war. Daher galt r als Korrekturfaktor für den Standard und konnte gewissermassen Indikator sein für die Qualität des markierten Peptids. [7] [8]

Diplomarbeit Fabienne Feurer Resultate
11
5. Resultate 5.1. Binding Assay nach Lindmo 5.1.1. Parameter zur Etablierung des Lindmo Im Laufe der Experimente nach Lindmo, die in der ersten Phase einer beträchtlichen Streuung unterlagen, wurden kontinuierlich einige wichtig erscheinende Parameter verändert und auf ihre Auswirkungen auf den Binding Assay nach Lindmo getestet. Die gesuchten Resultate waren Kd (Dissoziationskonstante) und r (reaktiver bindender Anteil des Standards). r hängt dabei deutlich von den Markierungsbedingungen ab. Der Kd-Wert jedoch hängt von wesentlich mehr Faktoren, nämlich der Peptidkonzentration, der Zellkonzentration, der Qualität und/oder Reinheit des markierten Peptids, der Intaktheit der Zellen und der Genauigkeit der Verdünnungsreihe ab. Ausserdem schienen die Grösse der wells (Well-Platte) sowie verschiedene Substanzen des Binding Buffers einen Einfluss zu haben. Peptidkonzentration vs. Zellkonzentration Die Peptidmenge / well sollte weit kleiner sein als die Menge Rezeptoren [mol] / well. Damit könnte jeweils die maximale Menge an Peptid binden und durch die Entstehung des Gleichgewichts zw. "freiem Peptid", "freien Rezeptoren" und Peptid-Rezeptorkomplex würde eine Sättigung bei der höchsten Zellkonzentration möglich. Natürlich musste erst der optimale Zellkonzentrationsbereich gefunden werden. Qualität und Reinheit des markierten Peptids Falls das Rohprodukt für einen Lindmo verwendet wurde, konnte durch entsprechende HPLC-Kontrolle der Anteil Pertechnetat und Carbonyl festgestellt werden. Die Kd-Werte waren dementsprechend höher (=schlechtere Bindung) und die Spezifische Bindung niedriger als bei gereinigtem markierten Peptid. Dies kam daher, dass im ungereinigten Produkt noch etwa gleichviel nicht-markiertes Peptid enthalten war, welches ebenso an spezifische Bindungsstellen binden konnte, aber nicht in der Messung enthalten war und selbstverständlich auch nicht von diagnostischem und therapeutischen Wert sein konnte. Für die Qualität des Produktes spielte auch der momentane Zustand des Generators, bzw. des Eluats eine Rolle. Falls z.B. längere Zeit nicht mehr eluiert worden war, konnte nach der Carbonylierung eine Menge Pertechnetat zur Folge auftreten. Zellen und Verdünnungsreihe Es versteht sich von selbst, dass die Herstellung der Verdünnungsreihe der Zellsuspension möglichst exakt erfolgen sollte. Fehler in der Reihe führten unweigerlich zu Fehlern der Bindungsresultate und zu unschönen Sättigungskurven, die für den Lindmo-Plot eine schlechte Regression bedeuteten und somit einen erhöhten Fehleranteil für Kd und den bindenden Anteil ergaben.

Diplomarbeit Fabienne Feurer Resultate
12
Bei höherer Zellkonzentration war es von Vorteil, grössere wells (Well-Platte) für das Ausplatieren zu wählen, da weniger Multilayer, d.h. mehrschichtige Zellcluster entstehen konnten. Dadurch würden nämlich gewisse Zellen bzw. Rezeptoren nicht mehr zugänglich sein. Da jedoch die Peptidkonzentration weit unter der Konzentration der Mole Bindungsstellen lag, sollten selbst bei Clusterbindung noch genügend Zellen für die Bindung zur Verfügung stehen. Binding Buffer mit BSA oder FBS ? Der verwendete Binding Buffer (BB) enthielt entweder 0.5% BSA oder 0.5% FBS. Nach einigen Vergleichen konnte festgestellt werden, dass der Binding Buffer mit BSA bessere Resultate lieferte als der BB mit FBS. Daher wurde nach einer ersten Phase der Verwendung von Binding Buffer mit FBS auf Binding Buffer mit BSA gewechselt. Beide Proteine dienen einerseits zur Absättigung von unspezifischen Bindungsstellen an der Zelloberfläche und andererseits zur Bildung von möglichst physiologisch vergleichbaren Bedingungen, wie sie in vivo (Blut) herrschen. Zusätzlich können Enzyme der HT-29 Zellen (endogene Peptidasen), die ev. das Peptid spalten würden, von diesen Proteinen blockiert werden. Aktivitätsmenge Neben der geeigneten Peptidkonzentration sollte auch die Menge total zugegebener Aktivität gewählt werden. Der Anteil, der tatsächlich binden würde, war zu Beginn nur ansatzweise (aus Scatchard-Daten) bekannt. Für eine gute Messbarkeit sollte die Aktivität des Standards ca. bei 1'000'000 cpm liegen, oder umgerechnet 16 kBq eingesetzt werden. Da bei der Halbwertszeit von 99mTc von 6 h auch während dem Binding Assay mit gewissen Verlusten zu rechnen war, wurde für den Standard 20 kBq gewählt. Dies entsprach Peptidkonzentrationen von durchschnittlich 1 bis 4 fmol pro well bei anfänglich 10-15 nmol des Peptids in der Reaktionslösung des 2. Markierungsschrittes. Bestimmung der Nicht-Spezifischen Bindung Zur Bestimmung von NSB/Standard sollte ein nicht-markiertes Peptid mit gleichen oder ähnlichen Bindungseigenschaften verwendet werden. Dieses, im voraus zugegeben, sättigt alle spezifischen Bindungsstellen ab, so dass das zugegebene markierte Peptid nur noch an die nicht spezifischen Bindungsstellen (z.B. an Stellen zwischen oder neben den spezifischen NTS1-Rezeptoren) binden kann. Aus früheren Experimenten war bekannt, dass z.B. das kalte NT-XI schlechter bindet als das mit 99mTc-Carbonyl markierte NT-XI. Da bei in vitro-Versuchen kein starker Abbau durch Peptidasen befürchtet werden musste, wurde für die Bestimmung der nichtspezifischen Bindung das ursprüngliche bindende Fragment NT 8-13 gewählt, weil es den Bindungseigenschaften (Kd) der markierten Neurotensin-Derivaten am nächsten kam.

Diplomarbeit Fabienne Feurer Resultate
13
Dauer der Inkubation bei 37°C Die zu Beginn während 2 h bei 22°C inkubierten Testsysteme zeigten nur ungenügende Resultate. Daraufhin wurde auf 37°C gewechselt, erstens wegen der Nähe zu physiologischen Bedingungen und zweitens, um die Reaktion bzw. die Einstellung des Gleichgewichts zu beschleunigen. Mehrere Experimente zeigten jedoch, dass nicht egal war, wie lange bei 37°C inkubiert wurde. Ein Anstieg der Spezifischen Bindung von 30 bis 120 Minuten von etwa 10 % wurde ersichtlich. Um zu sehen, um wieviel die Bindung nach 120 Minuten noch ansteigen konnte, wurden drei Binding Assays angesetzt und nach 90', 120' und 240' gestoppt. Der Anstieg der Spezifischen Bindung von 90' zu 120' betrug ca. 3 %, der Anstieg von 120' bis 240' noch knapp 1 %. Also schien die Bindung nach 2 h Inkubationsdauer bei 37°C die Gleichgewichtsgrenze langsam erreicht zu haben. 5.1.2. Bedingungen für den Binding Assay Die optimalen Bedingungen für den Binding Assay und die Kd-Bestimmung nach Lindmo waren: -Inkubation: 120 Minuten 37°C leichtes Schütteln -Aktivitätsmenge: 20 kBq / well -Binding Buffer: Binding Buffer mit BSA -Bestimmung der nicht-spezifischen Bindung mit cold NT 8-13 -beste "höchste" Zellkonzentration beim Ausplatieren der Zellen: 2-3.5 mio / ml in einer 4x6 Well-Platte Endmenge 1. Kolonne --> 1-1.75 mio Zellen / well (Endvolumen --> 0.5 ml / well) -Ablösen der Zellen mit NaOH 1N während 30 Minuten bei 37°C -Waschen der Zellen vor Versuch: mit Binding Buffer BSA je 2 x 0.2 ml / well -Waschen der Zellen nach Versuch: mit PBS 1x je 2 x 0.2 ml / well
Diplomarbeit Fabienne Feurer Resultate
14
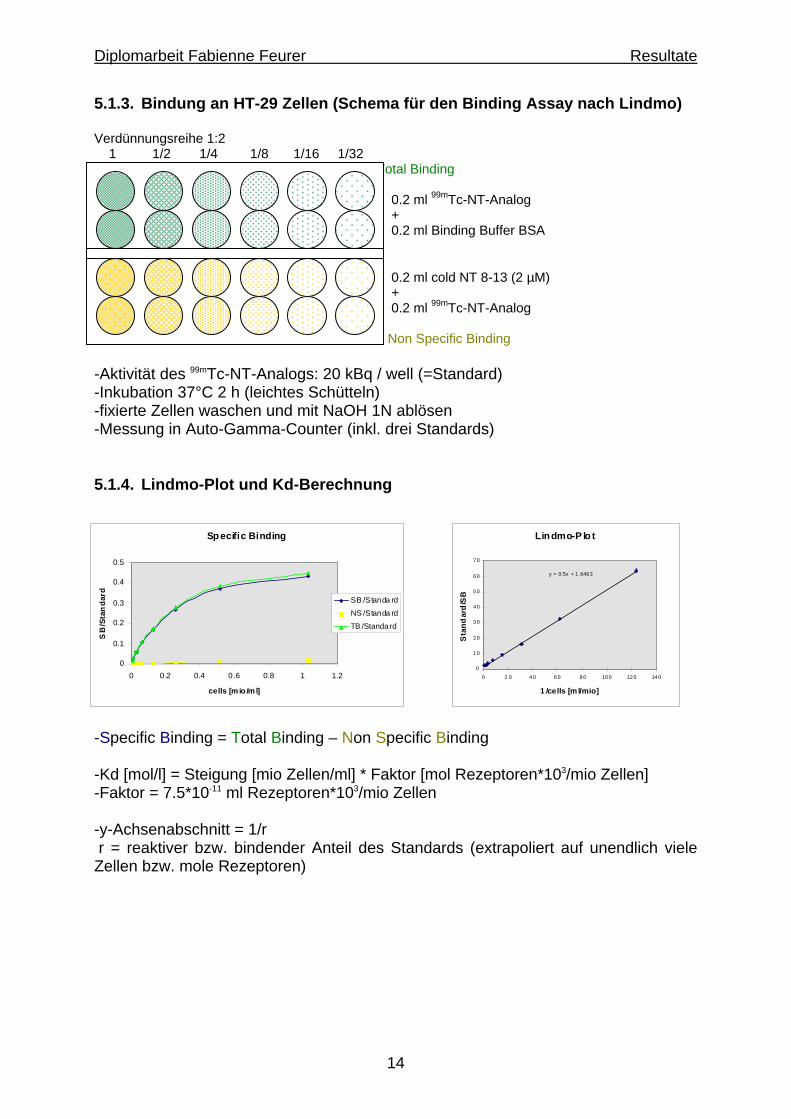
5.1.3. Bindung an HT-29 Zellen (Schema für den Binding Assay nach Lindmo) Verdünnungsreihe 1:2 1 1/2 1/4 1/8 1/16 1/32 Total Binding 0.2 ml 99mTc-NT-Analog + 0.2 ml Binding Buffer BSA 0.2 ml cold NT 8-13 (2 µM) + 0.2 ml 99mTc-NT-Analog Non Specific Binding -Aktivität des 99mTc-NT-Analogs: 20 kBq / well (=Standard) -Inkubation 37°C 2 h (leichtes Schütteln) -fixierte Zellen waschen und mit NaOH 1N ablösen -Messung in Auto-Gamma-Counter (inkl. drei Standards) 5.1.4. Lindmo-Plot und Kd-Berechnung
-Specific Binding = Total Binding – Non Specific Binding -Kd [mol/l] = Steigung [mio Zellen/ml] * Faktor [mol Rezeptoren*103/mio Zellen] -Faktor = 7.5*10-11 ml Rezeptoren*103/mio Zellen -y-Achsenabschnitt = 1/r r = reaktiver bzw. bindender Anteil des Standards (extrapoliert auf unendlich viele Zellen bzw. mole Rezeptoren)
Lindmo-P lo t
y = 0.5x + 1 .6493
0
10
20
30
40
50
60
70
0 2 0 4 0 60 8 0 10 0 120 14 0
1 /cells [ml/mio]
Sta
ndar
d/S
B
Specific Binding
0
0.1
0.2
0.3
0.4
0.5
0 0.2 0.4 0.6 0.8 1 1.2
cells [mio /ml]
SB
/Sta
nda
rd
SB /Standa rdNS /Standa rdTB /Standard
Diplomarbeit Fabienne Feurer Resultate
15
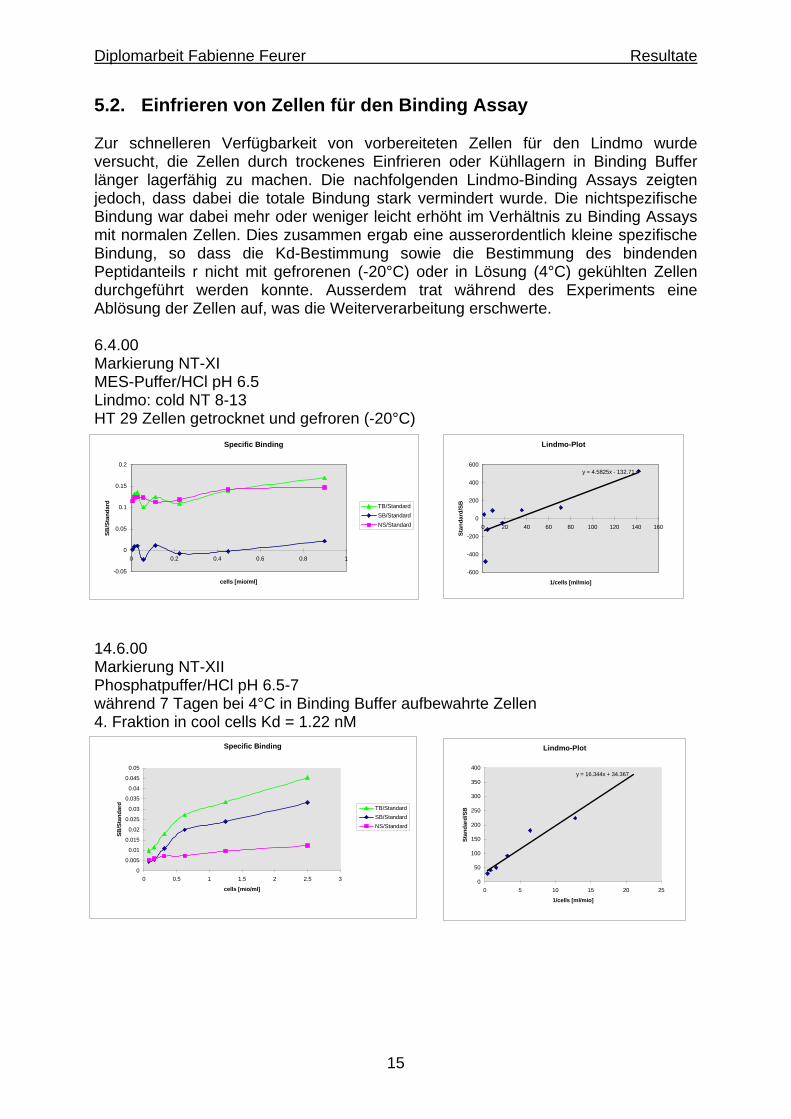
5.2. Einfrieren von Zellen für den Binding Assay Zur schnelleren Verfügbarkeit von vorbereiteten Zellen für den Lindmo wurde versucht, die Zellen durch trockenes Einfrieren oder Kühllagern in Binding Buffer länger lagerfähig zu machen. Die nachfolgenden Lindmo-Binding Assays zeigten jedoch, dass dabei die totale Bindung stark vermindert wurde. Die nichtspezifische Bindung war dabei mehr oder weniger leicht erhöht im Verhältnis zu Binding Assays mit normalen Zellen. Dies zusammen ergab eine ausserordentlich kleine spezifische Bindung, so dass die Kd-Bestimmung sowie die Bestimmung des bindenden Peptidanteils r nicht mit gefrorenen (-20°C) oder in Lösung (4°C) gekühlten Zellen durchgeführt werden konnte. Ausserdem trat während des Experiments eine Ablösung der Zellen auf, was die Weiterverarbeitung erschwerte. 6.4.00 Markierung NT-XI MES-Puffer/HCl pH 6.5 Lindmo: cold NT 8-13 HT 29 Zellen getrocknet und gefroren (-20°C)
14.6.00 Markierung NT-XII Phosphatpuffer/HCl pH 6.5-7 während 7 Tagen bei 4°C in Binding Buffer aufbewahrte Zellen 4. Fraktion in cool cells Kd = 1.22 nM
Specific Binding
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0.045
0.05
0 0.5 1 1.5 2 2.5 3
cells [mio/ml]
SB/S
tand
ard
TB/StandardSB/StandardNS/Standard
Lindmo-Plot
y = 16.344x + 34.367
0
50
100
150
200
250
300
350
400
0 5 10 15 20 25
1/cells [ml/mio]
Stan
dard
/SB
Specific Binding
-0.05
0
0.05
0.1
0.15
0.2
0 0.2 0.4 0.6 0.8 1
cells [mio/ml]
SB/S
tand
ard
TB/StandardSB/StandardNS/Standard
Lindmo-Plot
y = 4.5825x - 132.71
-600
-400
-200
0
200
400
600
0 20 40 60 80 100 120 140 160
1/cells [ml/mio]
Stan
dard
/SB
Diplomarbeit Fabienne Feurer Resultate
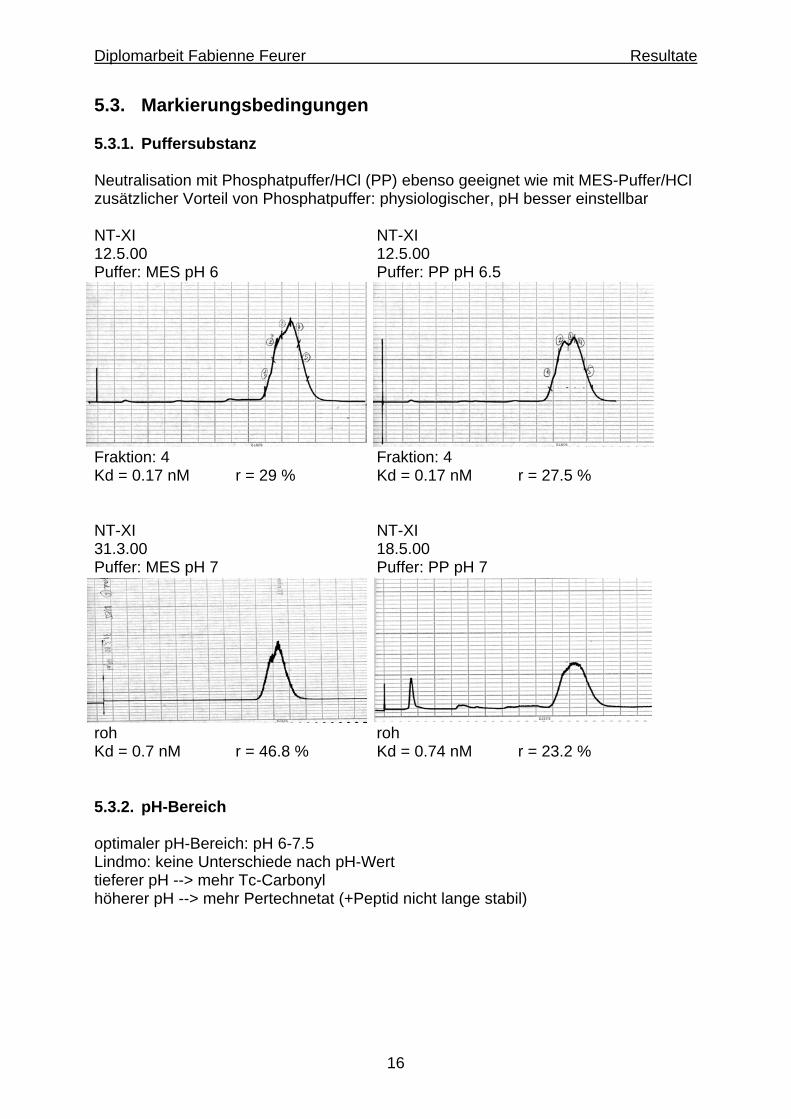
16
5.3. Markierungsbedingungen 5.3.1. Puffersubstanz Neutralisation mit Phosphatpuffer/HCl (PP) ebenso geeignet wie mit MES-Puffer/HCl zusätzlicher Vorteil von Phosphatpuffer: physiologischer, pH besser einstellbar NT-XI NT-XI 12.5.00 12.5.00 Puffer: MES pH 6 Puffer: PP pH 6.5
Fraktion: 4 Fraktion: 4 Kd = 0.17 nM r = 29 % Kd = 0.17 nM r = 27.5 % NT-XI NT-XI 31.3.00 18.5.00 Puffer: MES pH 7 Puffer: PP pH 7
roh roh Kd = 0.7 nM r = 46.8 % Kd = 0.74 nM r = 23.2 % 5.3.2. pH-Bereich optimaler pH-Bereich: pH 6-7.5 Lindmo: keine Unterschiede nach pH-Wert tieferer pH --> mehr Tc-Carbonyl höherer pH --> mehr Pertechnetat (+Peptid nicht lange stabil)
Diplomarbeit Fabienne Feurer Resultate
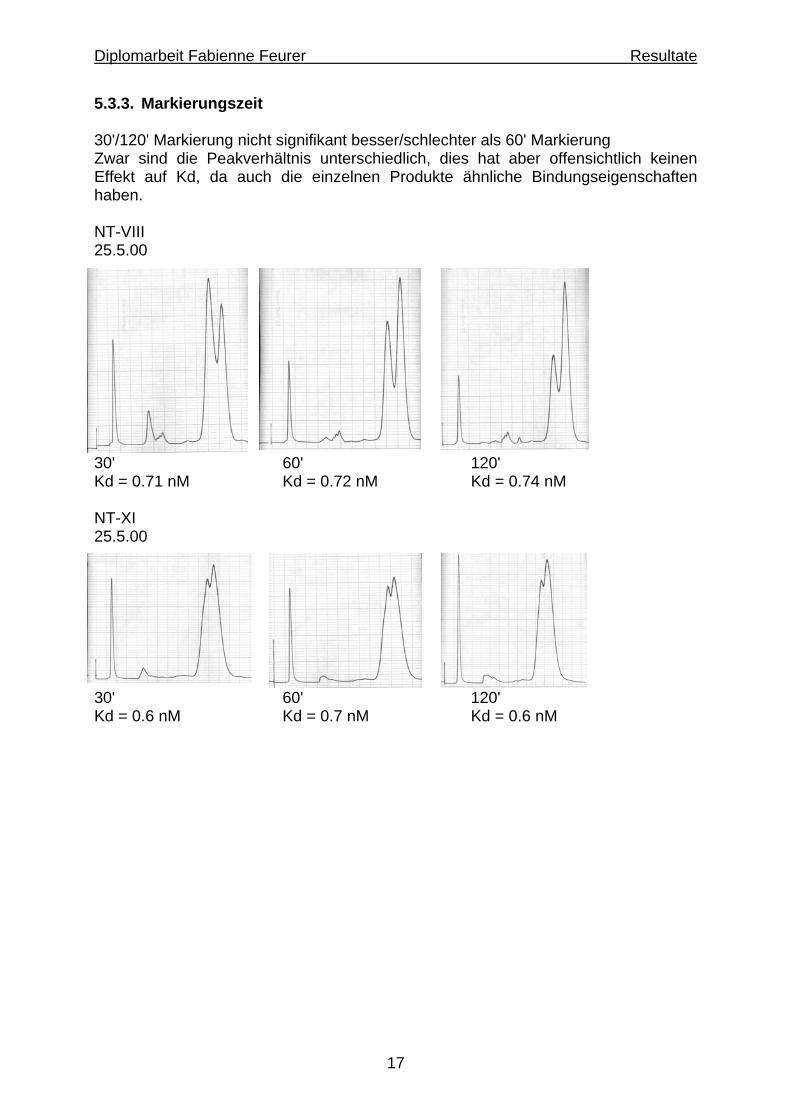
17
5.3.3. Markierungszeit 30'/120' Markierung nicht signifikant besser/schlechter als 60' Markierung Zwar sind die Peakverhältnis unterschiedlich, dies hat aber offensichtlich keinen Effekt auf Kd, da auch die einzelnen Produkte ähnliche Bindungseigenschaften haben. NT-VIII 25.5.00
30' 60' 120' Kd = 0.71 nM Kd = 0.72 nM Kd = 0.74 nM NT-XI 25.5.00
30' 60' 120' Kd = 0.6 nM Kd = 0.7 nM Kd = 0.6 nM
Diplomarbeit Fabienne Feurer Resultate
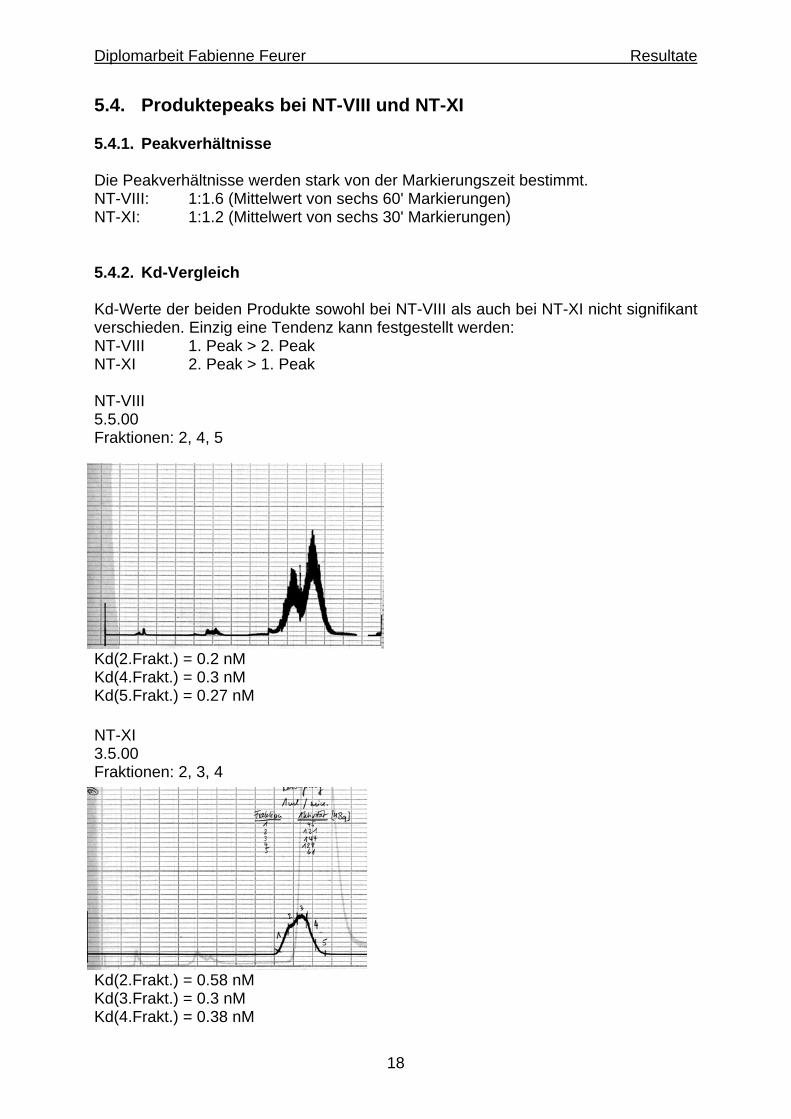
18
5.4. Produktepeaks bei NT-VIII und NT-XI 5.4.1. Peakverhältnisse Die Peakverhältnisse werden stark von der Markierungszeit bestimmt. NT-VIII: 1:1.6 (Mittelwert von sechs 60' Markierungen) NT-XI: 1:1.2 (Mittelwert von sechs 30' Markierungen) 5.4.2. Kd-Vergleich Kd-Werte der beiden Produkte sowohl bei NT-VIII als auch bei NT-XI nicht signifikant verschieden. Einzig eine Tendenz kann festgestellt werden: NT-VIII 1. Peak > 2. Peak NT-XI 2. Peak > 1. Peak NT-VIII 5.5.00 Fraktionen: 2, 4, 5
Kd(2.Frakt.) = 0.2 nM Kd(4.Frakt.) = 0.3 nM Kd(5.Frakt.) = 0.27 nM NT-XI 3.5.00 Fraktionen: 2, 3, 4
Kd(2.Frakt.) = 0.58 nM Kd(3.Frakt.) = 0.3 nM Kd(4.Frakt.) = 0.38 nM

Diplomarbeit Fabienne Feurer Resultate
19
roh (vor Inkubation)
Fraktion 2 (vor Inkubation)
Fraktion 5 (vor Inkubation)
roh (37°C 2 h)
Fraktion 2 (37°C 2 h)
Fraktion 5 (37°C 2 h)
roh (75°C 2 h)
Fraktion 2 (75°C 2 h)
Fraktion 5 (75°C 2 h)
5.4.3. Inkubationskontrollen Die einzelnen Produktepeaks (Fraktion 2 und 5) wurden gesammelt und einzeln eingespritzt. Es liess sich zeigen, dass die Peaks keine Artefakte darstellten und als vollständige Peaks mit derselben Retentionszeit wie im Rohprodukt eluiert wurden. Die Inkubation der beiden Fraktionen bei 37°C während 2 h führte bei NT-VIII zu praktisch keiner Veränderung des Peakverhältnisses, bei 75°C und 2 h zeigte sich aber bei beiden Substanzen das gleiche Peak-Muster wie beim Rohprodukt, was auf ein Gleichgewicht der beiden Produkte schliessen liess. Bei NT-XI hatte sich das Gleichgewicht bei einer Inkubation von 75°C nach 2 h noch nicht eingestellt und bei 2 h 37°C schienen die einzelnen Produkte stabil zu bleiben. NT-VIII
NT-XI roh (vor Inkubation)
Fraktion 2 (vor Inkubation)
Fraktion 4 (vor Inkubation)
roh (37°C 2 h)
Fraktion 2 (37°C 2 h)
Fraktion 4 (37°C 2 h)
roh (75°C 2 h)
Fraktion 2 (75°C 2 h)
Fraktion 4 (75°C 2 h)
Diplomarbeit Fabienne Feurer Resultate
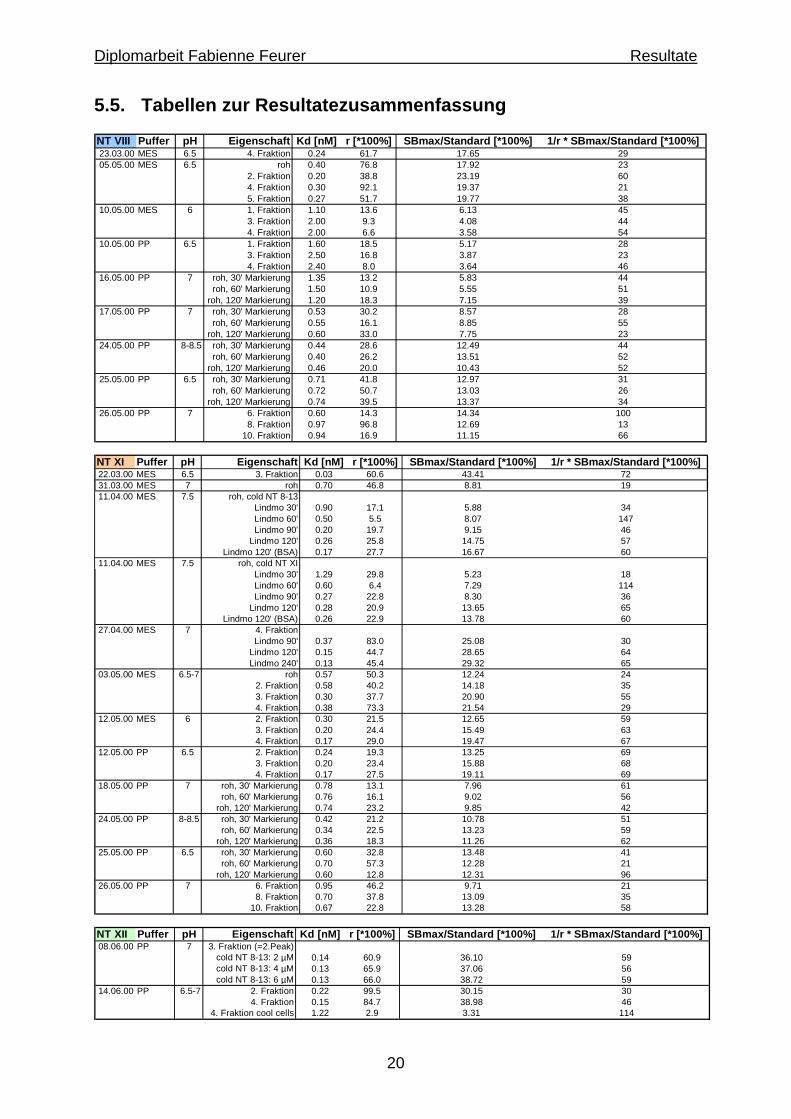
20
5.5. Tabellen zur Resultatezusammenfassung NT VIII Puffer pH Eigenschaft Kd [nM] r [*100%] SBmax/Standard [*100%] 1/r * SBmax/Standard [*100%]23.03.00 MES 6.5 4. Fraktion 0.24 61.7 17.65 2905.05.00 MES 6.5 roh 0.40 76.8 17.92 23
2. Fraktion 0.20 38.8 23.19 604. Fraktion 0.30 92.1 19.37 215. Fraktion 0.27 51.7 19.77 38
10.05.00 MES 6 1. Fraktion 1.10 13.6 6.13 453. Fraktion 2.00 9.3 4.08 444. Fraktion 2.00 6.6 3.58 54
10.05.00 PP 6.5 1. Fraktion 1.60 18.5 5.17 283. Fraktion 2.50 16.8 3.87 234. Fraktion 2.40 8.0 3.64 46
16.05.00 PP 7 roh, 30' Markierung 1.35 13.2 5.83 44roh, 60' Markierung 1.50 10.9 5.55 51
roh, 120' Markierung 1.20 18.3 7.15 3917.05.00 PP 7 roh, 30' Markierung 0.53 30.2 8.57 28
roh, 60' Markierung 0.55 16.1 8.85 55roh, 120' Markierung 0.60 33.0 7.75 23
24.05.00 PP 8-8.5 roh, 30' Markierung 0.44 28.6 12.49 44roh, 60' Markierung 0.40 26.2 13.51 52
roh, 120' Markierung 0.46 20.0 10.43 5225.05.00 PP 6.5 roh, 30' Markierung 0.71 41.8 12.97 31
roh, 60' Markierung 0.72 50.7 13.03 26roh, 120' Markierung 0.74 39.5 13.37 34
26.05.00 PP 7 6. Fraktion 0.60 14.3 14.34 1008. Fraktion 0.97 96.8 12.69 13
10. Fraktion 0.94 16.9 11.15 66 NT XI Puffer pH Eigenschaft Kd [nM] r [*100%] SBmax/Standard [*100%] 1/r * SBmax/Standard [*100%]22.03.00 MES 6.5 3. Fraktion 0.03 60.6 43.41 7231.03.00 MES 7 roh 0.70 46.8 8.81 1911.04.00 MES 7.5 roh, cold NT 8-13
Lindmo 30' 0.90 17.1 5.88 34Lindmo 60' 0.50 5.5 8.07 147Lindmo 90' 0.20 19.7 9.15 46
Lindmo 120' 0.26 25.8 14.75 57Lindmo 120' (BSA) 0.17 27.7 16.67 60
11.04.00 MES 7.5 roh, cold NT XILindmo 30' 1.29 29.8 5.23 18Lindmo 60' 0.60 6.4 7.29 114Lindmo 90' 0.27 22.8 8.30 36
Lindmo 120' 0.28 20.9 13.65 65Lindmo 120' (BSA) 0.26 22.9 13.78 60
27.04.00 MES 7 4. FraktionLindmo 90' 0.37 83.0 25.08 30
Lindmo 120' 0.15 44.7 28.65 64Lindmo 240' 0.13 45.4 29.32 65
03.05.00 MES 6.5-7 roh 0.57 50.3 12.24 242. Fraktion 0.58 40.2 14.18 353. Fraktion 0.30 37.7 20.90 554. Fraktion 0.38 73.3 21.54 29
12.05.00 MES 6 2. Fraktion 0.30 21.5 12.65 593. Fraktion 0.20 24.4 15.49 634. Fraktion 0.17 29.0 19.47 67
12.05.00 PP 6.5 2. Fraktion 0.24 19.3 13.25 693. Fraktion 0.20 23.4 15.88 684. Fraktion 0.17 27.5 19.11 69
18.05.00 PP 7 roh, 30' Markierung 0.78 13.1 7.96 61roh, 60' Markierung 0.76 16.1 9.02 56
roh, 120' Markierung 0.74 23.2 9.85 4224.05.00 PP 8-8.5 roh, 30' Markierung 0.42 21.2 10.78 51
roh, 60' Markierung 0.34 22.5 13.23 59roh, 120' Markierung 0.36 18.3 11.26 62
25.05.00 PP 6.5 roh, 30' Markierung 0.60 32.8 13.48 41roh, 60' Markierung 0.70 57.3 12.28 21
roh, 120' Markierung 0.60 12.8 12.31 9626.05.00 PP 7 6. Fraktion 0.95 46.2 9.71 21
8. Fraktion 0.70 37.8 13.09 3510. Fraktion 0.67 22.8 13.28 58
NT XII Puffer pH Eigenschaft Kd [nM] r [*100%] SBmax/Standard [*100%] 1/r * SBmax/Standard [*100%]08.06.00 PP 7 3. Fraktion (=2.Peak)
cold NT 8-13: 2 µM 0.14 60.9 36.10 59cold NT 8-13: 4 µM 0.13 65.9 37.06 56cold NT 8-13: 6 µM 0.13 66.0 38.72 59
14.06.00 PP 6.5-7 2. Fraktion 0.22 99.5 30.15 304. Fraktion 0.15 84.7 38.98 46
4. Fraktion cool cells 1.22 2.9 3.31 114

Diplomarbeit Fabienne Feurer Resultate
21
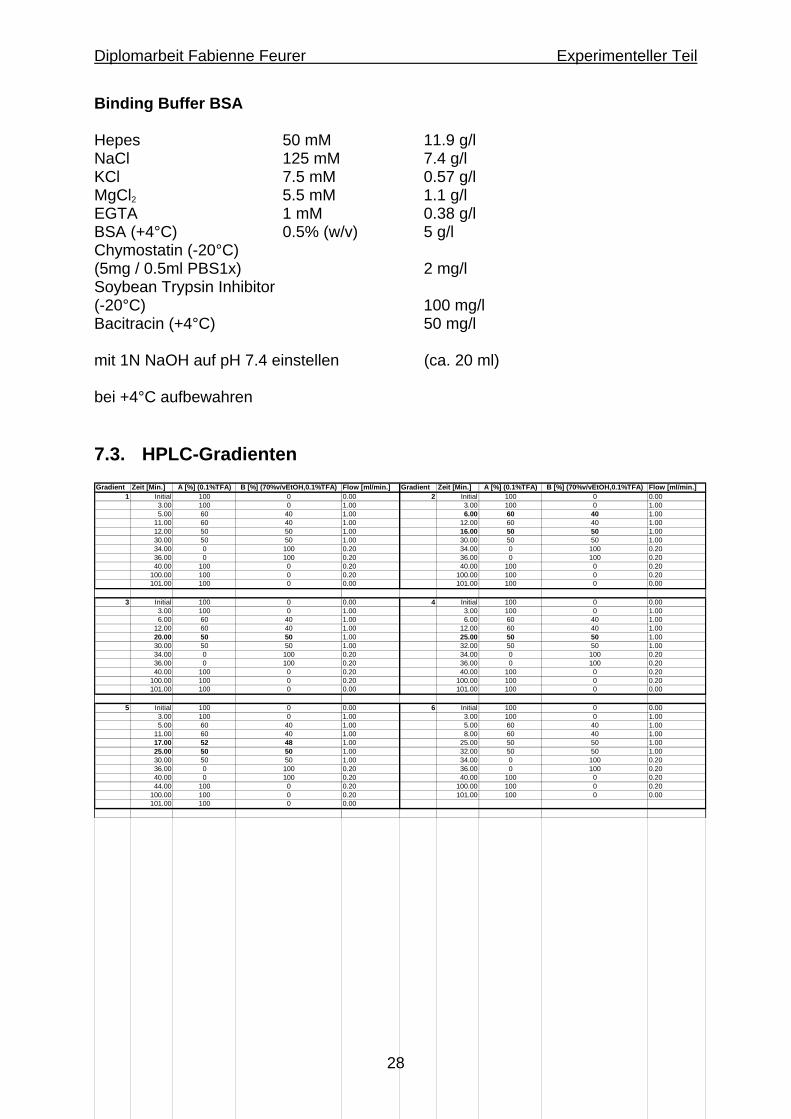
5.6. HPLC-Optimierung Ausgehend vom vorhergehend verwendeten Gradienten Nr. 1 wurde versucht, durch Veränderung der HPLC-Parameter (Zeit, Fluss und Lösungsmittelverhältnisse) eine bessere Trennung der markierten Peptide zu erhalten. Da beide markierten Peptide im Bereich der Lösungsmittelverhältnisse A 60-->50% B 40-->50% eluiert wurden, sollte eine langsame Veränderung dieses Verhältnisses eine Verbesserung ergeben. Daraus entstand Gradient 2 und seine Erweiterungen Gradienten 3 und 4. Nach rechnerischer und graphischer Ermittlung der exakten Verhältnisse der Lösungsmittel A und B während dem Auftreten von 99mTc-NT-VIII sollte mit Gradient Nr. 5 dieser Bereich für NT-VIII noch zusätzlich verlängert werden. Da dies nicht zur gewünschten Verbesserung der Trennung der beiden Produktepeaks führte, wurde wieder ausgehend von Gradient Nr. 4 eine nochmalige zeitliche Erweiterung dessen angesetzt. Dies führte zu einem Gradienten Nr. 6, mit dem sich sowohl NT-XI als auch NT-VIII noch besser trennen liessen. Die Retentionszeiten von NT-VIII lagen nun bei ca. 19.5-22 Min. für den 1. Peak und ca. 21-22.5 Min. für den zweiten Peak. Die Retentionszeiten von NT-XI lagen bei ca. 20-24 Min. für den ersten und bei ca. 21-24.5 Min. für den zweiten Peak. NT-XII zeigte folgende Retentionszeiten: ca. 20-21.5 Min. für den ersten und ca. 21-23 Min. für den zweiten Peak. Die Erfahrung zeigte ausserdem, dass die Retentionszeiten empfindlich auf Variationen im Ethanolgehalt von Lösungsmittel B reagierten. Es empfiele sich daher, das Lösungsmittel B (70% EtOH mit 0.1% TFA) zur besseren Reproduzierbarkeit durch Einwägen anstatt durch Abmessen im Messzylinder herzustellen.

Diplomarbeit Fabienne Feurer Diskussion
22
6. Diskussion 6.1. Binding Assay nach Lindmo 6.1.1. Parameter zur Etablierung des Lindmo Zu Beginn der Arbeit stellte sich die Frage, ob der Binding Assay nach Lindmo und seine Ergebnisse genügend Aussagekraft besässen, um erstens die Qualität der Markierung zu beurteilen und zweitens die Bindungsaffinität via Kd-Wert zu charakterisieren. Dazu musste versichert werden können, dass alle Parameter, die entweder eingreifend oder begleitend für den Assay eine Rolle spielten, bekannt und geklärt waren. Diese Parameter waren: -Peptidkonzentration -Zellkonzentration -Qualität des markierten Neurotensin-Derivates -Puffersubstanzen -Aktivitätsmenge -Wahl des nicht-markierten Peptids für die Bestimmung der NSB -Inkubation Das Erreichen einer Sättigung der Bindung von Peptid an Rezeptor ist an das Entstehen eines Gleichgewichts nach dem Massenwirkungsgesetz gebunden. Die Bestimmung des Kd-Wertes beruht auf folgender Gleichung: [Rez.] * [Peptid] Kd = -------------------------- [Rez.-Peptid] Dabei würde im Normalfall mit "freien" Rezeptoren gerechnet. Da die Rezeptoren aber in diesem Fall auf der Zelloberfläche sitzen, kann Kd nur bedingt als korrekter thermodynamischer Wert angenommen werden. Es zeigte sich aber, dass sowohl die Analyse nach Scatchard als auch nach Lindmo in der Praxis zuverlässige Werte lieferten. Schliesslich geht es vor allem darum, ein Mass für die Bindungseigenschaften des markierten Peptids zu haben, und dies wird auch durch den Einbezug der Bestimmung der Nicht Spezifischen Bindung erfüllt. Das gewählte Verhältnis zwischen Peptid und Zellen pro well (viel weniger Peptid und ansteigende Zellkonzentration) erwies sich als geeignet, wobei die Zellkonzentration den Wert 3.5 mio Zellen/ml möglichst nicht überschreiten sollte. Möglicherweise hat hier die erhöhte Clusterbildung im Zentrum der wells eine negative Auswirkung. Es zeigte sich nämlich, dass ab einer Zellkonzentration von 3.5 mio Zellen/ml (Konzentration für 1. Kolonne der Verdünnungsreihe) die Sättigungskurve (SB/Standard) zur höchsten Zellkonzentration hin deutlich abnahm. Der Grund dafür könnte sein, dass hier aufgrund des Clusters Rezeptoren schlechter zugänglich sind als bei der zweithöchsten Zellkonzentration. Es könnten aber auch zu hohe Werte erhalten werden: Eine erhöhte Bildung von Zellclustern im Well-Zentrum könnte eine schwer abschätzbare Diffusion von markiertem Peptid in dortige Zellzwischenräume nach sich ziehen. Es stellt sich die

Diplomarbeit Fabienne Feurer Diskussion
23
Frage, ob der Konzentrationsgradient von äusserer Umgebung nach den Zellzwischenräumen im Cluster genügend stark gegen die Diffusionsbarrieren der Zellverbände wäre. Falls ja, hätte das Peptid während der 2 h Inkubation die Möglichkeit, in die Zwischenräume zu gelangen. Bei der kurzen Waschzeit nach dem Binding Assay würde allerdings nicht mehr die gleiche Menge hinausgewaschen. Dies führte dann zu falsch hohen Werten der Totalen Bindung. Die Erfahrung zeigte, dass zuverlässige Resultate mit max. 2-3.5 mio Zellen / ml (1-1.75 mio Zellen / well) erhalten werden konnten. Der auf den Assay folgende Waschvorgang kann zu Zellverlusten führen. Hierbei ist der prozentuale Fehler umso grösser je weniger Zellen ausplatiert wurden. Dies würde zu falsch niedrigen Werten der Totalen Bindung führen. Aus diesem Grund und nach bisheriger Erfahrung sollte nicht mit weniger als 0.06-0.1 mio Zellen / ml (0.03-0.05 mio Zellen / well) gearbeitet werden. Grundsätzlich könnte eine Clusterbildung am ehesten vermieden werden, wenn noch grössere Wells gewählt würden, oder man verwendet suspendierte Zellen für den ganzen Test. Dann ist jedoch die Handhabung nach dem Test einiges aufwendiger (Zentrifugation, Waschen) und bei der Zentrifugation muss darauf geachtet werden, dass die Zellen dabei nicht zerstört werden und so internalisierte Peptid-Rezeptorkomplexe ungewollt in Lösung gingen. Die Entnahme des Überstandes muss praktisch gut durchführbar sein, da sonst experimentelle Fehler vorprogrammiert sind. Die Qualität der markierten Peptide hängt ab von der Ausbeute und dem Anteil Pertechnetat und Carbonyl. Falls diese Anteile sehr gross sind, sollte eine Reinigung durchgeführt werden. So wird ein optimaler Bindungsassay ermöglicht, da auch kein kaltes Peptid die Reaktion (Komplexbildung) zwischen Peptid und Rezeptor konkurrenzieren könnte. Der Lindmo wird in einem Binding Buffer (BB) mit FBS oder BSA durchgeführt. Der Binding Buffer enthält verschiedene Substanzen, die einen möglichst physiologischen Zustand ermöglichen. Der Trypsininhibitor verhindert, dass sich die Zellen während des Versuchs ablösen können und sorgt so für intakte Bindungsstellen. Die Antibiotika dienen zur Abwehr von Kontamination, welche während des Versuchs nicht ausgeschlossen werden kann und welche den Tod der Zellen zur Folge haben könnte (=Verlust von Rezeptoren). Zur Frage, warum der BB mit BSA bessere Resultate lieferte als der BB mit FBS, könnte folgendes eine Erklärung sein: FBS enthält im Unterschied zu BSA mehrere Wachstumsfaktoren, die auch gewisse zelleigene Enzyme induzieren oder inhibieren könnten. Ein weiterer Faktor ist, dass FBS teurer als BSA ist, und wenn BSA mindestens gleichgute Resultate liefert, dies dem FBS vorzuziehen wäre.

Diplomarbeit Fabienne Feurer Diskussion
24
Vor der Inkubation mit dem markierten Peptid werden die Zellen mit Binding Buffer BSA gewaschen. Damit werden verbliebene Reste des Mediums vollständig entfernt. Dies wäre auch mit PBS1x möglich. Nach dem Versuch wird mit einfachem PBS1x gewaschen und die Zellen mit 1N NaOH abgelöst. Eine Ablösung mit Trypsin 0.05 % ist ebenfalls erfolgreich, lässt aber anschliessend keine Proteinbestimmung mehr zu (Trypsin reagiert mit). Die Wahl des kalten Peptids für die Bestimmung der nichtspezifischen Bindung führte zu NT 8-13, da es zu NT-VIII und NT-XI die ähnlichsten Bindungs-eigenschaften besitzt. Der Grund warum z.B. kaltes NT-XI schlechter bindet als markiertes NT-XI ist noch unklar (Vergleich IC50- und Kd-Werte). Der freie Ligand mit (NαHis)Ac hat wegen der Protonierung eine Ladung von 2+ und 1- (netto 1+), währenddem der Komplex (=markiertes NT-Derivat) ungeladen ist. Es könnte sein, dass die Ladung die Bindung an den Rezeptor stört. Dass das nicht an die Rezeptoren bindende Prolin-7 die Bindung von NT 7-13 an Rezeptoren nicht stört, heisst nicht automatisch, dass sich das nicht komplexierte (NαHis)Ac (im Falle von kaltem NT-XI) ebenso verhält. Obwohl die bindende Einheit NT 8-13 ist, hat die Einführung von Liganden resp. Komplexen einen Einfluss auf die Bindung an Rezeptoren. Für die Experimente nach Lindmo mit ungereinigtem Produkt, wo neben markiertem noch kaltes Peptid vorhanden ist, erweist sich die schlechtere Bindungsfähigkeit des kalten Peptids zumindest als Vorteil. 6.1.2. Gamma-Counter Der messbare Bereich im y-Counter ergibt eine untere Grenze der Zellzahl (messbare Peptidkonzentration : Zellbindungsstellen) und die obere und untere Grenze der zu wählenden Aktivität. Die ausgewählte Aktivitätsmenge / well lag bei 20 kBq und würde ca. 1'200'000 cpm entsprechen. Bei mehr als ca 2'000'000 cpm verlässt man den linearen Bereich des Counters, bei weniger als ca. 1000 cpm erfolgt ein grösserer Fehler in der Zählstatistik. 6.1.3. Bedingungen für den Binding Assay nach Lindmo Die zum Schluss gewählten Bedingungen sollten nach einer guten Markierung und bei intaktem Peptid ein gutes Resultat nach Lindmo-Plot ergeben. Gewisse Parameter wie Peptidkonzentration, Zellkonzentration und Verdünnungsreihe, Aktivitätsmenge und Puffersubstanzen können und dürfen kleineren Schwankungen unterlegen sein. Eine Abweichung von diesen "optimalen" Parametern muss nicht unbedingt eine Verschlechterung der Resultate zur Folge haben. Eine gute Qualität der Markierung sowie die Einhaltung der Inkubationsbedingungen sollten aber vorausgesetzt bleiben.

Diplomarbeit Fabienne Feurer Diskussion
25
6.2. Einfrieren von Zellen für den Binding Assay HT-29 Zellen konnten nicht für längere Zeit fixiert gefroren oder gekühlt aufbewahrt werden. Probleme entstanden dadurch, dass die Zellen schon die Trocknung vor dem Einfrieren wahrscheinlich nicht überlebten. Die Zellform veränderte sich. Beim anschliessenden Einfrieren auf –20°C gingen vermutlich die Rezeptoren kaputt. Die zweite Methode sah eine Aufbewahrung in Binding Buffer bei 4°C vor. Doch die Zellen lösten sich auch hier nach einiger Zeit vom Boden d.h. waren nicht mehr konfluent. Die Vermutung liegt nahe, dass durch die Behandlung bei solch tiefen Temperaturen die Zellen ihre Rezeptoren und andere Oberflächenproteine entweder internalisiert hatten oder dass diese zerstört wurden. Während des Binding Assays lösten sich ausserdem die Zellen teilweise oder ganz vom Boden ab und schwammen obenauf. Die Rezeptoren waren nicht mehr intakt oder nicht mehr zugänglich für das markierte Peptid. Es war aber möglich, die Zellen fixiert vorbereitet einen Tag länger als normal aufzubewahren. Dabei könnte z.B. Medium mit nur 1% FBS verwendet werden (Zellen wachsen weniger schnell als mit 10% FBS). 6.3. Markierungsbedingungen 6.3.1. Puffersubstanz Die Vorteile von Phosphatpuffer/HCl überwiegen den zuvor längere Zeit verwendeten MES-Puffer/HCl. Die Löslichkeit von MES ist eingeschränkt. Oft gab es schon bei der Herstellung der MES-Neutralisationslösung das Problem der schnell übersättigten Lösung. Bei Phosphatpuffer/HCl ist nach der Neutralisation bereits eine gewisse Physiologie gegeben, so dass nach Qualitätskontrolle die Lösung direkt weiterverwendet werden kann. 6.3.2. pH-Bereich Mit der Phosphatpuffer/HCl-Neutralisationslösung können pH-Werte von pH 6 bis pH 8.5 eingestellt werden. Peptide bleiben im allgemeinen in saurer Lösung länger stabil als in leicht basischer. Ob das Peptid bei pH 8.5 schneller reagiert, wurde nicht weiter untersucht. Sicher ist, dass nach einigen Stunden das Peptid nicht mehr intakt war. Die HPLC-Spur zeigte mehrere Zerfallsprodukte.

Diplomarbeit Fabienne Feurer Diskussion
26
6.4. Zwei Produktepeaks bei NT-VIII und NT-XI Das Vorkommen von zwei Produktepeaks weist erstmal auf zwei verschiedene Konfigurationen hin. Inkubationskontrollen zeigten, dass beide Substanzen in ein Gleichgewicht wechseln, wenn genügend Energie über genügend lange Zeit zur Verfügung steht. Die Beobachtung, dass aus den isolierten Peaks bei 75°C dasselbe Peakmuster entsteht, beweist, dass ein Gleichgewicht vorhanden ist. Die wenig verschiedenen Retentionszeiten lassen auf Unterschiede in der Konformation (verschiedene Isomere der Komplexe) oder Polarität schliessen. Eine erste Hypothese war, dass ein Teil des Carbonyls nur zweifach (mit zwei Koordinationsbindungsstellen), ein anderer Teil jedoch dreifach die (NαHis)Ac-Bindungsstellen besetzte. Die zweifach gebundene markierte Form enthält möglicherweise eine zusätzliche positive Ladung, welche die Polarität beeinflusst (je nachdem ob die Carboxylatgruppe oder Amingruppe frei bleibt; bei einem pH-Wert von 6-7 ist das Amin protoniert). Beide Formen könnten dabei miteinander im Gleichgewicht liegen und zur Bildung der dreifach gebundenen markierten Form wäre mehr Energie nötig. Bei NT-VIII wurde dies deutlich, da dort zu Beginn mehr vom ersten Peak vorhanden war und dieser Teil nach 2 h deutlich abgenommen hatte. Bei NT-XI war dies allerdings schwerer ersichtlich zumal dort noch ein dritter Peak vermutet werden musste. Also konnte dies nicht die einzige Hypothese bleiben. Eine zweite Hypothese berücksichtigt mehr die Seite der unterschiedlichen Raumbeanspruchung der verschiedenen Produkte. Sie geht davon aus, dass grundsätzlich mehrere Peptidfaltungen möglich sind und zwar unabhängig von einer Markierung. Ein Gleichgewicht durch kontinuierliche Umlagerungen wäre auch hier möglich. Es wird vermutet, dass es mehr und weniger stabile Faltungen gibt. Die stabileren treten schliesslich in erhöhtem Masse auf als die weniger stabileren. Dass diese unterschiedlich gefalteten Peptidkonformationen auch unterschiedliche Bindungseigenschaften haben könnten, liegt nahe. Um die tatsächliche räumliche Anordnung zu verstehen, wären in diesem Fall zusätzliche Analysen nötig, z.B. MS und NMR. Da in den Bindungseigenschaften der beiden Formen des markierten Peptids nur kleine Unterschiede resultierten, wird vermutet, dass die Strukturunterschiede im Tc-(NαHis)Ac-Komplex lokalisiert sind, welche jedoch noch nicht bewiesen sind. 6.5. Schlussfolgerungen Der Binding Assay nach Lindmo eignet sich für die regelmässige Qualitätskontrolle bei einer geplanten Humananwendung. Das Bestehen des Gleichgewichts zwischen den zwei Formen des markierten Peptids und deren gleicher Bindungscharakter wird es erlauben, die Mischung beider Formen zur Injektion am Patienten zu verwenden.

Diplomarbeit Fabienne Feurer Experimenteller Teil
7. Experimenteller Teil 7.1. Chemikalien Reagentien und Materialien wurden aus folgenden Quellen erhalten: Kultur- medium McCoy's 5A-GLUTAMAX ITM, Foetales Rinder Serum (FBS), Antibiotische/Antimykotische Lösung (PSF), Trypsin/EDTA, Rinder Serum Albumin (BSA) und Soybean Trypsin Inhibitor wurden von GIBCO BRL Life Technologies AG (Basel, Schweiz) erhalten. HEPES, Chymostatin, Bacitracin und Neurotensin-Fragment 8-13 wurden von Sigma (Buchs, Schweiz) erhalten. EGTA (Ethylenglycol-bis(aminoethylether)-tetraessigsäure), Morpholinoethansulfonsäure (MES), Ethanol, Trifluoressigsäure (TFA), Na2CO3, NaBH4 sowie NaKTartrat wurden von Fluka (Buchs, Schweiz) erhalten. HCl, NaCl, MgCl2, NaOH, KCl, Na2HPO4 und NaH2PO4*H2O wurden bei Merck (Dietikon, Schweiz) eingekauft. Der Technetium-Generator "Ultratechnekow FM" wurde von Mallinckrodt Diagnostica (Dietzenbach, Deutschland) erhalten. Einmal täglich morgens wurde mit physiologischer Kochsalzlösung (0.9% NaCl) eluiert. Das frische Eluat enthielt eine Mischung von 99mTc und 99Tc in Form von Pertechnetat. Es wurde immer mit frischem Eluat gearbeitet. 7.2. Pufferlösungen PBS 5x NaCl 0.5 M 29.22 g/l Na2HPO4 0.21 M 29.81 g/l NaH2PO4*H2O 0.055 M 7.59 g/l mit 1N NaOH auf pH 7.4 einstellen (ca. 26.6 ml) (PBS1x = PBS5x 1:5 verdünnt) Phosphatpuffer/HCl Phosphatpuffer 1M: Na2HPO4 11.59 g NaH2PO4*H2O 2.53 g in 100 ml Neutralisationslösung: 2 Teile HCl 1M (Titrisol) + 1 Teil Phosphatpuffer 1M
Diplomarbeit Fabienne Feurer Experimenteller Teil
28
Binding Buffer BSA Hepes 50 mM 11.9 g/l NaCl 125 mM 7.4 g/l KCl 7.5 mM 0.57 g/l MgCl2 5.5 mM 1.1 g/l EGTA 1 mM 0.38 g/l BSA (+4°C) 0.5% (w/v) 5 g/l Chymostatin (-20°C) (5mg / 0.5ml PBS1x) 2 mg/l Soybean Trypsin Inhibitor (-20°C) 100 mg/l Bacitracin (+4°C) 50 mg/l mit 1N NaOH auf pH 7.4 einstellen (ca. 20 ml) bei +4°C aufbewahren 7.3. HPLC-Gradienten Gradient Zeit [Min.] A [%] (0.1%TFA) B [%] (70%v/vEtOH,0.1%TFA) Flow [ml/min.] Gradient Zeit [Min.] A [%] (0.1%TFA) B [%] (70%v/vEtOH,0.1%TFA) Flow [ml/min.]
1 Initial 100 0 0.00 2 Initial 100 0 0.003.00 100 0 1.00 3.00 100 0 1.005.00 60 40 1.00 6.00 60 40 1.00
11.00 60 40 1.00 12.00 60 40 1.0012.00 50 50 1.00 16.00 50 50 1.0030.00 50 50 1.00 30.00 50 50 1.0034.00 0 100 0.20 34.00 0 100 0.2036.00 0 100 0.20 36.00 0 100 0.2040.00 100 0 0.20 40.00 100 0 0.20
100.00 100 0 0.20 100.00 100 0 0.20101.00 100 0 0.00 101.00 100 0 0.00
3 Initial 100 0 0.00 4 Initial 100 0 0.003.00 100 0 1.00 3.00 100 0 1.006.00 60 40 1.00 6.00 60 40 1.00
12.00 60 40 1.00 12.00 60 40 1.0020.00 50 50 1.00 25.00 50 50 1.0030.00 50 50 1.00 32.00 50 50 1.0034.00 0 100 0.20 34.00 0 100 0.2036.00 0 100 0.20 36.00 0 100 0.2040.00 100 0 0.20 40.00 100 0 0.20
100.00 100 0 0.20 100.00 100 0 0.20101.00 100 0 0.00 101.00 100 0 0.00
5 Initial 100 0 0.00 6 Initial 100 0 0.003.00 100 0 1.00 3.00 100 0 1.005.00 60 40 1.00 5.00 60 40 1.00
11.00 60 40 1.00 8.00 60 40 1.0017.00 52 48 1.00 25.00 50 50 1.0025.00 50 50 1.00 32.00 50 50 1.0030.00 50 50 1.00 34.00 0 100 0.2036.00 0 100 0.20 36.00 0 100 0.2040.00 0 100 0.20 40.00 100 0 0.2044.00 100 0 0.20 100.00 100 0 0.20
100.00 100 0 0.20 101.00 100 0 0.00101.00 100 0 0.00

Diplomarbeit Fabienne Feurer Experimenteller Teil
29
7.4. Markierung von Neurotensin-Derivaten 7.4.1. Carbonylierung 1. Kit herstellen: 4 mg Na2CO3, 5.5 mg NaBH4 und 15 mg NaKTartrat in Penicillin
Vial mind. 60 Min. mit N2 spülen, dann verschliessen 2. Penicillin Vial mit CO während 30 Minuten spülen 3. 2-4 ml Generatoreluat mit 0.5 bis 40 GBq Natriumpertechnetat mit Spritze
zugeben 4. Reaktion bei 75°C während 45-60 Minuten 5. Neutralisation mit Phosphatpuffer/HCl auf pH 6-7.5
(Kontrolle mit pH-Stäbchen) 6. Auf RT abkühlen und HPLC-Kontrolle durchführen Allgemein: Zur Entnahme von Lösung wurde zusätzlich eine Spritze mit Aktivkohle und etwas Watte verschlossen aufgesetzt, damit der Druckausgleich stattfinden konnte, ohne dass zuviel Sauerstoff hineingelangen und ohne dass Dampf entweichen konnte. 7.4.2. Reaktion mit Neurotensin 7. 10-15 µl Peptidlösung 1 mM in Reactivial mit N2 spülen, gut verschliessen 8. 0.2-0.6 ml 99mTc-Carbonyl-Lösung (Produkt von Carbonylierung) zufügen 9. Reaktion bei 75°C während 30-120 Minuten 10. HPLC-Kontrolle 11. Reinigung 7.5. Reinigung und Sammeln von Fraktionen Zur Reinigung wurde möglichst viel des Produkts der Markierung durch die HPLC eluiert. Sobald das Peptid im Chromatogramm erschien, wurden jede Minute Fraktionen gesammelt. Dies ergab bei einem Fluss von 1 ml/min ein Volumen von 1 ml pro Fraktion. Anschliessend wurde die Aktivität der Fraktionen gemessen. Zur weiteren Verarbeitung mussten die Fraktionen häufig neutralisiert werden, da nach der Reinigung ein pH-Wert von 2-3 (TFA in Lösungsmitteln) vorlag. Es konnte mit 10 µl 1N NaOH und 100 µl PBS5x auf einen pH-Wert zwischen pH 6.5 und 7.5 neutralisiert werden. Der Alkoholgehalt war häufig zu hoch für eine Behandlung in vivo wie in vitro (v.a. Scatchard, stellte für Lindmo kein Problem dar, da Verdünnung für Assay gross genug), daher konnte die Fraktion mit Stickstoff gespült und dann leicht erwärmt werden, wobei Alkohol verdunstete und sich die Peptidkonzentration erhöhte (vorher und nachher wägen).
Diplomarbeit Fabienne Feurer Experimenteller Teil
30
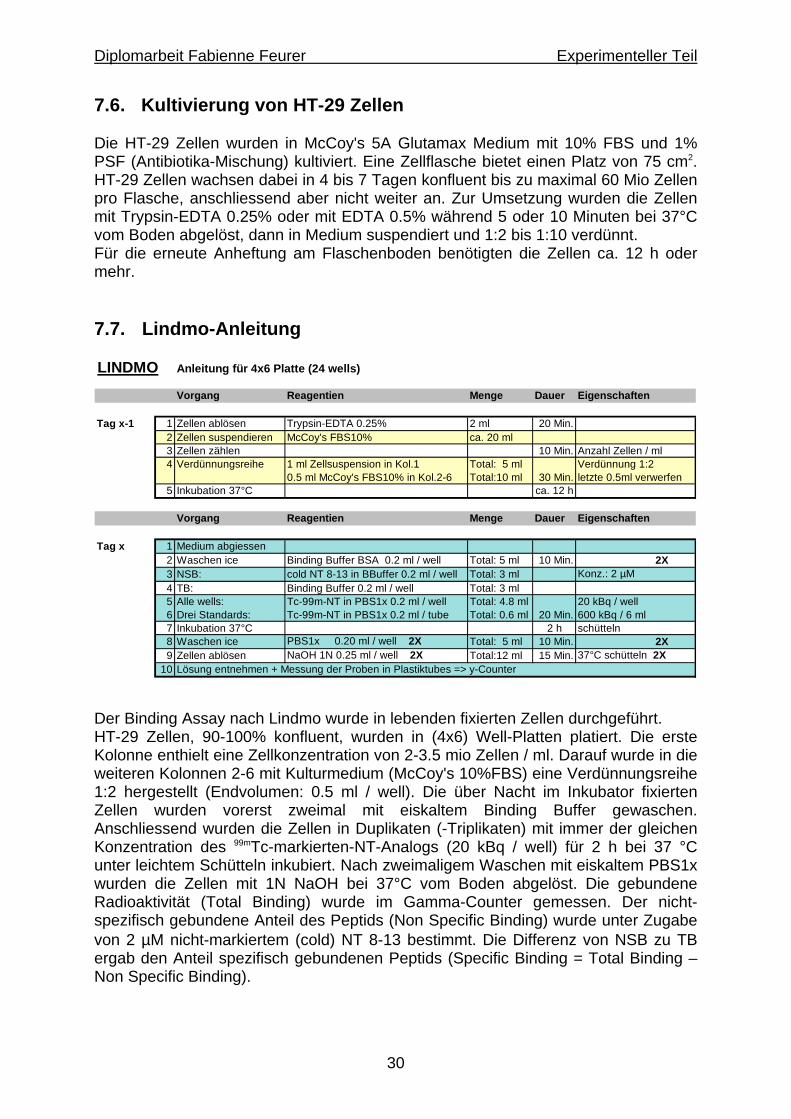
7.6. Kultivierung von HT-29 Zellen Die HT-29 Zellen wurden in McCoy's 5A Glutamax Medium mit 10% FBS und 1% PSF (Antibiotika-Mischung) kultiviert. Eine Zellflasche bietet einen Platz von 75 cm2. HT-29 Zellen wachsen dabei in 4 bis 7 Tagen konfluent bis zu maximal 60 Mio Zellen pro Flasche, anschliessend aber nicht weiter an. Zur Umsetzung wurden die Zellen mit Trypsin-EDTA 0.25% oder mit EDTA 0.5% während 5 oder 10 Minuten bei 37°C vom Boden abgelöst, dann in Medium suspendiert und 1:2 bis 1:10 verdünnt. Für die erneute Anheftung am Flaschenboden benötigten die Zellen ca. 12 h oder mehr. 7.7. Lindmo-Anleitung LINDMO Anleitung für 4x6 Platte (24 wells)
Vorgang Reagentien Menge Dauer Eigenschaften
Tag x-1 1 Zellen ablösen Trypsin-EDTA 0.25% 2 ml 20 Min.2 Zellen suspendieren McCoy's FBS10% ca. 20 ml3 Zellen zählen 10 Min. Anzahl Zellen / ml4 Verdünnungsreihe 1 ml Zellsuspension in Kol.1 Total: 5 ml Verdünnung 1:2
0.5 ml McCoy's FBS10% in Kol.2-6 Total:10 ml 30 Min. letzte 0.5ml verwerfen5 Inkubation 37°C ca. 12 h
Vorgang Reagentien Menge Dauer Eigenschaften
Tag x 1 Medium abgiessen2 Waschen ice Binding Buffer BSA 0.2 ml / well Total: 5 ml 10 Min. 2X3 NSB: cold NT 8-13 in BBuffer 0.2 ml / well Total: 3 ml Konz.: 2 µM4 TB: Binding Buffer 0.2 ml / well Total: 3 ml5 Alle wells: Tc-99m-NT in PBS1x 0.2 ml / well Total: 4.8 ml 20 kBq / well6 Drei Standards: Tc-99m-NT in PBS1x 0.2 ml / tube Total: 0.6 ml 20 Min. 600 kBq / 6 ml7 Inkubation 37°C 2 h schütteln8 Waschen ice PBS1x 0.20 ml / well 2X Total: 5 ml 10 Min. 2X9 Zellen ablösen NaOH 1N 0.25 ml / well 2X Total:12 ml 15 Min. 37°C schütteln 2X
10 Lösung entnehmen + Messung der Proben in Plastiktubes => y-Counter
Der Binding Assay nach Lindmo wurde in lebenden fixierten Zellen durchgeführt. HT-29 Zellen, 90-100% konfluent, wurden in (4x6) Well-Platten platiert. Die erste Kolonne enthielt eine Zellkonzentration von 2-3.5 mio Zellen / ml. Darauf wurde in die weiteren Kolonnen 2-6 mit Kulturmedium (McCoy's 10%FBS) eine Verdünnungsreihe 1:2 hergestellt (Endvolumen: 0.5 ml / well). Die über Nacht im Inkubator fixierten Zellen wurden vorerst zweimal mit eiskaltem Binding Buffer gewaschen. Anschliessend wurden die Zellen in Duplikaten (-Triplikaten) mit immer der gleichen Konzentration des 99mTc-markierten-NT-Analogs (20 kBq / well) für 2 h bei 37 °C unter leichtem Schütteln inkubiert. Nach zweimaligem Waschen mit eiskaltem PBS1x wurden die Zellen mit 1N NaOH bei 37°C vom Boden abgelöst. Die gebundene Radioaktivität (Total Binding) wurde im Gamma-Counter gemessen. Der nicht-spezifisch gebundene Anteil des Peptids (Non Specific Binding) wurde unter Zugabe von 2 µM nicht-markiertem (cold) NT 8-13 bestimmt. Die Differenz von NSB zu TB ergab den Anteil spezifisch gebundenen Peptids (Specific Binding = Total Binding – Non Specific Binding).

Diplomarbeit Fabienne Feurer Experimenteller Teil
31
7.8. Messung im Gamma-Counter Zusätzlich zu den einzelnen Proben wurden drei mal eine Menge Standard in die Messröhrchen transferiert. Die Messung erfolgte automatisch, wobei für eine lineare, d.h. genaue Messung die untere Grenze bei ca. 1000 cpm und die obere bei ca. 2'000'000 cpm liegt. 7.9. Auswertung der Resultate zum Lindmo Die gemessenen Werte enthielten den Standard, die Totale Bindung und die Nicht-Spezifische Bindung. Aus der Differenz konnte die Spezifische Bindung berechnet werden. In einem Diagramm wurde darauf der relative Anteil von Totaler, Nicht- Spezifischer und Spezifischer Bindung zum Standard (SB/Standard) gegen die Zellkonzentration in mio/ml dargestellt. Die Nicht Spezifische Bindung sollte dabei weit unter der Totalen Bindung liegen, bestenfalls <1%. Für den Lindmo Plot wurden die Werte der Spezifischen Bindung invertiert, so dass wir ein Diagramm von Standard/SB versus 1/Zellkonzentration erhielten. Die Punkte sollten auf einer Geraden liegen. Zur Anpassung an die Punkte wurde eine lineare Regression durchgeführt. Die gesuchten Daten erhielten wir aus der Geradengleichung. Gesucht waren: Steigung für Kd-Berechnung und y-Achsenabschnitt für Bestimmung des bindenden Anteils an total zugegebenem Peptid (% vom Standard). Berechnungen Kd [mol/l] = Steigung [mio Zellen/ml] * Faktor [mol Rezeptoren*103/mio Zellen] -Faktor = 7.5*10-11 mol Rezeptoren*103/mio Zellen -y-Achsenabschnitt = 1/r r = reaktiver bzw. bindender Anteil des Standards (extrapoliert auf unendlich viele Zellen bzw. mole Rezeptoren) Die Berechnung des Faktors beruhte auf Daten eines Binding Assays von 99mTc-NT-IX an HT-29 Zellen nach Scatchard, woraus durch Bmax (bei Kd = 1 nM) und durch Proteinbestimmungen (225*10-15 mol Rezeptoren / mg Protein und 3.3 * 10-7 mg Protein / Zelle) die Anzahl der Mole Rezeptoren pro Zelle berechnet werden konnten. Demnach besitzt eine HT-29 Zelle im Durchschnitt 45'000 Rezeptoren, was 7.5*10-
20 Molen Rezeptoren pro Zelle entspricht. (Division durch Loschmidtsche Zahl 6.023*1023 Teilchen/mol)

Diplomarbeit Fabienne Feurer Experimenteller Teil
32
7.10. Problembehandlung Mögliche Fehlerquellen bei unerwartet schlechtem Lindmo Die Nicht Spezifische Bindung ist zu hoch: -es wurde zuwenig cold NT 8-13 eingesetzt oder dieses ist nicht mehr intakt -Zellzahl in den betroffenen Reihen hat verhältnismässig stark zugenommen -die Anzahl der NTS1-Rezeptoren war aus irgendeinem Grund vermindert Die Totale Bindung ist zu niedrig: -ein Teil des markierten Neurotensin-Derivates ging bei der Herstellung kaputt -die Anteile an freiem Pertechnetat und/oder Carbonyl und/oder der Anteil an nicht markiertem Peptid waren überdurchschnittlich hoch -Die eingesetzte Peptidmenge / well war um mehrere Zehnerpotenzen grösser als die Menge Bindungsstellen / well Mögliche negative Einflüsse durch Zellen auf den Binding Assay nach Lindmo -unregelmässiges Wachstum nach Ausplatieren während 12 h als Funktion der Zellzahl (weniger Zellen-->schnelleres Wachstum und umgekehrt) -Wachstum während Inkubationszeit aufgrund Temperatur oder Neurotensin-induziert

Diplomarbeit Fabienne Feurer Literatur
33
8. Literatur [1] D. Block, R.I.J. Feitsma, P. Verneij, E.J.K. Pauwels (1999) Eur J. Nucl. Med 26, 1511-1519, "Peptide radiopharmaceuticals in nuclear medicine" [2] P. Kitabgi et al (1992) Ann. N.Y. Acad. Sci., 668, und T.P. Davis et al (1993) Crit. Rev. Neurobiol. 7 [3] J.P. Vicent, J. Mazella and P. Kitabgi (1999) TIPS 20 : 302-309 "Neurotensin and neurotensin receptors" [4] Elisa Garcia Garayoa, Lesley Allemann-Tannahill, Peter Bläuenstein, Martine Willmann, Nathalie Carrel-Remy, Dirk Tourwé, P. August Schubiger (1999) "Biological characterisation of new 99mTc-labelled neurotensin analogues" Publikation eingereicht [5] P.A. Schubiger, E. Garcia-Garayoa and Peter Bläuenstein (2000) "Design and Radiolabelling of stable Neurotensin derivatives" Publikation eingereicht [6] David B. Bylund and Henry I.Yamamura (1990) "Methods in Neurotransmitter receptor analysis" edited by Henry I.Yamamura et al., 1-35 "Methods for Receptor Binding" Raven Press New York [7] H. Stapfer, S. Schneebeli, P.A. Schubiger (1989) Zentrum für Radiopharmazie, PSI Villigen AG, "Kontrollvorschrift für I-123-Granuloszint" [8] T. Lindmo, E. Boven, F. Cuttitta, J. Fedorko and P.A. Bunn, Jr. (1984) J. of Immunolog. Methods 72, 77-89 "Determination of the Immunoreactive Fraction of Radiolabelled Monoclonal Antibodies by Linear Extrapolation to Binding at Infinite Antigen Excess" [9] R. Ian Freshney (1987) "Culture of Animal Cells – A Manual of Basic Technique" Wiley-Liss, New York





























