Embed Size (px)
Citation preview

Malaria is one of the most widespread infectious diseases of our time. The global malaria map has been shrinking over the past 50 years, but today more people are at risk of suffering from malaria than at any other time in his-tory: ~40% of the world’s population live in countries where the disease is endemic and ~247 million people suffer from the disease every year1.
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anaemia, cerebral malaria and, if untreated, death. Plasmodium falciparum is the domi-nant species in sub-Saharan Africa, and is responsible for the almost 1 million deaths each year1. The disease burden is heaviest in African children under 5 years of age (who have frequent attacks and little immunological protection) and in pregnant women1. Malaria is both a cause and a consequence of poverty: in countries with intense malaria transmission, the economic impact of the disease slows economic growth by 1.3% per year2, which translates into a gross domestic product cost in sub-Saharan Africa of US$12 billion per year3.
The global fight to control malaria requires a multi-faceted approach. At present, we have a wide range of effective tools, which include insecticide and larvicide spraying, the use of insecticide-impregnated bed nets to protect against infection by mosquitoes, and medicines to both treat the infection and prevent it in pregnant
women and in children under 5 years of age. However, long-term prophylaxis by vaccination has been especially challenging as the parasite has various sophisticated mechanisms to avoid the host immune system. Currently, no vaccine is available. The most effective first-generation vaccine in development, GlaxoSmithKline (GSK)’s RTSS, reduces the risk of clinical malaria, delays time to new infection and reduces episodes of severe malaria for at least 18 months4. It has recently entered Phase III clinical trials. Even with all available strategies combined, gaps remain in our armoury against malaria, and a substantial number of patients will suffer from this disease over the coming decades.
In November 2007, the Bill and Melinda Gates Foundation set an agenda with the final goal of the eradi-cation of malaria (that is, the extinction of all species of Plasmodium that can cause malaria in humans)5. This goal is also supported by the World Health Organization (WHO) through its Roll Back Malaria (RBM) partner-ship (see Further information), by the Global Fund to Fight AIDS, Tuberculosis and Malaria (see Further information), a related developing initiative — the Affordable Medicines Facility for malaria (AMFm) — and the Malaria Eradication Research Agenda (MalERA; see Further information). To achieve the goal of eradica-tion, antimalarial drug discovery must continue working on the development of new medicines to treat malaria
*Medicines for Malaria Venture, 20 Route de Pré-Bois, 1215 Geneva, Switzerland.‡Barcelona Centre for International Health Research, Hospital Clinic/IDIBAPS, Universitat de Barcelona, Barcelona, Spain, and Centro de Investigação em Saúde de Manhiça, Manhiça, Maputo, Mozambique. Correspondence to T.N.C.W.e-mail: [email protected]:10.1038/nrd2972Published online 16 October 2009
New medicines to improve control and contribute to the eradication of malariaTimothy N. C. Wells*, Pedro L. Alonso‡ and Winston E. Gutteridge*
Abstract | Despite being one of the most prevalent tropical diseases, for many years malaria was not a commercial priority for the pharmaceutical industry. However, in response to the emergence and spread of resistance to the available antimalarial drugs, there has been a renaissance in the discovery and development of new medicines to control the disease in the last few years. The persistent threat of resistance means that new molecules with novel mechanisms of action are continually required. Furthermore, the recent call for the elimination and eradication of malaria has prompted an extension of the stages of the life cycle of malaria parasites that should be targeted by new molecules. Recent advances in genome-based technologies and in in vitro screening of whole parasites have broadened the range of therapeutic targets and are accelerating the development of a new generation of treatments for both malaria control and eradication.
REVIEWS
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 879
© 2009 Macmillan Publishers Limited. All rights reserved

Artemisinin-based combination therapy(ACT). A treatment regimen for malaria, usually comprising two medicines, one of which is artemisinin or one of its derivatives. ACTs come in two varieties: co-packaged or the preferred co-formulated.
MerozoiteThe life cycle stage in which the malaria parasite has the ability to penetrate and infect red blood cells. The parasite expresses receptors and proteolytic enzymes on its surface to facilitate this process.
HypnozoiteThe dormant liver stage of P. vivax and P. ovale. The parasite remains in hepatocytes after the other liver-stage parasites have replicated and released their merozoites. Hypnozoites are the source of malaria relapses.
GametocyteThe male and female sexual stages of the malaria parasite. Formed in the red blood cells, it remains dormant unless taken up by a mosquito, in which it forms gametes, which fuse to form the ookinete.
episodes, mainly targeting the asexual blood stages of P. falciparum. However, an arsenal of drugs will also be needed to tackle emerging drug resistance, block the transmission of the parasite to other persons by the mosquito vector and, in the case of Plasmodium vivax infection, target the dormant liver stage of the parasite.
In this Review, we examine the currently available antimalarial drugs, their limitations and the need for new drug classes with improved product profiles. We also consider some of the challenges that are faced in the development and delivery of new medicines in a cost-effective and timely fashion, and the solutions required to overcome them.
The challenges of the Plasmodium speciesThere are four main species of the malaria parasite that are known to infect humans. P. falciparum is responsible for the majority of the malaria-linked deaths in sub-Saharan Africa and is therefore currently the most important target. P. vivax causes as much as 25–40% of the global malaria burden6, particularly in South and Southeast Asia, and Central and South America. It does not nor-mally progress to cerebral malaria, and has traditionally been labelled benign. However, P. vivax causes a greater increase in the host inflammatory response than P. falci-parum at equivalent parasitaemia7. Also, it is probable that mortality from P. vivax is under-reported, as recent analyses in Papua (Indonesia) have shown similar mor-tality figures in children infected with P. vivax to those infected with P. falciparum8. The other two main species that are known to infect humans are Plasmodium ovale and Plasmodium malariae. Currently, these are diagnosed by microscopy and represent a small percentage of infec-tions. In addition, some human malaria can be caused by the primate malaria parasite, Plasmodium knowlesi, but it has not been established whether human–mosquito–human transmission can occur.
Drugs that are active on the asexual blood stages of P. falciparum, such as the artemisinin-based combination therapies (ACTs), are assumed to be fully active against the other species. Mixed infections of P. falciparum and P. vivax are rarely reported, but PCR detection methods have shown that these can be as high as 30% of all Plasmodium infections9. Diagnosis based on PCR analysis will undoubtedly lead to a re-evaluation of the presence of mixed infections.
From a treatment and eradication perspective, there are three key differences between the species. The first difference occurs in the liver. Following infection by a mosquito (FIG. 1), P. falciparum and P. malariae rapidly progress through the hepatocytes, undergoing asexual division with the release of large numbers of merozoites into the host bloodstream. For P. vivax and P. ovale, some of the parasites in the hepatocytes become dormant hypno-zoites10. These forms can be reactivated after periods which vary depending on the species and the host, and can be as short as 3 weeks or as long as several years. Unless the hypnozoites are eliminated, the disease caused by these species will continue to relapse. As P. vivax transmission is rarely intense, activation of hypnozoites is thought to be an important contributor to disease frequency.
The second difference between the species is in the time taken for the parasite to replicate in the host. The time between febrile paroxysms varies from ~48 hours f or P. falciparum and P. vivax to 72 hours for the more benign P. malariae. There has been a recent interest in P. knowlesi11, as PCR methods show that it is often mis-diagnosed as P. malariae infection, which is usually uncom-plicated and has low parasitaemia. However, P. knowlesi replicates every 24 hours, and so is potentially life threat-ening if not treated properly12. In this case, developing a therapy with a rapid onset of action will be crucial.
The third difference between the species is the timing of the appearance of gametocytes in the bloodstream13. In P. falciparum infection, the gametocytes do not appear until several days after the initial parasitaemia and fever whereas, in P. vivax infection, they appear concurrently or even before asexual parasites. An ideal treatment for blood stages of P. vivax must be able to kill existing gametocytes, rather than simply prevent them from differentiating.
Currently available antimalarialsThe first widely used antimalarial drug was quinine, a natural product extracted from the bark of the tree Cinchona calisaya. Synthetic derivatives of quinine (FIG. 2), such as the 8-aminoquinoline primaquine and the 4-aminoquinoline chloroquine, can be traced back to the pioneering work of Ehrlich on methylene blue in the late nineteenth century. These drugs cause parasite death by blocking the polymerization of the toxic by-product of haemoglobin degradation, haem, into insoluble and non-toxic pigment granules, resulting in cell lysis and parasite cell autodigestion14.
Rationally designed therapies have followed this antimetabolite approach. For example, malaria para-sites are unable to salvage folate, but need this cofactor to synthesize tetrahydrofolate for methylation reactions. Inhibitors of dihydropteroate synthase (by sulphona-mides such as sulphadoxine) and dihydrofolate reductase (by 2,4-diaminopyrimidines such as pyrimethamine) are potent antimalarial drugs, especially when administered in combination. For most of the second half of the twen-tieth century, control of acute uncomplicated malaria caused by all four species of Plasmodium involved the use of chloroquine for first-line treatment and a combi-nation of sulphadoxine and pyrimethamine as second-line treatment.
In all cases, resistance to these drugs has emerged. Resistance to drugs such as sulphadoxine–pyrimeth-amine, for which there are specific enzyme targets, arose more rapidly than to chloroquine, for which the target is haem polymerization. Also, for reasons that are unclear, chloroquine resistance arose more rapidly in P. falciparum than in P. vivax. The initial strategy to address this was to modify the molecular structure of the existing drugs. Synthesis of pyrimethamine-like drugs did not result in an active replacement. However, the synthesis of chloroquine-like drugs led to the discovery of lumefantrine, piperaquine and pyronaridine in China, and amodiaquine, mefloquine and halofantrine in the United States. All of these molecules were active against chloroquine-resistant strains of P. falciparum.
R E V I E W S
880 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved
Nature Reviews | Drug Discovery
Salivary gland Midgut
wall
Zygote
Sporozoite
Sporozoite infectshepatocyte
Schizont
Ookinete
Schizont
Merozoite
Red blood cell
Micro-gametocyte
Macro-gametocyte
Mammal
Mosquito
Hypnozoite
1
2
35
6
7
8
9
10
11
4
Oocyst
Fever clearance timeThe time taken for body temperature to return to normal levels after the first dose of an antimalarial has been given.
Drugs targeting P. falciparum. The key event that led to the current era of treatment for P. falciparum infection can be dated back to the discovery of the sesquiter pene lactone artemisinin (known as Qing hao su) in 1972 by Chinese scientists15. It is an endoperoxide-containing natural product isolated from the leaves of the sweet wormwood, Artemisia annua. More potent derivatives of artemisinin were subsequently developed, including dihydroartemisinin (DHA; the main active metabo-lite), artemether, artemotil and artesunate (FIG. 2). Whereas both artemether and artemotil are insoluble, artesunate has good solubility and is therefore suit-able for intravenous and oral administration. The new artemisinin derivatives were fully active against all existing drug-resistant strains of P. falciparum. Unlike other antimalarial drugs, the artemisinins rapidly kill all
the blood stages of the parasite, resulting in the shortest fever clearances times of all antimalarials16. Furthermore, the artemisinins also kill gametocyte stages, thereby reducing transmission.
The risk of drug resistance can be reduced by com-bination therapy (BOx 1). The frequency of resistance to P. falciparum is estimated at 1 in 1010 parasites, and there is a parasite burden of ~1012 in severe malaria. Combining two medicines with different mechanisms of action lowers the probability that a resistant parasite will emerge17. In 2006, the WHO produced new treatment guidelines for uncomplicated P. falciparum malaria, advising that the treatment of choice should be a com-bination of two or more antimalarials with different mechanisms of action18. The gold standard has become fixed-dose ACTs, with both drugs in the same tablet.
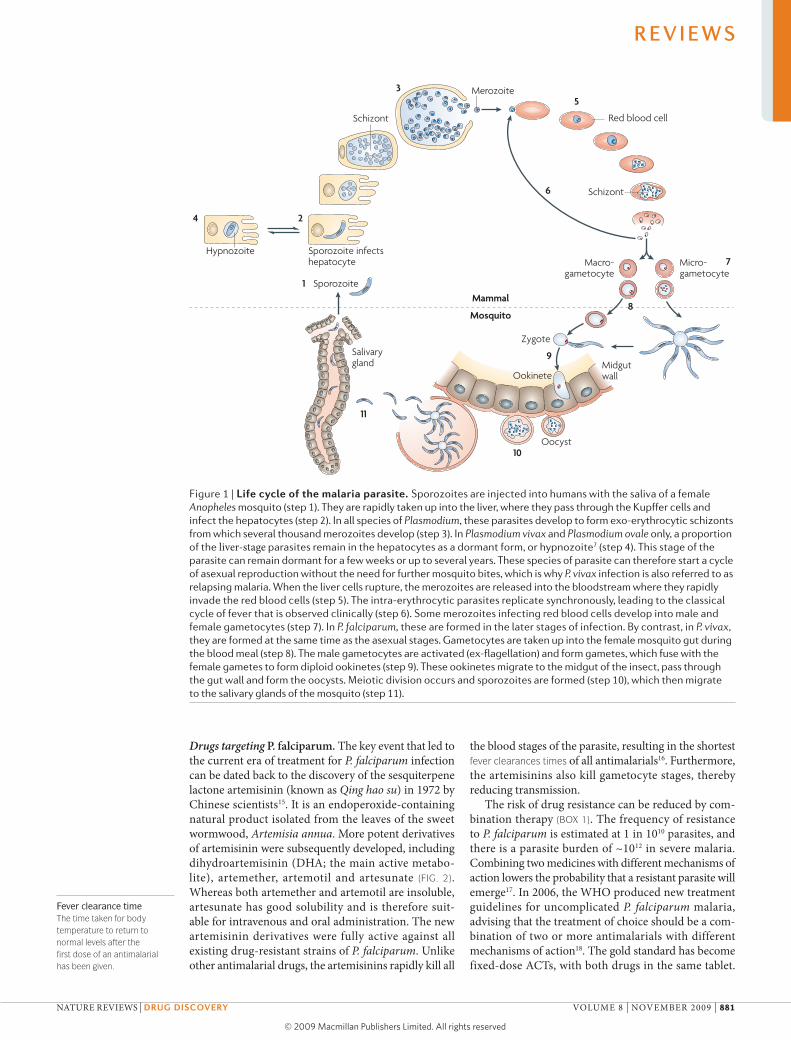
Figure 1 | Life cycle of the malaria parasite. Sporozoites are injected into humans with the saliva of a female Anopheles mosquito (step 1). They are rapidly taken up into the liver, where they pass through the Kupffer cells and infect the hepatocytes (step 2). In all species of Plasmodium, these parasites develop to form exo-erythrocytic schizonts from which several thousand merozoites develop (step 3). In Plasmodium vivax and Plasmodium ovale only, a proportion of the liver-stage parasites remain in the hepatocytes as a dormant form, or hypnozoite7 (step 4). This stage of the parasite can remain dormant for a few weeks or up to several years. These species of parasite can therefore start a cycle of asexual reproduction without the need for further mosquito bites, which is why P. vivax infection is also referred to as relapsing malaria. When the liver cells rupture, the merozoites are released into the bloodstream where they rapidly invade the red blood cells (step 5). The intra-erythrocytic parasites replicate synchronously, leading to the classical cycle of fever that is observed clinically (step 6). Some merozoites infecting red blood cells develop into male and female gametocytes (step 7). In P. falciparum, these are formed in the later stages of infection. By contrast, in P. vivax, they are formed at the same time as the asexual stages. Gametocytes are taken up into the female mosquito gut during the blood meal (step 8). The male gametocytes are activated (ex-flagellation) and form gametes, which fuse with the female gametes to form diploid ookinetes (step 9). These ookinetes migrate to the midgut of the insect, pass through the gut wall and form the oocysts. Meiotic division occurs and sporozoites are formed (step 10), which then migrate to the salivary glands of the mosquito (step 11).
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 881
© 2009 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
O
OH
H
O OO
H
OO
O
H
H
O
H
HO
OO
O
H
H
O
H
O
OO
O
H
H
O
H
O
OO
O
H
H
O
H
O
O
O
HO
O
H
H
O OO
H
N
S
O
O
OO
O NHO
NH2
N
HN
Cl
HN
OO
O
NCl
HNN
OH
N O
N
N
N
Cl
N
N
N
N
Cl
N
N
O
OH
H
Cl
HNN
N
N
FFF
F
FF
OH
NH
H
Cl
Cl
Cl
HO
N
N
O
HNN
N
O
HNNH2 N
O
HNNH2
O
O
FFF
O
O
OH
Cl
Cl
HN
NH
HN
HN
NHH2N
SNH
OO NN
O
O
N
N
H2N
NH2Cl
Cl
HNN
OH
N
Artemisinin DHA Artemether Artemotil/Arteether Artesunate
Artemisone
Chloroquine
Quinine
Atovaquone
Pamaquine Primaquine Tafenoquine
Proguanil Sulphadoxine Pyrimethamine
Mefloquine Lumefantrine
Amodiaquine Pyronaridine Piperaquine
OZ277/RBx11160 PA1103/SAR116242
a
c
b
d
e
R E V I E W S
882 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

Parasite clearance timeThe time taken for asexual parasites to no longer be detectable microscopically after the first dose of an antimalarial has been given.
Artemether–lumefantrine (Coartem; Novartis/Chinese Academy of Medical Military Sciences), the first such fixed-dose ACT, was launched in 2001. It has the advan-tage that lumefantrine had never been used as a mono-therapy, and so there has been no opportunity for drug resistance to develop against it. Subsequently developed ACTs, containing amodiaquine, mefloquine, piperaquine and pyronaridine, do not have this advantage.
Drugs targeting P. vivax. In contrast to P. falciparum malaria, P. vivax malaria is generally still treatable with chloroquine, which remains the WHO’s recommenda-tion for first-line treatment against this species. However, some areas in South and Southeast Asia now harbour chloroquine-resistant P. vivax parasites15. In these areas, the WHO recommends the use of an ACT. For P. vivax malaria, the particular challenge remains the prevention of relapse as patients who have had all blood stages of the parasite removed can still be re-infected by activation of the hypnozoite.
Among the first generation of synthetic drugs for malaria was the 8-aminoquinoline pamaquine (FIG. 2), which was introduced in 1926 (ReF. 19). It has anti-relapse activity but causes haemolysis in patients who have a deficiency in the enzyme glucose 6-phosphate 1 dehydrogenase (G6PD), meaning that ideally it should not be prescribed without patient screening. This is a considerable problem in disease-endemic areas, where up to 20% of the patients might have this deficiency20. A second-generation molecule, primaquine, was initially used to treat those returning from the Korean war, in whom long-term relapses were common. It is now the anti-relapse drug of choice for P. vivax malaria. However, primaquine also causes haemolysis in G6PD-deficient patients and must be taken daily for 14 days to be effec-tive; such compliance is unachievable in practice. A third-generation 8-aminoquinoline, tafenoquine, which could be taken as a 2–3-day treatment course, is currently in clinical development21. The mechanism of action of the 8-aminoquinolines against hypnozoites and the mecha-nism of haemolysis are not known, but they might be similar. Primaquine metabolism includes the formation
of a reactive quinone imine, and potentially a peroxy radical, which would be more toxic in the absence of G6PD as the cellular environment would be more oxidative.
Drugs targeting severe malaria. Severe malaria presents additional clinical challenges: patients are unconscious and need a rapidly acting intravenous or intramuscular medication. Traditionally quinine was used, but the rapid parasite clearance time (PCT) of artemisinin deriva-tives makes them clinically superior22. Developing new drugs for severe malaria requires large studies of up to 5,000 patients. Currently, the main challenge is to ensure a supply of drugs that are manufactured to international standards of good manufacturing practice (GMP). Not everyone has access to a health worker who can inject a drug, and so a rectal artesunate formulation has been developed as a prereferral treatment for use while the patient is rushed to the clinic23.
Prophylaxis. Prophylaxis for high-risk groups, particularly pregnant women and children, in countries in which malaria is endemic, was promoted in many African countries post-independence. In children under 5 years of age, this intervention has been shown to increase mean haemoglobin levels, reduce the frequency of clini-cal episodes and reduce overall mortality. The effects in pregnant women, particularly among those who are pregnant for the first time, include a significant increase in haemoglobin, a reduced prevalence of placental infec-tion, a reduction in the frequency of prematurity and low birth weight, and therefore an increase in life expectancy of the newborn.
Difficulties in maintaining compliance, together with an impaired acquisition of natural immunity in young children, led to a re-formulation of the approach of giving antimalarials for prophylaxis to these two high-risk groups. Intermittent preventive treatment (IPT) aims to deliver antimalarial prophylaxis at defined intervals, mostly using contacts that already exist between the target population and the health services for delivery. A regimen of two or three doses of sulphadoxine–pyrimethamine, chosen because of its long half-life, delivered during pregnancy has been shown to provide considerable benefit to both the mother and the foetus, and is currently implemented routinely as part of comprehensive malaria control strategies throughout most countries in which malaria is endemic. A similar approach is being developed for infants of up to 5 years of age. The medicine is administered during the routine vaccination contacts that are made during the first year of life, and there is strong evidence to suggest that the approach is safe and confers a considerable and cost-effective health benefit24. IPT is likely to be most benefi-cial in areas of high parasite transmission, and its policy implications are currently being reviewed by the WHO. Although sulphadoxine–pyrimethamine retains sub-stantial prophylactic efficacy, despite moderate to high levels of resistance to treatment, there is obviously con-cern that it may eventually lose its effectiveness. Many of the other antimalarial drugs developed so far also have long half-lives (that of mefloquine is 21 days, for
Figure 2 | structures of selected key antimalarial compounds. The artemisinin combination therapies now being launched are fixed-dose combinations of an artemisinin derivative (a) and a partner drug (b). a | The artemisinins include artemisinin, dihydroartemisinin (DHA; the main active metabolite), artemether (the methyl ether), artemotil (the ethyl ether) and artesunate. These are thought to work by activation of the endoperoxide group in the presence of free Fe2+, which is liberated in the infected erythrocyte when the parasite digests haemoglobin. b | The 4-aminoquinolines and amino-alcohols used in the treatment of uncomplicated malaria include chloroquine, amodiaquine, pyronaridine, piperaquine, quinine, mefloquine and lumefantrine. c | New generations of synthetic endoperoxides are being tested, which may overcome artemisinin resistance. These include the semi-synthetic derivative artemisone (under development by Hong Kong University of Science and Technology), the endoperoxides OZ277/RBx11160 (also known as arterolane; under development by Ranbaxy) and PA1103/SAR116242 (also known as trioxaquine; under development by Sanofi–Aventis). d | The current non-artemisinin-containing combinations include atovaquone–proguanil (Malarone; GlaxoSmithKline) and sulphadoxine–pyrimethamine (Fansidar; Roche). e | The 8‑aminoquinolines that target P. vivax hypnozoites include pamaquine, primaquine and tafenoquine (under development by GlaxoSmithKline).
▶
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 883
© 2009 Macmillan Publishers Limited. All rights reserved

Causal prophylactic activityeffect of antimalarial drugs that prevent sporozoite infection or replication of parasites in the liver, in contrast to suppressive prophylaxis in which replication of parasites in the blood is prevented.
example). Therefore, in addition to curing any existing parasitaemia, a single administration might prevent subsequent infections for several weeks. Clinically, a single course of treatment of artemether–lumefantrine or DHA–piperaquine protects 70% and 80% of patients, respectively, from re-infection over the following 6 weeks (M. Corsi, personal communications). The drug devel-opment challenge in both infants and pregnant women is to determine the correct dose. Not only are the phar-macokinetics different in these populations, but the drug exposure needed for prevention will be different from that required for cure.
Antimalarial medicines are used not only for treat-ment but also for prophylaxis in high-risk groups. Non-immune travellers, to malaria-endemic areas are an important group. Of the antimalarial drugs that have been developed, only pyrimethamine, proguanil and atovaquone are known to have causal prophylactic activity, and prevent the infection from reaching the blood stage. All other chemoprophylactics work solely as blood schizonticides. The drug of choice is GSK’s Malarone, a fixed combination of atovaquone–proguanil, which has causal prophylactic, blood schizonticidal and trans-mission-blocking activities25. It is relatively expensive compared with other antimalarials, owing to the diffi-culty in making atovaquone, and has to be administered daily. Mefloquine, which is less expensive and has a long half-life, provides good prophylaxis even when adminis-tered weekly, but it is associated with gastro intestinal and psychiatric side effects. It is a mixture of two diastero-isomers, and preclinical data suggest the psychiatric effects are due to the (–)-erythro diastereoisomer. Clinical studies are ongoing with the pure (+)-erythro diaster-eoisomer, to determine whether this has a better safety profile. Older drugs such as chloroquine, sulphadoxine–pyrimethamine and biguanides (such as proguanil) have reduced efficacy in a number of areas owing to increasing resistance.
The future of ACTsThere are currently five fixed-dose ACTs available (TABLe 1). The development of a medicine with two active ingredients poses unique challenges. First, there is the need to select two active ingredients with different mech-anisms of action that are therapeutically synergistic but do not adversely affect the uptake, distribution or safety of each other. The optimum dose for each ingredient needs to be confirmed in patients. Second, co-formulation is often a challenge, as the drugs must not interact with each other chemically. The final product has to be stable, pref-erably for 2–3 years with less than 5% degradation, under conditions of high temperature and relative humidity (30 ± 2°C and 65 ± 5% relative humidity) (details on the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use website; see Further information). With a cost limitation of $1 per treatment, formulation must not substantially increase the expense. Third, medicines may have to be dosed and formulated differently for small children and pregnant women.
One Achilles’ heel of ACTs is that they rely on artem-isinin derivatives, and are all susceptible to the same mechanism of drug resistance. Early warnings of resist-ance have come from the observed increased PCTs on the Thai–Cambodian border26. The mechanism of action of artemisinin involves activation of the endoperoxide moiety by Fe2+, which is liberated in the infected eryth-rocytes during haemoglobin digestion27. Specific molec-ular targets have been identified27, but such a reactive intermediate could attack many targets. Although small changes in the half-maximal inhibitory concentration (IC50) of artemisinin-based drugs have been reported28, no artemisinin-resistant isolate has been characterized. Potential mechanisms for resistance include mutations in genes encoding drug efflux pumps (as were seen for the 4-aminoquinolines) or mechanisms that alter the con-centration of free Fe2+ (required for endoperoxide activa-tion). Several research groups have developed synthetic artemisinin analogues, based on the endoperoxide motif, but with diverse structures (five- or six-membered rings29 and with three or four oxygen moieties30) (FIG. 2). Five are in the late stages of development, including OZ277/RBx11160 (ReF. 31), OZ439, CDRI97/78, PA1103/SAR114262 and a semi-synthetic molecule, artemisone. Will these share a common resistance pathway with artemisinin? This would be unlikely if resistance is due to an efflux pump, given the diversity of chemical struc-tures of the compounds. However, if resistance arises from controlled destruction of the endoperoxide, a consider-able portion of the drug development pipeline could be at risk. These compounds are currently being tested against clinical isolates to better understand this situation.
If all endoperoxide compounds eventually become compromised by resistance, how will we treat patients in the medium term? Malaria therapy could revert to more traditional antibiotics, some of which are in Phase II and Phase III clinical trials for this indication (FIG. 3). The most advanced product is the combination of azithromycin and chloroquine, which is currently in Phase III clinical development for IPT during pregnancy. Azithromycin is
Box 1 | Strengths and weaknesses of artemisinin combination therapies
strengths• Activity against all asexual blood stages of the parasite
• High parasite killing rates per intra-erythrocytic cycle
• Rapid parasite and fever clearance
• High efficacy against chloroquine-resistant and sulphadoxine–pyrimethamine-resistant strains
• Low adverse-event levels
• Gametocytocidal activity resulting in transmission-blocking activity
Weaknesses• Lack of activity against the hypnozoite stage of Plasmodium vivax, so they do not
result in a radical cure of the malaria caused by this species
• Lack of causal prophylactic activity and the short half-life of artemisinin derivatives means that their use in prophylaxis is contraindicated
• The potential for a mismatch in pharmacokinetics between partner drugs, which enhances the risk of resistance generation: the artemisinin derivative is protected from the rapid development of resistance as it circulates in the presence of the long-lived component, but the long-lived component is not protected by the artemisinin
R E V I E W S
884 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

only moderately effective against malaria parasites as a monotherapy, but its activity is enhanced in the presence of chloroquine32. Interestingly, the antibiotic reverses the clinically observed chloroquine-resistance phenotype, by a mechanism that is not yet known33. The fact that this is the only combination in Phase III trials that does not contain an endoperoxide underlines the importance of
representing a wide range of molecular targets in a drug discovery portfolio. The alternative line of defence against artemisinin resistance would be atovaquone–proguanil. However, the ease with which resistance to this combina-tion develops in the laboratory, combined with the cost, means that from a public-health perspective it would be advisable to save this medicine as a last resort.
Table 1 | Fixed-dose ACTs (already available or in late-stage development)
compound Launch date Key strengths Key weaknesses stability Formulation
Arthemether–lumefantrine (Coartem/Coartem-D*; Novartis/Chinese Academy of Medical Military Sciences/MMV)
First quarter (Q1) of 2001 (Coartem) and Q1 of 2009 (Coartem-D)
• Market leader with over 200 million treatments given
• Excellent safety data• Paediatric formulation• First-line therapy in
most of anglophone sub-Saharan Africa
• Twice per day treatment
• Low bioavailability and potentially causes a dependence on fatty foods
• Second dose 6 hours after first dose
• Pharmacokinetic mismatch
24 months
• Tablet (adult)
• Dispersible (child)
Dihydroartemisinin–piperaquine (known as eurartesim‡; Sigma-Tau/MMV/Pfizer)
Q3 of 2010 • Once per day therapy• Long terminal half-life
of piperaquine: 80% of patients are protected against re-infection at 42 days
• Potential for IPTi• Excellent safety data
of non-GMP product
• Dihydroartemisinin is the least stable of the artemisinins
• The two component drugs interact
• Paediatric formulation is still in development
• Pharmacokinetic mismatch
24 months?
• Tablet (adult)
• Crushed tablet (child)
• Dispersible (child, 2012)
Artesunate–pyronaridine (known as pyramax; Shin Poong/MMV)
Q1 of 2011 • Once per day therapy• Clinical data and
registration also for Plasmodium vivax malaria
• Potential to combine with primaquine for radical cure
• Excellent safety of pyronaridine monotherapy
• Limited safety data on combination (3,000 patients in clinical dossier)
• Pharmacokinetic mismatch
24+ months
• Tablet (adult)
• Sachet (child)
Artesunate–amodiaquine (Coarsucam; Sanofi-Aventis/DNDi/MMV)
Q4 of 2008 • Once per day dosing• First-line therapy in
francophone Africa
• Resistance (to amodiaquine) can compromise efficacy
• Increased nausea• No approval by
stringent regulatory authority
• Pharmacokinetic mismatch
24 months
Dispersible tablet for all ages
Artesunate–mefloquine (ASMQ; Farmanguinhos/DNDi/Cipla)
Q2 of 2008 • Once per day therapy• Satisfactory safety
record in Thailand of non-fixed-dose combination
• Successful treatment of P. vivax malaria in chloroquine-resistant areas
• Long half-life of mefloquine means potential for IPT
• Mixture of two diastereoisomers
• Psychiatric and gastrointestinal adverse events
• No approval by stringent regulatory authority
• No pre-qualification by the WHO
• Pharmacokinetic mismatch
• Expensive
No data reported
Crushed tablet for all ages
*Coartem-D is a child-friendly dispersible form of artemether–lumefantrine, launched this year. ‡Dihydroartemisinin–piperaquine is sold by Chongqing Holley as Artekin; this has not been prequalified by the World Health Organization (WHO) or approved by a stringent regulatory authority. ACTs, artemisinin-based combination therapies; DNDi, Drugs for Neglected Diseases initiative; GMP, good manufacturing practice; IPTi, intermittent preventive treatment in infants; MMV, Medicines for Malaria Venture.
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 885
© 2009 Macmillan Publishers Limited. All rights reserved

Nature Reviews | Drug Discovery
TrioxaquinePalumed/Sanofi–Aventis
Two compoundsNovartis
(+)-MefloquineTreague
Two pyridonesGSK
MK 4815MMV/Merck
AQ-13Immtech/Tulane University
CDRI 97/78 Ipca Laboratories & CDRI
4-pyridoneGSK/MMV
TafenoquineGSK/MMV
IsoquineLiverpool Schoolof TropicalMedicne
OZ 439Monash/UNMC/STI/MMV
SAR 97276CNRS/Sanofi–Aventis
Methylene blue–amodiaquineHeidelberg/DSM
TinidazoleWRAIR
FerroquineSanofi–Aventis
Fosmidomycin–clindamycinJomaa
ArtemisoneUHKST/MMV
Arterolane–piperaquineRanbaxy
a Translational
Preclinical Phase I
Azithromycin–chloroquinePfizer
ASMQFarminguinhos/Cipla
Pyramax Shin Poong/Universityof Iowa/MMV
EurartesimSigma-Tau/MMV
CoarsucamSanofi–Aventis/DNDi
Coartem-DNovartis/MMV
b Development
Phase III ApprovedPhase II
ICHInternational Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, which issues guidelines for good laboratory and clinical practice.
Antimalarial target product profilesSuccessful drug discovery involves starting with a clear idea of what the final product should ideally look like and being able to describe this in a target product pro-file (TPP). In the case of drug combinations, the TPP summarizes the properties of the combination medicine; a single molecule does not have to tick all the boxes. TABLe 2 summarizes the TPPs for antimalarial drugs.
As we consider our current efforts to control malaria and the goal of eradication, it is apparent that new oral antimalarial products for the treatment and prophylaxis of malaria ideally should have four important properties. First, they should be able to treat existing disease — that is, they should have activity against the blood parasites of the four key species that cause malaria in humans, including organisms that are resistant to existing thera-pies. Second, they should kill the hypnozoites of P. vivax and P. ovale and prevent relapse. Third, they should kill gametocytes to prevent re-infection of mosquitoes and subsequent transmission to other people. Fourth, they should have causal prophylactic activity, preventing initial infection following a mosquito bite by killing the sporozoites and exo-erythrocytic schizonts. It is unlikely that an individual molecule will have all of these attributes, and considerations of resistance generation suggest that a drug combination strategy is preferable.
An appropriate combination of drugs, formulated into a fixed-dose combination product, should be sufficient. In addition, if artemisinin resistance spreads, there will also be a need for a new fast-acting blood schizon-ticide that can be administered as an intramuscular or intravenous injection for severe malaria.
One of the most important aspects to consider at an early stage is the cost of the product: in disease-endemic countries, patients can often only afford $0.10 per treat-ment regimen. The target ex-factory price for new treat-ments is currently considered to be $1, which means that components must be relatively potent or very cheap. The Global Fund for HIv, Tuberculosis and Malaria is launching the AMFm, which should both subsidise the purchase of drugs and manage the price increases along the supply chain, allowing this price gap to be bridged in both the public and private sectors.
Another important aspect in malaria drug devel-opment is patient compliance. Current treatments for uncomplicated malaria require 3 days of dosing. As with antibiotics in the West, there is a risk that patients will not finish the course of treatment, as fever is cleared early in treatment. With hypnozoites the challenge is even greater: currently the treatment is 14 days of pri-maquine, with no symptomatic effect on patients. Short courses of treatments that can be directly monitored will be advantageous in an eradication programme.
Safety is also a crucial issue for new antimalarials. The current medicines have a good safety profile, with serious adverse events observed at a frequency lower than one patient in a thousand. The main reason for the high mortality associated with malaria is a lack of access to therapy in the short term and the threat of resistance in the long term. An important consideration is that the new medicines will be used by millions of patients in malaria-endemic countries where safety reporting is not well developed. It is therefore doubly important that the development of a new antimalarial product should follow the same guidelines as any therapy intended for use in Europe, the United States or Japan. The ICH guidelines ensure that all the work reported meets a certain stand-ard, particularly with regard to the efficacy, safety and stability of the drug. A key standard that is expected by the regulatory authorities in these areas is that an adequate number of patients, usually 2,000–3,000 in total, have been given the medicine during the clini-cal development programme. In addition, the develop-ment programme should effectively represent all the age ranges and patient types affected by the disease. Such studies should prevent the approval of medicines that cause high levels of serious adverse events and give a preliminary idea of the degree and frequency of non-life-threatening side effects. Further Phase Iv trials, which examine the seriousness of adverse events, as well as post-marketing surveillance, will enable an accurate assessment of the risks and benefits of a new medi-cine. This issue is particularly important in the case of malaria, as national malaria control programmes aim to change the status of such medicines from prescription-only to over the counter, to facilitate access to the whole population.
Figure 3 | The global antimalarial drug portfolio (June 2009). Antimalarial medicines that are currently in development, from the start of regulatory good laboratory practice preclinical development to registration with stringent regulatory authorities or the World Health Organization prequalification group. Data were obtained from public-domain sources in July 2009 (details of the individual projects are summarized in ReF. 44). CDRI, Central Drug Research Institute; CNRS, Centre Nationale de la Recherche Scientifique; DNDi, Drugs for Neglected Diseases initiative; GSK, GlaxoSmithKline; MMV, Medicines for Malaria Venture; STI, Swiss Tropical Institute; UHKST, University of Hong Kong Institute of Science and Technology; UNMC, University of Nebraska Medical Center; WRAIR, Walter Reed Army Institute of Research.
R E V I E W S
886 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved
Another important issue to consider when devel-oping antimalarial drugs is the quality of the medicine itself. Counterfeits of branded medicines, which often contain no active ingredients34 and medicines that con-tain less than the required active ingredient (owing to inappropriate standards of manufacture, formulation, packaging and stability), result in further morbidity and an increased risk of the development of resistance. Therefore, stringent international standards of GMP, set by regulatory authorities or the WHO, need to be fol-lowed. In addition, programmes for access to medicines must ensure that the medicines that are most readily available are those of the highest quality, with constant surveillance for counterfeits and local enforcement of standards in disease-endemic countries.
The arrival of new technologiesIn malaria drug discovery, two major technological breakthroughs in the past 7 years have greatly enhanced our discovery efficiency. First, the sequencing of the whole genomes of P. falciparum35 and P. vivax36, which has been important in identifying the full range of potential drug targets. Moreover, it has provided the basis for comparisons between the gene expression patterns at different stages of the parasite lifecycle and between different parasite species. This data set provides the ability to search for target classes that have not been pursued before in drug discovery37. It is still possible to prioritize novel targets based on the likelihood of find-ing a small-molecule inhibitor38 and on their similarity
to targets for which a drug has already been found. This opens up the possibility of ‘orthologue searching’: com-pounds in a company’s database can be selected from medicinal-chemistry programmes that are directed against a human target for which there is a close rela-tive in the parasite. A small collection of compounds, including structurally related, active and inactive com-pounds against the human target, can then be tested against the parasite enzyme. This has been successful with other pathogens, with hit rates as high as 10–25%, and can lead to the development of compounds that are selective for the parasite target39.
The second key development is high-content screen-ing: the ability to study the viability of the parasite in 384- and 1,536-well formats40. This means that large compound collections can be rapidly screened, with throughputs of 100,000 per month possible even in academic centres (v. Avery, personal communications). This has changed the dynamics of interactions between researchers in industry and academia, as compound col-lections can be screened blind at a cost of less than $1 per compound. In the assays to determine the viability of P. falciparum in erythrocytes, the hit rate is often as high as 0.5% of compounds showing activity at less than 1 μM. This gives several chemical series to work on: over 10,000 distinct hits from a major pharmaceutical collection can be prioritized at an early stage. Early prioritization can be based on potency against Plasmodium spp., half-life, lack of activity against human cell lines and a potentially low cost of goods. Identifying the molecular target is not
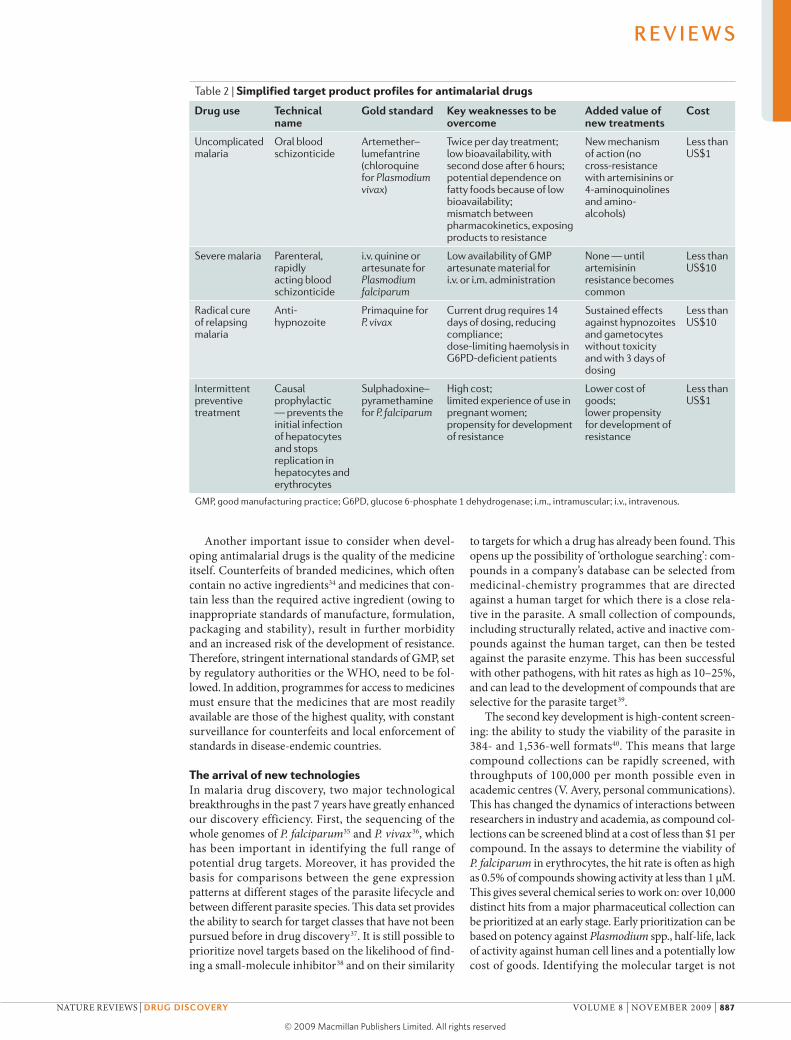
Table 2 | Simplified target product profiles for antimalarial drugs
Drug use Technical name
gold standard Key weaknesses to be overcome
Added value of new treatments
cost
Uncomplicated malaria
Oral blood schizonticide
Artemether–lumefantrine (chloroquine for Plasmodium vivax)
Twice per day treatment; low bioavailability, with second dose after 6 hours; potential dependence on fatty foods because of low bioavailability; mismatch between pharmacokinetics, exposing products to resistance
New mechanism of action (no cross-resistance with artemisinins or 4-aminoquinolines and amino- alcohols)
Less than US$1
Severe malaria Parenteral, rapidly acting blood schizonticide
i.v. quinine or artesunate for Plasmodium falciparum
Low availability of GMP artesunate material for i.v. or i.m. administration
None — until artemisinin resistance becomes common
Less than US$10
Radical cure of relapsing malaria
Anti- hypnozoite
Primaquine for P. vivax
Current drug requires 14 days of dosing, reducing compliance; dose-limiting haemolysis in G6PD-deficient patients
Sustained effects against hypnozoites and gametocytes without toxicity and with 3 days of dosing
Less than US$10
Intermittent preventive treatment
Causal prophylactic — prevents the initial infection of hepatocytes and stops replication in hepatocytes and erythrocytes
Sulphadoxine–pyramethamine for P. falciparum
High cost; limited experience of use in pregnant women; propensity for development of resistance
Lower cost of goods; lower propensity for development of resistance
Less than US$1
GMP, good manufacturing practice; G6PD, glucose 6-phosphate 1 dehydrogenase; i.m., intramuscular; i.v., intravenous.
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 887
© 2009 Macmillan Publishers Limited. All rights reserved

ReticulocyteAn immature red blood cell.
simple but the aim should be to identify the molecu-lar mechanism at the stage when a clinical candidate is selected.
The success of high-content screening in finding new chemotypes with activity against the erythrocytic stages suggests that this could be applied to the other stages of the parasite lifecycle. These assays exist for gameto-cytes and liver stages for both P. falciparum and P. vivax, with the exception of the hypnozoite. The challenge is to automate these screens and make them suitable for 384-well assays. For the hypnozoite stages of P. vivax, the problem is more complex41 as infective P. vivax sporo-zoites are needed, and so access to mosquito breeding facilities is required. To infect the mosquitoes requires access to blood from patients or experimentally-infected primates. New technologies for cryopreservation of spo-rozoites could facilitate this process42. The cell culture challenges are also more complex for this species, as P. vivax requires reticulocytes for culture. The hepato-cytes used in culture represent a further challenge: cell lines are available but they have low infection rates43. Most work is still focused on primary human hepato-cytes or infecting primate hepatocytes with the related species Plasmodium cynomolgi. The recent spotlight on eradication has served to increase the amount of funding for this area, and there are some initial results suggesting a cell-based assay for hypnozoites might be possible.
Having consistent tests for all of the stages of the parasite life cycle will be an important step forward in the agenda to find new medicines. A short-term objec-tive would be to test all the molecules that are currently in the malaria drug development pipeline in the available cell assays for all the key stages of the parasite lifecycle
Table 3 | Key challenges in early drug discovery for malaria
Key issue scientific challenges current approaches
New medicines that overcome artemisinin resistance
• Definition of the molecular mechanism of resistance
• Testing novel endoperoxide medicines against clinical isolates from areas of resistance
• Developing new rapidly acting medicines with novel modes of action
Culture systems for Plasmodium vivax in erythrocytes
• Achieve stable cultures reproducibly
• Experimenting with cell culture systems using reticulocytes and stem cell-derived reticulocytes
New medicines which safely eliminate hypnozoites from the liver
• Reproducible cellular assays for hypnozoites
• Simple rodent assays for in vivo activity
• Clinical trial paradigms that distinguish relapse from re-infection
• Infection of primate (Rhesus) liver cells from Plasmodium cynomolgi
• Searching for human cell lines in which hypnozoites will form
• Rebuilding human liver tissue in SCID mice, or with stem cell-derived material
• Treatment and follow-up of patients in a place where re-infection does not occur (but this is normally limited to 28 days)
New medicines which can block transmission
• Better understanding of gametocyte transmission
• Characterizing current therapies by in vivo feeding and dissection of mosquitoes
• Seeking better biomarkers of transmission potential
SCID, severe combined immunodeficient.
discussed above, and generate malaria ‘life cycle finger-prints’ for each molecule. This will enable side by side comparison of molecules at a reasonably early stage, and could be crucial for entering the right medicines into the appropriate clinical assays. The key challenges in early-stage discovery brought by the eradication agenda are summarized in TABLe 3.
The malaria research pipelineOver 90% of current malaria drug discovery projects are targeting the asexual blood stages of P. falciparum. Those which are close to clinical development have been recently reviewed44. The first group are those which aim to improve on drugs that have validated activity in human disease (traditionally known as ‘fast followers’). Strategies include improving inhibitors of dihydrofolate reductase to develop drugs that are active against resistant strains45, and developing new inhibitors of mitochondrial electron transport, which are active against atovaquone-resistant parasites with mutations in cytochrome bc1 (ReF. 46). Here, the TPP is reasonably simple, and the key advantage of a new-generation inhibitor will be action against resistant strains of the parasite. Improvements in the potency of 4-aminoquinolines or amino-alcohols is another potential approach. In this case, there is the opportunity to make a radical change in the way the drug is presented (for exam-ple, combining a 4-aminoquinoline with an endoper-oxide29 or combining an antimalarial with a resistance blocker47). However, with the prospect of 5 fixed-dose ACTs being available to patients over the next 2 years, any new 4-aminoquinoline-containing compound must show a substantial benefit in terms of cost or safety.
Another strategy is to develop drugs on the basis of newly defined molecular targets for which there are sup-porting data that an inhibitor will have an effect on the parasite, but as yet no clinical validation (TABLe 4). The project in question is usually supported by strong in vivo data, such as the finding that specific gene knockouts prevent the generation of viable cell lines in P. berghei. Although such validation is useful, confirmation is based on a negative result and there are substantial differences between P. berghei and P. falciparum36. For many of these targets, medicinal chemistry is aided by high-resolution structural information. Pathways such as nucleoside bio-synthesis have been highlighted by genome sequence analy sis41, leading to the identification of three potential drug targets. Dihydroorotate dehydrogenase (DHODH) has been targeted, as the parasite and mammalian forms differ considerably. Screening against DHODH has yielded potent and selective compounds that can now be optimized with the availability of high-resolution struc-tural data of the enzyme–ligand complex48,49. The power of the rational design approach has also been underlined by the design of inhibitors of adenosine deaminase based on detailed electronic calculations of the reaction transition state50. Also, inhibitors designed to be active against the transition state of purine nucleoside phosphorylase have been shown to be active against P. falciparum51,52. Some of these compounds are ready for testing in humans and have been shown to be safe, and so it should be possible to rapidly evaluate the target in human malaria.
R E V I E W S
888 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved
ApicoplastAn organelle that is present in malaria parasites and is thought to be a relic of the chloroplast. It contains DNA which codes for ~30 plant-like proteins and bacteria-like ribosomes. A further 500 nuclear-encoded proteins of unknown phylogenetic origin are predicted to be targeted to this organelle.
A potential set of key pathways to be used as drug targets occur in the apicoplast in Plasmodium spp. This organelle is not found in the human host, and contains metabolic pathways and enzymes that are similar to those seen in plant choloroplasts53. These pathways should offer targets that have a high degree of parasite selectivity. The most extensively studied is the 1-deoxy-d-xylulose 5-phosphate pathway for isoprenoid biosynthesis, which is the target of fosmidomycin, now in Phase II clinical trials in combination with clindamycin or azithromycin. In addition, the apicoplast has unique fatty-acid biosyn-thesis pathways54, suggesting that other parasite targets that are inhibited by herbicides should be considered. Unfortunately, inhibiting these pathways leads to long PCTs, with parasite death delayed until the next round of replication.
Another approach to drug discovery is to use the expertise on target families that have already been successfully targeted in other therapeutic areas — for example, kinases55 and histone deacetylases56. The chal-lenge is to identify molecules that can be sufficiently dis-criminated from the equivalent host targets to ensure safety. In the case of protease targets, such as the cysteine proteases, falcipain57 and the serine protease PfSUB1 (ReF. 58), inhibitors were difficult to develop as drug candidates because of selectivity issues.
Most of these approaches target the erythrocyte stages of P. falciparum. However, if the drugs could be delivered by an intramuscular or intravenous route, rather than orally, and were fast-acting, they could also be devel-oped for severe malaria, as has been shown for a specific choline channel blocker, albitiazolium bromide (known as TE3 and SAR97276; Centre Nationale de la Recherche Scientifique/Sanofi–Aventis), which is now in Phase II clinical trials59. Developing compounds that are active
against gametocytes, sporozoites, exo-erythrocytic liver schizonts and hypnozoites is considerably more chal-lenging, as a reliable cellular model is needed to define whether key metabolic pathways, such as mitochondrial respiration or nucleoside biosynthesis, are crucial for the reactivation of the hypnozoite. The lack of information in this area underlines how neglected the research into P. vivax hypnozoites has been.
Public–private partnershipsIn the past decade, there has been a fundamental shift in the way that new medicines are developed for neglected disease. Although the global antimalarial drug market is huge in terms of unmet medical need, it is small in terms of profit. Owing to the low price that can be charged for malaria medicines in disease-endemic countries, it is not possible to obtain a financial return on R&D investment from these drugs. To bridge this gap a new model has been developed, by which academia and the biotech-nology and pharmaceutical industry can collaborate to discover, develop and deliver new medicines. The new Medicines for Malaria venture (MMv) was established in 1999 as a public–private partnership (PPP), which provides much of the necessary finance, without expec-tation of repayment, so that emerging new products can be priced solely on the basis of manufacturing costs. Working in concert with another PPP — the Drugs for Neglected Disease initiative (DNDi) — and using finan-cial resources provided by the Bill and Melinda Gates Foundation, other private foundations and a number of governments, the MMv with the help of its partners has established a large portfolio of antimalarial drugs. By sharing knowledge and best practice across the industry, the discovery and development of the next generation of medicines will be greatly accelerated.
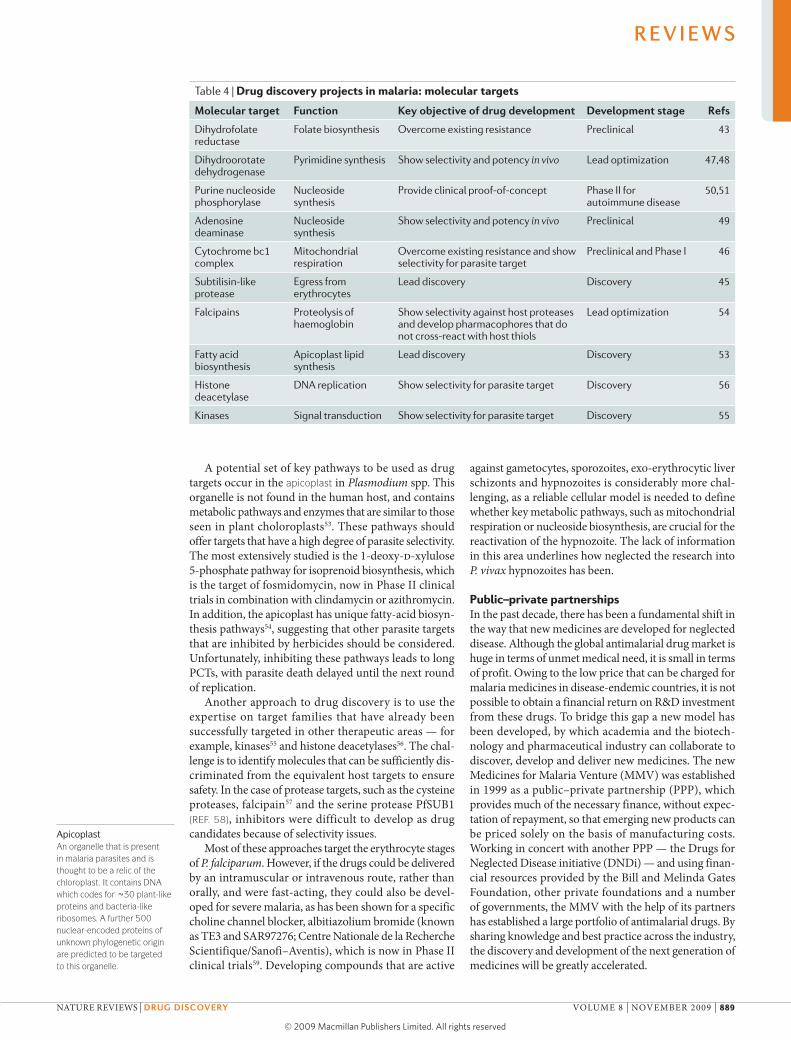
Table 4 | Drug discovery projects in malaria: molecular targets
Molecular target Function Key objective of drug development Development stage refs
Dihydrofolate reductase
Folate biosynthesis Overcome existing resistance Preclinical 43
Dihydroorotate dehydrogenase
Pyrimidine synthesis Show selectivity and potency in vivo Lead optimization 47,48
Purine nucleoside phosphorylase
Nucleoside synthesis
Provide clinical proof-of-concept Phase II for autoimmune disease
50,51
Adenosine deaminase
Nucleoside synthesis
Show selectivity and potency in vivo Preclinical 49
Cytochrome bc1 complex
Mitochondrial respiration
Overcome existing resistance and show selectivity for parasite target
Preclinical and Phase I 46
Subtilisin-like protease
Egress from erythrocytes
Lead discovery Discovery 45
Falcipains Proteolysis of haemoglobin
Show selectivity against host proteases and develop pharmacophores that do not cross-react with host thiols
Lead optimization 54
Fatty acid biosynthesis
Apicoplast lipid synthesis
Lead discovery Discovery 53
Histone deacetylase
DNA replication Show selectivity for parasite target Discovery 56
Kinases Signal transduction Show selectivity for parasite target Discovery 55
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 889
© 2009 Macmillan Publishers Limited. All rights reserved

1. World Health Organization. World Malaria Report 2008 (Geneva, Switzerland) The World Health Organization website [online] <http://www.who.int/malaria/wmr2008/malaria2008.pdf> (2008).
2. Gallup, J. L. & Sachs, J. D. The economic burden of malaria. Am. J. Trop. Med. Hyg. 64, 85–96 (2001).
3. The World Bank. World Bank Booster Programme for Malaria Control in Africa. The World Bank website [online] <http://siteresources.worldbank.org/EXTAFRBOOPRO/Resources/MALARIAREPORTfinalLOWRES.pdf>(2007).
4. Guinovart, C. et al. Insights into long-lasting protection induced by RTS,S/AS02A malaria vaccine: further results from a phase IIb trial in Mozambican children. PLoS ONE 4, e5165 (2009).
5. Roberts, L. & Enserink, M. Did they really say.. eradication? Science 318, 1544–1545 (2007).
6. Price, R. N. et al. Vivax malaria: neglected and not benign. Am. J. Trop. Med. Hyg. 77 (Suppl. 6) 79–87 (2007).A key reference that provides an update on the incidence of P. vivax malaria, showing that the incidence, severity and downstream complications of the disease are often underestimated.
7. Hemmer, C. J. et al. Stronger host response per parasitized erythrocyte in Plasmodium vivax or ovale, than in Plasmodium falciparum malaria. Trop. Med. Intl Health 11, 817–823 (2006).
8. Poespoprodjo, J. R. et al. Vivax malaria: a major cause of morbidity in early infancy. Clin. Infect. Dis. 48, 1704–1712 (2009).
9. Mayxay, M., Pukrittayakamee, S., Newton, P. N. & White, N. J. Mixed-species malaria infections in humans. Trends Parasitol. 20, 233–240 (2004).
10. Krotoski, W. A. et al. Relapses in primate malaria: discovery of two populations of exoerythrocytic stages. Preliminary note. BMJ 280, 153–154 (1980).
11. Chin, W., Contacos, P. G., Coatney, R. G. & Kimbal, H. R. Naturally acquired quotidian-type malaria in man transferable to monkeys. Science 149, 865 (1965).
12. Cox-Singh, J. et al. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin. Infect. Dis. 46, 165–171 (2008).
13. Sinden, R. E. in Malaria: Parasite Biology, Pathogenesis and Protection (ed. Sherman, I. W.) 25–48 (ASM Press, Washington, D. C., 1998).
14. Olliaro, P. L. & Yuthavong, Y. An overview of chemotherapeutic targets for antimalarial drug discovery. Pharmacol. Ther. 81, 91–110 (1999).
15. Luo, X. D. & Shen, C. C. The chemistry, pharmacology and clinical applications of qinghaosu (artemisinin) and its derivatives. Med. Res. Rev. 7, 29–52 (1987).
16. Meshnick, S. R., Taylor, T. E. & Kamchonwongpaisan, S. Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy. Microbiol. Rev. 60, 301–315 (1996).
17. White, N. J. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 354, 739–749 (1999).
18. World Health Organization. Guidelines for the Treatment of Malaria. The World Health Organization website [online] <www.who.int/bookorders/anglais/detart1.jsp?sesslan=1&codlan=1&codcol=15&codcch=662> (2006).
19. Muehlens, P. Die Behandlung der natuerlichen menschlichen Malaria-infektionen mit Plasmochin. Arch. Shiffs-u. Troppenhyg. 30, 25–32 (1926).
20. Beutler, E., Duparc, S. & G6PD Deficiency Working Group. Glucose-6-phosphate dehydrogenase deficiency and antimalarial drug development. Am. J. Trop. Med. Hyg. 77, 779–789 (2007).G6PD deficiency protects patients against malaria infection, but some key medicines in this group that are traditionally used in malaria treatment, such as dapsone and primaquine, cause haemolysis. This review summarizes the latest advances in the field.
21. Breukner, R. P., Lasseter, K. C., Lin, E. T. & Schuster, B. G. First-time-in-humans safety and pharmacokinetics of WR238605, a new antimalarial. Am. J. Trop. Med. Hyg. 58, 645–649 (1998).
22. Dondorp, A., Nosten, F., Stepniewska, K., Day, N. & White, N. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet 366, 717–725 (2005).
23. Gomes, M. F. et al. Pre-referral rectal artesunate to prevent death and disability in severe malaria: a placebo-controlled trial. Lancet 373, 522–523 (2009).
24. Aponte, J. J. et al. Efficacy and safety of intermittent preventive treatment with sulfadoxine-pyrimethamine for malaria in African infants: a pooled analysis of six randomised, placebo-controlled trials. Lancet 17 Sept 2009 (doi:10.1016/S0140-6736(09)61258-7).
25. Nakato, H., Vivancos, R. & Hunter, P. R. A systematic review and meta-analysis of the effectiveness and safety of atovaquone proguanil (Malarone) for chemoprophylaxis against malaria. J. Antimicrob. Chemother. 60, 929–936 (2007).
26. Noedl, H. et al. Evidence of artemisinin-resistant malaria in western Cambodia. New Engl. J. Med. 359, 2619–2620 (2008).
27. Meshnick, S. R. Artemisinin: mechanisms of action, resistance and toxicity. Int. J. Parasitol. 32, 1655–1660 (2002).
28. Dondorp, A. M. et al. Artemisinin resistance in Plasmodium falciparum malaria. New Engl. J. Med. 361, 455–467 (2009).This paper provides the most recent findings on artemisinin resistance in the Thai–Cambodia border region.
29. Cosledan, F. et al. Selection of a trioxaquine as an antimalarial drug candidate. Proc. Natl Acad. Sci. USA 105, 17579–17584 (2008).
30. Ellis, G. L. et al. Two-step synthesis of achiral dispiro-1,2,4,5-tetraoxanes with outstanding antimalarial activity, low toxicity, and high-stability profiles. J. Med. Chem. 51, 2170–2177 (2008).
31. Vennerstrom, J. L. et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430, 900–904 (2004).This study described the first synthetic endoperoxide to enter clinical development, arterolane (now known as RBx-11160; Ranbaxy).
This molecule showed that simpler versions of artemisinin can be produced and removed the dependence on plant-based cultivation for the next generation of antimalarials.
32. Ohrt, C. et al. Assessment of azithromycin in combination with other antimalarial drugs against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 46, 2518–2524 (2002).
33. Dunne, M. W. et al. A multicenter study of azithromycin, alone and in combination with chloroquine, for the treatment of acute uncomplicated Plasmodium falciparum malaria in India. J. Infect. Dis. 191, 1582–1588 (2005).
34. Green, M. D. Antimalarial drug resistance and the importance of drug quality monitoring. J. Postgrad. Med. 52, 288–290 (2006).
35. Gardner, M. J. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 (2002).
36. Carlton, J. M. et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455, 757–763 (2008).
37. Zhou, Y. et al. Evidence-based annotation of the malaria parasite’s genome using comparative expression profiling. PLoS ONE 3, e1570 (2008).
38. Aguero, F. et al. Genomic-scale prioritization of drug targets: the TDR Targets database. Nature Rev. Drug Discov. 7, 900–907 (2008).
39. Grundner, C. et al. Structural basis for selective inhibition of Mycobacterium tuberculosis protein tyrosine phosphatase PtpB. Structure 15, 499–509 (2007).
40. Plouffe, D. et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl Acad. Sci. USA 105, 9059–9064 (2008).This paper summarizes the results and analyses of the high-content cellular screen used to find new medicines against P. falciparum. This was the first large-scale (2 million compounds) antiparasitic compound screen, and arguably launched a new era for malaria screens and parasitology thinking.
41. Mazier, D., Rénia, L. & Snounou, G. A pre-emptive strike against malaria’s stealthy hepatic forms. Nature Rev. Drug Discov. (in the press).
42. Luke, T. C. & Hoffman S. L. Rationale and plans for developing a non-replicating metabolically active radiation attenuated Plasmodium falciparum sporozoite vaccine. J. Exp. Biol. 206, 3803–3808 (2003).
43. Udomsangpetch, R., Kaneko, O., Chotivanich, K. & Sattabongkot, J. Cultivation of Plasmodium vivax. Trends Parasitol. 24, 85–88 (2008).
44. Olliaro, P. & Wells, T. N. C. The global portfolio of new antimalarial medicines under development. Clin. Pharm. Ther. 85, 584–595 (2009).
45. Yuthavong, Y., Kamchonwongpaisan, S., Leasrtsakulpanich, U. & Chitnumsub, P. Folate metabolism as a source of molecular targets for antimalarials. Future microbiol. 1, 113–135 (2006).
ConclusionsSpurred on by the global spread of resistance to the cur-rent standard antimalarial drugs, such as chloroquine and sulphadoxine–pyrimethamine, the past 10 years have seen a considerable expansion in the research and development of new medicines to maintain and improve the clinical control of malaria. Now, with the re-introduction of a malaria eradication agenda, the next 10 years are set to be even more demanding. In the short term, there should be a group of new ACTs avail-able for use. The challenge will be to better understand how to deploy these for treatment and prophylaxis in the populations that are currently most at risk — young children and pregnant women. For the medium-term treatment of malaria, there is a strong clinical pipeline of new medicines that could deliver additional ACTs if
these are required, a new, shorter-course co-packaged radical cure for P. vivax malaria, a GMP parenteral for-mulation of artesunate for severe malaria to replace intravenous quinine, and the first of a series of inno-vative non-artemisinin-based combination therapies to deal with ACT resistance. For the long-term treat-ment and eventual eradication of malaria, there are further challenges — in particular, the urgent need for medicines to target the dormant hypnozoite stage of P. vivax, and for new products which will block the transmission of malaria by killing the gametocytes and inactivating sporozoites and exo-erythrocytic schizonts. At the same time, we must keep a watch-ful eye on the emergence of resistance and develop an understanding of its potential impact on priority needs for new medicines.
Radical cureThe elimination from patients infected with P. vivax or P. ovale of all parasites, including the dormant liver hypnozoites. Unless this is done, patients will subsequently relapse.
R E V I E W S
890 | NOvEMBER 2009 | vOlUME 8 www.nature.com/reviews/drugdisc
© 2009 Macmillan Publishers Limited. All rights reserved

46. Yeates, C. L. et al. Synthesis and structure–activity relationships of 4-pyridones as potential antimalarials. J. Med. Chem. 51, 2845–2852 (2008).
47. Kelly, J. X. et al. Discovery of dual function acridones as a new antimalarial chemotype. Nature 459, 270–273 (2009).
48. Baldwin, J. et al. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 280, 21847–21853 (2005).
49. Gujjar, R. et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 52, 1864–1872 (2009).
50. Larson, E. T. et al. Structures of substrate- and inhibitor-bound adenosine deaminase from a human malaria parasite show a dramatic conformational change and shed light on drug selectivity. J. Mol. Biol. 381, 975–988 (2008).
51. Taylor Ringia, E. A. & Schramm, V. L. Transition states and inhibitors of the purine nucleoside phosphorylase family. Curr. Top. Med. Chem. 5, 1237–1258 (2005).
52. Madrid, D. C. et al. Plasmodium falciparum purine nucleoside phosphorylase is critical for the viability of malaria parasites. J. Biol. Chem. 283, 35899–35907 (2008).
53. Waller, R. F. & McFadden, G. I. The apicoplast: a review of the derived plastid of apicomplexan parasites. Curr. Issues Mol. Biol. 7, 57–79 (2005).This review summarizes the identification of the apicoplast and the targets that have emerged.
54. Freundlich, J. S. et al. X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J. Biol. Chem. 282, 25436–25444 (2007).
55. Doerig, C. et al. Protein kinases of malaria parasites: an update. Trends Parasitol. 24, 570–577 (2008).
56. Patel, V. et al. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 52, 2185–2187 (2009).
57. Kerr, I. D. et al. Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: implications for substrate specificity J. Med. Chem. 52, 852–857 (2009).
58. Yeoh, S. et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131, 1072–1083 (2007).
59. Wengelnik, K. et al. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science 295, 1311–1314 (2002).
AcknowledgementsWe thank our colleagues on the staff and External Scientific Advisory Committee for MMV and our research and develop-ment partners, and the various governments, foundations and charities that have provided the finance required to operate this PPP. We apologize to those whose work was not cited due to space constraints.
DATABASESUniProtKB: http://www.uniprot.orgG6PD
FURTHER INFORMATIONMedicines for Malaria Venture: http://www.mmv.orgAffordable Medicines Facility for Malaria: http://www.rollbackmalaria.org/mechanisms/globalsubsidytaskforce.htmlGlobal Fund to Fight AIDS, Tuberculosis and Malaria: http://www.theglobalfund.org/en/The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use: http://www.ich.orgMalaria Eradication Research Agenda: http://malera.tropika.net/Roll Back Malaria: http://www.rollbackmalaria.org/
ALL LinKs Are AcTive in The onLine pDF
R E V I E W S
NATURE REvIEWS | Drug Discovery vOlUME 8 | NOvEMBER 2009 | 891
© 2009 Macmillan Publishers Limited. All rights reserved





























