Embed Size (px)
Citation preview

Presentation of an article from the British Journal of Haematology
By Dawn Myelle-WatsonJuly 27, 2010

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

DESCRIPTION
Note affected RBC are smaller, more dense, and round in shape compared to other cells
Hereditary spherocytosis (HS) is a structural defect of the red cell cytoskeleton
Membrane protein defects result in cytoskeleton instability
Spectrin deficiency leads to loss of surface area which produces spherical RBCs
•HS is the most common of the hereditary hemolytic anemia's among those of Northern European descent.
•Prevalence approximately 1/2000 – 1/5000 in the population

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

SYMPTOMS
Newborn baby undergoing phototherapy (bili-lights) for jaundice – a complication of HS
◦ Patients present with classic symptoms of HS: Anemia Jaundice Splenomegaly Gall Stones
◦ Aplastic anaemia due to concurrent infection with parvovirus B19
◦ Megaloblastic anaemia - folate deficiency due to the constant RBC regeneration

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

TESTING
Graphic organizer of how a RBC changes from the normal biconcave shape to the spherical shape found in HS patients
Family history Osmotic fragility (OF) blood
test (after 6 months old) Complete blood count
(CBC) Reticulocyte count Liver panel Other tests if inconclusive
data is found

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

CLINICAL CLASSIFICATIONClassification Trait Mild Moderate Severe
Haemoglobin (g/dl)
Normal 11–15 8–12 6–8
Reticulocyte count %
Normal (<3%) 3–6 >6 >10
Bilirubin (lmol/l)
<17 17–34 >34 >51
Spectrin* per erythrocyte(% of normal)
100 80–100 50–80 40–60
Splenectomy Not required Usually not necessaryduring childhoodand adolescence
Necessary during schoolage before puberty
Necessary – delay until6 years if possible

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

INHERITANCE PATTERNS
Top: location of spleen; Bottom: enlarged adult spleen
Most cases of HS are a result of an autosomal dominant mutation(s)
There are no documented cases of a homozygous dominant case (most likely fatal prior to birth)
Approximately 25% of cases are a result of a recessive mutation and is often more severe

BACKGROUND
Graphic of normal erythrocytes (red blood cells)
What is Hereditary Spherocytosis (HS)?
Description Symptoms Testing Clinical classification Inheritance patterns Treatment

TREATMENTPrior to splenectomy: Neonates with
hyperbilirubinemia caused by HS require phototherapy
Folic acid (1 ms/d) required to sustain erthropoiesis
Possible blood transfusions if hemoglobin count is <6
Vaccinations include Hib, meningococcal, & pneuomoccocal
Post-splenectomy Prophylactic antibiotics for 5
years post-surgery Update vaccinations per
protocol Possible cholecystectomy
(gall bladder removal) Precautions against fatal
sepsis caused by a capsulated organism infection

INTRODUCTION MATERIALS & METHODS RESULTS DISCUSSION/CONCLUSION FURTHER RESEARCH
“Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese
Hereditary Spherocytosis Patients”

Spectrin defects Ankryin defects often associated with spectrin defects;
translocatation of chromosome 8 or deletion of short arm of chromosome 8 where ankryin gene is located
Band 3 deficiency; suggests band 3 protein is unstable Protein 4.2 (pallidin) deficiency (mutation or complete absence) The molecular defect in one of the proteins underlying HS
causes a primary deficiency in that protein; however, in somecases more than one protein is deficient (although only oneprotein molecular defect exists)
Hypothesize - the primary protein deficiency triggers secondary proteins deficiencies in proteins with which that protein interacts
INTRODUCTION

Prx2 = Peroxiredoxin 2 – most abundant cytoplasmic proteins & when hemoglobin is denatured, Prx2 binds to cytoplasmic domain of band 3
G3PD = Glyceraldehyde-3-phosphate dehgydrogenase typically cytoplasmic, but in erythrocytes 90% of G3PD is bound to RBC membrane in the inactive form* becoming active when released into the cytoplasm after band 3 phosphorylation
INTRODUCTION
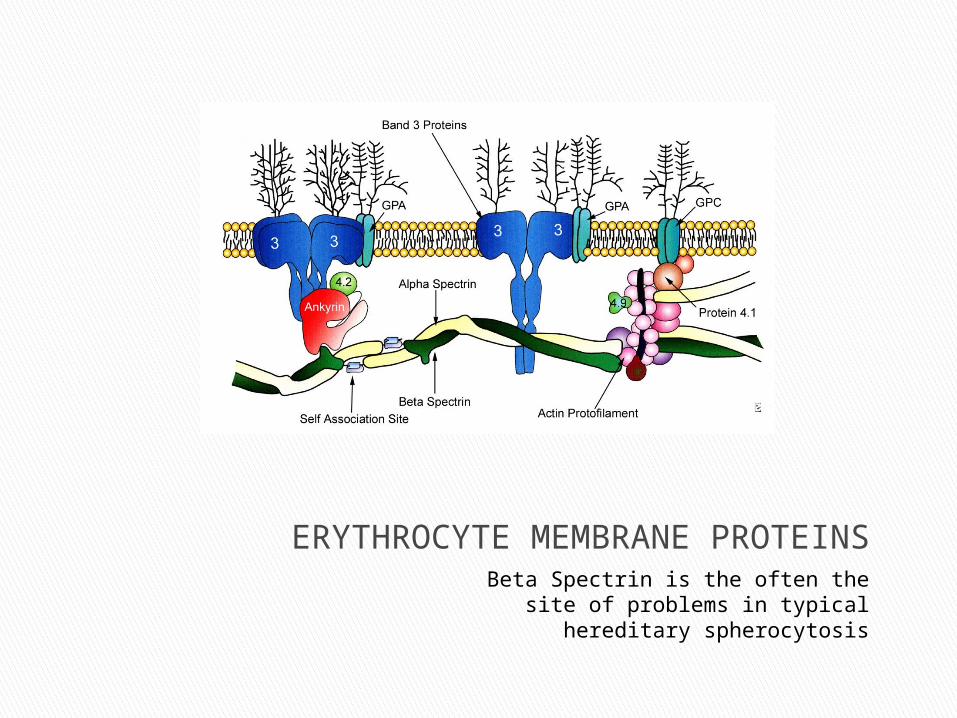
ERYTHROCYTE MEMBRANE PROTEINSBeta Spectrin is the often the site of problems
in typical hereditary spherocytosis
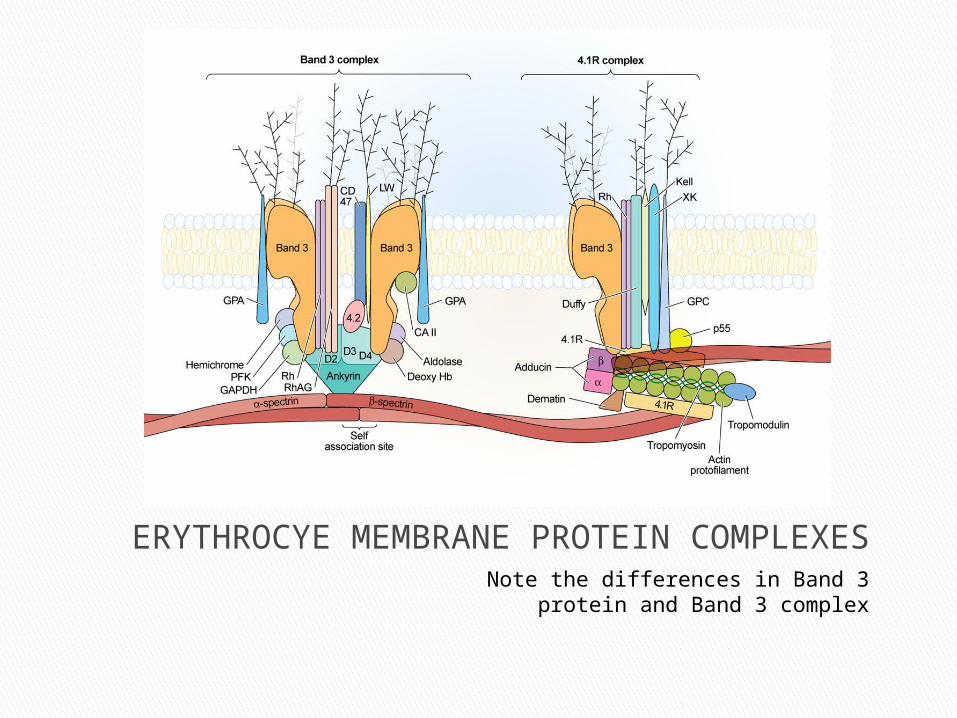
ERYTHROCYE MEMBRANE PROTEIN COMPLEXESNote the differences in Band 3 protein and
Band 3 complex

INTRODUCTION MATERIALS & METHODS RESULTS DISCUSSION/CONCLUSION FURTHER RESEARCH
“Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese
Hereditary Spherocytosis Patients”

Studied 203 individuals from 71 families and 12 unrelated subjects (total 215 people) in the north region of Portugal
160 were diagnosed with HS; classified as mild, moderate, or severe; identified as having had splenectomy or nonsplenectomized
Prepared erythrocytes membranes for electrophoretic anlysis; identified & quantified erythrocyte membrane proteins; Immunoblot analysis for Prx2
MATERIALS & METHODS

SPSS version 16.0 was used One-way ANOVA with Bonferroni’s Non-parametric Kruskal-Wallis H test Mann-Whitney U test Pearson’s correlation coefficient & Spearman’s rank
were used P value lower than 0.05 was considered as statistically
significant
STATISTICAL ANALYSIS

INTRODUCTION MATERIALS & METHODS RESULTS DISCUSSION/CONCLUSION FURTHER RESEARCH
“Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese
Hereditary Spherocytosis Patients”

RESULTS
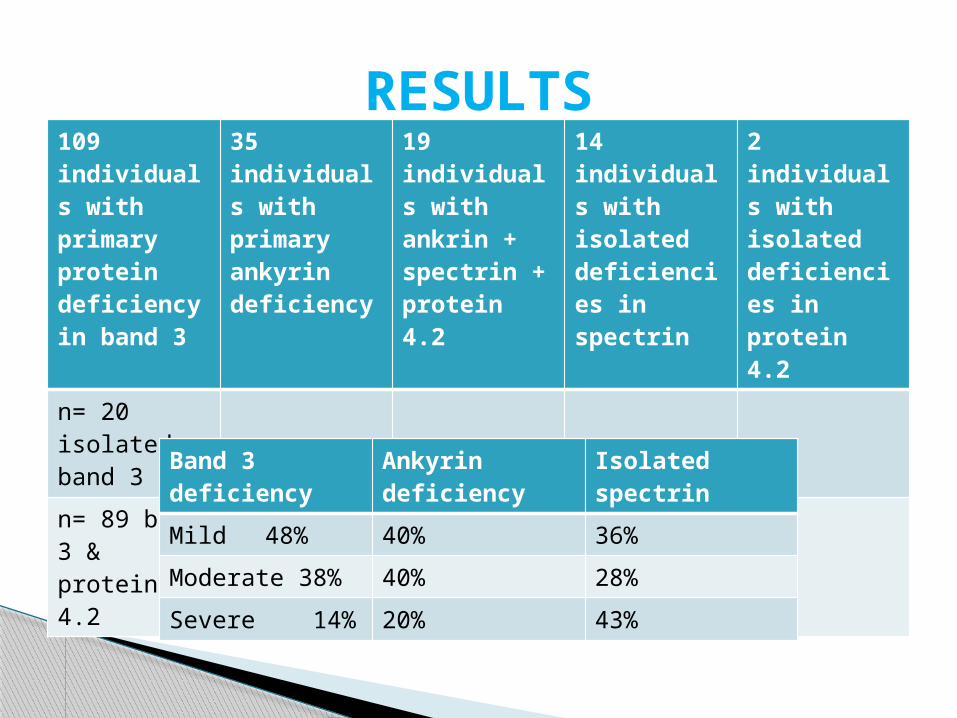
109 individuals with primary protein deficiency in band 3
35 individuals with primary ankyrin deficiency
19 individuals with ankrin + spectrin + protein 4.2
14 individuals with isolated deficiencies in spectrin
2 individuals with isolated deficiencies in protein 4.2
n= 20 isolated band 3
n= 89 band 3 & protein 4.2
RESULTS
Band 3 deficiency Ankyrin deficiency Isolated spectrin
Mild 48% 40% 36%
Moderate 38% 40% 28%
Severe 14% 20% 43%

RESULTS

INTRODUCTION MATERIALS & METHODS RESULTS DISCUSSION/CONCLUSION FURTHER RESEARCH
“Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese
Hereditary Spherocytosis Patients”

Northern European studies differ slightly in patterns of clinical outcomes and molecular defects underlying HS compared to this study (Southern Europeans).
Confirmed hypothesis that an imbalance between protein deficiency levels is responsible for increased membrane destabilization – explaining a more severe clinical pattern
Found statistically significant positive correlation between membrane-linked G3PD and reticulocyte count (r=0.500; p<0.001)
DISCUSSION/CONCLUSION

INTRODUCTION MATERIALS & METHODS RESULTS DISCUSSION/CONCLUSION FURTHER RESEARCH
“Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese
Hereditary Spherocytosis Patients”

Further & more extensive studies regarding cytoplasmic proteins that are able to link to the RBC membrane (like G3PD, metHb, & Prx2) to clarify their contribution to membrane destabilization.
Further molecular analysis can help make clear the genotype-phenotype interactions and the influence of genetic modifiers in HS clinical outcomes.
FURTHER RESEARCH…

Bolton-Maggs, P., Stevens, R., Dodd, N., Lamont, G., Tittensor, P. & King, M.-J. (2004) Guidelines for the diagnosis and management of hereditary spherocytosis. British Journal of Haematology, 126, 455-474.
Rocha, S., Costa, E., Rocha-Pereira, P., Ferreira, F., Cleto, E., Barbot, J., Quintanilha, A., Belo, L., & Santos-Silva, A. (2010) Erythrocyte membrane protein destabilization versus clinical outcome in 160 Portuguese Hereditary Spherocytosis patients. British Journal of Haematology, 149, 785-794.
Images used in the background portion were taken from Google Images searches
SOURCE CITATIONS





























