Embed Size (px)
Citation preview

www.elsevier.com/locate/gene
Gene 328 (2004) 1–16
Review
Nuclear retinoid receptors and the transcription of retinoid-target genes
Julie Bastien, Cecile Rochette-Egly*
Institut de Genetique et de Biologie Moleculaire et Cellulaire, CNRS/INSERM/ULP, UMR 7104, 1 rue Laurent Fries, BP 10142, Illkirch Cedex 67404, France
Received 14 October 2003; accepted 2 December 2003
Received by A.J. van Wijnen
Abstract
The pleiotropic effects of retinoids are mediated by nuclear retinoid receptors (RARs and RXRs) which are ligand-activated transcription
factors. In response to retinoid binding, RAR/RXR heterodimers undergo major conformational changes and orchestrate the transcription of
specific gene networks, through binding to specific DNA response elements and recruiting cofactor complexes that act to modify local
chromatin structure and/or engage the basal transcription machinery. Then the degradation of RARs and RXRs by the ubiquitin–proteasome
controls the magnitude and the duration of the retinoid response. RARs and RXRs also integrate a variety of signaling pathways through
phosphorylation events which cooperate with the ligand for the control of retinoid-target genes transcription. These different modes of
regulation reveal unexpected levels of complexity in the dynamics of retinoid-dependent transcription.
D 2004 Elsevier B.V. All rights reserved.
Keywords: Retinoids; Nuclear receptors; Transcription; Degradation; Kinases; Phosphorylation; Ubiquitin–proteasome
1. Introduction
Vitamin A and its active derivatives referred to as
retinoids are non-steroid hormones which play a critical
role in the development and homeostasis of virtually every
vertebrate tissues through their regulatory effects on cell
differentiation, proliferation and apoptosis (Ross et al.,
2000; Altucci and Gronemeyer, 2001a). It has long been
established that retinoids exert their action by regulating the
expression of specific subsets of genes within target tissues.
However, it is only during the last 15 years that the
understanding for retinoids action rapidly increased, subse-
quently to the cloning of nuclear retinoid receptors and the
0378-1119/$ - see front matter D 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.gene.2003.12.005
Abbreviations: RAR, retinoic acid receptor; RXR, retinoid X receptor;
RA, retinoic acid; DBD, DNA-binding domain; LBD, ligand-binding
domain; RARE, retinoic acid response element; HAT, histone acetyltrans-
ferase; HMT, histone methyltransferase; HDAC, histone deacetylase; CBP,
CREB-binding protein; Cdk, cyclin-dependent kinase; TBP, TATA-binding
protein; GTF, general transcription factor; MAPK, mitogen-activated
protein kinase; P13K, phosphoinositide 3-kinase; PTEN, phosphatase and
tensin homolog; PPAR, peroxisome proliferator activated receptor; LXR,
liver X receptor; APL, acute promyelocytic leukemia.
* Corresponding author. Tel.: +33-3-88-65-34-59; fax: +1-33-3-88-65-
32-01.
E-mail address: [email protected] (C. Rochette-Egly).
identification, within the promoters of retinoid-responsive
genes, of elements exhibiting a high affinity for these
receptors (for review, see (Chambon, 1996; Laudet and
Gronemeyer, 2001), and references therein). Then these
nuclear receptors have been shown to work as ligand-
activated transcription activators in a spatiotemporal specific
manner during embryonic development.
During the last decade, the molecular rationale for
retinoid receptors action has been facilitated by the identi-
fication of the DNA- and ligand-binding domains (DBD
and LBD, respectively) (Chambon, 1996), and by the
determination of their crystal structure (Renaud and Moras,
2000). Moreover, a number of studies demonstrated that
they have to contend with repressive chromatin structures
in order to activate gene expression. Indeed, as most target
genes are initially silent and packed in a dense chromatin
structure, liganded retinoid receptors recruit a battery of
intermediary proteins, including coactivators, chromatin
remodellers and modifyers which act in a coordinated
and/or combinatorial manner to decompact chromatin and
direct RNA polymerase II (RNA Pol II) and the general
transcription factors (GTFs) to the promoter (Dilworth and
Chambon, 2001).
Then an important question was what happens after the
retinoid-activated receptors have bound their DNA reponse

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–162
elements and recruited the transcription machinery. Now,
there is increasing evidence that the ubiquitin–proteasome
machinery degrades the retinoid receptors subsequently to
their activation (Zhu et al., 1999; Boudjelal et al., 2000;
Kopf et al., 2000; Gianni et al., 2002a). This degradation
process may either disrupt the transcription initiation com-
plex, allowing elongation to proceed, and/or terminate the
response to retinoids in order to allow rapidly other tran-
scriptional programs.
Finally, the last years have witnessed a new way of
regulation of retinoid receptors. Indeed, ongoing studies
revealed that they can integrate multiple signaling pathways
through their phosphorylation (Rochette-Egly, 2003). Alter-
natively, retinoids cross-talk with a number of signaling
pathways. All these processes converge towards finely
tuned transcriptional control.
This review will describe our current knowledge about
the molecular mechanisms through which retinoid receptors
regulate transcription, ranging from DNA binding, dynam-
ics of ligand binding and chromatin remodeling and finally
to their degradation. It will also focus on how sequential
and/or coordinated phosphorylation events regulate their
functionality.
Fig. 1. Schematic representation of the functional domains and the major phosphor
and the ligand-binding domain (LBD) are schematically represented (not to scale).
respectively, are depicted. The target sequences for phosphorylation are also shown
amino-terminal kinase.
2. Retinoid receptors contain a DNA-binding domain
and two activation functions AF-1 and AF-2
The retinoid signal is transduced by two families of
nuclear receptors, the retinoic acid receptors (RARs) and
the retinoid X receptors (RXRs), which work as RXR/RAR
heterodimers (Kastner et al., 1997; Mark et al., 1999). Each
family consists of three isotypes (a, h and g) encoded by
separate genes (Leid et al., 1992; Mangelsdorf and Evans,
1995; Chambon, 1996). RARs are activated by all-trans
retinoic acid (RA) and its 9-cis isomer, while RXRs are only
activated by 9-cis RA. For each isotype, there are at least
two isoforms that are generated by differential promoter
usage and alternative splicing and differ only in their N-
terminal regions.
As most nuclear hormone receptors, retinoid receptors
exhibit a modular structure composed of 6 conserved
regions designated A–F (Fig. 1). The highly conserved C
region harbors the DBD which confers sequence specific
DNA recognition. This domain is composed of two zinc-
nucleated modules, two a-helices and a COOH-terminal
extension (CTE) (Zechel et al., 1994a,b). Helix 1 and helix 2
cross at right angles to form the core of the DBD folding
ylation sites of nuclear retinoid receptors. The DNA-binding domain (DBD)
The functional AF-1 and AF-2 domains which lie in the A/B and E regions,
. MAPK, mitogen-activated protein kinase ; MKK, MAPK kinase; JNK, Jun

Fig. 2. (A) High resolution solution structure of the RXRa DNA-binding domain, NMR (mmdbId: 8588). (B) Structure of the RAR/RXR DBD heterodimer in
complex with a DR1 element (mmdbId: 13,928). The interfaces between both partners involve the CII finger surface of the RAR and the T box of the RXRa
partner.
J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 3
into a single globular domain (Fig. 2A) that has been
determined by nuclear magnetic resonance and crystallo-
graphic studies (Lee et al., 1993).
Region E is the second most conserved region and
corresponds to the LBD. It is functionally complex as it
contains the ligand-binding pocket, the main dimerization
domain and the ligand-dependent transactivation function
(AF-2). The structures of the LBDs of RARs and RXRs are
rather similar as demonstrated by the cristallographic studies
Fig. 3. Comparison of the crystal structures of the apo-RXRa (A, PDB1LBD; R
ligand-binding domains reveals the ligand-induced transconformation.
(Wurtz et al., 1996; Moras and Gronemeyer, 1998; Renaud
and Moras, 2000) (Fig. 3A). The LBDs are formed by 12
conserved alpha-helices and a beta-turn (situated between
H5 and H6) which are folded into a three-layered, antipar-
allel helical sandwich with H4, H5, H8 H9 and H11
sandwiched between H1, H2 and H3 on one side and H6,
H7 and H10 on the other. In this structure, the C-terminal
helix H12, which encompasses the AF-2 activation domain,
points away from the LBD core.
enaud et al., 1995) and holo-RARg (B, PDB2LBD; Bourguet et al., 1995)

Fig. 4. (A) The classical retinoid response element is a direct repeat of the
motif 5V-PuG(G/T)TCA spaced by 1 (DR1), 2 (DR2)or 5 (DR5) base pairs.
The natural retinoid response elements from the promoters of some RA-
target genes are shown. (B) On DR2 and DR5 elements, retinoid receptors
bind as RXR/RAR hetodimers with the RXR partner occupying the 5Vmotif. (C) DR1 elements bind either RXR/RAR hetodimers with the
reverse polarity, or RXR homodimers.
J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–164
The heterodimerization surface involves residues from
H7, H9, H10 and H11, as well as loops L8–9 and L9–10
(Bourguet et al., 2000). Helices H9 and H10 contribute to
more than 75% of the total dimerization surface and
constitute the core of the dimer interface. The ligand-
binding pocket comprises hydrophobic residues mainly
from helices H3, H5, H7 and the h-sheet. The precise
contacts with the ligands have been characterized and are
unique for each receptor–cognate ligand pair (Klaholz et al.,
1998; Gehin et al., 1999).
Importantly, the LBD also contains consensus phosphor-
ylation sites (see Fig. 1). Indeed, upon activation of PKA
signaling (Rochette-Egly et al., 1995), the LBD of RARs is
phosphorylated at a conserved serine residue, located be-
tween helices H9 and H10 (Ser 369 for RARa1 and Ser 360
for RARg2). The RXRa LBD can be also phosphorylated
by MAPKs, but at residues (Tyr 248, Ser 265) located in the
Omega loop (between H1 and H3) (Adam-Stitah et al.,
1999; Lee et al., 2000; Matsushima-Nishiwaki et al., 2001;
Adachi et al., 2002).
The N-terminal A/B region harbors a ligand-indepen-
dent transcriptional activation function (AF-1). The three-
dimensional structure of the AF-1 domains of RARs and
RXRs has not been solved yet and structure prediction is
not straightforward. However, the interesting feature of
this domain is that it contains several consensus phos-
phorylation sites (Rochette-Egly, 2003) for proline-depen-
dent kinases which include cyclin-dependent kinases
(CDKs) and MAP kinases (Fig. 1) (Morgan, 1997; Davis,
2000; Pearson et al., 2001). In that context, our laboratory
demonstrated that RARa1 and RARg2 are phosphorylated
in their B region at serines 77 and 68, respectively, by the
cyclin H-dependent kinase cdk7 associated to the general
transcription factor TFIIH (Rochette-Egly et al., 1997;
Bastien et al., 2000). In addition, RARg2 can be phos-
phorylated at the nearby serine residue (Ser66) by
p38MAPK (Gianni et al., 2002a,b). RARh2 is also
phosphorylated at similar residues (K. Drean and C.
Rochette-Egly, unpublished data), but the responsible
kinases remain to be determined. RXRa is phosphorylated
in its A region at Ser22 by a cdk that is distinct from
cdk7 (Adam-Stitah et al., 1999). Importantly, three addi-
tional residues (Ser61, Ser75 and Thr87) can be phos-
phorylated by stress kinases (JNKs) (Adam-Stitah et al.,
1999).
The D region serves as an hinge between the DBD and
the LBD, allowing rotation of the DBD. Therefore it might
allow the DBD and the LBD to adopt different conforma-
tions without creating steric hindrance problems. It also
harbors nuclear localization signals. The F region is absent
in RXRs and its role in RARs, if any, is still unknown.
However, this region is phosphorylated (Rochette-Egly et
al., 1997; Bastien et al., 2000) and it is tempting to speculate
that it may modulate the activation functions AF-1 and/or
AF-2 as in the case of the estrogen receptor (Montano et al.,
1995; Metivier et al., 2002).
3. First step: retinoid receptors binding to responsive
elements located in the regulatory sequences of target
genes
In the absence of ligand, retinoid receptors are found
primarily in the nucleus. They bind as asymetric, oriented
RAR/RXR heterodimers to specific DNA sequences or RA
response elements (RAREs) composed typically of two
direct repeats of a core hexameric motif, PuG(G/T)TCA
(Leid et al., 1992; Mangelsdorf and Evans, 1995) (Fig. 4A).
The classical RARE is a 5-bp-spaced direct repeat (referred
to as DR5). However, RAR/RXR heterodimers also bind to
direct repeats separated by 1 bp (DR1) or 2 bp (DR2). RXRs
also bind to DR1 as RXR/RXR homodimers.
RAREs have been identified in the promoters of a large
number of retinoid-target genes implicated in a wide variety
of functions (Fig. 4A). The classical DR5 elements are
found in the promoters of the RARh gene itself (de The
et al., 1990), CYP26 (Loudig et al., 2000), and several Hox
and HNF genes (Dupe et al., 1997; Qian et al., 2000). DR2
elements were identified in the CRBPI and CRABPII
promoters (Smith et al., 1991; Durand et al., 1992). The
only natural DR1 element binding RXR homodimers has
been found in the rat CRBPII promoter (Mangelsdorf et al.,
1991).

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 5
On DR2 and DR5 elements (Fig. 4B), RXR occupies the
5Vhexameric motif, whereas the RAR partner occupies the 3Vmotif (Chambon, 1996; Laudet and Gronemeyer, 2001). In
contrast, on DR1 elements (Fig. 4C), the polarity is re-
versed, with the RAR DBD binding upstream and the RXR
DBD downstream (5V-RAR/RXR-3V), switching the activity
of the heterodimer from an activator to a repressor of
retinoid-responsive genes.
The cristallographic structure of RXR homodimers and
RAR/RXR heterodimers in complex with DNA has been
solved (Rastinejad et al., 2000; Khorasanizadeh and Rasti-
nejad, 2001; Rastinejad, 2001) (Fig. 2B). Each DBD interacts
with the DNAmajor groove at the level of an half-site through
the P box of the first helix containing three exposed residues
responsible for discrimination between different half-sites
sequences. Then, they arrange in head to tail, with coopera-
tive contacts between the DBDs, leading to a mutual rein-
forcement of protein–protein and protein–DNA interactions.
Depending on the DR spacing, different regions of the DBD
of each receptor are used to create the dimerization interface,
in order to achieve the required binding to the response
elements. The heterodimeric DBD interface that is responsi-
ble for the binding of RXR/RAR heterodimers to DR5
elements involves the D box of the RXR second zinc finger,
and the tip of the RAR first zinc finger. However, when the
heterodimers bind with the reverse polarity to DR1 elements
(Fig. 2B), they associate through the second zinc finger of
RAR and the so-called T box (within the CTE) of RXR. The
same type of dimerisation interface (T box of the downstream
partner and second zinc finger of the upstream one) is
responsible for the cooperative binding of RXR homodimers
to DR1 elements. This implies that the DBDs must be
rotationally flexible with respect to the LBD dimerization
interface and that the DNA curvature is different.
In conclusion, the DBDs of each heterodimerization
partner dictate the specificity of RAREs recognition and
contribute through their dimerisation to increase DNA
binding efficiency.
4. Second step: ligand binding, coactivators recruitment
and chromatin decompaction
When genes are silent, DNA is packaged into a highly
organized and compact nucleoprotein structure known as
chromatin which impedes all the transcription steps. The
basic unit of chromatin is the nucleosome which consists of
DNA wrapped around a protein core containing two copies
each of four histone proteins. Protruding from the nucleo-
somes are the N-terminal «tails» of the core histones whose
interaction with DNA can be modulated upon covalent
modifications (acetylation, phosphorylation, methylation,
etc).
According to the current model of gene regulation by
retinoids established by Dilworth et al (Dilworth and
Chambon, 2001), in a context of chromatin where the
nucleosomes do not impede the binding of RAR/RXR
heterodimers to their DNA recognition sequences, unli-
ganded and DNA-bound retinoid receptors repress transcrip-
tion (Fig. 5A) through the recruitment of the corepressors
NCoR and SMRT (Glass and Rosenfeld, 2000; Aranda and
Pascual, 2001). A single corepressor molecule interacts
through two conserved box motifs, with the LBDs of both
heterodimeric partners. In fact, the corepressors reside in or
recruit high molecular weight complexes endowed with
histone deacetylase activity (HDACs) which increase the
interaction of the N-terminal histone tails with the nucleo-
somal DNA.
Thus, to activate gene expression, retinoid receptors will
have to contend with the repressive chromatin structures in
order to allow the recruitment of the transcription machin-
ery. In this regard, the ligand-induced conformational
changes in the receptors will cause the dissociation of
corepressors and the coordinated and/or combinatorial re-
cruitment of coactivators which upon association with larger
complexes with chromatin modifying and remodeling ac-
tivities will decompact repressive chromatin and facilitate
the positioning of the transcriptional machinery at the
promoter (Fig. 5B and C).
Indeed, ligand binding induces conformational changes
in the LBD (Bourguet et al., 1995; Renaud et al., 1995;
Wurtz et al., 1996; Moras and Gronemeyer, 1998; Egea et
al., 2001), including repositioning of helix H11 in the
continuity of H10. The most striking effect is the swinging
of helix 12 which moves in a «mouse trap model», being
tightly packed against H3 and H4 (Fig. 3B). H12 makes
direct contacts with the ligand and seals the «lid»of the
ligand-binding pocket, further stabilizing ligand binding.
Simultaneously, the Omega loop flips over underneath H6,
carrying along the N-terminal part of H3. According to
recent studies, ligand binding would be facilitated by
cellular retinoic acid-binding protein II (CRABPII) which
upon shuttling into the nucleus and interaction with RAR/
RXR heterodimers (Delva et al., 1999) channels retinoic
acid to RARs (Budhu and Noy, 2002). This interaction of
RARs with CRABPII is stabilized by cyclin D3 (Despouy et
al., 2003).
The ligand-induced conformational changes favor the
interactions between RAR and RXR and therefore increase
their DNA affinity (Rastinejad et al., 2000; Depoix et al.,
2001). They also cause corepressor release and create a new
hydrophobic cleft formed between H3, H4 and H12 which
constitutes a surface where coactivators can bind (Fig. 5B).
According to recent studies, the surfaces involved in core-
pressor and coactivator binding partially overlap and the
ligand-induced repositioning of H12 would result in a
switch from a corepressor to a coactivator-binding surface
(Nagy et al., 1999; Perissi et al., 1999; Glass and Rosenfeld,
2000). Within RAR/RXR heterodimers bound at DR5
elements, though both liganded partners are theoretically
able to recruit coactivators, RXR is «subordinated» to its
RAR partner (Roy et al., 1995; Willy and Mangelsdorf,

Fig. 5. Three-step mechanism of retinoid receptor action. (A) In the absence of ligand, retinoid receptors bound to response elements located in the promoter of
target genes are associated with histone deacetylase-containing (HDAC) complexes tethered through corepressors and repress transcription. (B) Upon ligand
binding, the corepressors dissociate, allowing the recruitment of coactivators associated with complexes displaying histone acetyltransferase (HAT),
methyltransferase, kinase or ATP-dependent remodeling (SWI/SNF) activities that decompact repressive chromatin. (C) In the third step, the coactivators
dissociate and the SMCC mediator complex assembles. Then the mediator expedites entry of the RNA Pol II and the general transcription factors to the
promoter, resulting in transcription initiation.
J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–166
1999). This phenomenon has been attributed to the fact that
liganded RXR cannot dissociate corepressors and therefore
coactivators cannot be recruited (Germain et al., 2002).
However, in the presence of both RAR and RXR ligands,
there is synergy originating from the RAR agonist-induced
dissociation of corepressors and the subsequent cooperative
binding of coactivators to the two partners. Most impor-
tantly, the two partners synergize with each other for
transcription, not only through their AF-2 domains but also
through their AF-1 domains (Gianni et al., 2003), very
likely via their cooperation for the recruitment of coactiva-
tors (Bommer et al., 2002).
The first identified family of ligand-recruited retinoid
receptors coactivators is the SRC/p160 family which
includes SRC-1/NCoA-1, TIF-2/GRIP-1/NCoA-2/SRC-2
and pCIP/ACTR/AlB1/TRAM1/RAC3/SRC-3 (Chen,
2000; McKenna and O’Malley, 2002). Other coactivators,
p300/CBP (Vo and Goodman, 2001) and CARM-1 (Chen et
al., 1999a; Kouzarides, 2002), have been characterized, but
they are structurally and functionally distinguishable from
the SRC/p160 family. A recurring structural feature of all
these coactivators is a highly conserved alpha-helical
LxxLL motif (where L is leucine and x is any amino acid)
from a single to several copies, which is implicated in their

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 7
ligand-dependent recruitment by the AF-2 domain of reti-
noid receptors. Cocrystal structures indicate that two LxxLL
motifs from a single p160 coactivator molecule interact with
the AF-2 domains of both partners. However, one molecule
of coactivator can be cooperatively recruited by each
member of the heterodimer. Most importantly, the coacti-
vators also contain domains which interact with other
coactivators. Accordingly, TIF2 possesses one domain
interacting with CBP/p300 and a second one which has
been recently shown to interact with CARM-1 (McKenna
and O’Malley, 2002).
p300/CBP and, to a lesser extent, the members of the
p160 family locally modify chromatin structure through
their histone acetyltransferase (HAT) activity which acety-
lates lysine residues located at the N-terminal tails of
histones, thereby weakening the interaction of the N-termi-
nal tails with the nucleosome DNA (Fig. 5B). Other
coactivators such as CARM-1 act through their histone
methyltransferase (HMT) activity which upon methylation
of specific arginine or lysine residues also change histone–
DNA and histone–histone contacts. Then the opening of the
chromatin environment is achieved by the recruitment
through pCIP and CBP, of larger complexes with histone
acetyltransferase (PCAF) or histone methyltransferase ac-
tivities (Kouzarides, 2000; Roth et al., 2001; Zhang and
Reinberg, 2001). Note that the efficiency of histone acety-
lation and methylation is regulated upon phosphorylation of
the nearby serines residues by associated kinases (Cheung et
al., 2000; Lo et al., 2001). Altogether, these histone mod-
ifications create tags or binding sites that form an «histone
code» read by a specialized bromodomain present in the
chromatin modifiers (Jeanmougin et al., 1997; Strahl and
Allis, 2000; Berger, 2002). This code would coordinate the
recruitment of additional HATs or HMTs for further chro-
matin decompaction. It would also allow the recruitment of
ATP-dependent chromatin remodelers (SWI/SNF) which
use the energy of ATP hydrolysis to reposition nucleosomes
at the promoter through sliding them in cis or displacing
them in trans, allowing the formation of nucleosome-free or
nucleosome-spaced regions (Kingston and Narlikar, 1999;
Vignali et al., 2000; Narlikar et al., 2002).
5. Third step: recruitment of the transcriptional
machinery
Once repressive chromatin has been decondensed, it has
been proposed that a coregulators exchange occurs, in order
to allow the RARE-bound heterodimers to participate in the
entry of RNA-Pol II and GTFs into the preinitiation com-
plex (Chen et al., 1999b; Malik and Roeder, 2000). The
current working hypothesis is that the p160 coactivators
dissociate, subsequent to their acetylation which decreases
their ability to interact with the receptors (Chen et al.,
1999b), or to their degradation by the proteasome (Yan et
al., 2003). Then the retinoid receptors become able to recruit
the transcription machinery via their association with the so-
called SMCC (Srb and Mediator protein containing com-
plex) mediator complex (Malik and Roeder, 2000; Dilworth
and Chambon, 2001; Woychik and Hampsey, 2002). The
subunit of the mediator complex that is responsible for
interaction with the AF-2 domain of liganded retinoid
receptors was identified as DRIP205 which is identical to
TRAP220 and contains a LxxLL nuclear receptor box motif.
Whether other subunits interact with the N-terminal AF-1
domain of RARs and RXRs, as described for the glucocor-
ticoid receptor (Hittelman et al., 1999), remains to be
determined.
Then the mediator expedites entry of the transcriptional
machinery to the promoter (Fig. 5C) through its interaction
(via other subunits) with the RNA Pol II holoenzyme
(Woychik and Hampsey, 2002). This process also involves
the six GTFs (Orphanides et al., 1996). The large multi-
subunit TFIID, which binds to the promoter through its
TBP, possesses associated factors or TAFIIs developing
kinase and acetylase activities (TAFII250). This will in-
crease chromatin remodeling at the promoter to permit tight
binding of the basal transcriptional machinery. Some TAFIIs
(May et al., 1996; Lavigne et al., 1999) and TFIIH also
interact with retinoid receptors (Rochette-Egly et al., 1997),
thus increasing the efficiency of the preinitiation complex
assembly (see below). Finally, the recruitment of GTFs is
also enhanced by p300/CBP and by components of the SWI/
SNF complex associated to the RNA Pol II holoenzyme
(Adelman and Lis, 2002; Orphanides and Reinberg, 2002).
Once transcription has been initiated, RNA Pol II traffics
along the gene to be transcribed. This process involves
chromatin remodeling and modifying activities endowed by
subunits of the elongation factors that track with elongating
RNA Pol II (Orphanides and Reinberg, 2000). Finally, the
equilibrium will shift in favour of histone tail deacetylation,
and methylation at residues leading to the rapid conversion
of chromatin to a repressed conformation.
In summary, in a context of chromatin where the nucle-
osomes do not impede the binding of RAR/RXR hetero-
dimers to their DNA recognition sequences (Dilworth and
Chambon, 2001), liganded RAR/RXR heterodimers recruit
first coactivators and HAT complexes resulting in histone
acetylation. Then ATP-dependent remodeling complexes are
recruited, leading to the displacement of impeding nucleo-
somes within the proximal promoter region, thus facilitating
access of the general transcription machinery to the pro-
moter. However, it cannot be excluded that the reverse order
of events, which is the recruitment of ATP-dependent
remodelers before HATs, could occur in a context of highly
condensed chromatin (Cosma, 2002). Therefore, the relative
timing and order of recruitment of chromatin modifyers and
remodellers would depend upon the nature of the promoter
and the chromatin structure in which it resides (Fry and
Peterson, 2001) in order to allow the most efficient solution
(Aalfs and Kingston, 2000; Cosma, 2002), each player
helping the other and each step facilitating another one.

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–168
The key is that the appropriate end stage, e.g. a properly
decondensed chromatin with a functional preinitiation com-
plex posed for transcription, be attained in a timely manner.
6. Control of RAR/RXR transactivation by the
ubiquitin–proteasome system
In recent years, it has become evident that the transcrip-
tional activity of retinoid receptors, as that of most tran-
scription factors, is also regulated by the ubiquitin–
proteasome pathway. Paradoxically, both the proteolytic
and non-proteolytic activities of this system appear to
modulate transcription at different levels (Ferdous et al.,
2001; Salghetti et al., 2001; Tansey, 2001; Conaway et al.,
2002; Muratani and Tansey, 2003).
One main role of the ubiquitin–proteasome system is to
degrade transcriptional activators. In this process, following
a signal, the substrate protein is multi-ubiquitylated at a
lysine group and then targeted for destruction by the 26S
proteasome. The 19S subcomplex of the proteasome recog-
nizes the multi-ubiquitylated substrate, removes the ubiq-
uitin groups, unfolds the substrate and feeds the resulting
unstructured chain into the 20S catalytic core of the protea-
some where it is degraded (DeMartino and Slaughter, 1999).
It has been recently demonstrated that, within RAR/RXR
heterodimers bound at response elements, both partners are
degraded by the proteasome in response to retinoids (Zhu et
al., 1999; Boudjelal et al., 2000; Kopf et al., 2000; Osburn et
al., 2001; Tanaka et al., 2001; Gianni et al., 2002a, 2003).
This process involves the ubiquitylation of RARs (Zhu et
al., 1999; Kopf et al., 2000) and the recruitment of the
proteasome at the AF-2 domain through SUG-1 (Gianni et
al., 2002a) which is one of the six ATPases in the base of the
19S regulatory complex of the 26S proteasome. It has been
proposed that this degradation process would provide a
mechanism to control the magnitude and the duration of
retinoid-mediated transcription.
The importance of the ubiquitin–proteasome system in
retinoid receptors transactivation came from the particular
case of the RARg isotype (Gianni et al., 2002a). Indeed,
blocking either the ubiquitin or the proteasome systems
abrogates not only the degradation of RARg, but also
RARg-mediated transcription. The paradoxal mechanism
of how the ubiquitin–proteasome system regulates RARg/
RXR transcriptional activity is currently unknown but
several lines of evidence indicate that the ubiquitin ligases
(Imhof and McDonnell, 1996; McKenna et al., 1998), as
well as some components of the proteasome system, such as
SUG-1 (vom Baur et al., 1996), are able to bind retinoid
receptors. In addition, ubiquitin ligases belong to complexes
that are integral components of the mammalian mediator
complex associated to the Pol II transcription machinery
(Brower et al., 2002; Conaway et al., 2002) and SUG-1 also
interact with the general transcription factor TFIIH (Fraser
et al., 1997; Weeda et al., 1997; Sandrock and Egly, 2001).
Finally, the 19S subcomplex of the proteasome has been
shown to associate with transcription activators (Gonzalez et
al., 2002) and to participate to elongation (Ferdous et al.,
2001). Therefore, the ubiquitin–proteasome machineries
may play a dual role, controlling on the one hand the
functionality of RARg/RXR heterodimers through helping
the recruitment of the transcription machinery (Lin et al.,
2002), and on the other hand the ubiquitylation and the
subsequent degradation of the heterodimers. Such a dual
role may regulate the dynamic assembly/disassembly of
retinoid receptors to the promoter of the target genes, as
recently demonstrated for other nuclear receptors such as the
estrogen and androgen receptors (Freeman and Yamamoto,
2002; Kang et al., 2002; Reid et al., 2003).
It must be noted that the same conclusions could not be
made for the other RAR isotype RARa, since inhibition of
the proteasome by specific inhibitors did not abrogate, but
amplified RARa-mediated transcription (Gianni and Roch-
ette-Egly, unpublished observations), as described for the
glucocorticoid receptor (Wallace and Cidlowski, 2001).
Why the two isotypes RARa and RARg are not regulated
similarly will require further investigations. Finally, the
ubiquitin–proteasome system also targets histones and other
coregulators (Wang et al., 2002; Yan et al., 2003), therefore
increasing the complexity of retinoid-dependent transcrip-
tional control (Muratani and Tansey, 2003).
7. Regulation of RAR/RXR-mediated transcription
through phosphorylation
RARs and RXRs are substrates for a multitude of kinases
(see Figs. 1 and 6) (Rochette-Egly, 2003). Importantly,
subsequent to their interaction with TFIIH, RARs (RARa
and RARg) are phosphorylated in their N-terminal A/B
region by the cdk7 subunit of TFIIH which has a cyclin-
dependent kinase activity (Rochette-Egly et al., 1997; Bas-
tien et al., 2000). This phosphorylation process which has
been extensively studied, especially in the case of RARa,
plays a critical role in the retinoid response. Indeed, upon
mutations in a TFIIH subunit resulting in an incorrect
positioning of the cdk7 kinase relative to its substrate,
RARa is underphosphorylated and retinoid-dependent tran-
scription is decreased (Keriel et al., 2002). If phosphoryla-
tion of the AF-1 domain by TFIIH occurs when the GTFs
are recruited at the promoter, the hypothesis that the
phosphorylation of the AF-1 domain helps the ligand-
dependent recruitment of coactivators and chromatin mod-
ifyers would be rather elusive. Instead, phosphorylation
could facilitate the recruitment of components of the tran-
scription machinery and therefore stabilize the formation of
the NR transcription complex. Future efforts will undoubt-
edly reveal new «coregulators» interacting with the phos-
phorylated motif of RARa and therefore regulating RARa-
mediated transcription. However, it is not excluded that
phosphorylation might rather facilitate the dissociation of

Fig. 6. Signaling pathways activating MAP kinases (Erks, JNKs and p38), PI3K, Akt, PKA and PKC are involved in the control of retinoid-mediated
transcription. Nuclear retinoid receptors are targeted by phosphorylations in response to signaling pathways. These phosphorylations may modulate their
potentiality to recruit cofactors associated with the trascription machinery. The coactivators are also phosphorylated, modulating their activity and/or their
potentiality to recruit the chromatin modifying (HAT, HMT) and remodeling complexes. Finally, histones, the general transcription factors and the RNA
Pol II can be also phosphorylated. RTK, receptor tyrosine kinase; PI3K, phosphatidylinositol 3-kinase; Erk, extracellular signal-related kinase; MAPK,
mitogen-activated protein kinase; MEKK, MAPK kinase kinase; ASK1, apoptosis-stimulating kinase 1; AC, Adenylate cyclase; JNK, Jun amino-terminal
kinase.
J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 9
RARa from transcription inhibitors or help the dissociation
of RARa from the transcription machinery in order to allow
elongation to proceed. Note that, in contrast to other
transcription activators, phosphorylation of the AF-1 do-
main of RARa does not influence the ubiquitylation and the
proteasomal degradation of RARa (Kopf et al., 2000).
It must be pointed out that in the particular case of the
RARg isotype, phosphorylation by TFIIH, though neces-
sary (Bastien et al., 2000), is not sufficient. Indeed, RARg
needs to be also phosphorylated at an additional nearby
residue by p38MAPK, subsequently to its activation by
retinoids (Gianni et al., 2002a,b). Phosphorylation of these
two residues is required for both the transactivation and the
degradation of RARg.
The critical role of RARg phosphorylation has been
further dissected in our group, by using F9 cells which
represent a cell-autonomous system for analyzing retinoid
signaling (for review, see Rochette-Egly and Chambon,
2001). In these cells, the retinoid signal is transduced by
RARg/RXR heterodimers and therefore the various RA
responses are abolished in RARg null cells. Taking advan-
tage that the RA responses can be restored upon reexpres-
sion of the receptor to wild-type levels, the same strategy
has been used with RARg mutated at the phosphorylation
sites located in the N-terminal AF-1 domain. It has been
demonstrated that the integrity of these phosphorylation
sites is indispensable to the activation of a subset of RA-
target genes, for RARg degradation and for RA-induced F9
cell differentiation (Taneja et al., 1997; Kopf et al., 2000;
Gianni et al., 2002a).
Though it is assumed that phosphorylation by both
TFIIH and p38MAPK is crucial for both the transcriptional
activity and the degradation of RARg, the paradoxal mech-
anism of how phosphorylation regulates these two processes
is currently unknown. Nevertheless, some speculative mod-
els can be proposed (see Fig. 7). As phosphorylation of the
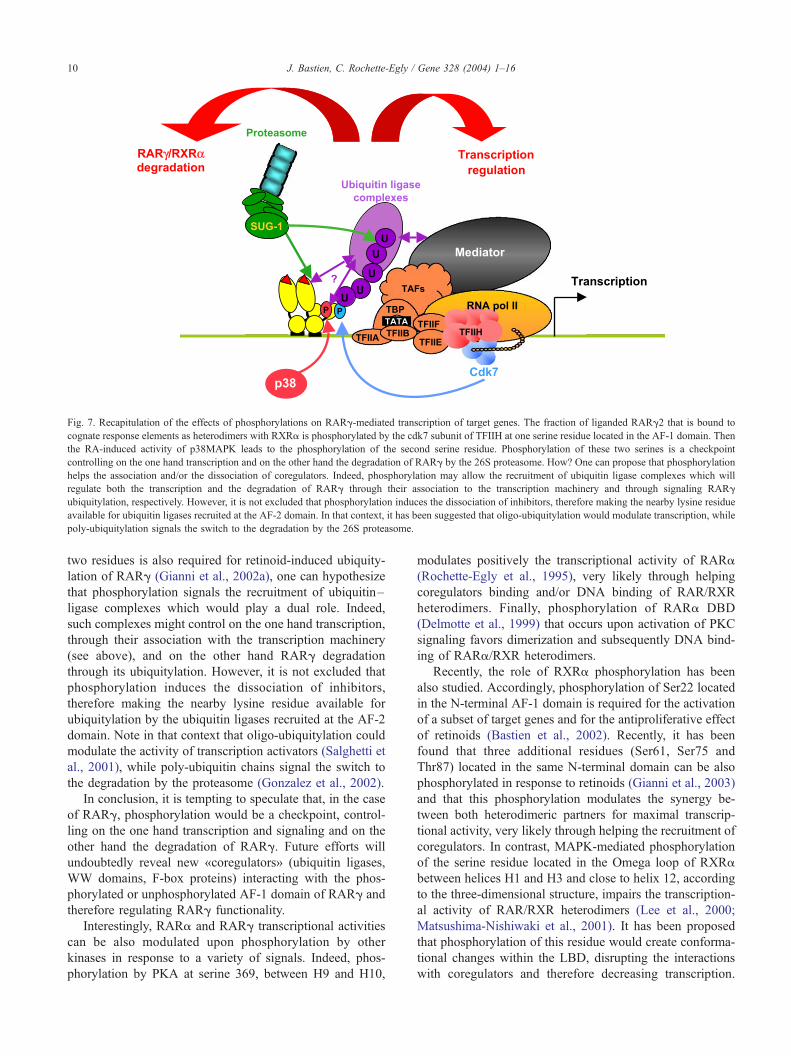
Fig. 7. Recapitulation of the effects of phosphorylations on RARg-mediated transcription of target genes. The fraction of liganded RARg2 that is bound to
cognate response elements as heterodimers with RXRa is phosphorylated by the cdk7 subunit of TFIIH at one serine residue located in the AF-1 domain. Then
the RA-induced activity of p38MAPK leads to the phosphorylation of the second serine residue. Phosphorylation of these two serines is a checkpoint
controlling on the one hand transcription and on the other hand the degradation of RARg by the 26S proteasome. How? One can propose that phosphorylation
helps the association and/or the dissociation of coregulators. Indeed, phosphorylation may allow the recruitment of ubiquitin ligase complexes which will
regulate both the transcription and the degradation of RARg through their association to the transcription machinery and through signaling RARg
ubiquitylation, respectively. However, it is not excluded that phosphorylation induces the dissociation of inhibitors, therefore making the nearby lysine residue
available for ubiquitin ligases recruited at the AF-2 domain. In that context, it has been suggested that oligo-ubiquitylation would modulate transcription, while
poly-ubiquitylation signals the switch to the degradation by the 26S proteasome.
J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–1610
two residues is also required for retinoid-induced ubiquity-
lation of RARg (Gianni et al., 2002a), one can hypothesize
that phosphorylation signals the recruitment of ubiquitin–
ligase complexes which would play a dual role. Indeed,
such complexes might control on the one hand transcription,
through their association with the transcription machinery
(see above), and on the other hand RARg degradation
through its ubiquitylation. However, it is not excluded that
phosphorylation induces the dissociation of inhibitors,
therefore making the nearby lysine residue available for
ubiquitylation by the ubiquitin ligases recruited at the AF-2
domain. Note in that context that oligo-ubiquitylation could
modulate the activity of transcription activators (Salghetti et
al., 2001), while poly-ubiquitin chains signal the switch to
the degradation by the proteasome (Gonzalez et al., 2002).
In conclusion, it is tempting to speculate that, in the case
of RARg, phosphorylation would be a checkpoint, control-
ling on the one hand transcription and signaling and on the
other hand the degradation of RARg. Future efforts will
undoubtedly reveal new «coregulators» (ubiquitin ligases,
WW domains, F-box proteins) interacting with the phos-
phorylated or unphosphorylated AF-1 domain of RARg and
therefore regulating RARg functionality.
Interestingly, RARa and RARg transcriptional activities
can be also modulated upon phosphorylation by other
kinases in response to a variety of signals. Indeed, phos-
phorylation by PKA at serine 369, between H9 and H10,
modulates positively the transcriptional activity of RARa
(Rochette-Egly et al., 1995), very likely through helping
coregulators binding and/or DNA binding of RAR/RXR
heterodimers. Finally, phosphorylation of RARa DBD
(Delmotte et al., 1999) that occurs upon activation of PKC
signaling favors dimerization and subsequently DNA bind-
ing of RARa/RXR heterodimers.
Recently, the role of RXRa phosphorylation has been
also studied. Accordingly, phosphorylation of Ser22 located
in the N-terminal AF-1 domain is required for the activation
of a subset of target genes and for the antiproliferative effect
of retinoids (Bastien et al., 2002). Recently, it has been
found that three additional residues (Ser61, Ser75 and
Thr87) located in the same N-terminal domain can be also
phosphorylated in response to retinoids (Gianni et al., 2003)
and that this phosphorylation modulates the synergy be-
tween both heterodimeric partners for maximal transcrip-
tional activity, very likely through helping the recruitment of
coregulators. In contrast, MAPK-mediated phosphorylation
of the serine residue located in the Omega loop of RXRa
between helices H1 and H3 and close to helix 12, according
to the three-dimensional structure, impairs the transcription-
al activity of RAR/RXR heterodimers (Lee et al., 2000;
Matsushima-Nishiwaki et al., 2001). It has been proposed
that phosphorylation of this residue would create conforma-
tional changes within the LBD, disrupting the interactions
with coregulators and therefore decreasing transcription.

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 11
However, according to recent studies, it would make RXRa
more resistant to proteolytic degradation, therefore exerting
dominant negative inhibition (Matsushima-Nishiwaki et al.,
2001).
It must be noted that the various signal transduction
pathways also cross-talk with retinoid receptors transactiva-
tion through the phosphorylation of the coactivators and
corepressors (see Fig. 6). The phosphorylation of corepres-
sors such as SMRT correlates with an inhibition of their
interaction with RARs and their redistribution from the
nucleus to the cytoplasm (Hong and Privalsky, 2000). In
contrast, the phosphorylation of p300/CBP, pCIP, SRC-1
and TIF-2 by a variety of kinases, including MAPKs or
PKA (Fig. 7), rather enhances their enzymatic activity as
well as their efficiency to interact with retinoid receptors
and/or the HAT complexes (Font de Mora and Brown, 2000;
Rowan et al., 2000a,b; Lopez et al., 2001; Vo and Goodman,
2001). Histones are also phosphorylated, increasing mark-
edly the efficiency of HATs and HMTs to acetylate or
methylate the nearby lysine residues. The same observation
has been made for the general transcription factors (GTFs)
and RNA Pol II (Orphanides and Reinberg, 2002). All these
phosphorylation processes converge towards the formation
of an efficient transcription initiation complex and a con-
trolled maximal response.
8. Retinoid receptors cross-talk with other signaling
pathways
Due to the ability of RXRs to serve as heterodimeric
partners not only to RARs, but also to several other nuclear
receptors (PPARs, LXR) (Willy and Mangelsdorf, 1999), it
is evident that retinoids can also control, the transcription of
a wider set of hormone-responsive genes (Leid et al., 1992;
Mangelsdorf and Evans, 1995; Chambon, 1996). Moreover,
one has to consider that, as is true for many other genes, the
promoters of retinoid-target genes contain, in addition to the
cognate response elements (RAREs), other regulatory
sequences which associate together with several transcrip-
tion activators in enhanceosomes. As an example, RARs
cooperate with SF1 and Sp1/Sp3 for the transactivation of
the Oct-3/4 and CYP26 promoters, respectively (Barnea and
Bergman, 2000; Loudig et al., 2000). Similarly, in the
presence of cytokines, STAT5 cooperates with RARs to
achieve maximum transcription of some RA-target genes (Si
and Collins, 2002). Such synergistic effects very likely
result from the cooperative recruitment of coregulators,
increasing chromatin remodeling and/or entry of the tran-
scription machinery to the promoter. This would explain
why, in vivo, the mechanisms of regulation differ from one
gene to the other and depend on the cell type (Nagpal et al.,
1992; Folkers et al., 1993).
However, retinoids are also able to antagonize the
activation of a subset of heterologous genes. The best
example is that of the AP-1-regulated genes (Shaulian and
Karin, 2002). The repressive effect of retinoids on AP-1
activity has been correlated with their antitumor activity and
the importance of this cross-talk for growth control is
increasingly recognized (Altucci and Gronemeyer, 2001b).
However, the mechanistic basis of the anti-AP-1 activity of
retinoid receptors remains still elusive, despite the proposal
of several distinct mechanisms. Competition for limiting
amounts of a common coactivator (Kamei et al., 1996) has
been proposed, as well as the inhibition of the JNKs
pathway (Caelles et al., 1997; Lee et al., 1999), disruption
of the Jun–Fos dimerization (Zhou et al., 1999) or exclusion
of some components (kinases, CBP) from the AP-1 com-
plexes (Benkoussa et al., 2002). Another example of repres-
sion by retinoid receptors has been demonstrated for
oncogenic h-catenin-mediated gene transcription (Easwaran
et al., 1999; Xiao et al., 2003) but whether liganded retinoid
receptors compete with h-catenin or induce its proteasomal
degradation is still elusive. Finally, retinoids have been
recently demonstrated to antagonize the activation of Smads
regulated genes that occurs in response to TGF-h, very
likely through the activation of a phosphatase, thereby
reducing the levels of phosphorylated Smads (Cao et al.,
2003). However, Pendaries et al. (2003) reported that, in the
presence of antagonists, RARg can potentiate the activation
of TGF-h target genes, via a direct interaction with Smads.
Such observations are of prime importance since it would
dictate whether retinoid agonists or antagonists should be
used in association with TGF-h in the treatment of abnormal
wound healing.
Finally, retinoids have been shown recently to inhibit the
PI3K/Akt pathway. Indeed, our group demonstrated recently
that retinoids target both the PI3K and the phosphatase
PTEN through transcriptional processes involving RAR/
RXR heterodimers (J. Bastien et al., manuscript submitted).
According to another recent study (del Rincon et al., 2003),
retinoids antagonize the mitogenic IGF-IR/Akt pathway
through the dephosphorylation and degradation of the
IRS-1 adaptor protein. This will undoubtedly have conse-
quences on the phosphorylation and therefore on the activity
of the Akt targets, therefore increasing the complexity of the
antiproliferative and differentiative action of retinoids.
9. Conclusion and perspectives
Retinoids are essential for the control of normal cell
differentiation and proliferation. Therefore they are morph-
ogens and essential regulators of embryogenesis. In adults,
they are required for the proper functioning of a number of
organs. All these functions involve the transcriptional con-
trol of a large number of genes by RXR/RAR heterodimers,
as gene-ablation experiments generate embryonic develop-
ment defects and abrogate the differentiative and antiproli-
ferative effects of RA.
According to the current model described in the present
review, retinoid-dependent gene-specific transcription is

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–1612
orchestrated by nuclear receptors which upon DNA and
retinoid binding trigger a cascade of dynamic events result-
ing in an appropriately remodelled chromatin template with
a functional preinitiation complex positioned for transcrip-
tion. At the final end, retinoid receptors are degraded by the
ubiquitin–proteasome pathway.
Though such a model improved efficiently our under-
standing of retinoids action, it is evident that the mecha-
nisms by which retinoid receptors regulate gene
transcription in vivo must be more complex and a number
of questions remain still unanswered. For instance, it is still
unclear how RARs phosphorylation controls the dynamic
recruitment and disassembly of complexes formed at the
promoters of retinoid-target genes during the initiation of
transcription. In addition, considering the complexity of the
promoters of the various retinoid-responsive genes, with
respect to response elements for other transcription activa-
tors, the nucleosomal architecture, the diversity of the cell
type specific coregulators and of the physiological signals, it
is evident that gene transcription is subject to multiple cell
specific combinatorial regulations.
Considering the importance of the regulatory networks
governing the normal transcription of a given gene, the
deregulation of any factor or of the signal transduction
pathways would contribute to the development of diseases
or tumoral processes. As an example, a number of cancers,
especially lung cancers (Picard et al., 1999), are character-
ized by the loss of RARh2 expression due to the hyper-
methylation of its promoter and its subsequent silencing.
Though the gene programs that are specifically regulated by
RARh are unknown, this receptor has been considered as a
tumor suppressor (Altucci and Gronemeyer, 2001b). Anoth-
er example is given by the PML-RARa and PLZF-RARa
fusion proteins (for review, see Altucci and Gronemeyer,
2001b) which, through aberrant recruitment of corepressors,
silence RA-target genes leading to a block of differentiation
of promyelocytic cells in acute promyelocytic leukemia
(APL).
Interestingly, alterations in the phosphorylation of RARs
may also account for some diseases or cancers. Indeed, a
defect in the phosphorylation of RARs has been observed in
cells from xeroderma pigmentosum patients (Keriel et al.,
2002) due to a decreased interaction with the transcription
factor TFIIH which harbours the kinase cdk7. This phos-
phorylation defect would account in part for the develop-
ment abnormalities of these patients (Egly, 2001). In
addition, as alterations in the phosphorylation of RARs
result in aberrant turnover and transactivation (Taneja et
al., 1997; Matsushima-Nishiwaki et al., 2001; Gianni et al.,
2002b), one can postulate that the retinoid resistance of
some cancer cells would result from aberrant kinome
signaling (Blume-Jensen and Hunter, 2001; Tari et al.,
2002).
Future research should focalize at understanding how
retinoid-mediated transcription is finely tuned in the context
of complex natural promoters. In addition, more studies
concerning the regulation of retinoid receptors activity
through phosphorylation should provide new insights in
the developmental processes and in cancer mechanisms.
Acknowledgements
We are particularly grateful to Gaetan Bour and Emilie
Gaillard for enthusiastic discussions and for critics. Many
thanks also to all the past members of the group for their
contribution to the work. We are also very grateful to Prof.
P. Chambon for constant support. Our studies mentioned in
the text have been supported by funds from the Centre
National de la Recherche Scientifique (CNRS), the Institut
National de la Sante et de la Recherche Medicale
(INSERM), the Hopital Universitaire de Strasbourg, the
Association pour la Recherche sur le Cancer, and Bristol-
Myers Squibb. JB was supported by the Ministere de la
Recherche et de l’Enseignement Superieur and by the Ligue
Nationale contre le Cancer.
References
Aalfs, J.D., Kingston, R.E., 2000. What does ‘chromatin remodeling’
mean? Trends Biochem. Sci. 25, 548–555.
Adachi, S., Okuno, M., Matsushima-Nishiwaki, R., Takano, Y., Kojima, S.,
Friedman, S.L., Moriwaki, H., Okano, Y., 2002. Phosphorylation of
retinoid X receptor suppresses its ubiquitination in human hepatocellu-
lar carcinoma. Hepatology 35, 332–340.
Adam-Stitah, S., Penna, L., Chambon, P., Rochette-Egly, C., 1999. Hyper-
phosphorylation of the retinoid X receptor alpha (RXRa) by activated c-
Jun N-terminal kinases (JNKs). J. Biol. Chem. 274, 18932–18941.
Adelman, K., Lis, J.T., 2002. How does Pol II overcome the nucleosome
barrier? Mol. Cell 9, 451–452.
Altucci, L., Gronemeyer, H., 2001a. Nuclear receptors in cell life and
death. Trends Endocrinol. Metab. 12, 460–468.
Altucci, L., Gronemeyer, H., 2001b. The promise of retinoids to fight
against cancer. Nat. Rev., Cancer 1, 181–193.
Aranda, A., Pascual, A., 2001. Nuclear hormone receptors and gene ex-
pression. Physiol. Rev. 81, 1269–1304.
Barnea, E., Bergman, Y., 2000. Synergy of SF1 and RAR in activation of
Oct-3/4 promoter. J. Biol. Chem. 275, 6608–6619.
Bastien, J., Adam-Stitah, S., Riedl, T., Egly, J.M., Chambon, P., Rochette-
Egly, C., 2000. TFIIH interacts with the retinoic acid receptor gamma
and phosphorylates its AF-1-activating domain through cdk7. J. Biol.
Chem. 275, 21896–21904.
Bastien, J., Adam-Stitah, S., Plassat, J.L., Chambon, P., Rochette-Egly, C.,
2002. The phosphorylation site located in the A region of RXRa is
required for the anti-proliferative effect of retinoic acid and the activa-
tion of RA-target genes in F9 cells. J. Biol. Chem. 24, 24.
Benkoussa, M., Brand, C., Delmotte, M.H., Formstecher, P., Lefebvre, P.,
2002. Retinoic acid receptors inhibit AP1 activation by regulating ex-
tracellular signal-regulated kinase and CBP recruitment to an AP1-re-
sponsive promoter. Mol. Cell. Biol. 22, 4522–4534.
Berger, S.L., 2002. Histone modifications in transcriptional regulation.
Curr. Opin. Genet. Dev. 12, 142–148.
Blume-Jensen, P., Hunter, T., 2001. Oncogenic kinase signalling. Nature
411, 355–365.
Bommer, M., Benecke, A., Gronemeyer, H., Rochette-Egly, C., 2002. TIF2
mediates the synergy between RARalpha 1 activation functions AF-1
and AF-2. J. Biol. Chem. 277, 37961–37966.

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 13
Boudjelal, M., Wang, Z., Voorhees, J.J., Fisher, G.J., 2000. Ubiquitin/pro-
teasome pathway regulates levels of retinoic acid receptor gamma and
retinoid X receptor alpha in human keratinocytes. Cancer Res. 60,
2247–2252.
Bourguet, W., Ruff, M., Chambon, P., Gronemeyer, H., Moras, D., 1995.
Crystal structure of the ligand-binding domain of the human nuclear
receptor RXR-alpha. Nature 375, 377–382.
Bourguet, W., Vivat, V., Wurtz, J.M., Chambon, P., Gronemeyer, H., Mo-
ras, D., 2000. Crystal structure of a heterodimeric complex of RAR and
RXR ligand-binding domains. Mol. Cell 5, 289–298.
Brower, C.S., Sato, S., Tomomori-Sato, C., Kamura, T., Pause, A., Stear-
man, R., Klausner, R.D., Malik, S., Lane, W.S., Sorokina, I., Roeder,
R.G., Conaway, J.W., Conaway, R.C., 2002. Mammalian mediator sub-
unit mMED8 is an Elongin BC-interacting protein that can assemble
with Cul2 and Rbx1 to reconstitute a ubiquitin ligase. Proc. Natl. Acad.
Sci. U. S. A. 99, 10353–10358.
Budhu, A.S., Noy, N., 2002. Direct channeling of retinoic acid between
cellular retinoic acid-binding protein II and retinoic acid receptor sen-
sitizes mammary carcinoma cells to retinoic acid-induced growth arrest.
Mol. Cell. Biol. 22, 2632–2641.
Caelles, C., Gonzalez-Sancho, J.M., Munoz, A., 1997. Nuclear hormone
receptor antagonism with AP-1 by inhibition of the JNK pathway.
Genes Dev. 11, 3351–3364.
Cao, Z., Flanders, K.C., Bertolette, D., Lyakh, L.A., Wurthner, J.U., Parks,
W.T., Letterio, J.J., Ruscetti, F.W., Roberts, A.B., 2003. Levels of phos-
pho-Smad2/3 are sensors of the interplay between effects of TGF-beta
and retinoic acid on monocytic and granulocytic differentiation of HL-
60 cells. Blood 101, 498–507.
Chambon, P., 1996. A decade of molecular biology ot retinoic acid recep-
tors. FASEB J. 10, 940–954.
Chen, J.D., 2000. Steroid/nuclear receptor coactivators. Vitam. Horm. 58,
391–448.
Chen, D., Ma, H., Hong, H., Koh, S.S., Huang, S.M., Schurter, B.T.,
Aswad, D.W., Stallcup, M.R., 1999a. Regulation of transcription by a
protein methyltransferase. Science 284, 2174–2177.
Chen, H., Lin, R.J., Xie, W., Wilpitz, D., Evans, R.M., 1999b. Regulation
of hormone-induced histone hyperacetylation and gene activation via
acetylation of an acetylase. Cell 98, 675–686.
Cheung, P., Allis, C.D., Sassone-Corsi, P., 2000. Signaling to chromatin
through histone modifications. Cell 103, 263–271.
Conaway, R.C., Brower, C.S., Conaway, J.W., 2002. Emerging roles of
ubiquitin in transcription regulation. Science 296, 1254–1258.
Cosma, M.P., 2002. Ordered recruitment: gene-specific mechanism of tran-
scription activation. Mol. Cell 10, 227–236.
Davis, R.J., 2000. Signal transduction by the JNK group of MAP kinases.
Cell 103, 239–252.
Delmotte, M.H., Tahayato, A., Formstecher, P., Lefebvre, P., 1999. Serine
157, a retinoic acid receptor alpha residue phosphorylated by protein
kinase C in vitro, is involved in RXR.RARalpha heterodimerization and
transcriptional activity. J. Biol. Chem. 274, 38225–38231.
del Rincon, S.V., Rousseau, C., Samanta, R., Miller Jr., W.H., 2003. Ret-
inoic acid-induced growth arrest of MCF-7 cells involves the selective
regulation of the IRS-1/PI 3-kinase/AKT pathway. Oncogene 22,
3353–3360.
Delva, L., Bastie, J.N., Rochette-Egly, C., Kraiba, R., Balitrand, N.,
Despouy, G., Chambon, P., Chomienne, C., 1999. Physical and func-
tional interactions between cellular retinoic acid binding protein II and
the retinoic acid-dependent nuclear complex. Mol. Cell. Biol. 19,
7158–7167.
DeMartino, G.N., Slaughter, C.A., 1999. The proteasome, a novel protease
regulated by multiple mechanisms. J. Biol. Chem. 274, 22123–22126.
Depoix, C., Delmotte, M.H., Formstecher, P., Lefebvre, P., 2001. Control
of retinoic acid receptor heterodimerization by ligand-induced struc-
tural transitions. A novel mechanism of action for retinoid antagonists.
J. Biol. Chem. 276, 9452–9459.
Despouy, G., Bastie, J.N., Deshaies, S., Balitrand, N., Mazharian, A.,
Rochette-Egly, C., Chomienne, C., Delva, L., 2003. Cyclin D3 is a
cofactor of retinoic acid receptors, modulating their activity in the pre-
sence of cellular retinoic acid-binding protein II. J. Biol. Chem. 278,
6355–6362.
de The, H., Vivanco-Ruiz, M.M., Tiollais, P., Stunnenberg, H., Dejean, A.,
1990. Identification of a retinoic acid responsive element in the retinoic
acid receptor beta gene. Nature 343, 177–180.
Dilworth, F.J., Chambon, P., 2001. Nuclear receptors coordinate the acti-
vities of chromatin remodeling complexes and coactivators to facilitate
initiation of transcription. Oncogene 20, 3047–3054.
Dupe, V., Davenne, M., Brocard, J., Dolle, P., Mark, M., Dierich, A.,
Chambon, P., Rijli, F.M., 1997. In vivo functional analysis of the
Hoxa-1 3V retinoic acid response element (3VRARE). Development
124, 399–410.
Durand, B., Saunders, M., Leroy, P., Leid, M., Chambon, P., 1992. All-
trans and 9-cis retinoic acid induction of CRABPII transcription is
mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated
motifs. Cell 71, 73–85.
Easwaran, V., Pishvaian, M., Salimuddin, Byers, S., 1999. Cross-regulation
of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr. Biol. 9,
1415–1418.
Egea, P.F., Rochel, N., Birck, C., Vachette, P., Timmins, P.A., Moras, D.,
2001. Effects of ligand binding on the association properties and con-
formation in solution of retinoic acid receptors RXR and RAR. J. Mol.
Biol. 307, 557–576.
Egly, J.M., 2001. The 14th datta lecture. TFIIH: from transcription to
clinic. FEBS Lett. 498, 124–128.
Ferdous, A., Gonzalez, F., Sun, L., Kodadek, T., Johnston, S.A., 2001. The
19S regulatory particle of the proteasome is required for efficient tran-
scription elongation by RNA polymerase II. Mol. Cell 7, 981–991.
Folkers, G.E., van der Leede, B.J., van der Saag, P.T., 1993. The retinoic
acid receptor-beta 2 contains two separate cell-specific transactivation
domains, at the N-terminus and in the ligand-binding domain. Mol.
Endocrinol. 7, 616–627.
Font de Mora, J., Brown, M., 2000. AIB1 is a conduit for kinase-mediated
growth factor signaling to the estrogen receptor. Mol. Cell. Biol. 20,
5041–5047.
Fraser, R.A., Rossignol, M., Heard, D.J., Egly, J.M., Chambon, P., 1997.
SUG1, a putative transcriptional mediator and subunit of the PA700
proteasome regulatory complex, is a DNA helicase. J. Biol. Chem.
272, 7122–7126.
Freeman, B.C., Yamamoto, K.R., 2002. Disassembly of transcriptional re-
gulatory complexes by molecular chaperones. Science 296, 2232–2235.
Fry, C.J., Peterson, C.L., 2001. Chromatin remodeling enzymes: who’s on
first? Curr. Biol. 11, R185–R197.
Gehin, M., Vivat, V., Wurtz, J.M., Losson, R., Chambon, P., Moras, D.,
Gronemeyer, H., 1999. Structural basis for engineering of retinoic acid
receptor isotype-selective agonists and antagonists. Chem. Biol. 6,
519–529.
Germain, P., Iyer, J., Zechel, C., Gronemeyer, H., 2002. Co-regulator re-
cruitment and the mechanism of retinoic acid receptor synergy. Nature
415, 187–192.
Gianni, M., Bauer, A., Garattini, E., Chambon, P., Rochette-Egly, C.,
2002a. Phosphorylation by p38MAPK and recruitment of SUG-1 are
required for RA-induced RARg degradation and transactivation. EMBO
J. 21, 3760–3769.
Gianni, M., Kopf, E., Bastien, J., Oulad-Abdelghani, M., Garattini, E.,
Chambon, P., Rochette-Egly, C., 2002b. Down-regulation of the phos-
phatidylinositol 3-Kinase/Akt pathway is involved in retinoic acid-in-
duced phosphorylation, degradation, and transcriptional activity of
retinoic acid receptor gamma 2. J. Biol. Chem. 277, 24859–24862.
Gianni, M., Tarrade, A., Nigro, E.A., Garattini, E., Rochette-Egly, C., 2003.
The AF-1 and AF-2 domains of RAR gamma 2 and RXR alpha coo-
perate for triggering the transactivation and the degradation of RAR
gamma 2/RXR alpha heterodimers. J. Biol. Chem. 278, 34458–34466.
Glass, C.K., Rosenfeld, M.G., 2000. The coregulator exchange in transcrip-
tional functions of nuclear receptors. Genes Dev. 14, 121–141.
Gonzalez, F., Delahodde, A., Kodadek, T., Johnston, S.A., 2002. Recruit-

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–1614
ment of a 19S proteasome subcomplex to an activated promoter. Sci-
ence 296, 548–550.
Hittelman, A.B., Burakov, D., Iniguez-Lluhi, J.A., Freedman, L.P., Gara-
bedian, M.J., 1999. Differential regulation of glucocorticoid receptor
trancriptional activation via AF-1 associated proteins. EMBO J. 18,
5380–5388.
Hong, S.H., Privalsky, M.L., 2000. The SMRT corepressor is regulated by a
MEK-1 kinase pathway: inhibition of corepressor function is associated
with SMRT phosphorylation and nuclear export. Mol. Cell. Biol. 20,
6612–6625.
Imhof, M.O., McDonnell, D.P., 1996. Yeast RSP5 and its human homolog
hRPF1 potentiate hormone-dependent activation of transcription by hu-
man progesterone and glucocorticoid receptors. Mol. Cell. Biol. 16,
2594–2605.
Jeanmougin, F., Wurtz, J.M., Le Douarin, B., Chambon, P., Losson, R.,
1997. The bromodomain revisited (letter). Trends Biochem. Sci. 22,
151–153.
Kamei, Y., Xu, L., Heinzel, T., Torchia, J., Kurokawa, R., Gloss, B., Lin,
S.C., Heyman, R.A., Rose, D.W., Glass, C.K., Rosenfeld, M.G., 1996.
A CBP integrator complex mediates transcriptional activation and AP-1
inhibition by nuclear receptors. Cell 85, 403–414.
Kang, Z., Pirskanen, A., Janne, O.A., Palvimo, J.J., 2002. Involvement of
proteasome in the dynamic assembly of the androgen receptor transcrip-
tion complex. J. Biol. Chem. 277, 48366–48371.
Kastner, P., Mark, M., Ghyselinck, N., Krezel, W., Dupe, V., Grondona,
J.M., Chambon, P., 1997. Genetic evidence that the retinoid signal is
transduced by heterodimeric RXR/RAR functional units during mouse
development. Development 124, 313–326.
Keriel, A., Stary, A., Sarasin, A., Rochette-Egly, C., Egly, J.M., 2002. XPD
mutations prevent TFIIH-dependent transactivation by nuclear receptors
and phosphorylation of RARalpha. Cell 109, 125–135.
Khorasanizadeh, S., Rastinejad, F., 2001. Nuclear-receptor interactions on
DNA-response elements. Trends Biochem. Sci. 26, 384–390.
Kingston, R.E., Narlikar, G.J., 1999. ATP-dependent remodeling and ace-
tylation as regulators of chromatin fluidity. Genes Dev. 13, 2339–2352.
Klaholz, B.P., Renaud, J.P., Mitschler, A., Zusi, C., Chambon, P., Grone-
meyer, H., Moras, D., 1998. Conformational adaptation of agonists to
the human nuclear receptor RAR gamma. Nat. Struct. Biol. 5, 199–202.
Kopf, E., Plassat, J.L., Vivat, V., de The, H., Chambon, P., Rochette-
Egly, C., 2000. Dimerization with retinoid X receptors and phosphor-
ylation modulate the retinoic acid-induced degradation of retinoic acid
receptors alpha and gamma through the ubiquitin–proteasome path-
way. J. Biol. Chem. 275, 33280–33288.
Kouzarides, T., 2000. Acetylation: a regulatory modification to rival phos-
phorylation? EMBO J. 19, 1176–1179.
Kouzarides, T., 2002. Histone methylation in transcriptional control. Curr.
Opin. Genet. Dev. 12, 198–209.
Laudet, V., Gronemeyer, H., 2001. Nuclear Receptor Factsbook Academic
Press, London.
Lavigne, A.C., Mengus, G., Gangloff, Y.G., Wurtz, J.M., Davidson, I.,
1999. Human TAF(II)55 interacts with the vitamin D(3) and thyroid
hormone receptors and with derivatives of the retinoid X receptor that
have altered transactivation properties. Mol. Cell. Biol. 19, 5486–5494.
Lee, M.S., Kliewer, S.A., Provencal, J., Wright, P.E., Evans, R.M., 1993.
Structure of the retinoid X receptor alpha DNA binding domain: a helix
required for homodimeric DNA binding. Science 260, 1117–1121.
Lee, H.Y., Sueoka, N., Hong, W.K., Mangelsdorf, D.J., Claret, F.X., Kurie,
J.M., 1999. All-trans-retinoic acid inhibits Jun N-terminal kinase by
increasing dual-specificity phosphatase activity. Mol. Cell. Biol. 19,
1973–1980.
Lee, H.Y., Suh, Y.A., Robinson, M.J., Clifford, J.L., Hong, W.K.,
Woodgett, J.R., Cobb, M.H., Mangelsdorf, D.J., Kurie, J.M., 2000.
Stress pathway activation induces phosphorylation of retinoid X re-
ceptor. J. Biol. Chem. 275, 32193–32199.
Leid, M., Kastner, P., Chambon, P., 1992. Multiplicity generates diversity
in the retinoic acid signalling pathways. Trends Biochem. Sci. 17,
427–433.
Lin, H.K., Altuwaijri, S., Lin, W.J., Kan, P.Y., Collins, L.L., Chang, C.,
2002. Proteasome activity is required for androgen receptor transcrip-
tional activity via regulation of androgen receptor nuclear translocation
and interaction with coregulators in prostate cancer cells. J. Biol. Chem.
277, 36570–36576.
Lo, W.S., Duggan, L., Tolga, N.C., Belotserkovskya, R., Lane, W.S., Shie-
khattar, R., Berger, S.L., 2001. Snf1—a histone kinase that works in
concert with the histone acetyltransferase Gcn5 to regulate transcription.
Science 293, 1142–1146.
Lopez, G.N., Turck, C.W., Schaufele, F., Stallcup, M.R., Kushner, P.J.,
2001. Growth factors signal to steroid receptors through mitogen-acti-
vated protein kinase regulation of p160 coactivator activity. J. Biol.
Chem. 276, 22177–22182.
Loudig, O., Babichuk, C., White, J., Abu-Abed, S., Mueller, C., Petkovich,
M., 2000. Cytochrome P450RAI(CYP26) promoter: a distinct compo-
site retinoic acid response element underlies the complex regulation of
retinoic acid metabolism. Mol. Endocrinol. 14, 1483–1497.
Malik, S., Roeder, R.G., 2000. Transcriptional regulation through Media-
tor-like coactivators in yeast and metazoan cells. Trends Biochem. Sci.
25, 277–283.
Mangelsdorf, D.J., Evans, R.M., 1995. The RXR heterodimers and orphan
receptors. Cell 83, 841–850.
Mangelsdorf, D.J., Umesono, K., Kliewer, S.A., Borgmeyer, U., Ong, E.S.,
Evans, R.M., 1991. A direct repeat in the cellular retinol-binding pro-
tein type II gene confers differential regulation by RXR and RAR. Cell
66, 555–561.
Mark, M., Ghyselinck, N.B., Wendling, O., Dupe, V., Mascrez, B., Kastner,
P., Chambon, P., 1999. A genetic dissection of the retinoid signalling
pathway in the mouse. Proc. Nutr. Soc. 58, 609–613.
Matsushima-Nishiwaki, R., Okuno, M., Adachi, S., Sano, T., Akita, K.,
Moriwaki, H., Friedman, S.L., Kojima, S., 2001. Phosphorylation of
retinoid X receptor alpha at serine 260 impairs its metabolism and func-
tion in human hepatocellular carcinoma. Cancer Res. 61, 7675–7682.
May, M., Mengus, G., Lavigne, A.C., Chambon, P., Davidson, I., 1996.
Human TAF(II28) promotes transcriptional stimulation by activation
function 2 of the retinoid X receptors. EMBO J. 15, 3093–3104.
McKenna, N.J., O’Malley, B.W., 2002. Combinatorial control of gene
expression by nuclear receptors and coregulators. Cell 108, 465–474.
McKenna, N.J., Nawaz, Z., Tsai, S.Y., Tsai, M.J., O’Malley, B.W., 1998.
Distinct steady-state nuclear receptor coregulator complexes exist in
vivo. Proc. Natl. Acad. Sci. U. S. A. 95, 11697–11702.
Metivier, R., Stark, A., Flouriot, G., Hubner, M.R., Brand, H., Penot, G.,
Manu, D., Denger, S., Reid, G., Kos, M., Russell, R.B., Kah, O.,
Pakdel, F., Gannon, F., 2002. A dynamic structural model for estrogen
receptor-alpha activation by ligands, emphasizing the role of interac-
tions between distant A and E domains. Mol. Cell 10, 1019–1032.
Montano, M.M., Muller, V., Trobaugh, A., Katzenellenbogen, B.S., 1995.
The carboxy-terminal F domain of the human estrogen receptor: role in
the transcriptional activity of the receptor and the effectiveness of anti-
estrogens as estrogen antagonists. Mol. Endocrinol. 9, 814–825.
Moras, D., Gronemeyer, H., 1998. The nuclear receptor ligand-binding
domain: structure and function. Curr. Opin. Cell Biol. 10, 384–391.
Morgan, D.O., 1997. Cyclin-dependent kinases: engines, clocks, and
microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291.
Muratani, M., Tansey, W.P., 2003. How the ubiquitin–proteasome system
controls transcription. Nat. Rev., Mol. Cell Biol. 4, 192–201.
Nagpal, S., Saunders, M., Kastner, P., Durand, B., Nakshatri, H., Chambon,
P., 1992. Promoter context- and response element-dependent specificity
of the transcriptional activation and modulating functions of retinoic
acid receptors. Cell 70, 1007–1019.
Nagy, L., Kao, H.Y., Love, J.D., Li, C., Banayo, E., Gooch, J.T., Krishna,
V., Chatterjee, K., Evans, R.M., Schwabe, J.W.R., 1999. Mechanism of
corepressor binding and release from nuclear hormone receptors. Genes
Dev. 13, 3209–3216.
Narlikar, G.J., Fan, H.Y., Kingston, R.E., 2002. Cooperation between com-
plexes that regulate chromatin structure and transcription. Cell 108,
475–487.

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–16 15
Orphanides, G., Reinberg, D., 2000. RNA polymerase II elongation
through chromatin. Nature 407, 471–475.
Orphanides, G., Reinberg, D., 2002. A unified theory of gene expression.
Cell 108, 439–451.
Orphanides, G., Lagrange, T., Reinberg, D., 1996. The general transcription
factors of RNA polymerase II. Genes Dev. 10, 2657–2683.
Osburn, D.L., Shao, G., Seidel, H.M., Schulman, I.G., 2001. Ligand-depen-
dent degradation of retinoid X receptors does not require transcriptional
activity or coactivator interactions. Mol. Cell. Biol. 21, 4909–4918.
Pearson, G., Robinson, F., Beers Gibson, T., Xu, B.E., Karandikar, M.,
Berman, K., Cobb, M.H., 2001. Mitogen-activated protein (MAP) ki-
nase pathways: regulation and physiological functions. Endocr. Rev. 22,
153–183.
Pendaries, V., Verrecchia, F., Michel, S., Mauviel, A., 2003. Retinoic acid
receptors interfere with the TGF-b/Smad signaling pathway in a ligand-
specific manner. Oncogene 22, 8212–8220.
Perissi, V., Staszewski, L.M., McInerney, E.M., Kurokawa, R., Krones, R.,
Rose, D.W., Lambert, M.H., Milburn, M.V., Glass, C.K., Rosenfeld,
M.G., 1999. Molecular determinants of nuclear receptor–corepressor
interaction. Genes Dev. 13, 3198–3208.
Picard, E., Seguin, C., Monhoven, N., Rochette-Egly, C., Siat, J., Borrelly,
J., Martinet, Y., Martinet, N., Vignaud, J.M., 1999. Expression of re-
tinoid receptor genes and proteins in non-small-cell lung cancer (see
comments) . J. Natl. Cancer Inst. 91, 1059–1066.
Qian, A., Cai, Y., Magee, T.R., Wan, Y.J., 2000. Identification of retinoic
acid-responsive elements on the HNF1alpha and HNF4alpha genes.
Biochem. Biophys. Res. Commun. 276, 837–842.
Rastinejad, F., 2001. Retinoid X receptor and its partners in the nuclear
receptor family. Curr. Opin. Struck. Biol. 11, 33–38.
Rastinejad, F., Wagner, T., Zhao, Q., Khorasanizadeh, S., 2000. Structure of
the RXR-RAR DNA-binding complex on the retinoic acid response
element DR1. EMBO J. 19, 1045–1054.
Reid, G., Hubner, M.R., Metivier, R., Brand, H., Denger, S., Manu, D.,
Beaudouin, J., Ellenberg, J., Gannon, F., 2003. Cyclic, proteasome-
mediated turnover of unliganded and liganded ERalpha on responsive
promoters is an integral feature of estrogen signaling. Mol. Cell 11,
695–707.
Renaud, J.P., Moras, D., 2000. Structural studies on nuclear receptors. Cell.
Mol. Life Sci. 57, 1748–1769.
Renaud, J.P., Rochel, N., Ruff, M., Vivat, V., Chambon, P., Gronemeyer,
H., Moras, D., 1995. Crystal structure of the RAR-gamma ligand-bind-
ing domain bound to all-trans retinoic acid. Nature 378, 681–689.
Rochette-Egly, C., 2003. Nuclear receptors: integration of multiple signal-
ling pathways through phosphorylation. Cell Signal. 15, 355–366.
Rochette-Egly, C., Chambon, P., 2001. F9 embryocarcinoma cells: a cell
autonomous model to study the functional selectivity of RARs and
RXRs in retinoid signaling. Histol. Histopathol. 16, 909–922.
Rochette-Egly, C., Oulad-Abdelghani, M., Staub, A., Pfister, V., Scheuer,
I., Chambon, P., Gaub, M.P., 1995. Phosphorylation of the retinoic acid
receptor-alpha by protein kinase A. Mol. Endocrinol. 9, 860–871.
Rochette-Egly, C., Adam, S., Rossignol, M., Egly, J.M., Chambon, P.,
1997. Stimulation of RAR alpha activation function AF-1 through bind-
ing to the general transcription factor TFIIH and phosphorylation by
CDK7. Cell 90, 97–107.
Ross, S.A., McCaffery, P.J., Drager, U.C., De Luca, L.M., 2000. Retinoids
in embryonal development. Physiol. Rev. 80, 1021–1054.
Roth, S.Y., Denu, J.M., Allis, C.D., 2001. Histone acetyltransferases. Ann.
Rev. Biochem. 70, 81–120.
Rowan, B.G., Garrison, N., Weigel, N.L., O’Malley, B.W., 2000a. 8-Bro-
mo-cyclic AMP induces phosphorylation of two sites in SRC-1 that
facilitate ligand-independent activation of the chicken progesterone re-
ceptor and are critical for functional cooperation between SRC-1 and
CREB binding protein. Mol. Cell. Biol. 20, 8720–8730.
Rowan, B.G., Weigel, N.L., O’Malley, B.W., 2000b. Phosphorylation of
steroid receptor coactivator: 1. Identification of the phosphorylation
sites and phosphorylation through the mitogen-activated protein kinase
pathway. J. Biol. Chem. 275, 4475–4483.
Roy, B., Taneja, R., Chambon, P., 1995. Synergistic activation of retinoic
acid (RA)-responsive genes and induction of embryonal carcinoma cell
differentiation by an RA receptor alpha (RAR alpha)-, RAR beta-, or
RAR gamma-selective ligand in combination with a retinoid X recep-
tor-specific ligand. Mol. Cell. Biol. 15, 6481–6487.
Salghetti, S.E., Caudy, A.A., Chenoweth, J.G., Tansey, W.P., 2001. Regu-
lation of transcriptional activation domain function by ubiquitin. Sci-
ence 293, 1651–1653.
Sandrock, B., Egly, J.M., 2001. A yeast four-hybrid system identifies Cdk-
activating kinase as a regulator of the XPD helicase, a subunit of tran-
scription factor IIH. J. Biol. Chem. 276, 35328–35333.
Shaulian, E., Karin, M., 2002. AP-1 as a regulator of cell life and death.
Nat. Cell Biol. 4, E131–E136.
Si, J., Collins, S.J., 2002. IL-3-induced enhancement of retinoic acid re-
ceptor activity is mediated through Stat5, which physically associates
with retinoic acid receptors in an IL-3-dependent manner. Blood 100,
4401–4409.
Smith, W.C., Nakshatri, H., Leroy, P., Rees, J., Chambon, P., 1991. A
retinoic acid response element is present in the mouse cellular retinol
binding protein I (mCRBPI) promoter. EMBO J. 10, 2223–2230.
Strahl, B.D., Allis, C.D., 2000. The language of covalent histone modifi-
cations. Nature 403, 41–45.
Tanaka, T., Rodriguez de la Concepcion, M.L., De Luca, L.M., 2001.
Involvement of all-trans-retinoic acid in the breakdown of retinoic acid
receptors alpha and gamma through proteasomes in MCF-7 human
breast cancer cells. Biochem. Pharmacol. 61, 1347–1355.
Taneja, R., Rochette-Egly, C., Plassat, J.L., Penna, L., Gaub, M.P.,
Chambon, P., 1997. Phosphorylation of activation functions AF-1
and AF-2 of RAR alpha and RAR gamma is indispensable for dif-
ferentiation of F9 cells upon retinoic acid and cAMP treatment.
EMBO J. 16, 6452–6465.
Tansey, W.P., 2001. Transcriptional activation: risky business. Genes Dev.
15, 1045–1050.
Tari, A.M., Lim, S.J., Hung, M.C., Esteva, F.J., Lopez-Berestein, G., 2002.
Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast
cancer cells. Oncogene 21, 5224–5232.
Vignali, M., Hassan, A.H., Neely, K.E., Workman, J.L., 2000. ATP-depen-
dent chromatin-remodeling complexes. Mol. Cell. Biol. 20, 1899–1910.
Vo, N., Goodman, R.H., 2001. CREB-binding protein and p300 in tran-
scriptional regulation. J. Biol. Chem. 276, 13505–13508.
vom Baur, E., Zechel, C., Heery, D., Heine, M.J., Garnier, J.M., Vivat, V.,
Le Douarin, B., Gronemeyer, H., Chambon, P., Losson, R., 1996. Dif-
ferential ligand-dependent interactions between the AF-2 activating do-
main of nuclear receptors and the putative transcriptional intermediary
factors mSUG1 and TIF1. EMBO J. 15, 110–124.
Wallace, A.D., Cidlowski, J.A., 2001. Proteasome-mediated glucocorticoid
receptor degradation restricts transcriptional signaling by glucocorti-
coids. J. Biol. Chem. 276, 42714–42721.
Wang, J., Barsky, L.W., Shum, C.H., Jong, A., Weinberg, K.I., Collins,
S.J., Triche, T.J., Wu, L., 2002. Retinoid-induced G1 arrest and differ-
entiation activation are associated with a switch to cyclin-dependent
kinase-activating kinase hypophosphorylation of retinoic acid receptor
alpha. J. Biol. Chem. 277, 43369–43376.
Weeda, G., Rossignol, M., Fraser, R.A., Winkler, G.S., Vermeulen, W.,
van’t Veer, L.J., Ma, L., Hoeijmakers, J.H., Egly, J.M., 1997. The
XPB subunit of repair/transcription factor TFIIH directly interacts with
SUG1, a subunit of the 26S proteasome and putative transcription fac-
tor. Nucleic Acids Res. 25, 2274–2283.
Willy, P.J., Mangelsdorf, D.J., 1999. Nuclear orphan receptors:the search
for novel ligands and signaling pathways. In: O’Malley, B.W. (Ed.),
Hormone and Signaling. Academic Press, San Diego, pp. 307–358.
Woychik, N.A., Hampsey, M., 2002. The RNA polymerase II machinery:
structure illuminates function. Cell 108, 453–463.
Wurtz, J.M., Bourguet, W., Renaud, J.P., Vivat, V., Chambon, P., Moras, D.,
Gronemeyer, H., 1996. A canonical structure for the ligand-binding do-
main of nuclear receptors (see comments). Nat. Struct. Biol. 3, 87–94
(published erratum appears in Nat Struct Biol 1996 Feb; 3(2):206).

J. Bastien, C. Rochette-Egly / Gene 328 (2004) 1–1616
Xiao, J.H., Ghosn, C., Hinchman, C., Forbes, C., Wang, J., Snider, N.,
Cordrey, A., Zhao, Y., Chandraratna, R.A., 2003. APC-independent
regulation of beta-catenin degradation via a retinoid X receptor-medi-
ated pathway. J. Biol. Chem. 278, 29954–29962.
Yan, F., Gao, X., Lonard, D.M., Nawaz, Z., 2003. Specific ubiquitin-con-
jugating enzymes promote degradation of specific nuclear receptor
coactivators. Mol. Endocrinol. 17, 1315–1331.
Zechel, C., Shen, X.Q., Chambon, P., Gronemeyer, H., 1994a. Dimeriza-
tion interfaces formed between the DNA binding domains determine the
cooperative binding of RXR/RAR and RXR/TR heterodimers to DR5
and DR4 elements. EMBO J. 13, 1414–1424.
Zechel, C., Shen, X.Q., Chen, J.Y., Chen, Z.P., Chambon, P., Gronemeyer,
H., 1994b. The dimerization interfaces formed between the DNA bind-
ing domains of RXR, RAR and TR determine the binding specificity
and polarity of the full-length receptors to direct repeats. EMBO J. 13,
1425–1433.
Zhang, Y., Reinberg, D., 2001. Transcription regulation by histone methy-
lation: interplay between different covalent modifications of the core
histone tails. Genes Dev. 15, 2343–2360.
Zhou, X.F., Shen, X.Q., Shemshedini, L., 1999. Ligand-activated retinoic
acid receptor inhibits AP-1 transactivation by disrupting c-Jun/c-Fos
dimerization. Mol. Endocrinol. 13, 276–285.
Zhu, J., Gianni, M., Kopf, E., Honore, N., Chelbi-Alix, M., Koken, M.,
Quignon, F., Rochette-Egly, C., de The, H., 1999. Retinoic acid induces
proteasome-dependent degradation of retinoic acid receptor a (RARa)
and oncogenic RARa fusion proteins. Proc. Natl. Acad. Sci. U. S. A.
96, 14807–14812.





























