Embed Size (px)
Citation preview

Copyright � 2007 by the Genetics Society of AmericaDOI: 10.1534/genetics.106.069963
Review
Applications of High-Throughput RNA Interference Screens toProblems in Cell and Developmental Biology
Norbert Perrimon1 and Bernard Mathey-Prevot
Department of Genetics, Howard Hughes Medical Institute, Harvard Medical School, Boston, Massachusetts 02175
ABSTRACT
RNA interference (RNAi) in tissue culture cells has emerged as an excellent methodology foridentifying gene functions systematically and in an unbiased manner. Here, we describe how RNAi high-throughput screening (HTS) in Drosophila cells are currently being performed and emphasize thestrengths and weaknesses of the approach. Further, to demonstrate the versatility of the technology, weprovide examples of the various applications of the method to problems in signal transduction and celland developmental biology. Finally, we discuss emerging technological advances that will extend RNAi-based screening methods.
MUCH of what we know today about the molecularmechanisms underlying fundamental cellular
and developmental processes in higher eukaryotes canbe traced back to a genetic screen in a model organism.Over the years, the development and application ofnew genetic tools have constantly provided new meansto discover gene function. Recently, this has been bestillustrated with the application of RNA interference(RNAi) to functional genomics, which relies on the abil-ity of double-stranded RNAs (dsRNAs), small interfer-ing RNAs (siRNAs), or small hairpin RNAs to silencea target gene through the specific destruction of thatgene’s mRNA (Fire et al. 1998; Elbashir et al. 2001;reviewed in Echeverri and Perrimon 2006).
Here, we focus on the application of RNAi to high-throughput screening (HTS) in Drosophila tissue cul-ture cells. This systematic and unbiased approach togene discovery was made possible with the breakthroughdiscovery by Hammond et al. (2000) and Clemens et al.(2000) that the addition of long dsRNAs to Drosophilacells can elicit a potent and specific RNAi effect. Further,the completion of the Drosophila genome sequence in2000 (Adams et al. 2000) made it feasible to extend thismethod to the genomewide scale. These developmentscreated an opportunity to investigate the function ofevery gene in tissue culture assays (cell-based assays),where each assay can be designed to address a specificbiological question. RNAi HTS in Drosophila tissue cul-ture is borrowed from systematic RNAi screens carried
out earlier in Caenorhabditis elegans. RNAi reagents weredelivered either by feeding bacteria expressing a specificdsRNA to animals or after dsRNA injection into embryos(Fraser et al. 2000; Gonczy et al. 2000; Sonnichsen et al.2005). Similar genomewide libraries are being imple-mented in mammalian cell culture systems (Echeverri
and Perrimon 2006; Moffat and Sabatini 2006).In each organism or system, RNAi-based screening is
being used to identify gene function rapidly, systemat-ically, and in an unbiased manner. Because full ge-nomes can be screened, two different philosophies haveemerged behind performing such genomewide screens.The first one is centered on ‘‘gene discovery’’ and stemsfrom the same rationale invoked in more traditionalgenetics screens and, as such, is concerned mainly withthe identification of one or several important missingcomponents in a biological process. The second one ismore in tune with a ‘‘systems biology’’ approach. Ratherthan focusing primarily on a small number of genes, thegoal here is to organize large sets of genes intofunctional subnetworks that carry out and modulate aspecific program or response, with the overall goal ofpredicting the biological response of this network toperturbation or stimulation.
RNAi HTS METHODS
Building on the finding that long dsRNA added toDrosophila cells results in specific gene expressionknockdown (Clemens et al. 2000; Hammond et al.2000), a number of laboratories, including our own,applied the approach to perform genome-scale func-tional screens in Drosophila cell-based assays (Kiger
1Corresponding author: Department of Genetics, Howard HughesMedical Institute, Harvard Medical School, 77 Ave. Louis Pasteur, Boston,MA 02175. E-mail: [email protected]
Genetics 175: 7–16 ( January 2007)

et al. 2003; Lum et al. 2003; Boutros et al. 2004; Foley
and O’Farrell 2004; Armknecht et al. 2005). Inour laboratory, we assembled a genomewide collectionof dsRNAs targeting all known Drosophila genes(Boutros et al. 2004). We then developed a robustexperimental platform (Armknecht et al. 2005) andbuilt a suitable infrastructure necessary to run large-scale RNAi screens in Drosophila cells. As this technol-ogy attracted a lot of outside interest, we made itavailable to the rest of the scientific community andopened the Drosophila RNAi Screening Center (DRSC)(see http://flyrnai.org) at Harvard Medical School(Flockhart et al. 2006).
The basic experimental design for HTS adopted inmany experimental settings involves three major steps(Figure 1): (1) gene-specific dsRNAs are arrayed into96- or 384-well assay plates using robotics, (2) cells areuniformly and rapidly dispensed into the plates using aliquid dispenser, and (3) after the appropriate incuba-tion time, cells are subjected to individual treatments ina highly parallel fashion, fixed, or directly processed forthe assay readout. The above experimental platformallows for a flexible screening methodology, where eachstep can be optimized to suit a wide range of assays, andis usually performed in 384-well plates to maximize thehigh-throughput nature of the screens (Armknecht
et al. 2005; Echeverri and Perrimon 2006). Many ofthe screens performed to date rely on the use of eithera plate reader (or a variation thereof, the automatedfluorometric imaging plate reader) (Vig et al. 2006) todetect fluorescence or luminescence signals or high-throughput wide-field (Kiger et al. 2003) and confocalmicroscopy (Pelkmans et al. 2005) (Figure 2).
Cell lines for cell-based assays: The design of cell-based assays is often dictated by the characteristics ofavailable cell lines, and whenever possible, has takenadvantage of their different attributes. Importantly,cell lines can be manipulated to expand the range ofpossible assays through either transient or stable trans-
fection of DNA constructs. The most commonly usedcell lines in Drosophila RNAi HTS are S2 (and itsderivative, S2R1) (Yanagawa et al. 1998) and Kc, whichare of embryonic hemocyte origin. Their chief advan-tage is that they require simple bathing in serum-freemedium to take up dsRNA efficiently (Clemens et al.2000; Armknecht et al. 2005) and can be readilyinfected by a variety of viruses or bacteria (Agaisse
et al. 2005; Cherry et al. 2005; Philips et al. 2005). Clone8 cells are another popular cell line as these cells aremore amenable for studying certain pathways (e.g.,Hedgehog signaling) (Lum et al. 2003; Nybakken et al.2005), as they are derived from the wing-disc epithelium(Yanagawa et al. 1997). Although clone 8 cells showpoor uptake with the bathing method, they are readilytransfectable using standard lipid-based dsRNA trans-fection methods (Lum et al. 2003; Armknecht et al.2005). Many other Drosophila cell lines of variousorigins can also be used for RNAi applications and areavailable from the Drosophila Genomics ResearchCenter (http://dgrc.cgb.indiana.edu/).
Transcriptional and enzymatic reporter screens:Many assays are based on transcriptional reportersexpressing various luciferases (e.g., Lum et al. 2003;Baeg et al. 2005; DasGupta et al. 2005; Muller et al.2005; Nybakken et al. 2005), detection of enzymaticactivity (e.g., Boutros et al. 2004; Bard et al. 2006), orfluorescent dyes (Vig et al. 2006; Zhang et al. 2006)whose overall chemiluminescence or fluorescence out-put is rapidly measured using a plate reader. The gen-eration of numerical readouts for each condition ineach well tested makes it possible to employ variousstatistical treatments to rigorously identify dsRNAs thatare the true positives.
Antibody- and microscopy-based screens: A powerfulapplication of antibody-based screens involves the use ofprotein-modification specific antibodies (such as anti-phospho-tyrosine/serine or methyl-lysine antibodies)directed against central components of a signaling
Figure 1.—RNAi HTS platform. The threemain steps in RNAi HTS are depicted: (1) array-ing of the library to 384-well plates; (2) additionof Drosophila cells (10,000–40,000/well) andtreatment with dsRNAs for 30 min in the absenceof serum (bathing method) or with transfectionreagents together with other DNA constructs (ifneeded); and (3) automated detection step (mostcommonly performed with a plate reader or an in-verted epifluorescence microscope) to quantitateor monitor the effects of the dsRNAs in each well.
8 N. Perrimon and B. Mathey-Prevot
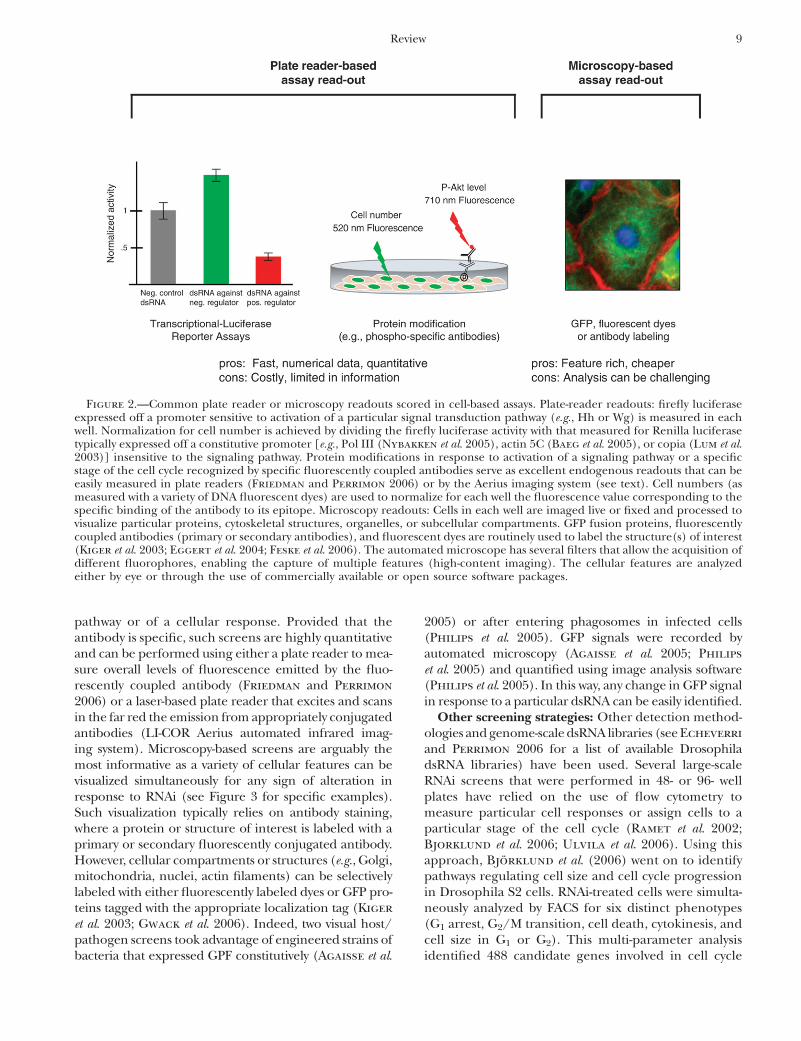
pathway or of a cellular response. Provided that theantibody is specific, such screens are highly quantitativeand can be performed using either a plate reader to mea-sure overall levels of fluorescence emitted by the fluo-rescently coupled antibody (Friedman and Perrimon
2006) or a laser-based plate reader that excites and scansin the far red the emission from appropriately conjugatedantibodies (LI-COR Aerius automated infrared imag-ing system). Microscopy-based screens are arguably themost informative as a variety of cellular features can bevisualized simultaneously for any sign of alteration inresponse to RNAi (see Figure 3 for specific examples).Such visualization typically relies on antibody staining,where a protein or structure of interest is labeled with aprimary or secondary fluorescently conjugated antibody.However, cellular compartments or structures (e.g., Golgi,mitochondria, nuclei, actin filaments) can be selectivelylabeled with either fluorescently labeled dyes or GFP pro-teins tagged with the appropriate localization tag (Kiger
et al. 2003; Gwack et al. 2006). Indeed, two visual host/pathogen screens took advantage of engineered strains ofbacteria that expressed GPF constitutively (Agaisse et al.
2005) or after entering phagosomes in infected cells(Philips et al. 2005). GFP signals were recorded byautomated microscopy (Agaisse et al. 2005; Philips
et al. 2005) and quantified using image analysis software(Philips et al. 2005). In this way, any change in GFP signalin response to a particular dsRNA can be easily identified.
Other screening strategies: Other detection method-ologies and genome-scale dsRNA libraries (see Echeverri
and Perrimon 2006 for a list of available DrosophiladsRNA libraries) have been used. Several large-scaleRNAi screens that were performed in 48- or 96- wellplates have relied on the use of flow cytometry tomeasure particular cell responses or assign cells to aparticular stage of the cell cycle (Ramet et al. 2002;Bjorklund et al. 2006; Ulvila et al. 2006). Using thisapproach, Bjorklund et al. (2006) went on to identifypathways regulating cell size and cell cycle progressionin Drosophila S2 cells. RNAi-treated cells were simulta-neously analyzed by FACS for six distinct phenotypes(G1 arrest, G2/M transition, cell death, cytokinesis, andcell size in G1 or G2). This multi-parameter analysisidentified 488 candidate genes involved in cell cycle
Figure 2.—Common plate reader or microscopy readouts scored in cell-based assays. Plate-reader readouts: firefly luciferaseexpressed off a promoter sensitive to activation of a particular signal transduction pathway (e.g., Hh or Wg) is measured in eachwell. Normalization for cell number is achieved by dividing the firefly luciferase activity with that measured for Renilla luciferasetypically expressed off a constitutive promoter [e.g., Pol III (Nybakken et al. 2005), actin 5C (Baeg et al. 2005), or copia (Lum et al.2003)] insensitive to the signaling pathway. Protein modifications in response to activation of a signaling pathway or a specificstage of the cell cycle recognized by specific fluorescently coupled antibodies serve as excellent endogenous readouts that can beeasily measured in plate readers (Friedman and Perrimon 2006) or by the Aerius imaging system (see text). Cell numbers (asmeasured with a variety of DNA fluorescent dyes) are used to normalize for each well the fluorescence value corresponding to thespecific binding of the antibody to its epitope. Microscopy readouts: Cells in each well are imaged live or fixed and processed tovisualize particular proteins, cytoskeletal structures, organelles, or subcellular compartments. GFP fusion proteins, fluorescentlycoupled antibodies (primary or secondary antibodies), and fluorescent dyes are routinely used to label the structure(s) of interest(Kiger et al. 2003; Eggert et al. 2004; Feske et al. 2006). The automated microscope has several filters that allow the acquisition ofdifferent fluorophores, enabling the capture of multiple features (high-content imaging). The cellular features are analyzedeither by eye or through the use of commercially available or open source software packages.
Review 9

regulation, ubiquitination, vesicular and nuclear trans-port, and regulation of four ligand-induced signalingpathways [Wnt/Wg, MAPK, FKBP12-rapamycin-associatedprotein/target of rapamycin, and just another kinase(JAK)/signal transducers and activators of transcription(STAT)]. Coupled with high-resolution imaging micros-copy, this approach was quite powerful as it allowedmultiple parameters to be analyzed simultaneously(Bjorklund et al. 2006). One limitation of monitoringcell cycle progression through DNA content only is thefact that many Drosophila cell lines are polyploid.Finally, a smaller-scale screen has been performed onRNAi cell microarrays where dsRNAs at first spotted athigh density on a microscope slide to which cells aresubsequently added (Wheeler et al. 2005).
CONSIDERATIONS WITH THE TECHNOLOGY
A major advantage of the RNAi HTS platform is that itis fast, versatile, and it enables screening in an unbiasedmanner. However, there are a number of importantconsiderations to keep in mind regarding this evolvingmethodology. All high-throughput approaches haveexperimental noise from both technical and biologicalartifacts. The vast number of dsRNAs present in ge-nomewide collections means that the primary screen
can be performed at best in duplicate because of highcost, which limits the ability to gauge reproducibility ofthe data with confidence. Thus, in most cases, dataanalysis methods are used to aid in the extraction ofrobust data sets. Further, variations in the level of geneknockdown may affect the ability to assay the function ofsome genes. RNAi is a method of ‘‘gene knockdown’’and not ‘‘gene knockout.’’ Thus, while a hypomorphicstate could be advantageous in cases where genes areassociated with cell lethality, RNAi may fail to identifygenes whose activities need to be completely eliminatedto show a phenotype. In addition, the time required toachieve optimal knockdown may not be the same forall genes analyzed. In general, a 3-day RNAi treatmentis chosen for most screens. However, if the proteinencoded by the targeted gene has a long half-life, a3-day treatment may be too short to cause a measurablephenotype. Conversely, when RNAi is rapid, secondaryeffects through feedback regulation may be scored.Finally, the large number of candidate genes (usually inthe hundreds) typically implicated in these screens makesit virtually impossible to confirm every single hit throughcareful biochemical or genetic validation experiments.
A critical aspect of an RNAi HTS platform is thequality and specificity of the RNAi library in use. It isimportant to keep in mind that each library is based on
Figure 3.—Example of visual cell-based assays.(A) Translocation of a nuclear factor of activatedT cells (NFAT)–GFP fusion transcription factor(in green) to the nucleus in response to theopening of Ca21 channels evoked by thapsigargin(TG). (A1) In the absence of TG, NFAT–GFP iscytoplasmic. (A2) Addition of TG to the cellsleads to the rapid translocation of NFAT–GFPto the nucleus. (A3) Addition of dsRNA targetingolf186-F abrogates the translocation of NFAT–GFP in response to TG (Feske et al. 2006) (photo-graphs are courtesy of Y. Gwack). (B) Cell-basedassays that monitor the intracellular proliferationof a pathogen. Mycobacterium fortuitum engi-neered to express the GFP protein from thepmap24 promoter (only active when the bacteriahas been internalized in the phagosome com-partment) was added to S2 cells. (B1) LittleGFP signal is detected 6 hr after infection as mostof the bacteria have not entered the phagosomecompartment. (B2) Successful infection by thebacteria and robust growth are observed 24 hr af-ter addition of the pathogen to the cells. The GFPsignal is quantitated using automated image anal-ysis software to monitor the effect of the variousdsRNAs on infection and growth of the pathogen(Philips et al. 2005). S2 nuclei are shown in redand bacteria in green (photographs are courtesyof J. Philips). (C) Round Kc cells (C1) and spread-out S2R1 cells (C2), shown in low and higher
magnifications, can be used to study various aspects of cell morphology and adhesion (Kiger et al. 2003). Tubulin (green)was visualized with FITC-conjugated antitubulin antibody, actin (red) with tetramethyl rhodamine isothiocyanate (TRITC)–phalloidin, and DNA (blue) with DAPI (photographs are courtesy of A. Kiger). (D) A simple DNA stain can be used to screenfor genes involved in cytokinesis. (D1) Untreated Drosophila Kc167 cells. (D2) Cells exposed to dsRNA targeting Aurora Bkinase for 4 days. The cells are shown at low and higher magnifications. Cytoplasm (red) is stained with N-hydroxysuccinimide–tetramethylrhodamine and DNA (green) with Hoechst dye (photographs are courtesy of U. Eggert).
10 N. Perrimon and B. Mathey-Prevot

the best annotation at the time that it was assembled. Asgenome annotations are still evolving, any change needsto be incorporated in a library to keep it up to date. Forinstance, the first library used at the DRSC (Boutros
et al. 2004) was designed to target all Drosophila genespredicted from two annotations: an early BDGP/Celeraannotation (13,672 genes) (Adams et al. 2000) and onefrom the Sanger Center (20,622 genes) (Hild et al.2003). Combining the two annotations resulted in atotal of 21,306 nonredundant possible transcripts in theDrosophila genome, with 14,556 dsRNAs targetingannotations present in both the BDGP and SangerCenter sets and 6750 dsRNAs targeting Sanger annota-tions not found in the BDGP set (Sanger-only dsRNAs).However, many of the dsRNAs targeting the Sanger-onlypredictions were eventually removed from the DRSCRNAi library as Stolc et al. (2004) could confirm ex-pression of only 291 of the Sanger-only predictionsusing a DNA oligonucleotide microarray approach,while Yandel et al. (2005) found that only 10% of theSanger-only predictions were likely to correspond togenes containing introns.
In addition to requiring periodic updates to reflectchanges introduced by more accurate and comprehen-sive annotations of the Drosophila genome, librariesneed to be controlled for off-target effects (OTEs)potentially associated with the use of dsRNAs. Althoughthis issue is familiar to investigators using siRNAs inmammalian systems (where it had been recognizedearly on) ( Jackson et al. 2003), its extent had beenunderestimated in the Drosophila and C. elegans commu-nities. However, two recent studies reported strong evi-dence for OTEs in Drosophila RNAi screens (Kulkarni
et al. 2006; Ma et al. 2006). Ma et al. (2006) focusedprimarily on OTEs caused by the presence of trinucle-otide CA[AGCT] or CAN repeats in some dsRNAs.Results from this study indicate that as few as five con-tiguous CAN repeats in dsRNAs can lead to toxicity ornonspecific effects in treated cells, resulting in likelyfalse positives in certain types of screens (Ma et al. 2006).In the second study, the prevalence of OTEs in Dro-sophila RNAi screens was examined through a review of30 genomewide screens (Kulkarni et al. 2006). Theauthors found that the presence of OTEs correlates withperfect homologies to genes (including but not re-stricted to CAN repeats) other than the intended targetsthat are present in some dsRNAs. A trend could alreadybe observed with 17-nt perfect homology, but homolo-gies of 19 nt or beyond were found to be highlysignificant. This analysis led to a complete overhaul ofthe library, where all dsRNAs predicted to have OTEswere replaced with new ones devoid of any homology toother genes (on the basis of a threshold of 19 nt).Regardless, one should keep in mind that we still donot fully understand all the rules governing OTEs inDrosophila cells. In fact, it has been shown in mamma-lian cells that short sequences in siRNAs identical to the
seed region of microRNAs represent the major source ofOTEs in mammalian cell-based screens (Birmingham
et al. 2006). It is not known yet to what extent thismechanism is a concern in either C. elegans or Drosoph-ila where long dsRNAs are used as RNAi reagents. Thus,because OTEs are particularly difficult to predict and toensure data reproducibility, the recommended practicein the field is now to simply use multiple independentRNAi reagents against the same gene (Echeverri et al.2006). In this regard, the practice of generating validationsets consisting of new dsRNAs that are distinct from anydsRNA tested in a primary screen should be encouraged.These dsRNAs should be devoid of predicted perfecthomologies to nontarget genes and should correspond toevery hit identified in a completed screen.
APPLICATIONS OF RNAi HTS TO GENE DISCOVERY
RNAi HTS in cell-based assays has proven to be arobust method for identifying gene function. Screenscarried out in cell lines have covered a wide range ofbiological questions, including cell viability (Boutros
et al. 2004), cell morphology (Kiger et al. 2003), cellcycle (Bjorklund et al. 2006), cytokinesis (Eggert et al.2004), susceptibility to DNA-damaging agents, RNAprocessing, general (Bard et al. 2006) and specializedsecretion, calcium stores (Feske et al. 2006; Vig et al.2006; Zhang et al. 2006), factors influencing polyQaggregation and toxicity, mitochondrial dynamics, circa-dian clock, cellular response to metals, hypoxia, phago-cytosis (Ramet et al. 2002; Kocks et al. 2005), innateimmunity (Foley and O’Farrell 2004; Gesellchen
et al. 2005; Kleino et al. 2005), cell susceptibility toinfection by viruses or other intracellular pathogens(Agaisse et al. 2005; Cherry et al. 2005, 2006; Philips
et al. 2005) as well as most of the major signalingpathways (Lum et al. 2003; Baeg et al. 2005; DasGupta
et al. 2005; Muller et al. 2005; Nybakken et al. 2005;Bartscherer et al. 2006; Friedman and Perrimon
2006; Gwack et al. 2006; Ma et al. 2006).Importantly, RNAi screens have identified important
components in a biological process that had eludedmore conventional approaches. A perfect example isillustrated by three independent screens, which werelargely designed to identify a long-sought transmem-brane channel regulating calcium stores in eukaryoticcells. All three screens identified olf186-F as the geneencoding either the elusive CRAC channel or a compo-nent of it (Feske et al. 2006; Vig et al. 2006; Zhang et al.2006). That all three screens, based on slightly differentcell-based assays, identified the same component vali-dates the notion that genomewide RNAi screens can bea powerful and specific approach for gene discovery. Ina different line of investigation, Philips et al. (2005)sought to identify in a visual screen host factors thatmight interfere with Mycobacterium infection andintracellular growth in S2 cells. They identified a new
Review 11

factor, Peste (Pes), a CD36 family member, which isrequired for uptake of mycobacteria (Philips et al.2005). To address a related question, Kocks et al. (2005)used a clever combination of specific RNAi and RNAprofiling, which led to the identification of eater, a geneencoding a scavenger receptor-like type I transmem-brane protein mediating phagocytosis of bacteria. RNAiscreens focusing on signaling pathways have also yieldednew components that are important effectors in thesepathways, such as Ihog (Yao et al. 2006) and PP2A (Hhpathway) (Nybakken et al. 2005), Evi (Wg pathway)(Bartscherer et al. 2006), PTP61F (JAK/STAT path-way) (Baeg et al. 2005; Muller et al. 2005), and dGCKIII(MAPK pathway). Kiger et al. (2003) performed a large-scale screen (targeting �1000 genes) to identify genesthat affected cell morphology when knocked down bytheir cognate dsRNAs. Using automated fluorescencemicroscopy to visualize actin filaments, microtubules,and DNA (see C1 and C2 in Figure 3), they classifiedphenotypes according to well-defined parameters thatwere applied to score each image by eye. Importantly,knockdown of genes known to be arranged within thesame pathway resulted in similar phenotypic profiles orphenoprints. The authors took advantage of this prop-erty to predict where previously uncharacterized geneswithin a phenotypic cluster might be acting, leadingthem, for example, to assign the role for the citronkinase as a Rho 1 effector required for cytokinesis(Kiger et al. 2003).
APPLICATIONS OF RNAi HTS TO SYSTEM BIOLOGY
As exemplified by the various screens already per-formed, RNAi HTS in cell-based assays is an excellentmethod for identifying gene functions involved in cellsignaling, cell biology, physiology, differentiation, andhost/pathogen interactions. However, rather than fo-cusing primarily on a few interesting new candidates, anexciting potential of RNAi HTS is to provide a globalsystems-wide understanding of the subcellular networksthat carry out and modulate a specific program orresponse, such as the propagation and integration ofinput signals through a signaling network. The largenumber of genes, usually in the hundreds, identified ingenomewide screens creates a challenge: it significantlyincreases the number of candidate genes that could beclassified as bona fide effectors in a signaling pathway orbiological process, but it could also lead to the inclusionof false positives as it is virtually impossible to confirmevery single hit through biochemical or genetic valida-tion experiments. Thus, the success of the approachrelies on low experimental rates of false positives andfalse negatives. Providing that RNAi data sets have beencurated to contain only high confidence hits, the infor-mation can be integrated with transcriptional profil-ing (RNA profiling), interactome data sets (proteomeand genetic interactomes), and published literature
(literature-mining tools) to support or confirm theconnections made between components of a network(Gunsalus et al. 2005; Mukherji et al. 2006).
One of the especially promising applications of RNAiHTS is to expand our knowledge of the structure ofsignaling networks. Indeed, analysis of the proteomefrom either two-hybrid or mass spectrometry ap-proaches has revealed that hundreds rather than tensof proteins are associated with a signaling network. Thiscomplexity in scale has been validated by recent studiesof synthetic lethality screens in yeast (Tong et al. 2004),and most recently in C. elegans (Lehner et al. 2006), andthus illustrates our partial knowledge of the complexityof signaling networks. A recent screen, which took intoaccount OTEs and controlled for them, deliveredappropriate data sets that can be used for a systemsbiology approach. The screen for modulators of MAPKactivity (Friedman and Perrimon 2006) monitored thestate of phosphorylated Drosophila ERK (dpERK) as amarker of MAPK activation upon insulin stimulation. Inthis way, small but significant changes in MAPK signal-ing could be measured by following dpERK levels,providing the ability for the first time to reveal quanti-tative changes, which by themselves might not beenough to lead to a measurable phenotype in vivo.Similar to the findings of other signal transductionscreens, the major known components were readilyidentified in this screen and .300 additional compo-nents were shown to affect the steady state of MAPKactivity in RNAi-treated cells. Importantly, the list ofgenes showed a highly significant enrichment in genesencoding pathway components, in genes conservedacross organisms utilizing this pathway, and finallyin genes encoding orthologous proteins dynamicallyphosphorylated in HeLa cells following EGF stimulus(Olsen et al. 2006), suggesting the existence of exten-sive crosstalks among different signaling pathways.Ultimately, it will be important to project the informa-tion derived from such screens on data sets obtainedfrom other ‘‘omics’’ approaches such as protein–proteininteraction maps, genetic interaction networks, andRNA-profiling experiments, to strengthen the connec-tivity links among all these components, and to derive acomprehensive depiction of signaling networks, wherethe flow of information is highly dynamic and dependson extensive feedback loops and crosstalks amongcoexisting signaling pathways.
FUTURE DEVELOPMENTS OF THE TECHNOLOGY
Since the most important requirement for a success-ful RNAi HTS is a robust cell-based assay that can be pro-cessed in a high-throughput manner, it is an excitingtime to think about where the major technological ad-vances will come from in the next few years in this area.
Design of more sophisticated cell-based assays: Inthe context of signaling, RNAi HTS designed to capture
12 N. Perrimon and B. Mathey-Prevot

the activity of multiple readouts rather than to measurethe activity of a single signaling pathway, as has beendone so far, will be extremely informative with regards tothe characterization of crosstalk and flow of informationthrough signaling networks. For example, RNAi HTSthat simultaneously monitors the state of two or morepathway activities would allow one to identify commonregulators and crosstalk regulation. Emerging technol-ogies such as multiplex FISH (Capodieci et al. 2005)and microsphere-based high-throughput gene expres-sion profiling (based on the Luminex xMAP system)(Naciff et al. 2005) provide cost-effective methods todetect gene expression signatures composed of a subsetof reporter genes of interest in a large number of sam-ples. While the multiplex FISH approach can measuretranscript levels of ,15 reporter genes on a single-cellbasis, the Luminex system can be employed to followthe levels of at least 100 genes simultaneously in cellextracts (Peck et al. 2006).
Similarly, multiplexing assays at the level of proteinmodification have great potential (Irish et al. 2004). Forexample, monitoring the phosphorylation state of bothMAPK and another key kinase regulated in response toreceptor tyrosine kinase activation (i.e., Raf or MEK)would provide insights into the structure of the MAPKnetwork, in particular of the feedback loops. The samekind of multi-sensors strategy can be applied directly tocell biological studies whereby the state of differentcellular structures or cellular processes is monitored(i.e., markers for different endosomal compartments).Similarly, in the context of the host/pathogen screens,screens can be designed whereby a single cell is infectedby two different pathogens (i.e., Mycobacterium andListeria). Providing that each pathogen can be visualizedindependently, RNAi perturbations that affect selectivelythe uptake or growth of Mycobacteria but not of Listeria(or vice versa) would be readily identified.
Perhaps the most significant advances in RNAi HTSwill come from high content screening (HCS). Cell-based HCS that rely on cellular phenotypes are becom-ing one of the preferred methods in RNAi HTS becausethey generate data sets that are rich in information. Forexample, the intensity and localization of fluorescentmarkers reflecting the number, size, and shape ofcellular compartments can be analyzed quantitatively.Further, as described below, the use of primary cellsoffers ample opportunities to carry out cell morphologyscreens in a biologically relevant context, as diversestructures such as muscles, neuromuscular junctions,tracheal tubes, etc., can form in these cultures. Thus,many of the most interesting screens that will beperformed in the future are likely to rely on microscopy.In the past few years, major advances have been maderegarding the development of both conventionaland confocal HTS microscopes, such that they nowoffer excellent image resolution and unparalleledimage acquisition speed (see review by Carpenter
and Sabatini 2004). One current limitation, however,is that the vast majority of commercial software packagesuse proprietary codes. As such, they perform quite wellin focused applications that require an accurate cellor nuclei count, a measure of pixel intensities corre-sponding to well-defined objects, or an accurate numer-ical representation of how much of a protein localizes tospecific subcellular compartments. However, they oftenfall short in the kind of flexibility and sophistication thatmany academic projects require. In this regard, majoradvancements in this field will come from the develop-ment of open source and sophisticated image analysismethods that are able to process quickly the largeamount of images generated, e.g., Cellprofiler (http://www.cellprofiler.org) (Carpenter et al. 2006).
Development of new cell lines and primary cells:Although advances in assay designs are poised to makemajor contributions to RNAi HTS, the success of thescreens ultimately relies on the relevance of the celltypes in which the questions are being studied. Asignificant caveat of using the established cell linesdescribed earlier is that they have adapted physiologi-cally to the culture medium, and most of the cell lineshave departed from diploidy or may harbor activatedsignaling pathways. To resolve these issues and expandthe repertoire of assays in more biologically relevantcells, RNAi HTS for axonal outgrowth and muscleintegrity has been conducted by simply deriving cellsfrom embryos that express a GFP marker in the cells ofinterest. The first screen focused on primary musclecells ( J. Bai and N. Perrimon, unpublished results).Muscle phenotypes were scored by visual inspection ofmicroscopic images acquired using an automated in-verted fluorescence microscope. A number of pheno-typic classes (collapsed muscle, small or large muscles,disrupted sarcomere structures with no striation, actinfilaments retracted or relaxed from the cell periphery)were identified. The second screen dealt with axonaloutgrowth of neuronal cells, whereby the morphologyof GFP-labeled neuronal cells in response to RNAi wasrecorded by automated microscopy and visually scoredaccording to various criteria that included the extentof excessive branching, defasciculation, axon blebbing,cell loss, and reduced outgrowth (K. J. Sepp, P. Hong,S. B. Lizarraga, J. S. Liu, L. A. Mejia, C. A. Walsh andN. Perrimon, unpublished results).
Altogether, screens of muscle and neuronal cells arereadily feasible and bode well for follow-up screens orother screen applications that require the use of pri-mary cell cultures. For instance, assays can be devisedwhereby a specific gene (e.g., a gene coding for a mu-tated form of a protein linked to a human disease)can be ectopically expressed in the cells of interest(Figure 4). If misexpression of that gene in the primarycells results in a phenotype, then an RNAi suppressor/enhancer screen can be conducted. Other promisingHTS applications in primary cells include assays
Review 13

designed to determine the mode of action of a drug. If,for example, a small molecule induces neurite out-growth, a suppressor/enhancer RNAi screen may iden-tify components of the pathway that are modulated bythe compound. Finally, culture of additional cell typesalso may be feasible. Indeed, there are reports in theliterature that fat body and tracheal cells as well asvarious epithelial cells types can be cultured (Shields
et al. 1975). In addition, Niki et al. (2006) recentlyreported their success at culturing germline stem cellsfrom adult Drosophila ovaries, thus opening up thepossibility of conducting RNAi screens in totipotentcells.
Novel RNAi screening platforms: Although RNAiHTS in multi-well plates have great applications, twoemerging technologies offer new opportunities in Dro-sophila functional genomics: cell arrays and transgenicRNAi.
Cell arrays and their applications to synthetic phenotypicscreens: As we have learned from yeast studies, manygenetic interactions can be revealed by unbiased anal-ysis of synthetic genetic interactions (see, for example,Tong et al. 2004). Although one can envision testing byRNAi HTS in 384-well plates all genomewide interac-
tions between a specific gene and others (�15,000pairwise combination), or alternatively, testing for allpairwise combinations in a set of 100 genes (4900combinations), it is clearly not realistic on a larger scale.Miniaturization of the methodology offered by theRNAi-cell microarray method, whereby dsRNAs can bespotted at high density on glass slides (RNAi micro-arrays) and assayed in visual screens (Wheeler et al.2004; Guertin et al. 2006), provides a solution as itreduces significantly the cost associated with the tech-nology. In this system, cells are cultured on a glass slideprinted at known locations with an array of 2–3 nl of saltsolution droplets containing the RNAi reagent. Becauseeach spot is only 200 mm in diameter, 6000 such spotscan easily be printed on one standard microscope slide.Cells (80–200) that settle on the printed spots take upthe dsRNA and induce over time, through RNAi, thedegradation of the targeted mRNAs, resulting in aconcomitant decrease in the encoded proteins. Thistechnique requires very little reagents, lends itself wellfor streamlining assay conditions, and, since it is basedon microscopy, allows for a variety of cellular pheno-types to be assayed (Wheeler et al. 2005).
In vivo validation using transgenic hairpin flies: In vivovalidation of the results obtained from RNAi tissueculture screens is an important and necessary step tofully establish the relevance of the observation madefrom RNAi HTS. In addition to collections of existingDrosophila mutations and the arsenal of existing toolsavailable to generate mutations (local excision of a Pelement inserted near the gene under investigation,homologous recombination, etc.) (Nagy et al. 2003;Venken and Bellen 2005; Beller and Oliver 2006),transgenic RNAi—whereby a hairpin construct againsteach gene of interest is expressed under upstreamactivating sequence (UAS) control (see, for example,Lee and Carthew 2003)—is emerging as the method ofchoice for in vivo validation. There are currently twomajor efforts to assemble large collections of transgenicflies for RNAi (http://www.imba.oeaw.ac.at/index.php?id¼252 and http://www.shigen.nig.ac.jp/fly/nigfly/rnaiListAction.do?browseOrSearch¼browse). One ad-vantage of using the UAS/Gal4 for expression of hairpinsused for knockdowns is the rich repertoire of cell-type-specific and heat-inducible Gal4 drivers available, whichcan be exploited to achieve spatially and temporally con-trolled RNAi knockdown of genes that may be essentialand/or pleiotropic. Importantly, the generation of suchlines would benefit from using the site-specific integra-tion method (Groth et al. 2004) to target transgenes toloci that are highly inducible.
Concluding remarks: The power of model systemsover the years has relied on the design of sophisticatedmethods to conduct genetic screens. The amazing ad-vances in RNAi in the past few years have added an evenmore comprehensive set of tools to the already powerfularmamentarium available to a geneticist. Although we
Figure 4.—Applications of primary cell cultures to RNAiHTS. Embryos (4–6 hr after egg laying) are mechanicallydissociated by douncing and the cell suspension cultured inthe presence of dsRNA (bathing method) ( J. Bai and N.Perrimon, unpublished results; K. J. Sepp, P. Hong, S. B.Lizarraga, J. S. Liu, L. A. Mejia, C. A. Walsh and N.Perrimon, unpublished results). Phenotypes in these culturesare typically read after 7–10 days. Cell-based assays in primarymuscle and neuronal cells can be easily engineered by express-ing a gene of interest using an appropriate Gal4 driver, e.g.,dmef2-Gal4 for muscle or elav-Gal4 for neuronal cells. Primarycells can be visualized using specific antibody labeling orTRITC–phalloidin staining for muscle cells that reveals thesarcomeric structure of differentiated muscles or, as shownhere for neurons, following the expression of GFP (elav-Gal4; UAS-GFP).
14 N. Perrimon and B. Mathey-Prevot

are already witnessing the impressive impact that RNAihas had on the field of developmental and cell biology,emerging improvements and technologies in RNAi-based screening methods will undoubtedly continueto extend our ability to access knowledge in unprece-dented ways.
We thank both past and current members of the lab for efforts indeveloping the various RNAi methods and screens discussed in thisreview, as well as for stimulating discussions. Our work is supportedby the National Institutes of Health (National Institute of GeneralMedical Sciences RO1 GM067761) and the Howard Hughes MedicalInstitute.
LITERATURE CITED
Adams, M. D., S. E. Celniker, R. A. Holt, C. A. Evans, J. D. Gocayne
et al., 2000 The genome sequence of Drosophila melanogaster.Science 287: 2185–2195.
Agaisse, H., L. S. Burrack, J. A. Philips, E. J. Rubin, N. Perrimon
et al., 2005 Genome-wide RNAi screen for host factors requiredfor intracellular bacterial infection. Science 309: 1248–1251.
Armknecht, S., M. Boutros, A. Kiger, K. Nybakken, B. Mathey-Prevot et al., 2005 High-throughput RNA interference screensin Drosophila tissue culture cells. Methods Enzymol. 392: 55–73.
Baeg, G. H., R. Zhou and N. Perrimon, 2005 Genome-wide RNAianalysis of JAK/STAT signaling components in Drosophila.Genes Dev. 19: 1861–1870.
Bard, F., L. Casano, A. Mallabiabarrena, E. Wallace, K. Saito
et al., 2006 Functional genomics reveals genes involved in pro-tein secretion and Golgi organization. Nature 439: 604–607.
Bartscherer, K., N. Pelte, D. Ingelfinger and M. Boutros,2006 Secretion of Wnt ligands requires Evi, a conserved trans-membrane protein. Cell 125: 523–533.
Beller, M., and B. Oliver, 2006 One hundred years of high-throughput Drosophila research. Chromosome Res. 14: 349–362.
Birmingham, A., E. M. Anderson, A. Reynolds, D. Ilsley-Tyree, D.Leake et al., 2006 39 UTR seed matches, but not overall identity,are associated with RNAi off-targets. Nat. Methods 3: 199–204.
Bjorklund,M.,M.Taipale,M.Varjosalo, J.Saharinen, J.Lahdenpera
et al., 2006 Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 439: 1009–1013.
Boutros, M., A. A. Kiger, S. Armknecht, K. Kerr, M. Hild et al.,2004 Genome-wide RNAi analysis of growth and viability inDrosophila cells. Science 303: 832–835.
Capodieci, P., M. Donovan, H. Buchinsky, Y. Jeffers, C. Cordon-Cardo et al., 2005 Gene expression profiling in single cellswithin tissue. Nat. Methods 2: 663–665.
Carpenter, A. E., and D. M. Sabatini, 2004 Systematic genome-wide screens of gene function. Nat. Rev. Genet. 5: 11–22.
Carpenter, A. E., T. R. Jones, M. R. Lamprecht, C. Clarke, I. H.Kang et al., 2006 CellProfiler: image analysis software for iden-tifying and quantifying cell phenotypes. Genome Biol. 7: R100.
Cherry, S., T. Doukas, S. Armknecht, S. Whelan, H. Wang et al.,2005 Genome-wide RNAi screen reveals a specific sensitivityof IRES-containing RNA viruses to host translation inhibition.Genes Dev. 19: 445–452.
Cherry, S., A. Kunte, H. Wang, C. Coyne, R. B. Rawson et al.,2006 COPI activity coupled with fatty acid biosynthesis is re-quired for viral replication. PLoS Pathog. 2: 10.
Clemens, J. C., C. A. Worby, N. Simonson-Leff, M. Muda, T.Maehama et al., 2000 Use of double-stranded RNA interferencein Drosophila cell lines to dissect signal transduction pathways.Proc. Natl. Acad. Sci. USA 97: 6499–6503.
DasGupta, R., A. Kaykas, R. T. Moon and N. Perrimon, 2005 Func-tional genomic analysis of the Wnt-Wingless signaling pathway.Science 308: 826–833.
Echeverri, C. J., and N. Perrimon, 2006 High-throughput RNAiscreening in cultured cells: a user’s guide. Nat. Rev. Genet. 7:373–384.
Echeverri, C. J., P. A. Beachy, B. Baum, M. Boutros, F. Buchholz
et al., 2006 Minimizing the risk of reporting false positives inlarge-scale RNAi screens. Nat. Methods 3: 777–779.
Eggert, U. S., A. A. Kiger, C. Richter, Z. E. Perlman, N. Perrimon
et al., 2004 Parallel chemical genetic and genome-wide RNAiscreens identify cytokinesis inhibitors and targets. PLoS Biol. 2:e379.
Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber
et al., 2001 Duplexes of 21-nucleotide RNAs mediate RNA inter-ference in cultured mammalian cells. Nature 411: 494–498.
Feske, S., Y. Gwack, M. Prakriya, S. Srikanth, S. H. Puppel et al.,2006 A mutation in Orai1 causes immune deficiency by abro-gating CRAC channel function. Nature 441: 179–185.
Fire, A., S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver et al.,1998 Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811.
Flockhart, I., M. Booker, A. Kiger, M. Boutros, S. Armknecht
et al., 2006 FlyRNAi: the Drosophila RNAi screening centerdatabase. Nucleic Acids Res. 34: D489–D494.
Foley, E., and P. H. O’Farrell, 2004 Functional dissection of aninnate immune response by a genome-wide RNAi screen. PLoSBiol. 2: E203.
Fraser, A. G., R. S. Kamath, P. Zipperlen, M. Martinez-Campos,M. Sohrmann et al., 2000 Functional genomic analysis of C.elegans chromosome I by systematic RNA interference. Nature408: 325–330.
Friedman, A., and N. Perrimon, 2006 A functional RNAi screen forregulators of receptor tyrosine kinase and ERK signalling. Nature444: 230–234.
Gesellchen, V., D. Kuttenkeuler, M. Steckel, N. Pelte and M.Boutros, 2005 An RNA interference screen identifies inhibi-tor of apoptosis protein 2 as a regulator of innate immune signal-ling in Drosophila. EMBO Rep. 6: 979–984.
Gonczy, P., C. Echeverri, K. Oegema, A. Coulson, S. J. Jones et al.,2000 Functional genomic analysis of cell division in C. ele-gans using RNAi of genes on chromosome III. Nature 408:331–336.
Groth, A. C., M. Fish, R. Nusse and M. P. Calos, 2004 Con-struction of transgenic Drosophila by using the site-specific inte-grase from phage phiC31. Genetics 166: 1775–1782.
Guertin, D. A., K. V. Guntur, G. W. Bell, C. C. Thoreen and D. M.Sabatini, 2006 Functional genomics identifies TOR-regulatedgenes that control growth and division. Curr. Biol. 16: 958–970.
Gunsalus, K. C., H. Ge, A. J. Schetter, D. S. Goldberg, J. D. Han
et al., 2005 Predictive models of molecular machines involvedin Caenorhabditis elegans early embryogenesis. Nature 436:861–865.
Gwack, Y., S. Sharma, J. Nardone, B. Tanasa, A. Iuga et al., 2006 Agenome-wide Drosophila RNAi screen identifies DYRK-familykinases as regulators of NFAT. Nature 441: 646–650.
Hammond, S. M., E. Bernstein, D. Beach and G. J. Hannon,2000 An RNA-directed nuclease mediates post-transcriptionalgene silencing in Drosophila cells. Nature 404: 293–296.
Hild, M., B. Beckmann, S. A. Haas, B. Koch, V. Solovyev et al.,2003 An integrated gene annotation and transcriptional profil-ing approach towards the full gene content of the Drosophila ge-nome. Genome Biol. 5: R3.
Irish, J. M., R. Hovland, P. O. Krutzik, O. D. Perez, O. Bruserud
et al., 2004 Single cell profiling of potentiated phospho-proteinnetworks in cancer cells. Cell 118: 217–228.
Jackson, A. L., S. R. Bartz, J. Schelter, S. V. Kobayashi, J. Burchard
et al., 2003 Expressionprofilingrevealsoff-targetgeneregulationby RNAi. Nat. Biotechnol. 21: 635–637.
Kiger, A., B. Baum, S. Jones, M. Jones, A. Coulson et al., 2003 Afunctional genomic analysis of cell morphology using RNA inter-ference. J. Biol. 2: 27.
Kleino, A., S. Valanne, J. Ulvila, J. Kallio, H. Myllymaki et al.,2005 Inhibitor of apoptosis 2 and TAK1-binding protein arecomponents of the Drosophila Imd pathway. EMBO J. 24:3423–3434.
Kocks, C., J. H. Cho, N. Nehme, J. Ulvila, A. M. Pearson et al.,2005 Eater, a transmembrane protein mediating phagocytosisof bacterial pathogens in Drosophila. Cell 123: 335–346.
Review 15

Kulkarni, M. M., M. Booker, S. J. Silver, A. Friedman, P. Hong
et al., 2006 Evidence of off-target effects associated with longdsRNAs in Drosophila melanogaster cell-based assays. Nat. Meth-ods 3: 833–838.
Lee, Y. S., and R. W. Carthew, 2003 Making a better RNAi vector forDrosophila: use of intron spacers. Methods 30: 322–329.
Lehner, B., C. Crombie, J. Tischler, A. Fortunato and A. G. Fraser,2006 Systematic mapping of genetic interactions in Caeno-rhabditis elegans identifies common modifiers of diverse signal-ing pathways. Nat. Genet. 38: 896–903.
Lum, L., S. Yao, B. Mozer, A. Rovescalli, D. Von Kessler et al.,2003 Identification of Hedgehog pathway components by RNAiin Drosophila cultured cells. Science 299: 2039–2045.
Ma, Y., A. Creanga, L. Lum and P. A. Beachy, 2006 Prevalence ofoff-target effects in Drosophila RNA interference screens. Nature443: 359–363.
Moffat, J., and D. M. Sabatini, 2006 Building mammalian signal-ling pathways with RNAi screens. Nat. Rev. Mol. Cell Biol. 7: 177–187.
Mukherji, M., R. Bell, L. Supekova, Y. Wang, A. P. Orth et al.,2006 Genome-wide functional analysis of human cell-cycle reg-ulators. Proc. Natl. Acad. Sci. USA 103: 14819–14824.
Muller, P., D. Kuttenkeuler, V. Gesellchen, M. P. Zeidler and M.Boutros, 2005 Identification of JAK/STAT signalling compo-nents by genome-wide RNA interference. Nature 436: 871–875.
Naciff, J. M., B. D. Richardson, K. G. Oliver, M. L. Jump, S. M.Torontali et al., 2005 Design of a microsphere-based high-throughput gene expression assay to determine estrogenic po-tential. Environ. Health Perspect. 113: 1164–1171.
Nagy, A., N. Perrimon, S. Sandmeyer and R. Plasterk, 2003 Tailor-ing the genome: the power of genetic approaches. Nat. Genet.33 (Suppl): 276–284.
Niki, Y., T. Yamaguchi and A. P. Mahowald, 2006 Establishment ofstable cell lines of Drosophila germ-line stem cells. Proc. Natl.Acad. Sci. USA 103: 16325–16330.
Nybakken, K., S. A. Vokes, T. Y. Lin, A. P. McMahon and N. Perrimon,2005 A genome-wide RNA interference screen in Drosophilamelanogaster cells for new components of the Hh signaling path-way. Nat. Genet. 37: 1323–1332.
Olsen, J. V., B. Blagoev, F. Gnad, B. Macek, C. Kumar et al.,2006 Global, in vivo, and site-specific phosphorylation dynam-ics in signaling networks. Cell 127: 635–648.
Peck, D., E. D. Crawford, K. N. Ross, K. Stegmaier, T. R. Golub
et al., 2006 A method for high-throughput gene expression sig-nature analysis. Genome Biol. 7: R61.
Pelkmans, L., E. Fava, H. Grabner, M. Hannus, B. Habermann et al.,2005 Genome-wide analysis of human kinases in clathrin- andcaveolae/raft-mediated endocytosis. Nature 436: 78–86.
Philips, J. A., E. J. Rubin and N. Perrimon, 2005 Drosophila RNAiscreen reveals CD36 family member required for mycobacterialinfection. Science 309: 1251–1253.
Ramet, M., P. Manfruelli, A. Pearson, B. Mathey-Prevot and R. A.Ezekowitz, 2002 Functional genomic analysis of phagocytosis
and identification of a Drosophila receptor for E. coli. Nature416: 644–648.
Shields, G., A. Dubendorfer and J. H. Sang, 1975 Differentiationin vitro of larval cell types from early embryonic cells of Drosoph-ila melanogaster. J. Embryol. Exp. Morphol. 33: 159–175.
Sonnichsen, B., L. B. Koski, A. Walsh, P. Marschall, B. Neumann
et al., 2005 Full-genome RNAi profiling of early embryogenesisin Caenorhabditis elegans. Nature 434: 462–469.
Stolc, V., Z. Gauhar, C. Mason, G. Halasz, M. F. van Batenburg
et al., 2004 A gene expression map for the euchromatic genomeof Drosophila melanogaster. Science 306: 655–660.
Tong, A. H., G. Lesage, G. D. Bader, H. Ding, H. Xu et al.,2004 Global mapping of the yeast genetic interaction network.Science 303: 808–813.
Ulvila, J., M. Parikka, A. Kleino, R. Sormunen, R. A. Ezekowitz
et al., 2006 Double-stranded RNA is internalized by scavengerreceptor-mediated endocytosis in Drosophila S2 cells. J. Biol.Chem. 281: 14370–14375.
Venken, K. J., and H. J. Bellen, 2005 Emerging technologies forgene manipulation in Drosophila melanogaster. Nat. Rev. Genet.6: 167–178.
Vig, M., C. Peinelt, A. Beck, D. L. Koomoa, D. Rabah et al.,2006 CRACM1 is a plasma membrane protein essential forstore-operated Ca21 entry. Science 312: 1220–1223.
Wheeler, D. B., S. N. Bailey, D. A. Guertin, A. E. Carpenter, C. O.Higgins et al., 2004 RNAi living-cell microarrays for loss-of-function screens in Drosophila melanogaster cells. Nat. Methods1: 127–132.
Wheeler, D. B., A. E. Carpenter and D. M. Sabatini, 2005 Cellmicroarrays and RNA interference chip away at gene function.Nat. Genet. 37: S25–S30.
Yanagawa, S., J. S. Lee, T. Haruna, H. Oda, T. Uemura et al.,1997 Accumulation of Armadillo induced by Wingless, Dishev-elled, and dominant-negative Zeste-White 3 leads to elevatedDE-cadherin in Drosophila clone 8 wing disc cells. J. Biol. Chem.272: 25243–25251.
Yanagawa, S., J. S. Lee and A. Ishimoto, 1998 Identification andcharacterization of a novel line of Drosophila Schneider S2 cellsthat respond to wingless signaling. J. Biol. Chem. 273: 32353–32359.
Yandell, M., A. M. Bailey, S. Misra, S. Shu, C. Wiel et al., 2005 Acomputational and experimental approach to validating annota-tions and gene predictions in the Drosophila melanogaster ge-nome. Proc. Natl. Acad. Sci. USA 102: 1566–1571.
Yao, S., L. Lum and P. Beachy, 2006 The ihog cell-surface proteinsbind Hedgehog and mediate pathway activation. Cell 125: 343–357.
Zhang, S. L., A. V. Yeromin, X. H. Zhang, Y. Yu, O. Safrina et al.,2006 Genome-wide RNAi screen of Ca21 influx identifiesgenes that regulate Ca21 release-activated Ca21 channel activity.Proc. Natl. Acad. Sci. USA 2: 2.
Communicating editor: A. Spradling
16 N. Perrimon and B. Mathey-Prevot




























![Medical-Genetics-ch5-2009 Medical Students [Compatibility Mode]](https://img.dokumen.tips/doc/110x75/577d35261a28ab3a6b8faacf/medical-genetics-ch5-2009-medical-students-compatibility-mode.jpg)
