Embed Size (px)
Citation preview

1893
Diminished Contractile Response to IncreasedHeart Rate in Intact HumanLeft Ventricular HypertrophySystolic Versus Diastolic Determinants
Chun-Peng Liu, MD; Chih-Tai Ting, MD; William Lawrence, MD; W. Lowell Maughan, MD;Mau-Song Chang, MD; David A. Kass, MD
Background. Experimental studies indicate that in addition to diastolic dysfunction, hypertrophiedmyocardium can display depressed contractile responses, particularly at rapid heart rates, compoundingreserve limitations. This study tests whether such abnormalities exist in intact human subjects atphysiological paced rates and, if so, whether they are linked to simultaneous rate-dependent deteriorationin diastolic function.Methods and Results. Ten subjects with left ventricular hypertrophy (LVH) and 8 normal control subjects
were studied. Most LVH patients presented with dyspnea and/or pulmonary edema and had concentrichypertrophy. Since rapid pacing simultaneously alters cardiac filling volumes and pressures, pressure-
volume relation analysis was used to better define changes in contractile response. Patients were
instrumented with a conductance catheter and micromanometer for pressure-volume data recording anda balloon occluder at the right atrial-inferior vena caval junction to vary filling and thus generate functionrelations. Data were obtained at baseline and at three atrial pacing rates (100, 120, 150 min'1). Inaddition, single-beat force-interval data were used to indirectly examine calcium cycling kinetics. LVHsubjects demonstrated baseline diastolic abnormalities, including prolonged relaxation, elevated end-di-astolic pressure, and reduced chamber compliance. However, systolic function was similar to that incontrol subjects. With rapid pacing, normal subjects displayed a positive contractile response, whereasthis was markedly diminished in LVH subjects. With abrupt termination of pacing and return to slowersinus rhythm, LVH subjects displayed greater initial potentiation followed by a more rapid decline thancontrol subjects, suggesting abnormalities of calcium handling. Despite contractile abnormalities,diastolic function did not further deteriorate with rapid pacing and thus did not appear to be tightly linkedto the systolic changes.
Conclusions. Pacing stress in intact human LVII can result in systolic impairment superimposed on
preexisting but not worsened diastolic dysfunction. Abnormal calcium handling probably contributesprominently to this response. (Circulation. 1993;88[part 11:1893-1906.)KEY WORDs * pressure-volume relation * pacing * hemodynamics
C linical left ventricular hypertrophy (LVH) is prin-cipally considered a disorder of diastolic func-tion. Resting isovolumic relaxation is prolonged,
early rapid filling is slowed, and chamber and myocardialstiffness are increased.'-4 These abnormalities in turn havebeen related to a variety of changes including alteredcalcium handling5-10 and collagen content."11'2 In contrastto diastole, resting systolic function often appears normalor even supranormal in humans with LVH, and a similarvariability has been found in animal models.5'13-17
Both diastolic and systolic function ofLVH hearts arereported to diminish at increased heart rate in intact
Received February 17, 1993; revision accepted June 16, 1993.From the Division of Cardiology, Department of Internal
Medicine, The Johns Hopkins Medical Institutions, Baltimore,Md, and the Department of Cardiology, Taipei Veterans GeneralHospital, Yang-Ming Medical College, and National DefenseMedical Center, Taipei, Taiwan, ROC.
Correspondence to Dr Kass, Carnegie 538, Johns HopkinsMedical Institutions, 600 N Wolfe St, Baltimore, MD 21287.
animals,14"8'19 isolated muscle,8 and single cells.20 Dia-stolic deterioration is commonly reported in the form ofmarked increases in end-diastolic pressure or tensionand in systolic dysfunction by reduced force generationor shortening. Systolic changes have been more difficultto quantify in intact preparations, since varying cyclelength simultaneously alters cardiac filling volume andpressures. This limits the interpretation of many fre-quently used systolic parameters such as maximal rate ofpressure rise (dP/dtm.), peak systolic pressure, or frac-tional shortening.14"9'21'22 While isolated tissue resultsare more direct, the kinetics of calcium cycling itselfappears to be slowed in these preparations comparedwith in situ23 hearts, and this could influence the results.Finally, it remains unknown whether similar systolicchanges exist in intact humans with LVH at physiolog-ical heart rates.A more heart-specific analysis of systolic reserve
during pacing is afforded by pressure-volume relations.Thus far, this approach has only been applied to the
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1894 Circulation Vol 88, No 4, Part 1 October 1993
TABLE 1. Baseline Clinical, Hemodynamic, and Echocardiographic Characteristics ofPatient GroupsPatient Age, y Sex HTN Hx Presenting Symptom ABP, mm Hg dWTh,, CON/ASYMControl patients
1 39 M + Atypical CP, borderline ETT 140/80 0.9 ...
2 40 F - Atypical CP 160/90 1.1 ...
3 46 M - Atypical CP, borderline EUr 140/94 1.0 ...
4 19 M + Borderline HTN 140/70 1.0 ...
5 41 M +/- Headache, transient HTN 142/85 1.0 ...
6 50 F - Atypical CP 128/88 1.0 ...
7 41 M +/- Dizziness, HTN workup 138/98 0.9 ...
8 25 M +/- Dizziness, HTN workup 140/88 0.8 ...
Mean 37.6 141/87 0.96
SD 10.4 8.8/8.6 0.09
LVH patients
9 62 M - Atypical CP, presyncope 104/60 2.1 ASYM10 47 F +/- CHF, acute pulmonary edema 170/100 1.8 CON1 1 57 F + CHF, acute pulmonary edema 120/70 1.4 CON
12 53 F + CHF, acute pulmonary edema 150/70 1.7 CON
13 29 F - CHF, dyspnea, atypical CP 110/80 2.2 ASYM
14 62 M + CP 172/80 1.8 CON
15 49 M + CP 110/70 1.5 CON
16 57 F + CHF, acute pulmonary edema 155/72 1.8 CON
17 63 M + CHF, acute pulmonary edema 196/102 1.6 CON
18 66 F - CHF, dyspnea, CP 145/85 2.8 CON
Mean 54.5* 143/79 1.9*
SD 10.9 31.3/13.6 0.4
HTN indicates hypertension; ABP, average blood pressure; dWrh,nx, maximal diastolic wall thickness; CON/ASYM, concentric orasymmetric hypertrophy; CP, chest pain; ETT, exercise tolerance test; and CHF, congestive heart failure.*P<.01 vs control subjects; +/- reflects an equivocal history.
normal canine left ventricle.24-26 However, recently de-veloped catheter-based techniques enable repeatedmeasurement of pressure-volume relations in the samehuman subject under a variety of acute conditions.27-29Since this method uses mechanical rather than pharma-cological changes in chamber load to generate therelations, it is ideally suited for studying pacing mechan-ics at multiple rates in the same subject.
In the present study, this method was used to test thehypothesis that heart rate-dependent inotropic reserveis limited in intact humans with significant LVH. Mech-anisms for systolic depression were further explored bytesting whether simultaneous deterioration of diastolicfunction was present and/or necessary and if alteredcalcium handling played a role. The latter was examinedby measuring postextrasystolic potentiation, potentia-tion decay, and the contractile response to variations inindividual-beat intracycle length.
MethodsSubject Group
Studies were conducted in 18 adult patients referredfor diagnostic cardiac catheterization. Summary clinicaldata are provided in Table 1. Ten patients (mean age,
54±11 years) had LVH documented by echocardiogra-phy, with a mean diastolic wall thickness of 1.9±0.4 cm(mean±SD). Most also had ECG evidence of LVH.LVH was concentric in 8 of the patients. The majority ofLVH patients (n=7) presented with exertional or restdyspnea (New York Heart Association [NYHA] class IIto IV), with 5 of the subjects experiencing severeparoxysmal pulmonary edema. The remaining 3 pa-tients presented with atypical chest pain and/or presyn-cope. Seven patients had a history of treated hyperten-sion. No patient had documented hypertrophy and/orsystolic murmurs from childhood or a family history ofLVH. All subjects had normal coronary arteriography.The control group comprised 8 patients aged 38±10
years, with normal ECGs, coronary arteriograms, con-trast ventriculography, and echocardiograms. Four ofthese patients presented with atypical chest pain, and 4had headache and/or dizziness and were admitted toevaluate possible early hypertension. These latter sub-jects had not received chronic treatment and werenormotensive in the hospital. All patients in both LVHand control groups were in normal sinus rhythm. Theresearch protocol was approved by the Joint Committeeon Clinical Investigation of the Johns Hopkins Medical
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

Liu et al Pacing Hemodynamics and Cardiac Hypertrophy 1895
Institutions and the Clinical Investigation Committee ofthe Veterans General Hospital, Taipei, Taiwan. Allpatients provided informed consent, and there were nocomplications as a consequence of the study.
ProcedureChronic medications were withheld for at least 24 to
48 hours before the study. Subjects were premedicatedwith benzodiazapam (10 mg) and diphenhydramine (50mg) and then underwent routine coronary angiography,left ventriculography, and right heart catheterization viafemoral access. This confirmed the absence of coronaryartery, valvular, and pericardial disease. Subjects inwhom concomitant disease was identified were ex-cluded. Nonionic contrast was used to minimize hemo-dynamic sequelae.
After right and left heart catheterization, specializedcatheters were placed for pressure-volume analysis.Details of this procedure have been reported else-where.27-29 Briefly, an 8F multielectrode conductance(volume) catheter (Webster Labs, Baldwin Park, Calif)was introduced via the femoral artery and advanced tothe left ventricular apex. A micromanometer-tippedcatheter (SPC-330A, or 320, Millar, Houston, Tex) wasadvanced through the full length of the conductancecatheter to measure cavity pressure. The volume cath-eter was connected to a stimulator/microprocessor (Sig-ma V, CardioDynamics, Rijnsburg, The Netherlands,n=8, or VCU, Cardiac Pacemaker Inc, St. Paul, MN,n= 10). These devices provide a low-amperage ACexcitation current (20 kHz or 1 to 2 kHz, respectively) atbase and apex electrodes. Measured voltages at inter-vening electrodes are converted to segmental conduc-tances, which are then added in real time to yield atime-varying signal proportional to total chamber bloodvolume. Proper placement of the catheter within theventricle is determined by fluoroscopic inspection andby examining each segmental pressure-volume loop.Moving up from the apex, the first electrode segmentwith a PV loop that is isovolumic or moves clockwisedefines the position of the aortic valve. Only segmentsbelow this level are combined into a total volume signal.
In addition to volume and micromanometer cathe-ters, a specially designed 7F balloon occlusion catheter(SP-9168, Cordis, Miami, Fla) was introduced through afemoral vein and placed within the right atrium. Infla-tion (15 to 25 mL of C02) and gentle withdrawal of thisballoon toward the inferior vena cava produced a tran-sient reversible decline in venous return and thus,volume. This generated pressure-volume relations ateach pacing rate. Atrial pacing was performed using a2.5F bipolar flexible tip pacing wire (98-100H, Baxter,Irvine, Calif) advanced through the lumen of the bal-loon catheter.
ProtocolAfter placement of all catheters, cardiac output,
simultaneous steady-state pressure-volume signals, andsystolic and diastolic pressure-volume relations duringtransient inferior vena cava (IVC) occlusion were re-corded. Right atrial pacing was then instituted at 90 to100 min' and increased by 20 to 30 min' increments toa maximum of 150 to 170. Two minutes were provided ateach steady-state heart rate before repeat data collec-
nation of pacing (ie, return to normal sinus rhythm).This provided evaluation of potentiation (first beat afterpacing cessation) and decay of potentiation (termedrecirculation fraction).30,31 Variation in baseline heartrate, Mobitz I second-degree heart block at rapid ratesin two patients, and occasional difficulty in maintainingsteady pacing during simultaneous preload reductionprevented all data from being obtained in every patientat each heart rate. However, generally, 80% or more ofsubjects were represented at each rate.The mechanical response to varying pacing rate is
known to be influenced by calcium cycling kinetics.Recent data in intact dogs23 suggest that these kinetics,as globally indexed by the mechanical restitution curve(MRC), appear much faster in intact preparations com-pared with isolated hearts32 or muscle.33,34 The MRCrelates the contractile response of a test contraction toits prior cycle length and is thought to index the kineticsof calcium cycling.31-34 Thus, to better clarify the systolicpacing response, MRCs were obtained in a subset ofpatients (LVH, 7; controls, 4). Atrial pacing was set at100 min', and test stimuli then were introduced, vary-ing the interval between the test beat and the laststeady-state paced beat. This test stimulus interval(TSI) started short at 375 milliseconds and was gradu-ally lengthened by 25 to 50 milliseconds until nativesinus activity preempted the stimulus. Each TSI wasseparated by at least 10 steady-state beats. The MRCwas generated by plotting the contractile response foreach test beat versus TSI.Throughout each study, ventricular pressure-volume
loops were continuously displayed in real time usingcustom designed data acquisition and display software.Analog signals were digitized at 200 Hz and stored onremovable hard drives for subsequent off-line analysis.This software also provided pacing triggers, which weretransmitted to an isolated pacing spike generator(Bloom Associates, Reading, Pa).
Data AnalysisOur current approach to calibrating the volume cath-
eter signal in humans has been reported recently.29 Thevolume signal is proportional to absolute left ventricularcavity volume with a nonzero offset and nonunity slope.The slope is determined from the ratio of thermodilu-tion (or ventriculographic) stroke volume to the strokevolume measured by catheter. The latter is defined asthe mean width of the pressure-volume loop. Thecalibration offset is determined by matching end-dia-stolic catheter volume or ejection fraction to ventricu-lography values. Calibration of the volume signal wasperformed at baseline only. Care was taken to assurethat catheter position remained stable throughout thestudy, minimizing calibration error.
Ventricular pressures, volumes, and other steady-state parameters were derived from 5 to 10 successivecardiac cycles, synchronized to the R wave of theelectrocardiogram, and signal averaged. End-diastolicpressure (Ped) was the pressure at the lower right cornerof the pressure-volume loop, determined by automatedalgorithm.29 End-systolic pressure (P,,) was measured atthe point of maximal elastance (maximal P/[V-V0J),where V. is volume axis intercept of the end-systolicpressure-volume relation (ESPVR). End-systolic and
ti-diastolic volumes (V and Ved) were determined bytion. Data also were recorded immediately after termi-
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1896 Circulation Vol 88, No 4, Part 1 October 1993
averaging five volume points centered at mean leftventricular pressure during isovolumic contraction andrelaxation, respectively. Stroke volume was the meanwidth of the pressure-volume loops, and stroke workwas the area within the loop. The first derivative ofpressure (dP/dtm.,) was digitally derived by a five-pointweighted slope and normalized to Ved to index contrac-tile function.35 A second systolic parameter was maximalpower (PWRma,,) divided by Ved, where power wasderived from the product of ventricular pressure andthe derivative of volume. The time constant of isovolu-mic relaxation was determined from pressures extend-ing from -dP/dtma. to pressure at the onset of filling.Two methods were used, neither of which assumed azero pressure asymptote at time= co. One was the in-verse slope of the linear regression of dP/dt versusP(t),36 and the second used nonlinear regression to fitthe model P(t)=P.+Poet". Arterial load was assessedby the effective arterial elastance Ea=PejSV and bytotal estimated resistance Rt=0.9* Ea * HR.37
Pressure-Volume RelationsIn addition to steady-state parameters, systolic and
diastolic chamber functions were assessed by pressure-volume relations derived from multiple cardiac cycles(13+±5, mean±SD) at varying preload during transientIVC occlusion. Two systolic relations were obtained.The ESPVR was determined by linear regression as theset of points at maximal P/(V-Vo) (average correlationcoefficient, r=.98+.02), with slope Ees (end-systolicelastance) and volume intercept V0. The latter wasobtained by an iterative method.38 The linear relationbetween stroke work and Ved was also derived fromthese same cardiac cycles, and the slope of this relation(M,,) also indexed contractile function.39 This latterrelation is measured over a broader range of values thanthe ESPVR and is independent of chamber size andvolume signal gain, as the volume terms in both numer-ator and denominator cancel.
Diastolic chamber compliance was determined fromthe end-diastolic pressure-volume relation (EDPVR).This was generated by combining end-diastolic pres-sure-volume points from the latter third of cardiacfilling from each beat during IVC occlusion. Data (meanof 31 points) were fit to a linear model29 to determinecompliance, with a mean linear correlation of 0.88. Alinear rather than monoexponential model was usedsince it fit the data virtually as well and was less sensitiveto precise data point selection.
Force-Interval RelationsTo better assess the role of altered calcium handling
toward steady-state pacing results, responses to single-beat variations in intercycle length were measured andused to characterize (1) maximal systolic potentiation,(2) recirculation fraction, and (3) mechanical restitu-tion. Although methods for measuring intracellularcalcium exist for isolated tissues and hearts, these arenot applicable to intact humans. Thus, at present,calcium handling can only be indirectly studied inhumans by means of force-interval relations.
Postextrasystolic potentiation was assessed by exam-ining the contractility of the first postpaced beat aftercessation of atrial pacing. To minimize effects of simul-
tile function was indexed by single-beat Bes (using the V.obtained during atrial pacing).The rate of decay of potentiation after pacing cessa-
tion is thought to index the percent of calcium recycledthrough the sarcoplasmic reticulum,30,40 termed therecirculation fraction. After the initial postpacing po-tentiation, subsequent beats display a gradual geometricdecline in contractility to baseline. Heart rate and Vedare nearly identical for these postpaced beats, allowingdP/dtm. to index contractile function.39 To obtain therecirculation fraction, the following parameter (Rn) wasdetermined for the first 6 postpaced beats:
Rn=[dP/dtma,(n)/dP/dtm.(ss)]- 1
where the numerator and denominator are the values ofdP/dtmax for beat n (n=1 to 6) and the fully decayedsteady state (ss), respectively. R. follows a geometricdecay, that is, R.= K' R,, where K iS the recirculationfraction, (K< 1.0). It follows that R,+1=R,- K, and ap-
plying linear regression to the R, versus Rn+l pointsyields the value for K.
Mechanical restitution curves were obtained fromsingle test cycle data described above. Contractile forcefor each test beat was estimated using single beat Ees(assuming constant V., determined from the ESPVR atthe steady-state cycle length, 600 milliseconds). The teststimulus interval (TSI) was directly measured betweensuccessive QRS complexes, varying slightly from theatrial stimulation interval due to AV delay. Ees of eachTSI was normalized to the value at the basic pacingcycle length (600 milliseconds). The resulting data werefit to a monoexponential in the form
Ees(TSI)/Ees(600)= CRm,, [1 -ee(-TSI-TS/Te]where TSIO is the longest stimulus duration at which nomechanical force develops, Tc is the time constant ofmechanical restitution, and CRm., is the relation plateaugiving contractile potentiation at long cycle lengths.
Validation of Single-Beat Contractility AnalysisPotentiation and mechanical restitution analysis re-
quired single-beat estimates of contractility that werelittle influenced by simultaneous loading change. Asused by Freeman and Colson,23 single-beat E,, servedthis purpose. The approach is supported by the follow-ing. E5, derived from multiple cycles during IVC inflowocclusion (Ee4ESPVR]) was compared with that mea-sured from single beats at the same heart rate assumingthe same V. (EJ5[SB]). The two values were nearlyidentical: Ees(ESPVR) =1.0 * Ees(SSB)+0.186 (r=.943,SEE=0.057, P<.0001). Second, 6 studies were per-formed in isolated blood-perfused canine ventriclescomparing MRCs derived under isovolumic conditions("gold standard") with those determined under physio-logical ejection.41 As shown by example in Fig 1, theresults were nearly identical. Under isovolumic condi-tions, developed pressure (DP) indexed contractility ateach cycle length (Fig 1A), whereas Ees was used whenhearts "ejected" into the computer-simulated vascularsystem (Fig 1, B and C). The two MRCs derived frommany such test beats are shown in panel D and werevirtually superimposable. Similar results were obtained
taneous changes in both preload and afterload, contrac- in all 6 experiments.
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

Liu et al Pacing Hemodynamics and Cardiac Hypertrophy 1897
A40 LV
E30
20
3W WLP
E loow
ECG S
C,)~
120.
go
0 40 60VOLUME (ml)
B
10
120 W P
ECG
D2.0
CL0.% 1.5
0cM 1.0.
N
0z
A-A EJCAN (em)*-* 5gUC (UP)
0.*1 i0 250 S5W 750 lo0 1250 1500
TSI (msec)
FIG 1. Validation of single-beat end-systolic elastance (EaJ analysis for de-termination of mechanical restitutioncurve (MRC) in intact heart. Data wereobtained in isolated blood-perfused ca-nine ventricles. MRC generated underejecting conditions was compared withthe "gold standard" MRC from thesame heart obtained under isovolumet-ric conditions. A, At constant volume, asingle test stimulus interval (TSI) isintroduced and results in lower devel-oped pressure (DP) (see arrow). B,When the samepacing sequence is usedunder ejecting conditions, varying cyclelength also results in marked changes inend-diastolic volume. Thus, to assesscontractility, a more load-insensitivemeasure is required. C, This is obtainedby plotting the data from B as pressure-volume loops and determining the E,=for each beat by connecting the end-systolic pressure with the volume axisintercept (solid lines). This is shown forthe steady-state beat (thick middleloop), the TSI, andpost-TSI beats. Thisslope is lower for the TSI beat. D, Bycombining many beats at varying TSIs,MRCs are generated. Isovolumic datausing DP are compared with ejectingdata usingE, The relations were virtu-ally identical This was observed in all 6hearts studied. LVW indicates left ven-tricular volume; LVP, left ventricularpressure.
Statistical AnalysisAll data in the text and tables are provided as
mean+SD. Figure data are displayed as mean±SEM toassist visual display of multiple mean trends and differ-ences between subject groups. The influence of heartrate on a given parameter was determined by repeated-measures ANOVA, with post hoc testing using Dun-nett's multiple comparisons test. Between-group com-parisons of heart rate influences were determined bycovariance analysis, with individual differences analyzedwith Dunnett's test. Statistical significance was acceptedat the P<.05 level.
ResultsBaseline Hemodynamic Data: ControlVersus HypertrophyTable 2 provides rest hemodynamic data for control
and hypertrophy groups. Significant disparities werepresent only in diastolic parameters. End-diastolic pres-sure was nearly double in LVH patients (P<.01) atsimilar diastolic volumes, and the time constant ofisovolumic relaxation was prolonged by 32% at similarmean end-systolic pressure, volume, and heart rate.Pressure-volume analysis revealed a reduced chambercompliance (3.2±0.7 vs 5.1±2.3 m~lmm Hg, P<.05) inLVH. In contrast to diastole, virtually all systolic func-
tion parameters were similar at baseline. The LVHgroup had a somewhat higher Ees, but this achievedborderline significance (P=.093). Other contractilitymeasures such as the slope of the stroke work to Vedrelation (Mw) and maximal power and dP/dtma. normal-ized to Ved were similar at baseline.
Systolic Response to Incremental PacingFig 2 summarizes mean systolic responses to pacing,
and Table 3 provides the ANOVA testing for effects ofheart rate and patient group (control vs LVH) on thepacing response. Stroke volume fell similarly in bothgroups with pacing, although cardiac output (not dis-played) was unchanged. Rate-dependent reductions inend-systolic pressure and increases in (dP/dtmux)/Vedwere similar in both groups. Increased pacing ratelowered Ves in control subjects but not LVH subjects,and ejection fraction was slightly lower in LVH (P<.05vs control subjects at fastest rate). Maximal ventricularpower normalized to Ved also demonstrated a markedblunted response in LVH subjects, rising by 86.1% incontrol subjects versus 40.9% in LVH (P=.011).The Ve,, EF, and maximal power data suggested an
abnormal contractile response to increased heart rate inLVH subjects. This was better defined by pressure-volume relation analysis. Fig 3 shows ESPVRs for
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1898 Circulation Vol 88, No 4, Part 1 October 1993
TABLE 2. Baseline Hemodynamic Values in Control and LVH SubjectsControl LVH P
Diastolic
End-diastolic pressure, mm Hg 12.3+4.9 20.2+6.2 <.01
End-diastolic volume, mL 108.1+±27.9 114.0+40.4 NSRelaxation (r), milliseconds 40.8±+10.6 52.6± 13.9 <.05
Peak filling rate/Ved, sec-1 3.6+0.6 3.6±0.9 NSDiastolic compliance, mL/mm Hg 5.1±2.3 3.2+0.7 <.05
HemodynamicHeart rate, min-1 71.1 +9.4 79.6±14.1 NSStroke volume, mL 64.9±19.2 58.2±17.8 NSCardiac output, L/min 4.6±1.3 4.6±1.1 NSTotal resistance, mm Hg . mL-1 * s-1 1.9±0.6 2.2±1.0 NSArterial elastance, mm Hg/mL 2.3±0.7 2.9±1.4 NS
SystolicEjection fraction, % 60.1±8.7 54.5±17.4 NSEnd-systolic pressure, mm Hg 137.4±16.2 150.2±29.2 NSEnd-systolic volume, mL 43.9±14.9 55.4±37.4 NS(dP/dtmax)/Ved, mm Hg * s-' * mL-1 18.7±6.1 19.0±7.3 NS
Powerm1/Ved, W/mL-*100 6.3±1.0 6.4±1.5 NSEnd-systolic elastance, mm Hg/mL 2.1±0.9 4.2±3.8 .09
Slope of SW-Ved relation, mm Hg 81.8±10.4 99.1±28.2 NS
LVH indicates left ventricular hypertrophy.
multiple pressure-volume loops in example control(panels a through d) and LVH (panels e through h)subjects. The baseline systolic relations are reproduced
in each subsequent panel. In the control subject, rapidpacing resulted in an increase in the slope of theESPVR (ie, positive inotropic response), consistent
* * CONTROL100 o-*0 LVH
60
(ml) 60+4Z~ *
40-
20 . , ,SO 90 120 150
Ef
200
P0s150 * *(mmHg)
100 *
50 . .60 90 120 150
60 *
/dtmox/Ved 4 * t(mmHg/ec/lnd) * *
20
60 5 120 150
60 14 *
V I t-m +---t..-4 PWRmox/Ved(ml) (0 tW ~ t100)
40
90 120 150 60 90 120 150
HEART RATE (min-1 )
FIG 2. Influence of heart rate onsystolic function parameters. Data areas displayed. From left to right startingat the top, panels are SV, stroke vol-ume; Pes, end-systolic pressure; EF,ejection fraction; dP/dtlVed, first deriv-ative of left ventricular pressure!V,&;Ves, end-systolic volume; and PWR,,,W,Ved, maximal ventricular power di-vided by Ved. *Significant differenceswithin groups at a given rate comparedwith baseline; #significant between-group differences. ANOVA resultscomparing groups are provided inTable 2.
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

Liu et al Pacing Hemodynamics and Cardiac Hypertrophy 1899
TABLE 3. Statistical Tests of Heart Rate Influence on Cardiac andHemodynamic Parameters
Heart Rate Effect
Repeated-MeasuresANOVA LVH Effect
Parameter Control LVH Offset Interaction
Diastolic
End-diastolic pressure .001 NS <.001 NS
End-diastolic volume <.001 <.001 NS NS
Relaxation (T) NS .01 NS <.05
Diastolic compliance NS NS <.01 NS
Hemodynamic
Stroke volume <.001 <.001 NS NS
Cardiac output NS NS NS NS
Arterial resistance NS NS NS NS
Arterial elastance <.001 <.001 NS NS
Systolic
Ejection fraction <.05 .09 <.05 NS
End-systolic pressure .01 <.05 NS NS
End-systolic volume .01 NS NS .01
(dP/dtw)/Vd <.05 <.02 NS NS
PWRmw/Ved <.01 NS NS <.05
End-systolic elastance (Ees) <.05 NS .01 * <.01 *
SW-Vd relation slope (MJ) <.01 .064 <.05* <.01*
Four sets of P values are provided. The first two columns give repeated-measures ANOVA results within eachsubject group. The last two columns provide multiple regression analysis results testing for an independent effectof left ventricular hypertrophy (LVH, patient group) on heart rate dependence for each parameter. This effect isexpressed as an offset (ie, LVH alters mean value of parameter but not dependence on rate) and interaction effect(LVH alters heart rate dependence). Raw data are provided in Figs 3 and 6.
*Analysis performed on data normalized to initial baseline to reduce effect of intersubject variability.
with the normal rate treppe. In contrast, the LVHsubject displayed a flattening of the relation and arightward shift, indicating worsened contractile functionwith increased heart rate.Group data for slopes of ESPVR (Ees) and SW-Ved
(M,,,) relations are displayed in Fig 4. Data are shownnormalized to baseline. In the control group, bothcontractility indexes significantly rose during rapid pac-ing, reaching a maximal percent increase of between79.4% for E,, and 20.1% for M,, (both P<.05). Incontrast, the inotropic response to pacing was severelyblunted in LVH subjects, with no significant increase ineither contractility index (P<.01 vs control).The indication from all ejection phase measurements
(EF, Ees, Ms^, and PWRmax/Ved) that the inotropicresponse to pacing was abnormal in LVH stood incontrast to the result for (dP/dtmax )/Ved, an isovolumet-ric index. To determine if differences in vascular loadingexplained this disparity, we compared total arterialresistance and effective arterial elastance for the twogroups. Neither differed at baseline (Table 2). Pacingdid not significantly alter resistance at any of the heartrates, and while Ea increased near linearly with rate, thisresponse was similar in both groups. For example, atpeak pacing rate, Ea was 82.1% above baseline incontrol subjects versus 75.3% in LVH subjects (P=NS).
Thus, there were no significant disparities in arterialload.
Relation Between Systolic and DiastolicPacing-Induced Abnormalities
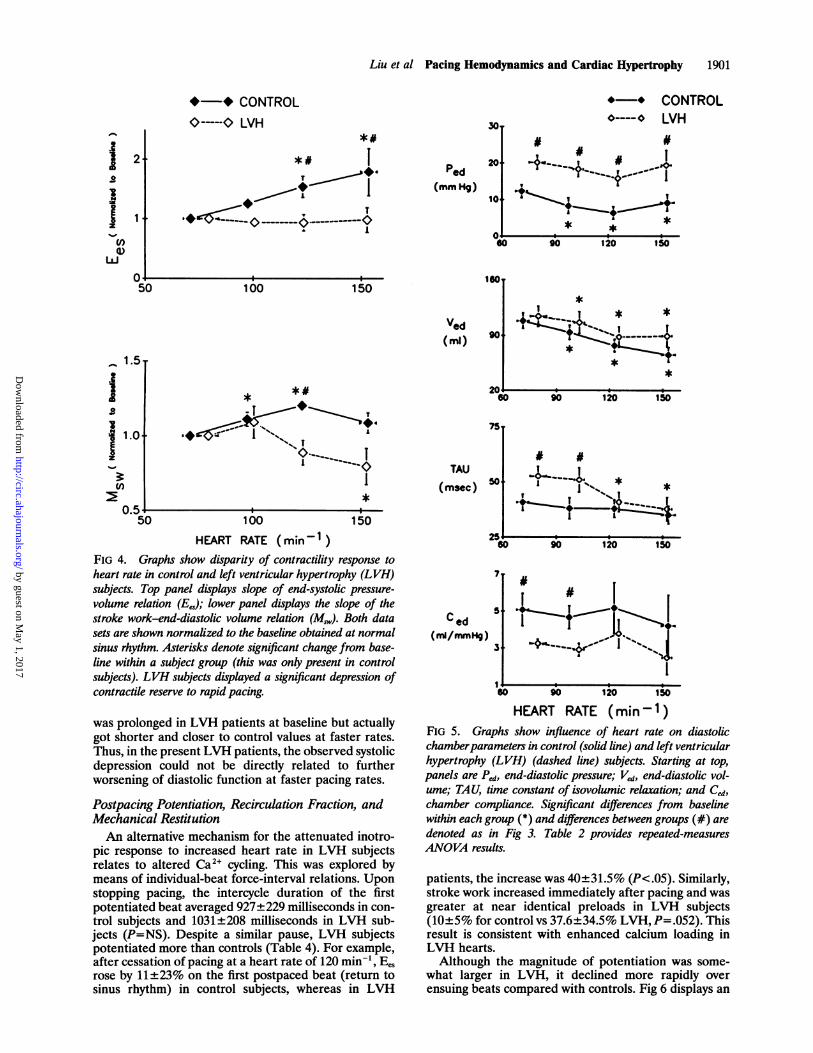
Several of the proposed mechanisms for a bluntedsystolic response to heart rate in LVH closely link it tosimultaneous worsening of diastolic properties.81419,22In general, deterioration is evidenced by a markedincrease in end-diastolic pressure or stress. To test forsuch interdependence in the present study, diastolicparameters were also examined (Fig 5 and Table 3).
Incremental pacing shortened diastolic filling period;thus, end-diastolic volume (Ved) fell with increasedheart rate. Interestingly, and somewhat unexpectedly,this reduction was similar in both patient groups, sug-gesting that despite LVH, net chamber filling was notdifferentially compromised, at least up to heart ratesnear 150. End-diastolic pressure was elevated at base-line in the LVH group, and it remained so at all heartrates (P<.05). However, it did not disproportionatelyrise in the LVH subjects at faster rates. Similar resultswere found for chamber compliance, which remainedsignificantly lower at each heart rate in the LVH groupbut demonstrated no change with pacing rate in eithergroup. The time constant of isovolumetric relaxation
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1900 Circulation Vol 88, No 4, Part 1 October 1993
CONTROL200
J150 EE
WWr
g
VOLUME (ml)
FIG 3. Pressure-volume loops and rela-tions in an example control (a-d) and leftventricular hypertrophy (LVH) (e-h) sub-ject at baseline (a and e, respectively) andwith incremental atrial pacing. The base-line end-systolic pressure-volume relation(ESPVR) is shown by the solid line and isreproduced in each subsequent panel atincreasing heart rate. Respective heartrates for each panel are a, 70; b, 100; c,130; d, 160; e, 70; f1 100; g, 120; and h,150 min'. For the control (non-LVH)patient, the slope of the ESPVR increasedwith each increment in heart rate. Incontrast, this slopefell atfaster rates in theLVH subject. Diastolic pressure-volumerelations (lower boundary) were generatedfrom these same data using the end-dia-stolic portion of each pressure-volumeloop measured during transient preloadreduction. See text for details.
VOLUME (ml)
VOLUME (ml)
:FE
3
200l
150
100
so.
EIWVOLUME (ml)
LVH
0.
VOLUME (ml)
IEIE
EE
g.O
VOLUME (ml) VOLUME (ml)
1501h
1004gEI
so/
CMI @ | | a0 50 100 150 200
VOLUME (ml)
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from
Liu et al Pacing Hemodynamics and Cardiac Hypertrophy 1901
*-* CONTROL
*#
*51* *IT si~~~
* <------- -------- >- -------
& 1~~~~
30T
20fPed
(mm Hg )10
(4a
*-* CONTROLo----o LVH
I# #
tA
A
* +*D 90 120 150
100160
150
Ved(ml)
* *TA
1 -v
I*
100
90
*
11 .-oqe ,
£
**i
690 120 1l0
7ST
TAU
(msec)
150
HEART RATE (min-1 )FIG 4. Graphs show disparity of contractility response toheart rate in control and left ventricular hypertrophy (LVH)subjects. Top panel displays slope of end-systolic pressure-
volume relation (EeJ; lower panel displays the slope of thestroke work-end-diastolic volume relation (MJ,). Both datasets are shown normalized to the baseline obtained at normalsinus rhythm. Asterisks denote significant change from base-line within a subject group (this was only present in controlsubjects). LVH subjects displayed a significant depression ofcontractile reserve to rapid pacing.
was prolonged in LVH patients at baseline but actuallygot shorter and closer to control values at faster rates.Thus, in the present LVH patients, the observed systolicdepression could not be directly related to furtherworsening of diastolic function at faster pacing rates.
Postpacing Potentiation, Recirculation Fraction, andMechanical RestitutionAn alternative mechanism for the attenuated inotro-
pic response to increased heart rate in LVH subjectsrelates to altered Ca2' cycling. This was explored bymeans of individual-beat force-interval relations. Uponstopping pacing, the intercycle duration of the firstpotentiated beat averaged 927+229 milliseconds in con-trol subjects and 1031±208 milliseconds in LVH sub-jects (P=NS). Despite a similar pause, LVH subjectspotentiated more than controls (Table 4). For example,after cessation of pacing at a heart rate of 120 min', Eesrose by 11+23% on the first postpaced beat (return tosinus rhythm) in control subjects, whereas in LVH
C ed
( m/mmHg)
Sw
# #
*t~~*N
Z5 l &I6B0 90 120 150
1
60 90 120 1soHEART RATE (min l1)
FIG 5. Graphs show influence of heart rate on diastolicchamberparameters in control (solid line) and left ventricularhypertrophy (LVH) (dashed line) subjects. Starting at top,panels are Ped, end-diastolic pressure; Ved, end-diastolic vol-ume; TAU, time constant of isovolumic relaxation; and Ced,
chamber compliance. Significant differences from baselinewithin each group (*) and differences between groups (#) are
denoted as in Fig 3. Table 2 provides repeated-measuresANOVA results.
patients, the increase was 40±31.5% (P<.05). Similarly,stroke work increased immediately after pacing and wasgreater at near identical preloads in LVH subjects(10+5% for control vs 37.6+34.5% LVH, P=.052). Thisresult is consistent with enhanced calcium loading inLVH hearts.Although the magnitude of potentiation was some-
what larger in LVH, it declined more rapidly overensuing beats compared with controls. Fig 6 displays an
.aI
~1U)
Ua50
'a 1.0
arsC
~1-
50
20 -
1&-
U.Z) i a i
1
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1902 Circulation Vol 88, No 4, Part 1 October 1993
TABLE 4. Potentiation After Terminationof Pacing
Control LVHSubjects Subjects ANOVA
End-systolic elastance
HR-1 +14.1±15.4 +19.6±20.5 1
HR-2 +11.0±23.3 +39.8±31.5* , P=.081
HR-3 +11.2±21.0 +20.3±34.4 J
Stroke workHR-1 +10.0±4.9 +37.5±34.5 1
HR-2 +41.8±22.2 +111.6±101.5* , P<.05
HR-3 +135.2±57.0 +160.1±88.3 J
End-diastolic volume
HR-1 +6.0±6.8 +11.9±6.2 1
HR-2 +26.9±15.0 +28.7±14.8 P=NS
HR-3 +59.6±29.9 +53.3±25.8 J
Data (percent change compared with baseline during pacing)are provided for each steady-state pacing rate for end-systolicelastance, stroke work, and end-diastolic volume. LVH indicatesleft ventricular hypertrophy; HR, heart rate.
*Significant change (P<.05) by multiple comparisons test.
individual example of the data used to determine thisdecay. Upon termination pacing, dP/dtmax (top panel)increased and then declined over successive beats in ageometric (monoexponential) manner. Plotting data forbeat n+ 1 versus n yielded a linear plot, whose slope isthe decay constant, or recirculation fraction (0.51, or
51% in this example). For the overall group (combiningdata after termination of each rapid heart rate), themean recirculation fraction was 46.3±2.5% for controlsubjects versus 30.1±3.7% for LVH subjects (P=.001).These results suggested that the portion of calciumrecycled via the sarcoplasmic reticulum was reduced byLVH. At faster heart rates, this could contribute to afall in available calcium and thus reduced contractileforce.
Analysis of mechanical restitution curves from bothpatient groups are provided in Table 4 and Fig 7.Relations for each patient comprised an average of21±3 different test cycles, with the test stimulus span-ning 400±54 to 930±170 milliseconds. Shorter cyclelengths were prevented by atrioventricular nodal block-ade and longer lengths by spontaneous sinus activation.There was virtually no difference in the MRCs obtainedfrom the control or LVH subjects. Furthermore, aspreviously reported in intact dogs,23 both sets of datademonstrated a rapid restitution time constant (TJ) of104.6±14.2 milliseconds in the control subjects and116.2±16.4 milliseconds in LVH subjects (P=NS). Thisis more than twice as fast as observed in isolated wholecanine hearts32 and as much as 6 to 10 times as fast asthat of isolated muscle.33,34 This suggests that Ca21recycling is fast in both groups and is a less likelycandidate to underlie the disparate contractile responseto pacing.
DiscussionThe primary goal of this study was to test whether the
inotropic response to increased heart rate is blunted in
dP/dt
2000 4000 6000
LVV
MAA'\AAVv22000 4o00 60C
c
0V
0-
xE
_0I c
0.00oo
k710
0.6 T
0.4+
0.2./
0.0 4I I0.0 0.2 0.4 0.6
(dP/dtmax)norm-1 (Beot n]
FIG 6. Graphs show recircula-tion fraction (RF) in an exam-ple subject. On the fifth cardiaccycle, rapid atrial pacing is ter-minated and the subsequent 6cardiac cycles at normal sinusrhythm recorded. Note that end-diastolic volume is nearly identi-cal for each of these postpacedbeats. The top panel displaysdPldt, and the maximal value isgreatest on the first postpacedcontraction (potentiation) andsubsequently declines. This de-cay follows a geometric decline.In the right-handpanel, dP/dt,,,,,of beat 1 is plot vs beat 2, beat 2vs beat 3, etc. The slope of thisrelation provides the RF (0.51 inthis control example). For thegroup overall, RF was 46.3±2.5% for control subjects vs36.5+3.7% for LVH (P<.001by ANCOVA).
TIME (msec)
2600
00 1300
.zEE.300
0
40
01 * "
i
E
EE
-26W ___ __ __-
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

Liu et al Pacing Hemodynamics and Cardiac Hypertrophy 1903
UI)Q1)
LLJ
J
LaiNJ
0
z
0.4
FIG 7. Mechanand left ventriculare shown normi
pacing cycle lenstions were superi(dotted and dasTable 4.
intact human sThe results frsteady-state oranalysis supportent with sever
ple, in canine
extent and vellpacing, suggest:LVH model stufast rates (>20(range (60 to 14ventricle displathe present stuwas diminishedin control subjobserved. Thisical relevance,;during exertion
It is intriguiindexes demonin the LVH paisovolumetric ithere were nobetween LVHejection loadunlikely that thmore complexmodynamic an;tion phase me;Furthermore, ]
measured at sttoo demonstralsupport derivesvelocity and exheart rates, w
controls.An alternatih
reflects fundanaspects of contiof enhanced %
internal ventricspecies, hypert
zyme shift favoring slower actin-myosin interaction andsustained force development.44 While this exact bio-
fT T A - * - ,chemical alteration does not occur in humans,45 similarkinetic changes have been observed. Slowing of cross-bridge cycling kinetics could inhibit force generationwhen shortening must be achieved rapidly.
- CONTROL Additional mechanisms for a blunted systolic re-sponse to rapid pacing in human LVH include inade-
- LVH quate myocardial perfusion, a dependence upon wors-ened diastolic function, and alterations of calciumhandling. Although the global nature of the present
| | § | l @ l data precludes a precise examination of these mecha-400 600 800 1000 1200 nisms, the results do lend support to some over others.FEST STIMULUS INTERVAL (msec) Reduced myocardial flow caused by markedly ele-tical restitution curves from control (n=4) vated diastolic pressures and/or reduced coronary flowtarhypertrophy (LVH) (n=7) subjects. Data reserve could result in myocardial ischemia at fast2lized to the E,s measured at the steady-state rates.19l2246,47 Arguing against this mechanism in theyth (600 milliseconds). The two mean rela- present data is the lack of simultaneous diastolic dete-rmposable, as were the monoexponential fits rioration and the shortening of isovolumic relaxation.shed lines). Fit parameters are provided in Diastolic abnormalities have been reported to precede
systolic ones with pacing-induced ischemia.47 Further-more, cardiodepression was not sustained after termi-nating pacing; rather, potentiation increased at the
subjects with clinically significant LVH. more rapid rates and then declined to the same steady*om ejection phase indexes based on state. These are admittedly indirect arguments, and theymultiple-beat pressure-volume relation do not exclude a role for ischemia.
rt this hypothesis. This finding is consis- In many LVH pacing studies, systolic and diastolical prior experimental results. For exam- deterioration occur simultaneously. For example, Fujiihypertrophy,14"16 systolic pressure and et al14 reported a near sixfold rise in end-diastolic wallocity of shortening are reduced during stress at very rapid pacing rates (270 min' in the dog)ing a systolic decrement. In these canine simultaneously with a significant decline in fractionalLdies, this decline is usually noted at very shortening and systolic pressure. Recently, Gwathmey) min'). This contrasts to the slower rate et a18 reported in isolated hypertrophied muscle strips$0 min') over which the normal canine that increasing the stimulation frequency resulted in atys a positive inotropic response.24 25 In decline of developed pressure primarily caused by a riseidy, the LVH force-frequency response in diastolic tension. The latter correlated with an in-even at such slower heart rates, whereas crease in measured intracellular calcium, and the au-jects, a positive inotropic response was thors raised the possibility that calcium overload withsuggests that the phenomenon has clin- LVH at rapid pacing rates simultaneously led to systolicas these are heart rates typically achieved and diastolic dysfunction. Similar simultaneous in-1. creases in systolic and diastolic calcium levels withing that all ejection phase contractile pacing were not found in a recent study performed in[strated an abnormal response to pacing isolated hypertrophied myocytes.20Ltients, whereas this was not true of the The present data question the notion that diastolicindex (dP/dtm.)/Vcd. As noted earlier, deterioration is required to observe diminished systolicsignificant differences in arterial loads function in hypertrophied hearts during pacing. Signif-and control subjects at each rate; thus, icant diastolic abnormalities existed at baseline; how-lid not explain the disparity. It is also ever, there was no significant further decline in diastolic1is represents an artifact of simple versus function with pacing. In fact, relaxation time, which was(pressure-volume relation-derived) he- prolonged at rest, shortened progressively more in thealysis. There was consistency for all ejec- LVH subjects than in control subjects. In neither groupasures despite their varying derivations. did it ever achieve a duration at which incompletePWRma/Ved and ejection fraction were relaxation would be anticipated. Interestingly, similarready state, like (dP/dtm.)/Ved, yet they shortening of relaxation time has been reported inted a blunted rate response. Additional subjects with dilated cardiomyopathy during pacing2's from a canine study of LVH14 in which and recently in isolated muscle strips from hypertro-ctent of shortening were reduced at fast phied human hearts.48rhereas dP/dtmix increased identical to In a prior pacing study of human hypertrophic car-
diomyopathy, Cannon et al'8 reported a large rise in Pedve explanation for this disparity is that it (>100% from a baseline of 13 mm Hg) at rates similarnental differences in ejection-dependent to the present investigation (maximum near 150 min').raction in LVH. This could be in the form These authors also demonstrated reduced coronaryshortening deactivation42 or a greater flow reserve at the maximal pacing rate and speculatedular resistance43 in LVH hearts. In some that reduced chamber compliance from ischemia re-
;rophied hearts undergo a myosin isoen- sulted in the elevated Ped. Perhaps importantly, the
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1904 Circulation Vol 88, No 4, Part 1 October 1993
TABLE 5. Mechanical Restitution Curves for Control and LVH SubjectsTime Constant (TJ), Plateau Intercept (T.),
milliseconds (CM) millisecondsControl 116.2+16.4 1.04+0.016 266.1 ±19.6LVH 104.6±14.2 1.06±0.013 268.6+16.8
LVH indicates left ventricular hypertrophy. Data are fit to a three-parameter monoexponential (see"Methods"). There are no significant differences between the parameters estimated from control vsLVH relations.
subjects of their study were younger, had asymmetrichypertrophy, and presented with chest pain as thedominant symptom. Typical pain was elicited in 90% oftheir subjects during rapid pacing. In contrast, subjectsin the present study were older, had mostly concentrichypertrophy, and primarily presented with dyspneaand/or pulmonary edema. None of the subjects devel-oped chest pain or ECG changes during pacing. Thesedifferences suggest that the interdependence betweensystolic and diastolic dysfunction during pacing in LVHmay vary, depending on the form and/or clinical pre-sentation of the disease. There are clearly situationswhere the existence of both are strongly temporallycorrelated. However, as the present data demonstrate,this is not always the case.
Reduction of systolic force could stem from abnor-malities of Ca2' transients and cycling. In isolatedhypertrophied myocytes,5,20 peak calcium transients arereduced at faster stimulation rates. Abnormalities ofcalcium handling are also suggested by enhanced extra-systolic potentiation and a more rapid decay of poten-tiation in hypertrophied compared with normal mus-cle.40,49 The former behavior has been ascribed to a netincrease in inward Ca21 current50,51 and the latter toreduced sarcoplasmic reticular Ca2' reuptake. Bothcould reduce systolic force generation at fast heartrates. The present data are consistent with several ofthese prior results, and, to our knowledge, represent thefirst demonstration of such phenomena in intact hu-mans with LVH. Seed et aP30 reported recirculationfractions of 52% in normal subjects and 37% in subjectswith dilated cardiomyopathy of various etiologies. Thisis strikingly similar to the results of the present investi-gation and may in part reflect the presence of congestivefailure symptoms in many of the subjects.Although these data are consistent with a role for
altered Ca2' handling toward systolic depression, theyseem at odds with the finding that Ped did not signifi-cantly rise and that relaxation time constant shortenedduring rapid pacing in LVH subjects. However, Ped iscritically influenced by passive as well as active chamberproperties. These stem from collagen content, intersti-tial fibrosis, and fiber architecture.1112 Changes in thesefactors with LVH may overwhelm changes caused byCa21 cycling abnormalities.52 Likewise, pressure decay isinfluenced by factors other than calcium reuptake suchas passive elastic recoil and systolic loading. Thesefactors might counter others related to active calciumcycling.
In addition to potentiation and potentiation decay,calcium handling is frequently assessed by the phenom-enon of mechanical restitution. This monoexponentialrelation links the variation of contractile response of asingle test beat to its preceding cycle length. Its time
constant (TJ) is thought to provide a measure of thetime required for complete Ca2' cycling, stemmingmostly from the restitution time of the SR-Ca2+ releasechannel.53 It is near 250 milliseconds in blood-perfusedisolated dog hearts32 and 700 milliseconds in isolatedmuscles.34 However, in intact dogs, Freeman and Col-son23 recently reported that T, was 64 milliseconds,close to the 100 milliseconds we found in both patientgroups. This suggests that intactness of the preparationcan have a profound influence on rates of Ca24 cycling.The short time constant found in both patient groupssuggests that only at heart rates faster than 400 milli-seconds (150 min') would one expect a decline incontractile force caused by inadequate diastolic time forfull mechanical restitution. It is thus less likely to haveexplained the disparity between LVH and controlresponses.
Study LimitationsMechanics data are presented as chamber indexes
(pressure-volume) rather than as estimated myocardialindexes (ie, stiffness or stress-strain). This was donebecause the echocardiographic data required to providemeaningful mass and wall geometry measurementswere of inconsistent quality to provide precise numbersand were not obtained during preload reduction tran-sients. However, since the study focused on relativechanges within a given subject and wall mass and overallgeometry were not acutely altered, the findings proba-bly parallel those that would be obtained based onstress-strain calculations.
Left ventricular hypertrophy is recognized to be aheterogeneous disorder. The present study results arebased on 10 patients, and while they all had substantialwall thickening, they no doubt represent a limited andmixed spectrum of disease. As noted above, there aresome important differences between the present studysubjects and those of prior human and animal modelstudies. For one, the present subjects were older andmost presented with exertional or rest dyspnea andpulmonary congestion. Their pacing response may dif-fer from younger subjects with genetic and asymmetricLVH. While several had a history of hypertension, therewas no significant difference in arterial pressures be-tween LVH and control subjects for the study. This ispotentially an important difference when comparingthese results with those from animal models using aorticbanding, in which the baseline left ventricular pressuresare often nearly twice those in the control group.14,19Such marked pressure differences may contribute toworsened diastolic properties.54There are limitations to the volume catheter method
for volume assessment that should be considered. Al-though prior studies have shown good linear correla-
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

Li et al Pacing Hemodynamics and Cardiac Hypertrophy 1905
tions between conductance catheter signals and othermeasures,55-57 there are concerns about potential non-linearity to the signal.56-" However, this limitation wasunlikely to have significantly influenced the results.First, identical methods were used over similar volumeranges in both subject groups; thus, calibration errorswould not underlie consistent disparities. Second, mul-tiple measures of chamber systolic and diastolic perfor-mance were used, some dependent and others indepen-dent of volume signal calibration. For example, neitherM,, or PWRmM/Ved are dependent on either cathetercalibration gain or offset (they have units of mm Hg andmm Hg/s, respectively), yet their results were analogousto Ee,, which is volume dependent. Much of the pacinganalysis compared each subject's response with theirrespective baseline, further reducing the effects of cal-ibration error.
ConclusionsThe principal new findings of the present investiga-
tion are that in subjects with significant symptomaticLVH, systolic contractile response to physiological heartrate increase is markedly diminished. Furthermore, thissystolic deterioration does not necessarily require simul-taneous worsening of diastolic abnormalities, specifi-cally elevations in diastolic pressure or further declinein relaxation or compliance. Analysis of force-intervalrelations suggests that abnormalities in calcium entryand reuptake probably contribute to the observed pac-ing-induced systolic dysfunction. In contrast, slowing ofmechanical restitution (overall calcium cycling time)does not appear to play a major role.
Loss of a rate-dependent systolic reserve in humanLVH suggests an additional mechanism beyond gradi-ent reduction and improved filling whereby rate controlby j-blocker therapy would be efficacious. Future stud-ies with this and other therapies, such as calciumchannel blockade and chronic AV sequential pacing,will be needed to determine if this abnormality canindeed be improved.
AcknowledgmentsThe authors gratefully thank Dr Yasuhiko Harasawa for his
assistance in performing the isolated heart studies. This studywas supported by US Public Health Service research grantsHL-01820, HL-33243, and HL-01610; Lao Yeo MedicalFoundation, 920; Academica Sinica (1988) (Visiting Fellow-ship of the Internal Medicine Department, National DefenseMedical Center, Taiwan, ROC [C.P.L.1); and an AmericanHeart Association Established Investigator Award (D.A.K).
References1. Lorell BH, Grossman W. Cardiac hypertrophy: the consequences
for diastole. JAm Coil CardioL 1987;9:1189-1193.2. Hess OM, Schneider J, Koch R, Bamert C, Grimm J, Krayenbuehl
HP. Diastolic function and myocardial structure in patients withmyocardial hypertrophy. Circulation. 1981;63:360-371.
3. Bonow RO, Ostrow HG, Rosing DR, Cannon RO III, Lipson LC,Maron BJ, Kent KM, Bacharach SL, Green MV. Effects ofverapamil on left ventricular systolic and diastolic function inpatients with hypertrophic cardiomyopathy: pressure-volumeanalysis with a nonimaging scintillation probe. Circulation. 1983;68:1062-1073.
4. Maron BJ, Bonow RO, Cannon RO III, Leon MB, Epstein SE.Hypertrophic cardiomyopathy: interrelations of clinical manifes-tations, pathophysiology, and therapy. N Engi J Med. 1987;316:780-789.
5. Bailey BA, Houser SR. Calcium transients in feline left ventricularmyocytes with hypertrophy induced by slow progressive pressureoverload. J Moll Cell CardioL 1992;24:365-373.
6. Bentivegna LA, Ablin LW, Kihara Y, Morgan JP. Altered calciumhandling in left ventricular pressure-overload hypertrophy asdetected with aequorin in the isolated, perfused ferret heart. CircRes. 1991;69:1538-1545.
7. Gwathmey JK, Morgan JP. Altered calcium handling in experi-mental pressure-overload hypertrophy in the ferret. Circ Res. 1985;57:836-843.
8. Gwathmey JK, Warren SE, Briggs GM, Copelas L, Feldman MD,Phillips PJ, Callahan M Jr, Schoen FJ, Grossman W, Morgan JP.Diastolic dysfunction in hypertrophic cardiomyopathy. J ClinInvest. 1991;87:1023-1031.
9. de la Bastie D, Levitsky D, Rappaport L, Mercadier J-J, MarotteF, Wisnewsky C, Brovkovich V, Schwartz K, Lompre A-M.Function of the sarcoplasmic reticulum and expression of its Ca2+-ATPase gene in pressure overload-induced cardiac hypertrophy inthe rat. Circ Res. 1990;66:554-564.
10. Ito Y, Suko J, Chidsey CA. Intracellular calcium and myocardialcontractility v calcium uptake of sarcoplasmic reticulum fractionsin hypertrophied and failing rabbit hearts. J Mol Cell CardioL1974;6:237-247.
11. Weber KT, Janicki JS, Shroff SG, Pick R, Chen RM, Bashey RI.Collagen remodeling of the pressure-overloaded, hypertrophiednonhuman primate myocardium. Circ Res. 1988;62:757-765.
12. Factor SM, Butany J, Sole MJ, Wigle ED, Williams WC, RojkindM. Pathologic fibrosis and matrix connective tissue in the subaorticmyocardium of patients with hypertrophic cardiomyopathy. JAmColl Cardiol 1991;17:1343-1351.
13. Mirsky I, Aoyagi T, Crocker VM, Fujii AM. Preload dependenceof fiber shortening rate in conscious dogs with left ventricularhypertrophy. JAm Coll Cardiol 1990;15:890-899.
14. Fujii AM, Gelpi RJ, Mirsky I, Vatner SF. Systolic and diastolicdysfunction during atrial pacing in conscious dogs with left ven-tricular hypertrophy. Circ Res. 1988;62:462-470.
15. Alyono D, Ring WS, Crumbley AJ, Schneider JR, O'Connor MJ,Parrish D, Bache RJ, Anderson RW. Global left ventricular con-tractility in three models of hypertrophy evaluated with E,,.J SurgRes. 1984;37:48-54.
16. Matsuno Y, Morioka S, Murakami Y, Kobayashi S, Moriyama K.Left ventricular end-systolic wall stress-dimension relationship inunanesthetized dogs with perinephritic hypertension. Jpn Circ J.1988;52:1370-1376.
17. Mirsky I, Pfeffer JM, Pfeffer MA, Braunwald E. The contractilestate as the major determinant in the evolution of left ventriculardysfunction in the spontaneously hypertensive rat. Circ Res. 1983;53:767-778.
18. Cannon RO III, Rosing DR, Maron BJ, Leon MB, Bonow RO,Watson RM, Epstein SE. Myocardial ischemia in patients withhypertrophic cardiomyopathy: contribution of inadequate vaso-dilator reserve and elevated left ventricular filling pressures. Cir-culation. 1985;71:234-243.
19. Bache RJ, Arentzen CE, Simon AB, Vrobel TR. Abnormalities inmyocardial perfusion during tachycardia in dogs with left ven-tricular hypertrophy: metabolic evidence for myocardial ischemia.Circulation. 1984;69:409-417.
20. Siri FM, Krueger J, Nordin C, Ming Z, Aronson RS. Depressedintracellular calcium transients and contraction in myocytes fromhypertrophied and failing guinea pig hearts. Am J Physiol 1991;261:H514-H530.
21. Feldman MD, Alderman JD, Aroesty JM, Royal HD, Ferguson JJ,Owen RM, Grossman W, McKay RG. Depression of systolic anddiastolic myocardial reserve during atrial pacing tachycardia inpatients with dilated cardiomyopathy. J Clin Invest. 1988;82:1661-1669.
22. Nakano K, Corin WJ, Spann JF Jr, Biederman RWW, Denslow S,Carabello BA. Abnormal subendocardial blood flow in pressureoverload hypertrophy is associated with pacing-induced suben-docardial dysfunction. Circ Res. 1989;65:1555-1564.
23. Freeman GL, Colston JT. Evaluation of left ventricular mechanicalrestitution in closed-chest dogs based on single-beat elastance. CurcRes. 1990;67:1437-1445.
24. Maughan WL, Sunagawa K, Burkhoff D, Graves WL, Hunter WC,Sagawa K. Effect of heart rate on the canine end-systolic pressure-volume relationship. Circulation. 1985;72:654-659.
25. Freeman GL, Little WC, O'Rourke RA. Influence of heart rate onleft ventricular performance in conscious dogs. Circ Res. 1987;61:455-464.
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

1906 Circulation Vol 88, No 4, Part 1 October 1993
26. Miura T, Miyazaki S, Guth BD, Kambayashi M, Ross J Jr.Influence of the force-frequency relation on left ventricularfunction during exercise in conscious dogs. Circulation. 1992;86:563-571.
27. Kass DA, Midei M, Graves W, Brinker JA, Maughan WL. Use ofa conductance (volume) catheter and transient inferior vena cavalocclusion for rapid determination of pressure-volume relationshipsin man. Cathet Cardiovasc Diagn. 1988;15:192-202.
28. Kass DA, Midei M, Brinker J, Maughan WL. Influence of coronaryocclusion during PTCA on end-systolic and end-diastolic pressure-volume relations in humans. Circulation. 1990;81:447-460.
29. Liu CP, Ting CT, Yang TM, Chen JW, Chang MS, Maughan WL,Lawrence W, Kass DA. Reduced left ventricular compliance inhuman mitral stenosis: role of reversible internal constraint. Cir-culation. 1992;85:1447-1456.
30. Seed WA, Noble MIM, Walker JM, Miller GAH, Pidgeon J,Redwood D, Wanless R, Franz MR, Schoettler M, Schaefer J.Relationships between beat-to-beat interval and the strength ofcontraction in the healthy and diseased human heart. Circulation.1984;70:799-805.
31. Schouten VJA, van Deen JK, de Tombe P, Veerveen AA. Force-interval relationship in heart muscle of mammals: a calcium com-partment model. Biophys J. 1987;51:13-26.
32. Burkhoff D, Yue DT, Franz MR, Hunter WC, Sagawa K.Mechanical restitution of isolated perfused canine left ventricles.Am J Physiol. 1984;246:H8-H16.
33. Cooper IC, Fry CH. Mechanical restitution in isolated mammalianmyocardium: species differences and underlying mechanisms.J Mol Cell Cardiol. 1992;22:439-452.
34. Wier WG, Yue DT. Intracellular calcium transients underlying theshort-term force-interval relationship in ferret ventricular myo-cardium. J Physiol. 1986;376:507-530.
35. Kass DA, Maughan WL, Guo ZM, Kono A, Sunagawa K, SagawaK. Comparative influence of load versus inotropic states on indexesof ventricular contractility: experimental and theoretical analysisbased on pressure-volume relationships. Circulation. 1987;76:1422-1436.
36. Raff GL, Glantz SA. Volume loading slows left ventricular iso-volumic relaxation rate. Circ Res. 1981;48:813-824.
37. Kelly RP, Ting CT, Yang TM, Liu CP, Maughan WL, Chang MS,Kass DA. Effective arterial elastance as index of arterial vascularload in humans. Circulation. 1992;86:513-521.
38. Kono A, Maughan WL, Sunagawa K, Kallman C, Sagawa K,Weisfeldt ML. Left ventricular end-ejection pressure and peakpressure to estimate the end-systolic pressure-volume relationship.Circulation. 1984;70:1057-1065.
39. Glower DD, Spratt JA, Snow ND, Kabas JS, Davis JW, Olsen CO,Tyson GS, Sabiston DC, Rankin JS. Linearity of the Frank-Starling relationship in the intact heart: the concept of preloadrecruitable stroke work. Circulation. 1985;71:994-1009.
40. Schouten VJA, Vliegen HW, van der Larse A, Huysmans HA.Altered calcium handling at normal contractility in hypertrophiedrat heart. J Mol Cell Cardiol. 1992;22:987-998.
41. Sunagawa K, Burkhoff D, Lim KO, Sagawa K. Impedance loadingServo pump system for excised canine ventricle. Am J Physiol.1982;243:H346-H350.
42. Suga H, Yamakoshi K. Effects of stroke volume and velocity ofejection on end-systolic pressure of canine left ventricle: end-systolic volume clamping. Circ Res. 1977;40:445-450.
43. Schroff SG, Janicki JS, Weber KT. Evidence and quantitation ofleft ventricle systolic resistance. Am J Physiol. 1985;249:H358-H370.
44. Alpert NR, Mulieri LA. Increased myothermal economy of iso-metric force generation in compensated cardiac hypertrophyinduced by pulmonary artery constriction in the rabbit. Circ Res.1982;50:491-500.
45. Mercadier JJ, Bouveret P, Gorza L, Shiaffino S, Clark WA, Zak R,Swynghedauw B, Schwartz K. Myosin isoenzymes in normal andhypertrophied human ventricular myocardium. Circ Res. 1983;53:52-62.
46. Jeremy RW, Fletcher PJ, Thompson J. Coronary pressure-flowrelations in hypertensive left ventricular hypertrophy. Circ Res.1989;65:224-236.
47. Aroesty JM, McKay RG, Heller GV, Royal HD, Als AV,Grossman W. Simultaneous assessment of left ventricular systolicand diastolic dysfunction during pacing-induced ischemia. Circu-lation. 1985;71:889-900.
48. Schumacher C, Becker H, Sigmund M, Hanrath P, Schondube F,Wendt G, Schulte HD, Preusse C. Inversed force-frequency rela-tionship in hypertrophic obstructive cardiomyopathy. Circulation.1992;86(suppl I):I-839. Abstract.
49. Anderson PAW, Manring A, Arentzen CE, Rankin JS, JohnsonEA. Pressure-induced hypertrophy of cat right ventricle. Circ Res.1977;41:582-588.
50. Keung EC. Calcium current is increased in isolated adult myocytesfrom hypertrophied rat myocardium. Circ Res. 1989;64:753-763.
51. Kleiman RB, Houser SR. Calcium currents in normal and hyper-trophied isolated feline ventricular myocytes. Am J Physiol. 1988;255:1434-1442.
52. Kass DA, Wolff MR, Ting CT, Liu CP, Chang MS, Lawrence W,Maughan WL. Diastolic compliance of hypertrophied ventricle isnot acutely altered by pharmacologic agents influencing activeprocesses. Ann Intern Med. In Press.
53. Banijamali HS, Gao W-D, MacIntosh BR, ter Keurs HEDJ. Force-interval relations of twitches and cold contractures in rat cardiactrabeculae: effect of Ryanodine. Circ Res. 1992;69:937-948.
54. Gelpi RJ, Pasipoularides A, Lader AS, Patrick TA, Chase N,Hittinger L, Shannon RP, Bishop SP, Vatner SF. Changes indiastolic cardiac function in developing and stable perinephritichypertension in conscious dogs. Circ Res. 1991;68:555-567.
55. Burkhoff D, van der Velde E, Baan J, Maughan WL, Sagawa K.Accuracy of volume measurement by conductance catheter inisolated, ejecting canine hearts. Circulation. 1985;72:440-447.
56. Applegate RJ, Chang CP, Little WC. Simultaneous conductancecatheter and dimension assessment of left ventricular volume inthe intact animal. Circulation. 1990;81:638-648.
57. Boltwood CM, Appleyard RF, Glantz SA. Left ventricular volumemeasurement by conductance catheter in intact dogs. Circulation.1989;80:1360-1377.
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from

C P Liu, C T Ting, W Lawrence, W L Maughan, M S Chang and D A Kasshypertrophy. Systolic versus diastolic determinants.
Diminished contractile response to increased heart rate in intact human left ventricular
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 1993 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.88.4.1893
1993;88:1893-1906Circulation.
http://circ.ahajournals.org/content/88/4/1893World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer available in the
Permissions in the middle column of the Web page under Services. Further information about this process isOnce the online version of the published article for which permission is being requested is located, click Request
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office.Circulation Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on May 1, 2017
http://circ.ahajournals.org/D
ownloaded from





























![Review Article Similarities and Differences between the … · 2019. 7. 31. · to diastolic heart failure [ , ]. Compared with systolic heart failure, diastolic heart failure is](https://img.dokumen.tips/doc/110x75/610a99d464112c7f8e47019f/review-article-similarities-and-differences-between-the-2019-7-31-to-diastolic.jpg)