Embed Size (px)
Citation preview

Inhibition of a New Differentiation Pathway in Neuroblastoma by
Copy Number Defects of N-myc, Cdc42, and nm23 Genes
Linda J. Valentijn, Arjen Koppen, Ronald van Asperen, Heather A. Root,Franciska Haneveld, and Rogier Versteeg
Department of Human Genetics, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands
Abstract
The best studied oncogenic mechanisms are inactivatingdefects in both alleles of tumor suppressor genes andactivating mutations in oncogenes. Chromosomal gains andlosses are frequent in human tumors, but for many regions,like 1p36 and 17q in neuroblastoma, no mutated tumorsuppressor genes or oncogenes were identified. Amplificationof N-myc in neuroblastoma is strongly correlated with loss of1p36 and gain of 17q. Here we report that N-myc down-regulates the mRNA expression of many genes with a role incell architecture. One of them is the 1p36 gene Cdc42 .Restoring the Cdc42 expression in neuroblastoma cellsstrongly induced differentiation. N-myc also inhibited Cdc42functioning at the protein level. This was mediated by nm23-H1 and nm23-H2, which are located in the amplified 17qregion. Nm23-H1 and nm23-H2 are strongly up-regulateddownstream targets of N-myc . Nm23-H1 was shown to bindCdc42 and prevented the induction of differentiation. Over-expression of Nm23 due to gain of 17q and induction by N-myccombined with weak expression of Cdc42 due to loss of 1p36and down-regulation by N-myc can thus block differentiation.Although this marks Cdc42 as a candidate tumor suppressorgene, no mutations were found. Further silencing of Cdc42 bysmall interfering RNA induced massive apoptosis, indicatingthat tumor cell survival requires a minimal Cdc42 activity.Three regions of chromosomal gain and loss thus affect genesfunctioning in one pathway in neuroblastoma. They convergeto bring the pathway out of balance and prevent Cdc42mediated differentiation. (Cancer Res 2005; 65(8): 3136-45)
Introduction
Neuroblastoma is a childhood tumor originating from neural crest–derived cells. Approximately 20% of neuroblastomas have N-mycamplification, which is usually accompanied by loss of heterozygosityof distal chromosome 1p and additional copies of chromosome 17q(1). Deletions of the short arm of chromosome 1 in N-mycamplified neuroblastomas are typically very large. The shortest 1pdeletion we observed in a series of N-myc amplified neuroblasto-mas was 24.4 Mb, from the telomere till the RUNX3 gene (ref. 2;http://genome.cse.ucsc.edu). The region is deleted in 90% to 95% ofall N-myc amplified tumors, whereas about 5% of N-myc amplifiedneuroblastomas have no apparent 1p deletions. In addition, N-mycsingle copy neuroblastomas can have 1p deletions. These deletions
are of variable length as well, but can be smaller with a moretelomeric SRO (3, 4). The different SROs in N-myc-amplifiedneuroblastomas have raised the idea that more than one tumorsuppressor gene maps on distal chromosome 1p (5).N-myc-amplified neuroblastomas follow a very aggressive course
(6). Overexpression of transfected N-myc genes in neuroblastomacell lines strongly increased proliferation rates (7, 8). Recent mRNAexpression profiling experiments, serial analysis of gene expression(SAGE) and microarray analysis, identified many myc-regulatedgenes (9–13). We used SAGE to identify genes regulated by N-mycin neuroblastoma. Genes up-regulated by N-myc were involved inrRNA processing and protein synthesis (9). Two recently identifiedN-myc targets, nm23-H1 and nm23-H2 , map to chromosomal band17q21, in the chromosome 17q region overrepresented inneuroblastoma (14).Additional copies of the distal chromosome 17q arm are present
in about 70% of neuroblastoma tumors, including all N-mycamplified cases. The copy number ranges between two and eightextra alleles and the minimal region of gain is 17q21-17qter. Themap position of the nm23-H1 and nm23-H2 genes at 17q21 makesthem interesting candidates for a role in neuroblastoma patho-genesis. The nm23 proteins encode nucleoside diphosphatekinases, the suppliers of nucleoside triphosphates. In severalhuman tumors, like breast cancer and melanoma, decreasedexpression was correlated with poor prognosis (15, 16). Up-regulation of nm23-H1 reverted melanoma cells from metastatic tononmetastatic (17). In contrast, in several other tumors includingneuroblastoma and osteosarcoma, elevated expression of nm23-H1and nm23-H2 was associated with a poor prognosis (18, 19). Theextra copies of 17q and the induction by N-myc result in a stronglyincreased expression of the nm23 genes in neuroblastoma cell linesand tumors (14). Besides the function of the nm23 proteins asdinucleoside kinases, growing evidence exists for additionalfunctions in signal transduction (20–22).In this article, we identified genes down-regulated by N-myc in
neuroblastoma. One of the key genes is Cdc42 . The Cdc42 genemaps on 1p36, and one copy is consistently deleted in N-mycamplified neuroblastoma. In addition, we provide evidence that theactivity of the Cdc42 protein is down-regulated by nm23-H1 andnm23-H2, which are induced by N-myc .
Materials and Methods
Cell culture. The SHEP-2 and SHEP-21N cell lines were cultured in
RPMI, the SKNAS-NmycER cells in DMEM. Both media were supplemented
with 10% FCS, 2 mmol/L glutamine, 50 units/mL penicillin, and 50 units/mL streptomycin. N-myc expression in SHEP-21N was repressed by
tetracycline ( final concentration, 50 ng/mL; Sigma, St. Louis, MO). The
NmycER protein in the SKNAS-NmycER cells was activated upon additionof 4-hydroxytamoxifen ( final concentration, 50 nmol/L; Sigma). The ER
domain has a point mutation, which enables transactivation of the
chimearic protein by 4-hydroxytamoxifen and not estrogens (23).
Requests for reprints: Linda J. Valentijn, Department of Human Genetics M1-134,Academic Medical Center, University of Amsterdam, P.O. Box 22700, 1100 DEAmsterdam, the Netherlands. Phone: 31-20-566-2386; Fax: 31-20-691-8626; E-mail:[email protected].
I2005 American Association for Cancer Research.
Cancer Res 2005; 65: (8). April 15, 2005 3136 www.aacrjournals.org
Research Article
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

Plasmids. The pCdc42flag plasmid was constructed by amplification ofcDNA with primers Cdc42Efor (5V-TATATAGAATTCATTTCAGCAATGCA-GACAATTAAG-3V) and Cdc42Xrev (5V-TATATACTCGAGTAGCAGCACA-CACCTGCG-3V). The amplified product was digested with EcoRI and XhoI
and cloned into the EcoRI and XhoI sites of pCMV-Tag4 vector (Stratagene,La Jolla, CA). The pCdc42G12Vflag and pCdc42T17Nflag constructs were
produced by amplification of Cdc42 from vector pCdc42flag using primer
T3 (5V-AATTAACCCTCACTAAAGGG-3V) in combination with mutant
primers CdcG12Vrev (5V-GTGTAGGATATCAGGAGACATGTTTTACCAA-CAGCAACATCGC-3V) or CdcT17Nrev (5V-GTGTAGGATATCAGGAGA-CAGTTTTTACC-3V). The PCR products were digested with BamHI and
EcoRV and ligated in the BamHI and EcoRV sites of pCdc42flag vector.
Northern blot analysis. Total RNA (20 Ag per lane) was separated on a1% agarose gel in the presence of 6.7% formaldehyde and blotted on
Hybond-N membranes (Amersham, Piscataway, NJ) in 10� SSC. Hybridiza-
tion was carried out overnight in 0.5 mol/L NaHPO4 (pH 7.0), 7% SDS,1 mmol/L EDTA at 65jC. Filters were washed in 40 mmol/L NaHPO4, 1%
SDS at 65jC. Probes were labeled by random priming of sequence-verified
PCR products.
Western blotting. Primary antibodies: anti-N-myc (PharMingen, SanDiego, CA), anti-Cdc42-B8 (Santa Cruz Biotechnology, Santa Cruz, CA),
anti--tubulin (Roche, Nutley, NJ), anti-Caspase-3 (Cell Signaling Technology,
Beverly, MA), and anti-poly(ADP-ribose) polymerase (Biomol Research
Labs, Plymouth Meeting, PA). Western blotting was done according tostandard procedures, secondary antibody anti-mouse linked to horseradish
peroxidase (Amersham), detected with Enhanced Chemiluminescence KIT
(Amersham).Transient transfection and immunofluoresence microscopy. For
transient expression, the cells were grown for 48 hours on glass slides in
medium supplemented with 1% FCS. Four micrograms of plasmid DNA and
4 AL DAC-30 (Eurogentec, Seraing, Belgium) in 1 mL medium, without FCS
and antibiotics, was incubated for 30 minutes at room temperature. For
double transfections, 4 Ag of each plasmid were incubated with 8 Ag DAC-
30. The DNA/DAC-30 mix was added dropwise to the cell cultures. After
16 hours, the medium was replaced by fresh medium with 1% FCS. Forty
hours after transfection, the slides were fixed with 4% paraformaldehyde in
PBS for 25 minutes. The cells were permeabilized with PBS/0.05% Triton X-
100. The slides were washed thrice with PBS/TX (PBS, 0.01% Triton X-100).
The slides were blocked for 30 minutes in Abdil (PBS, 0.01% Triton X-100,
1% bovine serum albumin). The antibodies were diluted in Abdil each
incubated for 30 minutes each incubation followed by four washes in PBS/
TX. Antibodies: 400 times diluted mouse-anti-flag M2 (Stratagene), 100
times diluted TRITC- or FITC-conjugated anti-mouse (Sigma), 200 times
diluted goat-anti-NFL (Santa Cruz Biotechnology), and 200 times diluted
TRITC-conjugated anti-goat (Sigma). The slides were rinsed with H2O, dried
and mounted in Vectashield (Vector Laboratories, Burlingame, CA) with
4V,6-diamidino-2-phenylindole (nuclear staining).
Glutathione S-transferase pulldown. Nm23H1 was amplfied with
adaptor oligonucleotides (5V-TATATAGGATCCATGGCCAACTGTGAGCG-TAC-3V and 5V-TATATGAATTCTCTGCCCTCCTGTCATTCAT-3V). The prod-
uct was digested with BamHI/EcoRI and cloned in the pGEX2TK vector
(Pharmacia, Piscataway, NJ) to yield GST-nm23H1.GST-nm23H1 transformed bacteria were lysed in PBS-0.01%Triton X-100.
Cell lysis and pulldown were done as described by Sander et al. (24).
RNA interference. The sequences of the single stranded RNA mole-
cules (Isogen, Maarsen, The Netherlands) are for Cdc42-a 5V-CUAUGCAGU-CACAGUUAUGTT-3Vand 5V-CAUACCUGUGACUGCAUAGTT-3V, for Cdc42-b5V-CUCACCACUGUCCAAAGACTT-3V and 5V-GUCUUUGGACAGUGGUGAG-TT-3V, for N-myc-a 5V-CACCAAGGCUGUCACCACATT-3Vand 5V-UGUGGU-GACAGCCUUGGUGTT-3V, for N-myc-b 5V-CCCAGACCUCGAGUUUGACTT-3Vand 5V- GUCAAACUCGAGGUCUGGGTT-3V, and for GFP 5V-GACCCGCGC-CGAGGUGAAGTT-3V and 3V-CUUCACCUCGGCGCGGGUCTT-3V.
Cells were grown to 30% confluency in 6-cm plates. RNA interferencewas done as described by Elbashir et al. (25). In short, 20 Amol/L per
single stranded RNA molecule in 30 AL annealing buffer [0.1 mol/L KAc,
30 mmol/L HEPES/KOH (pH 7.4), and 2 mmol/L MgAc] was heated at 95jCfor 5 minutes, followed by incubation at 37jC for 1 hour. The small
interfering RNA (siRNA) mixture was transfected serum-free with 30 ALLipofectamine (Invitrogen, Carlsbad, CA) as described by the manufacturer.
After 4 hours, the medium was supplemented with FCS to a final con-
centration of 10%.
For immunofluorescence analysis cells were grown on glass slides in6-well plates and transfected with 15 AL siRNA and 15 AL Lipofectamine.
After 72 hours, the cells were fixed with formaldehyde and stained with
TRITC-phalloidin (Sigma) and 4V,6-diamidino-2-phenylindole.
Results
Genes down-regulated by N-myc . We have applied the SAGEtechnique to identify downstream target genes of N-myc . Theneuroblastoma cell line SHEP has no N-myc amplification andexpression. A tetracycline-regulated N-myc expression vector hasbeen introduced into these cells, resulting in the SHEP-21N clone(8). SAGE libraries were constructed from N-myc expressingSHEP-21N cells and from SHEP-2 (empty vector control) cells (9).We identified 34,443 tags for SHEP-21N and 44,450 tags for SHEP-2, each representing one mRNA expressed in these cells. 112 Tagswere significantly (P < 0.01) down-regulated in the N-mycexpressing cells. We could reliably assign 78 of these tags to atotal of 64 genes (http://bioinfo.amc.uva.nl/HTMseq/controller;Table 1). We analyzed 14 genes by Northern blot, whichconfirmed the SAGE data (Fig. 1A). The overall expressionpattern showed that N-myc down-regulates many genes involvedin the architecture of the cell and the extracellular matrix.Cytoskeletal components like h-actin and vimentin are down-regulated, but also genes which regulate the structures, liketransgelin (TAGLN). The adhesion properties of cells depend onthe extracellular matrix and membrane molecules. Genes of bothgroups are repressed by N-myc . Several collagens are regulated byN-myc , but also connective tissue growth factor (CTGF) andSPARC , which regulate the build-up of the extracellular matrix. Inaddition, cadherin 11 (CDH11) and other genes encodingmembrane proteins are down-regulated.To investigate whether the genes were indeed down-regulated by
N-myc , we did Northern blot analysis of a time course experimentin which N-myc was switched off and on in the SHEP-21N cell line.The N-myc expression was switched off by tetracycline for 7 days,after which N-myc was switched on for another 7 days (Fig. 1B).Two distinct patterns of N-myc regulation emerged. Expression of alimited number of genes increased within 24 hours after N-myc wasturned off. This group includes galectin-1 (LGALS), S100A10 , andcaveolin-1 (CAV1 ; Fig. 1B). Reexpression of N-myc decreased theexpression of these genes (Fig. 1B). The majority of genes follow aslow N-myc dependent regulation. The expression of COL1A1 ,TAGLN , SPARC , FN1 , and SDC2 increased 4 days after N-myc wasswitched off (Fig. 1B). These analyses show that genes involved incell architecture and adhesion are important targets of down-regulation by N-myc . Most of these genes are slowly regulated andthus most likely represent secondary targets.Cdc42 expression is down-regulated by N-myc . One of the
genes identified as an N-myc downstream target is Cdc42 (Table 1).The SAGE-tag frequencies show that Cdc42 is expressed at a levelof 12.6 mRNAs per 20,000 mRNAs in SHEP-2 and 2.9 per 20,000 inSHEP-21N. Cdc42 is a member of the Rho GTPase subfamily andfunctions in a series of signaling pathways, which are particularlyimportant in cytoskeletal remodeling (26, 27). As many genes witha function in these processes are down-regulated by N-myc , Cdc42might be a key target of N-myc to control cytoskeletal remodeling.Northern blot analysis confirmed the decreased expression of
N-myc, Cdc42, and nm23 Cooperate in Neuroblastoma
www.aacrjournals.org 3137 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
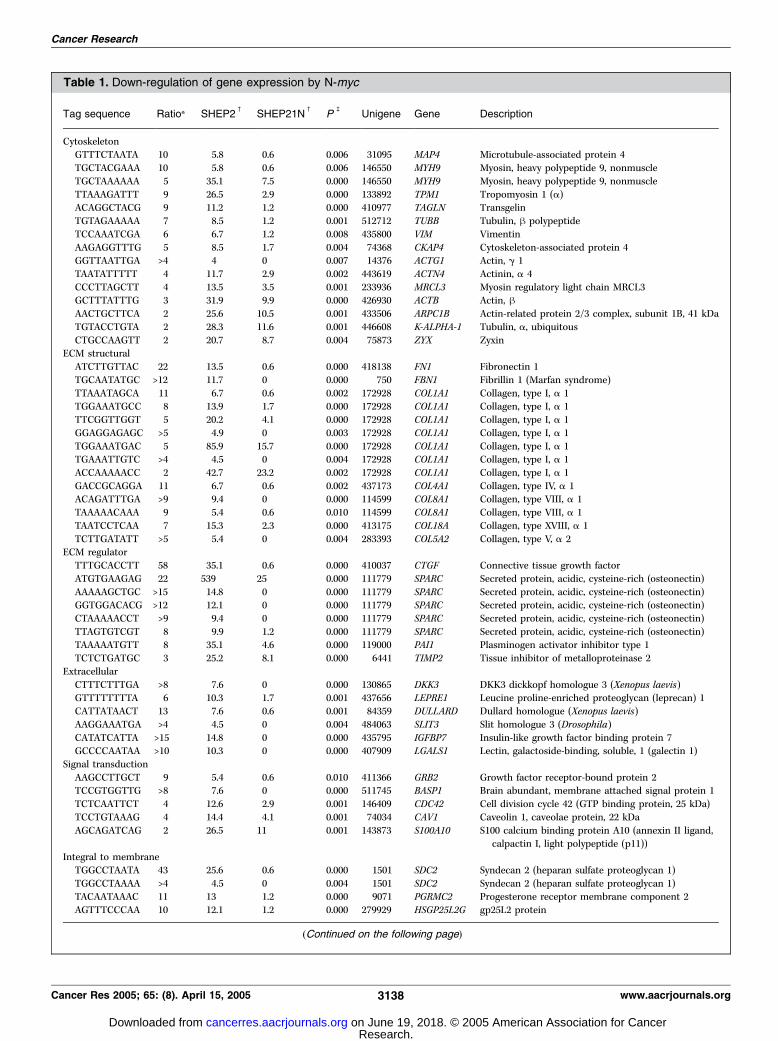
Table 1. Down-regulation of gene expression by N-myc
Tag sequence Ratio* SHEP2c
SHEP21Nc
Pb
Unigene Gene Description
CytoskeletonGTTTCTAATA 10 5.8 0.6 0.006 31095 MAP4 Microtubule-associated protein 4
TGCTACGAAA 10 5.8 0.6 0.006 146550 MYH9 Myosin, heavy polypeptide 9, nonmuscle
TGCTAAAAAA 5 35.1 7.5 0.000 146550 MYH9 Myosin, heavy polypeptide 9, nonmuscle
TTAAAGATTT 9 26.5 2.9 0.000 133892 TPM1 Tropomyosin 1 (a)ACAGGCTACG 9 11.2 1.2 0.000 410977 TAGLN Transgelin
TGTAGAAAAA 7 8.5 1.2 0.001 512712 TUBB Tubulin, h polypeptide
TCCAAATCGA 6 6.7 1.2 0.008 435800 VIM Vimentin
AAGAGGTTTG 5 8.5 1.7 0.004 74368 CKAP4 Cytoskeleton-associated protein 4GGTTAATTGA >4 4 0 0.007 14376 ACTG1 Actin, g 1
TAATATTTTT 4 11.7 2.9 0.002 443619 ACTN4 Actinin, a 4
CCCTTAGCTT 4 13.5 3.5 0.001 233936 MRCL3 Myosin regulatory light chain MRCL3GCTTTATTTG 3 31.9 9.9 0.000 426930 ACTB Actin, hAACTGCTTCA 2 25.6 10.5 0.001 433506 ARPC1B Actin-related protein 2/3 complex, subunit 1B, 41 kDa
TGTACCTGTA 2 28.3 11.6 0.001 446608 K-ALPHA-1 Tubulin, a, ubiquitous
CTGCCAAGTT 2 20.7 8.7 0.004 75873 ZYX ZyxinECM structural
ATCTTGTTAC 22 13.5 0.6 0.000 418138 FN1 Fibronectin 1
TGCAATATGC >12 11.7 0 0.000 750 FBN1 Fibrillin 1 (Marfan syndrome)
TTAAATAGCA 11 6.7 0.6 0.002 172928 COL1A1 Collagen, type I, a 1TGGAAATGCC 8 13.9 1.7 0.000 172928 COL1A1 Collagen, type I, a 1
TTCGGTTGGT 5 20.2 4.1 0.000 172928 COL1A1 Collagen, type I, a 1
GGAGGAGAGC >5 4.9 0 0.003 172928 COL1A1 Collagen, type I, a 1
TGGAAATGAC 5 85.9 15.7 0.000 172928 COL1A1 Collagen, type I, a 1TGAAATTGTC >4 4.5 0 0.004 172928 COL1A1 Collagen, type I, a 1
ACCAAAAACC 2 42.7 23.2 0.002 172928 COL1A1 Collagen, type I, a 1
GACCGCAGGA 11 6.7 0.6 0.002 437173 COL4A1 Collagen, type IV, a 1ACAGATTTGA >9 9.4 0 0.000 114599 COL8A1 Collagen, type VIII, a 1
TAAAAACAAA 9 5.4 0.6 0.010 114599 COL8A1 Collagen, type VIII, a 1
TAATCCTCAA 7 15.3 2.3 0.000 413175 COL18A Collagen, type XVIII, a 1
TCTTGATATT >5 5.4 0 0.004 283393 COL5A2 Collagen, type V, a 2ECM regulator
TTTGCACCTT 58 35.1 0.6 0.000 410037 CTGF Connective tissue growth factor
ATGTGAAGAG 22 539 25 0.000 111779 SPARC Secreted protein, acidic, cysteine-rich (osteonectin)
AAAAAGCTGC >15 14.8 0 0.000 111779 SPARC Secreted protein, acidic, cysteine-rich (osteonectin)GGTGGACACG >12 12.1 0 0.000 111779 SPARC Secreted protein, acidic, cysteine-rich (osteonectin)
CTAAAAACCT >9 9.4 0 0.000 111779 SPARC Secreted protein, acidic, cysteine-rich (osteonectin)
TTAGTGTCGT 8 9.9 1.2 0.000 111779 SPARC Secreted protein, acidic, cysteine-rich (osteonectin)TAAAAATGTT 8 35.1 4.6 0.000 119000 PAI1 Plasminogen activator inhibitor type 1
TCTCTGATGC 3 25.2 8.1 0.000 6441 TIMP2 Tissue inhibitor of metalloproteinase 2
Extracellular
CTTTCTTTGA >8 7.6 0 0.000 130865 DKK3 DKK3 dickkopf homologue 3 (Xenopus laevis)GTTTTTTTTA 6 10.3 1.7 0.001 437656 LEPRE1 Leucine proline-enriched proteoglycan (leprecan) 1
CATTATAACT 13 7.6 0.6 0.001 84359 DULLARD Dullard homologue (Xenopus laevis)
AAGGAAATGA >4 4.5 0 0.004 484063 SLIT3 Slit homologue 3 (Drosophila)
CATATCATTA >15 14.8 0 0.000 435795 IGFBP7 Insulin-like growth factor binding protein 7GCCCCAATAA >10 10.3 0 0.000 407909 LGALS1 Lectin, galactoside-binding, soluble, 1 (galectin 1)
Signal transduction
AAGCCTTGCT 9 5.4 0.6 0.010 411366 GRB2 Growth factor receptor-bound protein 2TCCGTGGTTG >8 7.6 0 0.000 511745 BASP1 Brain abundant, membrane attached signal protein 1
TCTCAATTCT 4 12.6 2.9 0.001 146409 CDC42 Cell division cycle 42 (GTP binding protein, 25 kDa)
TCCTGTAAAG 4 14.4 4.1 0.001 74034 CAV1 Caveolin 1, caveolae protein, 22 kDa
AGCAGATCAG 2 26.5 11 0.001 143873 S100A10 S100 calcium binding protein A10 (annexin II ligand,calpactin I, light polypeptide (p11))
Integral to membrane
TGGCCTAATA 43 25.6 0.6 0.000 1501 SDC2 Syndecan 2 (heparan sulfate proteoglycan 1)
TGGCCTAAAA >4 4.5 0 0.004 1501 SDC2 Syndecan 2 (heparan sulfate proteoglycan 1)TACAATAAAC 11 13 1.2 0.000 9071 PGRMC2 Progesterone receptor membrane component 2
AGTTTCCCAA 10 12.1 1.2 0.000 279929 HSGP25L2G gp25L2 protein
(Continued on the following page)
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3138 www.aacrjournals.org
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

Cdc42 mRNA in SHEP-21N compared with SHEP-2 (Fig. 2A).When N-myc expression in SHEP-21N was down-regulated bytetracycline, Cdc42 mRNA expression increased within 24 hours(Fig. 2A). Western blot analysis showed that also the Cdc42protein level increased after N-myc was switched off (Fig. 2B). Theregulation of Cdc42 was also investigated in cell line SKNAS-NmycER, which expresses a hybrid N-myc-estrogen receptorprotein that can be activated by 4-hydroxytamoxifen. TheSKNAS-NmycER cells were treated with 4-hydroxytamoxifen andthe Cdc42 mRNA expression decreased within 24 hours after N-mycactivation (Fig. 2C). The Cdc42 reduction was moderate (3-fold). Inaddition, we analyzed the expression of Cdc42 mRNA in a panel ofneuroblastoma tumors by Northern blot. Cdc42 expression wasreduced in 10 tumors with N-myc amplification compared with 10N-myc single copy tumors (Fig. 2D). The tumors show noexpression of c-myc (Fig. 2D). We conclude that Cdc42 mRNAexpression in neuroblastoma cells is down-regulated by N-myc ,resulting in decreased Cdc42 protein levels.The Cdc42 gene maps at 21.5 Mb from the 1p telomere, within
the 24.4 Mb region consistently deleted in N-myc amplifiedneuroblastomas (2). The net result is that Cdc42 is 50% reducedby deletion of one allele in N-myc amplified neuroblastomatumors, and the expression is further down-regulated by N-myc .
We therefore analyzed whether Cdc42 could function as a tumorsuppressor gene in neuroblastoma.Activated Cdc42 induces neuronal differentiation in SHEP-
21N cells. Cdc42 is a G-protein that is active in the GTP-boundstate and inactive in the GDP-bound state. We investigated the roleof Cdc42 in neuroblastoma by transient expression of the Cdc42protein coupled to a COOH-terminal FLAG tag (Cdc42flag). Wefirst transfected a mutant Cdc42 form that is permanently active(Cdc42-G12V). After 40 hours, 42% of the SHEP-21N cellsexpressing the Cdc42-G12Vflag protein showed neuronal differen-tiation, as evident by long neurite extensions of at least twice thelength of the cell body (Fig. 3A and B). The extensions wereanalyzed for expression of neurofilament, a marker for neuronaldifferentiation. The extensions in cells transfected with Cdc42-G12V show formation of neural fibers with strong expression of theneurofilament light chain (Fig. 3C). The differentiation was notobserved in untransfected cells. In addition, we transfected SHEP-21N with a wild-type Cdc42flag construct and with a dominant-negative Cdc42 mutant (Cdc42-T17N). Wild-type Cdc42flag andthe dominant-negative Cdc42-T17Nflag-transfected cells showedonly 3% and 6% differentiation, respectively (Fig. 3A and B),suggesting that the wild-type Cdc42 protein in SHEP-21N cells is inthe inactive GDP-bound state. However, when N-myc was switched
Table 1. Down-regulation of gene expression by N-myc (Cont’d)
Tag sequence Ratio* SHEP2c
SHEP21Nc
Pb
Unigene Gene Description
TCTTCTGCCA 6 9.4 1.7 0.002 86392 KIAA1402 CSGlcA-T KIAA1402 proteinTTCCTCCTTT >7 7.2 0 0.000 151155 TM6SF1 Transmembrane 6 superfamily member 1
TTACTTATAC >6 6.3 0 0.000 159 TNFRSF1A Tumor necrosis factor receptor superfamily, member 1A
TAAGAAAATG >6 5.8 0 0.001 443435 CDH11 Cadherin 11, type 2, OB-cadherin (osteoblast)
AGAACCTTAA >5 4.9 0 0.003 181244 HLA-A Major histocompatibility complex, class I, AGCCCTTTCTC 3 25.2 7.5 0.000 7835 MRC2 Mannose receptor, C type 2
GTTGTGGTTA 2 60.3 28.5 0.000 75415 B2M h-2-microglobulin
Transcription
TACAGAGGGA 8 9.4 1.2 0.000 406096 ZNF216 Zinc finger protein 216AAAAGATACT 6 7.6 1.2 0.003 82071 CITED2 Cbp/p300-interacting transactivator, with Glu/Asp-rich
COOH-terminal domain, 2
GCCATATTAT >5 5.4 0 0.002 170752 CRIM1 Cysteine-rich motor neuron 1CTCGAATAAA >4 4.5 0 0.004 34871 ZFHX1B Zinc finger homeobox 1b
Others
AATTTTCATT 10 5.8 0.6 0.006 339024 LOC253827 Hypothetical protein LOC253827
TTTTGTATTT 10 5.8 0.6 0.006 439190 C14orf31 Chromosome 14 open reading frame 31GGTGAAGACA 7 8.5 1.2 0.001 26951 RUSC2 RUN and SH3 domain containing 2
CCCTGATTTT 6 19.8 3.5 0.000 183684 EIF4G2 Eukaryotic translation initiation factor 4 g, 2
ATGTGAAGAA 6 6.7 1.2 0.008 13662 MGC5508 Hypothetical protein MGC5508
GAATAAATGT 6 7.2 1.2 0.005 497972 FKBP9 FK506 binding protein 9, 63 kDaGCTGTTTTGT >6 6.3 0 0.000 426312 AMOTL2 Angiomotin like 2
TTCCGGTTCC 5 9 1.7 0.003 172609 NUCB1 Nucleobindin 1
GTGGCTGAAA >5 4.9 0 0.003 401929 rpL10 ribosomal protein L10
CTTAATCCTG 4 30.6 8.1 0.000 298275 SLC38A2 Solute carrier family 38, member 2GAAATAATGG 4 18.4 4.6 0.000 343879 SPANXC SPANX family, member C
AAGGCAATTT 4 12.6 2.9 0.001 301626 FLJ11739 Homo sapiens cDNA FLJ11739 fis
AAGTGAAACA 4 10.3 2.9 0.006 93659 ERP70 Protein disulfide isomerase related protein(calcium-binding protein, intestinal-related)
TTAAAACAAA >4 4 0 0.01 370610 WAC WW domain containing adaptor with coiled coil
AAAACATTCT 2 67.5 32.5 0.00 323562 DKFZp564K142 DKFZp564K142 implantation-associated protein
*Fold down-regulation of SHEP-2/SHEP-21N.cThe expression in SHEP-2 and SHEP-21N is given in number of tags normalized per 20,000 tags.bPs were calculated with the Monte Carlo simulation of the SAGE software (40).
N-myc, Cdc42, and nm23 Cooperate in Neuroblastoma
www.aacrjournals.org 3139 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

off with tetracycline, also wild-type Cdc42flag-transfected cellsdisplayed neuronal differentiation (28%), whereas the dominant-negative Cdc42 construct remained inactive (Fig. 3B). This stronglysuggests that N-myc not only down-regulates the Cdc42 mRNAexpression level but also keeps the Cdc42 protein in its inactiveGDP-bound form.Nm23 suppresses the activity of Cdc42. Cycling between the
inactive GDP-bound and active GTP-bound state of Cdc42 isregulated by GTPase-activating proteins (GAP) and guaninenucleotide exchange factors. The inhibition of Cdc42-induceddifferentiation by N-myc indicated that N-myc controls a GAP orguanine nucleotide exchange factor of Cdc42. Previous experimentshave shown that N-myc strongly up-regulates the expression of thenm23-H1 and nm23-H2 genes in SHEP-21N (14). We confirmed theup-regulation of the nm23-H1 gene in the SKNAS-NmycER cell linetreated with 4-hydroxytamoxifen (Fig. 2C). In addition, in tumorswith N-myc amplification, nm23-H1 is also highly expressed(Fig. 2D).Nm23 proteins are primarily known to function as dinucleo-
side kinases, but a possible role for nm23-H1 in GDP and GTPexchange of Rad1 has been reported (20). We investigatedwhether the nm23-H1 and Cdc42 proteins can physically interactwith each other. For this purpose, we constructed theglutathione S-transferase (GST) fusion protein GST-nm23-H1.The GST-nm23-H1 protein was immobilized on glutatione-sepharose beads and incubated with a lysate of SHEP-21N cellstransfected with Cdc42. The beads were analyzed for boundproteins by Western blot. Cdc42 was pulled down by the GST-nm23-H1 protein and not by the GST protein alone (Fig. 3D). Asa control for unspecific protein binding in the pull-down assay,the Western blots were incubated with an N-myc antibody. N-myc did not bind to the GST or GST-nm23-H1 protein (Fig. 3D).We concluded that nm23-H1 and Cdc42 can physically interact.
We also analyzed the functional interaction of nm23-H1 andnm23-H2 with Cdc42. Transient transfection of a wild-typeCdc42-flag construct into SHEP-21N cells resulted in 8%differentiation of the transfected cells (Fig. 3E). When N-mycwas switched off in this experiment, the Cdc42flag-induceddifferentiation increased to 20% of the transfected cells. As N-myc down-regulation resulted in a decrease of nm23-H1 andnm23-H2 , we reconstituted these proteins by cotransfections.SHEP-21N cells in which N-myc expression was switched off bytetracycline were cotransfected with wild-type Cdc42flag andnm23-H1 and/or nm23-H2 expression vectors. Cotransfection ofeither nm23-H1 or nm23-H2 could completely replace the effectof N-myc , as the differentiation reduced from 20% to 6%(Fig. 3E). The percentage of differentiated cells is not furtherreduced by transfection of both nm23-H1 and nm23-H2 .Therefore, nm23-H1 and nm23-H2 prevent Cdc42-induceddifferentiation. As N-myc strongly up-regulates nm23-H1 andnm23-H2 , they are likely to mediate the effect of N-myc .Cdc42 RNA interference: Cdc42 is essential for cell division.
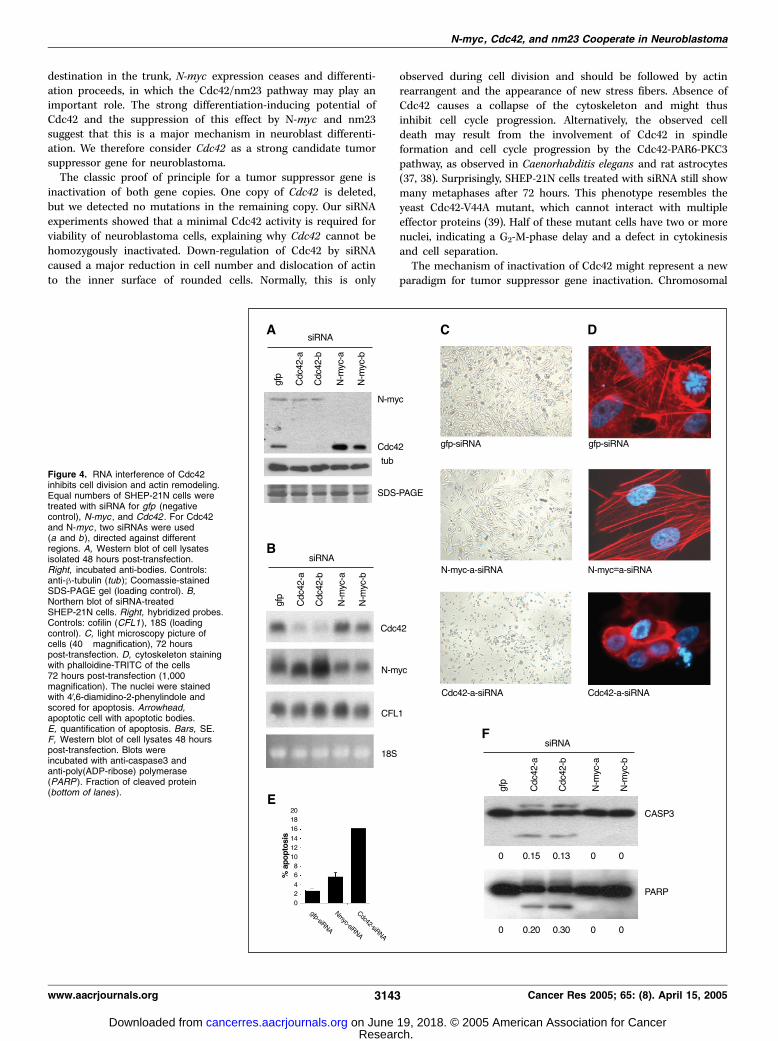
Neuroblastomas with N-myc amplification still have one Cdc42allele. The classic paradigm for tumor suppressor geneinactivation is deletion of one copy and mutation of the secondallele. We screened the coding region of Cdc42 for mutations inover 50 neuroblastoma tumors and cell lines but no mutationswere found. The Cdc42 expression in N-myc amplified tumorssuggests haplo-insufficiency as a potential mechanism fortumorigenesis. We further decreased the expression of Cdc42by RNA interference. The effect of two Cdc42-specific siRNAs wascompared with two N-myc-siRNAs, and siRNA for green fluores-cent protein, which has no target in the SHEP-21N cells. Westernblot and Northern blot analysis showed that 48 hours aftertransfection the expression of N-myc or Cdc42 was stronglyreduced by N-myc-siRNA and Cdc42-siRNA, respectively (Fig. 4A
Figure 1. Gene repression by N-myc . A, confirmation ofthe SAGE data. Northern blot analysis of 14 genes inSHEP-2 and SHEP-21N cells. Blot was also hybridizedwith N-myc (confirmation N-myc expression) andcofilin-1 (CFL1 , an equally expressed gene). The 28Sribosomal band as loading control. B, Northern blotanalysis of a time course experiment of N-myc .SHEP-21N cells were treated for 0 to 7 days withtetracycline, which switches off the ectopic N-mycexpression. After 7 days, the cells were grown for a weekin the absence of tetracycline (7+/7�). Right, analyzedgenes.
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3140 www.aacrjournals.org
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

and B). Cell growth of the N-myc-siRNA treated cells wasstrongly reduced compared with the gfp-siRNA control (Fig. 4C).Silencing of Cdc42 had an even more dramatic effect on cell growth.The majority of SHEP-21N cells treated with Cdc42-siRNA diedwithin 72 hours after transfection (Fig. 4C). In contrast, cells treatedwith gfp- or N-myc-siRNA survived the treatment. The experimentswere repeated on glass slides and the actin cytoskeleton and nucleiwere stained 72 hours after transfection. In the absence of Cdc42,the actin was organized in a peripheral membrane-associated ring,indicating that these cells were unable to rearrange the cytoskeletonafter cell division (Fig. 4D). The cells transfected with Cdc42-siRNAshowed 16% apoptotic nuclei, compared with 2% and 5% in thegfp-siRNA and N-myc-siRNA–treated cells, respectively (Fig. 4E).The apoptosis was further analyzed on Western blot. The cellswere treated for 48 hours with siRNAs and the lysates werescreened for cleaved proteins. In cells treated with Cdc42-siRNAs,both caspase3 and poly(ADP-ribose) polymerase showed theapoptosis-induced cleavage products (Fig. 4F ). No cleavedproducts were visible after treatment with gfp- or N-myc-siRNA.Therefore, Cdc42 is an essential gene for the growth and survival ofneuroblastoma cells. This provides an explanation for the absenceof Cdc42 mutations: a minimal amount of Cdc42 is required forcell survival.
Discussion
Cellular transformation involves increased proliferation, alter-ations in cell shape and adhesion. The N-myc protein regulatesgenes involved in these processes. Here, we have shown that N-myc represses genes determining the cell structure. The N-myctargets include structural genes but also one of the keyregulators Cdc42. Cdc42 is a member of the Rho GTPasesubfamily and functions in a series of signaling pathways,
which are particularly important in cytoskeletal remodeling (26,27). It has a major role in actin reorganization, as Cdc42-GTPbinds to WASP, which in turn binds and activates the Arp2/3complex (28). This induces the branching of actin filaments andthe formation of the cytoskeleton. Several genes functioning inthis process are regulated by N-myc . N-myc not only down-regulates Cdc42 , but also actin (ACTG1 and ACTB) and ARPC1B(Table 1). Furthermore, Cdc42 has been shown to mediatelaminin-dependent differentiation in PC12 cells and murineneuroblastoma cells (29–31). Here, we have shown thatreconstitution of active Cdc42 allowed differentiation of humanneuroblastoma cells. The Cdc42 expression and activity isdown-regulated at three different levels in N-myc amplifiedneuroblastoma cells (Fig. 5). First, the expression is 50%reduced by loss of heterozygosity of 1p36, as it maps in the1p region consistently deleted in N-myc amplified neuroblasto-mas. Second, a further 4- to 5-fold down-regulation of Cdc42mRNA is caused by the overexpression of N-myc . Together, thisresults in an estimated 8- to 10-fold down-regulation at themRNA level. Third, N-myc inhibits the differentiation inducedby the Cdc42 protein. This effect of N-myc is fully reconstitutedby transfection of nm23-H1 and nm23-H2 constructs, whichstrongly suggests that the N-myc effect is mediated by thenm23 proteins.Regulation of the nm23-H1 and nm23-H2 genes in neuroblas-
toma is a mirror image of the Cdc42 regulation. The nm23 genesmap in the minimal region of 17q gain and are present withtwo to eight extra copies. The expression of these nm23 copiesis 6- to 10-fold up-regulated by N-myc , resulting in a 12- to 80-fold overexpression of nm23-H1 and -H2 (14). Therefore, theratio between Cdc42 and nm23 protein levels is probably at least100-fold out of balance. The differentiation-blocking effect of N-myc is thus amplified by chromosomal imbalances of the
Figure 2. Cdc42 is down-regulated by N-mycin cell lines and tumors. A, Northern blot analysisof a time course experiment of N-myc . SHEP-21Ncells were treated for 0 to 6 days with tetracycline,which switches off the ectopic N-myc expression.Lane 1, total RNA of SHEP-2 cells. The 28Sribosomal band as loading control. B, Western blotanalysis of N-myc and Cdc42 proteins in a similartime course experiment in SHEP-21N. Part of theCoomassie-stained SDS-PAGE as loading control.C, Northern blot of SKNAS-N-mycER cells treatedfor 0 to 3 days with 4-hydroxytamoxifen (4OHT ),which activates the N-mycER protein. Expressionof Cdc42 and nm23-H1 mRNA. D, Northern blotanalysis of N-myc , nm23-H1 , Cdc42 , andc-myc expression of 10 N-myc -amplified and10 N-myc single-copy tumors.
N-myc, Cdc42, and nm23 Cooperate in Neuroblastoma
www.aacrjournals.org 3141 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

downstream mediators, Cdc42 and nm23-H1 and nm23-H2.Moreover, neuroblastomas frequently have an unbalancedtranslocation between chromosomes 1p and 17q, causing lossof heterozygosity of 1p and gain of 17q (32, 33). This would, by asingle hit, disturb the balance between Cdc42 and the nm23genes. The translocation event is often followed by duplicationof the derivative chromosome 1 with the extra 17q material,further increasing the imbalance. Amplification of N-myc wouldboost the effect of the imbalance, by down-regulation of theremaining Cdc42 copy and up-regulation of the overrepresentednm23 genes. The down-regulation of Cdc42 and up-regulation ofthe nm23 genes by N-myc is very consistent in neuroblastomacell lines and tumors. In the two cell lines in which we canmanipulate N-myc activity, Cdc42 is down-regulated and thenm23 genes are up-regulated. Furthermore, comparison of apanel of 10 N-myc amplified tumors and 10 N-myc single copyneuroblastomas strongly supports an identical effect of N-mycon Cdc42 and the nm23 genes in vivo (this article and ref. 14).The nm23 proteins could function as a GAP for Cdc42, keeping
Cdc42 in the inactive, GDP-bound state. Several lines of evidencesupport this role for nm23-H1. Direct GAP activity of nm23-H1was shown for the Rad1 GTPase in melanoma cells (20).Involvement in the regulation of Cdc42-family member Rac1was established as well. Nm23-H1 is recruited toward adhesivejunctions by ARF6-GTP (34). At these junctions, nm23-H1negatively regulates TIAM1 and subsequently Rac1 is inactivated
(21). In this study, we showed that nm23-H1 can bind to theCdc42 protein and nm23 can inhibit the Cdc42-induced differen-tiation. These data suggest that nm23-H1 and nm23-H2 also exertsGAP-activity for Cdc42.Is Cdc42 the elusive neuroblastoma suppressor gene at distal
chromosome 1p? If Cdc42 indeed functions as a tumorsuppressor gene, it would be unlikely to be the only 1p suppressorgene in neuroblastoma. Deletions of 1p in N-myc amplifiedneuroblastomas are at least 24.4 Mb. If Cdc42 , which maps at 21.5Mb from the telomere, would be the one and only suppressorgene, a certain frequency of interstitial deletions encompassingCdc42 would be expected. The length of the deletions haspreviously raised the idea that more than one suppressor gene isinvolved (5). Thus far, only one homozygous 1p36 deletion wasidentified in a neuroblastoma cell line (35). The scarcity ofhomozygous 1p36 deletions in N-myc amplified neuroblastomashas suggested that the postulated tumor suppressor gene(s) mayfunction by haploinsufficiency. Our data suggest that Cdc42 mayrepresent the first example of such a gene. The chromosome 17qregion gained in neuroblastoma tumors is also very large.Therefore, in addition to the nm23 genes, other 17q genes maycontribute to neuroblastoma pathogenesis as well.Neuroblastomas are embryonal tumors that originate from
neural crest–derived cells. During normal embryogenesis N-myc isexpressed in migrating neuroblasts (36) and probably preventspremature differentiation. When neuroblasts arrive at their
Figure 3. Neuronal differentiation ismediated by active Cdc42 andcounteracted by nm23-H1. SHEP-21Ncells were grown without and withtetracycline (tet) and transfected withflag-tagged Cdc42, Cdc42-G12V, orCdc42-T17N. Cells were fixed 40 hoursafter transfection and stained with ananti-FLAG antibody to identify transfectedcells. A, examples of an undifferentiated(Cdc42-T17N transfected) anddifferentiated (Cdc42-G12V transfected)cell. B, differentiation frequencies in over500 flag-positive cells per transfectedconstruct. Bars, SE. *, P < 0.01 (v2 test).C, expression of neurofilament (NFL ) upondifferentiation in SHEP-21N cells positivefor Cdc42-G12V-flag expression. Notethe nodules of the neural structures arenegative for neurofilament. D, Cdc42 andnm23-H1 physically interact. SHEP-21Ncells were transfected with Cdc42. Thelysates were incubated with fusionprotein GST-nm23H1 (lanes 1 and 2)or GST (lanes 3 and 4) linked togluthatione-sepharose beads andanalyzed for bound Cdc42 protein(pull-down) on Western blot. Part of thelysate was analyzed for Cdc42 expression.Blots were stripped and incubated withanti-N-myc . E, analysis of neuronaldifferentiation by Cdc42 in the presence orabsence of nm23. The SHEP-21N cellswere grown in the absence or presenceof tetracycline and transfected withflag-tagged Cdc42 in combination withempty vector, nm23-H1 or nm23-H2.Cells were fixed 40 hours after transfectionand stained with an anti-flag antibodyto identify transfected cells. Approximately1,000 flag-positive cells were scoredfor neuronal differentiation. Bars,SE. *, P < 0.01 (v2 test).
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3142 www.aacrjournals.org
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
destination in the trunk, N-myc expression ceases and differenti-ation proceeds, in which the Cdc42/nm23 pathway may play animportant role. The strong differentiation-inducing potential ofCdc42 and the suppression of this effect by N-myc and nm23suggest that this is a major mechanism in neuroblast differenti-ation. We therefore consider Cdc42 as a strong candidate tumorsuppressor gene for neuroblastoma.The classic proof of principle for a tumor suppressor gene is
inactivation of both gene copies. One copy of Cdc42 is deleted,but we detected no mutations in the remaining copy. Our siRNAexperiments showed that a minimal Cdc42 activity is required forviability of neuroblastoma cells, explaining why Cdc42 cannot behomozygously inactivated. Down-regulation of Cdc42 by siRNAcaused a major reduction in cell number and dislocation of actinto the inner surface of rounded cells. Normally, this is only
observed during cell division and should be followed by actinrearrangent and the appearance of new stress fibers. Absence ofCdc42 causes a collapse of the cytoskeleton and might thusinhibit cell cycle progression. Alternatively, the observed celldeath may result from the involvement of Cdc42 in spindleformation and cell cycle progression by the Cdc42-PAR6-PKC3pathway, as observed in Caenorhabditis elegans and rat astrocytes(37, 38). Surprisingly, SHEP-21N cells treated with siRNA still showmany metaphases after 72 hours. This phenotype resembles theyeast Cdc42-V44A mutant, which cannot interact with multipleeffector proteins (39). Half of these mutant cells have two or morenuclei, indicating a G2-M-phase delay and a defect in cytokinesisand cell separation.The mechanism of inactivation of Cdc42 might represent a new
paradigm for tumor suppressor gene inactivation. Chromosomal
Figure 4. RNA interference of Cdc42inhibits cell division and actin remodeling.Equal numbers of SHEP-21N cells weretreated with siRNA for gfp (negativecontrol), N-myc , and Cdc42 . For Cdc42and N-myc , two siRNAs were used(a and b), directed against differentregions. A, Western blot of cell lysatesisolated 48 hours post-transfection.Right, incubated anti-bodies. Controls:anti-h-tubulin (tub ); Coomassie-stainedSDS-PAGE gel (loading control). B,Northern blot of siRNA-treatedSHEP-21N cells. Right, hybridized probes.Controls: cofilin (CFL1), 18S (loadingcontrol). C, light microscopy picture ofcells (40� magnification), 72 hourspost-transfection. D, cytoskeleton stainingwith phalloidine-TRITC of the cells72 hours post-transfection (1,000�magnification). The nuclei were stainedwith 4V,6-diamidino-2-phenylindole andscored for apoptosis. Arrowhead,apoptotic cell with apoptotic bodies.E, quantification of apoptosis. Bars, SE.F, Western blot of cell lysates 48 hourspost-transfection. Blots wereincubated with anti-caspase3 andanti-poly(ADP-ribose) polymerase(PARP ). Fraction of cleaved protein(bottom of lanes ).
N-myc, Cdc42, and nm23 Cooperate in Neuroblastoma
www.aacrjournals.org 3143 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

regions that consistently show hemizygosity or moderate copynumber gains are frequent in human tumors. For many of them,no tumor suppressor genes or oncogenes have been identified asyet. Here we show that these relatively limited changes in copynumbers can dramatically amplify the oncogenic effect of anactivated pathway, in this case the N-myc pathway. Importantly,in this model the actual tumor suppressor gene does not haveto be completely inactivated. Cdc42 is not fully destroyed likeclassic tumor suppressor genes but can be reactivated. Thisoffers a clinical perspective, as innovative drugs may interfere
in the pathway and restore the function of the remaining Cdc42allele.
Acknowledgments
Received 7/10/2004; revised 1/4/2005; accepted 1/31/2005.Grant support: Dutch Cancer Society (L.J. Valentijn and A. Koppen).The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank K. Berns for providing the pBSK-ER plasmid and J. Collard for the GST-plasmid.
Figure 5. Model for the down-regulationof Cdc42 activity by N-myc , loss ofheterozygosity of chromosomal band 1p36,and gain of the chromosome 17q arm.
References1. Bown N. Neuroblastoma tumour genetics: clinical andbiological aspects. J Clin Pathol 2001;54:897–910.
2. Spieker N, Beitsma M, Van Sluis P, Chan A, Caron H,Versteeg R. Three chromosomal rearrangements inneuroblastoma cluster within a 300-kb region on1p36.1. Genes Chromosomes Cancer 2001;31:172–81.
3. Bauer A, Savelyeva L, Claas A, Praml C, Berthold F,Schwab M. Smallest region of overlapping deletion in1p36 in human neuroblastoma: a 1 Mbp cosmid andPAC contig. Genes Chromosomes Cancer 2001;31:228–39.
4. Caron H, Spieker N, Godfried M, et al. Chromosomebands 1p35-36 contain two distinct neuroblastomatumor suppressor loci, one of which is imprinted. GenesChromosomes Cancer 2001;30:168–74.
5. Versteeg R, Caron H, Cheng NC, et al. 1p36: everysubband a suppressor? Eur J Cancer 1995;3:538–41.
6. Schwab M, Alitalo K, Klempnauer KH, et al. AmplifiedDNA with limited homology to myc cellular oncogene isshared by human neuroblastoma cell lines and aneuroblastoma tumour. Nature 1983;305:245–8.
7. Bernards R, Dessain SK, Weinberg RA. N-mycamplification causes down-modulation of MHC classI antigen expression in neuroblastoma. Cell 1986;47:667–74.
8. Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A,Schwab M. Conditional expression of N-myc in humanneuroblastoma cells increases expression of a-prothy-mosin and ornithine decarboxylase and acceleratesprogression into S-phase early after mitogenic stimula-tion of quiescent cells. Oncogene 1996;13:803–12.
9. Boon K, Caron HN, van Asperen R, et al. N-mycenhances the expression of a large set of genesfunctioning in ribosome biogenesis and protein syn-thesis. EMBO J 2001;20:1383–93.
10. Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification
and analysis of c-MYC target genes. Proc Natl Acad SciU S A 2002;99:6274–9.
11. Coller HA, Grandori C, Tamayo P, et al. Expressionanalysis with oligonucleotide microarrays reveals thatMYC regulates genes involved in growth, cell cycle,signaling, and adhesion. Proc Natl Acad Sci U S A2000;97:3260–5.
12. Guo QM, Malek RL, Kim S, et al. Identification of c-myc responsive genes using rat cDNA microarray.Cancer Res 2000;60:5922–8.
13. Schuhmacher M, Kohlhuber F, Holzel M, et al.The transcriptional program of a human B cellline in response to Myc. Nucleic Acids Res 2001;29:397–406.
14. Godfried MB, Veenstra M, van Sluis P, et al. The N-myc and c-myc downstream pathways include thechromosome 17q genes nm23-H1 and nm23-H2.Oncogene 2002;21:2097–101.
15. Hennessy C, Henry JA, May FE, Westley BR,Angus B, Lennard TW. Expression of the antimeta-static gene nm23 in human breast cancer: anassociation with good prognosis. J Natl Cancer Inst1991;83:281–5.
16. Florenes VA, Aamdal S, Myklebost O, MaelandsmoGM, Bruland OS, Fodstad O. Levels of nm23 messengerRNA in metastatic malignant melanomas: inversecorrelation to disease progression. Cancer Res 1992;52:6088–91.
17. Steeg PS, Bevilacqua G, Kopper L, et al. Evidence for anovel gene associated with low tumor metastaticpotential. J Natl Cancer Inst 1988;80:200–4.
18. Oda Y, Naka T, Takeshita M, Iwamoto Y, TsuneyoshiM. Comparison of histological changes and changes innm23 and c-MET expression between primary andmetastatic sites in osteosarcoma: a clinicopathologicand immunohistochemical study. Hum Pathol 2000;31:709–16.
19. Leone A, Seeger RC, Hong CM, et al. Evidence fornm23 RNA overexpression, DNA amplification andmutation in aggressive childhood neuroblastomas.Oncogene 1993;8:855–65.
20. Zhu J, Tseng YH, Kantor JD, et al. Interaction of theRas-related protein associated with diabetes rad andthe putative tumor metastasis suppressor NM23provides a novel mechanism of GTPase regulation.Proc Natl Acad Sci U S A 1999;96:14911–8.
21. Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K,Sugimura H. Tumor metastasis suppressor nm23H1regulates Rac1 by interaction with Tiam1. Proc NatlAcad Sci U S A 2001;98:4385–90.
22. Fournier HN, Dupe-Manet S, Bouvard D, et al.Integrin cytoplasmic domain-associated protein 1a(ICAP-1a) interacts directly with the metastasissuppressor nm23-H2, and both proteins aretargeted to newly formed cell adhesion sitesupon integrin engagement. J Biol Chem 2002;277:20895–902.
23. Littlewood TD, Hancock DC, Danielian PS, ParkerMG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for theregulation of heterologous proteins. Nucleic Acids Res1995;23:1686–90.
24. Sander EE, van Delft S, ten Klooster JP, et al. Matrix-dependent Tiam1/Rac signaling in epithelial cellspromotes either cell-cell adhesion or cell migrationand is regulated by phosphatidylinositol 3-kinase. J CellBiol 1998;143:1385–98.
25. Elbashir SM, Harborth J, Lendeckel W, Yalcin A,Weber K, Tuschl T. Duplexes of 21-nucleotide RNAsmediate RNA interference in cultured mammalian cells.Nature 2001;411:494–8.
26. Price LS, Collard JG. Regulation of the cytoskeletonby Rho-family GTPases: implications for tumour cellinvasion. Semin Cancer Biol 2001;11:167–73.
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3144 www.aacrjournals.org
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

27. Etienne-Manneville S, Hall A. Rho GTPases in cellbiology. Nature 2002;420:629–35.
28. Welch MD. The world according to Arp: regulation ofactin nucleation by the Arp2/3 complex. Trends CellBiol 1999;9:423–7.
29. Van Leeuwen FN, Kain HE, Kammen RA, Michiels F,Kranenburg OW, Collard JG. The guanine nucleotideexchange factor Tiam1 affects the neuronal morphol-ogy; opposing roles for the small GTPases Rac and Rho.J Cell Biol 1997;139:797–807.
30. Sarner S, Kozma R, Ahmad S, Lim L. Phosphatidy-linosinositol 3-kinase, Cdc42, and Rac1 act downstreamof Ras in integrin-dependent neurite outgrowth in N1E-115 neuroblastoma cells. Mol Cell Biol 2000;20:158–72.
31. Weston CA, Anova L, Rialas C, Prives JM, Weeks BS.Laminin-1 activates Cdc42 in the mechanism oflaminin-1-mediated neurite outgrowth. Exp Cell Res2000;260:374–8.
32. Caron H, van Sluis P, van Roy N, et al.Recurrent 1;17 translocations in human neuroblas-toma reveal nonhomologous mitotic recombinationduring the S/G2 phase as a novel mechanism forloss of heterozygosity. Am J Hum Genet 1994;55:341–7.
33. Savelyeva L, Corvi R, Schwab M. Translocationinvolving 1p and 17q is a recurrent genetic alteration ofhuman neuroblastoma cells. Am J Hum Genet 1994;55:334–40.
34. Palacios F, Schweitzer JK, Boshans RL, D’Souza-Schorey C. ARF6-GTP recruits Nm23-H1 to facilitatedynamin-mediated endocytosis during adherens junc-tions disassembly. Nat Cell Biol 2002;4:929–36.
35. Ohira M, Kageyama H, Mihara M, et al. Identificationand characterization of a 500-kb homozygously deletedregion at 1p36.2-p36.3 in a neuroblastoma cell line.Oncogene 2000;19:4302–7.
36. Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H.Regulation of the neural crest cell fate by N-myc :promotion of ventral migration and neuronal differen-tiation. Development 1997;124:1953–62.
37. Gotta M, Abraham MC, Ahringer J. CDC-42 controlsearly cell polarity and spindle orientation in C. elegans .Curr Biol 2001;11:482–8.
38. Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3h and adenomatous polyposis coli to control cellpolarity. Nature 2003;421:753–6.
39. Richman TJ, Sawyer MM, Johnson DI. The Cdc42pGTPase is involved in a G2/M morphogenetic check-point regulating the apical-isotropic switch andnuclear division in yeast. J Biol Chem 1999;274:16861–70.
40. Velculescu VE, Zhang L, Vogelstein B, Kinzler KW.Serial analysis of gene expression. Science 1995;270:484–7.
N-myc, Cdc42, and nm23 Cooperate in Neuroblastoma
www.aacrjournals.org 3145 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:3136-3145. Cancer Res Linda J. Valentijn, Arjen Koppen, Ronald van Asperen, et al.
Genesnm23, and Cdc42, N-mycby Copy Number Defects of Inhibition of a New Differentiation Pathway in Neuroblastoma
Updated version
http://cancerres.aacrjournals.org/content/65/8/3136
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/8/3136.full#ref-list-1
This article cites 39 articles, 15 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/8/3136.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/65/8/3136To request permission to re-use all or part of this article, use this link
Research. on June 19, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from


















![Smoking and Drinking in Relation to ... - Cancer Researchcancerres.aacrjournals.org/content/canres/50/20/6502.full.pdf · [CANCER RESEARCH 50, 6502-6507. October 15, 1990] Smoking](https://img.dokumen.tips/doc/110x75/5b5c6a2d7f8b9ac6028c3131/smoking-and-drinking-in-relation-to-cancer-cancer-research-50-6502-6507.jpg)









