Embed Size (px)
Citation preview

PAPER www.rsc.org/dalton | Dalton Transactions
Rational design of 1-D metal–organic frameworks based on the novelpyrimidine-4,6-dicarboxylate ligand. New insights into pyrimidine throughmagnetic interaction†‡
Garikoitz Beobide, Oscar Castillo,* Antonio Luque, Urko Garcıa-Couceiro, Juan P. Garcıa-Teran andPascual Roman
Received 28th November 2006, Accepted 12th April 2007First published as an Advance Article on the web 24th April 2007DOI: 10.1039/b617348a
Single crystal X-ray analysis of compounds H2pmdc·2H2O (1), KHpmdc (2), and K2pmdc (3) showsthat the pyrimidine-4,6-dicarboxylate (pmdc) dianion presents an almost planar geometry whichconfers a potential capability to act as a bis-bidentate bridging ligand, and therefore, to construct 1-Dmetal complexes. Based on this assumption, we have designed the first six transition metal complexesbased on this ligand of formula {[M(l-pmdc)(H2O)2]·H2O}n [M(II) = Fe (4), Co (5), Ni (6), Zn (7), Cu(8)] and {[Cu(l-pmdc)(dpa)]·4H2O}n (9) (dpa = 2,2′-dipyridylamine). The crystal structure of all ofthese complexes has been determined by single crystal X-ray measurements, except for compound 6whose X-ray powder diffraction pattern reveals that it is isostructural to compounds 4–7. Thebis-chelating pmdc ligand bridges sequentially octahedrally coordinated M(II) centres leading topolymeric chains. The hexacoordination of the metal centres is completed by two water molecules incompounds 4–8 and by the two endocyclic-N atoms of a terminal dpa ligand in compound 9.Cryomagnetic susceptibility measurements show the occurrence of antiferromagnetic intrachaininteractions for compounds 4–6 and 8–9 (J = −2.5 (4), −5.2 (6), −32.7 (8), and −0.9 (9) cm−1). Modelcalculations and analyses of the available experimental data have been used to examine the influence ofseveral factors on the nature and magnitude of the magnetic coupling constants in pyrimidine bridgedcomplexes, showing that metal deviation from the pyrimidine mean plane could lead to ferromagneticbehaviour.
Introduction
In recent years the area of inorganic crystal engineering1 hasbecome one of intense research activity due to the growingneed for novel solid-state architectures with potential applicationsas functional materials in fields such as catalysis, conductivity,zeolitic behaviour and magnetism.2 The judicious choice of themetal ion, a good understanding of the coordination preferencesof the bridging entities, and a careful selection of the terminalligands are key steps for the rational design of metal–organiccoordination polymers with novel topologies and specific chemicaland physical properties.3 In this context, p-conjugated N-donorbridging ligands, such as diazines and their polycarboxylic deriva-tives have appeared to be a well suited tool for the construction of
Departamento de Quımica Inorganica, Facultad de Ciencia y Tecnologıa,Universidad del Paıs Vasco/Euskal Herriko Unibertsitatea, Apartado 644,E-48080, Bilbao, Spain. E-mail: [email protected]; Fax: +34 94 6013500; Tel: +34 94 601 5991† The HTML version of this article has been enhanced with colour images.‡ Electronic supplementary information (ESI) available: Experimentaland simulated XRPD patterns of compounds 5 and 6. Selected bondlengths and angles within the pmdc entity for 1–9. Hydrogen bondinginteraction data. Crystal packing of compounds 1–9. Thermogravimetriccurves (TG/DTA) of compound 8. vMT vs T plots for compounds 4–6 and8–12. Calculated magnetic coupling constants, structural parameters andspin density representations of the dimer models employed throughout thework. See DOI: 10.1039/b617348a
extended arrays of metal ions with interesting physical propertiesin molecular magnetism or selective guest adsorption fields.4,5
The synthesis and characterization of metal coordination poly-mers based on the pyrazine-2,3-dicarboxylato ligand has evolvedrather rapidly in recent years,5,6 yielding metal–organic frame-works (MOFs) of rather differing degrees of dimensionality. Theproperties and coordination features of metal complexes basedon the pyrazine-2,5-dicarboxylato ligand have been investigatedrather less .7,8 Recently, the use of the latter ligand has allowedus to achieve the controllable growth of discrete dimeric entitiesand one-dimensional polymeric chains in which the organic linkershows a bis-bidentate coordination mode.9 Nonetheless, crystalstructures showing covalent skeletons of higher dimensionalityhave been reported too.8 The judicious design of an organic-linkerby using an analogous pyrimidine derivative could open a newroute for the construction of multidimensional structures witha tailored topology based on the self-assembly approach. Withregard to this matter, the pyrimidine-4,6-dicarboxylate (pmdc)dianion appears to be an appropriate candidate. Carboxylationin the 4- and 6-positions of the pyrimidinic ring avoids the sterichindrance between the carboxylate groups and confers on thedianion a planar topology. This structural feature provides thepmdc with the potential capability to behave as a bis-chelatingligand, and therefore construct analogous systems to those basedon the pyrazine-2,5-dicarboxylato bridging ligand. As far as weare aware, since Hunt and coworkers10 reported the synthesis ofpyrimidine-4,6-dicarboxylic acid (H2pmdc) in 1959, no organic
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2669

or metal–organic crystal structure containing it in either dianion,anion or neutral form has been reported.
In the area of molecular magnetism, a lot of work has beendevoted to the study of the magnetic interactions between twoparamagnetic centres bridged by an aromatic ring.11,12 The analysisof the spin delocalization and polarization effects is helpful inorder to achieve a better understanding of the magnetic inter-actions and/or magneto-structural relationships in this kind ofmetal complexes. The spin polarization mechanism itself providesa key tool for predicting the kind of magnetic interactions inhigh-spin organic molecules and therefore, designing molecule-based magnets.13 Thus, in order to get the desired spin alignmentbetween two paramagnetic metal atoms, phenyl derivatives14 anddiazine-type bridging ligands15 have been widely studied due totheir analogy with the organic polyradicals based on m- or p-phenylene magnetic couplers. Nevertheless, the application of thisstrategy to transition metal complexes, which provide a more stablespin source than those based on organic radicals, is more tricky andthe spin delocalization effect must also be taken into account.16
Here we describe for the first time the crystal structure of thepmdc entity in its acid (H2pmdc), acid salt (KHpmdc), and neutralsalt (K2pmdc) forms along with the self-assembly, rational designand magnetic and structural characterization of a novel familyof one-dimensional metal complexes based on this dianion asbridging ligand. Hereafter, the influence of several factors onthe magnetic coupling between two paramagnetic metal centresthrough pyrimidine-type bridges is analyzed using theoreticalDFT calculations and available experimental data.
Experimental
Preparations
Synthesis of H2pmdc·2H2O (1), KHpmdc (2) and K2pmdc (3).The pmdc ligand was prepared strictly following the procedurespreviously reported,10 obtaining the anhydrous KHpmdc (2) acidsalt, instead of the diprotic species indicated by the authors.Recrystallization of compound 2 was carried out using a mixtureof water and 2-propanol (2 : 3) to give X-ray suitable colourlesscrystals. Compounds 1 and 3 were prepared by shifting the pHof an aqueous solution of 2 with HNO3 (pH = 1) and KOH(pH = 8.0), respectively. Single crystals of 1 were obtained by therecrystallization of the crude product in a mixture of water andmethanol (3 : 1), while compound 3 was crystallized by the slowdiffusion of 1-butanol into an aqueous solution of the neutral salt.
Compound 1. Yield: 60%. Anal. Calcd for C6H8N2O6: C, 35.29;H, 3.95; N, 13.73. Found: C, 35.25; H, 4.02; N, 13.79%. Main IRfeatures (cm−1, KBr pellet): 3480vs for m(O–H); 3100w for m(C–H); 1710vs for mas(O–C–O); 1549m, 1479w for m(C=C + C=N);1310vs, 1281vs for m(Car–C); 1166m, 1164m, 1052w for dip(C–H);919w for dring; 802w for d(O–C–O); 692m for dop(C–H); 534w forp(CO2).
Compound 2. Yield: 75%. Anal. Calcd for C6H3KN2O4: C,34.96; H, 1.47; N, 13.60; K, 18.92. Found: C, 34.90; H, 1.36; N,13.53; K, 18.85%. Main IR features (cm−1, KBr pellet): 3486vs form(O–H); 3108w for m(C–H); 1702vs for mas(O–C–O); 1541, 1477wfor m(C=C + C=N); 1372w for m(Car–C); 1308vs, 1276vs for ms(O–C–O); 1171m, 1035m for dip(C–H); 915w for dring; 818m for d(O–
C–O); 689m for sring; 624m for dop(C–H); 544w for p(CO2); 425 form(M–O + M–N).
Compound 3. Yield: 70%. Anal. Calcd for C6H2K2N2O4: C,29.52; H, 0.83; N, 11.48; K, 31.95. Found: C, 29.56; H, 0.94; N,11.53; K, 31.91%. Main IR features (cm−1, KBr pellet): 3440vs form(O–H); 3102w for m(C–H); 1627vs for mas(O–C–O); 1582s, 1529mfor m(C=C + C=N); 1449w for m(Car–C); 1364s for ms(O–C–O);1289w, 1195w, 1071w, 1004w for dip(C–H); 933w for dring; 818wfor d(O–C–O); 720s, 711m for sring; 498w for dop(C–H); 413w form(M–O + M–N).
Synthesis of {[M(l-pmdc)(H2O)2]·H2O}n [M = Fe (4), Co(5), Ni (6), Zn (7), Cu (8)]. Compounds 4–8 were preparedby the slow diffusion of a water–methanolic solution of 2(0.0515 g, 0.250 mmol) into an aqueous solution of FeCl2
(0.0317 g, 0.250 mmol), Co(NO3)2·6H2O (0.0728 g, 0.250 mmol),Ni(NO3)2·4H2O (0.0725 g, 0.250 mmol), ZnCl2 (0.0341 g,0.250 mmol), or Cu(NO3)2·3H2O (0.0604 g, 0.250 mmol). Crystalgrowth of compounds 4, 5, 7, and 8 was observed 7 d later.Compound 6 was obtained as a green polycrystalline sample.
Compound 4. Yield: 60% (based on metal). Anal. Calcd forC6H8FeN2O7: C, 26.09; H, 2.92; N, 10.15; Fe, 20.27. Found: C,25.98; H, 3.05; N, 10.10; Fe, 20.32%. Main IR features (cm−1, KBrpellet): 3410vs, 3270sh for m(O–H); 3110w for m(C–H); 1662vs formas(O–C–O); 1604s, 1542m for m(C=C + C=N); 1475w for m(Car–C); 1360vs for ms(O–C–O); 1208w, 1111w, 1049w for dip(C–H);915w for dring; 844w for d(O–C–O); 742m for sring; 689m for dop(C–H); 493w for m(M–O + M–N).
Compound 5. Yield: 75%. Anal. Calcd for C6H8CoN2O7: C,25.81; H, 2.89; N, 10.04; Co, 21.20. Found: C, 25.60; H, 2.71; N,10.15; Co, 21.16%. Main IR features (cm−1, KBr pellet): 3410vsfor m(O–H); 2960w for m(C–H); 1667vs for mas(O–C–O); 1609s,1546m for m(C=C + C=N); 1475w for m(Car–C); 1360s for ms(O–C–O); 1204w, 1115w, 1016w for dip(C–H); 920w for dring; 849w ford(O–C–O); 750m for sring; 693m for dop(C–H); 404w for m(M–O +M–N).
Compound 6. Yield: 70%. Anal. Calcd for C6H8N2NiO7: C,25.90; H, 2.90; N, 10.08; Ni, 20.84. Found: C, 25.99; H, 2.79; N,10.15; Ni, 20.76%. Main IR features (cm−1, KBr pellet): 3420vs,3320sh for m(O–H); 1671vs for mas(O–C–O); 1609s, 1546s for m(C=C+ C=N); 1476w for m(Car–C); 1364s for ms(O–C–O); 1209m, 1120w,1062w for dip(C–H); 911w for dring; 844w for d(O–C–O); 755m forsring; 689m for dop(C–H); 511w, 413w for m(M–O + M–N).
Compound 7. Yield: 75%. Anal. Calcd for C6H8N2O7Zn: C,25.36; H, 2.84; N, 9.86; Zn, 22.51. Found: C, 25.42; H, 2.95; N,9.79; Zn, 22.48%. Main IR features (cm−1, KBr pellet): 3415vs,3310sh for m(O–H); 3115w for m(C–H); 1673vs for mas(O–C–O);1610s, 1543s for m(C=C + C=N); 1443w for m(Car–C); 1360vs forms(O–C–O); 1204m, 1113w, 1057w for dip(C–H); 920w for dring;850w for d(O–C–O); 750m for sring; 690m for dop(C–H); 603w forp(CO2); 513w, 497w for m(M–O + M–N).
Compound 8. Yield: 80%. Anal. Calcd for C6H8CuN2O7: C,25.45; H, 2.85; N, 9.90; Cu, 22.24. Found: C, 25.56; H, 2.70; N,10.00; Cu, 22.27%. Main IR features (cm−1, KBr pellet): 3456vsfor m(O–H); 3111w for m(C–H); 1689vs for mas(O–C–O); 1617m,1551m for m(C=C + C=N); 1410w for m(Car–C); 1347s for ms(O–C–O); 1208w, 1098w, 1026w for dip(C–H); 920w for dring; 849wfor d(O–C–O); 760m for sring; 733w, 684m for dop(C–H); 595w forp(CO2); 520w, 436w for m(M–O + M–N).
2670 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007

Synthesis of {[Cu(l-pmdc)(dpa)]·4H2O}n (9). An aqueous so-lution (10 mL) of 3 (0.0610 g, 0.250 mmol) was added dropwiseinto a mixture of Cu(NO3)2·3H2O (0.0604 g, 0.250 mmol) anddpa (0.0428 g, 0.250 mmol) dissolved in 10 mL of water and10 mL of methanol to give an immediate green polycrystallinepowder of compound 9. After filtering off the precipitate, theresulting solution was allowed to stand at room temperature.Suitable prismatic crystals for X-ray analysis were obtained 2weeks later. Yield: 70%. Anal. Calcd for C16H19CuN5O8: C, 40.67;H, 4.06; N, 14.83; Cu, 13.33. Found: C, 40.73; H, 4.15; N, 14.91;Cu%, 13.24. Main IR features (cm−1, KBr pellet): 3430vs form(O–H + N–H); 3080w, 3020w for m(C–H); 1657vs, 1631vs formas(O–C–O); 1600m, 1587m, 1547m, 1476vs for m(C=C + C=N);1418w for m(Car–C); 1355s for ms(O–C–O); 1262w, 1235w, 1200w,1173w, 1155w for dip(C–H); 1013m for dring; 844w for d(O–C–O); 773w, 737m for sring; 697w for dop(C–H); 533w for m(M–O +M–N).
Physical measurements
Elemental analyses (C, H, N) were performed on a LECO CHNS-932 microanalytical analyzer. Metal content was determinedby absorption spectrometry. The IR spectra (KBr pellets) wererecorded on a FTIR Mattson 1000 spectrometer in the 4000–400 cm−1 spectral region. Thermal analyses (TG/DTG/DTA)were performed on a TA Instruments SDT 2960 thermal analyzerin a synthetic air atmosphere (79% N2 : 21% O2) with a heatingrate of 5 ◦C min−1. Magnetic measurements were performed onpolycrystalline samples of the compounds with a Quantum DesignSQUID susceptometer covering the temperature range of 5.0–300 K at 0.1 T. The susceptibility data were corrected for thediamagnetism estimated from Pascal’s tables,17 the temperature-independent paramagnetism, and the magnetization of the sampleholder. X-Ray powder diffraction data (XRPD) were collected ona Phillips X’PERT powder diffractometer with Cu-Ka radiation insteps of 0.03◦ over the 2h 10–70◦ range with a fixed-time countingof 40 s at 293 K.
Single-crystal X-ray data collection and structure determination
Data collections on single crystals of compounds 1–5 and 7–9 were carried out at 293 K with an Xcalibur diffractometerequipped with an area detector and graphite monochromatedMo-Ka radiation (k = 0.71073 A). The data processing was donewith the CrysAlis RED program.18 Information concerning datacollection is summarized in Table 1. The structures were solvedby direct methods using the SIR97 program.19 Full matrix least-squares refinements were performed on F 2 using SHELXL97.20
All non-hydrogen atoms were refined anisotropically. All cal-culations were performed using the WinGX crystallographicsoftware package.21 The final geometrical calculations and thegraphical manipulations were carried out with the PARST9522
and PLATON23 programs. Although compound 2 can be solvedin the centrosymmetric C2/c space group, the hydrogen atom ofthe carboxylic group can only be unequivocally located if the lowersymmetry Cc space group is employed. The C–O bond distancesare consistent with this assignment. Compound 8 suffers animmediate partial dehydration after filtering off the mother liquoras has been confirmed by thermogravimetric measurements (seeESI‡). Therefore, the occupation factor of the crystallization watermolecule has been refined and kept fixed at the final refinementcycles at a value of 0.2. The XRPD pattern of compound 6resembles nicely the simulated one from the single-crystal structureof the {[M(l-pmdc)(H2O)2]·H2O}n series (see ESI‡). However,because of the low crystallinity of compound 6 a successfulRietveld refinement on the basis of the space group, cell parametersand atomic coordinates found for the single-crystal structures ofthe {[M(l-pmdc)(H2O)2]·H2O}n series could not be performed.
CCDC reference numbers 629014–629048.For crystallographic data in CIF or other electronic format see
DOI: 10.1039/b617348a
Computational details
DFT methods have been shown to give good estimates of the mag-netic interactions.24 A detailed description of the computational
Table 1 Single-crystal data and structure refinement details of compounds 1–5 and 7–9
1 2 3 4 5 7 8 9
Empiricalformula
C6H8N2O6 C6H3KN2O4 C6H2K2N2O4 C6H8FeN2O7 C6H8CoN2O7 C6H8N2O7Zn C6H6.4CuN2O6.2 C16H19CuN5O8
Formula weight 204.14 206.21 244.30 275.99 279.07 285.51 269.28 472.90Crystal system Triclinic Monoclinic Orthorhombic Monoclinic Monoclinic Monoclinic Monoclinic MonoclinicSpace group P1 Cc Pnma P21/c P21/c P21/c C2/c P21/ca/A 6.0866(5) 4.3290(10) 6.8143(3) 7.3475(5) 7.279(1) 7.2850(10) 6.977(2) 9.928(1)b/A 7.3670(10) 12.360(2) 18.1270(8) 12.1276(4) 12.095(2) 12.167(2) 12.234(3) 16.232(1)c/A 10.6488(8) 14.060(2) 6.3397(2) 12.3570(6) 12.185(3) 12.254(2) 12.008(3) 13.412(1)a/◦ 99.559(9) 90 90 90 90 90 90 90b/◦ 94.174(7) 94.51(2) 90 120.973(4) 120.21(2) 120.290(10) 120.44(3) 115.14(1)c /◦ 111.730(13) 90 90 90 90 90 90 90V/A3 432.68(9) 750.0(2) 783.10(5) 944.10(9) 927.1(4) 937.9(3) 883.7(5) 1956.6(3)Z 2 4 4 4 4 4 4 4Dobs/g cm−3 1.56(1) 1.82(1) 2.06(1) 1.94(1) 1.99(1) 2.01(1) 2.02(1) 1.60(1)Dcalc/g cm−3 1.567 1.826 2.072 1.942 2.000 2.022 2.020 1.605l/mm−1 0.142 0.688 1.195 1.623 1.877 2.645 2.487 1.172R1a 0.0601 0.0341 0.0409 0.0368 0.0396 0.0334 0.0409 0.0348wR2b 0.1433 0.0633 0.0905 0.0870 0.0798 0.0694 0.0819 0.0755
a R1 = ∑(‖F o‖ − |F c‖)/
∑|F o|. b wR2 = [
∑w(‖F o| − |F c‖)2/
∑w|F o|2]1/2.
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2671

strategy adopted in this work to compute the magnetic couplingconstant (Jcalc.) values has been described elsewhere.25 Densityfunctional theory is used to carry out two separate calculationsto evaluate the coupling constant of each compound. The onecalculation is for determining the high-spin state and the otherone is for determinig the low-spin broken symmetry state. Thecorrectness of the latter state has been ensured by means of its spindensity distribution. The hybrid B3LYP method26 was used in allcalculations as implemented in Gaussian-03,27 so that the exactHartree–Fock-type exchange was mixed with Becke’s expressionfor the exchange functional28 and the Lee–Yang–Parr correlationfunctional29 was used. The Gaussian implemented 6-31G(d) basisset has been employed throughout this work.
Two different types of isolated dimer models have beenemployed. The first one uses coordinates taken directly fromthe X-ray structure solution removing the remaining atoms,whereas the second type is a simplified dimer model which hasthe terminal ligands replaced by ammonia or water molecules,and the pyrimidine-4,6-dicarboxylato bridging ligand by a non-substituted pyrimidine molecule. The distance and angle param-eters used in the latter model are rounded mean values obtainedfrom the CSD structural database.30 Calculated J values, graphicalspin density representations and employed structural parametersare gathered in the ESI.‡
Results and discussion
Description of the structures
H2pmdc·2H2O (1), KHpmdc (2) and K2pmdc (3). Single-crystalX-ray analyses of the acid form (1), acid salt (2) and neutral salt (3)of pmdc allowed us to study for the first time its structural featuresin the solid state. In all of them the molecular entity (Fig. 1) isbasically planar or deviates slightly from planarity (dihedral anglebetween the pyrimidine ring and carboxylate groups ranges from2.1 to 11.4◦) which confers a potential ability to behave as a bis-chelating ligand and therefore, form a five-membered chelate ringwith each coordinated metal atom by means of one pyrimidine-N atom and one carboxylate-O atom. Bond distances and angles
Fig. 1 ORTEP drawings of the pmdc entity in compounds 1 (a), 2 (b) and3 (c) showing the immediate environments (ellipsoids at 50% probabilitylevel).
within the pmdc entity (see ESI‡) are comparable with those foundin carboxylic derivatives of other diazines.31,32
{[M(l-pmdc)(H2O)2]·H2O}n [M = Fe (4), Co (5), Zn (7), Cu(8)]. Compounds 4, 5 and 7 are isostructural and crystallize inthe P21/c space group, while compound 8 crystallizes in the C2/cspace group. All of them consist of one-dimensional chains, inwhich trans-[M(H2O)2]2+ units are sequentially bridged by bis-bidentate pmdc ligands (Fig. 2). The metal centre exhibits adistorted octahedral coordination environment, and it forms twofive-membered chelate rings with two symmetrically related pmdcligands, showing a significantly greater bite angle value for theCu complex [83.4(1)◦] than for the Fe, Co and Zn complexes (ca.76–79◦) because of the shorter bond distances between the Cu(II)atom and the donor atoms of the pmdc ligand (Table 2). Themetal coordination sphere is completed by two water moleculeswith noticeably longer bond distances for compound 8 [2.465(3)A] than those found in compounds 4, 5 and 7 (ca. 2.11 A), dueto the usual Jahn–Teller effect of octahedral copper(II) complexes.The M · · · M distance through the l-pyrimidine bridge is greaterfor the Fe(II) complex (6.181 A) than for the Co(II), Zn(II) andCu(II) complexes (6.093, 6.129 and 6.004 A, respectively), as canbe expected due to its greater ionic radius. In all cases, the M(II)atom shows a small displacement from the mean plane of thebridging ligand (ca. 0.09 A). In fact, the pmdc ligand is essentiallyplanar with the dihedral angles between the pyrimidine ring andcarboxylate groups ranging from 2.5 to 5.5◦.
The crystal building of these compounds is sustained by anintricate network of hydrogen bonding interactions involving thewater molecules and the carboxylate-O atoms (see ESI‡).
{[Cu(l-pmdc)(dpa)]·4H2O}n (9). The crystal structure of com-pound 9 is comprised of crystallization water molecules and [Cu(l-pmdc)(dpa)]n polymeric chains in which the [Cu(dpa)]2+ units aresequentially linked by cis-arranged bis-bidentate pmdc bridgeswith a twisting angle of ca. 81.0◦ (Fig. 3). The Cu(II) atom is locatedat the centre of an elongated octahedron as result of a pronouncedJahn–Teller effect (Table 3). The equatorial plane consists of two Natoms from a chelating dpa ligand [bite angle = 91.23(7)◦] and twocarboxylate-O atoms from two symmetry related pmdc ligands,and it forms dihedral angles with the pmdc bridge of ca. 83.0 and62.3◦. The two apical positions are occupied by two pyrimidine-N atoms belonging to two consecutive bridging ligands. Thisaxial–axial bridging mode of the l-pyrimidine ring results in anintrachain M · · · M distance of 6.748 A, much greater than thosefound in the above described compounds 4–8. The presence of achelating bulky ligand around the Cu(II) atom induces the twistingof the carboxylate groups (12.29 and 11.56◦) and as a consequence,it promotes a significant deviation of the metal centre from themean plane of the pmdc ligand (0.739 and 0.346 A). As canbe observed, each copper atom forms two five-membered chelaterings with two surrounding pmdc ligands. Because of the Jahn–Teller effect, the bite angle value can vary substantially dependingon the direction of the elongation. In the case of compound 8,the elongation takes places in the direction of the terminal ligandsleading to a bite angle of 83.4(1)◦. On the contrary, in compound9 the elongation occurs in the direction of the donor atoms of thebridging ligand which results in a noticeable decrease of the biteangles [77.04(7) and 74.84(6)◦].
2672 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007

Fig. 2 Perspective drawings of a fragment of the one-dimensional chain, (a) for compounds 4, 5 and 7, and (b) for compound 8 (ellipsoids at 50%probability level).
Table 2 Selected bond lengths (A) and angles (◦) in compounds 4, 5, 7 and 8a
4 5 7 8
M1–O1W 2.103(1) 2.070(2) 2.113(2) Cu1–O1W 2.465(3)M1–O2W 2.115(1) 2.101(2) 2.129(2) Cu1–N1 1.978(3)M1–N3a 2.122(1) 2.070(2) 2.076(2) Cu1–O51 1.946(2)M1–O71 2.128(1) 2.080(2) 2.094(2) N1–Cu1–N1b 180M1–N1 2.135(1) 2.084(2) 2.089(2) N1–Cu1–O51 83.45(10)M1–O81a 2.163(1) 2.113(2) 2.125(2) N1–Cu1–O51b 96.55(10)O1w–M1–O2w 175.91(5) 177.17(8) 175.54(8) N1–Cu1–O1w 87.83(11)O1w–M1–N3a 90.94(5) 91.40(9) 91.90(9) N1–Cu1–O1wb 92.17(11)O2w–M1–N3a 92.63(5) 90.73(9) 91.55(9) O51–Cu1–O51b 180O1W–M1–O71 91.47(4) 91.34(9) 92.35(9) O51–Cu1–O1w 85.40(9)O2W–M1–O71 89.83(4) 90.12(8) 89.94(9) O51–Cu1–O1wb 94.60(9)N3–M1–O71 100.51(4) 100.02(8) 98.38(8) O1w–Cu1–O1wb 180O1w–M1–N1 86.73(5) 88.07(9) 86.40(9)O2w–M1–N1 89.79(5) 89.84(9) 90.27(9)N3a–M1–N1 176.51(4) 178.52(9) 176.93(10)O71–M1–N1 76.97(4) 78.61(8) 79.15(8)O1w–M1–O81a 94.31(4) 92.34(9) 92.00(9)O2w–M1–O81a 84.62(4) 86.31(8) 85.92(9)N3–M1–O81a 76.21(4) 77.61(8) 78.52(8)O71–M1–O81a 173.38(4) 175.67(8) 174.74(9)N1–M1–O81a 106.54(4) 103.78(8) 104.08(8)
a Symmetry code: (a) x, −y − 1/2, z − 1/2. (b) −x + 2, −y, −z + 1.
In the supramolecular structure, each polymeric chain issurrounded by another six resembling a two-dimensional closepacking model. Crystallization water molecules are arranged asfour-membered discrete chains. Each water cluster is comprisedof four crystallographically distinguishable water molecules and itacts as a bridge among three neighboring coordination polymericchains by means of hydrogen bonding interactions, providing thecohesiveness to the supramolecular architecture.
To conclude the structural section, we would like to discusssome of the most relevant results of the research work reportedherein. In a Cambridge Structural Database (CSD, release august2006)30 search of transition metal complexes based on diazineligands with at least one coordinated N atom, 401, 109 and80 entries were found for pyrazine, pyrimidine and pyridazine,respectively. Furthermore, when the diazine ring acts as bridgingligand by means of its two endocyclic N atoms the number
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2673
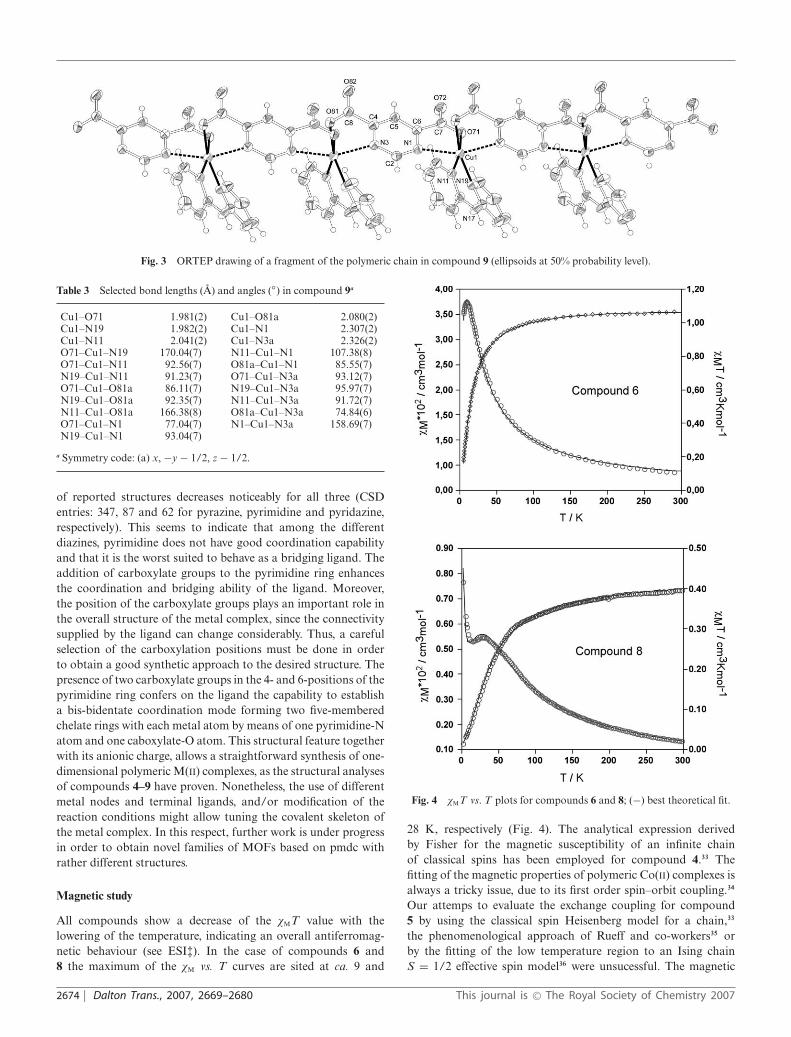
Fig. 3 ORTEP drawing of a fragment of the polymeric chain in compound 9 (ellipsoids at 50% probability level).
Table 3 Selected bond lengths (A) and angles (◦) in compound 9a
Cu1–O71 1.981(2) Cu1–O81a 2.080(2)Cu1–N19 1.982(2) Cu1–N1 2.307(2)Cu1–N11 2.041(2) Cu1–N3a 2.326(2)O71–Cu1–N19 170.04(7) N11–Cu1–N1 107.38(8)O71–Cu1–N11 92.56(7) O81a–Cu1–N1 85.55(7)N19–Cu1–N11 91.23(7) O71–Cu1–N3a 93.12(7)O71–Cu1–O81a 86.11(7) N19–Cu1–N3a 95.97(7)N19–Cu1–O81a 92.35(7) N11–Cu1–N3a 91.72(7)N11–Cu1–O81a 166.38(8) O81a–Cu1–N3a 74.84(6)O71–Cu1–N1 77.04(7) N1–Cu1–N3a 158.69(7)N19–Cu1–N1 93.04(7)
a Symmetry code: (a) x, −y − 1/2, z − 1/2.
of reported structures decreases noticeably for all three (CSDentries: 347, 87 and 62 for pyrazine, pyrimidine and pyridazine,respectively). This seems to indicate that among the differentdiazines, pyrimidine does not have good coordination capabilityand that it is the worst suited to behave as a bridging ligand. Theaddition of carboxylate groups to the pyrimidine ring enhancesthe coordination and bridging ability of the ligand. Moreover,the position of the carboxylate groups plays an important role inthe overall structure of the metal complex, since the connectivitysupplied by the ligand can change considerably. Thus, a carefulselection of the carboxylation positions must be done in orderto obtain a good synthetic approach to the desired structure. Thepresence of two carboxylate groups in the 4- and 6-positions of thepyrimidine ring confers on the ligand the capability to establisha bis-bidentate coordination mode forming two five-memberedchelate rings with each metal atom by means of one pyrimidine-Natom and one caboxylate-O atom. This structural feature togetherwith its anionic charge, allows a straightforward synthesis of one-dimensional polymeric M(II) complexes, as the structural analysesof compounds 4–9 have proven. Nonetheless, the use of differentmetal nodes and terminal ligands, and/or modification of thereaction conditions might allow tuning the covalent skeleton ofthe metal complex. In this respect, further work is under progressin order to obtain novel families of MOFs based on pmdc withrather different structures.
Magnetic study
All compounds show a decrease of the vMT value with thelowering of the temperature, indicating an overall antiferromag-netic behaviour (see ESI‡). In the case of compounds 6 and8 the maximum of the vM vs. T curves are sited at ca. 9 and
Fig. 4 vMT vs. T plots for compounds 6 and 8; (−) best theoretical fit.
28 K, respectively (Fig. 4). The analytical expression derivedby Fisher for the magnetic susceptibility of an infinite chainof classical spins has been employed for compound 4.33 Thefitting of the magnetic properties of polymeric Co(II) complexes isalways a tricky issue, due to its first order spin–orbit coupling.34
Our attemps to evaluate the exchange coupling for compound5 by using the classical spin Heisenberg model for a chain,33
the phenomenological approach of Rueff and co-workers35 orby the fitting of the low temperature region to an Ising chainS = 1/2 effective spin model36 were unsucessful. The magnetic
2674 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007

Table 4 Best-fit values for compounds 4–6, 8 and 9, and computed magnetic coupling constants
M(II) Compound vMT (300 K) Ja/cm−1 n2 × J g R × 105 Model Jcalc.b/cm−1 n2 × Jcalc.
Fe 4 3.27 −2.5 −40.0 2.15 0.03 Chain33 — —Co 5 2.34 — — — — — −1.7 −15.3Ni 6 1.07 −5.2 −20.8 2.07 0.01 Chain37 −5.0 −20.0Cu 8 0.39 −32.7 −32.7 2.14 0.01 Chain38 −19.5 −19.5Cu 9 0.46 −0.9 −0.9 2.22 0.25 Chain38 — —
a The spin Hamiltonian is defined by H = −J∑
Si × Si +1. b Convergence not reached for compounds 4 and 9.
data of compounds 6 and 8–9 have been fitted by numericalexpressions proposed for S = 1 and S = 1/2, respectively.37,38
The best fit values are shown in Table 4. Taking into accountthe structural features of the complexes, their magnetic datahave been fitted to the appropriate magnetic model. The modelused for each compound and the best fit values are shown inTable 4. The inclusion of a molecular field term to account forthe intermolecular interactions gave almost negligible values anddid not improve the fit. The magnetic interactions between theparamagnetic centres in the complexes described herein take placemainly through the pyrimidine ring. As occurs in the analogousdicarboxylic derivatives of the pyrazine,9,39 the contribution ofthe carboxylato groups can be considered negligible due to thelong magnetic pathway involving these groups. In order to getdeeper insight into the magnetic properties of our compounds,DFT UB3LYP calculations have been performed on isolated dimermodels built from their crystal structures keeping the structuralparameters unchanged. The presence of a great number of high-energy-lying non-bonding orbitals from the carboxylate groupsmakes the calculation of the magnetic coupling constants (Jcalc.)difficult. The dimer model for 6 was constructed using that of5, but replacing the metal atom and setting the coordinationbond distances to the rounded mean values obtained from theCSD structural database for octahedral Ni(II) complexes (Ni–Npym, Ni–Ow and Ni–Ocarb = 2.087 A). In the case of the Nidimer model the obtained value fit fairly well the experimentalone as can be observed in Table 4, and even in the case ofcompound 8 the deviation is only about 13 cm−1 which is anacceptable difference for this kind of calculation.24 The calculatedspin-density distributions for the ground state (S = 0) are shownin Fig. 5.
A lot of work has been devoted to the study of the magneticcoupling between two paramagnetic centres bridged by a diazine-type ligand.11,12 In the case of pyrimidine complexes the ferro-or antiferromagnetic nature and the magnitude of the couplingconstant can be tuned by means of the topology of the bridgingor terminal ligands and the nature of the paramagnetic centre.For instance, in copper(II) complexes the relative orientation ofthe magnetic orbitals can be easily controlled by the appropriateselection of the terminal ligands. In this way, copper(II) complexeswith axial–axial and axial–equatorial coordination modes canshow very weak antiferromagnetic and ferromagnetic behaviours,respectively.40 This explains fairly well the small J value ofcompound 9 with an axial–axial topology. Copper(II) complexeswith equatorial–equatorial (eq–eq) coordination geometry showan antiferromagnetic behaviour with values of the J magneticcoupling constant ranging from −20 to −40 cm−1.41–47 In typicallyelongated octahedral copper(II) complexes, the magnetic orbital
Fig. 5 Calculated spin-density distribution of the ground state (S = 0)of (a) [Co2(l-pmdc)(pmdc)2(H2O)4]2−, (b) [Ni2(l-pmdc)(pmdc)2(H2O)4]2−,and (c) [Cu2(l-pmdc)(pmdc)2(H2O)4]2− with a surface threshold level of0.0015.
(eg) corresponds to a dx2−y2 which affords r-type symmetry aroundthe metal–ligand bond and the resulting dr–nr orbital overlapbetween the metal and coordinated N-atoms is essential for therealization of the antiferromagnetic interaction. On this assump-tion, the J value of compound 8 is reasonably well explained.Nonetheless, in V(IV) complexes the unpaired electron (d1) residesin a t2g orbital and as a consequence, dp–pp orbital overlap canbe afforded leading to a ferromagnetic interaction.48 Thus, thepyrimidine can behave as ferro- or antiferromagnetic couplerdepending on the nature of the two bridged magnetic orbitals.In the former case, the magnetic coupling can be interpreted interms of spin delocalization to the diazine-N atoms which allowsan appreciable through-space orbital-interaction between the twor(N) orbitals of the pyrimidine, promoting an antiferromagneticcoupling.49 In the latter case the exchange mechanism can beunderstood on the basis of a spin polarization mechanism, whichinduces an alternating spin density throughout the p electronsystem (p-pathway) and therefore favors a parallel alignment of themetal spins.48 Consequently, when only t2g magnetic orbitals arepresent, spin polarization is dominant and ferromagnetic coupling
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2675

takes place through the p-pathway (dp–pymp–dp), whereas foronly eg magnetic orbitals the spin delocalization to the molecularorbitals of the bridging ligand results in an antiferromagneticcoupling mainly via the r-pathway (dr–pymr–dr).
For paramagnetic centres with different numbers of unpairedelectrons, it is more appropriate to compare the values of n2 × Jinstead of those of J (n being the number of magnetic orbitals).43,50
Previous research efforts have stated that although for pyrimidinebridged complexes antiferromagnetic terms (eg–eg) usually prevail,the influence of ferromagnetic terms (t2g–t2g, eg–t2g) promote thedecrease of the n2 × J absolute values as the number of t2g unpairedelectrons increases and therefore, in pyrimidine bridged complexesthe order of the absolute n2 × J value should be 52 × J(Mn) <
42 × J(Fe) < 32 × J(Co) < 22 × J(Ni) < 12 × J(Cu).43,50 However,in the compounds presented herein, this trend is not so clear(Table 4), and especially in the case of compound 4 which showsa greater n2 × J value than those for compounds 6 and 8 whichonly have eg unpaired electrons. This unusual behaviour for theiron(II) complex (4) seems to indicate that in this compound t2g–t2g terms are also antiferromagnetic. This striking fact inducedus to analyze the structural features which could explain it. Theonly significant difference within the chain, compared with theisomorphous compounds 5–8, is the greater M–N distance for theFe(II) complex (ca. 2.13 A for 4 vs. 1.978–2.084 A for 5–8). Inorder to analyze the effect of this M–N bond elongation in themagnetic interactions between the t2g orbitals, DFT calculationshave been performed on Cr(III) and V(IV) pyrimidine dimer modelsof formulas [Cr2(l-pym)(H2O)10]6+ and [(VO)2(l-pym)(H2O)6]4+,respectively (see ESI‡). The results are graphically representedin Fig. 6 and indicate that both ferromagnetic and antiferro-magnetic behaviours can be achieved depending on the M–Ndistance. Short distances allow the ferromagnetism to prevail andlonger ones give a weak antiferromagnetic coupling. Thereforethe greater |n2 × J| of compound 4 could be attributable tothe longer coordination bond distance of the pyrimidine-typebridge.
Fig. 6 Variation of the calculated coupling constants as a func-tion of the M–Npym bond distance for the [Cr2(l-pym)(H2O)10]6+ and[(VO)2(l-pym)(H2O)6]4+ dimer models.
The latter conclusion can be rationalized on the basis of theexpression proposed by Hay et al.51 for the singlet–triplet splitting
in a binuclear system with two unpaired electrons:
ES − ET = 2J = 2Kab − (e1 − e2)2
Jaa − Jab
(1)
where J is the coupling constant, and e1 and e2 are the energiesof φ1 and φ2, the single occupied molecular orbitals (SOMOs) ofthe complex. Kab, Jaa and Jab are two electron integrals involvingtwo localized versions of these orbitals, φA = N(φ1 + φ2) andφB = N(φ1 − φ2). The first term of eqn (1) is the ferromagneticcontribution to the magnetic exchange constant that accounts forthe stability of the triplet state, while the second one represents anantiferromagnetic term favouring a singlet ground state. The valueof Kab largely depends on the number of atoms (spacers) betweenthe two magnetic centres.16 When the bridging ligand has an evennumber of spacers Kab is close to zero as occurs for pyrazine-typemagnetic couplers, while for an odd number of spacers (pyrimidinefor example), Kab will be important and therefore ferromagneticbehaviour can be obtained if the antiferromagnetic term remainscomparatively small. Based on this second assumption, the mag-netic behaviour of pyrimidine bridged complexes has been usuallycompared with m-phenylene based organic radicals where the twoorbitals of the molecule interacting with the magnetic atomicorbitals are almost degenerate and the energy gap between thetwo SOMO’s will remain negligible. Therefore, the Kab exchangeintegral prevails and a ferromagnetic coupling is achieved. Thisis not the case of pyrimidine ligand, because the two molecularorbitals symmetry available to overlap with the metal centre showa substantial energy gap and therefore, the antiferromagnetic termcan not be disregarded and the net magnetic behaviour will resultfrom the balance of the two terms. Thus, taking into accounteqn (1) in the V(IV) pyrimidine dimeric model, a shorter V–N bond distance yields a greater value of the exchange integralKab,34 leading to a strengthening of the ferromagnetic interactions.When the distance is long enough, a marked lowering of Kab
takes place, and the antiferromagnetic term can prevail despitebeing relatively small. The calculated J values are comparable tothe reported experimental data48 (J = +3.1 cm−1, d(V–Npym) =2.163 A and J = +4.1 cm−1, d(V–Npym) = 2.145 A for compoundsof formulas [VO2(l-pym)(hfac)4] and [VO2(l-mpym)(hfac)4],respectively).
On the other hand, in a previous paper we analyzed the effectof the displacement of the metal atom from the pyrazine meanplane on the magnetic coupling constant.39 It was concludedthat the metal deviation allows the dr–pp overlap between themagnetic eg orbitals and the pyrazine bridge p-orbitals enablingthe p-pathway, which in this case leads to an increase of the anti-ferromagnetic coupling. Herein, we have analyzed the possibilityof an analogous behaviour for pyrimidine bridged complexes.Experimental data available nowadays indicate that those com-pounds with high metal atom deviations from the mean planeof the pyrimidine ring show weaker antiferromagnetic exchangeand in some cases even ferromagnetic couplings are observed(Table 5). Anyway, the available magnetic data for structurallycharacterized complexes having only pyrimidine-type bridgingligands are scarce. Therefore, in order to get better experimentalevidence for this behaviour, we have magnetically characterizeda family of compounds, with a high metal deviation from thepyrimidine mean plane, of formula [M(l-pym)(NO3)2(H2O)2]n
[M(II) = Mn (10), Co (11) and Ni (12)] (see ESI‡) whose synthesis
2676 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007

Table 5 J values, n2 × J values, and mean deviations of the metal atomsfrom the plane of the pyrimidine bridge (dM) for several complexes
Complexa J/cm−1 n2 × J/cm−1 dM/A Ref.
[Mn2(l-dppm)(hfac)4] −0.3 −7.5 0.108 43[Mn(l-pym)(NO3)2(H2O)2]n −0.2 −5.0 0.292 b
[Fe(l-pmdc)(H2O)2]n −2.5 −40.0 0.060 b
[Co2(l-dppm)(hfac)4] −2.2 −19.8 0.021 43[Co(l-pym)(NO3)2(H2O)2]n −1.0 −9.0 0.278 b
[Co(l-pym)(NCS)2]n >0 >0 0.429 44[Ni2(l-dppm)(hfac)4] −6.3 −25.2 0.027 43[Ni(l-pmdc)(H2O)2]n −5.2 −20.8 0.091c b
[Ni(l-pym)(NO3)2(H2O)2]n −4.0 −16. 0 0.232 b
[Cu(l-pman)(NO3)2(H2O)2]n −41.0 −41.0 0.094 45[Cu(l-pman)(NO3)2(MeOH)]n −35.6 −35.6 0.150 45[Cu(l-pmdc)(H2O)2]n −32.7 −32.7 0.041 b
[Cu2(l-dppm)(hfac)4] −32.0 −32.0 0.010 43[Cu(l-pym)(NO3)(dca)(H2O)]n −29.2 −29.2 0.154 46[Cu(l-pym)(NO3)2(H2O)2]n −25.0 −25.0 0.280 47
a dppm = bis(2-pyridyl)-4,6-pyrimidine; pman = 9-(5-pyrimidinyl) an-thracene b This work. c The dM value of [Ni(l-pmdc)(H2O)2]n correspondsto the mean value of the [M(l-pmdc)(H2O)2]n family of compounds.
and structure has been recently reported by Champness and co-workers.52 The magnetic behaviour of these compounds is thatof antiferromagnetically coupled M(II) centres with J (n2 × J) =−0.2 (−5.0), −1.0 (9.0) and −4.0 (16.0) cm−1 for compounds 10,11 and 12, respectively. In order to quantify the effect of the metaldeviation on the magnetic coupling constant, DFT calculationshave been performed on pyrimidine bridged homonuclear dimericmetal complexes of formulas [Cu2(l-pym)(H2O)6]4+ and [M2(l-pym)(H2O)10]4+ [M(II) = Mn, Fe, Co, Ni] with square planar andoctahedral coordination environments, respectively. The analyzedrange of metal deviation and the structural parameters employedin these models are rounded values close to those obtained asmean values from a CSD search. Fig. 7 shows the trend of n2 × Jobtained from the DFT calculations on dimer models togetherwith the available experimental data.
The calculated J values (see ESI‡) are comparable to the previ-ously reported experimental data40–44 and they indicate a continu-ous decrease of the antiferromagnetism, achieving a ferromagneticbehaviour for Mn, Fe, Co and Ni when deviations are greater thanca. 0.28–0.38 A (Fig. 7b). Experimental evidence for this trend isgiven by the complexes 10–12 and by the only structurally char-acterized ferromagnetic cobalt(II) pyrimidine bridged compoundof formula [Co(l-pym)2(NCS)2].44 In the case of 10–12 the metaldeviation ranges between 0.232 and 0.292 A, being still within theantiferromagetic region predicted by the DFT calculations, andthey show the lowest antiferromagnetic J values (Table 5). Onthe other hand, the deviation of compound [Co(l-pym)2(NCS)2](0.429 A) lies well within the predicted ferromagnetic region(Fig. 7b) as has also been experimentally observed. There is alsoanother example of a ferromagnetic pyrimidine-bridged cobalt(II)complex with formula [Co(l-pym)2(NCO)2] which is isostructuralto the previous one, but unfortunately no structural data areavailable.50
For the copper(II) model complex the decrease is less pro-nounced and ferromagnetic coupling has not been found withinthe analyzed range (Fig. 7a). In fact two different slopes areobserved, the first one corresponds to the Cu(II) model, and thesecond one, which is significantly steeper, is followed by Mn, Fe,
Fig. 7 Variation of the calculated coupling constants and distribution ofthe experimental data as function of the metal displacement from the meanplane of the pyrimidine ring (dM/A) for the (a) [Cu2(l-pym)(H2O)6]4+ and(b) [M2(l-pym)(H2O)10]4+ [M(II) = Mn, Fe, Co, Ni] dimer models.
Co, and Ni models. This seems to indicate that different magneticorbitals are playing a prominent role. In the case of the Cu(II)complexes it is obvious that the magnetic orbital implied is ofdx2−y2 character which is allowed by the deviation to establishdr–pp overlap, enabling the p-pathway (Fig. 8a). In the Ni(II)case the significantly greater slope is attributable to the presenceof a second eg magnetic orbital perpendicular to the pyrimidinemean plane which due to symmetry restrictions can not interactwith the pyrimidine p-orbital. However, the deviation of the metalatom relaxes this restriction allowing a net interaction with thep-pathway to be added to that described for copper(II). In thecase of Mn, Fe, and Co, one of the t2g magnetic orbitals (dxz inthe reference system of Fig. 9) can overlap effectively with the p-orbitals of the pyrimidine ring, but for this magnetic orbital theeffect of the metal lifting should not be so acute because althoughthe interaction increases on the upper side (in the direction ofthe deviation) it decreases on the bottom side (Fig. 8b). In the
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2677

Fig. 8 Schematic representation of the orbital interaction betweenpyrimidine p-orbitals and (a) eg and (b) t2g type orbitals showing the effectof the metal displacement.
Fig. 9 Arbitrary orientation of the magnetic orbitals in pyrimidinebridged compounds.
case of the other two t2g magnetic orbitals, due to their relativeorientation, metal elevation would lead to a negligible overlap withthe p-pathway (dxy, dyz). Therefore, the t2g magnetic orbitals shouldnot have a great influence on the observed trend of the magneticcoupling with the lifting of the metal center. It deserves a notethat a comparative study among the metal trends presents addeddifficulties due to the different orbital sizes, possible magneticorbital orientations, and coordination bond distances.
In this respect, we have analyzed the effect of the M–Npym
coordination bond distance together with the metal elevation onthe J value for dimer models of formulas [Ni2(l-pym)(H2O)10]4+
and [Cr2(l-pym)(H2O)10]6+. The results gathered in Fig. 10 showthat for the Ni(II) complex shorter bond distances yield greaterabsolute J values as can be expected for the r-pathway in theabsence of any deviation. Nonetheless, the elevation of the metalatom gives rise to an increasing slope with the shortening of thebond distance. This can be attributable to a more effective overlap
Fig. 10 Variation of the calculated coupling constants as a function ofthe metal displacement from the mean plane of the pyrimidine ring (dM)for [Ni2(l-pym)(H2O)10]4+ at different Ni–Npym bond distances (d1, d2, d3).
of the magnetic orbitals with the p-pathway for shorter bonddistances when the metal atom is lifted from the mean plane of thepyrimidine ring. The consequent strengthening of the p-pathwaypromotes a greater contribution of the ferromagnetic term, andthus the overall antiferromagnetism decays more rapidly forshorter coordination bond distances. In fact, in Cu(II) and Ni(II)complexes based on N,N ′-1,3-phenylenebis(oxamate) because ofthe almost perpendicular dihedral angle between the phenylenering and the oxamate groups, the exchange interaction takesplaces mainly through the p-system and thus, a ferromagneticcoupling is achieved as expected on the basis of a spin polarizationmechanism.53 The odd number of spacers involving the latterbridging ligand allows us to establish a certain analogy withpyrimidine, and thus it can be stated that the decrease of theoverall antiferromagnetism is in part due to the ferromagnetic con-tribution resulting from the interaction between the eg magneticorbitals and the p-system, although a weakening of the r-pathwaycould be involved as well. On the other hand, in the case of the[Cr2(l-pym)(H2O)10]6+ model where only t2g orbitals are present,shorter bond distances increase the ferromagnetism (Fig. 6), butthe elevation of the metal atom does not produce any significantvariation of the calculated J value nor a marked trend (dCr–N =1.900 A, |DJ| = 0.20 cm−1; dCr–N = 2.100 A, |DJ| = 0.01 cm−1;dCr–N = 2.300 A, |DJ| = 0.05 cm−1 for a metal displacement fromthe mean plane of the pyrimidine ring of ca. 0.37 A), being in goodconcordance with the above explanation for t2g magnetic orbitals(dxz, dxy and dyz).
In addition, DFT calculations over a dimer model of formula[Ni2(l-pym)(NH3)6]4+ show that the replacement of the watermolecules by ammonia molecules leads to a calculated J value of−10.2 cm−1, noticeably smaller than that for [Ni2(l-pym)(H2O)10]4+
(J = −17.3 cm−1). This is due to the greater withdrawal ofspin density from the metal centre by the ammonia ligandsthan that of the water molecules. Finally, the functionalizationof the pyrimidine bridging ligand can slightly modify the sym-metry adapted molecular orbitals of the diazine ring, and asa consequence the J value will vary. In this respect, we haveanalyzed the effect of the methylation (electron donor group)and carboxylation (electron withdrawing group) of pyrimidine inthe J value by using the [Cu2(l-5mpym)(H2O)6]4+ and [Cu2(l-5pymcH)(H2O)6]4+ dimer models (5mpym: 5-methylpyrimidine;5pymcH: 5-pyrimidinecarboxylic acid). The calculated J valuefor the methyl derivative (J = −33.5 cm−1) is greater than thatfor [Cu2(l-pym)(H2O)6]4+ (J = −31.5 cm−1), whereas for thecarboxylic derivative the calculated value is somewhat smaller (J =−28.5 cm−1). Therefore many of the disagreements between thecalculated theoretical trends and the experimental values observedin Fig. 7 can be attributed not only to the metal coordination bonddistances, but also to its coordination environment and to thefunctionalization of the pyrimidine bridge. In the case of the Cu(II)trend, there are two experimental values45 that lie considerablyhigher than the expected theoretical value for the model complex(Fig. 7a). This difference is probably due to the presence of ananthracene substituent in position 5 of the pyrimidine ring. Onthe other hand, for Mn, Fe, Co and Ni (Fig. 7b) there are fiveexperimental points that lie far below the theoretical trends. Inthese cases the coordination environments differ from that of thedimer models, and in addition in three of them43 the pyrimidinebridge is functionalized in positions 4 and 6 with two pyridine
2678 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007

groups, while the remaining two cases correspond to compounds 5and 6 based on the pyrimidine-4,6-dicarboxylato bridging ligand.It must be noted that in compounds 10–12, in which the pyrimidineis not functionalized, the experimental values agree fairly well withthe theoretical trends calculated for the modeled complexes andthat in all cases the trend is retained.
To conclude, it deserves to be emphasize that to rationalizethe nature and magnitude of the magnetic interaction, all theparameters studied above are necessary. First of all, it must betaken into account that in general the overall magnetic interactionresults from the balance between the r- and p-pathways when eg
and t2g magnetic orbitals are present, or when structural distortionallows the eg orbitals to interact with the pyrimidine p-system. Ithas been shown that the greater the metal deviation the smaller theantiferromagnetic interaction, and that ferromagnetic couplingcan be achieved if the deviation is high enough. Moreover, theshortening of the coordination bond distance plays an importantrole because it strengthens both the antiferromagnetic (eg–eg) andferromagnetic (t2g–t2g) interactions but also increases the ferro-magnetic contribution arising from the metal deviation. Thus, thepyrimidine ring can act as a potential ferro- or antiferromagneticcoupler depending on the structural features of the complex andthe nature of the magnetic orbitals involved. Nonetheless, theinfluence of the nature and position of the terminal ligands,and functionalization of the pyrimidine ring have an appreciablecontribution to the fine tuning of the overall magnetic interaction.
Acknowledgements
This work was supported by the Spanish Ministerio de Edu-cacion y Ciencia (MAT2005–03047). G. B. thanks the EuskoJaurlaritza/Gobierno Vasco for a doctoral fellowship (BF102.79).The SGI/IZO-SGIker UPV/EHU (supported by the NationalProgram for the Promotion of Human Resources within the Na-tional Plan of Scientific Research, Development and Innovation,Fondo Social Europeo and MCyT) is gratefully acknowledged forgenerous allocation of computational resources.
References
1 L. Brammer, Chem. Soc. Rev., 2004, 33, 476; D. Braga, L. Brammerand N. R. Champness, CrystEngComm, 2005, 7, 1.
2 S. L. James, Chem. Soc. Rev., 2003, 32, 276; C. Janiak, Dalton Trans.,2003, 2781.
3 B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40,2022; D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen and S. L.Price, CrystEngComm, 2002, 4, 500.
4 C. J. O’Connor, C. L. Klein, R. J. Majeste and L. M. Trefonas, Inorg.Chem., 1982, 21, 64.
5 M. Kondo, T. Okubo, A. Asami, S. I. Noro, T. Yoshitomi, S. Kitagawa,T. Ishii, H. Matsuzaka and K. Seki, Angew. Chem., Int. Ed., 1999, 38,140; R. Kitaura, K. Fujimoto, S. Noro, M. Kondo and S. Kitagawa,Angew. Chem., Int. Ed., 2002, 41, 133; D. Li and K. Kaneko, J. Phys.Chem. B, 2000, 104, 8940; T. K. Maji, K. Uemura, H.-C. Chang, R.Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 3269.
6 M. Gryz, W. Starosta, H. Ptasiewciz-Bak and J. Leciejewicz, J. Coord.Chem., 2003, 56, 1575; T. Premkumar and S. Govindarajan, Inorg.Chem. Commun., 2003, 6, 1385; J.-Z. Zou, Z. Xu, W. Chen, K. M. Loand X.-Z. You, Polyhedron, 1999, 18, 1507; R. Kitaura, S. Kitagawa,Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M.Kobayashi, H. C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M.Takata, Science, 2002, 298, 2358; Y. Kubota, M. Takata, R. Matsuda,R. Kitaura, S. Kitagawa, K. Kato, M. Sakata and T. C. Kobayashi,Angew. Chem., Int. Ed., 2005, 44, 920.
7 L. R. Harlow and S. H. Simonsen, Acta Crystallogr., Sect. B, 1974, 30,1370; H. Ptasiewicz-Bak and J. Leciejewicz, J. Coord. Chem., 1998, 44,299; H. Ptasiewicz-Bak and J. Leciejewicz, J. Coord. Chem., 1998, 44,237; G. Suss-Fink, L. Gonzalez-Cuervo, B. Therrien, H. Stoeckli-Evansand G. B. Shul’pin, Inorg. Chim. Acta, 2004, 357, 475.
8 G. Beobide, O. Castillo, A. Luque, U. Garcıa-Couceiro, J. P. Garcıa-Teran and P. Roman, Inorg. Chem. Commun., 2003, 6, 1224; H. T. Xu,N. W. Zheng, R. Y. Yang, Z. Q. Li and X. J. Jin, Inorg. Chim. Acta,2003, 349, 265; X.-J. Zheng and L.-P. Jin, J. Chem. Crystallogr., 2005,35, 865.
9 G. Beobide, O. Castillo, A. Luque, U. Garcıa-Couceiro, J. P. Garcıa-Teran and P. Roman, Inorg. Chem., 2006, 45, 5367.
10 R. R. Hunt, J. F. W. McOmie and E. R. Sayer, J. Chem. Soc., 1959, 525.11 M. Yasui, R. Takayama, N. Akiyama, D. Hashizume and F. Iwasaki,
Mol. Cryst. Liq. Cryst., 2002, 376, 519; Y. Takano, Y. Kitagawa, T.Onishi, Y. Yoshioka, K. Yamaguchi, N. Koga and H. Iwamura, J. Am.Chem. Soc., 2002, 124, 450.
12 J. S. Haynes, S. J. Rettig, J. R. Sams, R. C. Thompson and J. Trotter,Can. J. Chem., 1987, 65, 420; L. C. Francesconi, D. R. Corbin, A. W.Clauss, D. N. Hendrickson and G. D. Stucky, Inorg. Chem., 1981, 38,365; H. W. Richardson, J. R. Wasson and W. E. Hatfield, Inorg. Chem.,1977, 16, 484.
13 S. K. Silverman and D. A. Dougherty, J. Phys. Chem., 1993, 97, 13273;H. Iwamura, Adv. Phys. Org. Chem., 1990, 26, 179; A. Rajca, Chem.Rev., 1994, 94, 871.
14 T. Glaser, M. Gerenkamp and R. Frohlich, Angew. Chem., Int. Ed.,2002, 41, 3823; E. Pardo, J. Faus, M. Julve, F. Lloret, M. C. Munoz, J.Cano, S. Ottenwaelder, Y. Journaux, R. Carrasco, G. Blay, I. Fernandezand R. Ruiz-Garcıa, J. Am. Chem. Soc., 2003, 125, 10770.
15 Y. Takano, T. Onishi, Y. Kitagawa, T. Soda, Y. Yoshioka and K.Yamaguchi, Int. J. Quantum Chem., 2000, 80, 681; Y. Takano, H. Isobe,T. Kawakami and K. Yamaguchi, Mol. Cryst. Liq. Cryst., 2002, 379,531; S. Mitsubori, T. Ishida, T. Nogami, H. Iwamura, N. Takeda andM. Ishikawa, Chem. Lett., 1994, 685; A. M. W. Cargill Thompson,D. Gatteschi, J. A. McCleverty, J. A. Navas, E. Rentschler and M. D.Ward, Inorg. Chem., 1996, 35, 2701.
16 J. Cano, E. Ruiz, S. Alvarez and M. Verdaguer, Comments Inorg. Chem.,1998, 20, 27.
17 A. Earnshaw, Introduction to Magnetochemistry, Academic Press,London, 1968.
18 CrysAlis RED, version 1.170, Oxford Diffraction, Wroclaw, Poland,2003.
19 A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giaco-vazzo, A. Guagliardi, A. G. G. Moliterni and R. Spagna, J. Appl.Crystallogr., 1999, 32, 115.
20 G. M. Sheldrick, SHELXS97 and SHELXL97, University ofGottingen, Germany, 1997.
21 L. J. Farrugia, WINGX. A Windows program for crystal structureanalysis, University of Glasgow, UK, 1998.
22 M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659.23 A. L. Spek, PLATON, a multipurpose crystallographic tool, Utrecht
University, The Netherlands, 1998.24 J. Cano, P. Alemany, S. Alvarez, M. Verdaguer and E. Ruiz, Chem.–
Eur. J., 1998, 4, 476; M. D. Santana, G. Garcıa, M. Julve, F. Lloret,J. Perez, M. Liu, F. Sanz, J. Cano and G. Lopez, Inorg. Chem., 2004,43, 2132; E. Ruiz, A. Rodrıguez-Fortea and S. Alvarez, Inorg. Chem.,2003, 42, 4881.
25 E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999,20, 1391; E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem.Soc., 1997, 119, 1297; E. Ruiz, A. Rodrıguez-Fortea, J. Cano, S. Alvarezand P. Alemany, J. Comput. Chem., 2003, 24, 982; E. Rudberg, P. Salek,Z. Rinkevicius and H. Agren, J. Chem. Theory Comput., 2006, 2, 981.
26 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.27 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.
Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N.Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone,B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H.Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X.Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo,R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi,C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth,P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D.Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K.Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S.
This journal is © The Royal Society of Chemistry 2007 Dalton Trans., 2007, 2669–2680 | 2679

Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y.Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W.Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, RevisionB.04, Gaussian, Inc., Pittsburgh, PA, 2003.
28 A. D. Becke, Phys. Rev. A, 1998, 38, 3098.29 C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1998, 37, 785.30 F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380.31 P. Vishweshwar, A. Nangia and V. M. Lynch, J. Org. Chem., 2002, 67,
556.32 M. Tombul, K. Guven and N. Alkis, Acta Crystallogr., Sect. E, 2006,
62, 945.33 M. E. Fisher, Am. J. Phys., 1964, 32, 343; M. E. Fisher, J. Math. Phys.
(Melville, NY, U. S.), 1963, 4, 124.34 O. Kahn, Molecular Magnetism, VCH Publishers, Inc., New York,
1993.35 J.-M. Rueff, N. Masciocchi, P. Rabu, A. Sironi and A. Skoulios,
Eur. J. Inorg. Chem., 2001, 2843; J.-M. Rueff, N. Masciocchi, P. Rabu,A. Sironi and A. Skoulios, Chem.–Eur. J., 2002, 8, 1813.
36 M. E. Fisher, J. Math. Phys. (Melville, NY, U. S.), 1963, 4, 124.37 C. Y. Weng, Ph.D. Thesis, Carnegie Institute of Technology, 1968; A.
Meyer, A. Gleizes, J. J. Girerd, M. Verdaguer and O. Kahn, Inorg.Chem., 1982, 21, 1729.
38 J. W. Hall, Ph.D. Dissertation, University of North Carolina, 1977; J.Bonner and M. E. Fisher, Phys. Rev., 1964, 135, A640.
39 G. Beobide, O. Castillo, U. Garcıa-Couceiro, J. P. Garcıa-Teran, A.Luque, M. Martınez-Ripoll and P. Roman, Eur. J. Inorg. Chem., 2005,2586.
40 M. Yasui, Y. Ishikawa, T. Ishida, T. Nogami and F. Iwasaki, ActaCrystallogr., Sect. B, 2001, 57, 772.
41 T. Ishida, L. Yang and T. Nogami, Chem. Lett., 2003, 32, 1018.42 J. L. Manson, T. Lancaster, L. C. Chapon, S. J. Blundell, J. A. Schlueter,
M. L. Brooks, F. L. Pratt, C. L. Nygren and J. S. Qualls, Inorg. Chem.,2005, 44, 989.
43 T. Ishida, T. Kawakami, S. Mitsubori, T. Nogami, K. Yam-aguchi and H. Iwamura, J. Chem. Soc., Dalton Trans., 2002,3177.
44 F. Lloret, G. De Munno, M. Julve, J. Cano, R. Ruiz and A. Caneschi,Angew. Chem., Int. Ed., 1998, 37, 135.
45 T. Ezhura, K. Endo, K. Matsuda and Y. Aoyama, New J. Chem., 2000,24, 609.
46 J. L. Manson, J. Gu, J. A. Schlueter and H. H. Wang, Inorg. Chem.,2003, 42, 3950.
47 M. Yasui, Y. Ishikawa, N. Akiyama, T. Ishida, T. Nogami and F.Iwasaki, Acta Crystallogr., Sect. B, 2001, 57, 288.
48 T. Ishida, S. Mitsubori, T. Nogami, N. Takeda, M. Ishikawa and H.Iwamura, Inorg. Chem., 2001, 40, 7059.
49 F. Mohri, K. Yoshizawa, T. Yamabe, T. Ishida and T. Nogami, Mol.Eng., 1999, 8, 357.
50 F. Lloret, M. Julve, J. Cano and G. De Munno, Mol. Cryst. Liq. Cryst.,1999, 334, 569.
51 P. J. Hay, J. C. Thibeault and R. Hoffmann, J. Am. Chem. Soc., 1975,97, 4884.
52 S. A. Barnett, N. R. Champness and C. Wilson, Eur. J. Inorg. Chem.,2005, 1572.
53 I. Fernandez, R. Ruiz, J. Faus, M. Julve, F. Lloret, X. Ottenwaelder, Y.Journaux and M. C. Munoz, Angew. Chem., Int. Ed., 2001, 40, 3039;E. Pardo, I. Morales-Osorio, M. Julve, F. Lloret, J. Cano, J. Pasan, C.Ruiz-Perez, X. Ottenwaelder and Y. Journaux, Inorg. Chem., 2004, 43,7594.
2680 | Dalton Trans., 2007, 2669–2680 This journal is © The Royal Society of Chemistry 2007




























