Embed Size (px)
Citation preview

1
Rational Design and Structure Based
Investigation of Tyrosinase from Bacillus
megaterium
Research Thesis
In partial fulfillment of the requirements for the degree of Doctor of
Philosophy
Mor Goldfeder
Submitted to the Senate of
The Technion – Israel Institute of Technology
Nisan, 5774 Haifa March 2014

2
The research thesis was done under the supervision of Assoc. Prof.
Ayelet Fishman from the Dept. of Biotechnology and Food
Engineering, and Prof. Noam Adir from the Schulich Faculty of
Chemistry in the Interdepartmental Program of Biotechnology.
The generous financial help of the Leonard and Diane Sherman
Interdisciplinary Fellowship and the Miriam and Aaron Gutwirth Memorial
Fellowship is gratefully acknowledged
Publications:
1. V. Shuster Ben-Yosef, M. Sendovski, A. Fishman, (2010) Directed evolution of tyrosinase
for enhanced monophenolase/diphenolase activity ratio, Enzyme and Microbial
Technology, 47 (7), 372-376.
2. M. Sendovski, M. Kanteev, V. Shuster Ben-Yosef, N. Adir, A. Fishman, (2010)
Crystallization and preliminary X-ray crystallographic analysis of a bacterial tyrosinase
from Bacillus megaterium, Acta Crystallographica F: Structural Biology and
Crystallization Communication, 66 (9), 1101-1103.
3. M. Sendovski, M. Kanteev, V. Shuster Ben-Yosef, N. Adir, A. Fishman, (2011) First
structures of an active bacterial tyrosinase reveal copper plasticity, Journal of Molecular
Biology, 405 (1), 227-237.
4. M. Goldfeder, M. Egozy, V. Shuster Ben-Yosef, N. Adir, A. Fishman, (2013) Changes in
tyrosinase specificity by ionic liquids and sodium dodecyl sulfate, Applied Microbiology
and Biotechnology, 97 (5), 1953-1961.
5. M. Goldfeder, M. Kanteev, N. Adir, A. Fishman, (2013) Influencing the
monophenolase/diphenolase activity ratio in tyrosinase, Biochimica et Biophysica Acta –
Proteins and Proteomics, 1834 (3), 629-633.
6. M. Kanteev*, M. Goldfeder*, N. Adir, A. Fishman, (2013) The mechanism of copper
uptake by tyrosinase from Bacillus megaterium, Journal of Biological Inorganic
Chemistry,18 (8), 895-903. *Equally contributed.
7. M. Goldfeder and A. Fishman (2014) Modulating enzyme activity using ionic liquids or
surfactants, Applied Microbiology and Biotechnology, 98 (2), 545-554.

3
Table of contents
1. Introduction 11
1.1 Biocatalysis 11
1.2 Protein Engineering 11
1.3 X-ray crystallography 12
1.3.1 Protein crystal formation 13
1.3.2 The nature of protein crystals 13
1.3.3 Crystal diffraction 14
1.3.4 Data reduction 15
1.3.5 Structure determination 16
1.3.6 Calculation of electron density maps 17
1.3.7 Model refinement 17
1.4 Tyrosinase 18
1.4.1 Activity and abundance of tyrosinases 18
1.4.2 Tyrosinase and related type 3 copper proteins 19
1.4.3 Catalytic mechanism 20
1.4.4 Biotechnological applications of tyrosinase 22
2. Research objectives and significance 24
2.1 Research objective 24
2.2 Research significance 24
3. Materials and Methods 25
3.1 Materials 25
3.2 Bacterial strain and vector 26
3.3 Antibiotics 26
3.4 Growth media 26
3.4.1 LB medium 26
3.4.2 Terrific Broth (TB medium) 26
3.5 Buffers and solutions 26

4
3.5.1 Buffers for purification of TyrBm 26
3.5.1.1 Tris buffer 1M, pH=7.5 26
3.5.1.2 2M NaCl 27
3.5.1.3 2M Imidazole 27
3.5.1.4 Binding buffer 27
3.5.1.5 Elution buffer 27
3.5.2 Solutions for protein electrophoresis 27
3.5.2.1 Tris-SDS stock, pH 8.8 27
3.5.2.2 Tris-SDS stock, pH 6.8 27
3.5.2.3 Separating gel 12% (amounts for 2 gels) 27
3.5.2.4 Stacking gel 4% (quantity for 2 gels) 28
3.5.2.5 Tris-glycine electrode buffer, pH 8.3 28
3.5.2.6 Sample buffer × 4 28
3.5.2.7 Stain 28
3.5.2.8 De-stain buffer 28
3.6 Methods 28
3.6.1 Protein expression and purification 28
3.6.2 Protein determination using sodium dodecyl sulfate – polyacrylamide gel electrophoresis
(SDS-PAGE) 29
3.6.3 Tyrosinase activity assay with tyrosine and L-Dopa and as substrates 29
3.6.4 Tyrosinase activity in the presence of SDS or IL analyzed using high performance liquid
chromatography (HPLC) 30
3.6.5 Tyrosinase activity assay with phenol and catechol as substrates 30
3.6.6 Kinetic characterization of TyrBm wild-type and variants 31
3.6.7 Site-directed mutagenesis 31
3.6.8 Crystallization 32
3.6.9 TyrBm activity in crystal and crystal soak in additives, ligands and metal ions 32
3.6.10 Data collection and structure determination 33
3.6.11 Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) 33
3.6.12 Differential Scanning Calorimetry (DSC) 33

5
3.6.13 Bicinchoninic acid based assay (BCA) for copper uptake measurements 33
4. Articles 34
4.1 Crystallization and preliminary X-ray crystallographic analysis of a bacterial tyrosinase from
Bacillus megaterium 34
4.2 First structures of an active bacterial tyrosinase reveal copper plasticity 34
4.3 Changes in tyrosinase specificity by ionic liquids and sodium dodecyl sulfate 34
4.4 Influencing the monophenolase/diphenolase activity ratio in tyrosinase 34
4.5 The mechanism of copper uptake by tyrosinase from Bacillus megaterium 34
5. Unpublished results 35
5.1 TyrBm structures with ligands at the active site elucidate the catalytic mechanism 35
5.1.1 Abstract 35
5.1.2 Introduction 36
5.1.3 Results and Discussion 37
5.1.3.1 TyrBm structures with substrates in the active site elucidate reaction mechanism 37
2.3.1.5 Substrate deprotonation scenario 39
5.1.3.3 TyrBm structure with p- tyrosol in the active site 40
5.1.3.4 Pathway for ligand entrance based on structures with kojic acid 42
6. Discussion 44
6.1 Determination of active TyrBm structures 44
6.2 Copper binding and uptake in TyrBm 44
6.3 Mechanistic study of TyrBm and implication on protein engineering 46
6.4 Tyrosinase activity and selectivity in the presence of additives 49
7. References 51

6
List of Tables
Table 3.1: The ILs used in this study 25
Table 3.2: Primers used in this study. 32
Table 5.1: Data collection and refinement statistics. 43
Table 6.1: Studied second shell residues in TyrBm 47
List of Figures
Figure 1.1: Growing crystals using vapor diffusion hanging drop or sitting
drop methods. 13
Figure 1.2: Geometric condition fulfilling Bragg’s law. 14
Figure 1.3: Reaction scheme of tyrosinase presenting both the monophenolase
and diphenolase activities. 18
Figure 1.4: Placeholder residues at the active sites of type 3 copper proteins. 20
Figure 1.5: Proposed mechanisms of tyrosinase catalytic cycle. 21
Figure 5.1: Tyrosine and L-Dopa observed in the active site of TyrBm 37
Figure 5.2: Monophenol hydroxylation mechanism as suggested by Deeth and
Dietrich 38
Figure 5.3: Conserved water molecule and residues proposed to be
responsible for substrate deprotonation in TyrBm 40
Figure 5.4: p-Tyrosol is observed in the active site of TyrBm 41
Figure 5.5: Structure of kojic acid in the active site of TyrBm 42
Figure 6.1: TyrBm residues studied throughout this research 47
Figure 6.2: Tyrosinase monophenolase mechanism based on this research 48

7
Abstract
The goal of this research was to structurally investigate tyrosinase from Bacillus megaterium
through the determination of its crystal structure for the elucidation of its structure-function
properties.
Tyrosinase from the soil bacterium Bacillus megaterium (TyrBm) was previously isolated and
characterized in our lab. This work initiated with finding conditions for crystallization of TyrBm.
Crystals were obtained and the enzyme was shown to be active in crystal. Structures were
determined to a resolution of 2.0-2.3 Å. The active site copper ions, coordinated by six conserved
histidine residues, varied in occupancy and in position. This apparent mobility in copper binding
modes indicates that there is a pathway by which copper is accumulated or lost by the enzyme.
Second shell residues surrounding the active site were investigated for their influence on
activity and selectivity. Residues R209 and V218 were shown to play a role in substrate
orientation, due to their flexibility and proximity to the di-copper center.
Investigating copper accumulation in TyrBm, it was found that copper concentration has a
more significant effect on the diphenolase activity. Therefore, by decreasing the concentration of
copper, the monophenolase/diphenolase ratio was increased. Using a rational design approach, five
variants were identified as having an impact on copper uptake. For example, it was shown that a
major role of the highly conserved N205 is to stabilize the orientation of H204, thereby promoting
the correct coordination of CuB. A mechanism for copper accumulation by the enzyme was
proposed.
For the first time, structures of a tyrosinase with substrates in the active site were obtained by
replacing copper ions with zinc. Structures with L-tyrosine and L-Dopa determined to a resolution
of 2.2 Å, show that both substrates bind identically at the active site, towards CuA, as opposed to
the most prevalent models found in the literature. Furthermore, a structure was determined with the
monophenolic substrate p-tyrosol and copper ions, and the same orientation was observed. Two
structures with the inhibitor kojic acid were determined which show a pathway for the entrance of
ligands. Altogether, the determined structures elucidate parts of the catalytic mechanism of
tyrosinase and related proteins.
The effect of ionic liquids (ILs) and sodium dodecyl sulphate (SDS) on the activity of TyrBm
was investigated. In the presence of two water miscible ILs the monophenolase/diphenolase
activity ratio increased up to 5-fold. The addition of up to 50 mM SDS increased the activity 15-20
fold towards the non-native substrates phenol and catechol. A structure determined in the presence
of an SDS molecule shows it affects residue R209 and enables less polar substrates such as phenol
and catechol to penetrate more efficiently into the enzyme catalytic pocket.
In summary, in this work, structural data was obtained for the wild type enzyme and various
mutants, thus providing significant new information about tyrosinases. The copper uptake

8
mechanism was established; the role of second shell residues and their influence on activity and
selectivity was understood, as well as the effect of additives. This new information can be further
used to engineer and tailor TyrBm for various biotechnological applications.

9
Abbreviations
[BMIM][BF4] 1-Butyl-3-methylimidazolium BF4
[BMIM][Cl] 1-Butyl-3-methylimidazolium Cl
[EMIM][EtSO4] 1-Ethyl-3-methylimidazolium ethyl sulfate
BCA Bicinchoninic acid based assay
CMC Critical micelle concentration
DMSO Dimethyl sulfoxide
DSC Differential Scanning Calorimetry
EAN Ethylammonium nitrate
EDTA Ethylenediaminetetraacetic acid
ESRF European Synchrotron Radiation Facility
HPLC High performance liquid chromatography
ICP-AES Inductively Coupled Plasma Atomic Emission Spectroscopy
IL Ionic liquids
LB Luria Bertani medium
LBK LB medium with kanamycin (25 µg/ml)
L-Dopa L-3,4-dihydroxyphenylalanine
MAD Multiple wavelength anomalous dispersion
MBTH 3-methyl-2-benzothiazolinone hydrazone
MIR Multiple isomorphous replacement
MR Molecular replacements
PCR Polymerase chain reaction
PEG Polyethylene glycol
PTU Phenylthiourea
SDS Sodium dodecyl sulfate
SDS-PAGE SDS-polyacrylamide gel electrophoresis
TB Terrific Broth
TEMED N,N,N′,N′-tetramethylethylenediamine

10
TyrBm Tyrosinase from Bacillus megaterium
TyrSc Tyrosinase from Streptomyces castaneoglobisporus
WT Wild type

11
1. Introduction
1.1 Biocatalysis
Biocatalysis is the utilization of biological entities, either isolated enzymes or whole cell
systems, for the synthesis of chemicals (1). The great motivation for biocatalytic processes stems
from a number of important issues; these processes usually have a lower effect on the environment
compared to their chemical equivalents, they enable the use of mild conditions and allow to lower
operation costs, they improve the purity of products due to exquisite regio- and stereoselectivity,
and they allow the synthesis of otherwise inaccessible chemicals (1-5). Enzymes are typically used
for hydrolytic or isomerization reactions and whole cells are often used for synthetic reactions that
require cofactors which must be regenerated. Both isolated enzymes and whole cells are used in
industry today, and are an active area of research (4, 5). However, biocatalysis has some
limitations, especially in terms of development time scale which is longer than that of chemical
processes (1). In many cases there is a need for a process of identification and isolation of the
enzyme. Furthermore, natural enzymes usually do not perform efficiently in the conditions required
for industrial processes: high substrate concentration, extreme pH and temperature, nonaquaous
solvents and more (1). Thus, the path to a robust biocatalyst for practical application, poses a great
challenge (6). A number of solutions have been employed to deal with this challenge. The use of
immobilized enzymes is one option; another is to make use of enzymes isolated from
extremophiles, microorganisms living at extreme temperature, pressure, pH, or osmolarity (1).
However, the main strategy in the past decade consists of generating enzyme variants which are
more efficient in catalyzing the reaction under the specific conditions of the industrial process, in
other words employment of protein engineering.
1.2 Protein Engineering
Protein engineering describes the process of altering the structure of an existing protein to
improve its properties (7). This important technology can increase our basic understanding of how
enzymes function and have evolved, and it is the key method for improving enzyme properties for
applications in pharmaceuticals, green chemistry and biofuels. The use of protein engineering has
led to many successful industrial solutions such as proteases engineered to tolerate the bleach in
laundry detergents and improve cleaning of clothes (8), or the engineering of enzymes for
enhanced oil recovery and for cellulosic ethanol manufacture (9). There are three main approaches
for designing a desired protein: rational design, directed evolution and computational/statistical
methods. The combination of them has been shown to be most valuable (10).
Rational design is based on structural information, such as the protein’s crystal structure or a
homolog model designed on the basis of genetically-related enzymes. Such knowledge enables to
rationally identify specific residues that can be altered to obtain the desired property. Site-directed

12
mutagenesis can be used to understand the function of specific residues and to introduce changes in
the selectivity and activity of the enzyme (1). Nonetheless, it is usually difficult to predict the effect
of a particular mutation on enzyme parameters, even when the crystal structure is known. To
enhance the productivity of the mutagenesis process, and thus to increase the probability of finding
positive mutations, many studies have used site-directed saturation mutagenesis, which introduces
a full diversity of amino acids in one desired position (1). This strategy may be referred to as a
semi-rational approach.
Directed evolution, known as “Darwinian evolution in the test tube”, relies on the simple yet
powerful Darwinian algorithm of mutation and selection (11), and does not rely on the structure-
function relationship of an enzyme. It refers to an ensemble of technologies aiming at optimizing
existing biomolecules or creating new ones by first creating a diversity of mutant genes and then
sorting them based on their corresponding phenotype (1). One of the most common and convenient
methods is random mutagenesis by error-prone PCR (ep-PCR). A starting gene is amplified over a
million fold in an imperfect copying process that generates uncontrolled errors (1). The position
and nature of the mutations are spread over the amplified sequence. This method allows identifying
"hot spots" in the gene of interest that can be further improved by site-directed saturation
mutagenesis. The most challenging issue when using directed evolution is the identification of the
positive or improved variants. Since only a very small portion of the variants show improvement, a
large number of mutants must be tested. Ideally, the screening assay must be quantitative, fast,
cheap, sensitive, robust and highly reproducible.
This project focuses on rational design of a bacterial tyrosinase based on its crystal structure.
1.3 X-ray crystallography
X-ray crystallography is continues to be the most favored techniques for structure
determination of proteins and biological macromolecules (12). Structure determination can provide
detailed information about enzymatic mechanism, specificity of protein–ligand interactions and
drug design.
An object will diffract light only when its wavelength () is at the same order as the
dimensions of the object, or smaller (13). Visible light, which is electromagnetic radiation with
wavelengths of 400-700 nm, cannot produce an image of individual atoms in protein molecules, in
which the constituent atoms are only about 0.15 nm (1.5 Å) apart. Electromagnetic radiation of
this wavelength falls into the range of X-rays.
A single molecule is a very weak diffractor of X-rays, most of the X-rays will pass through a
molecule without being diffracted, so the diffracted beams are too weak to be detected (13).
Analyzing diffraction from crystals of macromolecules solves this problem. A crystal of a protein
contains many ordered molecules, so each molecule diffracts identically, and the diffracted beams
of all molecules can interact destructively canceling each other or constructively producing a

13
detectable X-ray beam (13). Single crystal diffraction, especially of cryogenically preserved
crystals also helps to overcome the damage caused by the interaction between the molecules and
the highly energetic X-rays.
1.3.1 Protein crystal formation
The determination of a three-dimensional structure by X-ray diffraction requires a relatively
large amount of high quality purified material. The protein molecules should be both chemically
and structurally homogeneous. The growth of protein crystals of sufficient quality for structure
determination is the rate limiting step in most protein crystallographic work, and is the least
understood (12). Crystals of proteins grow by slow, controlled precipitation from an aqueous
solution under conditions that do not denature the protein (13). In the common method of growing
protein crystals, purified protein is dissolved in an aqueous buffer containing a precipitant, such as
ammonium sulfate or polyethylene glycol, at a concentration just below that necessary to
precipitate the protein. The water is removed by controlled evaporation to produce precipitating
conditions, which are maintained until crystal growth ceases. One widely used technique is vapor
diffusion, in which the protein/precipitation solution is allowed to equilibrate in a closed container
with a large aqueous reservoir containing precipitant in an optimal concentration. Purified protein
is mixed with an equal amount of the reservoir solution and this mixture is deposited as a droplet
above the reservoir (Figure 1). Because the precipitant is the major solute present, vapor diffusion
in this closed system results in the net transfer of water from the protein solution to the reservoir.
Many factors influence the formation of protein crystals. These include protein purity, protein
concentration, precipitant, pH and temperature. The challenge of developing crystals of sufficient
quality entails controlling and testing a large number of parameters. For diffraction analysis,
protein crystals are usually required to be at least 0.1 mm in the longest dimension, to provide a
sufficient volume of crystal lattice that can be exposed to the beam.
1.3.2 The nature of protein crystals
Crystals are ordered three dimensional arrays of molecules (atoms or ions) that may be
characterized by a set of determinants that define the periodicity of fundamental units of which the
crystals are composed. The basic unit of a crystal that repeats in three dimensions by simple
translations to form the whole crystal is termed the unit cell. The dimensions of a unit cell are
designated by six numbers: three lengths (a, b, c) representing the edges of the cell and three angles
A B
Figure 1.1: Growing crystals using
vapor diffusion hanging drop (A) or
sitting drop (B) methods.

14
(α, β, γ) between these edges (13). Each unit cell is composed of asymmetric units. The asymmetric
unit is the smallest part of the crystal which, by symmetry operations, can generate the contents of
the unit cell. It is the asymmetric unit which we solve in protein structure determination.
Crystals are grouped into seven crystal systems based on the unit cell symmetry. The
combination of the unit cell symmetry and the number of lattice points which the unit cell contains,
produce 14 lattices which are known as Bravais lattices. There are 230 different combinations of
symmetry elements in crystals; each of this is called a space group (13). Since protein molecules
are inherently asymmetric, being composed of chiral amino-acid residues, some symmetry
operations are not allowed in space groups of protein crystals. For that reason, there are only 65
available space groups for protein crystals.
1.3.3 Crystal diffraction
When X-ray beams are directed on the atoms comprising a crystal, the electric field of the X-
radiation will cause the electrons of those atoms to oscillate at the same frequency (13). As a
consequence they will emit scattered radiation. The radiation scattered by all of the individual
atomic centers of the entire crystal will lead to constructive interference in only a very few well
defined directions. Bragg, in the early 20th century, showed that the angles at which diffracted
beams emerge from a crystal can be computed by treating diffraction as if it were reflection from
sets of equivalent, parallel planes of atoms in a crystal. A set of planes is designated by a set of
three numbers h, k, l, which are called Miller indices (13). Miller indices are the three intercepts
that a plane makes with the cell axes. Such planes exist as a consequence of the regular array of
unit cells in the protein crystal. Bragg showed that a set of parallel planes with index hkl produces a
diffracted beam when x-rays of wavelength λ impinge on the planes at an angle θ and are reflected
at the same angle, only when the following equation (Bragg’s Law) is satisfied: 2dhklsinθ=nλ,
where d is the distance between the planes in a set of equivalent parallel planes, θ is the angle of
diffraction, n is an integer and λ is the wavelength of the radiation (Figure 2).
The French mathematician Fourier showed that a periodic function can be described as the sum
of sine and cosine functions. Such a sum is called a Fourier series. Each set of parallel planes in the
crystal produces one reflection h, k, l, or one term in the Fourier series that describes the electron
density within the unit cell (13). The electron density describes the surface features and overall
Figure 1.2: Geometric
condition fulfilling
Bragg’s law.

15
shape of all constituents of the unit cell. The Fourier transform describes the mathematical
relationship between an object and its diffraction pattern. We use the Fourier transform in order to
convert the diffraction patterns to electron density maps and vice versa.
The ability to distinguish two close objects as separate entities rather than as a single, blurred
object is a measure of the resolution of the image under study. The minimum value of d recordable
in the diffraction pattern, corresponding to the highest θ angle reflection that can be observed, is
referred to as the resolution of the data set (13). It approximately indicates the minimum separation
between atoms that will be resolvable in the final electron density map. The smaller the numeric
values of the resolution, the larger the level of detail observable in the structure. In practice, protein
crystals rarely diffract to better than 1.5 Å, however for most purposes, a resolution of between 1.5
to 3.0 Å is sufficient.
Every atom of the crystal contributes to the scattering that gives rise to every diffraction spot
and similarly every diffraction spot receives a contribution from each atom. Each diffracted beam
is defined by three properties: its amplitude, its wavelength (which is fixed by the experimenter)
and its phase angle. The amplitude and phase of a given reflection (h) are described by a quantity
termed the structure factor F(h), whose amplitude is proportional to the square root of the intensity
of the diffraction spot, which can be readily measured. The phase component of the structure factor
cannot be experimentally measured, but knowledge of it is an essential requirement for structure
determination.
1.3.4 Data reduction
The three-dimensional crystal lattice gives rise to a three-dimensional lattice of diffraction
spots called a reciprocal lattice (13). The reciprocal lattice describes where on the detector the
diffraction spots will occur. Each individual reflection of the reciprocal lattice is associated with a
specific Miller indice and termed h, k and l which are integers (either positive or negative). The
Miller indices give the number of lattice spacings from the origin of the reciprocal lattice to the
reflection in the directions of the basis vectors a*, b* and c*, respectively.
After the collection of the diffraction data, the reflections are indexed (their Miller indices are
determined) and their intensities placed on the same scale (due to the inherent decrease in
diffraction power due to exposure to the X-rays, as well as other factors). An evaluation of the
quality of the processed data can be made using several objective criteria. One such parameter is
called Rmerge, which measures the agreement between equivalent reflections (13). The intensities of
the reflections should be identical and deviations from the average value (merged value) can be
used to estimate the quality of the data. A good value for Rmerge depends on the redundancy of the
data, i.e. how many equivalent reflections have been measured and how often. Another parameter
used in the evaluation of the processed data, denominated completeness, is the relationship between
the observed number of reflections (i.e. those actually measured) and those theoretically

16
measurable. Clearly the redundancy and completeness should be as great as possible and Rmerge as
small as possible. For data sets collected with synchrotron-produced radiation, the Rmerge values are
typically in the range of 0.05-0.1, redundancies are 3-20 (depending also on the degree of
symmetry of the crystal lattice) and the completeness is greater than 90% for all reflections.
1.3.5 Structure determination
The determination of the three-dimensional structure of a protein by X-ray diffraction implies
finding the phase angles and the intensities for all of the reflections generated by the incidence of
X-ray on the crystal.
The measured intensity for a reflection is proportional to |F(hkl)|2:
2
hklhkl FI where
|F(hkl)| is the structure factor amplitude of reflection (hkl). The structure factor amplitude is
atoms
j
jjjjhkl lzkyhxifF1
)( )(2exp , where f(j) is the atomic scattering factor for X-rays for
the j atom of coordinate (xj, yj, zj). The electron density in a crystal can be obtained by calculating
the Fourier summation:
)(2exp)(
1
),,( hkljjjhkllkhzyx lzkyhxiFV , where x,y,z are the
coordinates of the unit cell, and hkl is the phase angle per reflection (hkl) (13). As mentioned
above, the phase is not available through the diffraction experiment and hence exists the “phase
problem”. In protein crystallography there are basically three methods for solving this “phase
problem” which are used in accordance with the type of problem to be solved: multiple
isomorphous replacement (MIR), molecular replacement (MR) and multiple wavelength anomalous
dispersion (MAD) (12, 13). Variations and mixtures of these methods have been used, and for
ultra-high resolution data, the “direct method” used in small molecule crystallography can be
applied.
In multiple isomorphous replacement (MIR) the phases of the reflections are determined from
the knowledge of the position (and therefore, of the phases) of heavy atoms inserted into the
protein crystal.
Molecular replacement (MR) is based on the observation that homologous proteins share
identical folds and, depending on the degree of sequence identity (usually at least 30%), similar
tertiary structures. In this method, the molecule of a known structure is considered as an initial
model from which one can calculate a first estimate of the phase angles. The search model must be
correctly positioned within the unit cell of the structure determined. The search model is positioned
in the cell of the unknown structure by the application of six variables, three rotational and three
translational. The solution to the problem is to use two mathematical functions, termed the rotation
and translation functions, which do not depend on knowledge of the unknown structure. Atomic
positions in unknown structure are replaced by a map of interatomic vectors. The advantage of such

17
a map (a Patterson map) is that it can be calculated directly from the diffraction intensities without
any knowledge of the phase angles. Even though it is a vector map, it can still be used to initially
orient the search model, via the rotation function and then superpose it on the unknown structure,
via the translation function. Such functions can be readily computed nowadays with the help of
computer programs. This is the main method which was used in the present work.
The multiple wavelength anomalous dispersion (MAD) method is based on the fact that the
scattering of X-rays due to the valence electrons of atoms differs in phase by exactly 180° with
respect to the incident beam. However, the phase difference between the incident beam and the
beam scattered by electrons of atoms can be different from 180° at specific wavelengths; a property
called anomalous scattering. Instead of using different derivative crystals to measure several data
sets, as in MIR, in MAD all the data can be collected from the same crystal (or crystal type) but
using several different X-ray wavelengths. The MAD method requires tunable X-rays, which are
available only at synchrotron facilities. Anomalous scattering can also be used in order to
determine the position of atoms in the protein when the X-ray wavelength is near the atom
absorption edge. It can also unequivocally identify an atom (for instance identifying the presence of
bound copper as opposed to bound zinc) within the crystal lattice.
1.3.6 Calculation of electron density maps
After phase determination, an electron density map of the asymmetric unit may be calculated
by Fourier transformation of the structure factors (F(hkl)) (12, 13). The degree of detail to be
observed in the structure depends on the quality of the X-ray data and particularly their maximum
resolution.
1.3.7 Model refinement
The initial structural model will contain errors that can be minimized through model
refinement. This is a process of adjustment of the atomic coordinates of the model in order to
minimize the difference between the experimentally observed structure factor amplitudes (Fobs) and
those calculated from a hypothetical crystal containing the model (Fcalc) (13).
In general, the refinement process is not totally automated but must be interrupted periodically
for the calculation of new electron density maps based on better phases resulting from the new
atomic positions. Such maps need to be inspected using molecular graphics and manual alterations
made to the structure.
During refinement the quality of the resulting model may be evaluated by use of the
discordance index (R-factor) between the observed structure amplitudes (Fobs) and those calculated
from the model (Fcalc),
hkl
obs
calc
hkl
obs
F
FF
R (13). R-factors are measures of the extent to which a
crystallographic model accounts for the original experimental data, specifically, the measured

18
intensities of reflections in the diffraction pattern. An initial model will typically have an R factor
of around 0.4 – 0.5 while the refined model of a high resolution structure (< 2A) should have an R
factor near or less than 0.2 (13). The accuracy of the model obviously depends on the resolution
and so lower resolution structures may have higher R factors. Mathematical refinement methods
could drive the R-factor lower and lower, producing increasingly better agreement between
observed and calculated structure amplitudes, even while the model persisted in exhibiting
recognizably incorrect features. To prevent this problem, Brunger introduced the R free factor, Rfree
(14). The crystallographic R-factor was used as the target function, but 5-10% of the diffraction
data was withheld in formulating the observational equations emerging into the least squares
procedure. This prevented blind minimization since an Rfree based on just the 10% of reflections
that were saved is calculated to evaluate whether the differences between observed and calculated
│F(hkl)│were really minimized by true improvement of the model, or whether they were an
artifact of the least squares procedure.
1.4 Tyrosinase
1.4.1 Activity and abundance of tyrosinases
Tyrosinases (EC 1.14.18.1) are copper containing enzymes ubiquitously distributed in all
domains of life (15). They can be found in various prokaryotes as well as in plants, fungi,
arthropods, and mammals. They are mainly responsible for the formation of melanin as in skin
pigmentation and in fruit and vegetable browning, but have various other functions in different
species such as components in wound healing and primary immune response and protection against
radiation (15-17). Using molecular oxygen, tyrosinases perform two successive enzymatic
reactions: (i) the ortho-hydroxylation of monophenols to o-diphenols called the monophenolase or
cresolase activity, and (ii) the oxidation of o-diphenols to o-quinones called the diphenolase or
catecholase activity (Figure 3). The reactive quinones then polymerize spontaneously to melanins.
Figure 1.3: Reaction scheme of tyrosinase presenting both the monophenolase and diphenolase
activities.
Tyrosinases belong to the family of copper oxidases, and having a pair of copper ions in their
active site associates them to the "type-3-copper" protein super family along with catechol oxidases
and haemocycanins. The members of this family all have a conserved active site of six histidine
residues, which are provided by a four helical bundle, coordinating the two copper ions (CuA and
OH
R R
OH
OH
Tyrosinase
monophenolase
activity
O2
Tyrosinase
diphenolase
activity
O2
R
O
O
Monophenol o-Diphenol o-Quinone

19
CuB) (18). Because of their role in skin pigmentation, tyrosinases are perhaps the most intensely
studied enzymes of this family.
In the bacterial kingdom there are some examples of well characterized tyrosinases. They were
first described in several species of Streptomyces (19), but the enzyme has also been reported in
other species such as Rhizobium, Symbiobacterium thermophilum, Pseudomonas maltophilia,
Sinorhizobium meliloti, Marinomonas mediterranea, Thermomicronium roseum, Bacillus
thuringiensis, and Pseudomonas putida F6 (20-22). A unique tyrosinase with a high tyrosine-
hydroxylation/dopa-oxidation ratio was discovered in Ralstonia solanacearum by Solano and co-
workers (19). A tyrosinase from the soil bacterium Bacillus megaterium was isolated and
characterized in our lab, showing activity at high temperatures and in the presence of miscible
organic solvents (23).
Although tyrosinases have been intensively investigated biochemically for many years the
information on the residues involved in catalysis and structure-function correlations is limited. In
2006 the first and only crystal structure of tyrosinase from Streptomyces castaneoglobisporus in
complex with a caddie protein was determined by Matoba et al. at a resolution of 1.4 Å (24). The
caddie protein, expressed from orf378, is necessary for obtaining a functional form of the enzyme,
and is believed to perform a chaperone like function in providing the copper ions to the tyrosinase.
The determination of the first crystal structure of tyrosinase introduced a large amount of
information to the field, which will be described below, but at the same time left many open
questions and a need for more structural data and further molecular understanding of tyrosinases.
1.4.2 Tyrosinase and related type 3 copper proteins
Type 3 copper proteins, to which tyrosinase belongs, is a family of proteins with varying
structures, amino acid sequences and functions that possess a practically identical active site. This
conundrum has been the focus of many studies, with the recent structural studies providing major
contributions to our understanding of functional aspects of this family’s function. The two other
members are catechol oxidase (EC 1.10.3.1) which only performs diphenolase activity and is found
mostly in plants, and hemocyanins, which are oxygen carriers from the hemolymph of many
molluscs and arthropods (25). Catechol oxidase and hemocyanin structures were determined more
than a decade ago, and the first structure of a tyrosinase was determined more recently. When this
study initiated there were two crystal structures of catechol oxidase (26, 27), three of hemocyanins
(28-30), one of an arthropod prophenoloxidase (31), and one structure of a tyrosinase from
Streptomyces castaneoglobisporus (TyrSc) crystallized in complex with a caddie protein (24).
In hemocyanins, no enzymatic activity has been identified, and indeed, the active site is
blocked by the protein itself. A leucine (Leu2830) or phenylalanine (Phe49) (Figure 4A) residue
extends into the substrate binding site (28, 30) therefore no substrate can reach the active site and
unless these residues are removed no activity can be obtained. This was substantiated by the work

20
of Decker et al. in which hemocyanin from tarantula was turned into a weak phenoloxidase after in
vitro cleavage of an N-terminal peptide including a conserved Phe49 which serves as an allosteric
trigger during the oxygenation process (32). The blocking residue is considered the “placeholder”.
A similar placeholder was observed in the TyrSc structure in complex with a caddie protein.
Tyr98 of the caddie protein is oriented in a fashion similar to that of Phe49 in hemocyanin from
Limulus, blocking the active site (Figure 4B) (18). The tyrosine residue is just far enough from the
dicopper center for it not be hydroxylated itself. Detachment of the caddie protein will enable the
exposure of the active site to phenolic substrates and for mono- or diphenolase activity to occur. In
the structure of catechol oxidase from Ipomoea batatas, the lack of monophenolase activity was
attributed to the presence of Phe261 near CuA (Figure 4C) (27). This residue was also considered
to be a gate-keeper residue, controlling the entrance to the active site, similar to the placeholders
described in other proteins, since a structure determined with an inhibitor (phenylthiourea) in the
active site shows a significant movement of the residue (33). Based on the structures of the
different members of the type 3 copper protein family, it is evident that the differences in function
are due to variations in the residues surrounding the substrate-binding pocket.
Figure 1.4: Placeholder residues at the active sites of type 3 copper proteins. (A) Residue Phe49 reaching
into the active site of hemocyanin from Limulus in green (PDB code 1LLA), (B) Residue Tyr98 of the caddie
protein (ORF378) in dark blue reaching into the active site of tyrosinase from Streptomyces in red (PDB code
1WX2), (C) Phe261 blocking CuA in the active site of catechol oxidase from batatas in cyan (PDB code
1BT3).
1.4.3 Catalytic mechanism
The catalytic cycle of tyrosinase is based on the different functional forms the copper center
can adopt; the oxygenated oxy form (Eox, [CuII-O2
2--Cu
II]), the oxidized Cu(II) containing met form
(Em, [CuII-Cu
II]), and the reduced deoxy form (Ed, [Cu
I-Cu
I]) (15, 34, 35). Eox is able to catalyze the
hydroxylation of monophenolic substrates as well as the oxidation of diphenols, whereas Em results
in a dead-end complex with monophenols and cannot bind oxygen (36). In the oxy-form, molecular
oxygen is proposed to be bound as a peroxide between the two copper atoms in a side-on
conformation as visualized in previously determined crystal structures (37). In the absence of any
substrate, Em is the primary form (15).
C B
Phe49 Tyr98
Phe261
A

21
Although tyrosinase and its related proteins have been intensively studied, its catalytic
mechanism is still a subject of debate due to its complexity; the existence of two catalytic activities
at the same active site and the apparent lag period associated with monophenolase activity (38).
Based on the lack of monophenolase activity in catechol oxidase (that has a large aromatic residue
blocking CuA) it has been suggested that in tyrosinases (that lack this blocking residue)
monophenols bind to CuA and o-diphenols bind to CuB (39). The evidence provided by the
structure of TyrSc has led to two possible hydroxylation mechanisms as proposed by Matoba el al.
(24) and Decker et al. (18) (Figure 5A and B, respectively). Matoba et al. suggested that the
deprotonated monophenol binds to CuB, followed by the addition of oxygen to the ring in the ortho
position through a connection to CuA, enabled by the release of the flexible His54. This residue has
also been suggested to be involved in the deprotonation of the monophenolic substrate (31, 38). In
catechol oxidase the mechanism is not possible because of the fixed conformation of the
corresponding His residue by a thioether bond with an adjacent cysteine residue. Decker et al. (18,
37) suggested that the monophenol substrate is oriented towards CuA through hydrophobic
interactions with His194, in analogy to the orientation of Tyr98 (in TyrSc). Solano and co-workers
also proposed that monophenols dock to CuA but o-diphenols dock to CuB, based on site specific
mutations of mouse tyrosinase (38, 39). According to Decker et al., hydroxylation occurs via an
electrophilic substitution mechanism while the substrate is positioned in trans to His63 (His69 in
TyrBm). In order to allow electrophilic attack of the Cu2O2 moiety on the aromatic system in ortho
position to the hydroxyl group, the O-O axis of the peroxo ligand has to rotate to point towards the
substrate. Newer studies suggest that an initial butterfly distortion of the Cu2O2 core occurs and
subsequently the substrate reorients at an axial/equatorial fashion (40, 41). In these mechanisms,
the diphenolic intermediate generated by hydroxylation of the substrate presumably binds
asymmetrically to CuA and to the O2 (41). In the last step of the catalytic cycle, the bound catechol
is two-electron oxidized and released as a quinone, reforming the Cu(I)–Cu(I) deoxy site. The
bridging hydroxo ligand is thereby converted to water.
As recently described by Muñoz-Muñoz et al., the diphenolase activity of tyrosinase can go
through two different pathways; the prevalent pathway which results in the release of quinone and
water, leaving the active site of tyrosinase in the reduced deoxy form, and the suicide inactivation
pathway, in which one of the copper ions is reduced to Cu(0) and released from the active site (42,
43). This will cause the enzyme to inactivate, unless a new copper ion is introduced into the active
site.
Tyrosinases show a much higher specific activity for oxidation of o-diphenols (diphenolase
activity) than for hydroxylation of monophenols (monophenolase activity) (19). This low
monophenolase/diphenolase ratio is understandable, as chemical oxidation of o-diphenols is much
easier than hydroxylation of monophenols. It has been suggested that the catalytic cycle directly
leads to the quinone product and no catechol is released in between (18, 38). However, Tudela and

22
co-workers have demonstrated experimentally that o-diphenol is released into the reaction medium
during the enzymatic oxidation of monophenols to o-quinone by gas chromatography-mass
spectrometry analysis (44).
1.4.4 Biotechnological applications of tyrosinase
The monophenolase and diphenolase activities of tyrosinase are the basis for many industrial
biotechnological applications (16, 17). Environmental applications include the detoxification of
phenol-containing waste waters, contaminated soils, and as a biosensor for monitoring phenols (15,
20, 45). Synthetic melanin produced by tyrosinases has numerous applications as well: it can be
used to protect against radiation, serve as a cation exchanger as well as a carrier of drugs,
antioxidants, antiviral agents and immunogens (21, 45, 46). Due to its ability to react on tyrosine
residues, tyrosinase has also been used for the cross-linking of proteins in order to improve their
emulsification and gelation properties for various food applications (47, 48). The ability of
tyrosinases to convert monophenols into diphenols has motivated studies regarding the production
of various ortho-diphenols (also referred to as substituted catechols). Catechols, are important
intermediates for the synthesis of pharmaceuticals, agrochemicals, flavors, polymerization
inhibitors, and antioxidants (46, 49). For instance, L-dopa (3,4-dihydroxyphenylalanine) is a
common drug for Parkinson’s disease (50), while 3-methoxycatechol is an intermediate for the
antivascular agents combretastatin A-1 and combretastatin B-1 (51). Manufacture of these
substituted dihydroxylated compounds by chemical routes is difficult due to the employment of
aggressive reagents, expensive and complicated starting materials, multiple reaction steps, and low
yields (19, 52). Furthermore, despite the great potential, the synthesis of diphenols by tyrosinase
has been restricted due to the rapid oxidation of diphenols to quinones by the enzyme. There are
A B
Figure 1.5: Proposed mechanisms of tyrosinase
catalytic cycle. (A) Mechanism by Matoba et al. (24),
in which deprotonated monophenols bind to CuB. (B)
Mechanism by Decker et al. (18), in which
deprotonated monophenols bind to CuA.

23
only few reports in the literature on utilization of tyrosinase for production of catechols, such as the
synthesis of the antioxidant hydroxytyrosol by Espin et al. (53). The diphenol was successfully
produced in 100% yield when ascorbic acid was added in two-fold molar quantities for reduction of
the respective quinone back to the desired diphenol. In addition, Burton and co-workers (54)
showed that immobilization of mushroom tyrosinase on hydrophilic supports favors the production
of catechols over quinones. Similarly, the production of L-dopa using tyrosinase immobilized on
magnetic beads, was shown efficient by Tuncagil et al. (50). Study of the catalytic mechanism will
enable the protein engineering of a better catalyst for the various biotechnological applications.

24
2. Research objectives and significance
2.1 Research objective
The main objective of this research is to use structural data for engineering tyrosinase from
Bacillus megaterium and for elucidation of its structure-function properties.
The specific goals are:
a. To develop the conditions to obtain crystals of Bacillus megaterium tyrosinase (TyrBm) and to
determine the structure of tyrosinase at a high resolution.
b. To obtain structures of TyrBm with substrates and inhibitors in the active site.
c. To employ rational design in order to study the mechanism of tyrosinase and to obtain variants
with altered selectivity in reactions of interest. To then determine the structures of promising
variants.
d. To examine the activity and structure of TyrBm in the presence of additives such as surfactants
and ionic liquids.
2.2 Research significance
Although tyrosinase is one of the most studied enzymes of its type, much of its biological
functions are not fully understood due to lack of structural information. The crystal structure of
only one tyrosinase was determined before this study was initiated, leaving many questions
regarding its activity unanswered. Determining more tyrosinase structures under different
conditions (e.g. in the presence of a substrate or an inhibitor) will provide better understanding of
its characteristics and mechanism. Furthermore, the obtained knowledge will enable the rational
design of biocatalysts for various environmentally friendly industrial applications.
On a more general note, this structural and biochemical study may contribute to the
understanding of other metalloenzymes in terms of the relationship with their bound metals, of
enzyme stability in the presence of additives, and of ligands binding in crystal.

25
3. Materials and Methods
3.1 Materials
Chemicals
L-Dopa, ammonium persulfate and N,N,N′,N′-tetramethylethylenediamine (TEMED) were
purchased from Acros (Geel, Belgium). Trizma base, sodium cacodylate trihydrate, polyethylene
glycol (PEG) 8000, ZnAc, kanamycin, sodium dodecyl sulfate (SDS), imidazole,
acrylamide/methylenebisacrylamide, catechol, kojic acid, L-tyrosine disodium salt and 3-methyl-2-
benzothiazolinone hydrazone (MBTH) were purchased from Sigma-Aldrich (Rehovot, Israel).
Phenol, CuSO4 and dimethyl sulfoxide (DMSO) were purchased from Merck (Whitehouse Station,
N.J., USA). β-Mercaptoethanol was purchased from Spectrum (Gardena, Calif., USA). Methanol
and ethanol were purchased from Bio Labs (Jerusalem, Israel). Bio-Rad protein reagent was
purchased from Bio-Rad Laboratories (Richmond, Ca., USA). The ionic liquids (ILs) used in this
research (Table 3.1) were purchased from Iolitec (Denzlingen, Germany). All materials used were
of the highest purity available and were used without further purification.
Table 3.1: The ILs used in this study
IL Structure
1-Butyl-3-methylimidazolium BF4
([BMIM][BF4]) N N
+
CH3
CH3
BF4
-
1-Butyl-3-methylimidazolium Cl
([BMIM][Cl]) N N
+
CH3
CH3
-
Cl
1-Ethyl-3-methylimidazolium ethyl sulfate
([EMIM][EtSO4])
N N++
CH3CH3
S
O
O
O-
OCH3
Ethylammonium nitrate (EAN) N+
H
H
H
CH3 N+
O-
O-
O
Enzymes
Restriction enzymes, polymerases and other enzymes used in this work for molecular biology were
purchased from the following companies: Fermentas (Vilnius, Lithuania), New England Biolabs
(NEB) (Ipswich, Massachusetts). Mushroom (Agricus bisporus) tyrosinase was purchased from
Sigma-Aldrich (Rehovot, Israel).
All primers were synthesized by Sigma-Aldrich (Rehovot, Israel). Plasmid purification kits were
purchased from Qiagen (Hilden, Germany).

26
3.2 Bacterial strain and vector
The strain used in this research is BL21 DE3 purchased from Novagen (Darmstadt, Germany) and
contains the genotype: F– ompT gal dcm lon hsdSB(rB
- mB
-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1
sam7 nin5]). E. coli BL21 was used as a host for the plasmid pET9d, which was purchased from
Novagen (Darmstadt, Germany) and contains resistance to kanamycin.
3.3 Antibiotics
The antibiotic that was used in this work is kanamycin (Sigma-Aldrich, Rehovot, Israel). The
concentration of the stock solution sterilized by filtration (0.22 μm) was 25 mg∙ml-1
in water, and
the final concentration in the medium was 25 μg∙ml-1
. The stock solution was stored at -20°C.
3.4 Growth media
All the quantities are represented as percentage of weight to volume.
3.4.1 LB medium
Tryptone 1.0%
NaCl 1.0%
Yeast-extract 0.5%
For solid agar-plates 1.5% of agar was added to the medium. The sterilized mixture was poured
into plates. For LB medium with kanamycin (LBKV), kanamycin stock solution (25 mg/ml) was
added to a final concentration of 25μg∙ml-1
.
3.4.2 Terrific Broth (TB medium)
Tryptone 1.2%
Yeast-extract 2.4%
Glycerol 0.4%
0.72 M K2HPO4, 0.17 M KH2HPO4 solution 10%
For TB medium with kanamycin (TBK), kanamycin stock solution (25 mg∙ml-1
) was added to a
final concentration of 25 μg∙ml-1
.
3.5 Buffers and solutions
The majority of buffers and solutions that were used in this study, except for the ones that are
specified, were prepared according to Sambrook et al (55).
3.5.1 Buffers for purification of TyrBm
3.5.1.1 Tris buffer 1M, pH=7.5
121.4 g of Trizma base were dissolved in 800 ml of dH2O and the pH was adjusted by adding
concentrated HCl. After the HCl addition dH2O was added to a final volume of 1000ml.

27
3.5.1.2 2M NaCl
116.9 g of NaCl were dissolved in 800 ml of dH2O. After the dissolution, dH2O was added to a
final volume of 1000 ml.
3.5.1.3 2M Imidazole
136.2 g of Imidazole were dissolved in 800 ml of dH2O. After the dissolution, dH2O was added to a
final volume of 1000 ml.
3.5.1.4 Binding buffer
20mM Tris –HCl (40 ml of Tris HCl 1M)
500mM NaCl (500 ml of NaCl 2M)
20mM imidazole (20 ml of imidazole 2M)
dH2O was added to a final volume of 2000 ml. The solution was filtered through vacuum driven
filtration system (0.45μ).
3.5.1.5 Elution buffer
20mM Tris–HCl (40 ml of Tris HCl 1M)
500mM NaCl (500 ml of NaCl 2M)
500mM imidazole (500 ml of imidazole 2M)
dH2O was added to a final volume of 2000 ml. The solution was filtered through vacuum driven
filtration system (0.45μ).
3.5.2 Solutions for protein electrophoresis
3.5.2.1 Tris-SDS stock, pH 8.8
Tris (Trizma base) 1.5 M
SDS 0.4%
The adjustment of pH was carried out with HCl 32% (Frutarom).
3.5.2.2 Tris-SDS stock, pH 6.8
Tris 0.5 M
SDS 0.4%
The adjustment of pH was carried out with HCl 32%.
3.5.2.3 Separating gel 12% (amounts for 2 gels)
Tris-SDS stock, pH 8.8 4.95 ml
Acrylamide/methylenebisacrylamide 40% 6 ml
Glycerol 50% 400 μl
H2O 8.6 ml
After mixing and de-aeration the following ingredients are added:

28
TEMED 15 μl
Ammonium persulfate 10% 100 μl
3.5.2.4 Stacking gel 4% (quantity for 2 gels)
Tris-SDS stock, pH 6.8 1.75 ml
Acrylamide/methylenebisacrylamide 40% 0.695 ml
H2O 4.5 ml
After mixing and de-aeration the following ingredients are added:
TEMED 8 μl
Ammonium persulfate 10% 30 μl
3.5.2.5 Tris-glycine electrode buffer, pH 8.3
Tris 0.025 M
SDS 0.1%
Glycine 0.192 M
3.5.2.6 Sample buffer × 4
SDS (w/v) 10%
Glycerol (v/v) 20%
β-mercaptoethanol (v/v) 10%
Bromophenol blue (v/v) 2.5%
Tris-HCl, pH 6.8 0.5 M
3.5.2.7 Stain
Coomassie Brilliant Blue 2.5 g
Ethanol 500 ml
Acetic acid glacial 100 ml
dH2O is added to a final volume of 1000ml. The solution is filtered through Whatman paper.
3.5.2.8 De-stain buffer
Ethanol absolute (or methanol) 200 ml
Acetic acid 100 ml
dH2O is added to a final volume of 1000ml, the solution is stored at room temperature
3.6 Methods
3.6.1 Protein expression and purification
A tyrosinase producing Bacillus megaterium strain was previously isolated in our lab from soil
and the gene (accession no. ACC86108) was cloned with a His-tag at the C-terminus into
Escherichia coli BL21 (23). E. coli BL21 (DE3) cells harboring pET9d/tyr were grown in 0.5 L TB

29
medium over night at 37C and recombinant tyrosinase was expressed. The cells were harvested by
centrifugation, suspended in binding buffer and then broken by a French pressure cell (Spectronic
Instruments Inc., Rochester, N.Y., USA). The cell debris was removed by centrifugation. The
supernatant was applied to a Ni(II)-bound affinity column (previously charged with Ni2+
ions and
equilibrated with the binding buffer) (GE healthcare, Buckinghamshire, United Kingdom), and
elution was performed with an appropriate buffer (which includes 500mM imidazole). Protein
concentration of the eluted fractions were measured using Nanodrop (Thermo Scientific, MA,
USA) considering the following parameters MW=35.28 and ɛ=75.39. The fractions were run in an
SDS-PAGE gel and the clean fractions that contained only tyrosinase were combined and dialyzed
against 50 mM Tris-HCL buffer. Finally, the activity of the tyrosinase on 1 mM L-Dopa was
determined.
3.6.2 Protein determination using sodium dodecyl sulfate – polyacrylamide gel
electrophoresis (SDS-PAGE)
The samples were diluted with dH2O to a concentration of 0.5 µg·µl-1
. 10 μl of sample
buffer×4 were added to 30 μl of the sample and heated for 10 min at 95°C. The electrophoresis was
carried out with a mini-gel device (Bio-Rad, Richmond, California). The gel was prepared and
transferred following polymerization to the vertical mini-gel devise (Bio-Rad). The prepared
samples (40μl) were loaded onto the gel wells and were run in the presence of running buffer at a
current voltage of 150V. When the first bands reached the bottom of the gel, the voltage was
stopped, and gels were stained with Coomassie Blue for 20 min and de-stained overnight with the
de-stain solution. Molecular weight of the proteins was estimated according to a commercial
marker – Dual Color (Bio-Rad).
3.6.3 Tyrosinase activity assay with tyrosine and L-Dopa and as substrates
Tyrosinase activity assay is based on the determination of monophenolase and diphenolase
activity by monitoring the formation of L-dopachrome from 1 mM L-tyrosine or L-Dopa at a
wavelength of 475 nm (56). The reaction was measured at 25°C using 96-well plates with a final
volume of 200µl. Tris-HCL buffer (50 mM pH=7.5) and 0.01 mM CuSO4 were added to all of the
wells. The enzyme solution was added to a final concentration of 0.006 or 0.01 mg ml-1
. In some
cases the activity of mushroom tyrosinase was compared to that of TyrBm, and the same protein
concentration was used. Finally, the substrate was added to the wells at a final concentration of 1
mM to initiate the reaction. Negative control experiments were performed without the substrate as
well as without the enzyme. The plate was read by a multi-plate reader (OPTImax tunable
microplate reader; Molecular Devices, Sunnyvale, CA., USA) at a wavelength of 475nm for 10 min
with a measurement of absorption every 12 sec. The rate of dopachrome formation was defined as
the slope of the linear zone of absorbance versus the time plot. All measurements were carried out

30
in duplicates. Specific activity was calculated as the ratio of the conversion rate and the total
protein content.
Similarly, the monophenolase and diphenolase activity was measured in the presence of ILs or
SDS on L-tyrosine and L-Dopa. In order to do so, the additive was introduced in a range of
concentrations into the wells in which the reaction was performed. The activity was measured
without incubation time with the additive.
In order to examine the effect of different copper concentrations on the activity of TyrBm, the
assay was performed with CuSO4 in a wide range of concentrations (0-100 µM) and with the apo-
enzyme at a concentration of 6 g/ml. To obtain enzyme samples devoid completely of copper
(apo-enzyme), protein samples were incubated with 50 mM EDTA and dialyzed against Tris-HCl
buffer (50 mM pH=7.5) several times. Removal of Cu was confirmed by an inductively coupled
plasma atomic emission spectrometer (ICP-AES) (see below).
3.6.4 Tyrosinase activity in the presence of SDS or IL analyzed using high performance
liquid chromatography (HPLC)
The conversion of L-tyrosine to L-DOPA was determined using HPLC (Agilent 1100, Agilent
Technologies, Santa Clara, Calif., USA) by measuring the decrease in tyrosine concentration using
an Eclipse XDB C18 column (5 μm, 4.6 × 150 mm; Agilent Technologies, Santa Clara, Calif.,
USA). Two mM L-tyrosine was added to a 6 ml reaction volume containing 50 mM Tris-HCl at pH
7.5, 0.01 mM CuSO4, 30mM SDS or 10% EAN and 0.006 mg ml-1
purified enzyme. The reaction
was stopped periodically by adding 0.5 ml of the reaction mixture to 0.1 ml of 2 M HCl. The
samples were filtered using PVDF 0.45 µm filters (Millex HV, Millipore, Cork, Ireland) and
analyzed with a method comprising 2% acetonitrile in water (with 0.1% formic acid) at a flow rate
of 1 ml/min. A diode array detector was used at a fixed wavelength of 215 and 275 nm to monitor
the reaction in the presence of SDS and EAN respectively. Twenty µl of filtered samples were
injected into the column and under these conditions L-tyrosine eluted at 3.3 min. A calibration
curve was made with a commercial standard at 215 and 275 nm.
3.6.5 Tyrosinase activity assay with phenol and catechol as substrates
The monophenolase and diphenolase activity on phenol and catechol was determined by
monitoring the formation of an MBTH-quinone adduct from 1 mM phenol or catechol at a
wavelength of 500 nm. MBTH is a potent nucleophile that traps o-quinones to form a soluble
MBTH-quinone adduct with a high molar absorption coefficient (56). The reaction was performed
in 96-well plates for 10 min at 25°C. In addition to the reaction components mentioned in section
3.6.3, MBTH was added at a concentration of 1.5 mM. The rate of MBTH-quinone adduct
formation was defined as the slope of the linear zone of absorbance versus the time plot. All

31
measurements were carried out in duplicates. Specific activity was calculated using the absorption
coefficient for the MBTH-quinone adduct at 500 nm of 32500 M-1
cm-1
(56).
3.6.6 Kinetic characterization of TyrBm wild-type and variants
The values of Km and Vmax for TyrBm wild-type and variants were determined by a
colorimetric assay (as described in section 3.6.3) with the following conditions: 200 µl 50 mM Tris
HCl buffer pH 7.5, 0.01 mM CuSO4, 25ºC, employing 3 μg ml-1
of purified enzyme, and substrate
concentrations ranging from 0.1-6.0 mM for L-Dopa, and 0.02-6.0 mM for L-tyrosine. The
formation of L-dopachrome (ε = 3600 M-1
cm-1
) was monitored by measuring the absorbance at 475
nm. All measurements were performed in triplicates in 96-well plates at 25°C and monitored with a
multi-plate reader (OPTImax tunable microplate reader; Molecular Devices, Sunnyvale, CA.,
USA). The light path was determined as 0.68 cm.
3.6.7 Site-directed mutagenesis
Site-directed mutagenesis at the TyrBm gene was performed via QuickChange site-directed
mutagenesis kit PCR using the primers listed in Table 3-2. The wild-type TyrBm plasmid was used
as a template to create the variants. The first stage was to amplify the gene with the appropiate
primers that contain the desirable amino acid replacement by using pfu DNA polymerase. The PCR
reaction contained the following ingredients:
DNA template 100 ng
P1 forward, 100 ng·μl-1
2.5 μl
P2 Rear, 100 ng·μl-1
2.5 μl
dNTPs (1:1:1:1), 20 mM each 2 μl
Pfu buffer+Mg 5 μl
Pfu DNA polymerase 1 μl
H2O up to 50 μl
The PCR program consisted of an initial denaturation at 95ºC for 30 s, followed by 18 cycles of
95ºC for 30 s, 55ºC for 1 min, and 68ºC for 11 min. The PCR reaction was performed in 0.2 ml
tubes (ABgene, Epsom, UK) in a thermocycler (Biometra-GmbH, Goettingen, Germany). The
second stage was to digest the template by DpnI in order to get rid of the parental mathylated DNA.
The digestion was done by addition of 2 µl DpnI to the PCR solution and incubation for 2 h at
37ºC. The third stage was to transform the mutated plasmid into compotent E-coli BL21(DE3) by
electroporation. Verification of the mutations was obtained by sequencing.

32
Table 3.2: Primers used in this study.
Sequence 5’ 3’ Primer name
GACAGATGGGCGTTTTTCCTACTGCTCCGAATGAT V218F Val-Phe Forward
ATCATTCGGAGCAGTAGGAAAAACGCCCATCTGTC V218F Val-Phe Rear
GACAGATGGGCGTTGGGCCTACTGCTCCGAATGAT V218G Val-Gly Forward
ATCATTCGGAGCAGTAGGCCCAACGCCCATCTGTC V218G Val-Gly Rear
GCCACAGCTTCACGCTCGCGTACACCGTTG N205A Asp-Ala Forward
CAACGGTGTACGCGAGCGTGAAGCTGTGGC N205A Asp-Ala Rear
GCTTGAAGGAGCTATTAACGGGC F197A Phe-Ala Forward
GCCCGTTAATAGCTCCTTCAAGC F197A Phe-Ala Rear
CGATCGAAATGCAGCACATCTGAGTTCTGCTTTTTTACC M61L Met-Leu Forward
GGTAAAAAAGCAGAACTCAGATGTGCTGCATTTCGATCG M61L Met-Leu Rear
TGATACGCCGCCTTGGGATCTGACCAGCCAAAACAGCTTTCGT M184L Met-Leu Forward
ACGAAAGCTGTTTTGGCTGGTCAGATCCCAAGGCGGCGTATCA M184L Met-Leu Rear
*The specific codons are marked by a shaded background.
3.6.8 Crystallization
Crystallization of TyrBm was performed using the hanging-drop vapor diffusion method at 293
K. At first, 600 µl of the crystallization condition (reservoir), which includes 18% PEG 8000 and
0.1M sodium cacodilate trihydrate pH 6.5, were added to each of the 24 wells. Two microliters of
the enzyme solution (2 mg/ml) was placed on a siliconized glass circle cover slide. Following, 2 µl
of the reservoir was added to the enzyme solution on the cover slide. The slide was then placed
upside down on the well so that the drop is hanging above the reservoir. Finally, the plate was
placed in an incubator at 20C for at least 2 weeks until crystals were obtained.
3.6.9 TyrBm activity in crystal and crystal soak in additives, ligands and metal ions
To present activity in crystal mature TyrBm crystals were soaked in 0.5 mM L-tyrosine for 48
h. As opposed to L-Dopa, L-tyrosine in a solution does not oxidize spontaneously thus the hanging
drop remained clear and the reaction occurred only inside the crystals, turning them brown.
In order to see the effect of additives on the structure of TyrBm, a mature crystal was soaked
for 1-5 min in 2 or 10 mM SDS, 10% DMSO, and 10 or 20% ILs. At the end of the soak time the
crystal was frozen immediately in liquid nitrogen.
In order to trap ligands in the active site, a mature crystal was soaked overnight in 1 mM of
either CuSO4 or ZnCl2 and in 1 mM of the ligand (kojic acid, anacardic acid, phenylthiourea, L-
tyrosine, p-tyrosol, phenol, L-Dopa, hydroxytyrosol and catechol). Prior to freezing the crystal, it
was resoaked in the ligand for 1-5 min.

33
3.6.10 Data collection and structure determination
X-ray diffraction data was collected at the European Synchrotron Radiation Facility (ESRF),
Grenoble, France, at beamlines ID14-1 and ID 23-1. All data were indexed, integrated, scaled and
merged using Mosflm and Scala (57). The initial structure of TyrBm was solved by molecular
replacement (MR) using Phaser (58) and the coordinates of TyrSc (PDB code 1WX2) which has a
sequence identity of 40% to TyrBm served as the search model. A single solution was obtained for
two monomers in the asymmetric unit. Refinement was performed using CNS (59) Phenix (60) and
Refmac5 (61, 62) and manual model building, real-space refinement and structure validation was
performed using COOT (63). After determination of the first structure of TyrBm (PDB code
3NM8), all following data sets obtained were solved by molecular replacement using its
coordinates.
3.6.11 Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES)
The copper content of TyrBm was measured using an inductively coupled plasma atomic
emission spectrometer (ICP-AES), (ICP Spectrometer, iCap 6000 Series, Thermo Scientific, MA,
USA). CuSO4 was added to the protein samples, at a molar ratio of 2 moles of Cu2+
per 1 mole of
protein, for incubation periods of 5 min, 30 min and 6 hours. Each sample was subsequently
dialyzed 3 times, against 4 liters of Tris-HCl buffer (50 mM pH=7.5) in order to remove non-
specifically bound metal ions. The amount of bound Cu2+
was subsequently measured by ICP-AES.
3.6.12 Differential Scanning Calorimetry (DSC)
DSC measurements were carried out on a MicroCal VP-DSC. The reference cell contained a
dialysis buffer and the reaction cell contained 1 ml (2 mg/ml) of apo-protein in Tris-HCl buffer (50
mM pH=7.5), 1 mM CuSO4 was added to the protein sample in order to check the effect of Cu ions
on protein stability. To obtain a base line, buffer versus buffer (with and without CuSO4) was first
run, and then then subtracted from the sample curves. The measurements were performed by
scanning from low to high temperature at 1ºC/min and the data was processed with Origin.
3.6.13 Bicinchoninic acid based assay (BCA) for copper uptake measurements
The affinity of TyrBm towards copper was determined by the rate of copper uptake from the
BCA-Cu complex. BCA is highly sensitive and specific for Cu(I), which rapidly forms an intense
purple complex with BCA (ref). The reaction was performed in 96-well plates for 5 min at 25°C
and monitored with a multi-plate reader (OPTImax tunable microplate reader; Molecular Devices,
Sunnyvale, CA, USA) at 355 nm. Each reaction well contained Hepes buffer (50 mM pH 8), 0.07%
NaOH, 0.1% (w/v) ascorbic acid, 0.004% (w/v) BCA and 15µM CuSO4. The reaction was initiated
by the addition of 19 µg of the apo-enzyme into the reaction well and decrease in absorption was
measured. The rate of Cu uptake was defined as the slope of the linear zone of absorbance versus
the time plot. All measurements were carried out in triplicates, and repeated four times.

34
4. Articles
4.1 Crystallization and preliminary X-ray crystallographic analysis of a bacterial
tyrosinase from Bacillus megaterium
(2010, Acta Crystallographica F: Structural Biology and Crystallization Communication, 66
(9), 1101-1103)
4.2 First structures of an active bacterial tyrosinase reveal copper plasticity
(2011, Journal of Molecular Biology, 405 (1), 227-237)
4.3 Changes in tyrosinase specificity by ionic liquids and sodium dodecyl sulfate
(2013, Applied Microbiology and Biotechnology, 97 (5), 1953-1961)
4.4 Influencing the monophenolase/diphenolase activity ratio in tyrosinase
(2013, Biochimica et Biophysica Acta – Proteins and Proteomic, 1834 (3), 629-633)
4.5 The mechanism of copper uptake by tyrosinase from Bacillus megaterium
(2013, Journal of Biological Inorganic Chemistry, 18 (8), 895-903)

35
5. Unpublished results
5.1 TyrBm structures with ligands at the active site elucidate the catalytic
mechanism
Mor Goldfeder1, §
, Margarita Kanteev1, §
, Sivan Isaschar1, Noam Adir
2, Ayelet Fishman
1
1Department of Biotechnology and Food Engineering, Technion-Israel Institute of
Technology, Haifa 32000, Israel
2Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel
§ These authors equally contributed to this study.
5.1.1 Abstract
Tyrosinase is a copper containing oxidase ubiquitously distributed among all domains of life. It
is reactive towards both monophenols and diphenols, but the mechanism of the reaction has been a
subject of debate in the past two decades as more structural data became available. Structures of
tyrosinase from Bacillus megaterium with both tyrosine, the monophenol substrate, and L-Dopa,
the diphenol substrate, in the active site were determined to a resolution of 2.2 Å in the presence of
zinc ions. This is the first report on structures of any type 3 copper protein with substrates in the
active site. The structures show that both substrates bind identically at the active site, towards CuA,
as opposed to the prevalent models found in the literature. In a structure determined with the
monophenolic substrate p-tyrosol and copper ions, the same orientation is observed, and the
electron density map shows indications of the formation of the product. Furthermore, a new
structure with the inhibitor kojic acid in the active site was determined. Based on the new and
previously determined structures, a pathway of the entrance for ligands can be suggested.
Altogether, the determined structures elucidate some of the steps of the catalytic mechanism of
tyrosinase and related proteins.

36
5.1.2 Introduction
Tyrosinases (EC 1.14.18.1) are type 3 copper proteins able to perform two successive reactions
in the presence of molecular oxygen; the hydroxylation of phenols to form ortho-diphenols
(monophenolase activity), and the oxidation of o-diphenols to o-quinones (diphenolase activity)
(15, 54). Tyrosinases have a highly conserved active site albeit their wide distribution throughout
evolution (37, 52, 53, 64). The active site comprises six histidine residues which coordinate the two
copper ions CuA and CuB (15, 37). Type 3 copper proteins, to which tyrosinase belongs, is a
family of proteins with varying structures, amino acid sequences and functions that possess a
practically identical active site. The two other members are catechol oxidase (EC 1.10.3.1) which
performs only diphenolase activity and is found mostly in plants, and hemocyanins, which are
oxygen carriers from the hemolymph of many molluscs and arthropods (25). In hemocyanins, no
enzymatic activity has been identified, and indeed, the active site is blocked by the protein itself. A
leucine (Leu2830) or phenylalanine (Phe49) residue extends into the substrate binding site (28, 30)
therefore no substrate can reach the active site and unless these residues are removed no activity
can be obtained. This was substantiated by the work of Decker et al. in which hemocyanin from
tarantula was turned into a weak phenoloxidase after in vitro cleavage of an N-terminal peptide
including a conserved Phe49 which serves as an allosteric trigger during the oxygenation process
(32). The blocking residue is considered the “placeholder”.
A similar placeholder was observed in the TyrSc structure in complex with a caddie protein.
Tyr98 of the caddie protein is oriented in a fashion similar to that of Phe49 in hemocyanin from
Limulus, blocking the active site (18). The tyrosine residue is just far enough from the dicopper
center for it not be hydroxylated itself. In the structure of catechol oxidase from Ipomoea batatas,
the lack of monophenolase activity was attributed to the presence of Phe261 near CuA (27). This
residue was also considered to be a gate-keeper residue, controlling the entrance to the active site,
similar to the placeholders described in other proteins, since a structure determined with an
inhibitor (phenylthiourea) in the active site shows a significant movement of the residue (33).
Based on the structures of the different members of the type 3 copper protein family, it is evident
that the differences in function are due to variations in the residues surrounding the substrate-
binding pocket.
The catalytic cycle of tyrosinase is based on the different functional forms the copper center
can adopt; the oxygenated oxy form (Eox, [CuII-O2
2--Cu
II]), the oxidized Cu(II) containing met form
(Em, [CuII-Cu
II]), and the reduced deoxy form (Ed, [Cu
I-Cu
I]) (15, 34, 35). Eox is able to catalyze the
hydroxylation of monophenolic substrates as well as the oxidation of diphenols, whereas Em results
in a dead-end complex with monophenols and cannot bind oxygen (36). In the oxy-form, molecular
oxygen is proposed to be bound as a peroxide between the two copper atoms in a side-on
conformation as visualized in previously determined crystal structures (37). In the absence of any
substrate, Em is the primary form (15).

37
Although tyrosinase and its related proteins have been intensively studied, its catalytic
mechanism is still a subject of debate due to its complexity; the existence of two catalytic activities
at the same active site and the apparent lag period associated with monophenolase activity (38). In
the present study we show direct structural evidence that the mode of binding of substrates and
inhibitors in the active site are identical, leading to the identification of the more likely mechanism
of catalysis.
5.1.3 Results and Discussion
5.1.3.1 TyrBm structures with substrates in the active site elucidate reaction mechanism
Many attempts to determine a crystal structure of TyrBm with its substrates have been
performed using various soaking conditions. However, since the enzyme is highly active in its
crystal form (65) these attempts usually failed and the obtained structure was lacking the substrate
electron density. We have previously proposed that Zn2+
ions can bind similarly to Cu2+
within the
active site composed of six histidines (65). Furthermore, their presence inhibits the activity of
tyrosinase on both monophenol and diphenol substrates (65, 66). Based on these observations, we
soaked the crystals in Zn ions in order to enable binding of substrates to the active sites of TyrBm
in crystal such that they can be observed in the obtained electron density. The Zn and the substrate
were added, followed by an overnight soak and an additional short soak prior to freezing the
crystal. This approach was successful and for the first time high resolution structures with both
tyrosine and L-Dopa in the active site of tyrosinase were obtained (Fig 5.1) (Data collection and
refinement statistics are presented in Table 5.1).
Figure 5.1: Tyrosine and L-Dopa observed in the active site of TyrBm. (A) Tyrosine, colored in green
(oxygen atoms are presented in red and nitrogen atoms in dark blue) is observed in the active site oriented
through hydrophobic π-π interactions with H208. Its carboxyl side chain forms hydrogen bonds with
R209. Zinc ions are presented as grey spheres. (B) L-Dopa, colored in pink, is observed in the active site
in the same position as tyrosine.

38
In the structure determined to a resolution of 2.2 Å, tyrosine was observed bound to the active
site in met form. As previously suggested (18, 40), the hydroxyl is directed towards CuA (in this
case ZnA) and oriented through hydrophobic π-π interactions with His208, one of the six di-copper
coordinating histidine residues (Fig. 5.1A). The diphenol substrate, however, has usually been
proposed to bind to CuB (38, 41), based on catechol oxidase in which CuA is shielded by a bulky
phenylalanine residue and only the diphenolase activity is possible. Furthermore, our previous
results employing directed evolution on TyrBm, showed that when access to CuB is hindered by
mutation R209H, the diphenolase activity decreases while the monophenolase activity increases
(67). The crystal structure of TyrBm with L-Dopa at the active site proves unequivocally that
unlike previous assumptions, the diphenol substrate too binds to CuA in the same orientation
through interactions with His208 (Fig. 5.1B). The question that arises is therefore what
differentiates the activity of homologous catechol oxidase as well as TyrBm variants with altered
active sites? In a superposition of the obtained structures with substrates, and sweet potato catechol
oxidase (PDB code 1BUG, which includes the inhibitor PTU), it seems that, spatially, both tyrosine
and L-Dopa can enter and be positioned in the active site, considering the position of Phe261. It is,
however, too close to allow for any substrate rotation during the reaction (Figure 5.2).
During the monophenolase activity mechanism of tyrosinase, a rotation of the attached
substrate (tyrosine) is suggested to occur, allowing the electrophilic attack which leads to the
hydroxylation (18, 41). This was suggested by Decker et al. and further confirmed using molecular
dynamics study by Deeth and Dietrich (40). These studies, however, have not dealt with the
Figure 5.2: Monophenol
hydroxylation mechanism
as suggested by Deeth and
Dietrich (40). The red arrow
shows the rotation of the
substrate which is necessary
for the electrophilic attack.

39
molecular mechanism of reaction in case that the diphenol enters the active site. We suggest that
since the reaction on L-Dopa does not involve an electrophilic attack but only the oxidation of the
two hydroxyl groups, the rotation in this case is not necessary and does not occur. Thus enabling
the diphenolase reaction in catechol oxidase where the active site is partially blocked. Nonetheless,
it is very well possible that the orientation of substrates is not universal for all type-3-copper
proteins and for all active site forms (met vs. oxy) (38).
The positions of both tyrosine and L-Dopa are directed by residue R209 through their carboxyl
side chain (Fig. 5.1A). This position has been thoroughly investigated through directed evolution,
structure determination and with regards to its behavior in the presence of SDS (65, 67, 68). We
have found it has a major effect on the activity and substrate specificity of TyrBm. As mentioned
above, a mutation to histidine decreased the activity towards L-Dopa, but furthermore it has caused
the activity on the more hydrophobic substrates phenol and catechol in the presence of SDS to
increase 40-fold (68). The obtained structure confirms the role of R209 in substrate orientation in
the active site. To further examine its role we created variant R209F. Similarly to R209H, the
presence of a bulky residue at this position resulted in a decrease in activity towards L-Dopa by
20%. Moreover, the activity on the smaller hydrophobic substrates phenol and catechol improved 7
and 2-fold, respectively. These results, along with the previous studies mentioned above, further
confirm R209’s role in substrate specificity, and suggest a hot spot for mutagenesis within related
enzymes for varying the accessibility of different substrates. Indeed, this residue is not at all
conserved within type-3-copper proteins.
5.1.3.2 Substrate deprotonation scenario
In light of the obtained structures with substrates at the active site, an important element of the
reaction mechanism can be further elucidated. It has been shown using small-molecule model
systems that the substrate binds to the active site as a phenolate, and therefore a mechanism of
deprotonating by the enzyme is expected (41). This proton needs to then be returned to the active
site for the generation of water and the release of the quinone product. A number of options have
been previously suggested for the base that performs this initial step of deprotonation (24, 41).
Residue E195, a highly conserved glutamate located 7 Å away from the active site, was one of the
suggestions (33). However, it is too distant from the di-copper ions for deprotonation to occur.
Examination of the vicinity of this residue in TyrBm structures showed that almost invariably, a
water molecule is located bound equidistant between the E195 carboxyl group and the conserved
N205 carbonyl group (Fig. 5.3) previously shown to be vital for activity and copper uptake (69).
The same water molecule, in the same position, can also be observed in the structures of sweet
potato catechol oxidase (1BT3), Streptomyces tyrosinase (1WX2), Agaricus bisporus tyrosinase
(2Y9W), and Manduca sexta tyrosinase (3HHS). We propose that this conserved water molecule is
activated by E195 and N205 and serves as the base, that deprotonates the phenol. The involvement

40
of water molecules or hydroxide ions in enzyme catalysis has been previously shown in several
studies with other enzymes (70-72). In a QM
computational study performed by Siegbahn and
Borowski, the reaction mechanism was examined
and suggested a similar deprotonation scenario
(73). They showed that it is energetically feasible to
move the proton from tyrosine through the water
molecule to the glutamate residue (E195). They did
however consider the involvement of three more
residues in this scenario which are also highly
conserved, R178, N179 and D166 (Fig. 5.3).
Attempts to mutate E195 to arginine or leucine
resulted in insoluble proteins. This suggests that
E195 has an important role in the folding/stability
of the enzyme.
It is important to note that the suicide
inactivation pathway of diphenols is suggested to
be facilitated by the incorrect proton transfer to the
peroxide instead of to a close-by base (74). This
will then cause one copper ion to become reduced
to Cu(0) and the enzyme to be inactivated.
5.1.3.3 TyrBm structure with p- tyrosol in the active site
Tyrosinases in general are relatively non-specific and can accept many types of mono- and
diphenolic substrates (17). One of these substrates is p-tyrosol, which is the monophenolic
precursor for the commercially valuable antioxidant hydroxytyrosol. Hydroxytyrosol is present in
olives and in virgin olive oil, and it is linked to many of the benefits of the Mediterranean diet.
Espin et al. presented the enzymatic production of hydroxytyrosol using mushroom tyrosinase (53),
and this activity has also been optimized with TyrBm (unpublished results). Thus, we attempted to
Figure 5.3: Conserved water molecule and residues
proposed to be responsible for substrate deprotonation in
TyrBm. The water molecule, presented as a small red sphere, is
shown oriented by E195 and N205, colored in orange, with their
2Fo-Fc electron density map (grey wire) contoured at 2.2σ.
Conserved residues R191 N192 and D178 (orange) are also assumed
to be involved in the deprotonation scenario. All dashed lines
represent a distance of about 3.0 Å.

41
trap p-tyrosol in the active site. Using a similar method to that used for tyrosine and L-Dopa, but
using copper ions instead of zinc, we obtained a structure of TyrBm with p-tyrosol and Cu at the
active site determined to a resolution of 2.5 Å (Fig. 5.3A) (Data collection and refinement statistics
are presented in Table 5.1). We assume this structure was obtainable in the presence of copper ions
due to the lower activity rates of the enzyme on p-tyrosol, in comparison to tyrosine. Tyrosol can
be observed in both subunits, bound to CuA and oriented by His208, as was the case for the other
ligands. In one of the subunits, however, when modeling the p-tyrosol molecule into the electron
density map, we were surprised to find an unaccounted for electron density in the ortho position of
the monophenol (Fig. 5.3A).
Considering the presence of copper ions and that the enzyme is active in crystal, we assume that
what is observed is in fact the formation of the diphenol, hydroxytyrosol, or perhaps even the final
product, quinone. The latter is more probable since no oxygen/water molecules were observed at
the active site as would be expected in the deoxy state obtained at the end of the catalytic cycle.
When comparing the structure to a non-bound state of the enzyme (PDB code 3NQ0) a number of
differences can be seen. The residue in the loop above the active site, V218, moves 1.4 Å away,
and CuA moves 1.3 Å towards the hydroxyl of the bound substrate (Fig. 5.3B). Similarly, in
alcohol dehydrogenase, the active site zinc ion was studied during single substrate turnover, and
alternative coordination modes of the metal ion were observed along with changes in ligand metal
bond distances (75). This indicates that during different steps along the catalytic cycle, metal ions
may accommodate alternative locations.
Figure 5.4: p-Tyrosol is observed in the active site of TyrBm (A) p-Tyrosol is shown colored in teal with its 2Fo-
Fc electron density map (grey wire) contoured at 1.3σ. Copper ions are shown as brown spheres. p-Tyrosol is
observed in the active site oriented through hydrophobic π-π interactions with H208. A green arrow points at the
unaccounted for electron density which we propose shows hydroxylation has occurred in crystal. (B) Superposition
with a TyrBm structure without p-tyrosol (PDB code 3NM8) shows CuA and residues V218 and H60 have moved,
while H208 has not (stick representation and sphere in green for the structure without p-tyrosol, and stick
representation in white for structure with p-tyrosol).

42
5.1.3.4 Pathway for ligand entrance based on structures with kojic acid
Kojic acid is a commonly used inhibitor of tyrosinase. We have previously determined a
structure of WT TyrBm in complex with kojic acid (PDB code: 3NQ1) (65). In that structure, kojic
acid is oriented with the hydroxymethyl towards the active site at a relatively far distance of 7 Å,
and is bound strongly by interactions with residues in the active site entrance: Phe196, Pro201,
Asn205 and Arg209 (Fig. 5.4C). These residues have been since shown to have a major effect on
substrate specificity, activity, and copper uptake (67-69). This distant position was suggested to be
either a blocking site in which certain ligands bind and cause inhibition, or a step in the way into
the active site, a "resting point" for entering ligands. Taking into account that this structure was
obtained after a short 3 min soak of the crystal in kojic acid, our tendency was to the latter
suggestion.
In order to further examine ligand entrance to the active site, we used the long term soak
protocol described above, was successful affording a second crystal structure with kojic acid inside
the active site itself (Fig. 5.4A). The structure was determined to a resolution of 2.5 Å (Data
collection and refinement statistics are presented in Table 5.1). The kojic acid molecule appears in
both subunits of TyrBm in the same position, in which its ortho-positioned hydroxyl and carbonyl
groups are directed towards CuA and the ring is parallel to the imidazole ring of His208. Very
recently, a small molecule model and QM/MM study showed similar positioning of kojic acid at
the active site (76). Furthermore, this position is also identical to that of Tyr98 in the caddie protein
of TyrSc, which acts as a placeholder of the active site (24) (Fig. 5.4B). Considering the two
structures, a pathway for the entrance of ligands can be assumed (Fig. 5.4C). This would suggest
the molecule rotates while entering the active site.
C
uA
K
A
H2
08
Figure 5.5: Structure of kojic acid in the active site of TyrBm. (A) Kojic acid (KA), in stick representation colored in pink
(oxygen atoms in red), is observed in the active site so that its ortho-positioned hydroxyl and carbonyl groups are directed
towards CuA and the ring is parallel to the imidazole ring of His208. 2Fo-Fc electron density map (blue wire) contoured at 1.0σ
is presented around KA. Copper ions are shown as brown spheres, and water molecule as a small red sphere. (B) Superposition
of the structure with KA (TyrBm in grey, KA in pink stick representation) and the structure of TyrSc in complex with a caddie
protein (in cyan, PDB code 1WX2). The position of KA is identical to that of Tyr98 (cyan stick representation) of the caddie
protein. (C) Superposition of KA as observed in the two available structures, in the active site of TyrBm and at a 7Å distance
from it (PDB code 3NQ1). Observing the two structures, a pathway for the entrance of ligands can be assumed. Residues that
interact with KA in the enterance to the active site are colored in light blue. Nitrogen atoms are colored in dark blue.
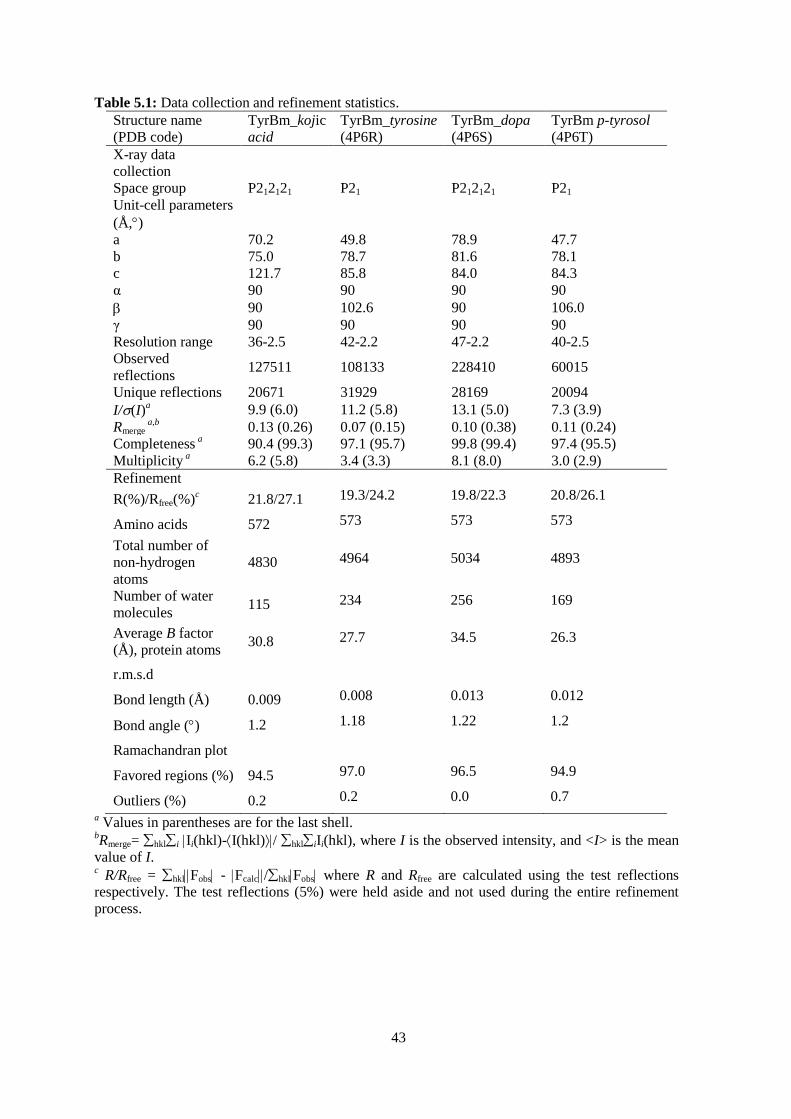
43
Table 5.1: Data collection and refinement statistics.
Structure name
(PDB code)
TyrBm_kojic
acid
TyrBm_tyrosine
(4P6R)
TyrBm_dopa
(4P6S)
TyrBm p-tyrosol
(4P6T)
X-ray data
collection
Space group P212121 P21 P212121 P21
Unit-cell parameters
(Å,)
a 70.2 49.8 78.9 47.7
b 75.0 78.7 81.6 78.1
c 121.7 85.8 84.0 84.3
α 90 90 90 90
90 102.6 90 106.0
γ 90 90 90 90
Resolution range 36-2.5 42-2.2 47-2.2 40-2.5
Observed
reflections 127511 108133 228410 60015
Unique reflections 20671 31929 28169 20094
I/(I)a 9.9 (6.0) 11.2 (5.8) 13.1 (5.0) 7.3 (3.9)
Rmerge a,b
0.13 (0.26) 0.07 (0.15) 0.10 (0.38) 0.11 (0.24)
Completeness a 90.4 (99.3) 97.1 (95.7) 99.8 (99.4) 97.4 (95.5)
Multiplicity a 6.2 (5.8) 3.4 (3.3) 8.1 (8.0) 3.0 (2.9)
Refinement
R(%)/Rfree(%)c
21.8/27.1 19.3/24.2 19.8/22.3 20.8/26.1
Amino acids 572 573 573 573
Total number of
non-hydrogen
atoms
4830 4964 5034 4893
Number of water
molecules 115 234 256 169
Average B factor
(Å), protein atoms 30.8 27.7 34.5 26.3
r.m.s.d
Bond length (Å) 0.009 0.008 0.013 0.012
Bond angle () 1.2 1.18 1.22 1.2
Ramachandran plot
Favored regions (%) 94.5 97.0 96.5 94.9
Outliers (%) 0.2 0.2 0.0 0.7
a Values in parentheses are for the last shell.
bRmerge= hkli Ii(hkl)-I(hkl)/ hkliIi(hkl), where I is the observed intensity, and <I> is the mean
value of I. c R/Rfree = hklFobs - Fcalc/hklFobs where R and Rfree are calculated using the test reflections
respectively. The test reflections (5%) were held aside and not used during the entire refinement
process.

44
6. Discussion
6.1 Determination of active TyrBm structures
The determination of protein structure has had a vast contribution to the understanding of
enzymatic mechanisms, protein stability, protein-protein and ligand-protein interactions, and to
drug design. Furthermore, knowledge of enzyme structures has an important impact on
advancements in the field of biocatalysis and protein engineering (77, 78). Structural data on
tyrosinase was unavailable until 2006, when the structure of a Streptomyces castaneoglobisporus
tyrosinase was determined in complex with a caddie protein by Matoba et al. (24). This structure
facilitated progress of many studies in the field, but it was clear that more tyrosinase structures in
different conditions are necessary. The presence of the caddie, indeed its very necessity in the
crystallization process, alluded that the enzyme was not in its totally active state.
Tyrosinase from the soil bacteria Bacillus megaterium (TyrBm) was previously isolated,
cloned and characterized in our lab (23). As it was easily produced and purified, as well as being
stable at high temperatures or in the presence of surfactants, it made a good candidate for crystal
structure determination. Indeed, conditions for crystallization were optimized relatively quickly,
and high quality crystals were obtained in two different conditions, with and without the presence
of zinc ions (65, 79). Mature crystals soaked in the monophenol substrate, tyrosine, turned brown
which indicated that hydroxylation and oxidation of the substrate into o-quinone had occurred and
melanin was accumulated. We can safely assume that all obtained TyrBm structures are that of
active enzyme molecules. The very activity of the crystals made structure determination of the
complex with native substrates difficult.
6.2 Copper binding and uptake in TyrBm
Metal ion binding sites and especially those involved in catalysis generally exhibit high
structural conservation (80, 81). As in the case of type 3 copper proteins, the locations of metal ions
and their ligating residues are identical, even among distant superfamily members.
In the case of TyrBm however, the determination of crystal structures in different conditions
revealed plasticity in the binding of copper. High resolution structures were determined from
crystal types obtained with and without zinc ions in the crystallization medium. The overall
structure was very similar, but the two crystal types varied significantly in their copper ion
occupancy. Studying a number of structures obtained in different conditions, the following
conclusions could be drawn: (i) Zinc ions can bind to the active site in the same way as copper ions
and cause activity inhibition. (ii) Either one, two or no copper ions may be coordinated at the active
site. (iii) When only one copper ion is present, it will always be CuA, indicating the greater
strength of binding of this ion. (iv) The binding position of CuA may vary (by 2 Å). To date no

45
other type 3 copper protein has been reported to contain less than two copper ions, or to present
varied positioning of its copper ions.
There are a number of examples in the literature for changes in configuration and binding of
metal ions. Serum paraoxonase-1 (PON1) is one such example. Its catalytic Ca2+
can accommodate
alternative locations and coordination modes allowing it to catalyze different reactions (80).
Another well studied case is that of xylose isomerase, in which the position of the catalytic Mg2+
may vary significantly (70). Similarly to TyrBm, xylose isomerase has two metal ions which are
involved in the reaction. In a structure with its substrate, a 1.8 Å movement of one of the two Mg2+
ions was observed. Furthermore, mutation in the metal’s ligating residues changed the specificity
of the enzyme towards substrates (82). In alcohol dehydrogenase, the active site zinc ion was
studied during single substrate turnover using time-resolved X-ray absorption spectroscopy, pre-
steady state kinetics and computational quantum chemistry (75). Alternative coordination modes of
the metal ion were observed along with changes in ligand metal bond distances. This indicates that
during different steps along the catalytic cycle, metal ions may accommodate alternative locations.
We have observed this in the recent TyrBm structure with p-tyrosol and copper ions, where CuA
moves towards the substrate 1.3 Å in comparison to a structure without the substrate. Overall, it has
been suggested that metal ion plasticity may contribute to the catalytic versatility of enzymes (80).
This would explain the fact that type 3 copper proteins are relatively non-specific and can accept
many types of mono- and diphenolic substrates (17).
The intriguing plasticity of copper ions in TyrBm has led to the study of its uptake in the
enzyme. In many of the studied tyrosinases a caddie protein is responsible for providing the active
site copper ions, and therefore it is necessary for activity (15, 17, 83); for example tyrosinase from
Streptomyces castaneoglobisporus (TyrSc) whose structure was determined in complex with a
caddie protein (24). TyrBm, however, does not require such a protein for its activity and therefore
the mechanism of copper accumulation apparently occurs within the enzyme and is important to
understand. Using a rational design approach, residues in the vicinity of the active site which
participate in copper binding and contribute to the enzyme selectivity were identified: N205, F197,
M61 and M184 (69). Methionine residues have been known to have the ability to ligate Cu ions
and are highly important for Cu transport (84, 85). In the structure of TyrSc with the caddie protein,
two methionine residues, spatially similar to TyrBm M61 and M184, were revealed to be important
for Cu binding (83). In TyrBm, residues F197, M61 and M84, which are located at the entrance to
the binding site, not only play a role in Cu uptake but are also important for enhancing the
diphenolase activity. Furthermore, a major role of the highly conserved N205 residue is to stabilize
the orientation of the H204 imidazole ring in the binding site, thereby promoting the correct
coordination of CuB. By mapping out and mutating these residues we showed that it is possible to
control TyrBm activity, and proposed a mechanism for copper accumulation in TyrBm.

46
Independent copper uptake by TyrBm is also important for its ability to cope with suicide
inactivation. Suicide inactivation may occur during tyrosinase diphenolase activity and is suggested
to result in the release of one of the copper ions (42, 74, 86). Many of the determined TyrBm
structures contain only one copper ion, CuA, which indicates that a copper ion can be expelled
easily from the enzyme. Furthermore, we have shown that the diphenolase activity of TyrBm
requires the presence of higher concentrations of copper than its monophenolase activity. It can
therefore be concluded that when copper is released from TyrBm during diphenolase activity, it can
be accumulated by the enzyme if it is present in the medium. In a copper depleted environment the
monophenolase/diphenolase activity ratio is therefore increased.
Overall, we have promoted the knowledge on TyrBm and its metal ions. Less is known,
however, about the enzyme in its native host and could be studied in the future in order to further
understand its copper uptake in Bacillus megaterium, its secretion to the environment, and its
importance to the host.
6.3 Mechanistic study of TyrBm and implication on protein engineering
This study sought to structurally investigate the mechanism of tyrosinase in order to engineer it
for biotechnological applications. The structural information collected and rational design which
was performed, along with the advancing research on type 3 copper proteins performed by other
groups, aided in elucidation of important mechanistic questions. The second shell residues in this
enzyme play a major role in substrate orientation and specificity, in copper ion accumulation and
orientation, and in regulation of all of these processes. Figure 6.1 presents the second shell residues
which were studied throughout this research, and table 6.1 describes the mutations performed and
the assumed role of each residue. The full presumed mechanism according to our findings is
described in Figure 6.2.
Based on these findings, tyrosinase may be engineered and tailored for a required process. For
example, increased monophenolase activity can be achieved by employing changes to residues
V218, M61, and R209. In order to affect tyrosinase accessibility to different substrate types,
changes to residue R209 should be performed. On the other hand, residue N205 should remain
untouched in order to retain an active enzyme. This is also true for the conserved residues M215
and E195. In general, as long as its conserved residues are not changed, the enzyme is quite stable
towards mutations in the second shell residues and therefore engineering it is relatively
uncomplicated.

47
Table 6.1: Studied second shell residues in TyrBm.
Residue Variants Study conclusions and assumed role
R209 R209H, R209A,
R209F
Affects monophenolase/diphenolase activity ratio
Affects substrate specificity
Role in substrate orientation
Affected by sodium dodecyl sulfate (SDS)
V218 V218G, V218F Affects monophenolase/diphenolase activity ratio
Role in substrate orientation
N205 N205A, N205D Conserved residue and vital for activity
Stabilizes H204 which coordinates CuB
Coordinates conserved water molecule assumingly involved in
substrate deprotonation
F197 F197A Regulates entrance of copper to the active site
M61 M61L Role in TyrBm copper uptake
Important for activity in low copper concentrations
Affects monophenolase/diphenolase activity ratio
M184 M184L Role in TyrBm copper uptake
Important for activity in low copper concentrations
M215 M215L, M215A Conserved residue
Structural role, since mutations resulted in an insoluble enzyme
E195 E195L, E195R Conserved residue
Coordinates conserved water molecule assumingly involved in
substrate deprotonation
Structural role, since mutations resulted in an insoluble enzyme
Figure 6.1: TyrBm residues studied throughout this research. The surface of the full structure of TyrBm dimer is
observed on the left in grey, while the surface of the investigated residues is colored in cyan. On the right hand side, an
enlargement of the active site is shown. Active site histidines are shown in stick representation colored in grey. Studied
second shell residues are colored in cyan. Oxygen atoms are presented in red, nitrogen atoms in dark blue and sulfur in
yellow. Copper ions are shown as brown spheres.

48
The determined structures with ligands, e.g. inhibitors and substrates, were most significant for
the study of the difference between reaction on mono- and diphenolic substrates. We have shown
that in contrast to prior assumptions, monophenol and diphenol substrates bind very similarly at the
active site, and both are directed towards CuA and oriented through hydrophobic π-π interactions
with H208. This understanding has emphasized the important role of other mechanistic processes
such as the monophenol rotation during hydroxylation, and the suicide inactivation by the diphenol
substrate. In any case, once a monophenol substrate goes through hydroxylation, the further
oxidation to quinone is apparently inevitable. Taking this into account, while our initial aim to
diminish the diphenolase activity for commercial production of catechols is no longer reasonable,
the activity can be manipulated towards different types of substrates and improved towards
monophenols.
Figure 6.2: Tyrosinase monophenolase mechanism based on this research. Tyrosine enters the active site through
a preliminary binding site oriented by P201 and F197. On its way to the active site it will then be deprotonated by the
conserved hydroxide held by E195. Tyrosine will then orient towards CuA through hydrophobic π-π interactions with
H208 and through electrostatic bonds with R209. Residue V218 will move first to allow entrance (proper positioning?)
and second and then to allow the necessary substrate rotation for the electrophilic attack on the oxygen. The product
will be released after both hydroxylation and oxidation occur.

49
6.4 Tyrosinase activity and selectivity in the presence of additives
The vast potential in using enzymes for industrial processes has raised the need to fine-tune or
modulate their activity according to the required process. While the main strategy used to achieve
this in the current research is protein engineering (7, 87), the modification of the solvent in the
immediate vicinity of the enzyme, e.g. medium engineering, affords an additional range of
possibilities (88). In this research both ILs and SDS were used in order to modify the activity and
selectivity of TyrBm.
In the presence of two water miscible ionic liquids, [BMIM][BF4] and ethylammonium nitrate,
the selectivity of the enzyme toward its substrates significantly changed; the commercially desired
activity on the monophenol improved while the activity on the diphenol decreased. The 5-fold
increase in monophenolase/diphenolase activity ratio was obtained while no change or decreased
activity was observed in tyrosinase from Agaricus bisporus. A similar decrease in the diphenolase
activity in the presence of [BMIM][BF4] was previously shown by Yang et al. on mushroom
tyrosinase while the monophenolase activity was not examined (89). It can therefore be concluded
that the substrate solubility could not be the main cause for the changed selectivity. Beyond the
contribution to substrate solubility, there are a number of more complex effects that lead to the
unexpected improvements in enzyme activities which are reported in the literature. The IL
interaction with the enzyme itself should be considered, as should the effect of the ions on the
water properties and on the protein-water interactions (90).
While the Hofmeister series is often used to explain the influence of water miscible ILs on
enzymes, it was shown to be occasionally reversed depending on the enzyme. Furthermore, since
we observed different results depending on the substrate in the case of TyrBm, the IL effect is even
more complex. It may therefore be suggested that the effect of IL on the water molecules, which is
not fully understood, causes a structural change in TyrBm which translates differently towards each
substrate.
Another type of additive which may be used as a solvent modifier is surfactants. Ionic
surfactants, usually denature proteins even in concentrations below their CMC due to their ability
to bind and interact with both native and denatured proteins (91). However, there are cases in
which these interactions cause enzyme activation rather than denaturation. The reports on the
enhanced activity of tyroinases in the presence of SDS are available for quite some time (92-95).
The observed improvements in activity are contributed to conformational changes caused by the
binding of SDS molecules in monomer form to the enzyme (92). The changes apparently involve
improvement of the active site accessibility to substrates. In most studied tyrosinases, the activation
does not continue above the CMC which is in the range of 0.8 to 1.1 mM depending on system
parameters. In this study however, the improvement in activity was obtained for the substrates L-
tyrosine and L-Dopa, in the presence of SDS concentrations above the CMC and up to 50 mM

50
SDS. When the monophenol substrate, phenol, and the diphenol substrate, catechol, typically poor
substrates of this enzyme, were assayed at similar conditions, an unprecedented 15-20 fold
improvement in the activity of TyrBm was obtained. The specific activity improved so significantly
that TyrBm was more active on phenol than on its native substrate, L-tyrosine. The crystal structure
determined in complex with one SDS molecule showed the movement of two residues, which
apparently enabled the accessibility of the hydrophobic substrates. Once again residue R209 was
found to be involved, as SDS caused its movement and its effect on the activity of variant R209H
was even more pronounced.
This study has highlighted the increasing possibilities and potential in modulating enzyme
activity and selectivity with the use of surfactants or ILs as co-solvents (96). There are various
mechanisms by which ILs in aqueous reaction systems can manipulate enzymatic performance, and
recent studies have shown that they are a complex combination of effects depending on the specific
system. Future advancement in basic research dealing with IL-enzyme interactions may expedite
rational design of beneficial ILs and promote their application in biocatalysis. The use of
surfactants to modulate enzyme activity is not straightforward as well. Usually known as
denaturing substances, surfactants can promote positive changes in some enzymes. These
modulations caused mainly by conformational changes can lead to important new applications.

51
7. References
1. Sylvestre J, Chautard H, Cedrone F, Delcourt M. 2006. Directed evolution of biocatalysts. Org. Process Res. Dev. 10:562-571.
2. Woodley JM. 2008. New opportunities for biocatalysis: making pharmaceutical processes greener. Trends Biotechnol. 26:321-327.
3. Tao J, Xu J-H. 2009. Biocatalysis in development of green pharmaceutical processes. Curr. Opin. Chem. Biol. 13:43-50.
4. Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B. 2001. Industrial biocatalysis today and tomorrow. Nature 409:258-268.
5. Pollard DJ, Woodley JM. 2007. Biocatalysis for pharmaceutical intermediates: the future is now. Trends Biotechnol. 25:66-73.
6. Turner NJ. 2009. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5:567-573.
7. Kazlauskas RJ, Bornscheuer UT. 2009. Finding better protein engineering strategies. Nat Chem Biol 5:526-529.
8. Estell DA, Graycar TP, Wells JA. 1985. Engineering an enzyme by site-directed mutagenesis to be resistant to chemical oxidation. J. Biol. Chem. 260:6518-6521.
9. Gray KA, Zhao L, Emptage M. 2006. Bioethanol. Curr. Opin. Chem. Biol. 10:141-146. 10. Brouk M, Nov Y, Fishman A. 2010. Improving Biocatalyst Performance by Integrating
Statistical Methods into Protein Engineering. Appl. Environ. Microbiol. On line. 11. Johannes TW, Zhao H. 2006. Directed evolution of enzymes and biosynthetic pathways.
Curr. Opin. Microbiol. 9:261-267. 12. Smyth MS, Martin JH. 2000. x ray crystallography. Mol Pathol 53:8-14. 13. Rhodes G. 1993. Crystallography made crystal clear - A guide for users of macromolecular
models first ed. Academic Press. 14. Brunger AT. 1992. Free R value: a novel statistical quantity for assessing the accuracy of
crystal structures. Nature 355:472-475. 15. Claus H, Decker H. 2006. Bacterial tyrosinases. Syst. Appl. Microbiol. 29:3-14. 16. Faccio G, Kruus K, Saloheimo M, Thöny-Meyer L. 2012. Bacterial tyrosinases and their
applications. Process Biochemistry 47:1749-1760. 17. Fairhead M, Thöny-Meyer L. 2012. Bacterial tyrosinases: old enzymes with new relevance
to biotechnology. New Biotechnology 29:183-191. 18. Decker H, Schweikardt T., Tuczek F. 2006. The first crystal structure of tyrosinase: All
questions answered? Angew. Chem., Int. Ed. Engl. 45:4546-4550. 19. Hernández-Romero D, Sanchez-Amat A, Solano F. 2006. A tyrosinase with an abnormally
high tyrosine hydroxylase/dopa oxidase ratio. FEBS Journal 273:257-270. 20. Dalfard AB, Khajeh K, Soudi MR, Naderi-Manesh H, Ranjbar B, Sajedi RH. 2006. Isolation
and biochemical characterization of laccase and tyrosinase activities in a novel melanogenic soil bacterium. Enzyme Microb. Technol. 39:1409-1416.
21. Liu N, Zhang T, Wang YJ, Huang YP, Ou JH, Shen P. 2004. A heat inducible tyrosinase with distinct properties from Bacillus thuringiensis. Lett. App. Microbiol. 39:407-412.
22. McMahon AM, Doyle EM, Brooks S, O'Connor KE. 2007. Biochemical characterisation of the coexisting tyrosinase and laccase in the soil bacterium Pseudomonas putida F6. Enzyme Microb. Technol. 40:1435-1441.
23. Shuster V, Fishman A. 2009. Isolation, cloning and characterization of a Tyrosinase with improved activity in organic solvents from Bacillus megaterium. J. Mol. Microb. Biotech. 17:188-200.
24. Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, M. S. 2006. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 281:8981-8990.

52
25. Itoh S, Fukuzumi S. 2007. Monooxygenase activity of type 3 copper proteins. Acc. Chem. Res. 40:592-600.
26. Virador VM, Reyes Grajeda JP, Blanco-Labra A, Mendiola-Olaya E, Smith GM, Moreno A, Whitaker JR. 2010. Cloning, sequencing, purification, and crystal structure of Grenache (Vitis vinifera) polyphenol oxidase. J. Agric. Food Chem. 58:1189-1201.
27. Eicken C, Krebs B, Sacchettini JC. 1999. Catechol oxidase -- structure and activity. Curr. Opin. Struct. Biol. 9:677-683.
28. Cuff ME, Miller KI, van Holde KE, Hendrickson WA. 1998. Crystal structure of a functional unit from Octopus hemocyanin. J. Mol. Biol. 278:855-870.
29. Gaykema WPJ, Hol WGJ, Vereijken JM, Soeter NM, Bak HJ, Beintema JJ. 1984. 3.2 A structure of the copper-containing, oxygen-carrying protein Panulirus interruptus haemocyanin. Nature 309:23-29.
30. Hazes B, Magnus KA, Bonaventura C, Bonaventura J, Dauter Z, Kalk KH, Hol WG. 1993. Crystal structure of deoxygenated Limulus polyphemus subunit II hemocyanin at 2.18 A resolution: clues for a mechanism for allosteric regulation. Protein Sci. 2:597-619.
31. Li Y, Wang Y, Jiang H, Deng J. 2009. Crystal structure of Manduca sexta prophenoloxidase provides insights into the mechanism of type 3 copper enzymes. Proc. Natl. Acad. Sci. U. S. A. 106:17002-17006.
32. Decker H, Rimke T. 1998. Tarantula hemocyanin shows phenoloxidase activity. J. Biol. Chem. 273:25889-25892.
33. Klabunde T, Eicken C, Sacchettini JC, Krebs B. 1998. Crystal structure of plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 5:1084-1090.
34. Decker H, Tuczek F. 2000. Tyrosinase/catecholoxidase activity of hemocyanins: structural basis and molecular mechanism. Trends Biochem. Sci. 25:392-397.
35. van Gastel M, Bubacco L, Groenen EJJ, Vijgenboom E, Canters GW. 2000. EPR study of the dinuclear active copper site of tyrosinase from Streptomyces antibioticus. FEBS Lett. 474:228-232.
36. Fenoll LG, Penalver MJ, Rodriguez-Lopez JN, Varon R, Garcia-Canovas F, Tudela J. 2004. Tyrosinase kinetics: discrimination between two models to explain the oxidation mechanism of monophenol and diphenol substrates. Int. J. Biochem. Cell B. 36:235-246.
37. Decker H, Schweikardt T, Nillius D, Salzbrunn U, Jaenicke E, Tuczek F. 2007. Similar enzyme activation and catalysis in hemocyanins and tyrosinases. Gene 398:183-191.
38. Olivares C, Solano F. 2009. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigm. Cell Melanoma R. 22:750-760.
39. Olivares C, Garcia-Borron JC, Solano F. 2002. Identification of active site residues involved in metal cofactor binding and stereospecific substrate recognition in mammalian tyrosinase. Implications to the catalytic cycle. Biochemistry 41:679-686.
40. Deeth RJ, Diedrich C. 2010. Structural and mechanistic insights into the oxy form of tyrosinase from molecular dynamics simulations. J. Biol. Inorg. Chem. 15:117-129.
41. Rolff M, Schottenheim J, Decker H, Tuczek F. 2011. Copper-O2 reactivity of tyrosinase models towards external monophenolic substrates: molecular mechanism and comparison with the enzyme. Chem. Soc. Rev. 40:4077-4098.
42. Muñoz-muñoz JL, García-molina F, García-ruiz PA, Molina-alarcón M, Tudela J, García-cánovas F, Rodríguez-lópez JN. 2008. Phenolic substrates and suicide inactivation of tyrosinase: kinetics and mechanism. Biochem. J. 416:431-440.
43. Muñoz-Muñoz JL, del Mar García-Molina M, Garcia-Molina F, Garcia-Ruiz PA, Garcia-Sevilla F, Rodriguez-Lopez JN, Garcia-Canovas F. 2013. Deuterium isotope effect on the suicide inactivation of tyrosinase in its action on o-diphenols. IUBMB Life 65:793-799.
44. Rodríguez-López JN, Fenoll LG, Peñalver MJ, García-Ruiz PA, Varón R, Martínez-Ortíz F, García-Cánovas F, Tudela J. 2001. Tyrosinase action on monophenols: evidence for direct enzymatic release of o-diphenol. BBA-Protein Struct. M. 1548:238-256.

53
45. Halaouli S, Asther M, Kruus K, Guo L, Hamdi M, Sigoillot JC, Asther M, Lomascolo A. 2005. Characterization of a new tyrosinase from Pycnoporus species with high potential for food technological applications. J. Appl. Microbiol. 98:332-343.
46. Halaouli S, Asther M, Sigoillot JC, Hamdi M, Lomascolo A. 2006. Fungal tyrosinases: new prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 100:219-232.
47. Lantto R, Puolanne E, Kruus K, Buchert J, Autio K. 2007. Tyrosinase-aided protein cross-linking: effects on gel formation of chicken breast myofibrils and texture and water-holding of chicken breast meat homogenate gels. J. Agric. Food Chem. 55:1248-1255.
48. Thalmann C, Lötzbeyer T. 2002. Enzymatic cross-linking of proteins with tyrosinase. Eur Food Res Technol 214:276-281.
49. Nolan L, O’Connor K. 2007. Use of Pseudomonas mendocina , or recombinant Escherichia coli cells expressing toluene-4-monooxygenase, and a cell-free tyrosinase for the synthesis of 4-fluorocatechol from fluorobenzene. Biotechnol. Lett. 29:1045-1050.
50. Tuncagil S, Kayahan SK, Bayramoglu G, Arica MY, Toppare L. 2009. L-Dopa synthesis using tyrosinase immobilized on magnetic beads. J. Mol. Catal. B-Enzym. 58:187-193.
51. Bui VP, Hansen TV, Stenstrøm Y, Hudlicky T. 2000. Direct biocatalytic synthesis of functionalized catechols: a green alternative to traditional methods with high effective mass yield. Green Chem. 2:263-265.
52. Held M, Suske W, Schmid A, Engesser K-H, Kohler H-PE, Witholt B, Wubbolts MG. 1998. Preparative scale production of 3-substituted catechols using a novel monooxygenase from Pseudomonas azelaica HBP 1. J. Mol. Catal. B-Enzym. 5:87-93.
53. Espin JC, Soler-Rivas C, Cantos E, Tomas-Barberan FA, Wichers HJ. 2001. Synthesis of the antioxidant hydroxytyrosol using tyrosinase as biocatalyst. J. Agric. Food Chem. 49:1187-1193.
54. Burton SG. 2003. Oxidizing enzymes as biocatalysts. Trends Biotechnol. 21:543-549. 55. Sambrook J, Green MR. 2001. Molecular cloning: a laboratory manual, 4th ed. Cold Spring
Harbor Laboratory Press, New York. 56. Rodriguez-lopez JN, Escribano J, Garciacanovas F. 1994. A continuous
spectrophotometric method for the determination of monophenolase activity of tyrosinase using 3-methyl-2-benzothiazolinone hydrazone. Anal. Biochem. 216:205-212.
57. Leslie AGW. 1992. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography:No. 26. 58. McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with
Phaser. Acta Crystallogr. D 63:32-41. 59. Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS,
Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54:905-921.
60. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66:213-221.
61. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240-255.
62. Skubak P, Murshudov GN, Pannu NS. 2004. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D 60:2196-2201.
63. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60:2126-2132.

54
64. Selinheimo E, NiEidhin D, Steffensen C, Nielsen J, Lomascolo A, Halaouli S, Record E, O'Beirne D, Buchert J, Kruus K. 2007. Comparison of the characteristics of fungal and plant tyrosinases. J. Biotechnol. 130:471-480.
65. Sendovski M, Kanteev M, Ben-Yosef VS, Adir N, Fishman A. 2011. First structures of an active bacterial tyrosinase reveal copper plasticity. J. Mol. Biol. 405:227-237.
66. Han HY, Zou HC, Jeon JY, Wang YJ, Xu WA, Yang JM, Park YD. 2007. The inhibition kinetics and thermodynamic changes of tyrosinase via the zinc ion. Biochim. Biophys. Acta 1774:822-827.
67. Shuster V, Sendovski M, Fishman A. 2010. Directed evolution of tyrosinase for enhanced monophenolase/diphenolase activity ratio. Enzyme Microb. Technol. In revision.
68. Goldfeder M, Egozy M, Shuster Ben-Yosef V, Adir N, Fishman A. 2013. Changes in tyrosinase specificity by ionic liquids and sodium dodecyl sulfate. Appl. Microbiol. Biotechnol. 97:1953-1961.
69. Kanteev M, Goldfeder M, Chojnacki M, Adir N, Fishman A. 2013. The mechanism of copper uptake by tyrosinase from Bacillus megaterium. J Biol Inorg Chem 18:895-903.
70. Lavie A, Allen KN, Petsko GA, Ringe D. 1994. X-ray crystallographic structures of D-xylose isomerase-substrate complexes position the substrate and provide evidence for metal movement during catalysis. Biochemistry 33:5469-5480.
71. Bailey LJ, McCoy JG, Phillips GN, Fox BG. 2008. Structural consequences of effector protein complex formation in a diiron hydroxylase. Proc. Natl. Acad. Sci. U. S. A. 105:19194-19198.
72. Walter NG. 2007. Ribozyme Catalysis Revisited: Is Water Involved? Molecular Cell 28:923-929.
73. Siegbahn PE, Borowski T. 2011. Comparison of QM-only and QM/MM models for the mechanism of tyrosinase. Faraday discussions 148:109-117; discussion 207-128.
74. Muñoz-Muñoz JL, Berna J, Garcia-Molina F, Garcia-Ruiz PA, Tudela J, Rodriguez-Lopez JN, Garcia-Canovas F. 2012. Unravelling the suicide inactivation of tyrosinase: A discrimination between mechanisms. J. Mol. Catal. B-Enzym. 75:11-19.
75. Kleifeld O, Frenkel A, Martin JML, Sagi I. 2003. Active site electronic structure and dynamics during metalloenzyme catalysis. Nat Struct Mol Biol 10:98-103.
76. Bochot C, Gouron A, Bubacco L, Milet A, Philouze C, Reglier M, Serratrice G, Jamet H, Belle C. 2014. Probing kojic acid binding to tyrosinase enzyme: insights from a model complex and QM/MM calculations. Chemical Communications 50:308-310.
77. Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. 2012. Engineering the third wave of biocatalysis. Nature 485:185-194.
78. Dror A, Fishman A. 2012. Engineering Non-Heme Mono- and Dioxygenases for Biocatalysis. Computational and Structural Biotechnology Journal 2.
79. Sendovski M, Kanteev M, Shuster V, Adir N, Fishman A. 2010. Primary x-ray crystallographic analysis of a bacterial tyrosinase from Bacillus megaterium. Acta Crystallogr. D Accepted.
80. Ben-David M, Wieczorek G, Elias M, Silman I, Sussman JL, Tawfik DS. 2013. Catalytic metal ion rearrangements underline promiscuity and evolvability of a metalloenzyme. J. Mol. Biol. 425:1028-1038.
81. Torrance JW, MacArthur MW, Thornton JM. 2008. Evolution of binding sites for zinc and calcium ions playing structural roles. Proteins: Struct., Funct., Genet. 71:813-830.
82. Karimäki J, Parkkinen T, Santa H, Pastinen O, Leisola M, Rouvinen J, Turunen O. 2004. Engineering the substrate specificity of xylose isomerase. Protein Engineering Design and Selection 17:861-869.
83. Matoba Y, Bando N, Oda K, Noda M, Higashikawa F, Kumagai T, Sugiyama M. 2011. A molecular mechanism for copper transportation to tyrosinase that is assisted by a metallochaperone, caddie protein. J. Biol. Chem. 286:30219-30231.

55
84. Guo Y, Smith K, Lee J, Thiele DJ, Petris MJ. 2004. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human ctr1 copper transporter. J. Biol. Chem. 279:17428-17433.
85. Singh SK, Roberts SA, McDevitt SF, Weichsel A, Wildner GF, Grass GB, Rensing C, Montfort WR. 2011. Crystal structures of multicopper oxidase CueO bound to copper(I) and silver(I): Functional role of a methionine-rich sequence J. Biol. Chem. 286:37849-37857.
86. Ramsden CA, Riley PA. 2010. Mechanistic studies of tyrosinase suicide inactivation. ARKIVOC 1:260-274.
87. Bommarius AS, Blum JK, Abrahamson MJ. 2011. Status of protein engineering for biocatalysts: how to design an industrially useful biocatalyst. Curr. Opin. Chem. Biol. 15:194-200.
88. Domingez de Maria P. 2011. Ionic liquids in biotransformations and organocatalysis: solvents and beyond. Wiley & Sons, Hoboken, New Jersey.
89. Yang Z, Yue YJ, Huang WC, Zhuang XM, Chen ZT, Xing M. 2009. Importance of the ionic nature of ionic liquids in affecting enzyme performance. J. Biochem. 145:355-364.
90. Yang Z. 2009. Hofmeister effects: an explanation for the impact of ionic liquids on biocatalysis. J. Biotechnol. 144:12-22.
91. Otzen D. 2011. Protein–surfactant interactions: A tale of many states. Biochim. Biophys. Acta, Proteins Proteomics 1814:562-591.
92. Gandia-Herrero F, Jimenez-Atienzar M, Cabanes J, Garcia-Carmona F, Escribano J. 2005. Differential activation of a latent polyphenol oxidase mediated by sodium dodecyl sulfate. J. Agric. Food Chem. 53:6825-6830.
93. Lopez-Serrano D, Sanchez-Amat A, Solano F. 2002. Cloning and molecular characterization of a SDS-activated tyrosinase from Marinomonas mediterranea. Pigm. Cell Res. 15:104-111.
94. Saeidian S, Keyhani E, Keyhani J. 2007. Effect of ionic detergents, nonionic detergents, and chaotropic agents on polyphenol oxidase activity from dormant saffron (Crocus stivus L.) corms. J. Agric. Food Chem. 55:3713-3719.
95. Espín JC, Wichers HJ. 1999. Activation of a latent mushroom (Agaricus bisporus) tyrosinase isoform by sodium dodecyl sulfate (SDS). Kinetic properties of the SDS-activated isoform. J. Agric. Food Chem. 47:3518-3525.
96. Goldfeder M, Fishman A. 2013. Modulating enzyme activity using ionic liquids or surfactants. Appl. Microbiol. Biotechnol.





























