Embed Size (px)
Citation preview

1990 Research Article
IntroductionPoly(ADP-ribosyl)ation is a NAD+-dependent post-translational
modification of proteins mediated by poly(ADP-ribose) polymerases
(PARPs). It is involved in various biological processes such as DNA
repair, transcription, mitotic segregation, telomere homeostasis and
cell death (Schreiber et al., 2006). PARP1, the founding member
of the PARP family, can detect DNA strand breaks and can become
activated and poly(ADP-ribosyl)ate itself or acceptor proteins
involved in chromatin structure and DNA metabolism. PARP1
activation and PAR synthesis have several outcomes: (1) labelling
of the damaged site by the PAR molecules; (2) local opening of the
chromatin structure by the transient heteromodification of histones;
(3) recruitment to the damage site of repair factors with strong
affinity for PAR, such as the X-ray repair cross-complementing
protein 1 (XRCC1); and (4) signaling of the severity of the DNA
insult for the cell to adapt its response accordingly (reviewed by
Schreiber et al., 2006).
PAR accumulation is transient. PAR is rapidly degraded by
poly(ADP-ribose) glycohydrolase (PARG), an enzyme with both
exo- and endoglycosidase activities that generate large amounts of
free ADP-ribose. In contrast to the 17 members of the human PARP
family (Amé et al., 2004; Schreiber et al., 2006), human PARG is
encoded by a single gene, but is present within the cell through
different isoforms displaying various subcellular localizations:
nuclear (PARG111), cytoplasmic (PARG102 and PARG99) and
mitochondrial (PARG60) (Meyer et al., 2007). Study of the
functional role of PARG through the generation of mutant mouse
models has been hampered by the existence of these isoforms. A
hypomorphic mutation targeting exons 2 and 3 (PARG�2/�3 mice)
is viable, but the mice are sensitive to ionizing radiation and
alkylating agents (Cortes et al., 2004). However, these mutant mice
displayed a 3.3-fold increase of the mitochondrial PARG activity,
which could partially compensate for the absence of the longer
PARG isoforms (Cortes et al., 2004). Targeting exon 4 of the PARGgene abolished any isoform production and led to early embryonic
lethality (Koh et al., 2004). Trophoblast PARG–/– cells were shown
to be highly sensitive to alkylating agents, to accumulate PAR and
to die by apoptosis, highlighting the crucial role of PARG in the
response to DNA damage. Similarly, a loss-of-function mutation in
the Drosophila melanogaster PARG gene led to increased lethality
associated with PAR-accumulation-induced neurodegeneration at
the larval stages (Hanai et al., 2004). Attempts to transiently silence
PARG expression with siRNA led to controversial results: either
protection (Blenn et al., 2006) or sensitivity (Fisher et al., 2007)
towards H2O2-induced cell death, which might be explained by
differences in cell types analyzed (HeLa versus A549 cells).
Many studies revealed the importance of PARP activity and PAR
levels in the control of the life-and-death balance following DNA
damage. PAR synthesis in response to DNA strand breaks allows
a rapid cellular evaluation of the damage range, with the outcome
depending on the cell type and the intensity of the DNA insult. In
neuronal cells, DNA damage triggers cell death by a caspase-
independent mechanism involving the mitochondrial apoptosis-
inducing factor (AIF) (Yu et al., 2002). PAR synthesis, which might
Poly(ADP-ribosyl)ation is a post-translational modification of
proteins involved in the regulation of chromatin structure, DNA
metabolism, cell division and cell death. Through the hydrolysis
of poly(ADP-ribose) (PAR), Poly(ADP-ribose) glycohydrolase
(PARG) has a crucial role in the control of life-and-death
balance following DNA insult. Comprehension of PARG
function has been hindered by the existence of many PARG
isoforms encoded by a single gene and displaying various
subcellular localizations. To gain insight into the function of
PARG in response to irradiation, we constitutively and stably
knocked down expression of PARG isoforms in HeLa cells.
PARG depletion leading to PAR accumulation was not
deleterious to undamaged cells and was in fact rather beneficial,
because it protected cells from spontaneous single-strand breaks
and telomeric abnormalities. By contrast, PARG-deficient cells
showed increased radiosensitivity, caused by defects in the
repair of single- and double-strand breaks and in mitotic
spindle checkpoint, leading to alteration of progression of
mitosis. Irradiated PARG-deficient cells displayed centrosome
amplification leading to mitotic supernumerary spindle poles,
and accumulated aberrant mitotic figures, which induced either
polyploidy or cell death by mitotic catastrophe. Our results
suggest that PARG could be a novel potential therapeutic target
for radiotherapy.
Supplementary material available online at
http://jcs.biologists.org/cgi/content/full/122/12/1990/DC1
Key words: NAD+ metabolism, PARP, DNA repair, Mitosis, Genome
stability
Summary
Radiation-induced mitotic catastrophe in PARG-deficient cellsJean-Christophe Amé1,*, Elise Fouquerel1,*, Laurent R. Gauthier2, Denis Biard3, François D. Boussin2,Françoise Dantzer1, Gilbert de Murcia1 and Valérie Schreiber1,‡
1IREBS-FRE3211 du CNRS, Université de Strasbourg, ESBS, Bd Sébastien Brant, BP 10413, 67412 Illkirch Cedex, France2Laboratoire de Radiopathologie/INSERM U967, CEA-DSV-IRCM, 92265 Fontenay aux Roses, Cedex 6, France3CEA-DSV-IRCM/INSERM U935, Institut A. Lwoff-CNRS, BP 8, 94801 Villejuif cedex, France*These authors contributed equally to this work‡Author for correspondence (e-mail: [email protected])
Accepted 24 February 2009Journal of Cell Science 122, 1990-2002 Published by The Company of Biologists 2009doi:10.1242/jcs.039115

1991Radiosensitivity of PARG-deficient cells
generate free polymers through the action of PARG, is required for
AIF to translocate from the mitochondria to the nucleus where it
induces large-scale DNA fragmentation (Andrabi et al., 2006;
Moubarak et al., 2007; Yu et al., 2006). The subsequent PARP1
overactivation leads to massive PAR synthesis, NAD+/ATP
depletion and, finally, cell death (Yu et al., 2002). PARP1–/– mice
and mice treated with PARP inhibitors are thus protected from many
pathophysiological situations where DNA is the target of massive
attack by reactive oxygen species, such as acute and chronic
inflammatory diseases, ischemia, as well as neurodegenerative
disorders (Jagtap and Szabo, 2005). In contrast to PARG+/– mice,
transgenic mice overexpressing PARG showed similar protection
against cerebral ischemia as observed in PARP1–/– mice (Andrabi
et al., 2006), confirming the beneficial effect of decreasing PAR
levels. Taken together, these data underscore how the fine-tuning
of PARP1 and PARG activity must precisely control the level of
PAR and the balance between life and death decisions.
In this study, we have generated a cellular model deficient in all
PARG isoforms by the stable and constitutive expression of a
shRNA, to gain insight into the function of PARG in the cell
response to irradiation. In normal conditions, PARG depletion
conferred protection against spontaneous single-strand breaks
(SSBs) and telomere aberrations, and showed a spontaneous
activation of ATM (ataxia telangiectasia mutated) along with
γH2AX phosphorylation. However, PARG-depleted cells were
radiosensitive, accumulated radioinduced PAR and showed slower
rates of repair of SSBs and double-strand breaks (DSBs). In
addition, they displayed radioinduced centrosome amplification and
fragmentation, and alteration of mitotic progression, with the
accumulation of aberrant mitotic figures. These aberrant mitotic cells
resulted in aneuploidy, polyploidy or cell death by mitotic
catastrophe.
ResultsAccumulation of PAR in shRNA-mediated stable knockdown ofPARG in HeLa cellsWe generated a stable HeLa cell line PARGKD, which constitutively
expresses a shRNA directed against PARG from an episomal
plasmid (Biard, 2007). The BD650 control line expresses a non
functional shRNA (Biard et al., 2005). The sequence of the PARG
shRNA maps to the catalytic domain of PARG, thus preventing the
synthesis of all PARG isoforms, which was confirmed by RT-PCR
(Fig. 1A). Downregulation of PARG111 was confirmed by western
blot using an antibody raised against the N-terminal part of PARG
(Fig. 1B) and by a zymogram, which showed that the PARG activity
detected at 111 kDa was dramatically decreased in the PARGKD
Fig. 1. Efficient shRNA-mediated stable and constitutive knockdown of PARG in HeLa cells leads to accumulation and persistence of PAR. (A) RT-PCR analysison total RNA extracted from BD650 control (lane 1) or PARGKD (lane 2) cells with primers specific for PARG or, as a negative control, Ogg1 transcripts.(B) Western blot analysis of BD650 control (lane 1) or PARGKD (lane 2) cell lysates probed successively with an anti-PARG N-terminal antibody and an anti-actinantibody to control loading. (C) Zymogram performed by running lysates from BD650 control (lane 1) or PARGKD (lane 2) cells on a 10% SDS-PAGE gelcontaining 32P-labelled automodified PARP1. The gel was renaturated before autoradiography. (D) Western blot analysis of PARGKD (lanes 1-3) or BD650 control(lanes 4-6) cell lysates successively probed with the 10H monoclonal anti-PAR antibody and with an anti-actin antibody to control loading. Lanes 1 and 4:untreated; lanes 2 and 5: 1 mM H2O2, 10 minutes; lanes 3 and 5: 1 mM H2O2 10 minutes, then incubated at 37°C in fresh medium for the indicated time.(E) Immunodetection of PAR (10H) in BD650 control or PARGKD cell either untreated or irradiated at 6 Gy and fixed at the indicated times. DNA is counterstainedwith DAPI. Scale bar: 20 μm.

1992
cell extract (Fig. 1C). No PARG activity was detected at sizes
corresponding to low molecular mass PARG isoforms, in either the
BD650 or PARGKD cell extracts. Altogether, these results validate
the efficiency of PARG silencing by this constitutive shRNA
approach. The efficiency of PARGKD was unaffected even after
long-term culture of the cells for more than 6 months (data not
shown).
Downregulation of PARG expression led to genotoxic-stress-
independent accumulation of PAR in PARGKD cells observed by
western blot (Fig. 1D, compare lanes 1 and 4) and
immunofluorescence microscopy (Fig. 1E). After treatment with 1
mM H2O2, more PAR accumulated in PARGKD cells than in BD650
control cells. Ninety minutes after H2O2 treatment, the amount of
PAR returned to its basal level in BD650 control cells but remained
high in the PARGKD cells (Fig. 1D, lanes 3 and 6), confirming the
lack of PARG activity. A similar persistence of PAR following H2O2
treatment of PARGKD cells was observed by immunofluorescence
microscopy using an anti-PAR antibody, and by measuring PARG
activity in permeabilized cells incubated with 32P-labelled NAD+
(data not shown). Accumulation and persistence of PAR was also
observed by immunofluorescence microscopy following X-
irradiation of PARGKD cells at 6 Gy: whereas PAR returned to basal
levels within 60 minutes after X-irradiation in control cells, PARGKD
cells still displayed high levels of PAR 90 minutes after irradiation
(Fig. 1E), which remained high for up to 3 hours (data not shown).
Altogether, these results indicate that PARG expression is
efficiently knocked down in PARGKD cells, resulting in both
genotoxic-stress-independent PAR synthesis and DNA-damage-
induced accumulation and persistence of PAR.
PARGKD cells display repair defect of radio-induced SSBs andDSBsThe observation of PAR synthesis in untreated PARGKD cells led
us to hypothesize that spontaneous DNA breaks might accumulate
in the absence of PARG, which could reflect a repair defect. To test
this hypothesis, we monitored the level of DNA strand breaks using
the alkaline COMET assay, which detects both SSBs and DSBs,
and the neutral COMET assay, which detects only DSBs. Alkaline
COMET assay (Fig. 2A; supplementary material Fig. S1A) revealed
a slight and reproducible, although statistically not significant,
smaller number of breaks in undamaged PARGKD cells compared
with those in control cells. This probably corresponds to SSBs,
because neutral COMET assay showed no spontaneous DSBs in
each cell line (Fig. 2B; supplementary material Fig. S1B), and
suggests first that PARG-silenced cells could be protected from
spontaneous SSBs, and second, that the PAR detected in the absence
of genotoxic insult is probably not caused by the accumulation of
spontaneous SSBs. In addition, a 6 Gy X-irradiation generated fewer
SSBs and DSBs in the PARGKD cells compared with the control
cells – a reproducible, although not statistically significant,
observation. This might be attributed to a quenching effect of the
PAR molecules towards free radicals generated by ionization. Time-
course evaluation of strand break resealing by alkaline COMET
assay revealed an almost complete repair of breaks as early as 30
minutes after irradiation in the BD650 control cells, whereas
PARGKD cells required more than 90 minutes to reach a similar
level of repair, indicating the presence of a repair defect in these
cells (Fig. 2A; supplementary material Fig. S1A). Neutral COMET
assay revealed a defect in the repair of radioinduced DSBs in
PARGKD cells compared with BD650 control cells (Fig. 2B;
supplementary material Fig. S1B). Taken together, these results
Journal of Cell Science 122 (12)
indicate that PARG is required for efficient repair of both SSBs and
DSBs.
We then examined the recruitment of the SSB repair factor
XRCC1, which normally accumulates within seconds at damaged
sites as a result of its strong affinity for PAR (El-Khamisy et al.,
2003; Mortusewicz et al., 2007; Mortusewicz and Leonhardt, 2007;
Okano et al., 2003; Pleschke et al., 2000). DNA-damaged sites were
introduced locally by UVA laser microirradiation in the presence
Fig. 2. SSB and DSB repair defect in PARGKD irradiated cells. Kinetics ofreligation of SSBs (A) or DSBs (B) in 6-Gy-irradiated BD650 control (bluecircles) and PARGKD (red squares) cells visualized by the alkaline (A) orneutral (B) COMET assay. The % of tail DNA versus total DNA is indicated asa function of time. Fifty cells were scored for each time point.(C) Immunostaining of PAR (left panels) and XRCC1 (middle panels) atdamaged sites locally introduced by laser microirradiation in BD650 control(upper panels) and PARGKD (lower panels) cells. DNA is stained with DAPI.Scale bar: 20 μm.
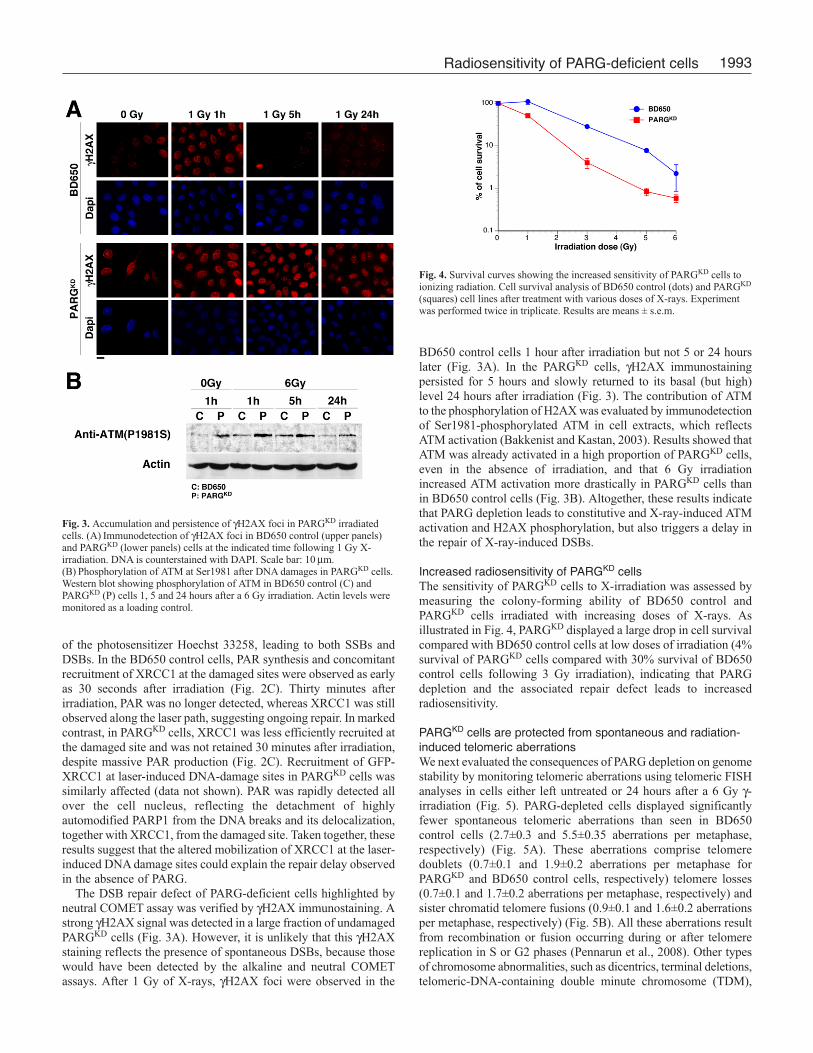
1993Radiosensitivity of PARG-deficient cells
of the photosensitizer Hoechst 33258, leading to both SSBs and
DSBs. In the BD650 control cells, PAR synthesis and concomitant
recruitment of XRCC1 at the damaged sites were observed as early
as 30 seconds after irradiation (Fig. 2C). Thirty minutes after
irradiation, PAR was no longer detected, whereas XRCC1 was still
observed along the laser path, suggesting ongoing repair. In marked
contrast, in PARGKD cells, XRCC1 was less efficiently recruited at
the damaged site and was not retained 30 minutes after irradiation,
despite massive PAR production (Fig. 2C). Recruitment of GFP-
XRCC1 at laser-induced DNA-damage sites in PARGKD cells was
similarly affected (data not shown). PAR was rapidly detected all
over the cell nucleus, reflecting the detachment of highly
automodified PARP1 from the DNA breaks and its delocalization,
together with XRCC1, from the damaged site. Taken together, these
results suggest that the altered mobilization of XRCC1 at the laser-
induced DNA damage sites could explain the repair delay observed
in the absence of PARG.
The DSB repair defect of PARG-deficient cells highlighted by
neutral COMET assay was verified by γH2AX immunostaining. A
strong γH2AX signal was detected in a large fraction of undamaged
PARGKD cells (Fig. 3A). However, it is unlikely that this γH2AX
staining reflects the presence of spontaneous DSBs, because those
would have been detected by the alkaline and neutral COMET
assays. After 1 Gy of X-rays, γH2AX foci were observed in the
BD650 control cells 1 hour after irradiation but not 5 or 24 hours
later (Fig. 3A). In the PARGKD cells, γH2AX immunostaining
persisted for 5 hours and slowly returned to its basal (but high)
level 24 hours after irradiation (Fig. 3). The contribution of ATM
to the phosphorylation of H2AX was evaluated by immunodetection
of Ser1981-phosphorylated ATM in cell extracts, which reflects
ATM activation (Bakkenist and Kastan, 2003). Results showed that
ATM was already activated in a high proportion of PARGKD cells,
even in the absence of irradiation, and that 6 Gy irradiation
increased ATM activation more drastically in PARGKD cells than
in BD650 control cells (Fig. 3B). Altogether, these results indicate
that PARG depletion leads to constitutive and X-ray-induced ATM
activation and H2AX phosphorylation, but also triggers a delay in
the repair of X-ray-induced DSBs.
Increased radiosensitivity of PARGKD cellsThe sensitivity of PARGKD cells to X-irradiation was assessed by
measuring the colony-forming ability of BD650 control and
PARGKD cells irradiated with increasing doses of X-rays. As
illustrated in Fig. 4, PARGKD displayed a large drop in cell survival
compared with BD650 control cells at low doses of irradiation (4%
survival of PARGKD cells compared with 30% survival of BD650
control cells following 3 Gy irradiation), indicating that PARG
depletion and the associated repair defect leads to increased
radiosensitivity.
PARGKD cells are protected from spontaneous and radiation-induced telomeric aberrationsWe next evaluated the consequences of PARG depletion on genome
stability by monitoring telomeric aberrations using telomeric FISH
analyses in cells either left untreated or 24 hours after a 6 Gy γ-irradiation (Fig. 5). PARG-depleted cells displayed significantly
fewer spontaneous telomeric aberrations than seen in BD650
control cells (2.7±0.3 and 5.5±0.35 aberrations per metaphase,
respectively) (Fig. 5A). These aberrations comprise telomere
doublets (0.7±0.1 and 1.9±0.2 aberrations per metaphase for
PARGKD and BD650 control cells, respectively) telomere losses
(0.7±0.1 and 1.7±0.2 aberrations per metaphase, respectively) and
sister chromatid telomere fusions (0.9±0.1 and 1.6±0.2 aberrations
per metaphase, respectively) (Fig. 5B). All these aberrations result
from recombination or fusion occurring during or after telomere
replication in S or G2 phases (Pennarun et al., 2008). Other types
of chromosome abnormalities, such as dicentrics, terminal deletions,
telomeric-DNA-containing double minute chromosome (TDM),
Fig. 3. Accumulation and persistence of γH2AX foci in PARGKD irradiatedcells. (A) Immunodetection of γH2AX foci in BD650 control (upper panels)and PARGKD (lower panels) cells at the indicated time following 1 Gy X-irradiation. DNA is counterstained with DAPI. Scale bar: 10 μm.(B) Phosphorylation of ATM at Ser1981 after DNA damages in PARGKD cells.Western blot showing phosphorylation of ATM in BD650 control (C) andPARGKD (P) cells 1, 5 and 24 hours after a 6 Gy irradiation. Actin levels weremonitored as a loading control.
Fig. 4. Survival curves showing the increased sensitivity of PARGKD cells toionizing radiation. Cell survival analysis of BD650 control (dots) and PARGKD
(squares) cell lines after treatment with various doses of X-rays. Experimentwas performed twice in triplicate. Results are means ± s.e.m.

1994
resulting respectively from fusions, deletions or recombinations in
G1, were found at similar frequencies in PARGKD and BD650
control cells (Fig. 5C). Interestingly, a 6 Gy irradiation increased
the frequency of all types of aberrations in BD650 control cells,
whereas PARGKD cells still displayed significant protection against
telomere losses (0.9±0.2 and 3.2±0.7 aberrations per metaphase for
PARGKD and BD650 control cells, respectively) and sister chromatid
fusions (1.2±0.2 and 2.0±0.3 aberrations per metaphase,
respectively), leaving other types of aberrations comparable with
the irradiated BD650 control cells (Fig. 5). These compelling results
Journal of Cell Science 122 (12)
indicate that PARG depletion protects telomeres from spontaneous
and radiation-induced aberrations occurring in S-G2 phases, but has
no apparent effect on telomeres during G1 phase.
Irradiated PARGKD cells display mitotic abnormalitiesTo evaluate the consequence of PAR and DNA-damage persistence
in PARGKD cells on cell cycle progression, FACS analyses were
performed on PARGKD and BD650 control cells, 24 hours and 48
hours after 3 or 6 Gy X-irradiation. The cell cycle distribution of
both cell lines was comparable in the absence of treatment or after
3 Gy of X-rays (Fig. 6A; and data not shown). Twenty-four hours
after a 6 Gy X-irradiation, PARGKD cells accumulated at the G2-
M phase of the cell cycle. To determine whether cells accumulated
at G2 phase or were blocked in mitosis, we scored the proportion
of mitotic cells identified by immunodetection of phosphorylated
H3 Serine 10 (Ser10-H3-P) and DAPI staining. Results showed
accumulation of mitotic cells in 6 Gy-irradiated PARGKD cells
compared to BD650 control cells (13.4% vs. 5.9%) (Fig. 6B). We
then scored by immunofluorescence microscopy the number of cells
in the different phases of mitosis, by immunodetecting P-Ser10-H3
and staining microtubules with anti-α tubulin, together with DAPI
staining. Results revealed that 6 Gy irradiation led to a reduction
of the proportion of PARGKD cells in anaphase (6% vs 18% for
irradiated BD650 control cells) and an accumulation of irradiated
PARGKD cells at prometaphase or metaphase (71% vs 62% for
irradiated BD650 control cells) (Fig. 6C), suggesting that PARG is
required for mitotic progression of damaged cells. Forty-eight hours
after a 6 Gy irradiation, a higher proportion of PARGKD cells
(13.02% vs 5.79 % for BD650 control cells) displayed polyploidy
(8n chromosomes) (Fig. 6A), suggesting a defective chromosome
segregation or cytokinesis.
To elucidate the cause of the mitotic arrest in irradiated PARGKD
cells, we examined the integrity of the mitotic spindle by
immunofluorescence microscopy, staining microtubules with anti-
α tubulin, spindle poles with anti-Cdk1, centromeres with CREST
antibody and DNA with DAPI. Twenty-four hours after a 6 Gy
irradiation, we observed many aberrant mitotic figures throughout
all stages of mitosis in irradiated PARGKD and particularly
misaligned chromosomes on the equatorial plaque in cells in
prometaphase or metaphase and mis-segregated chromosomes in
anaphase (Fig. 7A). This suggests that there is a defect in complete
chromosome congression to the metaphase plate and a defect in the
kinetochore function in the absence of PARG. In addition, we
observed numerous cells with supernumerary spindle poles from
Fig. 5. PARGKD cells show protection from spontaneous and γ-irradiation-induced telomeric aberrations. Percentage of telomeric aberrations per cell inmetaphase spreads of PARGKD and control HeLa cells (BD650KD) 24 hoursafter a 0 or 6 Gy γ-irradiation. Metaphase spreads were hybridizedsuccessively with telomeric PNA probe (in red) and a centromeric DNA probe(in green) and then counterstained with DAPI (blue). Total (A) or specifictypes of aberrations (B and C) were quantified per metaphase.(B) Quantification of telomeric aberrations occurring during or after telomerereplication in S or G2 phases. Telemeric aberrations were assigned to one ofthree groups: one extra telomere signal or ‘telomere doublet’, ‘one telomereloss’, or ‘sister chromatid telomere fusion’. (C) Quantification of telomericaberrations resulting from fusions, deletions or recombinations occurring inG1: ‘dicentric chromosome’, chromosome with ‘terminal deletion’ ortelomeric DNA-containing double minutes chromosomes (TDM). Resultswere obtained from three independent experiments. The number of metaphasesanalyzed is indicated in supplementary material Table S1. Results are means ±s.e.m. *P<0.05; **P<0.01; ***P<0.001 using the Student’s t-test; ns, non-significant difference.

1995Radiosensitivity of PARG-deficient cells
which some microtubules emanate to bind to kinetochores (Fig.
7Ab). We therefore monitored the number of multipolar versus
bipolar metaphases after 3 Gy (data not shown) and 6 Gy X-
irradiation (Fig. 7B). The proportion of multipolar metaphases in
PARGKD cells increased dramatically with the irradiation dose 24
hours after irradiation, and this proportion was even higher at 48
hours after irradiation (Fig. 7B). To confirm our observations with
live-cell videomicroscopy, we generated HeLa cell lines that
constitutively and stably expressed GFP-tagged histone H2B (H2B-
GFP) together with either control shRNA (H2B-GFP/BD650) or
PARG shRNA (H2B-GFP/PARGKD). Progression into mitosis was
followed 24 hours after 6 Gy irradiation for a period of 10 hours.
Results displayed in supplementary material Fig. S2, Table S2 and
Movies 1-6 showed that the duration of mitosis for unirradiated
H2B-GFP/PARGKD cells was increased compared with H2B-
GFP/BD650 cells (115 minutes versus 80 minutes, respectively)
(supplementary material Fig. S2A), owing to cycles of compaction
and decompaction of the metaphase plate. Twenty-four hours after
a 6 Gy irradiation, the duration of mitosis for H2B-GFP/PARGKD
dramatically increased to more than 400 minutes (compared with
96 minutes for H2B-GFP/BD650 cells) (supplementary material Fig.
S2A), with cells arrested at metaphase. All mitoses were abnormal
for both cell lines after irradiation (supplementary material Fig.
S2B), with a high proportion (more than 65%) of mitoses displaying
anaphase bridges (supplementary material Fig. S2D,E and Movie
1). However, in contrast to H2B-GFP/BD650, irradiation of H2B-
Fig. 6. Irradiation of PARGKD cells induces G2-M cell cycle arrest andaccumulation of cells in metaphase. (A) FACS analysis of BD650control and PARGKD cells 24 hours and 48 hours after irradiation: yaxis, cell numbers; x axis, relative DNA content based on propidiumiodide staining. (B) Percentage of BD650 control and PARGKD cells inmitosis (mitotic index) determined 24 hours after irradiation byimmunodetection of Ser10-H3-P and DNA staining with DAPI. 2700cells were counted. Cells in G2 that show a weak Ser10-H3-P nuclearsignal were not considered. (C) Distribution of the cells in the variousphases of mitosis in mock-irradiated and irradiated BD650 control andPARGKD cells 24 hours after treatment, by immunodetecting Ser10-H3-P, anti-α-tubulin and staining DNA with DAPI. The number of cellscounted is reported on the pie charts.
Fig. 7. Mitotic abnormalities accumulate in PARGKD cellsafter irradiation. (A) Projection of deconvolved merged imagestacks showing various stages (a, prometaphase/metaphase; b,anaphase; c, late anaphase; d, telophase), of the mitosis ofPARGKD cells 24 hours after a 6 Gy irradiation, stained withDAPI (blue), anti-α-tubulin (red) and anti-centromere (green)antibodies. A supernumerary spindle pole is indicated with anarrow. Scale bar: 5 μm. (B) Projection of deconvolved mergedimage stacks showing (a) multipolar metaphase PARGKD cell24 hours after a 6 Gy irradiation, stained with DAPI (blue),anti-α-tubulin (red) and CREST anti-centromere (green)antibodies, (b) multipolar metaphase PARGKD cell 24 hoursafter a 6 Gy irradiation, revealing centrosome multiplicationstained with DAPI (blue), anti-α-tubulin (red) and anti-Cdk1(green). Scale bar: 5 μm. (C) Irradiated PARGKD cells showedgradual increase of the percentage of multipolar metaphasesobserved by immunofluorescence microscopy. The number ofmitoses scored is reported on the bar chart.

1996
GFP/PARGKD cells led to a high proportion of mitoses displaying
chromosome misalignment (44%) (supplementary material Fig.
S2D,E and Movie 2) or multipolar mitoses (34%) (supplementary
material Fig. S2D,E and Movies 3 and 4). In addition, 24% of these
aberrant H2B-GFP/PARGKD mitoses ended in cell death
(supplementary material Fig. S2C,E and Movie 5; and see below).
Taken together, these results indicate that PARG impairment leads
to alterations of mitosis in irradiated cells leading to improper or
incomplete cell division that favours aneuploidy and/or polyploidy
or mitotic cell death. We hypothesized that these alterations could
result from both a defective kinetochore function and from the
presence of supernumerary spindle poles.
Irradiated PARGKD cells display centrosomal abnormalitiesThe presence of supernumerary spindle poles in irradiated PARGKD
cells prompted us to score the number of centrosomes, which were
identified by Cdk1/p34cdc2 (Fig. 8A,B,C) or pericentrin (Fig. 8C)
immunostaining. Unirradiated PARGKD cells revealed an increased
number of cells with more than two centrosomes (10.7% compared
with 2.8% for BD650 control). This proportion raised 24 hours after
irradiation with 3 or 6 Gy, and even higher 48 hours after irradiation
(52.0% of PARGKD cells compared with 25.3% of BD650 control
cells irradiated at 6 Gy). In addition, we observed an increased
frequency of cells showing fragmented centrosomal material (Fig.
8Aa,C) observed both with Cdk1 and pericentrin immunostaining,
reaching 48.3% in the PARGKD cells but only 17.9% in the BD650
control cells, 24 hours after a 6 Gy irradiation. However, 48 hours
after irradiation, the proportion of cells with fragmented centrosomes
was similar to the level observed for both unirradiated cell lines.
Taken together, these results indicate that depletion of PARG
exacerbates the centrosome amplification and fragmentation that
has been already described in irradiated cells, in cells deficient in
proteins involved in DSB repair or in cells overexpressing PARP3
and treated with an alkylating agent (Augustin et al., 2003; Date et
al., 2006; Griffin et al., 2000).
Irradiated PARGKD cells display kinetochore dysfunctionThe observation of defective kinetochore function in the absence
of PARG prompted us to monitor the functionality of the
kinetochore checkpoint. We immunodetected MAD2 checkpoint
protein that normally transiently localizes to the kinetochore of
misaligned chromosomes and delocalizes following proper
alignment (Chen et al., 1996). As expected, in unirradiated and 6
Gy irradiated BD650 control cells, MAD2 transiently colocalized
to the unattached kinetochores, but was no longer observed in
metaphase when chromosomes were properly aligned (Fig. 9A).
By contrast, MAD2 staining could still be detected in metaphase
and anaphase kinetochores in unirradiated PARGKD cells, with a
more-pronounced staining 24 hours after a 6 Gy irradiation.
Quantification of MAD2-positive anaphases revealed that 65% of
anaphases from unirradiated PARGKD cells and 85% from
irradiated cell displayed residual MAD2 staining at kinetochores.
Taken together, these results suggest that the kinetochore
checkpoint is activated in PARGKD cells by the relocalization of
MAD2 at unattached kinetochores, which could account for the
prolonged metaphases observed (supplementary material Fig. S2).
However, the PARGKD cells seem to inappropriately escape the
arrest and enter anaphase with a persistent MAD2 staining
suggesting a spindle assembly checkpoint (SAC) dysfunction. To
test this hypothesis, we incubated the two cell lines with
nocodazole, which prevents microtubule formation, triggers SAC
Journal of Cell Science 122 (12)
and blocks mitosis progression in prometaphase. Fig. 9B shows
that after 18 hours of treatment with 100 nM nocodazole, the
PARGKD cells have overcome the checkpoint, exiting mitosis
without correctly partitioning their genome and thus accumulating
Fig. 8. Irradiation of PARGKD cells induces abnormal centrosome morphologyand number. (A) Immunodetection of Cdk1 (green), α-tubulin (red) and DAPIstaining of DNA (blue) 24 hours (a) and 48 hours (b) after 6 Gy irradiation inPARGKD cells. Scale bar: 10 μm. (B,C) Bar charts indicate cells stained as in Ascored for centrosome amplification (B) and centrosome fragmentation (C) inBD650 control and PARGKD cells 24 hours and 48 hours after 6 Gyirradiation. For centrosome amplifications, cells were categorized asdisplaying 1 or 2 (≤2) or more than 2 (>2) centrosomes. The number of cellscounted is reported on the bar chart. (C) Immunodetection of Cdk1 (a, green),pericentrin (b, red) and DAPI staining of DNA (blue) 24 hours after 6 Gyirradiation in PARGKD cells. CDk1 and pericentrin colocalization signal isshown in inset c. Scale bar: 10 μm and 5 μm (insets a, b and c).

1997Radiosensitivity of PARG-deficient cells
as multinucleated cells (Fig. 9C). This result confirmed the SAC
deficiency observed in PARGKD cells.
Irradiation of PARGKD cells leads to mitotic catastropheNext, we aimed to explain the radiosensitivity of the PARGKD cells
and to determine by which mechanism the death occurred. Our first
hypothesis was that PARGKD cells would die by AIF-dependent cell
death, owing to the massive production of PAR following a 6 Gy
irradiation. However, no AIF release from mitochondria could be
observed under these conditions (data not shown) indicating that
PARGKD cells died by an AIF-independent cell death mechanism.
Scoring the proportion of necrotic and apoptotic cells with Annexin
V and propidium iodide staining by FACS showed only slight increase
of apoptotic cells in irradiated PARGKD cells compared with control
cells (data not shown). This was in agreement with the slight increase
of PARP1 cleavage observed in PARGKD cells compared with
control cells, observed at 48 hours and 72 hours after 6 Gy irradiation
(data not shown). This suggested that only a limited number of cells
underwent cell death, at each cell division. To examine the possibility
that these dying cells resulted from mitotic catastrophe, we looked
for the mitotic release of cytochrome c, a hallmark of apoptosis, by
immunofluorescence microscopy 48 hours after a 6 Gy irradiation
(Fig. 10A). Results showed that irradiated PARGKD cells displayed
two types of metaphase cells, one showing multipolar metaphase,
with no release of cytochrome c and normal staining of Ser10-H3-
P (Fig. 10Af-i). We assumed that these cells generate the polyploid
cells frequently observed in irradiated PARGKD cells (Fig. 10Aj; Fig.
6A). The second type of metaphases observed showed the release of
cytochrome c that is found associated with chromatin, probably
because of the high affinity of cytochrome c for DNA (Kleinschmidt
and Zahn, 1959). These metaphase cells showed non-aligned
chromosomes stained by DAPI and Ser10-H3-P (Fig. 10Ak-n), and
probably died by mitotic catastrophe, leading to the characteristic
formation of large nuclear bodies stained with DAPI (Fig. 10Ao)
(Castedo et al., 2004). Scoring the proportion of cells dying by either
interphasic apoptosis or mitotic catastrophe after irradiation revealed
that irradiated PARGKD cells essentially died by mitotic catastrophe
(Fig. 10B,C). This hypothesis was confirmed by live-cell
videomicroscopy using the H2B-GFP/BD650 and H2B-
GFP/PARGKD cell lines. Following mitotic progression for 10 hours,
24 hours after a 6 Gy irradiation, we observed that 24% of mitotic
H2B-GFP/PARGKD cells died by mitotic catastrophe (supplementary
material Fig. S2C,E and Movie 5). Taken together, these results
suggest that the increased radiosensitivity of PARGKD cells is likely
to be a consequence of mitotic catastrophe.
DiscussionIn this study, we have shown that PARG deficiency protects HeLa
cells from spontaneous SSBs and telomere aberrations, whereas
irradiation of these PARG-deficient cells leads to the accumulation
Fig. 9. Persistence of MAD2 staining inirradiated PARGKD cells and alteration of themitotic checkpoint after nocodazoletreatment. (A) Prometaphase, metaphase andanaphase cells from PARGKD and BD650control cell lines were stained with DAPI(blue), anti-MAD2 (red) and CREST anti-centromere (green) antibodies 24 hours aftermock irradiation or 6 Gy irradiation.Colocalization of MAD2 and CREST signalsis shown in the merge column. Thepercentage of cells with centromericdistribution of MAD2 in anaphases isindicated on the right. Scale bar: 10 μm.(B) PARGKD and BD650 control cell lineswere treated with 100 nM nocodazole for 18hours or mock-treated then stained withDAPI (blue). Scale bar: 30 μm. (C) Theaccumulation of multinucleated PARGKD
cells treated 18 hours with 100 nMnocodazole suggests a mitotic checkpointalteration. The number of cells counted isreported on the bar chart.

1998
of PAR, a delay in the repair of DNA strand breaks, centrosome
amplification and mitotic defects, generating polyploid cells or
resulting in cell death by mitotic catastrophe.
PARG depletion leads to accumulation and persistence of PARHigh levels of PAR were detected within PARGKD cells in the
absence of exogenously introduced DNA damage; however,
alkaline COMET assays revealed rather fewer SSBs in these cells
than in control cells, suggesting that PAR accumulation is not
solely due to spontaneous SSBs. Therefore, these PAR molecules
could result from PARP1 activation triggered by any of the recently
described DNA-damage-independent mechanisms of PARP1
activation observed during gene expression regulation or cell
signaling, such as binding to particular DNA structures or
chromatin states or post-translational modifications (for reviews,
see Cohen-Armon, 2007; Kraus, 2008). Alternatively, these PAR
molecules could reflect the accumulation of the reaction product
of any of the other active PARP family members. A comparable
genotoxic-stress-independent accumulation of PAR has been
reported in PARG–/– mice (Koh et al., 2004), in a Drosophila lack-
Journal of Cell Science 122 (12)
of-function PARG mutant (Hanai et al., 2004) and in an
Arabidopsis mutant that affects the catalytic activity of the PARG
homologue, tej (Panda et al., 2002). Although delayed, the
disappearance of PAR observed after irradiation could result either
from residual PARG molecules or from the activity of the recently
identified 39 kDa protein ARH3, which has PAR-degrading
activity (Oka et al., 2006).
Protective effect of PAR spontaneously produced in PARG-depleted cellsPrevious studies have proposed that the absence of PARG is
detrimental to the cells, because Parg–/– trophoblast cells showed
decreased proliferation and increased cell death (Koh et al., 2004).
In our shRNA approach, which efficiently silenced PARG
expression in HeLa cells, the basal and constitutive PAR synthesis
did not dramatically hamper cell behaviour. We rather observed a
beneficial effect, at least for genome integrity and telomere
stability. We cannot rule out the idea that a residual amount of
PARG might be sufficient for this protection. Alternatively, this
protection might occur only in tumour cells (HeLa) and not in
Fig. 10. Irradiation of PARGKD cells leads to mitoticcatastrophe. (A) Immunofluorescence microscopy analysis ofthe morphology of cells undergoing or not mitotic catastrophe.Forty-eight hours following 6 Gy irradiation, BD650 controland PARGKD cells were fixed with 4% formaldehyde andstained with DAPI (blue), anti-cytochrome c (red) and anti-Ser10-H3-P (green) antibodies. The insets in i and n detail thespecific distribution of the cytochrome c signal at highermagnification. The images shown in the right column arerepresentative of the postmitotic cell types that can be found inirradiated PARGKD cells: normal interphase cell (e),multinucleated cell (j) and dead cell after mitotic catastrophe(o). Scale bar: 10 μm. (B) Comparison of the morphology ofthe nucleus of cells undergoing apoptosis (BD650 control cells48 hours after 6 Gy irradiation) or mitotic catastrophe(PARGKD cells 48 hours after 6 Gy irradiation). Cells werefixed with 4% formaldehyde and stained with DAPI (blue) andanti-cytochrome c antibody (red). Scale bar: 10 μm.(C) Relative quantification of cell death by apoptosis andmitotic catastrophe. BD650 control and PARGKD cells wereirradiated with the indicated dose and fixed with 4%formaldehyde 24 hours or 48 hours following treatment andDNA stained with DAPI. Cells with the morphology shown inB were counted using immunofluorescence microscopy (3518cells were counted). The graphical representation expresses therelative percentage of cells in apoptosis and mitoticcatastrophe.

1999Radiosensitivity of PARG-deficient cells
untransformed cells (trophoblast cells or ES cells), but this remains
to be determined.
The spontaneous γH2AX staining detected in unirradiated
PARGKD cells correlates with the observed constitutive ATM
activation. It is, however, unlikely that it reflects the presence of
spontaneous DSBs, because this would certainly have repercussions
on cell viability, which is not the case. ATM activation has been
proposed to result from changes in chromatin structure,
independently of DNA damage (Bakkenist and Kastan, 2003;
Soutoglou and Misteli, 2008), a phenomenon that probably occurs
in PARGKD cells, supported by the established role of PAR in the
modulation of chromatin superstructure. In addition, a functional
interplay between PARP1 activity and ATM activity has been
reported (Aguilar-Quesada et al., 2007; Haince et al., 2007),
supporting the correlation between PAR accumulation and ATM
activation observed in PARG-depleted cells.
Most of the spontaneous and radioinduced telomere aberrations
found at lower frequency in PARGKD cells result from the lack (sister
chromatid fusions), improper (telomere doublets) or unstable
(telomere losses) formation of protected telomere structures during
or after replication (Pennarun et al., 2008). Interestingly, these type
of aberrations are observed at higher frequency in ATM-deficient
cells (Pennarun et al., 2008). The constitutive activation of ATM
observed in PARGKD cells could thus contribute to the observed
protection of telomeres from aberrations. It is also conceivable that
one of the PARPs reported to localize at telomeres, such as PARP1,
PARP2 or tankyrase 1 (TNKS1), could be directly implicated in
telomere protection. Indeed, we have previously proposed that
poly(ADP-ribosyl)ation of the telomeric factor TRF2 by PARP1 or
PARP2, leading to its detachment from the telomeric DNA, could
favour the maintenance of telomere integrity (Dantzer et al., 2004;
Gomez et al., 2006).
Depletion of PARG leads to SSB and DSB repair delayThe delay in the repair of SSBs and DSBs generated by X-
irradiation, observed in the absence of PARG, is consistent with
previous observations of a reduced rate of repair of oxidative lesions
in PARG-depleted A549 cells (Fisher et al., 2007). XRCC1 was
shown to relocalize in more distinct nuclear foci introduced by H2O2
treatment, and these foci persisted longer (Fisher et al., 2007). By
contrast, using laser microirradiation to locally introduce DNA
lesions, we observed that XRCC1 was less efficiently mobilized at
these damaged sites, and was then rapidly delocalized throughout
the nucleus. Since XRCC1 interacts noncovalently, but with high
affinity, to PAR through its BRCT1 domain (Pleschke et al., 2000),
it is likely that the genotoxic-stress-independent PAR produced in
the PARGKD cells binds to XRCC1, thus preventing its further
mobilisation to the laser-induced DNA breaks. Since PARG is not
present to tightly regulate the poly(ADP-ribosyl)ation status of
PARP1 at the site of massive DNA damage (Keil et al., 2006), as
introduced by the laser microirradiation, XRCC1 might get rapidly
delocalized together with the highly automodified PARP1
molecules. This premature delocalization of XRCC1 from the
damage site could account for the repair defect observed in
irradiated PARGKD cells.
PARG depletion increases radioinduced mitotic aberrationsMitotic aberration is a common response to X-irradiation, but
PARGKD cells displayed a higher proportion of aberrant mitoses
than irradiated control cells. In addition, a high proportion of cells
displayed supernumerary spindles, an observation that correlated
with the increased proportion of aneuploid and polyploid cells in
irradiated PARG-depleted cells.
Tankyrase 1, vPARP and PARP2 were found to be associated
with the mitotic spindle (Chang et al., 2005; Kickhoefer et al., 1999;
Schreiber et al., 2004) and PAR was detected in the spindle in
normal conditions. Tankyrase 1 activity was shown to be required
for the formation and maintenance of bipolarity of the mitotic
spindle but also for the separation of telomeres during anaphase
(Chang et al., 2005; Dynek and Smith, 2004). In Xenopus egg
extracts, PAR hydrolysis by the addition of PARG rapidly led to
misalignment of chromosomes and disruption of bipolar spindle
structure (Chang et al., 2004). These reports clearly define PAR
as a molecule that directly controls spindle function. In PARGKD
cells, we could detect PAR at the spindle at similar levels to those
in control cells (data not shown). In addition, formation of the
spindle was apparently not altered in the absence of PARG, even
after irradiation.
PARGKD cells showed a defective spindle assembly checkpoint:
whereas these cells normally activate the kinetochore checkpoint,
they escape this checkpoint prematurely, as shown by the persistence
of MAD2 at kinetochores in metaphases and anaphases. It is,
however, unlikely that the majority of the kinetochores remained
unattached at anaphase.
At least two PARPs, PARP1 and PARP2, could be important for
kinetochore function and mitotic progression of damaged cells.
PARP1 and PARP2 accumulate transiently at centromeres (during
S-G2 for PARP1 and pro-metaphase for PARP2) and interact with
the constitutive centromeric proteins CENPA, CENPB, the
kinetochore protein Bub3 and the mitotic kinase Aurora B
(AURKB) (Monaco et al., 2005; Saxena et al., 2002a; Saxena et
al., 2002b). These centromeric proteins were found to be poly(ADP-
ribosyl)ated after X-irradiation or oxidative damage, and poly(ADP-
ribosyl)ated Aurora B is no longer able to phosphorylate H3 Ser10
(Monaco et al., 2005). In addition, treatment of PARP2–/– cells with
a monofunctional alkylating agent leads to G2-M arrest, polyploidy
and increased cell death (Menissier de Murcia et al., 2003). These
results suggest that PARP1 and PARP2 are required for centromere
and/or kinetochore function when DNA integrity is challenged.
However, in PARGKD cells, Aurora B kinase activity does not seem
to be impaired, because phosphorylation of H3 Ser 10 was observed
to be efficient.
PARG-depleted cells are radiosensitive and die by mitoticcatastropheThe observed radiosensitivity of PARGKD cells is in agreement with
previous observations in Caenorhabditis elegans in which
expression of the two PARG homologues Pme-3 and Pme-4 has
been knocked down (St-Laurent et al., 2007), and with the observed
radiosensitivity of PARG�2/�3-deficient mice (Cortes et al., 2004).
Excessive amount of PAR is known to be cytotoxic, because
BioPORTER-mediated delivery of PAR induced AIF-dependent cell
death (Andrabi et al., 2006). However, no AIF activation and
translocation could be detected in irradiated control or PARGKD
cells (data not shown). This suggests that massive PAR production
is not sufficient per se to activate the AIF-dependent cell-death
pathway. However, we cannot completely exclude the notion that
the HeLa cells used in this study are impaired in the AIF-dependent
cell death pathway, because treatment with 100 μM MNNG was
able to trigger AIF-dependent cell death in only a small but similar
proportion of cells (1%; data not shown) for both BD650 control
and PARGKD cell lines.

2000 Journal of Cell Science 122 (12)
Our results show that irradiated PARGKD cells die mostly during
mitosis. Mitotic catastrophe is a cell death pathway arising in many
tumour cells when damaged DNA enters mitosis, or when bipolar
spindle assembly is prevented by centrosome amplification and the
establishment of supernumerary spindle poles (Dodson et al.,
2007). Both situations could synergize to increase the proportion
of mitotic catastrophe observed in irradiated PARGKD cells. The
centrosome was proposed to have a direct role in the DNA-damage
response as a checkpoint regulator or effector, with centrosomal
amplification being the endpoint leading to mitotic catastrophe to
eliminate the damaged cell (Loffler et al., 2006). The latter
hypothesis is probable, explaining the outcome of irradiated PARG-
depleted cells as shown in this study.
Whereas the significance of centrosome fragmentation is still under
debate (Dodson et al., 2004; Hut et al., 2003), centrosome
amplification could result from the uncoupling of centrosome
duplication from DNA duplication, a process observed in PARP1–/–
mouse fibroblasts (Kanai et al., 2003). Alternatively, defective
cytokinesis can also lead to unequal partition of spindle poles. PAR
and several PARPs were detected at centrosomes, such as PARP1,
PARP3 and tankyrase 1 (Augustin et al., 2003; Kanai et al., 2003;
Smith and de Lange, 1999). In addition, treatment of cells with PARP
inhibitors leads to centrosomal amplification, indicating that PAR is
involved in the regulation of centrosome duplication (Kanai et al.,
2003). Our results suggest that PAR levels must be tightly regulated,
because uncontrolled PAR synthesis can also deregulate centrosome
duplication. However, whether PAR acts at the centrosome itself, or
centrosome amplification is an endpoint consequence of deregulated
nuclear PAR synthesis remains to be determined. ATM and ATR
activities have been reported to be involved in centrosome
amplification following irradiation (Bourke et al., 2007). Thus, the
increased ATM activity in irradiated PARGKD cells is consistent with
the observed centrosome amplification.
Taken together, our results suggest that the absence of PARG is
beneficial for undamaged cells, but detrimental to irradiated cells,
and this radiosensitivity is the consequence of repair defects,
centrosome amplification and mitotic spindle checkpoint defects,
leading to either polyploidy or cell death by mitotic catastrophe.
Thus, a fine tuning of PAR synthesis and degradation is essential
to regulate the fate of the damaged cell, because prevention of either
PAR synthesis in PARP1- or tankyrase-1-deficient cells or of PAR
degradation in PARG-depleted cells can both lead to cell death, but
probably via different mechanisms. Whether the increased
radiosensitivity observed in the absence of PARG is cell-type
dependent remains to be determined, but our results suggest that
PARG could be a novel potential therapeutic target for radiotherapy.
Materials and MethodsCell linessiRNA design and cloning into pEBVsiRNA vectors and establishment of stable
knockdown and control HeLa clones were carried out as previously described (Biard,
2007; Biard et al., 2005). The RNAi sequence for PARG (NM_003631) stretched
nucleotides 2325-2343. HeLa cells expressing constitutively H2B-GFP were described
elsewhere (Kanda et al., 1998). Establishment of stable PARG knockdown and control
H2B-GFP expressing HeLa clones was carried out as previously described (Biard,
2007; Biard et al., 2005), leading to H2B-GFP/PARGKD and H2B-GFP/BD650 cell
lines, respectively. All cell lines were maintained in Dulbecco’s modified Eagle’s
medium (DMEM) (Gibco/BRL, Invitrogen) supplemented with 10% fetal bovine
serum (FBS; AdGenix) and 1% gentamicin (Gibco/BRL, Invitrogen) and 125 μg/ml
hygromycin B under 5% CO2.
Induction of DNA damagesFor X-irradiation, cells grown on glass coverslips in 35-mm culture dishes or in P100
Petri dishes were subjected to ionizing radiations using a Pantak Seifert X-ray systems
(Cegelec, France) operating at 100 kV, 4.5 mA. The dose was measured with a PTW-
UNIDOS E Universal Dosemeter. For telomere FISH experiments, γ-irradiation was
performed with a IBL637 irradiator (CisBio International). For local DNA damages,
cells were grown onto 55-μm-square CELLocate coverslips (Eppendorf, Hamburg,
Germany). The cells were incubated with 10 μg/ml Hoechst dye 33258 in DMEM
medium for 10 minutes at 37°C under 5% CO2. Laser microirradiation was performed
with a Leica DM LMD microscope (Leica Microsystems) fitted with a 337.1 nm
laser focused through a �40 or a �63 objective.
ZymogramPARG activity gel assay was performed as described by Amé et al. (Amé et al.,
2009).
Western blotting and cell lysatesCells (5�106) were lysed by three cycles of freeze-thaw in 20 mM Tris-HCl (pH
7.5) 400 mM KCl, 2 mM DTT, 20% glycerol, 1 mM Pefabloc and protease inhibitor
(Roche Diagnostic) then centrifuged for 40 minutes at 4°C. Twenty micrograms of
the supernatant were analyzed by western blot with the following antibodies: a mouse
monoclonal anti-poly(ADP-ribose) 10H (1:100, Sugimura, Tokyo, Japan), a rabbit
monoclonal anti-ATM (P1981S), (1:10,000, Epitomics), or a rabbit polyclonal anti-
actin (rAb, 1:10,000, Sigma) and an anti-Nter-PARG (rAb against N-terminal domain
of PARG, 1:3000).
Indirect immunofluorescence microscopyCells grown on glass coverslips were irradiated as described in the figure legends,
washed with PBS and fixed either 15 minutes with ice-cold methanol:acetone (v:v)
or 15 minutes with 4% formaldehyde in PBS at room temperature. Cells were washed
three times with PBS, 0.1% Tween (v:v). Cells were incubated overnight at 4°C
with PBS, 0.1% Tween (v:v), containing 1 mg/ml BSA and a primary antibody:
rabbit polyclonals anti-XRCC1 (1:1000, Alexis, Lausen), anti-Ser10-H3-P (1:2000,
Upstate) or anti-pericentrin (1:1000, Babco), and mouse monoclonals anti-poly(ADP-
ribose) 10H (IgG3κ, 1:1000), anti-γH2AX (IgG1, 1:2000, Upstate), anti-α-tubulin
(DM 1A) (IgG1k, 1:600, Sigma), anti-MAD2 (17D10) (IgG1, 1:500, Santa Cruz
Biotechnology), anti-Cdk1 (p34cdc2) (IgG2, 1:1000, Santa Cruz Biotechnology), an
anti-cytochrome c (IgG2, 1:800, Pharmingen) or a human polyclonal CREST
antibody (hAb, 1:800, kindly donated by K. H. Andy Choo, Royal Children’s
Hospital, Parkville, Australia). After washing, cells were incubated for 2 hours at
room temperature with the appropriate secondary antibodies: an Alexa Fluor 488
or 568 goat anti-mouse IgG or IgG1 or IgG2 or IgG3 (1:2000, Molecular Probes,
Invitrogen), an Alexa Fluor 488 goat anti-human IgG (1:600, Molecular Probes,
Invitrogen), an Alexa Fluor 488 or 568 goat anti-rabbit IgG. After three washes with
PBS, 0.1% Tween (v:v), DNA was counterstained with DAPI. Immunofluorescence
microscopy was performed using a Leica DMRA2 equipped with an Orca-ER CCD
camera (Hammamatsu) and the capture software OpenLab 4.1 (Improvision). 3D
deconvolution analysis and imaging of image stacks was performed using Volocity
4.0 (Improvision) when indicated in the figure legend. Merging images was done
using Photoshop CS3 (Adobe).
Identification and scoring of mitotic figuresCells were stained with anti-Ser10-H3-P, anti-α-tubulin antibodies and DAPI and
observed by fluorescence microscopy. Classification of prometamaphase and abnormal
metaphase was difficult to make due to the defect in chromosome congression
phenotype observed in PARGKD cells. These two mitotic stages were therefore
combined in the various scoring and noted as prometaphase/metaphase.
Single cell gel electrophoresis (COMET) assayCells were trypsinized and resuspended in low melting point agarose at 0.5% (104
cells/ml) and dropped onto 1% agarose in PBS coated slides. The alkaline COMET
assay was used to detect both SSBs and DSBs. Slides were irradiated as described
in figure legend, and immediately processed according to Trucco et al. (Trucco et
al., 1998). To detect DSBs, a neutral COMET assay was performed: the slides,
following irradiation and DNA damage recovery, were treated in a non-denaturating
lysis solution (25 mM EDTA (pH 9.5), 34 mM sodium lauryl sarcosinate, 87 mM
SDS) for 2 hours at RT, then washed 5 minutes in water. Electrophoresis was performed
in 1� TBE (pH 8.4) at 16 mA, 2.5 V/cm for 4 minutes. After dehydratation in 100%
ethanol at –20°C for 10 minutes, the slides were dried. The migrated DNA was stained
with ethidium bromide and visualized using a Leica DMRA2 fluorescent microscope
with the 20� lens. For each time point, several fields as those represented in
supplementary material Fig. S1 were captured by the Hammamatsu Orca-ER CCD
camera and used for quantitative assessment of DNA damage using the visCOMET
software (Impuls).
Cell cycle analysisFlow cytometry analysis (FACS) was performed as follows: 24 and 48 hours after
irradiation, cells were collected by trypsinization, washed with PBS and fixed in 75%
ethanol at 4°C for at least 24 hours. Cells were washed twice in PBS and nuclear
DNA was stained with propidium iodide (4 μg/ml; Sigma, St Louis, MO) in the
presence of RNase A (10 μg/ml; Sigma) in PBS for at least 30 minutes. Stained cells

2001Radiosensitivity of PARG-deficient cells
were analyzed on a FACScalibur (Becton Dickinson, Franklin Lakes, NJ) using
CellQuest software. Ten thousand cells gated as single cells were analyzed.
Colony-forming assayCells cultivated on 150-mm culture dishes were irradiated at various doses as indicated
above, trypsinized and counted. After appropriate dilution, 3�103 cells were seeded
on P60 culture dishes in triplicate and left to grow for 13 days. The number of colonies
for each dish was counted using ImageJ (NIH, Bethesda) after crystal violet staining.
Fluorescent in situ hybridization (FISH)Metaphase spreads and analysis of telomere aberrations were performed as previously
described (Pennarun et al., 2008). Numbers of chromosomes and metaphases
analyzed in FISH experiments is shown in supplementary material Table S1.
Live videomicroscopyCells were grown on glass coverslips mounted in a Ludin Chamber (LIS). Live
microscopy was carried out using an inverted microscope (Olympus IX81) placed
in an incubator chamber (LIS) maintained at 37°C, and coupled with a CoolSNAP
HQ camera (Princeton Instruments) controlled by Metamorph software (Universal
Imaging). Fluorescent images were taken on 10-15 fields using a 20� objective every
2 minutes for 10 hours.
We thank Geoffrey M. Wahl (SIBS, La Jolla, CA) for the HeLa H2B-GFP cell line. We acknowledge the Centre National de la RechercheScientifique, Université de Strasbourg, Electricité de France and Liguecontre le Cancer (Comité du Bas-Rhin) for their support.
ReferencesAguilar-Quesada, R., Munoz-Gamez, J. A., Martin-Oliva, D., Peralta, A., Valenzuela,
M. T., Matinez-Romero, R., Quiles-Perez, R., Menissier-de Murcia, J., de Murcia,
G., de Almodovar, M. R. et al. (2007). Interaction between ATM and PARP-1 in response
to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMCMol. Biol. 8, 29.
Amé, J. C., Spenlehauer, C. and de Murcia, G. (2004). The PARP superfamily.
BioEssays 26, 882-893.
Amé, J. C., Hakme, A., Quenet, D., Fouquerel, E., Dantzer, F. and Schreiber, V. (2009).
Detection of the nuclear poly(ADP-ribose)-metabolizing enzymes and activities in
response to DNA damage. Methods Mol. Biol. 464, 267-283.
Andrabi, S. A., Kim, N. S., Yu, S. W., Wang, H., Koh, D. W., Sasaki, M., Klaus, J. A.,
Otsuka, T., Zhang, Z., Koehler, R. C. et al. (2006). Poly(ADP-ribose) (PAR) polymer
is a death signal. Proc. Natl. Acad. Sci. USA 103, 18308-18313.
Augustin, A., Spenlehauer, C., Dumond, H., Menissier-De Murcia, J., Piel, M., Schmit,
A. C., Apiou, F., Vonesch, J. L., Kock, M., Bornens, M. et al. (2003). PARP-3 localizes
preferentially to the daughter centriole and interferes with the G1/S cell cycle progression.
J. Cell Sci. 116, 1551-1562.
Bakkenist, C. J. and Kastan, M. B. (2003). DNA damage activates ATM through
intermolecular autophosphorylation and dimer dissociation. Nature 421, 499-506.
Biard, D. S. (2007). Untangling the relationships between DNA repair pathways by silencing
more than 20 DNA repair genes in human stable clones. Nucleic Acids Res. 35, 3535-
3550.
Biard, D. S., Despras, E., Sarasin, A. and Angulo, J. F. (2005). Development of new
EBV-based vectors for stable expression of small interfering RNA to mimick human
syndromes: application to NER gene silencing. Mol. Cancer Res. 3, 519-529.
Blenn, C., Althaus, F. R. and Malanga, M. (2006). Poly(ADP-ribose) glycohydrolase
silencing protects against H2O2-induced cell death. Biochem. J. 396, 419-429.
Bourke, E., Dodson, H., Merdes, A., Cuffe, L., Zachos, G., Walker, M., Gillespie, D.
and Morrison, C. G. (2007). DNA damage induces Chk1-dependent centrosome
amplification. EMBO Rep. 8, 603-609.
Castedo, M., Perfettini, J. L., Roumier, T., Andreau, K., Medema, R. and Kroemer,
G. (2004). Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825-
2837.
Chang, P., Jacobson, M. K. and Mitchison, T. J. (2004). Poly(ADP-ribose) is required
for spindle assembly and structure. Nature 432, 645-649.
Chang, P., Coughlin, M. and Mitchison, T. J. (2005). Tankyrase-1 polymerization of
poly(ADP-ribose) is required for spindle structure and function. Nat. Cell Biol. 7, 1133-
1139.
Chen, R. H., Waters, J. C., Salmon, E. D. and Murray, A. W. (1996). Association of
spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science274, 242-246.
Cohen-Armon, M. (2007). PARP-1 activation in the ERK signaling pathway. TrendsPharmacol. Sci. 28, 556-560.
Cortes, U., Tong, W. M., Coyle, D. L., Meyer-Ficca, M. L., Meyer, R. G., Petrilli, V.,
Herceg, Z., Jacobson, E. L., Jacobson, M. K. and Wang, Z. Q. (2004). Depletion of
the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to
genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 24, 7163-7178.
Dantzer, F., Giraud-Panis, M. J., Jaco, I., Ame, J. C., Schultz, I., Blasco, M., Koering,
C. E., Gilson, E., Menissier-de Murcia, J., de Murcia, G. et al. (2004). Functional
interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity
negatively regulates TRF2. Mol. Cell. Biol. 24, 1595-1607.
Date, O., Katsura, M., Ishida, M., Yoshihara, T., Kinomura, A., Sueda, T. and
Miyagawa, K. (2006). Haploinsufficiency of RAD51B causes centrosome fragmentation
and aneuploidy in human cells. Cancer Res. 66, 6018-6024.
Dodson, H., Bourke, E., Jeffers, L. J., Vagnarelli, P., Sonoda, E., Takeda, S., Earnshaw,
W. C., Merdes, A. and Morrison, C. (2004). Centrosome amplification induced by
DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. 23,
3864-3873.
Dodson, H., Wheatley, S. P. and Morrison, C. G. (2007). Involvement of centrosome
amplification in radiation-induced mitotic catastrophe. Cell Cycle 6, 364-370.
Dynek, J. N. and Smith, S. (2004). Resolution of sister telomere association is required
for progression through mitosis. Science 304, 97-100.
El-Khamisy, S. F., Masutani, M., Suzuki, H. and Caldecott, K. W. (2003). A requirement
for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative
DNA damage. Nucleic Acids Res. 31, 5526-5533.
Fisher, A. E., Hochegger, H., Takeda, S. and Caldecott, K. W. (2007). Poly(ADP-ribose)
polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose)
glycohydrolase. Mol. Cell. Biol. 27, 5597-5605.
Gomez, M., Wu, J., Schreiber, V., Dunlap, J., Dantzer, F., Wang, Y. and Liu, Y. (2006).
PARP1 is a TRF2-associated poly(ADP-Ribose)polymerase and protects eroded
telomeres. Mol. Biol. Cell 17, 1686-1696.
Griffin, C. S., Simpson, P. J., Wilson, C. R. and Thacker, J. (2000). Mammalian
recombination-repair genes XRCC2 and XRCC3 promote correct chromosome
segregation. Nat. Cell Biol. 2, 757-761.
Haince, J. F., Kozlov, S., Dawson, V. L., Dawson, T. M., Hendzel, M. J., Lavin, M. F.
and Poirier, G. G. (2007). Ataxia telangiectasia mutated (ATM) signaling network is
modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to
DNA-damaging agents. J. Biol. Chem. 282, 16441-16453.
Hanai, S., Kanai, M., Ohashi, S., Okamoto, K., Yamada, M., Takahashi, H. and Miwa,
M. (2004). Loss of poly(ADP-ribose) glycohydrolase causes progressive
neurodegeneration in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 101, 82-86.
Hut, H. M., Lemstra, W., Blaauw, E. H., Van Cappellen, G. W., Kampinga, H. H. and
Sibon, O. C. (2003). Centrosomes split in the presence of impaired DNA integrity during
mitosis. Mol. Biol. Cell 14, 1993-2004.
Jagtap, P. and Szabo, C. (2005). Poly(ADP-ribose) polymerase and the therapeutic effects
of its inhibitors. Nat. Rev. Drug Discov. 4, 421-440.
Kanai, M., Tong, W. M., Sugihara, E., Wang, Z. Q., Fukasawa, K. and Miwa, M.
(2003). Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation
in regulation of centrosome function. Mol. Cell. Biol. 23, 2451-2462.
Kanda, T., Sullivan, K. F. and Wahl, G. M. (1998). Histone-GFP fusion protein enables
sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8,
377-385.
Keil, C., Grobe, T. and Oei, S. L. (2006). MNNG-induced cell death is controlled by
interactions between PARP-1, poly(ADP-ribose) glycohydrolase, and XRCC1. J. Biol.Chem. 281, 34394-34405.
Kickhoefer, V. A., Siva, A. C., Kedersha, N. L., Inman, E. M., Ruland, C., Streuli, M.
and Rome, L. H. (1999). The 193-kD vault protein, VPARP, is a novel poly(ADP-
ribose) polymerase. J. Cell Biol. 146, 917-928.
Kleinschmidt, A. and Zahn, R. K. (1959). Ueber Desoxyribonukleinsaure-Molekiile in
Protein-Mischfilmen. Z. Naturf. B 14, 770-779.
Koh, D. W., Lawler, A. M., Poitras, M. F., Sasaki, M., Wattler, S., Nehls, M. C., Stoger,
T., Poirier, G. G., Dawson, V. L. and Dawson, T. M. (2004). Failure to degrade
poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic
lethality. Proc. Natl. Acad. Sci. USA 101, 17699-17704.
Kraus, W. L. (2008). Transcriptional control by PARP-1: chromatin modulation, enhancer-
binding, coregulation, and insulation. Curr. Opin. Cell Biol. 20, 294-302.
Loffler, H., Lukas, J., Bartek, J. and Kramer, A. (2006). Structure meets function-
centrosomes, genome maintenance and the DNA damage response. Exp. Cell Res. 312,
2633-2640.
Menissier de Murcia, J., Ricoul, M., Tartier, L., Niedergang, C., Huber, A., Dantzer,
F., Schreiber, V., Ame, J. C., Dierich, A., LeMeur, M. et al. (2003). Functional
interaction between PARP-1 and PARP-2 in chromosome stability and embryonic
development in mouse. EMBO J. 22, 2255-2263.
Meyer, R. G., Meyer-Ficca, M. L., Whatcott, C. J., Jacobson, E. L. and Jacobson, M.
K. (2007). Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-
ribose) glycohydrolase (PARG) activity. Exp. Cell Res. 313, 2920-2936.
Monaco, L., Kolthur-Seetharam, U., Loury, R., Menissier-de, Murcia, J., de Murcia,
G. and Sassone-Corsi, P. (2005). Inhibition of Aurora-B kinase activity by poly(ADP-
ribosyl)ation in response to DNA damage. Proc. Natl. Acad. Sci. USA 102, 14244-14248.
Mortusewicz, O. and Leonhardt, H. (2007). XRCC1 and PCNA are loading platforms
with distinct kinetic properties and different capacities to respond to multiple DNA lesions.
BMC Mol. Biol. 8, 81.
Mortusewicz, O., Ame, J. C., Schreiber, V. and Leonhardt, H. (2007). Feedback-regulated
poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in
living cells. Nucleic Acids Res. 35, 7665-7675.
Moubarak, R. S., Yuste, V. J., Artus, C., Bouharrour, A., Greer, P. A., Menissier-de
Murcia, J. and Susin, S. A. (2007). Sequential activation of poly(ADP-ribose)
polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated
programmed necrosis. Mol. Cell. Biol. 27, 4844-4862.
Oka, S., Kato, J. and Moss, J. (2006). Identification and characterization of a mammalian
39-kDa poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 281, 705-713.
Okano, S., Lan, L., Caldecott, K. W., Mori, T. and Yasui, A. (2003). Spatial and temporal
cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 23, 3974-3981.

Panda, S., Poirier, G. G. and Kay, S. A. (2002). tej defines a role for poly(ADP-
ribosyl)ation in establishing period length of the arabidopsis circadian oscillator. Dev.Cell 3, 51-61.
Pennarun, G., Granotier, C., Hoffschir, F., Mandine, E., Biard, D., Gauthier, L. R.
and Boussin, F. D. (2008). Role of ATM in the telomere response to the G-quadruplex
ligand 360A. Nucleic Acids Res. 36, 1741-1754.
Pleschke, J. M., Kleczkowska, H. E., Strohm, M. and Althaus, F. R. (2000). Poly(ADP-
ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem.275, 40974-40980.
Saxena, A., Saffery, R., Wong, L. H., Kalitsis, P. and Choo, K. H. (2002a). Centromere
proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein
and are poly(ADP-ribosyl)ated. J. Biol. Chem. 277, 26921-26926.
Saxena, A., Wong, L. H., Kalitsis, P., Earle, E., Shaffer, L. G. and Choo, K. H. (2002b).
Poly(ADP-ribose) polymerase 2 localizes to mammalian active centromeres and interacts
with PARP-1, Cenpa, Cenpb and Bub3, but not Cenpc. Hum. Mol. Genet. 11, 2319-
2329.
Schreiber, V., Ricoul, M., Amé, J. C., Dantzer, F., Meder, V. S., Spenlehauer, C., Stiegler,
P., Niedergang, C., Sabatier, L., Favaudon, V. et al. (2004). PARP-2: structure-function
relationship. In Poly(ADP-ribosyl)ation (ed. A. Burkle). Austin, TX: Landes.
Schreiber, V., Dantzer, F., Ame, J. C. and de Murcia, G. (2006). Poly(ADP-ribose):
novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 7, 517-528.
Smith, S. and de Lange, T. (1999). Cell cycle dependent localization of the telomeric
PARP, tankyrase, to nuclear pore complexes and centrosomes. J. Cell Sci. 112, 3649-
3656.
Soutoglou, E. and Misteli, T. (2008). Activation of the cellular DNA damage response in
the absence of DNA lesions. Science 320, 1507-1510.
St-Laurent, J. F., Gagnon, S. N., Dequen, F., Hardy, I. and Desnoyers, S. (2007). Altered
DNA damage response in Caenorhabditis elegans with impaired poly(ADP-ribose)
glycohydrolases genes expression. DNA Repair (Amst.) 6, 329-343.
Trucco, C., Oliver, F. J., de Murcia, G. and Menissier-de Murcia, J. (1998). DNA repair
defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 26, 2644-
2649.
Yu, S. W., Wang, H., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier,
G. G., Dawson, T. M. and Dawson, V. L. (2002). Mediation of poly(ADP-ribose)
polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259-263.
Yu, S. W., Andrabi, S. A., Wang, H., Kim, N. S., Poirier, G. G., Dawson, T. M. and
Dawson, V. L. (2006). Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR)
polymer-induced cell death. Proc. Natl. Acad. Sci. USA 103, 18314-18319.
Journal of Cell Science 122 (12)2002





























