Embed Size (px)
Citation preview

Quantum-Mechanical Modeling
of High-Entropy Alloys
SHUO HUANG
Doctoral Thesis
School of Industrial Engineering and Management, Department of
Materials Science and Engineering, KTH, Sweden, 2018

Materialvetenskap
KTH
SE-100 44 Stockholm
ISBN 978-91-7729-765-9 Sweden
Akademisk avhandling som med tillstånd av Kungliga Tekniska högskolan framlägges
till offentlig granskning för avläggande av doktorsexamen tisdag den 12 juni 2018 kl
10:00 i B2, Kungliga Tekniska högskolan, Brinellvägen 23, Stockholm.
© Shuo Huang , 2018
Tryck: Universitetsservice US AB

iii
Abstract
High-entropy alloys (HEAs) consisting of multi-principal elements open up a
near-infinite compositional space for materials design. Extensive attention has
been put on HEAs, and interesting structural, physical and chemical properties
are being continuously revealed. Based on first-principle theory, here we focus
on the fundamental characteristics of HEAs, as well as on the optimization and
prediction of alternative alloy with promising technological applications.
The relative phase stability of different-types of HEAs is investigated from
the minimum of structural energy, and the composition-, temperature-, and
ordering-induced phase transformations are presented. The elastic properties
are discussed through the single-crystal and polycrystalline elastic moduli by
making use of a series of phenomenological models. The competition between
full slip, twinning, and stacking fault in face-centered cubic HEAs is analyzed
by studying the generalized stacking fault energy. The magnetic characteristics
are provided through the Heisenberg Hamiltonian model in connection with
Monte-Carlo simulation, and the Curie temperature of a large number of cubic
HEAs is mapped out with the help of mean-filed approximation. The thermal
expansion behavior is estimated by using the Debye-Grüneisen model.
This work provides some fundamental and pioneering theoretical points of
view to understand the intrinsic physical mechanisms in HEAs, and reveals
alternative opportunities for optimizing and designing properties of materials.
The challenge of comprehending the observed complex behavior behind the
multi-component nature of HEAs is great, on the other hand, the potential to
enhance the underlying theoretical understanding is remarkable.

iv
Sammanfattning
Högentropilegeringar, som består av flera viktiga grundämnen, är nuförtiden
ett av de mest spännande forskningsområdena inom materialvetenskap.
Baserat på första princips teori, fokuserar vi på de fundamentala egenskaperna
hos högentropilegeringar samt optimering och förutsägelse av alternativ
legeringar med lovande tekniska applikationer.
Det elastiska beteendet tillhandahålls från enkelkristall och polykristallin
elasticitetsmoduler genom användning av en rad fenomenologiska modeller.
Tävlingen mellan full glidning, tvillinggränser och staplingsfel under plastisk
deformation i ytcentrerat kubiskt högentropilegeringar analyseras från den
generella staplingsfelenergin. Magnetiska egenskaperna undersöks genom en
modell för den Hamiltonoperatoren i samband med Monte-Carlo simulering.
Curietemperaturen kartläggs för ett stort antal kubiska ekvimolära
högentropilegeringar med hjälp av medelfältapproximation. Temperatur-
variationen av termisk utvidgningskoefficient beräknas baserat på Debye-
Grüneisen modellen.
Det här arbetet ger några grundläggande och banbrytande teoretiska
synvinklar för att förstå de inneboende fysiska mekanismerna bakom den
multifunktionella naturen hos högentropilegeringar, och avslöjar alternativa
möjligheter att utforma och optimera materialets egenskaper.

v
Preface
List of included publications:
I. Temperature dependent stacking fault energy of FeCrCoNiMn high entropy alloy
Shuo Huang, Wei Li, Song Lu, Fuyang Tian, Jiang Shen, Erik Holmström, and
Levente Vitos, Scripta Materialia, 108, 44-47 (2015).
II. Phase stability and magnetic behavior of FeCrCoNiGe high-entropy alloy
Shuo Huang, Ádám Vida, Dávid Molnár, Krisztina Kádas, Lajos Károly Varga, Erik
Holmström, and Levente Vitos, Applied Physics Letters, 107, 251906 (2015).
III. Mechanism of magnetic transition in FeCrCoNi-based high entropy alloys
Shuo Huang, Wei Li, Xiaoqing Li, Stephan Schönecker, Lars Bergqvist, Erik
Holmström, Lajos Károly Varga, and Levente Vitos, Materials and Design, 103, 71-74
(2016).
IV. Thermal expansion in FeCrCoNiGa high-entropy alloy from theory and experiment
Shuo Huang, Ádám Vida, Wei Li, Dávid Molnár, Se Kyun Kwon, Erik Holmström,
Béla Varga, Lajos Károly Varga, and Levente Vitos, Applied Physics Letters, 110,
241902 (2017).
V. Thermal expansion, elastic and magnetic properties of FeCoNiCu-based high-entropy
alloys using first-principle theory
Shuo Huang, Ádám Vida, Anita Heczel, Erik Holmström, and Levente Vitos, JOM,
69, 2107-2112 (2017).
VI. Mechanical performance of FeCrCoMnAlx high-entropy alloys from first-principle
Shuo Huang, Xiaoqing Li, He Huang, Erik Holmström, and Levente Vitos, Materials
Chemistry and Physics, 210, 37-42 (2018).
VII. Mapping the magnetic transition temperatures for medium- and high-entropy alloys
Shuo Huang, Erik Holmström, Olle Eriksson, and Levente Vitos, Intermetallics, 95,
80-84 (2018).
VIII. Strengthening induced by magneto-chemical transition in Al-doped Fe-Cr-Co-Ni high-
entropy alloys
Shuo Huang, Wei Li, Erik Holmström, Levente Vitos, submitted to Phys. Rev. Appl.
IX. Phase-transition assisted mechanical behavior of TiZrHfTax high-entropy alloys
Shuo Huang, Wei Li, Erik Holmström, Levente Vitos, submitted to Sci. Rep.

vi
X. Elasticity of high-entropy alloys from ab initio theory
Shuo Huang, Fuyang Tian, Levente Vitos, submitted to J. Mater. Res.
Comment on my own contribution:
Literature survey, research planning, data analysis, and manuscript preparation
done jointly. My personal contributions to the above research steps are on the
average 75%, 80%, 70%, and 75%, respectively. Calculations were done
almost entirely by me (papers I, II, IV, V, VII, VIII, IX 90%, papers III and VI
70%). My contribution to Paper X is about 80 %.
List of publications not included in the thesis:
XI. C.H. Zhang, S. Huang, J. Shen, N.X. Chen, Chin. Phys. B, 21 (2012) 113401.
XII. S. Huang, C.H. Zhang, J. Sun, J. Shen, Chin. Phys. B, 22 (2013) 083401.
XIII. C.H. Zhang, S. Huang, J. Shen, N.X. Chen, J. Mater. Res., 28 (2013) 2720-2727.
XIV. S. Huang, C.H. Zhang, J. Sun, J. Shen, Mod. Phys. Lett. B, 27 (2013) 1350195.
XV. J. Sun, P. Qian, J. Shen, J.C. Li, S. Huang, J. Alloys Compd., 580 (2013) 522-526.
XVI. J. Sun, J. Shen, P. Qian, S. Huang, Physica B, 427 (2013) 110-117.
XVII. Z.F. Zhang, P. Qian, J.C. Li, Y.P. Li, S. Huang, J. Sun, J. Shen, Y. Liu, N.X. Chen,
Chin. J. Phys., 51 (2013) 606-618.
XVIII. C.H. Zhang, S. Huang, R.Z. Li, J. Shen, N.X. Chen, Int. J. Mod. Phys. B, 27 (2013)
1350147.
XIX. S. Huang, R.Z. Li, C.H. Zhang, J. Shen, Chin. J. Phys., 52 (2014) 891-902.
XX. S. Huang, R.Z. Li, S.T. Qi, B. Chen, J. Shen, Phys. Scr., 89 (2014) 065702.
XXI. S. Huang, R.Z. Li, S.T. Qi, B. Chen, J. Shen, Int. J. Mod. Phys. B, 28 (2014) 1450087.
XXII. S. Huang, R.Z. Li, S.T. Qi, B. Chen, J. Shen, Solid State Commun., 184 (2014) 52-55.
XXIII. S. Huang, C.H. Zhang, R.Z. Li, J. Shen, N.X. Chen, Intermetallics, 51 (2014) 24-29.
XXIV. C.H. Zhang, S. Huang, J. Shen, N.X. Chen, Intermetallics, 52 (2014) 86-91.
XXV. Á. Vida, L.K. Varga, N.Q. Chinh, D. Molnár, S. Huang, L. Vitos, Mater. Sci. Eng. A,
669 (2016) 14-19.
XXVI. Á. Vida, Z. Maksa, D. Molnár, S. Huang, J. Kovac, L.K. Varga, L. Vitos, N.Q. Chinh,
J. Alloys Compd., 743 (2018) 234-239.
XXVII. H. Huang, X. Li, Z. Dong, W. Li, S. Huang, D. Meng, X. Lai, T. Liu, S. Zhu, L. Vitos,
Acta Mater., 149 (2018) 388-396.
XXVIII. S. Huang, H. Huang, W. Li, D. Kim, S. Lu, X. Li, E. Holmström, S.K. Kwon, L. Vitos,
Twinning in metastable high-entropy alloys, submitted to Nat. Commun.
XXIX. S. Huang, W. Li, E. Holmström, S.K. Kwon, L. Vitos, Plastic deformation transition
in FeCrCoNiAlx high-entropy alloys, in manuscript.

vii
Contents
Abstract ........................................................................................................ iii
Sammanfattning ........................................................................................... iv
Preface ........................................................................................................... v
Contents ....................................................................................................... vii
1 Introduction .............................................................................................. 1
2 Theoretical methodology .......................................................................... 3
2.1 Schrödinger equation ............................................................................ 3
2.2 Density functional theory ..................................................................... 4
2.3 Exact muffin-tin orbital method ........................................................... 5
2.4 Coherent potential approximation ........................................................ 6
3 Structural properties ................................................................................ 8
3.1 Phase stability ....................................................................................... 8
3.1.1 Composition dependence .............................................................. 8
3.1.2 Temperature effect .......................................................................10
3.1.3 Ordering behavior ........................................................................11
3.2 Lattice parameter .................................................................................13
4 Deformation performance .......................................................................15
4.1 Elasticity ..............................................................................................15
4.1.1 Single-crystal elastic constants ....................................................15
4.1.2 Polycrystalline elastic moduli ......................................................16
4.2 Plasticity ..............................................................................................17
4.2.1 Generalized stacking fault energy ................................................17
5 Magnetic behavior ...................................................................................19
5.1 Magnetic moment ................................................................................19

viii
5.2 Curie temperature ................................................................................ 20
5.2.1 Monte-Carlo simulation ............................................................... 20
5.2.2 Mean-field approximation ........................................................... 23
6 Thermo-physical characteristics ............................................................ 26
6.1 Debye temperature .............................................................................. 26
6.2 Thermal expansion .............................................................................. 27
7 Summary and outlook ............................................................................. 30
Acknowledgement ....................................................................................... 31
Bibliography ................................................................................................ 32

1
Chapter 1
Introduction
High-entropy alloys (HEAs), first brought to general attention in 2004 [1, 2],
represent an exciting research area in materials science, since it is characterized
by many new findings, unexplained results, and vigorous controversies.
Being different from the conventional alloys, Yeh [3] defined HEAs based
on composition and configurational entropy: i, containing at least five principle
elements with the concentration of each element being between 5 and 35 at. %;
ii, having configurational entropy at a random state larger than 1.5 𝑅 (𝑅 is the
gas constant, ~ 8.314 Jmol-1K-1), despite showing single- or multi-phase at
room temperature. Notice that the definitions of HEAs are just guidelines, not
laws, that the basic principle is having high mixing entropy to enhance the
formation of solid solution phases, and avoid complicated structures [4].
In this scenario, a large number of different-types of HEAs has been reported
[4]. Interestingly, HEAs consisting of late 3d transition metals tend to form a
face-centered cubic (fcc) phase [5], those composed of refractory metals often
have a body-centered cubic (bcc) phase [6], while HEAs based on rare-earth
metals usually crystalize in a hexagonal closed-packed (hcp) phase [7]. In an
attempt to understand the phase selection, several parametric approaches have
been proposed by using physiochemical parameters, such as mixing enthalpy,
mixing entropy, melting point, atomic size mismatch, and valence electron
concentration [8-11]. On the other hand, the potential to enhance the empirical
rules to scientific theories is remarkable to tackle challenges in the family of
multi-component alloys.
Along with the appearance of the special microstructure, interesting physical
and chemical properties are being continuously revealed over the recent years.
In the case of the equiatomic fcc CrMnFeCoNi, for example, the strength and
ductility increase simultaneously at cryogenic temperatures [12, 13], which run

2
counter to many other materials where an inverse dependence of strength and
ductility is invariably seen [14].
The physical metallurgy principles might be different for HEAs as compared
to the conventional alloys. Meanwhile, although a large body of work has been
carried out since 2004, there are still many open questions that concern people
in the academic and metallurgic research communities [4]. For instance, are
HEAs thermodynamically stable or unstable in nature? How about the phase
diagrams of HEAs? What features are crucial for producing HEAs with
exceptional properties? How to accelerate the exploration of high-performance
single- or multi-phase HEAs? All these facts making HEAs attractive.
In this thesis, we present a theoretical study of fundamental characteristics
for a variety of HEAs, including the phase stability, elastic/plastic deformation,
magnetic behavior, and thermo-physical properties. This work provides some
theoretical points of view to study the intrinsic physical mechanisms behind
the complex behavior as observed in HEAs, and reveals some opportunities for
optimizing and designing properties of materials.

3
Chapter 2
Theoretical methodology
Many physical and chemical properties of materials depend on the interactions
of electrons and nuclei, which require a quantum-mechanical treatment. In this
scenario, one should find the solution of Schrödinger wave equation for multi-
particle system. However, it is incredible complicate to solve such equation in
practice. Thus, reasonable approximations and simplifications are necessary.
This chapter briefly presents some key approaches used in the present work.
2.1 Schrödinger equation
In quantum mechanics, the state of system is described by the multi-particle
Schrödinger equation
ℋΨ = 𝐸Ψ, (2.1)
where 𝐸 is the ground state energy, Ψ = Ψ(𝐫1, … , 𝐫N, 𝐑1, … , 𝐑M) is the wave-
function for N electrons and M nuclei system with positions 𝐫𝑖 (i = 1, …, N)
and 𝐑𝑗 (j = 1, …, M), respectively. The many-body Hamiltonian
ℋ = −ℏ2
2𝑚𝑒
∑ ∇𝐫𝑖
2𝑖 −
ℏ2
2∑
∇𝐑𝑗2
𝑀𝑗𝑗 − ∑ ∑
𝑒2𝑍𝑗
|𝐫𝑖−𝐑𝑗|𝑗𝑖 +1
2∑
𝑒2
|𝐫𝑖−𝐫𝑗|𝑖≠𝑗 +1
2∑
𝑒2𝑍𝑖𝑍𝑗
|𝐑𝑖−𝐑𝑗|𝑖≠𝑗 , (2.2)
consisting of electrons with mass 𝑚𝑒 and charge 𝑒, and nuclei with mass 𝑀𝑗
and charge −𝑍𝑗𝑒. Here ℏ = ℎ 2𝜋⁄ is the reduced Planck constant. The first two
terms denote the kinetic energy operators for electrons and nuclei, respectively.
The last three terms represent the corresponding Coulomb interactions of
electron-nuclei, electron-electron, and nuclei-nuclei, respectively. The above
Hamiltonian expresses the full theory. However, in real materials, solving Eq.
2.1 within Eq. 2.2 is a Herculean task.

4
Nuclei are much more massive than electrons, which implies that electrons can
respond to the nuclei moving instantaneously. Hence, one may neglect the
nuclei kinetic energy term in Eq. 2.2, i.e., Born-Oppenheimer approximation.
The nuclei-nuclei interactions are neglected, which shift the eigenvalues only.
Then the Hamiltonian is simplified as
ℋ = −ℏ2
2𝑚𝑒
∑ ∇𝐫𝑖
2𝑖 − ∑ ∑
𝑒2𝑍𝑗
|𝐫𝑖−𝐑𝑗|𝑗𝑖 +1
2∑
𝑒2
|𝐫𝑖−𝐫𝑗|𝑖≠𝑗 = 𝑇 + 𝑉 + 𝑈, (2.3)
where 𝑇 is the kinetic energy operator, 𝑉 represents the external potential from
the static nuclei background, and 𝑈 represents the internal potential from the
electron-electron interaction.
2.2 Density functional theory
Density functional theory (DFT) is one of the most popular approaches to the
theory of electronic structure, in which the electronic density plays the central
role, rather than the many-electron wave function [15].
In the framework of DFT, there are two important theorems [16]: i, the
external potential 𝑉(𝒓) is a unique functional of the electronic density 𝑛(𝒓),
apart from a trivial additive constant; ii, there exists a universal functional,
𝐹[𝑛(𝒓)], independent of 𝑉(𝒓), such that the expression
𝐸[𝑛(𝒓)] ≡ ∫ 𝑉(𝒓)𝑛(𝒓)𝑑𝒓 + 𝐹[𝑛(𝒓)], (2.4)
has its minimum value for the correct 𝑛0(𝒓). Based on the variational principle,
the minimum of 𝐸 equals the total energy of the electronic system. The 𝐹[𝑛] is usually represented as
𝐹[𝑛] =1
2∬
𝑛(𝒓)𝑛(𝒓′)
|𝒓−𝒓′|𝑑𝒓𝑑𝒓′ + 𝐺[𝑛], (2.5)
where 𝐺[𝑛] ≡ 𝑇𝑠[𝑛] + 𝐸xc[𝑛] is a universal functional like 𝐹[𝑛]. Here, 𝑇𝑠[𝑛]
is the kinetic energy of a system of non-interacting electrons, and 𝐸xc[𝑛] is the
exchange-correlation energy representing all electron-electron interactions
omitted in the other terms.
Based on the Kohn-Sham (KS) scheme [17], 𝑛(𝒓) is expressed as 𝑛(𝒓) =
∑ |𝜓𝑖(𝒓)|2𝑁𝑖=1 , where 𝜓𝑖(𝒓) is the KS orbitals describing electrons moving in
an effective potential, which contains all types of interactions. Therefore, one
can obtain 𝑛(𝒓) by solving the single-particle Schrödinger equation
[−1
2∇𝑖
2 + 𝑉(𝒓) + ∫𝑛(𝒓′)
|𝒓−𝒓′|𝑑𝒓′ + 𝑉xc] 𝜓𝑖(𝒓) = ϵ𝑖𝜓𝑖(𝒓), (2.6)

5
where ϵ𝑖 denotes the eigenvalues of the hypothetical KS particles, and 𝑉xc =
𝛿𝐸xc[𝑛] 𝛿𝑛⁄ represents the corresponding exchange-correlation potential.
As the exact form of 𝐸xc is not known, although the existence of exchange-
correlation is guaranteed by the basic theorem, one needs to resort to various
approximations. Nowadays, the local density approximation (LDA) [18] and
the generalized gradient approximation (GGA) [19] are two most widely used
approximations for the exchange-correlation term in computational materials
science based on DFT.
2.3 Exact muffin-tin orbital method
Developing accurate and efficient numerical approaches for solving the KS
equation challenges the computational materials science community. Several
techniques were introduced: i, full-potential methods, in which the wave-
function is taken into account with high accuracy, while at the price of very
expensive computational effort; ii, pseudopotential methods, in which a full-
potential description is kept in the interstitial region, and the true Coulomb-
like potential is replaced with a weak pseudopotential in the region near the
nuclei; iii, muffin-tin potential methods, in which the Kohn-Sham potential is
substituted by spherically symmetric potentials centered on atoms plus a
constant potential in the interstitial region.
In the present work, the exact muffin-tin orbitals (EMTO) method is applied.
The details about this method and its self-consistent implementation can be
found in previous work [20]. This section briefly introduces some key points.
According to the overlapping muffin-tin approximation, the effective single-
electron potential is approximated by the spherical potential wells 𝑉𝑅(𝑟𝑅) − 𝑉0
centered on lattice sites 𝑅 with notation 𝒓𝑅 ≡ 𝑟𝑅�̂�𝑅 = 𝒓 − 𝑹, plus a constant
potential 𝑉0, viz.,
𝑉eff = 𝑉(𝒓) + ∫𝑛(𝒓′)
|𝒓−𝒓′|𝑑𝒓′ + 𝑉xc ≈ 𝑉mt(𝒓) = 𝑉0 + ∑ [𝑉𝑅(𝑟𝑅) − 𝑉0]𝑅 , (2.7)
where 𝑉𝑅(𝑟𝑅) becomes equal to 𝑉0 outside the potential sphere of radius 𝑠𝑅.
The Eq. 2.6 within Eq. 2.7 are solved by expanding the KS orbital 𝜓𝑖(𝒓) in
terms of exact muffin-tin orbitals �̅�𝑅𝐿𝛼 (𝜖𝑖 , 𝒓𝑅), viz.,
𝜓𝑖(𝒓) = ∑ �̅�𝑅𝐿𝛼 (𝜖𝑖 , 𝒓𝑅)𝑣𝑅𝐿,𝑖
𝛼𝑅𝐿 , (2.8)

6
where the expansion coefficients 𝑣𝑅𝐿,𝑖𝛼 are determined from the condition that
the above expansion should be a solution for Eq. 2.6 in the entire space, and
the multi-index 𝐿 = (𝑙, 𝑚) represents the set of the orbital (𝑙) and magnetic
(𝑚) quantum numbers, respectively. The �̅�𝑅𝐿𝛼 (𝜖𝑖 , 𝒓𝑅) contains different basis
functions in different regions
�̅�𝑅𝐿𝛼 (𝜖𝑖 , 𝒓𝑅) = 𝜙𝑅𝐿
𝛼 (𝜖𝑖 , 𝑟𝑅) + 𝜓𝑅𝐿𝛼 (𝜖𝑖 − 𝑣0, 𝒓𝑅) − 𝜑𝑅𝐿
𝛼 (𝜖𝑖, 𝑟𝑅)𝑌𝐿(𝑟𝑅), (2.9)
where 𝜙𝑅𝐿𝛼 (𝜖𝑖 , 𝑟𝑅) is the partial wave inside the potential sphere (𝑟𝑅 ≤ 𝑠𝑅),
𝜓𝑅𝐿𝛼 (𝜖𝑖 − 𝑣0, 𝒓𝑅) is the screened spherical wave in the interstitial region, i.e.,
outside of the non-overlapping sphere 𝑎𝑅 (𝑎𝑅 ≤ 𝑠𝑅), and 𝜑𝑅𝐿𝛼 (𝜖𝑖 , 𝑟𝑅)𝑌𝐿(𝑟𝑅) is
the backward extrapolated free-electron wave function, matched continuously
and differentiable to the partial waves at 𝑠𝑅 and continuously to the screened
spherical waves at 𝑎𝑅. From the so-called kink-cancellation equation, which is
related to the boundary condition in the region 𝑎𝑅 ≤ 𝑟𝑅 ≤ 𝑠𝑅, one can find the
solution of the KS equation.
2.4 Coherent potential approximation
The main difficulty in the application of DFT to real system is the presence of
various kinds of disorder. One powerful technique is the coherent potential
approximation (CPA) [21-23]. It is based on the assumption that the alloy may
be replaced by an ordered effective medium, the parameters of which are
determined self-consistently. The single-site approximation is applied to the
impurity problem, i.e., one single impurity is embedded in an effective medium
and no information is provided about the individual potential and charge
density beyond the sphere or polyhedron around the impurity.
Basically, two main approximations are involved within the CPA: i, assume
that the local potentials around a certain type of atom from the alloy are the
same, i.e., the effect of local environment is neglected (all atoms belonging to
one particularly type are the same); ii, the system is replaced by a monoatomic
set-up described by the site independent coherent potential �̃� . In terms of
Green functions, one approximates the real Green function 𝑔 by a coherent
Green function �̃�. A single-site Green function 𝑔𝑖 is introduced for each alloy
component i = A, B, C, … in substitutional alloy AaBbCc…, where a, b, c, …
stand for the atomic fractions of the A, B, C, … atoms, respectively.
First, the coherent Green function �̃� is derived from the coherent potential
�̃� using an electronic structure method
�̃� = [𝑆 − �̃�]−1, (2.10)

7
where 𝑆 denotes the structure constant (slope) matrix corresponding to the
underlying lattice. Next, the Green functions of the alloy components, 𝑔𝑖, are
calculated by substituting the coherent potential of the CPA medium �̃� by the
real atomic potentials 𝑃𝑖 , viz.,
𝑔𝑖 = �̃� + �̃�(𝑃𝑖 − �̃�)𝑔𝑖 , 𝑖 = A, B, C …, (2.11)
where the condition is expressed via real-space Dyson equation. Finally, the
average of the individual Green functions should reproduce the single-site part
of the coherent Green function
�̃� = 𝑎𝑔A + 𝑏𝑔B + 𝑐𝑔C + ⋯. (2.12)
The above three equations are solved iteratively, and the output �̃� and 𝑔𝑖 are
used to determine the electronic structure, charge density and total energy of
the random alloy.

8
Chapter 3
Structural properties
Structural properties often hold the key to understand the material properties
from a microscopic point of view. For many reported HEAs, despite containing
multiple components with different crystal structures in their ground states,
there exists a preference for the formation of simple yet chemically disordered
solid solution phase rather than complex intermetallic structures. This chapter
briefly presents some fundamental structural characteristics of HEAs.
3.1 Phase stability
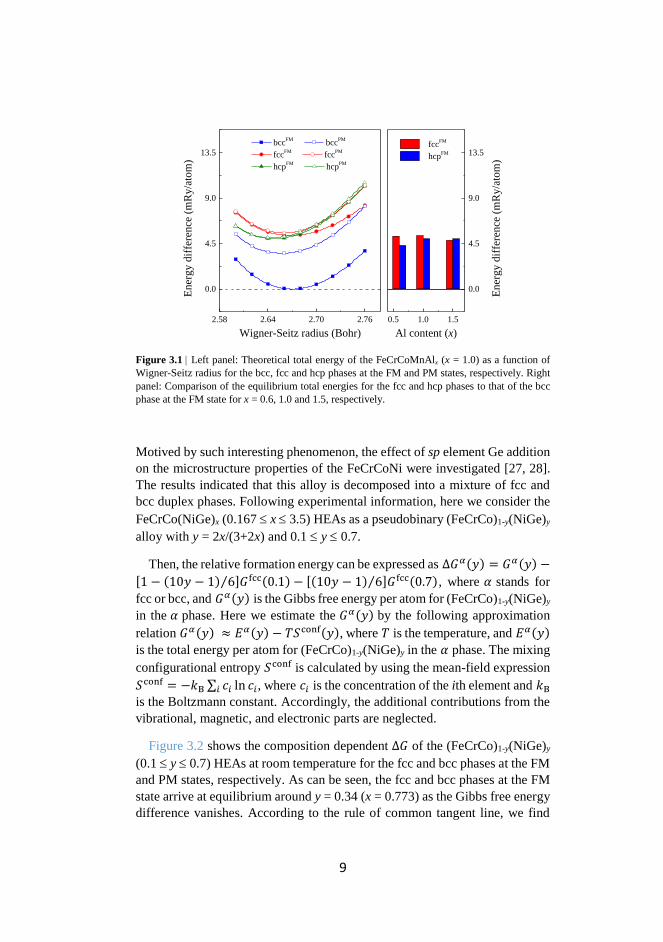
Experiments indicated that the equiatomic FeCrCoMnAl adopts a single bcc
solid solution phase [24]. Figure 3.1 (left panel) shows the total energy of the
FeCrCoMnAl as a function of Wigner-Seitz radius for the bcc, fcc, and hcp
phases at the ferromagnetic (FM) and paramagnetic (PM) states, respectively.
Here, all energies are plotted with respect to the equilibrium total energy of
FM bcc. As can be seen, the bcc structure is energetically favorable over the
fcc and hcp structures irrespective of the magnetic state. Extending the above
study to other compositions, as shown in Fig. 3.1 (right panel), we find that the
FeCrCoMnAlx (0.6 ≤ x ≤ 1.5) HEAs prefer the bcc structure rather than the
other two close-packed structures.
3.1.1 Composition dependence
In the widely studied FeCrCoNiAlx HEAs, the as-cast structure evolves from
the initial single fcc phase to a mixture of fcc and bcc duplex phases, and then
a single bcc phase with the increase of Al concentration [25, 26].
9
2.58 2.64 2.70 2.76
0.0
4.5
9.0
13.5
Ener
gy
dif
fere
nce
(m
Ry/a
tom
)
Wigner-Seitz radius (Bohr)
bccFM
bccPM
fccFM
fccPM
hcpFM
hcpPM
0.5 1.0 1.5
0.0
4.5
9.0
13.5
Ener
gy
dif
fere
nce
(m
Ry/a
tom
)
Al content (x)
fccFM
hcpFM
Figure 3.1 Left panel: Theoretical total energy of the FeCrCoMnAlx (x = 1.0) as a function of
Wigner-Seitz radius for the bcc, fcc and hcp phases at the FM and PM states, respectively. Right
panel: Comparison of the equilibrium total energies for the fcc and hcp phases to that of the bcc
phase at the FM state for x = 0.6, 1.0 and 1.5, respectively.
Motived by such interesting phenomenon, the effect of sp element Ge addition
on the microstructure properties of the FeCrCoNi were investigated [27, 28].
The results indicated that this alloy is decomposed into a mixture of fcc and
bcc duplex phases. Following experimental information, here we consider the
FeCrCo(NiGe)x (0.167 x 3.5) HEAs as a pseudobinary (FeCrCo)1-y(NiGe)y
alloy with y = 2x/(3+2x) and 0.1 y 0.7.
Then, the relative formation energy can be expressed as ∆𝐺𝛼(𝑦) = 𝐺𝛼(𝑦) −
[1 − (10𝑦 − 1) 6⁄ ]𝐺fcc(0.1) − [(10𝑦 − 1) 6⁄ ]𝐺fcc(0.7), where 𝛼 stands for
fcc or bcc, and 𝐺𝛼(𝑦) is the Gibbs free energy per atom for (FeCrCo)1-y(NiGe)y
in the 𝛼 phase. Here we estimate the 𝐺𝛼(𝑦) by the following approximation
relation 𝐺𝛼(𝑦) ≈ 𝐸𝛼(𝑦) − 𝑇𝑆conf(𝑦), where 𝑇 is the temperature, and 𝐸𝛼(𝑦)
is the total energy per atom for (FeCrCo)1-y(NiGe)y in the 𝛼 phase. The mixing
configurational entropy 𝑆conf is calculated by using the mean-field expression
𝑆conf = −𝑘B ∑ 𝑐𝑖 ln 𝑐𝑖𝑖 , where 𝑐𝑖 is the concentration of the ith element and 𝑘B
is the Boltzmann constant. Accordingly, the additional contributions from the
vibrational, magnetic, and electronic parts are neglected.
Figure 3.2 shows the composition dependent ∆𝐺 of the (FeCrCo)1-y(NiGe)y
(0.1 y 0.7) HEAs at room temperature for the fcc and bcc phases at the FM
and PM states, respectively. As can be seen, the fcc and bcc phases at the FM
state arrive at equilibrium around y = 0.34 (x = 0.773) as the Gibbs free energy
difference vanishes. According to the rule of common tangent line, we find

10
that at room temperature the (FeCrCo)1-y(NiGe)y is likely to form bcc phase for
y 0.25 (x 0.5), fcc phase for y 0.42 (x 1.1), and a mixture of fcc and bcc
duplex phases between the above limits.
0.1 0.2 0.3 0.4 0.5 0.6 0.7
-3.0
-1.5
0.0
1.5
3.0
-3.0
-1.5
0.0
1.5
3.0 T = 300 K
x=1.1x=0.5
Gib
bs
free
ener
gy
G (
mR
y)
Gib
bs
free
ener
gy
G (
mR
y)
NiGe content (y)
fccPM
bccPM
fccFM
bccFM
Figure 3.2 Comparison of the Gibbs free energies of the (FeCrCo)1-y(NiGe)y (0.1 y 0.7) HEAs
as a function of NiGe content at room temperature for the fcc and bcc phases at the FM and PM
states, respectively. Here y = 2x/(3+2x), where x is the atomic fraction of NiGe in FeCrCo(NiGe)x.
3.1.2 Temperature effect
Generally, the relative phase stability at ambient pressure and as a function of
temperature can be investigated from the free energies calculated for various
structures. Here, we decompose the free energy as 𝐹 = 𝐸 + 𝐹conf + 𝐹mag +
𝐹vib + 𝐹el, where 𝐸 is the internal energy, and 𝐹conf, 𝐹mag, 𝐹vib and 𝐹el are
the additional temperature-dependent contributions for the configurational,
magnetic, vibrational and electronic free energies, respectively.
The configurational free energy for an ideal solid solution can be estimated
by 𝐹conf = −𝑇𝑆conf. For a disordered paramagnetic state, the magnetic free
energy is expressed as 𝐹mag = −𝑇𝑆mag = −𝑘B𝑇 ∑ 𝑐𝑖 ln(1 + 𝜇𝑖)𝑖 , where 𝜇𝑖 is
the local magnetic moment of the 𝑖th element. The vibrational free energy can
be derived from the Debye-Grüneisen model 𝐹vib = 9𝑘B𝜃D 8⁄ + 3𝑘B𝑇 ln(1 −
𝑒−𝜃D 𝑇⁄ ) − 𝑘B𝑇𝐷(𝜃D 𝑇⁄ ), where 𝜃D is the Debye temperature, and 𝐷 is the
Debye integral [29]. The electronic free energy is defined as 𝐹el = 𝐸el − 𝑇𝑆el,
where 𝐸el and 𝑆el are the electronic energy and entropy, respectively, which
are obtained directly from the EMTO calculations with the finite-temperature
Fermi distribution [30].

11
Figure 3.3 shows the temperature-dependent free energies of the FeCrCoNiGa
for the fcc and bcc phases at the FM and PM states, respectively. For clarity,
all free energies are plotted with respect to the free energy of FM bcc. As can
be see, the fcc and bcc phases at the FM state arrive in equilibrium around
room temperature as the free energy difference vanishes. When comparing all
four free energies, we find that the bcc phase at the FM state is energetically
stable at low temperatures (cryogenic conditions), and the fcc phase at the PM
state becomes favorable at high temperatures.
0 600 1200-5.0
-2.5
0.0
2.5
5.0
0 600 1200-5.0
-2.5
0.0
2.5
5.0
Ener
gy
dif
fere
nce
(m
Ry)
Temperature (K)
fcc
FM
PM
643 K
691 K
Ener
gy
dif
fere
nce
(m
Ry)
bcc
FM
PM
Figure 3.3 Temperature-dependent free energies of the FeCrCoNiGa for the fcc and bcc phases
at the FM and PM states, respectively. All energies are plotted with respect to the corresponding
energy of FM bcc.
3.1.3 Ordering behavior
Previous experimental work indicated that in the Al-doped FeCrCoNi HEAs,
disordered bcc precipitates embedded in ordered B2 matrix appear at high Al
concentrations [31-33].
To investigate this ordering behavior, here we consider two alloy categories
with bcc underlying lattice: (i) the completely disordered phase (type I), which
stands for the reference structure where the alloy components randomly
occupy all sites, and (ii) the B2 ordered phase (type II), where the corner sites
(denoted by A) and the body-center sites (denoted by B) are occupied by
different types of atoms. We treat the equiatomic FeCrCoNiAl as a pseudo-
binary 𝐴0.8𝐵0.2 alloy, where 𝐴 represents the sub-system containing four
selected elements with equal concentration, and 𝐵 denotes the rest of the alloy
components. Then, the degree of chemical order can be controlled by changing

12
the composition at the corner positions as 𝐴0.8+𝑐𝐵0.2−𝑐 and at the body-center
positions as 𝐴0.8−𝑐𝐵0.2+𝑐, represented by (𝐴0.8+𝑐𝐵0.2−𝑐)(𝐴0.8−𝑐𝐵0.2+𝑐).
Accordingly, we change 𝑐 from 0 (i.e., 𝐵 atoms occupy both sites with equal
probability) to 0.2 (i.e., the probability of finding 𝐵 atoms on the corner site is
zero). The degree of chemical disorder can be expressed as 𝜂 = 5𝑐. We should
notice that although 𝑐 = 0 (𝜂 = 0) describes chemically equivalent A and B
sites, the magnetic difference is still allowed and thus this set up accounts for
a possible magnetic transition from B2-type to bcc-type magnetism.
Figure 3.4 (left panel) plots the equilibrium total energy difference between
type I and type II as a function of order parameter 𝜂 for situations when one
particular element is gradually moved from random distribution to sublattice
B. The obtained results reveal that Cr ordering is energetically unfavorable,
whereas Al ordering is always preferred, independent of 𝜂. Namely, Al is more
likely to occupy a particular sublattice rather than being randomly distributed.
Since there are still four alloy components which are randomly distributed,
one can go further and investigate the simultaneous ordering of two or more
elements. Below we consider only the case of Al co-ordering with Al order
parameter equal 1. For example, we move all available Cr atoms from the Al-
sublattice to the other one, while keeping the others randomly distributed. This
case is represented as Cr/Al-FeCoNi.
By studying a series of different kinds of Al co-ordering configurations in
the present work, as shown in Figure 3.4 (right panel), we find that Ni and Co
are prefer to be located on the Al-free site.
0.0 0.5 1.0-4.2
-2.1
0.0
2.1
4.2
6.3
A
B
A
B
En
erg
y d
iffe
ren
ce (
mR
y)
Order parameter ()
Fe
Cr Ni
Co AlII
chemical
transition
0 -10 -20
NiCo/FeAl-Cr
NiCo/CrAl-Fe
Ni/CoAl-FeCr
Ni/CrAl-FeCo
Ni/FeAl-CrCo
NiCo/Al-FeCr
NiCr/Al-FeCo
NiFe/Al-CrCo
Ni/Al-FeCrCo
Co/Al-FeCrNi
Cr/Al-FeCoNi
Fe/Al-CrCoNi
Al
co-o
rder
ing
co
nfi
gu
rati
on
Energy difference (mRy)
Figure 3.4 Equilibrium total energy of the FeCrCoNiAl for type II phase (with respect to type I)
as a function of order parameter for each alloy component in one-atom partition configurations
(left panel), and for a series of different kinds of Al co-ordering configurations (right panel).

13
3.2 Lattice parameter
Table 3.1 shows the calculated equilibrium Wigner-Seitz radius of a large
number of equiatomic medium- and high-entropy alloys with solid solution
phases, along with the corresponding values converted from the experimental
lattice parameters. The majority of selected alloys are based on 3d transition
metals. It is found that the theoretical values are in reasonable agreement with
the available experimental data.
For instance, the calculated equilibrium Wigner-Seitz radius of the FM fcc
FeCoNiCu is 2.641 bohr, which compares well with the experimental value of
2.648 bohr [34]. Alloying with Cr decreases the volume of the FeCoNiCu host,
in line with the experimental observation [1, 34].
Table 3.1. List of equiatomic medium- and high-entropy alloys crystallizing in the fcc and/or bcc
phases, respectively. The calculated equilibrium Wigner-Seitz radius for the fcc and bcc phases at
the FM (PM) state are presented, respectively, along with the available experimental data.
Alloy Phase fcc bcc
Cal. Expt. Cal. Expt. Ref.
AlNiCu fcc + bcc 2.711 2.690 2.710 2.767 [35]
CrFeNi fcc 2.626 (2.619) 2.636 (2.632) [5]
CrCoNi fcc 2.609 (2.604) 2.636 2.622 (2.618) [36]
MnFeNi fcc 2.617 (2.624) 2.646 (2.652) [5]
FeCoNi fcc 2.629 (2.606) 2.658 2.635 (2.624) [37]
AlFeCoNi bcc 2.668 (2.654) 2.669 (2.663) 2.682 [37] AlCoNiCu fcc + bcc 2.674 (2.670) 2.661 2.676 (2.674) 2.737 [35]
VCrFeMo bcc 2.790 (2.789) 2.771 (2.769) [38]
CrFeCoNi fcc 2.619 (2.604) 2.644 2.630 (2.624) [26] MnFeCoNi fcc 2.616 (2.606) 2.657 2.655 (2.640) [39]
FeCoNiCu fcc 2.641 (2.632) 2.648 2.647 (2.642) [34]
AlTiFeNiCu fcc + bcc 2.767 (2.765) 2.762 (2.760) [40] AlCrMnFeCo bcc 2.667 (2.660) 2.670 (2.658) 2.680 [24]
AlCrFeCoNi fcc + bcc 2.661 (2.653) 2.667 2.664 (2.659) 2.683 [26]
AlCrFeNiCu fcc + bcc 2.689 (2.688) 2.686 (2.683) [41] AlCrCoNiCu fcc + bcc 2.671 (2.671) 2.650 2.673 (2.671) 2.726 [35]
AlFeCoNiCu fcc + bcc 2.672 (2.665) 2.670 2.674 (2.670) 2.668 [42]
TiVCrFeMo bcc 2.836 (2.835) 2.799 (2.794) [38] TiCrFeCoNi fcc 2.690 (2.682) 2.691 (2.684) [43]
VMnFeCoNi fcc 2.638 (2.636) 2.664 (2.652) [44]
VFeCoNiCu fcc 2.662 (2.652) 2.666 (2.661) [8] CrMnFeCoNi fcc 2.607 (2.604) 2.659 2.640 (2.630) [45]
CrMnFeCoCu fcc 2.647 (2.630) 2.656 (2.647) [44]
CrMnFeNiCu fcc + bcc 2.664 (2.647) 2.664 (2.655) [41] CrFeCoNiCu fcc 2.638 (2.626) 2.642 2.643 (2.638) [1]
CrFeCoNiPd fcc 2.716 (2.706) 2.694 2.717 (2.723) [46]
CrFeNiCuMo fcc 2.743 (2.740) 2.740 (2.739) [41] MnFeCoNiCu fcc 2.646 (2.643) 2.671 (2.657) [47]
MnFeCoNiGa fcc + bcc 2.676 (2.673) 2.715 2.695 (2.685) 2.654 [39]
MnFeCoNiMo fcc 2.721 (2.717) 2.735 (2.731) [44]

14
FeCoNiCuMo fcc 2.731 (2.726) 2.736 (2.733) [47]
FeCoNiCuAg fcc 2.752 (2.750) 2.757 (2.756) [47] FeCoNiCuPt fcc 2.748 (2.745) 2.757 (2.755) [47]
AlSiFeCoNiCu fcc + bcc 2.673 (2.666) 2.693 2.677 (2.672) 2.638 [42]
AlTiCrFeCoNi fcc + bcc 2.721 (2.716) 2.718 (2.715) [43] AlTiFeCoNiCu fcc + bcc 2.730 (2.724) 2.681 2.729 (2.723) 2.709 [42]
AlCrFeCoNiCu fcc + bcc 2.669 (2.661) 2.669 2.670 (2.665) 2.671 [42]
TiCrMnFeCoNi fcc + bcc 2.686 (2.677) 2.688 2.687 (2.678) [2] VCrMnFeCoNi fcc 2.637 (2.637) 2.644 2.654 (2.645) [2]
CrMnFeCoNiCu fcc 2.652 (2.630) 2.651 2.656 (2.646) [2]
CrMnFeCoNiGe fcc + bcc 2.679 (2.663) 2.644 2.682 (2.676) [2] AlSiCrFeCoNiCu fcc + bcc 2.670 (2.664) 2.647 2.672 (2.668) 2.723 [35]
Table 3.2. Prediction of Wigner-Seitz radius of some equiatomic medium- and high-entropy alloys
for the bcc phase at the FM (PM) state. The “average” values obtained according to Vegard’s rule
by using the corresponding experimental data of the alloy components.
Alloy EMTO-CPA average Alloy EMTO-CPA average
AlVCr 2.775 2.829 AlVCrMn 2.717 2.797 AlVMn 2.742 2.835 AlVCrFe 2.723 (2.716) 2.789
AlVFe 2.756 (2.747) 2.824 AlVMnFe 2.708 (2.701) 2.793
AlCrMn 2.690 2.792 AlVNbMo 2.947 2.951 AlCrFe 2.704 (2.699) 2.781 AlVNbW 2.959 2.953
TiVNb 2.972 2.979 AlCrMnFe 2.672 (2.667) 2.760
TiVMo 2.909 2.931 TiVCrMn 2.741 2.812 TiCrMo 2.866 2.888 TiVCrMo 2.848 2.870
TiNbMo 3.003 3.017 TiVNbMo 2.956 2.966
VCrMn 2.668 2.732 VCrMnFe 2.660 (2.656) 2.716 VCrFe 2.678 (2.672) 2.721 VCrNbMo 2.881 2.874
VMnFe 2.666 (2.658) 2.727 AlTiVCrMn 2.776 2.848
VNbMo 2.943 2.937 AlVCrMnFe 2.692 (2.688) 2.771 AlTiVMn 2.813 2.889 AlVCrNbW 2.905 2.899
AlTiVFe 2.821 (2.811) 2.881 AlVCrNbMoW 2.911 2.904
AlTiCrMn 2.773 2.857
Table 3.2 lists some promising candidate alloy compositions, which are
suggested to adopt a single bcc solid solution phase at 873 K [48]. The Wigner-
Seitz radius of the AlVCrNbMoW, for example, is 2.904 bohr as determined
from Vegard’s rule by using the corresponding experimental data of the alloy
components [20, 49]. This estimated value is close to the EMTO-CPA result
of 2.911 bohr. From the detailed comparison of the two sets of data, we find
that there is a general good agreement between these theoretical predictions.

15
Chapter 4
Deformation performance
There exists a shape change in material when a sufficient load is applied, which
is called deformation. This chapter briefly presents two types of deformation:
i, elastic deformation, a temporary shape change that is self-reversing after the
load is removed; ii, plastic deformation, a permanent shape change that without
fracture under a sufficient load.
4.1 Elasticity
The elastic properties govern the stress-strain relation before yielding, and they
are sensitive to various internal and external conditions, e.g., ordering, pressure,
temperature. On an atomic scale, macroscopic elastic strain is manifested as
subtle changes in the interatomic spacing.
4.1.1 Single-crystal elastic constants
For a cubic lattice, the elastic properties can be fully characterized by three
independent elastic constants: 𝐶11, 𝐶12, and 𝐶44. Here, the bulk modulus 𝐵 =(𝐶11 + 2𝐶12) 3⁄ is derived from the equation of state fitted to the ab initio total
energies for a series of different volumes. The tetragonal shear modulus 𝐶′ =(𝐶11 − 𝐶12) 2⁄ and the cubic elastic modulus 𝐶44 are derived from the volume-
conserving orthorhombic and monoclinic deformations, respectively [20].
Experiments indicated that the FeCoNiCu and FeCoNiCuX (X = V, Cr, Mn)
HEAs adopt a single fcc solid solution phase [1, 8, 34, 50-53]. The present ab
initio data confirm the stability of the fcc structure relative to the bcc and hcp

16
structures [54]. Table 4.1 lists the calculated single-crystal elastic constants of
the FeCoNiCu and FeCoNiCuX (X = V, Cr, Mn) HEAs at the FM and PM
states, respectively. Usually, the mechanical stability requirement in a cubic
lattice leads to the following restrictions [55]: 𝐶11 > 0, 𝐶44 > 0, 𝐶11 − 𝐶12 >
0 and 𝐶11 + 2𝐶12 > 0. As shown in Table 4.1, the considered alloys all satisfy
the above criteria.
Table 4.1. Theoretical single-crystal elastic constants (𝐶11, 𝐶12, 𝐶44, and 𝐶′ = (𝐶11 − 𝐶12) 2⁄ , in
units of GPa) of the FeCoNiCu and FeCoNiCuX (X = V, Cr, Mn) HEAs for the fcc phase at the
FM and PM states, respectively.
Alloy 𝑪𝟏𝟏 𝑪𝟏𝟐 𝑪𝟒𝟒 𝑪′
FeCoNiCu FM 211.9 157.8 126.9 27.0
PM 213.1 147.7 138.2 32.7
FeCoNiCuV FM 196.3 158.2 124.1 19.1 PM 207.2 158.0 132.8 24.6
FeCoNiCuCr FM 209.2 149.6 142.2 29.8
PM 223.7 155.3 151.3 34.2 FeCoNiCuMn FM 189.8 123.8 139.2 33.0
PM 187.1 122.8 139.3 32.2
4.1.2 Polycrystalline elastic moduli
In general, the bulk modulus 𝐵 represents the resistance to volume change by
applied pressure, and the shear modulus 𝐺 indicates the resistance to reversible
deformations upon shear stress. The Young’s modulus 𝑌 is defined as the ratio
of the tensile stress to the corresponding tensile strain [56]. The 𝐵/𝐺 ratio has
often been used to describe the ductile/brittle behavior of materials. According
to the Pugh criterion [57], a high 𝐵/𝐺 ratio indicates a tendency for ductility,
while a small one for brittleness with the critical value around 1.75.
Here, 𝐺 is estimated from the arithmetic Hill average 𝐺 = (𝐺V + 𝐺R) 2⁄ ,
where 𝐺V and 𝐺R are the Voigt and Reuss bounds, respectively [58]. The
Young’s modulus 𝑌 and Poisson ratio 𝜐 are connected to 𝐵 and 𝐺 by the
relations 𝑌 = 9𝐵𝐺 (3𝐵 + 𝐺)⁄ and 𝜐 = (3𝐵 − 2𝐺) (6𝐵 + 2𝐺)⁄ , respectively.
Figure 4.1 shows the theoretical polycrystalline elastic moduli (𝐵, 𝐺, and 𝑌)
of the FeCoNiCu and FeCoNiCuX (X = V, Cr, Mn) HEAs at the FM and PM
states, respectively. The elastic moduli characterized by using the respective
properties of the pure elements are plotted for comparison [49]. As can be seen,
the simple rule of mixture of the selected property of HEAs can provide useful
first-level insight of the general trend as compared to the present ab initio data.

17
0 100 2000
50
100
150
200
250
0 100 200 0 100 2000
50
100
150
200
250
B
Ela
stic
mod
uli
(G
Pa)
Y
FM PM
FeCoNiCu
X = V
X = Cr
X = Mn
G
Average elastic moduli (GPa)
Ela
stic
mod
uli
(G
Pa)
Figure 4.1 Theoretical polycrystalline elastic moduli (𝐵 , 𝐺 , and 𝑌 ) of the FeCoNiCu and
FeCoNiCuX (X = V, Cr, Mn) HEAs for the fcc phase at the FM and PM states, respectively. The
“average” values obtained according to Vegard’s rule by using the corresponding experimental
data of the alloy components.
4.2 Plasticity
The activations of plastic deformation mechanism play an important role in
determining the mechanical performance of crystalline materials.
According to phenomenological model, the competition between the leading
deformation mechanisms in fcc metals, i.e., stacking fault (SF), twinning (TW),
and full-slip (SL), is related to the size of stacking fault energy (SFE) [59]. For
example, austenitic steels with SFE higher than 10-16 mJ/m2 are suggested to
show twinning [60-62], whereas alloys with SFE below this critical value
should exhibit deformation-induced phase transformation.
In the following, we briefly introduce an alternative approach [63] based on
the effective energy barriers (EEBs) in combination with the intrinsic material
parameters that obtained from the generalized stacking fault energy (𝛾-surface).
4.2.1 Generalized stacking fault energy
In fcc metals, the active deformation mode is decided by the competitions
between corresponding EEBs [63]: 𝛾SF
(𝜃) = 𝛾usf/ cos 𝜃 , 𝛾TW
(𝜃) = (𝛾utf −

18
𝛾isf)/ cos 𝜃, and 𝛾SL
(𝜃) = (𝛾usf − 𝛾isf)/ cos(60° − 𝜃) for SF, TW, and SL,
respectively. Here, 𝜃 reflects the effect of grain orientation relative to the
resolved shear direction, 𝛾isf is the intrinsic staking fault energy, 𝛾usf is the
unstable stacking fault energy, and 𝛾utf is the unstable twin fault energy.
Twinning is possible for 𝛾TW
≤ 𝛾SF
, otherwise martensitic transformation
occurs. Particularly, SF and SL or TW and SL can co-exist in material, whereas
SF and TW are exclusive to each other independently of 𝜃.
Figure 4.2 shows the room-temperature 𝛾-surfaces of the Fe40Mn40Co10Cr10,
CrMnFeCoNi, and CrCoNi alloys. As can be seen, all three alloys have 𝛾usf ≈
𝛾utf with small (or negative) 𝛾isf . This particular shape of 𝛾-surface differs
from that of the conventional fcc alloys [63, 64], and has important implication
on the deformation mechanisms.
Here, the 𝛾-surface is obtained from alias shear, i.e., by shifting the upper
part of the fcc {111} layers along the ⟨112̅⟩ direction. In practice, this process
requires a critical resolved shear stress, which raises a small but finite affine
shear, i.e., all atomic planes are actually elastically strained along the shear
direction [65]. Obviously, increasing affine shear strain increases (𝛾SF
− 𝛾TW
),
and thus favors twin nucleation for all considered alloys.
0.0 0.5 1.0 1.5 2.0
0
200
400
esfisffcc
isf
utf
usf
(
mJ/
m2)
Fault pathway (b)
Fe40
Mn40
Co10
Cr10
CrMnFeCoNi CrCoNi
0.0 0.8 1.6
alias
Strain (%)
affine
0
20
40
SF -
T
W (m
J/m2)
Figure 4.2 Generalized stacking fault energy curves of the Fe40Mn40Co10Cr10, CrMnFeCoNi, and
CrCoNi alloys at room-temperature. b =1
6⟨112⟩ is the Burgers vector of the partial dislocation
(in units of fcc lattice parameter). 𝛾isf, intrinsic stacking fault energy; 𝛾usf, unstable stacking fault
energy; 𝛾utf, unstable twining fault energy. Right panel shows (𝛾SF
− 𝛾TW
) as a function of strain.

19
Chapter 5
Magnetic behavior
Magnetism is one of the fundamental features of matter in solid state physics.
In general, the magnetization decreases with increasing temperature until the
long-range magnetic order disappears at Curie temperature (𝑇C). The bcc Fe,
for example, is characterized by the long-range ferromagnetic order below 𝑇C
1043 K. Notice that HEAs could be exciting candidate materials for magnetic
refrigeration applications based on the recent experimental work. This chapter
briefly presents some fundamental magnetic characteristics of HEAs.
5.1 Magnetic moment
Table 5.1 summarizes the calculated total and partial magnetic moments of the
FeCrCoNiAlx (0 x 2) HEAs at corresponding equilibrium volumes. Here,
the fcc phase is considered for x 1 and the bcc phase for x 0.5 based on the
microstructure observations [25, 26] and previous theoretical predictions [66].
Table 5.1. Total and partial magnetic moments (units of B per atom) of the FeCrCoNiAlx HEAs
as a function of Al fraction x for the fcc and bcc phases at the FM state, respectively.
fcc bcc
x = 0 x = 0.5 x = 1 x = 0.5 x = 1 x = 1.5 x = 2
Fe 1.96 1.97 1.98 2.23 2.17 2.13 2.08
Cr -0.72 -0.64 -0.60 -0.09 -0.07 -0.05 -0.03
Co 1.12 1.04 0.97 1.44 1.34 1.26 1.19 Ni 0.30 0.23 0.19 0.31 0.27 0.24 0.21
Al - -0.05 -0.04 -0.03 -0.03 -0.03 -0.03
Total 0.66 0.57 0.50 0.86 0.74 0.64 0.57

20
The changes of magnetic moments depend on the Al concentration and crystal
structure. For instance, the magnetic moment per Co atom decreases from 1.12
B at x = 0 to 0.97 B at x = 1 in the fcc phase, and in the bcc phase it varies
from 1.44 B to 1.19 B when x changes from 0.5 to 2. The total magnetic
moment per atom of the FeCrCoNiAl in the bcc phase is 0.74 B, which is 48 %
larger than the one in the fcc phase. Irrespective of the structure, Al addition is
found to decrease the total magnetic moments, which is consistent with the fact
that the Al is a non-magnetic metal.
5.2 Curie temperature
The Heisenberg model is a statistical mechanical model, which is widely used
to study the critical point and phase transition. Here, the effective Heisenberg-
like Hamiltonian is expressed as 𝐻 = − ∑ 𝐽𝑖𝑗𝐦𝑖 ∙ 𝐦𝑗𝑖,𝑗 , where Jij denotes the
strength of magnetic exchange interaction between atomic sites i and j with
magnetic moments mi and mj. Several approaches, such as Monte-Carlo (MC)
simulation and mean-field approximation (MFA), can be applied to evaluate
𝑇C of magnetic system within the Heisenberg Hamiltonian.
5.2.1 Monte-Carlo simulation
Figure 5.1 plots the magnetic exchange interactions between alloy components
of the equiatomic FeCrCoNiAl for the fcc and bcc phases, respectively. Here,
a reduced exchange interaction parameter 𝐽𝑖𝑗′ = 𝑧𝑝𝐽𝑖𝑗𝒎𝑖𝒎𝑗 , where zp is the
coordination number of the pth coordination shell, is employed to illustrate the
crystal structural effect.
As can be seen, the interactions show long-range oscillatory behavior, e.g.,
the Fe-Fe interaction is predominantly ferromagnetic but may have anti-
ferromagnetic contribution depending on the distance between the Fe atoms.
The ferromagnetic interactions in the fcc phase are mainly from the nearest-
neighbor Fe-Fe, Fe-Co, and Co-Co pairs, and anti-ferromagnetic interactions
from the nearest-neighbor Fe-Cr and Cr-Cr pairs. Notice that Cr is anti-parallel
with Fe/Co/Ni as shown in Table 5.1, hence, the positive interactions between
Cr and Fe/Co/Ni indicate an anti-ferromagnetic coupling. In the case of the bcc
phase, the dominating ferromagnetic interactions (Fe-Fe, Fe-Co and Co-Co
pairs at the nearest-neighbor shell) are much stronger than those in the fcc
phase. These interesting features demonstrate that the crystal structure has a
strong impact on the ferromagnetic behavior of the FeCrCoNiAl.

21
0 5 10 15 20-5.5
0.0
5.5
11.0
16.5
0 5 10 15 20-5.5
0.0
5.5
11.0
16.5
bcc
Fe-Fe
Fe-Cr
Fe-Co
Fe-Ni
Fe-Al
Cr-Cr
Cr-Co
Mag
net
ic e
xch
ange
inte
ract
ion
(m
Ry
)
pth coordination shell
fcc
Mag
enti
c ex
chan
ge
inte
ract
ion
(m
Ry
)
Cr-Ni
Cr-Al
Co-Co
Co-Ni
Co-Al
Ni-Ni
Ni-Al
Al-Al
Figure 5.1 Magnetic exchange interactions of the FeCrCoNiAl as a function of coordination shell
for the fcc and bcc phases, respectively.
Starting from the computed magnetic exchange interactions, here we perform
MC simulation implemented in the UppASD program [67] to estimate 𝑇C of
the FeCrCoNiAl for the fcc and bcc phases, respectively.
The MC simulation boxes contained up to 108000 atoms (subject to periodic
boundary conditions) for the fcc underlying lattice and up to 128000 atoms for
the bcc one. Random distributions of alloy components in the supercells were
generated, and 20000 MC steps have been used for equilibration, then followed
by 20000 steps for obtaining thermodynamic averages.
Figure 5.2 (a) shows the normalized magnetization (M/M0, with M and M0
being the magnetization at T and 0 K, respectively) of the FeCrCoNiAl as a
function of temperature for the fcc and bcc phases, respectively. The crystal
structure effect is clearly observed, i.e., the magnetic transition temperature in
the bcc phase is much higher than that in the fcc phase.
Figure 5.2 (b) shows the magnetic susceptibility (𝜒 = [⟨𝑀2⟩ − ⟨𝑀⟩2]/𝑘𝐵𝑇)
of the FeCrCoNiAl as a function of temperature for the fcc and bcc phases,
respectively. Results obtained from MC simulation with varying system size
are presented for comparison. Notice that 𝜒 diverges at the critical temperature
in the thermodynamic limit at the absence of external magnetic field. From the
robust susceptibility peak, we find that 𝑇C of the FeCrCoNiAl is 205 5 K in
the fcc phase, and 355 5 K in the bcc phase.

22
0 100 200 300 400 500 600
0.00
0.25
0.50
0.75
1.00
0.00
0.25
0.50
0.75
1.00
(a)
No
rmal
ized
mag
net
izat
ion
Norm
aliz
ed m
agnet
izat
ion
Temperature (K)
fcc
bcc
120 160 200 240 280
0.00
0.05
0.10
0.15
0.20
280 320 360 400 440
0.00
0.05
0.10
0.15
0.20
bcc
N = 32000
N = 62500
N = 108000
Mag
net
ic s
usc
epti
bil
ity
(a.
u.)
Temperature (K)
fcc(b)
Mag
net
ic s
usc
epti
bil
ity
(a.
u.)
N = 54000
N = 85750
N = 128000
Figure 5.2 Temperature dependent (a) normalized magnetization and (b) magnetic susceptibility
of the FeCrCoNiAl for the fcc and bcc phases, respectively. The magnetic susceptibilities are
shown for different simulation boxes (number of atoms N is indicated in the legend).
By applying the above procedure, in Fig. 5.3 we present the concentration
dependent 𝑇C for the fcc and bcc phases, respectively, along with the available
experimental values [68]. Clearly, 𝑇C of the considered HEAs depends on the
chemical composition, and especially on the crystal structure. For the alloys
with a single fcc or bcc phase, as well as a mixture of fcc and bcc duplex phases
(assuming equal phase fractions), 𝑇C decreases with increasing Al content,
which is in line with the experimental observation.

23
0.0 0.5 1.0 1.5 2.0
0
100
200
300
400
500
0
100
200
300
400
500
0 200 400 600
0.0
0.5
1.0
bccfcc + bccfcc
Curi
e te
mper
ature
(K
)
Curi
e te
mper
ature
(K
)
Al content (x)
fcc
bcc
Ref. a
Temperature (K)
Normalized
Magnetization
Figure 5.3 Theoretical Curie temperature of the FeCrCoNiAlx HEAs as a function of Al content
for the fcc and bcc phases, respectively. The values from Ref. a [68] are presented for comparison.
Experiments indicated that the equiatomic FeCrCoNi adopts a single fcc solid
solution phase, and has 𝑇C far below the room temperature. Combining the
previous results on the relative phase stability, here we may conclude that at
room temperature, the appearance of the bcc phase (as Al is gradually added)
causes the magnetic state change from paramagnetic to ferromagnetic.
5.2.2 Mean-field approximation
Alternatively, with the help of MFA, one can evaluate 𝑇C from the ab initio
energies, i.e., 3𝑘B𝑇C = 2(𝐸PM − 𝐸FM) (1 − 𝑐)⁄ , where 𝑐 is the concentration
of nonmagnetic alloy components, and 𝐸PM and 𝐸FM are the equilibrium total
energies per atom at the PM and FM states, respectively.
Considering the documented success and high efficiency, here we carry out
the 𝑇C mapping of a large number of HEAs by using the above approach. Table
5.2 summarizes the calculated 𝑇C of the selected equiatomic medium- and
high-entropy alloys. Results for the fcc and bcc phases are given to bring to
light the related properties as a function of crystal structure. The detailed phase
information can be found in Table 3.1. Here, the available experimental and
theoretical data are listed for comparison.

24
Table 5.2. List of equiatomic medium- and high-entropy alloys crystallizing in the fcc and/or bcc
phases, respectively. The calculated Curie temperature (K) for the fcc and bcc phases are presented,
respectively, along with the available experimental and other theoretical data.
Alloy fcc bcc Expt./cal. Ref. Phase
AlNiCu - - fcc + bcc
CrFeNi 80 225 fcc CrCoNi 25 120 fcc
MnFeNi 43 71 fcc
FeCoNi 804 1147 995 [69] fcc AlFeCoNi 497 763 bcc
AlCoNiCu 197 245 fcc + bcc
VCrFeMo 21 251 bcc CrFeCoNi 155 414 120-130 [68, 70] fcc
MnFeCoNi 166 580 fcc
FeCoNiCu 796 987 826 [71] fcc AlTiFeNiCu 213 286 fcc + bcc
AlCrMnFeCo 32 463 bcc
AlCrFeCoNi 136 334 fcc + bcc AlCrFeNiCu 124 159 fcc + bcc
AlCrCoNiCu 25 70 fcc + bcc AlFeCoNiCu 493 663 fcc + bcc
TiVCrFeMo 31 97 bcc
TiCrFeCoNi 95 339 fcc VMnFeCoNi 27 470 fcc
VFeCoNiCu 246 404 fcc
CrMnFeCoNi 27 397 20 [72] fcc CrMnFeCoCu 59 477 fcc
CrMnFeNiCu 60 191 fcc + bcc
CrFeCoNiCu 251 382 172 [51] fcc
CrFeCoNiPd 440 414 440 [46] fcc
CrFeNiCuMo 102 144 fcc
MnFeCoNiCu 146 540 400 [47] fcc MnFeCoNiGa 77 567 fcc + bcc
MnFeCoNiMo 68 540 fcc
FeCoNiCuMo 328 508 657 [47] fcc FeCoNiCuAg 805 881 fcc
FeCoNiCuPt 837 879 864 [47] fcc
AlSiFeCoNiCu 281 438 fcc + bcc AlTiCrFeCoNi 93 264 fcc + bcc
AlTiFeCoNiCu 274 438 fcc + bcc
AlCrFeCoNiCu 176 300 fcc + bcc TiCrMnFeCoNi 37 380 fcc + bcc
VCrMnFeCoNi 10 288 fcc
CrMnFeCoNiCu 124 371 fcc CrMnFeCoNiGe 97 346 fcc + bcc
AlSiCrFeCoNiCu 104 213 fcc + bcc
The calculated 𝑇C of the FeCoNiCu is 796 K, compares well with the previous
theoretical value of 826 K [71]. A significant decrease in 𝑇C is revealed when
adding equimolar V, Cr, and Mn to the FeCoNiCu host. One possible reason
for this decrease is the appearance of antiferromagnetic coupling between the

25
alloying elements (V, Cr, and Mn) and the Fe-Co-Ni matrix, as demonstrated
in previous work focused on the FeCrCoNi-based HEAs [27, 73-75].
Furthermore, according to MC simulation, 𝑇C of the CrMnFeCoNi is 10
10 K in the fcc phase, and 340 10 K in the bcc phase. We recall that 𝑇C
obtained with the help of MFA in the fcc and bcc phases are 27 K and 397 K,
respectively. The good agreement between these two approaches gives support
for the 𝑇C maps as shown in Table 5.2.
For most of the alloys considered here, 𝑇C is predicted to be larger in the bcc
phase than in the fcc phase, as shown in Figure 5.4. Here, we would like to
highlight a particularly experimental fact which gains theoretical support in the
mirror of the present ab initio data.
Experiments indicated that the equiatomic CrMnFeCoNi adopts a single fcc
solid solution phase, and in a paramagnetic state down to a temperature of 93
K (i.e., the magnetic transition temperature should be below 93 K) [76]. During
mechanical alloying and spark plasma sintering process, a minority bcc phase
formed in the fcc solid solution matrix. Measuring the magnetic hysteresis
curves at room temperature indicated that the as-milled powder possesses a
characteristic feature of soft magnet [77], which is fully supported by the
presently predicted TC in the bcc phase.
0 600 1200
0
300
600
900
1200
0
100
200
300
400
Curie temperature (K)
bcc
Curi
e te
mper
atu
re (
K)
Crystal structure
MFA
MC
CrMnFeCoNi
fcc
In t
he
bcc
ph
ase
In the fcc phase
Figure 5.4 Left panel: Comparison of Curie temperature obtained via MFA and MC approaches
of the CrMnFeCoNi for the fcc and bcc phases, respectively. Right panel: Curie temperature
obtained via MFA approach of the selected systems listed in Table 5.2 for the fcc and bcc phases.

26
Chapter 6
Thermo-physical characteristics
Thermo-physical performance can be simply viewed as material properties that
vary with the state variables, e.g., pressure and temperature, without altering
the chemical identity. Notice that some relevant parameters which today are
directly accessible from the ab initio calculations. This chapter briefly presents
some fundamental thermo-physical characteristics of HEAs.
6.1 Debye temperature
The Debye temperature 𝜃𝐷 is an important parameter that correlates with many
thermal characteristics of materials. It can be derived from elastic modulus,
electrical resistivity, thermal expansion, specific heat measurement, melting
point, X-ray and neutron diffraction intensity data, and so on. Since 𝜃𝐷 vary
with temperature, one should be careful when comparing them that obtained
by different methods.
Here, we calculate 𝜃𝐷 based on the average sound velocity 𝑣𝑚, viz., 𝜃D =
(ℏ 𝑘B⁄ )(6𝜋2 𝑉⁄ )1/3𝑣m with 𝑣m = [(1 𝑣L3⁄ + 2 𝑣T
3⁄ ) 3⁄ ]−1/3 , where 𝑉 is the
atomic volume, 𝑣L and 𝑣T are the longitudinal and transverse sound velocities,
respectively. The sound velocities are related to the bulk modulus 𝐵, shear
modulus 𝐺, and density 𝜌, viz., 𝑣L = √(𝐵 + 4𝐺 3⁄ ) 𝜌⁄ and 𝑣T = √𝐺 𝜌⁄ .
Figure 6.1 shows the calculated 𝑣L, 𝑣T, 𝑣m, and 𝜃D of the FeCrCoNiGa for
the fcc and bcc phases at the FM and PM states, respectively. As can be seen,
𝑣L, 𝑣T, and 𝑣m are about 5.37-5.71 km/s, 2.69-2.84 km/s, and 3.02-3.19 km/s,
respectively, which compare well with the corresponding experimental values
of 5.22 km/s, 2.82 km/s, and 3.15 km/s. In the case of 𝜃D, we get the values of

27
394 K (417 K) and 416 K (396 K) for the fcc and bcc phases at the FM (PM)
state, respectively. These values are close to 410 K as estimated in experiment.
0.0
2.5
5.0
7.5
So
un
d v
elo
city
(km
/s)
vL
vT
vm
0
200
400
600
fccFM
fccPM
bccFM
bccPM
Expt.
Deb
ye
tem
per
atu
re (
K)
Figure 6.1 Sound velocity (left panel) and Debye temperature (right panel) of the FeCrCoNiGa
for the fcc and bcc phases at the FM and PM states, respectively. The experimental results are
shown by the shaded bars. The experimental error bars for the sound velocities are ~2.5%.
Note: the present experimental data are from my co-authors as list in preface.
6.2 Thermal expansion
The Debye-Grüneisen model [29] is applied in the present work to estimate the
thermal expansion behavior of HEAs. The Grüneisen parameter 𝛾 describes
the anharmonic effects and gives the volume dependent Debye temperature by
𝜃D(𝑉) = 𝜃D(𝑉0)(𝑉0 𝑉⁄ )𝛾. Notice that 𝛾 can be expressed as 𝛾 = −𝑓 + 𝐵′ 2⁄ ,
where 𝐵′ is the pressure derivative of the bulk modulus. For pure metals, the
factor 𝑓 = 1/2 provides a good agreement with the experimental data [20, 29].
The obtained total energy as a function of volume in the static approximation
is carried out to determine the structural parameters at ground state, and then
to derive the macroscopic properties as a function of temperature.
Figure 6.2 presents the calculated lattice parameter 𝐿 and linear thermal
expansion coefficient 𝛼 of the FeCrCoNiGa in the temperature range of 0 −
900 K. The values of 𝐿 at 300 K are 3.637 (3.627) Å and 2.885 (2.884) Å for
the fcc and bcc phases at the FM (PM) state, respectively, and they increase by
1.11 (1.33) % and 0.68 (1.08) % when the temperature reaches 900 K. It is
evident that the temperature gives a larger influence in the fcc phase than in

28
the bcc phase. In addition, the calculated 𝛼 for all considered phases increase
rapidly at low temperatures and gradually turn towards a linear trend at high
temperatures. For the considered highest temperature region, the propensity of
increment becomes moderate, especially for the FM bcc.
0 300 600 9003.60
3.63
3.66
3.69
0 300 600 9002.85
2.88
2.91
2.94
Temperature (K)
L i
n t
he
fcc
ph
ase
(Å)
fcc
FM
PM
(a)
L i
n t
he
bcc
ph
ase
(Å)
bcc
FM
PM
0 300 600 9000.0
6.5
13.0
19.5
26.0
0.0
6.5
13.0
19.5
26.0(b)
Ther
mal
expan
sio
n
(
10
-6 K
-1)
Ther
mal
expan
sio
n
(
10
-6 K
-1)
Temperature (K)
fccFM
bccFM
fccPM
bccPM
Expt.
Figure 6.2 Temperature dependent (a) lattice parameter 𝐿 and (b) linear thermal expansion
coefficient 𝛼 of the FeCrCoNiGa for the fcc and bcc phases at the FM and PM states, respectively.
The experimental results are depicted by circles with error bars.
Note: the present experimental data are from my co-authors as list in preface.
We recall that according to experiments, this alloy possesses a mixture of fcc
and bcc duplex phases [28]. Hence, the actual mean 𝛼 should be estimated by
averaging over the individual phases. For example, assuming equal fractions
for the two phases around room temperature, as indicated from Fig. 3.3, we get

29
the thermal expansion coefficient 𝛼300K ≈ 13.3×10-6 K-1, which compares well
with the experimental data shown in Fig. 6.2. On the other hand, this value is
close to that of several different HEAs, e.g., ~ 14 ×10-6 K-1 for the FeCoNiCr
and ~ 15 ×10-6 K-1 for the FeCoNiCrMn [45, 69].
Interestingly, the measured 𝛼 increases sharply around Curie temperature.
Theory predicts 𝛼 values which increase in the order of FM bcc, PM bcc, FM
fcc and PM fcc in the entire temperature range. As a consequence, a mixture
of fcc and bcc duplex phases of the FeCrCoNiGa at the FM state results in a
small 𝛼 derived as the mean value below the critical temperature. Furthermore,
the measured 𝛼 is close to the theoretical value obtained for the FM bcc phase
at cryogenic conditions and to that for the PM fcc phase at high temperatures.
Hence, the sizable difference of the calculated 𝛼 values between the FM and
PM states can explain the observed anomalous thermal expansion behavior.

30
Chapter 7
Summary and outlook
The structural, deformation, magnetic, and thermos-physical characteristics of
different-types of HEAs are investigated in the present work by using first-
principle theory in combination with a series of phenomenological models.
The present calculations confirm and predict the relative phase stability of
several HEAs, e.g., a mixture of fcc and bcc duplex phases of the FeCrCoNiGa
at room temperature. The single-crystal elastic constants obtained from the
equation of state and the volume-conserving orthorhombic and monoclinic
deformations are presented and discussed. The elastic moduli characterized by
using the respective properties of the pure elements are shown for comparison.
An atomic-level transparent approach based on the effective energy barriers
are adopted to shed light on the origin of twinning in metastable fcc HEAs.
Analysis of magnetic characteristics of the FeCrCoNiAlx through Monte-Carlo
simulation indicates that the alloy’s magnetic state is controlled by the stability
of the Al-induced single- and/or dual-phase. The fact that certain HEAs have
drastically different ordering temperature for the fcc and bcc phases offers a
way to identify alloys with a very sharp change in the magnetic properties close
to a structural transition. The combined experimental and theoretical results
indicate that the FeCrCoNiGa has an anomalous thermal expansion behavior.
The results show that engineering the structural and/or magnetic transition,
offers rich opportunities for optimizing and designing HEAs with interesting
properties. The revealed strong coupling between the two degrees of freedom
brings the reported/predicted HEAs into the focus of technological including
magneto-caloric applications.
I plan to continue the research by involving multi-scale materials modeling
tools, e.g., hybrid Monte-Carlo/molecular dynamics simulations.

31
Acknowledgement
Thanks to my supervisor Prof. Levente Vitos for his continuous and invaluable
support, and professional guidance during my PhD work.
Thanks to my co-supervisor Dr. Erik Holmström, and Prof. Nanxian Chen, for
their ongoing encouragement and guidance.
Thanks to our group members: Andreas, Dávid, Dongyoo, Fuyang, Guijiang,
Guisheng, He, Henrik, Liyun, Raquel, Ruihuan, Ruiwen, Song, Stephan, Wei,
Wenyue, Xiaojie, Xiaoqing, Xun, Zhihua, and Zongwei for helpful discussion
and collaboration.
Thanks to Ádám Vida, Anita Heczel, Béla Varga, Chuanhui Zhang, Jiang Shen,
Krisztina Kádas, Lajos Károly Varga, Lars Bergqvist, Olle Eriksson, and Se
Kyun Kwon for helpful discussion and collaboration.
The China Scholarship Council, the Swedish Research Council, the Swedish
Foundation for Strategic Research, the Swedish Foundation for International
Cooperation in Research and Higher Education, the Carl Tryggers Foundation,
the Sweden’s Innovation Agency, the Swedish Energy Agency, the National
973 Project of China, the National Natural Science Foundation of China, and
the Hungarian Scientific Research Fund are acknowledged for financial
support. The Swedish National Infrastructure for Computing at the National
Supercomputer Centers in Linköping and Stockholm are acknowledged for
computational facilities.
Last but not the least, I would like to thank my family, relatives, and friends
for their continuous support and encouragement.

32
Bibliography
[1] J.W. Yeh, S.K. Chen, S.J. Lin, J.Y. Gan, T.S. Chin, T.T. Shun, C.H. Tsau, S.Y. Chang,
Nanostructured high-entropy alloys with multiple principal elements: novel alloy design
concepts and outcomes, Adv. Eng. Mater. 6 (2004) 299-303. [2] B. Cantor, I.T.H. Chang, P. Knight, A.J.B. Vincent, Microstructural development in
equiatomic multicomponent alloys, Mater. Sci. Eng. A 375-377 (2004) 213-218.
[3] J.W. Yeh, Alloy design strategies and future trends in high-entropy alloys, JOM 65 (2013) 1759-1771.
[4] M.C. Gao, J.W. Yeh, P.K. Liaw, Y. Zhang, High-entropy alloys: fundamentals and
applications (Springer, Switzerland, 2016). [5] Z. Wu, H. Bei, F. Otto, G.M. Pharr, E.P. George, Recovery, recrystallization, grain
growth and phase stability of a family of FCC-structured multi-component equiatomic
solid solution alloys, Intermetallics 46 (2014) 131-140. [6] O.N. Senkov, G.B. Wilks, D.B. Miracle, C.P. Chuang, P.K. Liaw, Refractory high-
entropy alloys, Intermetallics 18 (2010) 1758-1765.
[7] A. Takeuchi, K. Amiya, T. Wada, K. Yubuta, W. Zhang, High-entropy alloys with a hexagonal close-packed structure designed by equi-atomic alloy strategy and binary
phase diagrams, JOM 66 (2014) 1984-1992. [8] Y. Zhang, Y.J. Zhou, J.P. Lin, G.L. Chen, P.K. Liaw, Solid-solution phase formation rules
for multi-component alloys, Adv. Eng. Mater. 10 (2008) 534-538.
[9] X. Yang, Y. Zhang, Prediction of high-entropy stabilized solid-solution in multi-component alloys, Mater. Chem. Phys. 132 (2012) 233-238.
[10] S. Guo, C.T. Liu, Phase stability in high entropy alloys: formation of solid-solution phase
or amorphous phase, Prog. Nat. Sci. Mater. Int. 21 (2011) 433-446. [11] S. Guo, C. Ng, J. Lu, C.T. Liu, Effect of valence electron concentration on stability of fcc
or bcc phase in high entropy alloys, J. Appl. Phys. 109 (2011) 103505.
[12] B. Gludovatz, A. Hohenwarter, D. Catoor, E.H. Chang, E.P. George, R.O. Ritchie, A fracture-resistant high-entropy alloy for cryogenic applications, Science 345 (2014) 1153-
1158.
[13] A. Gali, E.P. George, Tensile properties of high- and medium-entropy alloys, Intermetallics 39 (2013) 74-78.
[14] R.O. Ritchie, The conflicts between strength and toughness, Nat. Mater. 10 (2011) 817-
822. [15] W. Kohn, Nobel Lecture: Electronic structure of matter-wave functions and density
functionals, Rev. Mod. Phys. 71 (1999) 1253-1266.
[16] P. Hohenberg, W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136 (1964) B864-B871. [17] W. Kohn, L.J. Sham, Self-consistent equations including exchange and correlation effects,
Phys. Rev. 140 (1965) A1133-A1138.
[18] J.P. Perdew, Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B 45 (1992) 13244-13249.

33
[19] J.P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77 (1996) 3865-3868.
[20] L. Vitos, Computational quantum mechanics for materials engineers (Springer, London,
2007). [21] P. Soven, Coherent-potential model of substitutional disordered alloys, Phys. Rev. 156
(1967) 809-813.
[22] D.W. Taylor, Vibrational properties of imperfect crystals with large defect concentrations, Phys. Rev. 156 (1967) 1017-1029.
[23] B.L. Győrffy, Coherent-potential approximation for a nonoverlapping-muffin-tin-
potential model of random substitutional alloys, Phys. Rev. B 5 (1972) 2382-2384. [24] A. Marshal, K.G. Pradeep, D. Music, S. Zaefferer, P.S. De, J.M. Schneider,
Combinatorial synthesis of high entropy alloys: introduction of a novel, single phase,
body-centered-cubic FeMnCoCrAl solid solution, J. Alloys Compd. 691 (2017) 683-689. [25] Y.F. Kao, T.J. Chen, S.K. Chen, J.W. Yeh, Microstructure and mechanical property of
as-cast, -homogenized, and -deformed AlxCoCrFeNi (0≤x≤2) high-entropy alloys, J.
Alloys Compd. 488 (2009) 57-64.
[26] H.P. Chou, Y.S. Chang, S.K. Chen, J.W. Yeh, Microstructure, thermophysical and
electrical properties in AlxCoCrFeNi (0≤x≤2) high-entropy alloys, Mater. Sci. Eng., B
163 (2009) 184-189.
[27] S. Huang, Á. Vida, D. Molnár, K. Kádas, L.K. Varga, E. Holmström, L. Vitos, Phase
stability and magnetic behavior of FeCrCoNiGe high-entropy alloy, Appl. Phys. Lett. 107 (2015) 251906.
[28] Á. Vida, L.K. Varga, N.Q. Chinh, D. Molnár, S. Huang, L. Vitos, Effects of the sp element
additions on the microstructure and mechanical properties of NiCoFeCr based high entropy alloys, Mater. Sci. Eng. A 669 (2016) 14-19.
[29] V.L. Moruzzi, J.F. Janak, K. Schwarz, Calculated thermal properties of metals, Phys. Rev.
B 37 (1988) 790-799. [30] L. Vitos, P.A. Korzhavyi, B. Johansson, Evidence of large magnetostructural effects in
austenitic stainless steels, Phys. Rev. Lett. 96 (2006) 117210. [31] W.R. Wang, W.L. Wang, S.C. Wang, Y.C. Tsai, C.H. Lai, J.W. Yeh, Effects of Al
addition on the microstructure and mechanical property of AlxCoCrFeNi high-entropy
alloys, Intermetallics 26 (2012) 44-51. [32] T. Yang, S. Xia, S. Liu, C. Wang, S. Liu, Y. Zhang, J. Xue, S. Yan, Y. Wang, Effects of
Al addition on microstructure and mechanical properties of AlxCoCrFeNi high-entropy
alloy, Mater. Sci. Eng. A 648 (2015) 15-22. [33] S.G. Ma, P.K. Liaw, M.C. Gao, J.W. Qiao, Z.H. Wang, Y. Zhang, Damping behavior of
AlxCoCrFeNi high-entropy alloys by a dynamic mechanical analyzer, J. Alloys Compd.
604 (2014) 331-339. [34] L. Liu, J.B. Zhu, C. Zhang, J.C. Li, Q. Jiang, Microstructure and the properties of
FeCoCuNiSnx high entropy alloys, Mater. Sci. Eng. A 548 (2012) 64-68.
[35] J.W. Yeh, S.Y. Chang, Y.D. Hong, S.K. Chen, S.J. Lin, Anomalous decrease in X-ray diffraction intensities of Cu–Ni–Al–Co–Cr–Fe–Si alloy systems with multi-principal
elements, Mater. Chem. Phys. 103 (2007) 41-46.
[36] B.C. Sales, K. Jin, H. Bei, G.M. Stocks, G.D. Samolyuk, A.F. May, M.A. McGuire, Quantum critical behavior in a concentrated ternary solid solution, Sci. Rep. 6 (2016)
26179.
[37] T.T. Zuo, R.B. Li, X.J. Ren, Y. Zhang, Effects of Al and Si addition on the structure and properties of CoFeNi equal atomic ratio alloy, J. Magn. Magn. Mater. 371 (2014) 60-68.
[38] J. Guo, X. Huang, W. Huang, Microstructure and room-temperature mechanical
properties of FeCrMoVTix high-entropy alloys, J. Mater. Eng. Perform. 26 (2017) 3071–3078.
[39] T. Zuo, M.C. Gao, L. Ouyang, X. Yang, Y. Cheng, R. Feng, S. Chen, P.K. Liaw, J.A.
Hawk, Y. Zhang, Tailoring magnetic behavior of CoFeMnNiX (X = Al, Cr, Ga, and Sn) high entropy alloys by metal doping, Acta Mater. 130 (2017) 10-18.
[40] S. Jiang, D. sun, Y. Zhang, S. Wang, C. Zhao, Plastic deformation mechanisms of
equiatomic Ni20Ti20Fe20Al20Cu20 high-entropy alloy at high temperatures, J. Mater. Sci. 52 (2017) 3199-3207.

34
[41] C. Li, J.C. Li, M. Zhao, Q. Jiang, Effect of alloying elements on microstructure and properties of multiprincipal elements high-entropy alloys, J. Alloys Compd. 475 (2009)
752-757.
[42] Y.X. Zhuang, W.J. Liu, Z.Y. Chen, H.D. Xue, J.C. He, Effect of elemental interaction on microstructure and mechanical properties of FeCoNiCuAl alloys, Mater. Sci. Eng. A 556
(2012) 395-399.
[43] K.B. Zhang, Z.Y. Fu, J.Y. Zhang, W.M. Wang, H. Wang, Y.C. Wang, Q.J. Zhang, J. Shi, Microstructure and mechanical properties of CoCrFeNiTiAlx high-entropy alloys, Mater.
Sci. Eng. A 508 (2009) 214-219.
[44] F. Otto, Y. Yang, H. Bei, E.P. George, Relative effects of enthalpy and entropy on the phase stability of equiatomic high-entropy alloys, Acta Mater. 61 (2013) 2628-2638.
[45] G. Laplanche, P. Gadaud, O. Horst, F. Otto, G. Eggeler, E.P. George, Temperature
dependencies of the elastic moduli and thermal expansion coefficient of an equiatomic, single-phase CoCrFeMnNi high-entropy alloy, J. Alloys Compd. 623 (2015) 348-353.
[46] M.S. Lucas, L. Mauger, J.A. Muñoz, Y. Xiao, A.O. Sheets, S.L. Semiatin, J. Horwath, Z.
Turgut, Magnetic and vibrational properties of high-entropy alloys, J. Appl. Phys. 109 (2011) 07E307.
[47] M. Kurniawan, A. Perrin, P. Xu, V. Keylin, M. McHenry, Curie temperature engineering
in high entropy alloys for magnetocaloric applications, IEEE Magn. Lett. 7 (2016) 1-5. [48] O.N. Senkov, J.D. Miller, D.B. Miracle, C. Woodward, Accelerated exploration of multi-
principal element alloys with solid solution phases, Nat. Commun. 6 (2015) 6529.
[49] K.A. Gschneidner, Physical properties and interrelationships of metallic and semimetallic elements, Solid State Phys. 16 (1964) 275-426.
[50] C.J. Tong, Y.L. Chen, J.W. Yeh, S.J. Lin, S.K. Chen, T.T. Shun, C.H. Tsau, S.Y. Chang,
Microstructure characterization of AlxCoCrCuFeNi high-entropy alloy system with multiprincipal elements, Metall. Mater. Trans. A 36 (2005) 881-893.
[51] X.F. Wang, Y. Zhang, Y. Qiao, G.L. Chen, Novel microstructure and properties of
multicomponent CoCrCuFeNiTix alloys, Intermetallics 15 (2007) 357-362. [52] L. Liu, J.B. Zhu, L. Li, J.C. Li, Q. Jiang, Microstructure and tensile properties of
FeMnNiCuCoSnx high entropy alloys, Mater. Des. 44 (2013) 223-227.
[53] S. Praveen, B.S. Murty, R.S. Kottada, Alloying behavior in multi-component AlCoCrCuFe and NiCoCrCuFe high entropy alloys, Mater. Sci. Eng. A 534 (2012) 83-
89.
[54] S. Huang, Á. Vida, A. Heczel, E. Holmström, L. Vitos, Thermal expansion, elastic and magnetic properties of FeCoNiCu-based high-entropy alloys using first-principle theory,
JOM 69 (2017) 2107-2112.
[55] D.C. Wallace, Thermodynamics of Crystals (Wiley, New York, 1972). [56] J.F. Nye, Physical properties of crystals: their representation by tensors and matrices
(Oxford University Press, New York, 1985). [57] S.F. Pugh, XCII. Relations between the elastic moduli and the plastic properties of
polycrystalline pure metals, Philos. Mag. 45 (1954) 823-843.
[58] R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc. Sect. A 65 (1952) 349-354.
[59] B.C. De Cooman, K.G. Chin, J.K. Kim, High Mn TWIP steels for automotive applications,
in New Trends and Developments in Automotive System Engineering, edited by M. Chiaberge (InTech, Rijeka, 2011).
[60] G. Frommeyer, U. Brüx, P. Neumann, Supra-ductile and high-strength manganese-
TRIP/TWIP steels for high energy absorption purposes, ISIJ Int. 43 (2003) 438-446. [61] S. Allain, J.P. Chateau, O. Bouaziz, S. Migot, N. Guelton, Correlations between the
calculated stacking fault energy and the plasticity mechanisms in Fe-Mn-C alloys, Mater.
Sci. Eng. A 387-389 (2004) 158-162. [62] A. Dumay, J.P. Chateau, S. Allain, S. Migot, O. Bouaziz, Influence of addition elements
on the stacking-fault energy and mechanical properties of an austenitic Fe-Mn-C steel,
Mater. Sci. Eng. A 483-484 (2008) 184-187. [63] M. Jo, Y.M. Koo, B.J. Lee, B. Johansson, L. Vitos, S.K. Kwon, Theory for plasticity of
face-centered cubic metals, Proc. Natl. Acad. Sci. USA 111 (2014) 6560-6565.
[64] W. Li, S. Lu, Q.M. Hu, S.K. Kwon, B. Johansson, L. Vitos, Generalized stacking fault energies of alloys, J. Phys.: Condens. Matter 26 (2014) 265005.

35
[65] S. Ogata, J. Li, S. Yip, Ideal pure shear strength of aluminum and copper, Science 298 (2002) 807.
[66] F.Y. Tian, L. Delczeg, N.X. Chen, L.K. Varga, J. Shen, L. Vitos, Structural stability of
NiCoFeCrAlx high-entropy alloy from ab initio theory, Phys. Rev. B 88 (2013) 085128. [67] B. Skubic, J. Hellsvik, L. Nordström, O. Eriksson, A method for atomistic spin dynamics
simulations: implementation and examples, J. Phys.: Condens. Matter 20 (2008) 315203.
[68] Y.F. Kao, S.K. Chen, T.J. Chen, P.C. Chu, J.W. Yeh, S.J. Lin, Electrical, magnetic, and Hall properties of AlxCoCrFeNi high-entropy alloys, J. Alloys Compd. 509 (2011) 1607-
1614.
[69] K. Jin, S. Mu, K. An, W.D. Porter, G.D. Samolyuk, G.M. Stocks, H. Bei, Thermophysical properties of Ni-containing single-phase concentrated solid solution alloys, Mater. Des.
117 (2017) 185-192.
[70] M.S. Lucas, D. Belyea, C. Bauer, N. Bryant, E. Michel, Z. Turgut, S.O. Leontsev, J. Horwath, S.L. Semiatin, M.E. McHenry, C.W. Miller, Thermomagnetic analysis of
FeCoCrxNi alloys: magnetic entropy of high-entropy alloys, J. Appl. Phys. 113 (2013)
17A923. [71] F. Körmann, D. Ma, D.D. Belyea, M.S. Lucas, C.W. Miller, B. Grabowski, M.H.F. Sluiter,
“Treasure maps” for magnetic high-entropy-alloys from theory and experiment, Appl.
Phys. Lett. 107 (2015) 142404. [72] D. Ma, B. Grabowski, F. Körmann, J. Neugebauer, D. Raabe, Ab initio thermodynamics
of the CoCrFeMnNi high entropy alloy: importance of entropy contributions beyond the
configurational one, Acta Mater. 100 (2015) 90-97. [73] S. Huang, W. Li, X. Li, S. Schönecker, L. Bergqvist, E. Holmström, L.K. Varga, L. Vitos,
Mechanism of magnetic transition in FeCrCoNi-based high entropy alloys, Mater. Des.
103 (2016) 71-74. [74] S. Huang, E. Holmström, O. Eriksson, L. Vitos, Mapping the magnetic transition
temperatures for medium- and high-entropy alloys, Intermetallics 95 (2018) 80-84.
[75] C. Niu, A.J. Zaddach, A.A. Oni, X. Sang, J.W. Hurt, J.M. LeBeau, C.C. Koch, D.L. Irving, Spin-driven ordering of Cr in the equiatomic high entropy alloy NiFeCrCo, Appl. Phys.
Lett. 106 (2015) 161906.
[76] O. Schneeweiss, M. Friák, M. Dudová, D. Holec, M. Šob, D. Kriegner, V. Holý, P. Beran, E.P. George, J. Neugebauer, A. Dlouhý, Magnetic properties of the CrMnFeCoNi high-
entropy alloy, Phys. Rev. B 96 (2017) 014437.
[77] W. Ji, W. Wang, H. Wang, J. Zhang, Y. Wang, F. Zhang, Z. Fu, Alloying behavior and novel properties of CoCrFeNiMn high-entropy alloy fabricated by mechanical alloying
and spark plasma sintering, Intermetallics 56 (2015) 24-27.



![TCS High Entropy Alloys Database (TCHEA4)€¦ · TCS High Entropy Alloys Database (TCHEA) is a thermodynamic database for high entropy alloys (HEA) [2004, Yeh; 2006, Yeh]. HEAs are](https://img.dokumen.tips/doc/110x75/60628f270c7c55437c6a10ae/tcs-high-entropy-alloys-database-tchea4-tcs-high-entropy-alloys-database-tchea.jpg)

























