Embed Size (px)
Citation preview

Quality and Regulatory Aspects in Early Phase Development of Pharmaceutical Products
PDA Israel
Key areas for strategic drug development
planning
Ramat Gan, 24th October 2018
Rivka Zaibel

והאיכותהרגולציה יסודות
מערך איכות –הביורפואיותאיכות בתעשיות אבטחת •
?cGMPמה זה •
•21 CFR 210, 21 CFR 211, 21 CFR 630
•ISO 13495, Quality System Regulation21 CFR 820
•EudraLex - Volume 4 - Good Manufacturing Practice (GMP) guidelines
•ICH Quality Guidelines
2

?איזה מערך איכות מתאים לי
:הגדרת המוצר
.השלב הראשון בקביעת הרגולציה ומערך האיכות המתאים הוא הגדרת המוצר
תרופה
מוצר ביולוגי
תרפיה תאית
תרפיה גנטית
אביזר רפואי
(Combination product)מוצר משולב
:מאפיינים אחרים שמשפיעים על מערך האיכות הם
תהליך הייצור
('השתלה וכו, משאף, קפסולה למתן אוראלי, משחה, זריקה)דרך מתן המוצר
3

? מה ניתן לעשות ומה צריך לעשות בתקופת החממה
First in manלהגיע עד : מטרה
לעשות את הדבר הנכון והנדרש ולא את מה שקל לעשות
הוכחת היתכנות
תהליך ייצור
פיתוח שיטות אנליטיות
כימות )ולידציה של שיטות אנליטיות התומכות בטוקסיקולוגיה ובקליניקה
(בדיקת הווצרות נוגדנים, חומר בפלסמה
GLPלא כולם מחייבים תנאי , מבחנים פרה קליניים
מתווה לניסוי קליני פאזה ראשונה
4

דרישות מיוחדות עבור מוצרים ביולוגיים
:ביולוגי/הביוטכנולוגימורכבות המוצר
, תאים מן הצומח, תאי יונקים, חיידקים)תלות במערכת ביולוגית בייצור
(תאים הומניים, מוצרי דם
ביולוגיות לקביעת פעילות, תלות בשיטות אנליטיות אימונולוגיות
חיידקים ומזהמים נוספים בלתי ידועים ומוגדרים , סכנת המצאות וירוסים
מראש
שימוש בחומרי גלם ממקור ביולוגי
מחסור במודלים לאיפיון המוצר ולפיתוח הפרה קליני שלו
ל מהווה אתגר למפתחים ומעלה את הדרישות הרגולטוריות הקיימות "כל הנ
לפני ביצוע ניסוי קליני כלשהו
5

הגדרה-אביזרים רפואיים
510K, PMA, CE mark
דהיינו אין , אביזר לשימוש ברפואה אשר אינו תרופה/אביזר רפואי הינו מוצר
.פיזיולוגית, לא פעילות מטבולית
קיימות שלוש רמות של אביזרים רפואיים לפי מידת המורכבות והנגיעה בגוף
Class 1, Class 2, Class 3, האדם
510K , דרך רישום של אביזרים רפואיים אשר נמצאוsubstantially equivalentבדרך כלל בדרך רישום זו . ב"לאביזרים רפואיים רשומים בארה
ניתן להמנע מניסויים קליניים
PMAב"רישום של אביזר רפואי חדש בארה.
CE mark–אישור שיווק אביזרים רפואיים באירופה
6

ISO 9001-2000, ISO 13495, Quality System
Regulation
ר על פי הרגולציה האירופאית הוא "אחד ההבדלים העיקריים בין תרופות לאמ
. שלא ניתן להתחיל ניסוי בבני אדם לפני שמכינים תיק טכני
ISOלפי תקן Biocompatibilityלצורך כך יש להשלים מבחני בטיחות
.וכן הקמה של מערך איכות, 10993
7

אתגרים של טכנולוגיות מתקדמות
Combination Productsו
ההתפתחות שחלה בשנים האחרונות בחקר הרפואה
המותאמת אישית מעודדת גם פיתוח של טכנולוגיות
השילוב הזה . דיאגנוסטיות שלא היו ידועות עד היום
מעמיד בפני המפתח והרגולטור אתגרים נוספים
. בהקשר של הגדרת המוצר
יש לקיים דו שיח , עד אשר יכתבו הנחיות רלוונטיות
עם הרשויות הרגולטוריות על מנת להגיע להבנות
.לגבי מה שנדרש ומה יהיה מקובל
8

9
Development Phases
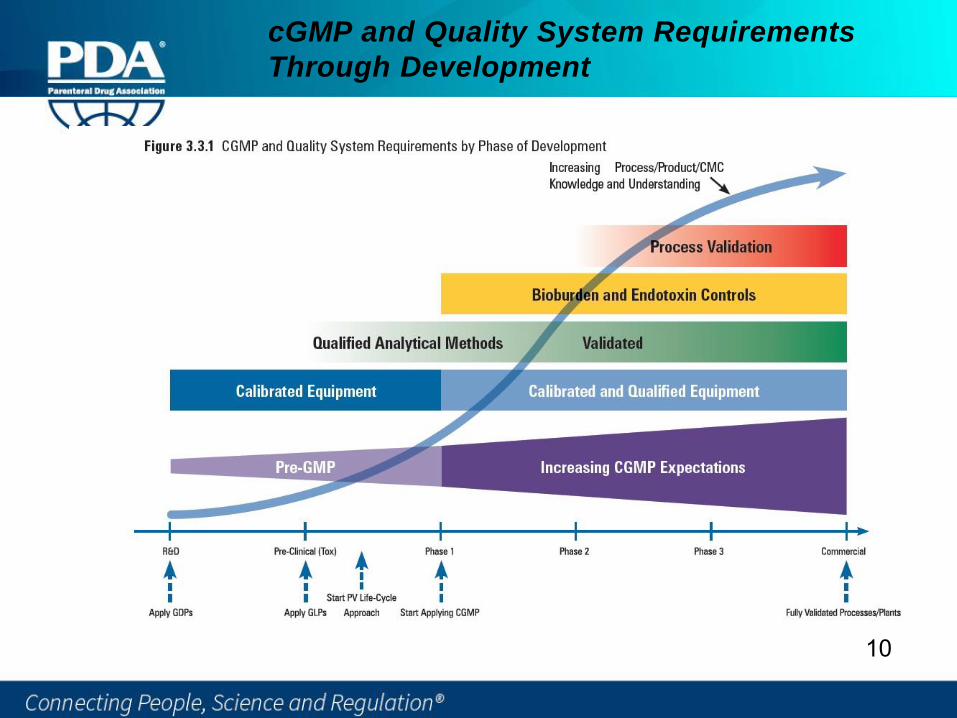
cGMP and Quality System Requirements
Through Development
10

cGMP Regulations
11

Definition of cGMP
“…the minimum current good manufacturing practice for
methods to be used in, and the facilities or controls to be
used for, the manufacture, processing, packing and holding
of a drug to assure that such drug meets the requirements of
the act as to safety, and has the identity and strength and
meets the quality and purity characteristics that it purports
or is represented to possess.”
-(21 CFR 210.1)
12

cGMP
current Good Manufacturing Practices
GMP is a dynamic concept and practice.
Staying “current” is driven by technology, improved practices
and regulatory issues.
13

Principles of cGMP
Essential Elements and Concepts of cGMP
• Good Documentation Practices
• Quality Systems (deviations; investigations; CAPA
and OOS)
• Facilities and Equipment Management
• Change Control Systems
• Materials Control
14

Compliance
Compliance with cGMP regulations is
the responsibility of the entire company
AND specifically the top management
15

Guidance for Industry
CGMP for Phase 1 Investigational Drugs
July 2008
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
Office of Regulatory Affairs (ORA)
cGMP for Phase 1 Investigational Drugs
16

Special Manufacturing Situations
Multi-Product Facilities
Biological and Biotechnological Products
Sterile Products/Aseptically Processed Products
cGMP for Phase 1 Investigational Drugs
17

Improve analytical methods, characterization and additional
aspects of product development
For Phase 3, The information amendment should include evidence
to support the elucidation and characterization of the structure.
This information can include elemental analysis, conformational
analysis, molecular weight determination, spectra from IR, NMR (1
H & 13C), UV, MS, optical activity, and if available, single crystal X-
ray diffraction data, if not previously provided. Analytical
procedures used to characterize the primary reference material
should also be provided in an information amendment.
Stress studies
cGMP for Phase 2 and 3 Investigational Drugs
18

EMA Guideline on the Requirements for the chemical and
pharmaceutical quality documentation concerning investigational
medicinal products in clinical trials
Final
May 2018
19

Information on chemical and pharmaceutical quality documentation
concerning investigational
medicinal products in clinical trials
IMPs should be produced in accordance with the principles and
the detailed guidelines of good manufacturing practices for
medicinal products (The rules governing medicinal products in
the European Community, Volume IV).
20

S.2.2.
A brief summary of the synthesis process, a flow chart of the
successive steps including, for each step the starting materials,
intermediates, solvents, catalysts and critical reagents used
should be provided. The manufacturing process and process
controls should be adequately described in such extent so it is
understood how impurities are introduced in the process, and
why the proposed control strategy is suitable.. During
development, as additional process knowledge is gained, further
details of IPCs should be provided and acceptance criteria
reviewed
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
21

S.2.4.
Control of Critical Steps and Intermediates
Tests and acceptance criteria for the control of critical steps in the synthesis process should be briefly summarised.
S.2.5.
Process Validation
Data on process validation should normally be collected throughout the development by the company, although they are not required to be submitted in the IMPD
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
22

S.2.6.
Manufacturing Process Development
It should be documented if the manufacturing process significantly
differs from that used for the production of the batches used in the
non-clinical studies.
Significant changes in the manufacturing process, which may
impact on quality, should be discussed.
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
23

S.4.
Control of the Drug Substance
3.4.1 Specification
The specifications, the tests and their acceptance criteria should be
specified for the batches of drug substances to be used in the clinical
trial.
Tests for identity, impurity and assay are mandatory
Upper limits should be set for impurities and they should be supported
by the impurity profile of the batches used in non-clinical studies.
The microbiological quality for drug substances used in aseptically
manufactured products should be specified.
Specifications and acceptance criteria should be reviewed and where appropriate adjusted to the current stage of development.
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
24

S.4.3.
Validation of Analytical Procedure
•Validation of analytical procedures during clinical development is seen
as an evolving process.
•Analytical procedures, which are either described in Ph.Eur., the
pharmacopoeia of a Member State, USP or JP general chapter, or are linked to a product specific monograph, are considered as validated.
25
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials

•For phase 1, the suitability of the analytical methods used should be
confirmed. The acceptance limits (e.g. acceptance limits for the control
of impurities, where relevant) and the parameters (specificity, linearity,
range, accuracy, precision, quantification and detection limit, as
appropriate) for performing validation of the analytical methods should
be presented in a tabulated form. Information for phase 2 and 3 clinical
trials. the suitability of the analytical methods used should be
demonstrated. Validation of the analytical methods used for release
and stability testing is expected. A tabulated summary of the results of
the validation carried out should be provided (e.g. results or values
found for specificity, linearity, range, accuracy, precision, quantification
and detection limit, as appropriate). It is not necessary to provide a full
validation report.
•In case of major changes in analytical methods, cross-validation data
should be presented. Re-analysis of preclinical batch with the new method should be considered
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
26

P. Drug Product
P.1 Description and composition of the investigational medicinal
product
The qualitative and quantitative composition of the IMP should be stated.
The information provided should include:
• a short statement or a tabulation of the dosage form and the function of
each excipient should be included.
• composition, i.e. list of all components of the dosage form and their amount
on a per-unit basis (including overages, if any), the function of the
components, and a reference to their quality standards (e.g. compendial
monographs or manufacturer’s specifications)
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
27

P.2 Pharmaceutical development
A short description of formulation development, including justification of
any new pharmaceutical form or excipient should be provided.
If changes in the formulation or dosage form compared to the IMP used
in earlier trials have been made, the relevance of the earlier material
compared to the product under testing should be described.
P.2.1 Manufacturing process development
Changes in the current manufacturing process compared to the ones
used in earlier trials should be explained, special consideration for changes in quality parameters with clinical relevance.
Information on chemical and pharmaceutical quality
documentation concerning investigationalmedicinal products in clinical trials
28

Process and Method Validation During
Development
FDA and EMA expect that the aseptic process is validated prior
to use of a sterile product in human. Validation is done by media
fill process simulation.
Bioburden, sterility and endotoxin should also be validated prior
to use of a sterile product in human.
For biological products, as relevant, viral clearance studies
should also be conducted prior to use in human.
29

Guideline on the requirements for quality documentation
concerning biological investigational medicinal products in
clinical trials
September 2017
30

International Conference on Harmonisation
“International Conference on Harmonisation (ICH) of
Technical Requirements for Registration of Pharmaceuticals
for Human Use”
Established in 1990
Documents issued through a 4-step process
Regulatory authorities and experts from the
pharmaceutical industry in Europe, Japan and the United
States are the official governing bodies.
Other world regulatory bodies recognize the ICH
documents.
31

International Conference on Harmonisation
Objectives
Achieve more economical use of human, animal and material
resources
Eliminate unnecessary delay in the global development and
availability of new medicines
Maintain safeguards on quality, safety and efficacy, and
regulatory obligations to protect public health
http://www.ich.org
32

International Conference on Harmonisation
Guidelines
The ICH process results in recommendations (issue
guidelines) on ways to achieve greater harmonization in the
interpretation and application of technical issues and
requirements for product registration and compliance.
These guidelines provide more detail regarding “how” to
achieve appropriate levels of technical standards, quality
oversight and level of detail for regulatory filings.
33

ICH Topics and Guidelines
Q = Quality: Product and testing quality
S = Safety: Topics relating to pre-clinical studies
E = Efficacy: Clinical studies in humans
M = Multidisciplinary: Cross-cutting topics
34

ICH Guidance Documents
ICH “Q” Guidance Documents to Note
Q1: StabilityQ2: Analytical ValidationQ3: ImpuritiesQ4: PharmacopeiasQ5: Biotech Products
Q6: Specifications
Q7: Active Pharmaceuticals Ingredients
Q8: Pharmaceutical Development
Q9: Quality Risk Management
Q10: Pharmaceutical Quality Systems
Q11: Development And Manufacture Of Drug Substances
35

36
משרד הבריאות הישראלי

תיק איכות
נדרש להגיש תיק איכות עבור מוצרים שמוגדרים
כתרפיה תאית
של אתרי GMPלכל שאר המוצרים נדרשת הצהרת
הייצור
עבור מוצרים להזרקה יידרש עכשיו גם הגשת תיק
ולידציה של , איכות עם דגש על שמירת הסטריליות
ואנדוטוקסיןסטריליטי, ביוברדןשיטות
משרד הבריאות הישראלי אימץ את הדרישות
האירופאיות מתוקף הסכמי ההכרה ההדדית עם EMA .37

History of FDA
The following slides present key points in the history of the FDA. It is interesting to understand that the development of FDA is directly related to failures or misuse of products.
38

History of FDA
Regulatory actionYear
Creation of the U.S. Pharmacopeia. Eleven physicians meet in Washington, D.C. to compile
the first list of standard drugs for the United States.
1820
Creation of the Bureau of Chemistry, precursor to the FDA. President Lincoln appoints chemist
C.M. Wetherill to the new Department of Agriculture.
1862
Biologics control act (virus Toxin Lax) later called the Public Health Service Act – Required
licensing of manufacturers and establishments, provided inspection authority and regulated
interstate sale of serum, vaccines and related products
1902
Congress passes the Biologics Control Act to ensure purity and safety of serums and vaccines.
That same year congress approved the study of chemical preservatives and color.
1902
First biologics regulations by publich health service Hygienic Laboratory- "Poison Squad," effect
of food preservatives and artificial colors on public health
1903
Congress passes The Food and Drug Act prohibiting interstate commerce of misbranded food
and beverages.
The Meat Inspection Act is passed on the same day due to insanitary conditions and the use of
poisonous preservatives and dyes.
1906
Pure Food and Drugs Act-Prohibited "misbranding and adulteration"1906
39

History of FDA
Regulatory actionYear
The Harrison Narcotic Act requires prescriptions for products exceeding allowable limits of
narcotics.
1914
U.S. v. 95 Barrels of Apple Cider Vinegar. The Supreme Court rules the Food and Drugs Act
allows condemning misleading labels
1924
The official name is given to the Food and Drug Administration.
1937: The Sulfanilamide Elixir Tragedy. 107 people, including many children, die as a result of
diethylene glycol contamination.
1930
PHS Hygienic Lab became NIH1930
NIH reorganized, Hygienic Lab became division of biologics Standardization 1937
Congress passes the Federal Food, Drug and Cosmetic Act (FDC). This law provides for
control of cosmetic devices, drug safety before marketing, standards of identity and quality,
authorizing factory inspections and adding the remedy of court injunctions for penalties.
1938
Food, Drug and Cosmetics Act (FD&C Act)- products must be shown to be 'safe'; authorized
factory inspections
1938
The U.S. v. Dotterweich. The Supreme Court rules that responsible officials and the
corporation itself may be prosecuted for violations of applicable laws. It is not necessary to
prove the violation was intended! Important GLP ramifications.
1943
40

History of FDA
Regulatory actionYear
PHS Act- Required Product License Application/ Establishment License Application (PLA/ELA,
precursor to the BLA); gave seizure power
1944
The FDA provides their first guidance document “Procedures for the Appraisal of the Toxicity
of Chemicals in Food”.
1949
The Division of Biologics Control becomes an independent entity of in the NIH after improperly
inactivated polio vaccines resulted in approximately 260 cases of polio.
1955
The Kefauver-Harris Drug Amendments are enacted to ensure drug efficacy and safety is
proven before marketing.
Thalidomide. The FDA did not allow this in the US and as a result there was increased
support for stronger drug regulations
1962
Division of Biologics Standardization transferred from NIH to FDA, became what is now called
CBER
1972
The FDA inspects Searle Laboratories and finds significant deficiencies in data recording,
training, analysis and data review. These findings resulted in the drafting of a Good
Laboratory Practice document. Adherence was voluntary.
1975
Food and Drug Administration Modernization Act (FDAMA)- Revised PHS Act and eliminated
the ELA
1997
Review of "well-characterized" proteins transferred to CDER2003
Patient Protection and Affordable Care Act (Obamacare)2010
41

THANKS
Rivka Zaibel





























