Embed Size (px)
Citation preview

Quality and Compliance Challenges for Biopharmaceutical Products
Ramon Rivera Gonzalez, Ph.D.
Director Quality Assurance
Amgen Manufacturing, Limited

2
Outline Background Information
Regulatory and Process Challenges
Quality by Design
Risk Management
Process Analytical Technology
Key Quality Systems• Deviations/Nonconformities• Corrective and Preventive Actions
Biological Product Deviations
FDA and EMEA Observations

3
Biotechnology
Biotechnology is a set of scientific techniques used to derive valuable products from living
organisms
Applications
biopharmaceutical drugs
agriculture
waste management

4
Background information
FDA- “sterile drugs should be manufactured by aseptic processing only when terminal sterilization is not feasible”.
Parenterals -Drug administration other than by the mouth or rectum- ex. Injection, infusion or implantation.– Biological products (vials or syringes)
•solubility, stability, maintain activity

5
Biotechnology
BIOLOGY
CHEMISTRY
ENGINEERING
6
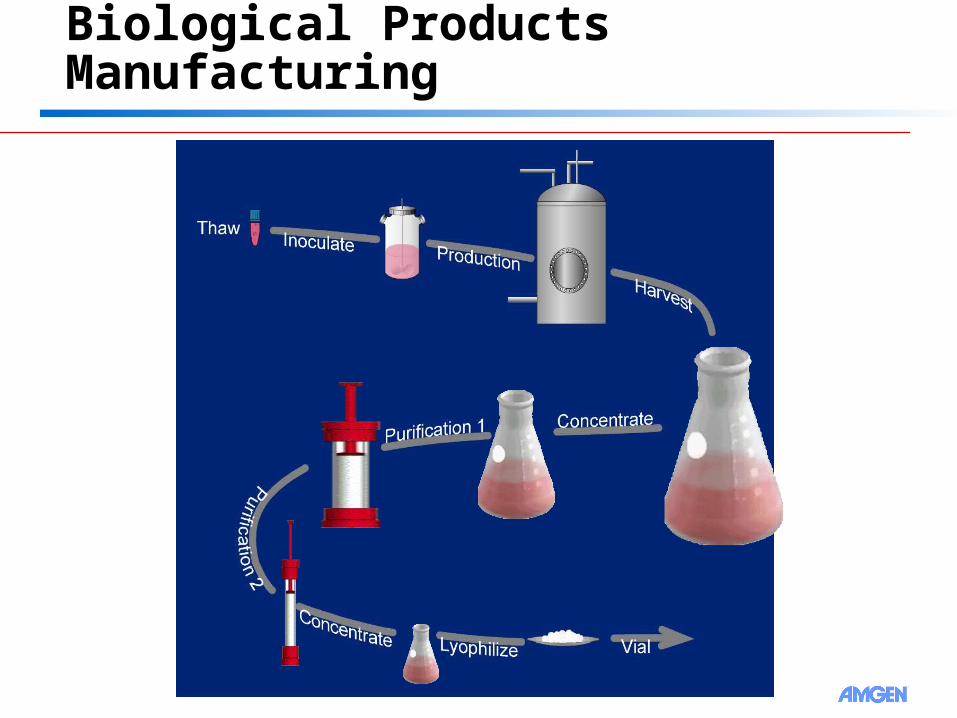
Biological Products Manufacturing

7
Biological Products Manufacturing

8
Bacterial Systems
(+)
– Grow fast
– Easy to maintain
– High yield
(-)
– Endotoxins
– Low expression or extracellular secretion
– No post-translational modifications machinery
E. coliE. coli

9
Mammalian Cells
(+)
– Adequate conformation
– Post-translational modifications
– Preferred for complex proteins
(-)
– Grow slower than bacteria
– Maintenance is expensive
– Usually lower yield
– Limited manufacturing applications
CHOCHO
MammalianMammalian

10
Quality and the Product Life Cycle Adherence to regulations
Control and maintenance of documentation
Quality of suppliers, components and raw materials
Reliability and consistency
Monitor/audit of the manufacturing process
Deviations, unexpected situations - product impact
Correction and prevention - CAPA
Lot release/rejection decision
Customer complaints
Continuous improvement

11
Quality Systems Emphasis
Quality Management
Quality Assurance
Risk Management– Evaluation analysis and quality risk management
tools
Preventive Action
Promote product and process improvement (i.e., continuous improvement)
“Continuous Improvement” of the Quality System

12
Regulatory Environment Challenges
Transfer of CBER products to CDER
GMPs for the 21st. Century
Aseptic Processing Guideline
Risk management
Increased scrutiny of product insert claims
Focus on patient safety
Process Analytical Technology (PAT)

13
Regulatory Environment Challenges
Quality by Design
Bioterrorism
Animal-derived materials
Country-specific regulatory requirements– Mexico, Brazil, Saudi Arabia, Japan
Biogenerics in EU
State of the art technology– Isolators

14
Major Process Challenges
• Sterile vs. Aseptic• Requires the application of microbiological contamination
control to prevent infectious organisms to be present in the sterile product
• Demonstrate “CONTROL” of the process, while technical complexity increases• Characterization to identify variability components• Application of science and new technologies
• Maintenance of the cell lines• Contamination risks
• Personnel as “incubators”• Source of microbial load

15
“Quality can not be tested into products; it has to
be built in by design”
Product quality and performance requires efficient design of manufacturing processes
Product specifications based on deep understanding of how formulation and process factors impact product performance
It provides a framework for continuous "real time" assurance of quality and continuous Improvement
Quality by Design

16
The importance of Design
Multidimensional combination and interaction of input variables and process parameters that have been demonstrated to provide an assurance of quality
Operating within design parameters will produce a product meeting designed quality attributes
Working within the design parameters is not considered a reportable change
Movement outside of design space is considered a change – subject to regulatory approval

17
Risk Management
What are the potential hazards to process and product ?
• Identify potential hazards both prospectively and in a reactive mode
Applies to components, container closure, raw materials, dosing devices, manufacturing process, drug substance, intermediates
How these factors influence variability of process, product performance, product safety and efficacy?

18
Risk Management
Risk management is a regulatory expectation for a modern quality system
Risk assessment, risk control, risk communication and risk review throughout product lifecycle
Decisions should be based upon process and product understanding
• Balance between the use of risk management and compliance with GMPs
Always include intended use, patient safety and availability

19
Process Analytical Technology
“…any system for continuous analysis and/or control of manufacturing processes based on real-time measurements, or rapid measurements during processing.”
• Source; FDA Human Drug cGMP Notes, Q1 2002

20
Process Analytical Technology
“At line” - the sample is removed, isolated from, and analyzed in close proximity to the process stream
“On-line” - the sample is diverted from the manufacturing process, and may be returned to the process stream
“In-line” – the sample is not removed from the process stream and can be invasive or non-invasive
• Source: FDA PAT Guidance

21
PAT - Benefits
Enhancement of process understanding Sources of variability identified and
explained Meet requirements for validating and
controlling process Quality attributes can be accurately and
reliably predicated Continuous improvement Integration of development, manufacturing,
QA and knowledge management Acceptability of in-process materials and
final product based on process data

22
Management Controls
Written quality policy and objectives
Management reviews – regulations and quality objectives– attendance documented– results, action plans/corrective actions
documented
Internal audits – auditors no direct responsibility for matters
being audited

23
Deviations/Nonconformities
Classification based on impact/risk
Investigations need to be thorough, stand-alone– Root cause analysis– Product impact
• Stability data, intrinsic vs. extrinsic, historical data
– Toxicological/health hazard evaluations based on route of administration to patient
– Corrective and Preventive actions

24
Corrective and Preventive Actions(CAPA)
Corrective = correct existing nonconformity
Prevention = potential recurrence of nonconformity
Regulatory expectations:– Identify sources of problems/nonconformities
• Unfavorable trends
– Prioritize based on risk– Defined action plans– Timely implementation– Measure and document effectiveness– Reviewed by Management

25
If a deviation/nonconformity involves distributed product…
Evaluate per 21CFR600.14 – Biological Product Deviation (BPD)
“must report any event and any information relevant to the event associated with the manufacturing, to include testing, processing, packing, labeling or storage, or with the holding or distribution, of a licensed biological product if the event meets the following criteria:
(1) Either;(i) Represents a deviation from cGMP, applicable regulations,
applicable standards, or established specifications that may affect the safety, purity or potency of that product; or
(ii)Represents an unexpected or unforeseeable event that may affect the safety, purity, or potency of that product; and
(2) Occurs in your facility or a facility under contract with you; and(3) Involves a distributed biological product

26
Some points regarding BPDs
Distributed = biological product has left the control of the licensed manufacturer
• Contract manufacturing
The decision to report should be based on whether the event had the potential to affect safety, purity, or potency of the product
The license holder is required to file at a date not to exceed 45 calendar days from the date that you, your agent, or another person, acquire information reasonably suggesting that a reportable event has occurred
Must report even if the investigation determined that there was no impact

27
BPD- Reportable Examples
Raw materials that failed specifications used in manufacturing
Aseptic processing not performed according to procedures
Stability testing not performed at required interval
Missing information in label (product type, lot number, storage temperature, concentration)
Distributed product prior to completion of required testing
Product release prior to validation of manufacturing process
Biological Drug Substance stored at the incorrect temperature

28
BPD – Non-reportable Examples
Raw material did not meet specification and was rejected prior to its use
Testing performed incorrectly, invalidated and repeated and found acceptable prior to distribution
Product labeled with a shortened expiration date (not due to failure to meet specification)
Product shipped to the incorrect facility
Customer order filled incorrectly (wrong product, wrong amount), but labeled correctly

29
EMEA Observations
Critical = give rise to product that could be harmful to patient
Major = result in product not meeting marketing authorization, major deviation from EU GMP
Other = usually lack information to be classified

30
EMEA 1995 - 2005
Critical• Design and maintenance of premises • Potential for microbiological or
chemical/physical contamination
Major/Other• Potential for microbial contamination• Documentation- Quality system
elements/procedures • Unauthorized activities requiring regulatory
filing• Design and maintenance of equipment and
premises

31
FDA Observations (1996-2006)
Failure Investigations (Nonconformities)
– Failure to conduct investigations– Failure to extend investigations to related
batches and products– Averaging OOS results with in-specification
results to support release– Delays in starting the investigation– Incomplete investigation into root cause– Failure to take immediate corrective action– Failure to address product impact

32
FDA Observations (1996-2006)
Record keeping– Batch Production and Control records do not
include complete information– “post-it” notes with hand-written original data– Lack of procedures– No revision history for procedures
Validations– Discrepancies against Master Validation Plan– Inadequate cleaning validation – Inadequate process validation– Inadequate validation of analytical methods

33
In Summary
The successful application of quality systems for the manufacturing of biological products requires the comprehensive synergy between: regulations, science/new technologies, process knowledge, Management accountability and efficient risk management

34
Questions?





























