Embed Size (px)
Citation preview

Psychopharmacology
Pharmacokinetics and Pharmacodynamics

Pharmacokinetics
• The time course of a particular drug’s action.
• Along with knowledge of the dosage taken, allows determination of:
– concentration of a drug at its receptors.
– intensity of drug effect on the receptors over time.

PharmacokineticsFour Processes: ADME
1. Absorption: movement of drug from site of administration to the blood
2. Distribution: movement of drug from blood to rest of body
3. Metabolism: breakdown of drug 4. Elimination of drug’s metabolic
waste products from the body

Absorption
Major routes of administration:1. Oral administration2. Rectal administration3. Inhalation4. Absorption through mucous membranes5. Absorption through the skin6. Injection

Oral Administration
• Drug must be soluble and stable in stomach fluid.
• Absorbed through upper intestine through passive diffusion.
• Drugs must generally be somewhat lipid soluble.
• Disadvantages: – Vomiting and stomach distress– Hard to know how much of drug will be
absorbed due to genetic differences.– Stomach acid destroys some drugs.

Rectal Administration
• Used if patient is vomiting, unconscious, or unable to swallow.
• Absorption is often irregular, unpredictable, and incomplete.

• Popular for recreational drugs (e.g., tobacco, marijuana, cocaine, heroin).
• Lung tissues’ large surface area allows for rapid absorption into blood.
• Pulmonary capillaries carry drug directly into left side of heart and then directly into the aorta and arteries going to the brain.
• Even faster onset than injection.
Inhalation

• Absorbed through membranes in mouth or nose.
• Examples: heart patient’s nitroglycerine, cocaine, nasal decongestants, nicotine gum.
Mucus!

Pharmacokinetics
5. Administration Through the Skin– Transdermal patches provide
continuous, controlled release.– Allow for slow, continuous
absorption over hours or days, minimizing side effects.
– Examples: nicotine, fentanyl (for chronic pain), nitroglycerine (for angina pectoris), estrogen (hormone replacement therapy).

• Intravenous Administration– Drug introduced directly into bloodstream.– Dosage can be extremely precise.– Fastest onset of pharmacological action
(most dangerous route). • Intramuscular Administration
• Drugs injected into skeletal muscle.• More rapid than absorption from stomach,
but slower than intravenous.• Type 1: Rapid onset/short duration of
action- Drug dissolved into aqueous solution.
• Type 2: Slow onset/prolonged duration- Drug suspended in oily solution.
• Subcutaneous Administration• Injected under the skin.
Injection

Drug molecules may be found in different places in the blood.
1. Plasma–more likely with water soluble drugs
2. Platelets–more likely with lipid soluble drugs
3. Attached to proteins (e.g., albumin)–bound vs. free
Distribution

• Before the blood goes to the rest of the body from the gastrointestinal (GI) tract, it passes through the liver.
• The liver is the major organ that breaks down drugs.
• Therefore, a certain amount of the drug will be inactivated or metabolized as it goes through the liver.
• Other routes may not be subject to this “first pass” effect.
Taking the First Pass

• The brain must protect neurons from toxins.
• But the brain has a great need for nutrients and oxygen (it has a high blood flow), which increases the risk of toxic danger.
• Solution = the blood-brain barrier (BBB)
• Capillaries in brain do not allow drugs to pass as easily as capillaries in rest of body.
The BBB

• Definition: Chemical changes that usually reduce the effect of drugs and increase their excretion.
• Kidneys filter waste from blood, collect it in bladder.
• Lipid-soluble drugs are hard for kidneys to hold onto; after collection, the molecules cross back into the circulation before they are excreted.
Metabolism

• The liver protects the body from toxic substances.– Contains enzyme systems that can
detoxify harmful substances.– Changes from highly lipid-soluble to
less lipid-soluble.– More likely to ionize.– May produce another lipid-
soluble molecule, an “active metabolite”
The Liver

Pharmacokinetics

• Past experience with drugs will affect the enzyme systems.
• Levels of enzymes can be increased by previous exposure to a specific drug.
• Called enzyme induction.
Enzyme Induction

Examples and Consequences:St. John's Wort: (with active ingredient hyperforin) stimulates a receptor (SXR in humans, PXR in nonhumans) in the liver to induce CYP3A, which breaks down many other drugs: Theophylline (asthma), warfarin (anticlotting), birth control pills, and immunosuppressant cyclosporin.
St. John and Your Liver

• Some drugs inhibit CYP enzymes and increase their own levels, as well as levels of any other drug metabolized by that enzyme. Can produce toxicities.
• Example: Inhibition of antipsychotic medication by SSRIs.
Enzyme Inhibition

• Lithium • Mushroom amanita muscaria
– In large doses it is toxic and lethal; small amounts are hallucinogenic.
– Hallucinogenic ingredients are not greatly metabolized and are passed to the urine. Siberian tribespeople discovered this and recycled the drug by drinking their urine.
Intact Excretion

• Primarily accomplished by
kidneys.2 organs (about the size of a fist)
located on either side of the spine in the back.
Keep the right balance of water and salt in the body
Filter everything out of blood and then selectively reabsorb what is required.
pH of urine can be manipulated to alter excretion of drugs.
Can be useful for eliminating certain drugs in overdose.
Excretion

The rate of excretion for most drugs can be described in terms of a half-life: time taken for the body to eliminate half of a given blood level of a drug.
Half-Life (T-1/2)
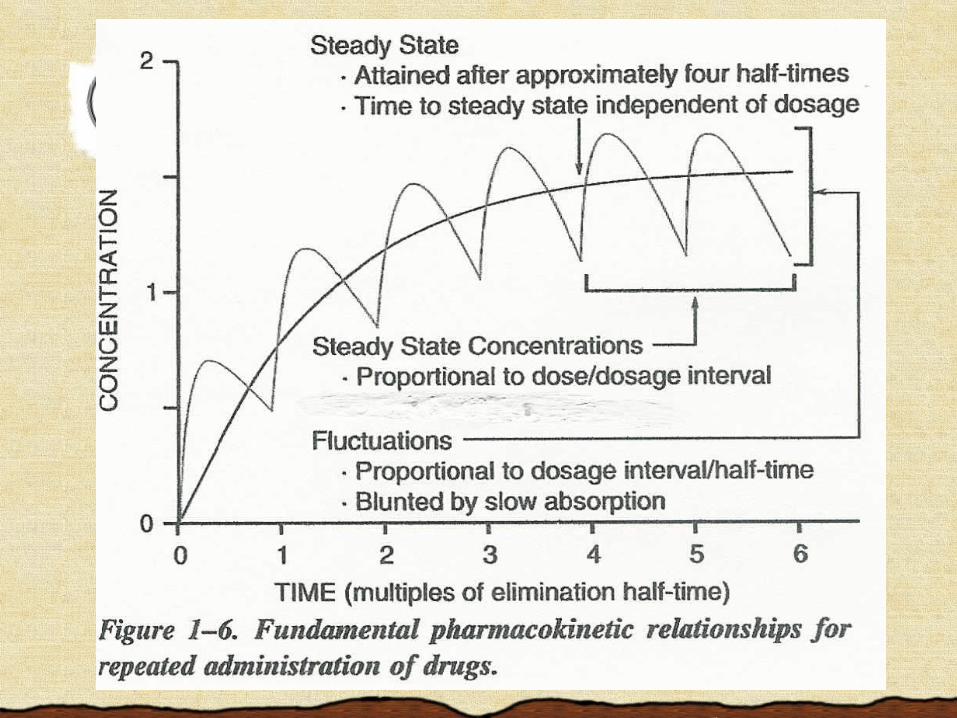

The Therapeutic Window
• For medical treatment, it is important that the right amount of drug is maintained in the blood.– If the amount is too high, the therapeutic effect
will not be any better, but there will be more undesirable side effects.
– If the amount is too low, it won’t have any beneficial effect.
• Drugs should be given in such a way that the blood concentration stays between a level that is too high and too low. This is called the Therapeutic Window.

Pharmacodynamics
• In contrast to pharmacokinetics (the study of what the body does to drugs), pharmacodynamics is the study of what a drug does to the body.

Receptors for Drug Action
1. Receptors are usually membrane-spanning proteins.
2. Not a simple globule, but a continuous series of amino acid loops embedded in the membrane.
3. Drugs and neurotransmitters attach inside between coils; held in place by ionic attractions. Classic concept of a receptor
Under Lock and Key

4. Reversible ionic binding activates the receptor by altering the structure of the protein.
5. Intensity of signal partly determined by percentage of receptors active.
6. Drugs can affect signal by binding either to receptor
7. site or to nearby site.Schematic of 5-HT1A receptor
Binding

Receptors for Drug ActionBinding results in 1 of 3 actions:1. Binding to site of normal endogenous
neurotransmitter initiates similar cellular response (agonistic action).
2. Binding to nearby site to facilitate transmitter binding (allosteric agonistic action).
3. Binding to receptor site, blocking access of transmitter to binding site (antagonistic action).
Pharmacodynamics
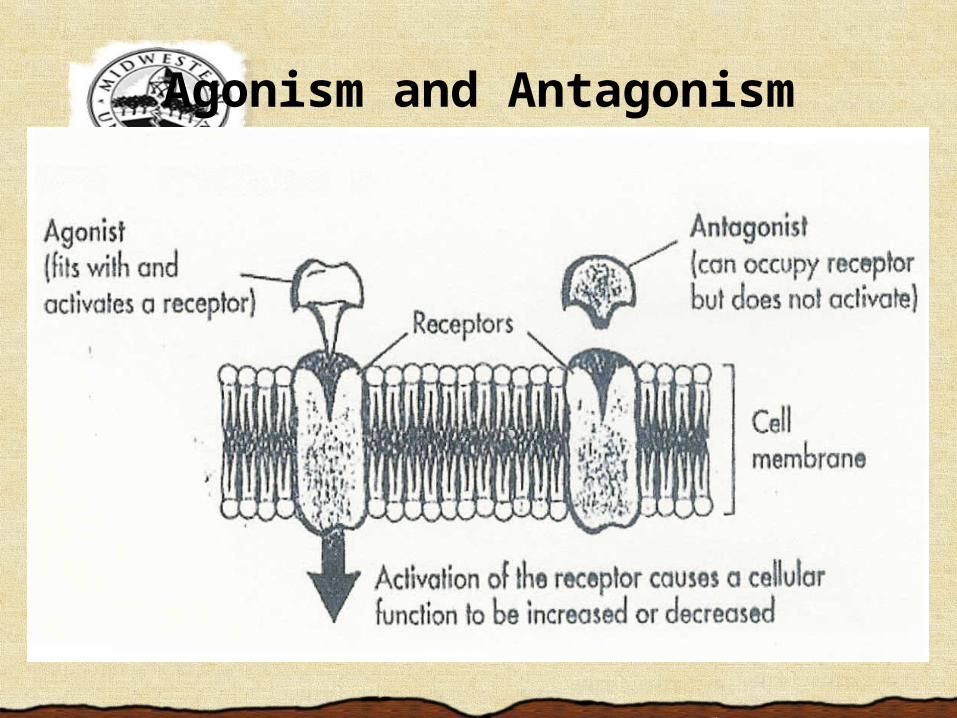
Agonism and Antagonism

• Ion Channel Receptors: Activation opens channel, allowing flow of ions to trigger or inhibit neural firing. – Benzodiazepines are GABA receptor
allosteric agonists.• Bind to nearby sites and facilitate GABA, flooding
neurons with Cl-, inhibiting neural actions.• Used as sedative, antianxiety, amnestic,
antiepileptic.
– Flumazenil binds to benzodiazepine site but does not interfere with GABA.
• Technically a benzodiazepine antagonist, used to treat benzodiazepine overdose.
Receptor Structure

• G Protein-Coupled Receptors: Induce release of intracellular protein, trigger second messengers.– Also called metabotropic receptors. – Control many cellular processes (e.g., ion
channel function, energy metabolism, cell division/differentiation, neuron excitability).
G Protiens

• Carrier Proteins: Bind to neurotransmitters to transport them back to presynaptic neuron.– Many drugs block carrier proteins for a specific
neurotransmitter (e.g., SSRIs).
• Enzymes: Function to break down neurotransmitters in synaptic cleft.– Inhibition by drugs increases transmitter
availability.– Irreversible acetylcholine esterase inhibitors are
used as pesticides and nerve gases. – Monoamine oxidase inhibitors inhibit breakdown
of NE and DA (used as antidepressants).
Acting Indirectly

Dose Response

• Potency = how well drug molecules attach to their sites of action (receptors).– More potent drugs usually attach better than
less potent drugs (binding); bind more tightly than less potent drugs.
– Binding = affinity. A more potent drug has greater affinity for its receptor (binds more tightly).• A less potent drug has less affinity for its receptor;
does not bind so tightly, can be more easily knocked off the receptor.
– Different drugs may bind to the same receptor, but with different affinities.
Potency

• Refers to the mostly linear central part of the curve; how sharply the effect changes with each change in dose.
• If a small change in dose produces a large change in effect, the slope is steep.
• If even large changes in dose produce small changes in effect, the slope
is shallow.
A Slippery Slope

• The maximum effect obtainable, with additional doses producing no effect.
– Some drugs may be potent, but they might never be able to produce a peak response no matter how much is given.
– A drug that is more efficacious (effective) can produce a greater peak, or maximum, effect than a drug that is less efficacious.
Efficacy
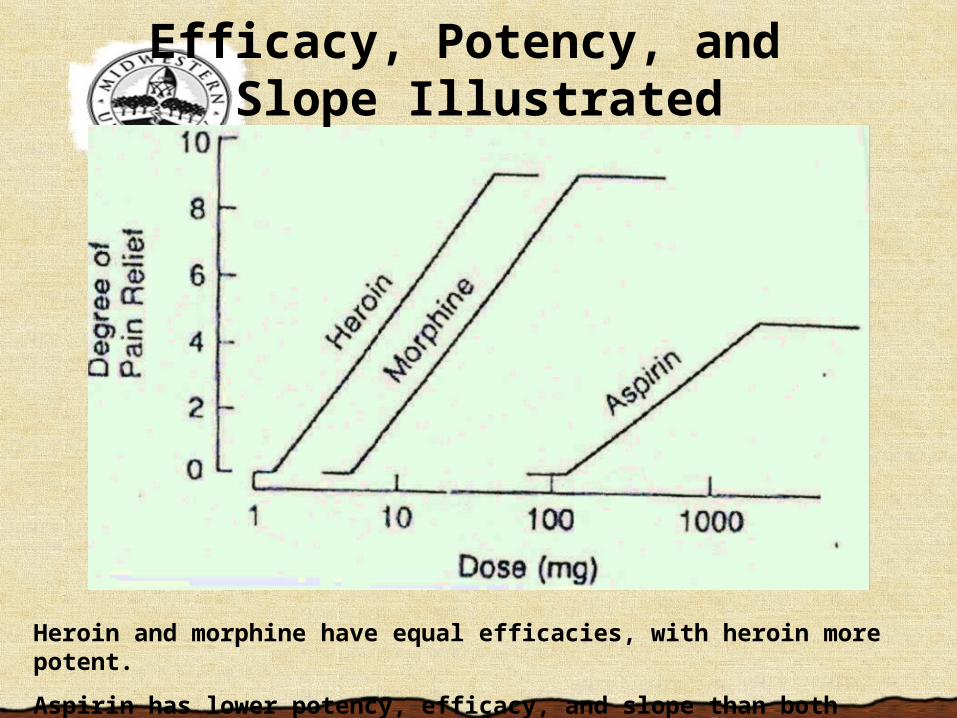
Heroin and morphine have equal efficacies, with heroin more potent.
Aspirin has lower potency, efficacy, and slope than both heroin and morphine.
Efficacy, Potency, and Slope Illustrated

• ED50
– The drug dose that produces desired effect in 50 percent of test subjects.
• LD50
– The lethal dose for 50 percent of test subjects.
– Tested using experimental animals.
• Therapeutic Index = ratio of LD50 to ED50
The Therapeutic Index

• Many studies today use double-blind, randomized, controlled clinical trials. – However, even these trials can misrepresent the
placebo effect. – Notably, the placebo effect in antidepressant studies
has risen dramatically between 1980 and 2000. • Double-blind tests with a placebo run-in period
may eliminate much of the placebo effect.– In these tests, every patient is administered a placebo
for a week. Patients who respond positively are removed from study.
I Shall Please





























