Embed Size (px)
Citation preview

Proton-conducting polymer electrolyte membranes based onhydrocarbon polymers
M. Rikukawa*, K. Sanui
Department of Chemistry, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102, Japan
Received 11 May 2000; accepted 18 July 2000
Abstract
This paper presents an overview of the synthesis, chemical and electrochemical properties, and polymer
electrolyte fuel cell applications of new proton-conducting polymer electrolyte membranes based on hydrocarbon
polymers. Due to their chemical stability, high degree of proton conductivity, and remarkable mechanical proper-
ties, per¯uorinated polymer electrolytes such as Na®onw, Aciplexw, Flemionw, and Dow membranes are some of
the most promising electrolyte membranes for polymer electrolyte fuel cells. A number of reviews on the synth-
esis, electrochemical properties, and fuel cell applications of per¯uorinated polymer electrolytes have also
appeared during this period. While per¯uorinated polymer electrolytes have satisfactory properties for a successful
fuel cell electrolyte membrane, the major drawbacks to large-scale commercial use involve cost and low proton-
conductivities at high temperatures and low humidities. Presently, one of the most promising ways to obtain high
performance proton-conducting polymer electrolyte membranes is the use of hydrocarbon polymers for the
polymer backbone. The present review attempts for the ®rst time to summarize the synthesis, chemical and
electrochemical properties, and fuel cell applications of new proton-conducting polymer electrolytes based on
hydrocarbon polymers that have been made during the past decade. q 2000 Elsevier Science Ltd. All rights
reserved.
Keywords: Proton; Electrolyte; Poly(4-phenoxybenzoyl-1,4-phenylene); Poly(ether-etherketone); Fuel cell; Sulfonation;
Conductivity
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1464
1.1. Polymer electrolyte fuel cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1464
1.2. Solid polymer electrolyte membranes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1465
2. Proton-conducting polymer electrolyte membranes based on sulfonated aromatic polymers . . . . . . . . .1468
2.1. Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1468
Prog. Polym. Sci. 25 (2000) 1463±1502
0079-6700/00/$ - see front matter q 2000 Elsevier Science Ltd. All rights reserved.
PII: S0079-6700(00)00032-0
* Corresponding author. Tel.: 181-3-3238-4250; fax: 181-3-3238-4198.
E-mail address: [email protected] (M. Rikukawa).

2.2. Thermal stability of sulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . . . . . . . .1471
2.3. Water uptake in sulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . . . . . . . . . .1473
2.4. Proton conductivity of sulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . . . . . .1476
3. Proton-conducting polymer electrolyte membranes based on alkylsulfonated aromatic polymers . . . . .1478
3.1. Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1478
3.2. Thermal stability of alkylsulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . . . .1480
3.3. Water uptake in alkylsulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . . . . . . .1483
3.4. Proton conductivity of alkylsulfonated aromatic polymer electrolyte membranes . . . . . . . . . . . .1484
4. Proton-conducting polymer electrolyte membranes based on acid±base polymer complexes . . . . . . . .1486
4.1. Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1486
4.2. Thermal stability of acid±base polymer complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1491
4.3. Conductivity of acid±base polymer complex . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1492
5. Other proton-conducting polymer electrolyte membranes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1496
6. Fuel cell applications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1499
7. Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1500
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1500
1. Introduction
1.1. Polymer electrolyte fuel cells
The idea of using an organic cation exchange membrane as a solid electrolyte in electrochemical cellswas ®rst described for a fuel cell by Grubb in 1959. At present the polymer electrolyte fuel cell (PEFC) isthe most promising candidate system of all fuel cell systems in terms of the mode of operation andapplications. As shown in Fig. 1, a PEFC consists of two electrodes and a solid polymer membrane,
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021464
Fig. 1. PEFC schematic.

which acts as an electrolyte. The polymer electrolyte membrane is sandwiched between two platinum-porous electrodes such as carbon paper and mesh. Some single cell assemblies can be mechanicallycompressed across electrically conductive separators to fabricate electrochemical stacks. In general,PEFCs require humidi®ed gases, hydrogen and oxygen (or air) as a fuel for their operation. Theelectrochemical reactions that occur at both electrodes are as follows:
Anode : H2 ! 2H1 1 2e2
Cathode : 1=2O2 1 2H1 1 2e2 ! H2O
Overall : H2 1 1=2O2 ! H2O 1 Electrical Energy 1 Heat Energy
In recent years, PEFCs have been identi®ed as promising power sources for vehicular transportationand for other applications requiring clean, quiet, and portable power. Hydrogen-powered fuel cells ingeneral have a high power density and are relatively ef®cient in their conversion of chemical energy toelectrical energy. Exhaust from hydrogen-powered fuel cells is free of environmentally undesirablegases such as nitrogen oxides, carbon monoxide, and residual hydrocarbons that are commonly producedby internal combustion engines. Carbon dioxide, a greenhouse gas, is also absent from the exhaust ofhydrogen-powered fuel cells. Thus, transportation uses, especially fuel cell electric vehicles (FCEV), areon attractive and effective application because of not only clean exhaust emissions and high-energyef®ciencies but also effective solution to the coming petroleum shortage. While FCEV might provide thegreatest societal bene®ts, its total impact would be small if only a few FCEVs are sold due to lack offueling infrastructure or due to high vehicle cost. The major obstacles for the commercial use of FCEVare expensive materials and low performances at high temperatures (over 1008C) and low humidities.
1.2. Solid polymer electrolyte membranes
In general, proton-conducting polymers are usually based on polymer electrolytes, which have nega-tively charged groups attached to the polymer backbone. These polymer electrolytes tend to be ratherrigid and are poor proton conductors unless water is absorbed. The proton conductivity of hydratedpolymer electrolytes dramatically increases with water content and reaches values of 1022±1021 S cm21.
The ®rst PEFC used in an operational system was the GE-built 1 kW Gemini power plant [1]. Thissystem was used as the primary power source for the Gemini spacecraft during the mid-1960s. Theperformances and lifetimes of the Gemini PEFCs were limited due to the degradation of poly(styrenesulfonic acid) membrane employed at that time. The degradation mechanism determined by GE wasgenerally accepted until the present time. It was postulated that HO2 radicals attack the polymerelectrolyte membrane. The second GE PEFC unit was a 350 W module that powered the Biosatellitespacecraft in 1969. An improved Na®onw membrane manufactured by DuPont was used as the electro-lyte. Fig. 2 shows the chemical structures of Na®onw and other per¯uorinated electrolyte membranes.The performance and lifetime of PEFCs have signi®cantly improved since Na®onw was developed in1968. Lifetimes of over 50,000 h have been achieved with commercial Na®onw120.
Na®onw 117 and 115 have equivalent repeat unit molecular weights of 1100 and thicknesses in the drystate of 175 and 125 mm, respectively. Na®onw 120 has an equivalent weight of 1200 and a dry statethickness of 260 mm. Ballard Technologies Corporation showed the possibility of the application of
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1465

PEFC for electric vehicles by using experimental per¯uorinated membranes developed by Dow Chemi-cal. Development of PEFC has been accelerated year by year after the report of Ballard Tech-nologies Corporation. The Dow membrane has an equivalent weight of approximately 800 and athickness in the wet state of 125 mm. In addition, Flemionw R, S, T, which have equivalentrepeat unit molecular weights of 1000 and dry state thicknesses of 50, 80, 120 mm, respectively,were also developed by Asahi Glass Company [2]. Asahi Chemical Industry manufactured aseries of Aciplexw-S membranes, which have equivalent repeat unit molecular weights of1000±1200 and dry state thicknesses of 25±100 mm.
These per¯uorinated ion exchange membranes including Neosepta-Fw (Tokuyama) and Gore-Selectw
(W. L. Gore and Associates, Inc.) have been developed for chlor-alkali electrolysis. The water uptakeand proton transport properties of this type of membrane have signi®cant effects on the performance ofPEFCs. These membranes have water uptakes of above 15 H2O/±SO3H, and maximizing membranewater uptake also maximizes the proton conductivity. In general, conductivities can reach values of1022±1021 S cm21. All of these membranes possess good thermal, chemical, and mechanical propertiesdue to their per¯uorinated polymer backbones.
A limiting factor in PEFCs is the membrane that serves as a structural framework to supportthe electrodes and transport protons from the anode to the cathode. The limitations to large-scalecommercial use include poor ionic conductivities at low humidities and/or elevated temperatures,a susceptibility to chemical degradation at elevated temperatures and ®nally, membrane cost.These factors can adversely affect fuel cell performance and tend to limit the conditions underwhich a fuel cell may be operated. For example, the conductivity of Na®onw reaches up to1022 S cm21 in its fully hydrated state but dramatically decreases with temperature above theboiling temperature of water because of the loss of absorbed water in the membranes. Conse-quently, the development of new solid polymer electrolytes, which are cheap materials andpossess suf®cient electrochemical properties, have become one of the most important areas forresearch in PEFC and FCEV.
Proton-conducting polymer electrolyte membranes for high performance PEFCs have to meet thefollowing requirements, especially for electric vehicle applications [3]:
1. low cost materials;2. high proton conductivities over 1008C and under 08C;3. good water uptakes above 1008C;4. durability for 10 years.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021466
Fig. 2. Chemical structures of per¯uorinated polymer electrolyte membranes.

The challenge is to produce a cheaper material that can satisfy the requirements noted above. Somesacri®ce in material lifetime and mechanical properties may be acceptable, providing cost factors arecommercially realistic. Good electrochemical properties over a wide temperature range may help theearly marketing of PEFCs. Presently, one of the most promising routes to high-performance proton-conducting polymer electrolyte membranes is the use of hydrocarbon polymers for polymerbackbones. The use of hydrocarbon polymers as polymer electrolytes was abandoned in theinitial stage of fuel cell development due to the low thermal and chemical stability of thesematerials. However, relatively cheap hydrocarbon polymers can be used for polymer electrolytes,since the lifetime of electrolytes required in FC vehicles are shorter when compared to use inspace vehicles. Also, catalyst and FC assembly technologies have improved and brought advan-tages to the lifetimes of PEFCs and related materials.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1467
Fig. 3. Chemical structure of polymer electrolyte membranes based on hydrocarbon polymers.

There are many advantages of hydrocarbon polymers that have made them particularly attractive:
1. Hydrocarbon polymers are cheaper than per¯uorinated ionomers, and many kinds of materials arecommercially available.
2. Hydrocarbon polymers containing polar groups have high water uptakes over a wide temperaturerange, and the absorbed water is restricted to the polar groups of polymer chains.
3. Decomposition of hydrocarbon polymers can be depressed to some extent by proper moleculardesign.
4. Hydrocarbon polymers are easily recycled by conventional methods.
Based on numerous works by other authors and our own research group, we review the syntheses ofnew proton-conducting polymer electrolyte membranes based on hydrocarbon polymers. The character-istics of these new materials which determine their potential applications, are discussed in detail. Areview of electrochemical properties, water uptake, and thermal stability makes possible a comprehen-sive understanding of the proton conduction mechanism and physical state of absorbed water in thesesystems.
2. Proton-conducting polymer electrolyte membranes based on sulfonated aromatic polymers
2.1. Materials
Over the last decade new proton-conducting polymer electrolyte membranes have been developed.These new membrane concepts include partially ¯uorinated membranes, composite membranes, and
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021468
Fig. 4. Levels of sulfonation of PEEK and PPBP as a function of reaction time at room temperature.

also aromatic polymer membranes. This section will give a brief overview on sulfonated aromaticpolymer membranes.
Poly(styrene sulfonic acid) is a basic material in this ®eld. In practice, poly(styrene sulfonic acid) andthe analogous polymers such as phenol sulfonic acid resin and poly(tri¯uorostyrene sulfonic acid), werefrequently used as polymer electrolytes for PEFCs in the 1960s. Chemically and thermally stablearomatic polymers such as poly(styrene) [4,5], poly(oxy-1,4-phenyleneoxy-1,4-phenylenecarbonyl-1,4-pheneylene) (PEEK) [6±9], poly(1,4-phenylene) [10±13], poly(oxy-1,4-phenylene) [14], poly(phe-nylene sul®de) [15], and other aromatic polymers [16±19], can be employed as the polymer backbonefor proton-conducting polymer electrolytes. These chemical structures are illustrated in Fig. 3. Thesearomatic polymers are easily sulfonated by concentrated sulfuric acid [20], by chlorosulfonic acid [21],
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1469
Fig. 5. FT-IR spectra of PEEK, PPBP, and their sulfonated polymers (S-PEEK and S-PPBP).

by pure or complexed sulfur trioxide [16,22,23], and by acetylsulfate [24]. Sulfonation with chlorosul-fonic acid or fuming sulfuric acid sometimes causes chemical degradation in these polymers. Accordingto Bishop et al. [25], the sulfonation rate of PEEK in sulfuric acid can be controlled by changing thereaction time and the acid concentration and can thereby provide a sulfonation range of 30±100%without chemical degradation or crosslinking reactions [26]. However, this direct sulfuric acid proce-dure cannot be used to produce truly random copolymers at sulfonation levels of less than 30%, becausedissolution and sulfonation in sulfuric acid occur in a heterogeneous environment due to the increase ofviscosity of reactant solutions. For this reason, the dissolution process was kept short, for less than 1 h, inorder to produce a more random copolymer. Sulfonation levels in PEEK and poly(4-phenoxybenzoyl-1,4-phenylene) (PPBP) [13] as a function of reaction time at room temperature are presented in Fig. 4.Since sulfonation is an electrophilic reaction, its application depends on the substituents present on thering. Electron-donating substituents will favor the reaction, whereas electron-accepting substituents willnot. Indeed, with PPBP the terminal phenyl ring of the substituent can be sulfonated under mild condi-tions as good as PEEK, although poly(4-benzoyl-1,4-phenylene) containing an electron acceptingsubstituent in the ring cannot be sulfonated under the same conditions. The sulfonation of PPBP andPEEK occurred to almost 80 mol% within 100 h. The level of sulfonation of PPBP became saturated at85 mol% per repeat unit, whereas the level of sulfonation of PEEK reached 100 mol%. This is becausethe viscosity of PPBP in sulfuric acid was so high that entanglements of pendant groups preventedfurther sulfonation. Bailly et al. [9] and Jin et al. [20] showed that the sulfonation of PEEK in sulfuricacid cannot produce a random copolymer especially below sulfonation of 30 mol% due to the hetero-geneous nature of the reaction. In our studies, heterogeneous reactions were also con®rmed in the case ofboth PPBP and PEEK. Since it is important for fuel cell applications to produce highly sulfonatedpolymers, this sulfonation method with sulfuric acid is one of the most suitable routes for preparingpolymer electrolytes of PEFCs.
In the case of sulfonated PEEK (S-PEEK), S-PEEK having 30% or more sulfonation wassoluble in N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or N-methyl-2-pyrrolidi-none (NMP); above 70%, the polymer were soluble in methanol and at 100%, in water. PPBPwas dissolved in common chlorine containing solvents such as chloroform and dichloromethane,whereas sulfonated PPBP (S-PPBP) was insoluble in the same solvents, but was soluble in DMF,DMSO, or NMP above 30% sulfonation. Above 65% sulfonation, S-PPBP swelled in methanoland water.
Fourier Transform Infrared (FT-IR) spectra for PEEK, PPBP, and their sulfonated polymers areshown in Fig. 5. The sulfonation of PEEK at room temperature in concentrated sulfuric acid places alimit of one sulfonic group per repeat unit [21,23,27]. It is also clear from the FT-IR data that PEEK issulfonated at one position on the phenylene ring between the ether groups. For S-PPBP, the intensity ofthe absorption band at 1070 cm21 assigned to the mono-substituted benzene ring decreased with anincreasing sulfonation level, while the intensities of absorption bands at 1020 and 730 cm21 attributed toa para-substituted benzene ring, and the S±O stretching vibration,respectively, also increased withincreasing sulfonation level. These FT-IR spectral data indicate that the sulfonation of PPBP in sulfuricacid takes place at the para-position of the terminal phenoxy group.
Tsuchida et al. [18,19] have synthesized poly(thiophenylenesulfonic acid) possessing up to 2.0 sulfo-nic acid groups per phenylene ring, as shown in Fig. 3. They polymerized 4-(methylsul®nyl)diphenylsul®de in sulfuric acid upon heating or in the presence of SO3 to yield a sulfonated poly(sulfoniumcation), which can be converted to the corresponding sulfonated poly(thiophenylene). The sulfonation
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021470

can be controlled by the reaction time, the temperature, and/or the addition of SO3. The resultingpolymer electrolytes are soluble in water and methanol and can form transparent ®lms.
Composite polymer electrolytes of Te¯on±FEP ®lms and poly(styrenesulfonic acid) were alsoprepared by simultaneous radiation grafting of styrene onto Te¯on±FEP ®lms and subsequent sulfona-tion to provide low cost proton-conducting polymers [4]. They immersed a Te¯on±FEP ®lm (DuPont,50 ^ 2 mm) into styrene monomer at 608C and irradiated at a dose rate of 0.5 Gy/min for variable timesup to 15 h with g-rays from a Co60 source. The radicals created by radiation in the system induce thepolymerization, and styrene monomers were grafted to the per¯uorinated matrix. The radiation-grafted®lms were sulfonated with chlorosulfonic acid. A linear dependence of degree of grafting on time wasobserved for grafting times up to 15 h. This sulfonated grafted-®lm was thermally stable up to 3108C asdetermined by thermogravimetric analysis.
Some patent applications in regard to new proton-conducting polymer electrolytes have beensubmitted by several industrial research groups [28,29]. Iwasaki et al. claimed proton-conducting poly-mer electrolytes for PEFC by sulfonating a precursor polyethersulfone having a 0.6±1.5 dl g21 reducedviscosity (in 1% w/v DMF). The sulfonated polyethersulfone has a structural repeating unit containingbenzene or naphthalene and has equivalent weights of 500±2500 g mol21. The fuel cell measurementsusing these materials were carried out in order to study the electrochemical properties.
Konishi et al. also claimed similar proton-conducting polymer electrolytes based on polyethersulfone.The protons of the sulfonic acid groups in their polymers are partly exchanged by metal ions such asmagnesium, titanium, aluminum, and lanthanum ions to improve durability. Their polymers have highconductivities of more than 1022 S cm21. The results of fuel cell tests using their membranes showed acell voltage of 550 mV and a current density of 700 mA cm22 under 1 atm humidi®ed H2/O2 at 708C. Nosigni®cant loss of performance was seen after 1000 h.
2.2. Thermal stability of sulfonated aromatic polymer electrolyte membranes
Polymer electrolyte membranes that exhibit fast proton transport at elevated temperatures are neededfor PEFCs and other electrochemical devices operating in the 100±2008C range. Operation of a PEFC atelevated temperatures has several advantages. It increases the kinetic rates for the fuel cell reactions, itreduces problems of catalyst poisoning by absorbed carbon monoxide in the 150±2008C range, itreduces the use of expensive catalysts, and it minimizes problems due to electrode ¯ooding. Thus,the thermal stability of proton-conducting polymer electrolyte membranes is a very important factorfor fuel cell applications. Several studies have addressed the issue of thermal stability for Na®onw andanalogous polymers. Chu et al. used IR spectroscopy to study the effect of heating Na®onw at tempera-tures of 22, 110, 160, 210, and 3008C [30]. These samples, which had an equivalent weight of 1100, werecoated onto a platinum ®lm, and heated in air. The study concluded that Na®onw loses its sulfonic acidgroups after being heated at 3008C for 15 min. Surowiec et al. conducted a study using thermogravi-metric analysis (TGA), differential thermal analysis (DTA), and FT-IR spectroscopy [31]. Their studiesconcluded that Na®onw loses only water below 2808C, while above 2808C the sulfonic acid groups arealso lost. Wilkie et al. reported using TGA and FT-IR spectroscopy that Na®onw produced water as wellas small amounts of sulfur dioxide and carbon dioxide between 35 and 2808C. Between 280 and 3558C,the evolution of sulfur dioxide and carbon dioxide increased while the evolution of water decreased. At3658C, the amounts of sulfur dioxide and carbon dioxide decreased dramatically, and above thistemperature absorbencies in FT-IR spectra of evolution gas were thought to be due to hydrogen ¯uoride,
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1471

M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021472
Fig. 6. TG-DTA curve of S-PPBP with 80 mol% sulfonation level.
Fig. 7. Degradation temperature of S-PPBP and S-PEEK as a function of sulfonation level.

silicon ¯uoride, and carbonyl¯uorides. Another study was conducted by Samms et al. in simulated fuelcell environments [32]. To simulate the conditions in a fuel cell, Na®onw samples were loaded with fuelcell grade platinum black and heated under atmospheres of nitrogen, 5% hydrogen, or air in a thermo-gravimetric analyzer. The products of decomposition were taken directly into a mass spectrometer foridenti®cation. In all cases, Na®onw was found to be stable up to 2808C, at which temperature the sulfonicacid groups began to decompose. They concluded that the addition of platinum to the polymer electro-lyte had no detectable effect on the temperature at which the sulfonic acid groups decomposed. The long-term stability in PEFCs cannot be accurately predicted by these results.
In our group, the thermal stabilities of our polymer electrolyte membranes were studied by heatingsamples in a TGA and analyzing the resulting residues by elemental analysis. For example, data for S-PPBP investigated by TGA at a heating rate of 108C min21 under nitrogen are displayed in Fig. 6. In theTGA data, S-PPBP showed an initial weight loss of about 20% of the original weight between 250 and4008C, which corresponds to a loss of sulfonic acid groups. The thermal stabilities of dry S-PEEK and S-PPBP are shown in Fig. 7, which shows thermal decomposition temperatures (Td) as a function of thelevel of sulfonation. In contrast to what was observed for PEEK and PPBP, degradation steps wereobserved for the sulfonated polymers between 350 and 2508C. The Td of S-PEEK and S-PPBP decreasedfrom 500 to 3008C for S-PEEK and 2508C for S-PPBP with increasing levels of sulfonation. Elementalanalysis of the residues indicated that the sulfur contents of S-PEEK and S-PPBP decreased from 7.62 to0.86% after heating at temperatures above 4008C. These results suggest that thermal decompositionoccurs by desulfonation. However, such thermal decomposition was not detected below 2008C. Suf®-cient thermal stabilities for fuel cell applications is retained in these polymer electrolytes even if the Td
values drop with sulfonation levels.Other proton-conducting polymer electrolytes based on sulfonated aromatic polymers also show
thermal degradation between 200 and 4008C. As arylsulfonation is known to be a reversible reaction,desulfonation of arylsulfonic acid easily occurs on heating acidic aqueous solution of arylsulfonic acid at100±1758C. Thus, the desulfonation of arylsulfonic acid provides fatal limitations of the thermal stabi-lity of sulfonated aromatic polymer electrolytes. The substituents present on the phenyl ring can besomewhat effective at depressing the thermal degradation process.
According to Tsuchida et al. [18], highly sulfonated poly(phenylene sul®de) possesses higher thermalstability than other sulfonated aromatic polymer electrolytes. They studied the thermal stabilities ofpoly(phenylene sul®de sulfonic acid) with different sulfonation levels using TGA. The decompositiontemperature of highly sulfonated polymer (degree of sulfonation per repeat unit; m � 2:0� is 265 and1258C higher than that for low sulfonated polymer �m � 0:6�: They suggest that the C±S bond of thehighly sulfonated polymer is stronger due to the two electron withdrawing sulfonic acid substituents onone phenyl ring. In addition, the initial weight loss for the higher sulfonated polymer between 265 and3808C is only 13%, which corresponds to the loss of two H2O molecules per repeat unit. Thus, thedesulfonation reaction in this polymer is depressed by substitution of electron withdrawing groups.
2.3. Water uptake in sulfonated aromatic polymer electrolyte membranes
Water is carried into the fuel cell via the humidi®ed H2/O2 gas streams entering the gas diffusionelectrodes. Some combination of water vapor and liquid water passes through each electrode to theelectrode/electrolyte interface. Water crossing this interface assists in the hydration of the electrolytemembrane. An additional source of water involves oxygen reduction occurring at the cathode. Water in
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1473

the membrane is transported in two main ways: electro-osmotic drag of water by proton transport fromanode to cathode and diffusion down concentration gradients that build up. Thus, adequate hydration ofelectrolyte membranes is critical to fuel cell operation. If the electrolyte membrane is too dry, itsconductivity falls, resulting in reduced cell performance. An excess of water in the fuel cell can leadto cathode ¯ooding problems, also resulting in less than optimal performances.
In the case of per¯uorinated polymer electrolytes, the structure of the electrolyte membrane in variousstates of hydration, the water uptake characteristics when exposed to liquid or vapor phase water, and theproperties of water in the electrolyte membrane have all been investigated in detail. The extremehydrophobicity of the per¯uorinated polymer backbone and the extreme hydrophilicity of the terminalsulfonic acid groups lead to a spontaneous hydrophilic/hydrophobic nano-separation [33,34]. In thepresence of water only the hydrophilic domain of the nano structure is hydrated. The absorbed wateralso acts as a plasticizer, which leads to further phase separation. Maximizing the membrane wateruptake maximizes the proton conductivity, while the hydrophobic domains provide relatively goodmechanical properties even in the presence of water. The activation enthalpy of water diffusion insuch membranes is almost identical to that in pure water for water contents higher than H2O/SO3H� 3. The per¯uorinated polymer electrolyte acts as a nano-porous inert sponge for the absorbedwater, which in fact shows very little interaction with the polymer except for the ®rst three watermolecules per sulfonic acid group.
Water uptake from the vapor phase is likely to be the principal mode of external hydration forthe membrane in PEFCs. We have investigated the absorption of water vapor in S-PEEK and
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021474
Fig. 8. Water uptake of S-PEEK and S-PPBP at room temperature as a function of relative humidity.

S-PPBP. S-PEEK and S-PPBP ®lms were placed under various humidities to measure their equilibriumwater contents. It is quite interesting to compare the water uptake and proton conductivities of S-PEEKand S-PPBP in terms of the structural effect of hydration on the sulfonic acid moieties because PEEK is astructural isomer of PPBP. The water uptake of S-PEEK and S-PPBP at room temperature is shown inFig. 8. The results are similar to those reported with Na®onw membranes [1]. Over the entire range ofrelative humidity, the activity coef®cient of water in the polymer is greater than unity if one assumes asthe ideal behavior a Rault's law relationship between water activity and membrane water content. Theequilibrium water contents of S-PEEK and S-PPBP increased sigmoidally with increasing levels ofsulfonation. Two regions are discriminated in the following way. Relatively little increase of wateruptake is observed for the region of relative humidity� 0±50%, and a signi®cantly greater increase ofwater uptake is noted with increasing relative humidities in the range of 50±100%. The former regioncorresponds to water uptake due to solvation of the proton and sulfonate ions. The latter region corre-sponds to water involved in polymer swelling. In the former region, the water in the polymer is engagedin strong interactions with ionic components of the polymer, and these interactions overcome the strongtendency of polymer to exclude water due to its hydrophobic nature and resistance to swelling. In thelatter region, these hindrances to water uptake are surmounted once the water in the bathing gas hassuf®cient chemical potential. The water content of S-PPBP with 65 mol% sulfonation was larger thanthat of S-PEEK with the same sulfonation over the entire relative humidity range, and these valuesincreased to 8.7 molecules of water per sulfonic acid group for S-PPBP and 2.5 for S-PEEK at 100%relative humidity and room temperature. The densities of S-PEEK and S-PPBP with 65 mol%
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1475
Fig. 9. Proton conductivity of S-PPBP and S-PEEK with various sulfonation levels as a function of relative humidity at room
temperature.
sulfonation level, which were measured by a pycnometric method, were 1.373 for S-PPBP and 1.338 forS-PEEK. The surfaces and fracture surfaces for both sulfonated polymers were also found to be nearlythe same by using scanning electron microscopy. Thus, the difference in the water uptake between S-PEEK and S-PPBP might be due to the ¯exible pendant phenoxybenzoyl groups in S-PPBP, whichenhance plasticity, water penetration, and water absorption in the terminal sulfonic groups. The wateruptakes of S-PPBP are nearly equal to that of Na®onw membranes. The hydrophilic domains only absorbwater in per¯uorinated polymer electrolytes that have a spontaneous hydrophilic/hydrophobic nano-separation, while the water molecules of hydration are completely dispersed in polymer electrolytesbased on hydrocarbon polymers. In fact, the water molecules in sulfonated hydrocarbon polymers showrelatively strong interactions with their sulfonic acid groups as estimated by differential thermal analysisand consequently result in high proton conductivities at high temperatures and low humidities.
2.4. Proton conductivity of sulfonated aromatic polymer electrolyte membranes
The proton conductivity of sulfonated poly(thiopheneylene) is about 1025 S cm21 at 30% RH and itincreases exponentially with relative humidity. The conductivity at 94% RH reaches up to 1022 S cm21,where the water content of the polymer is 10.3 H2O per sulfonic acid group.
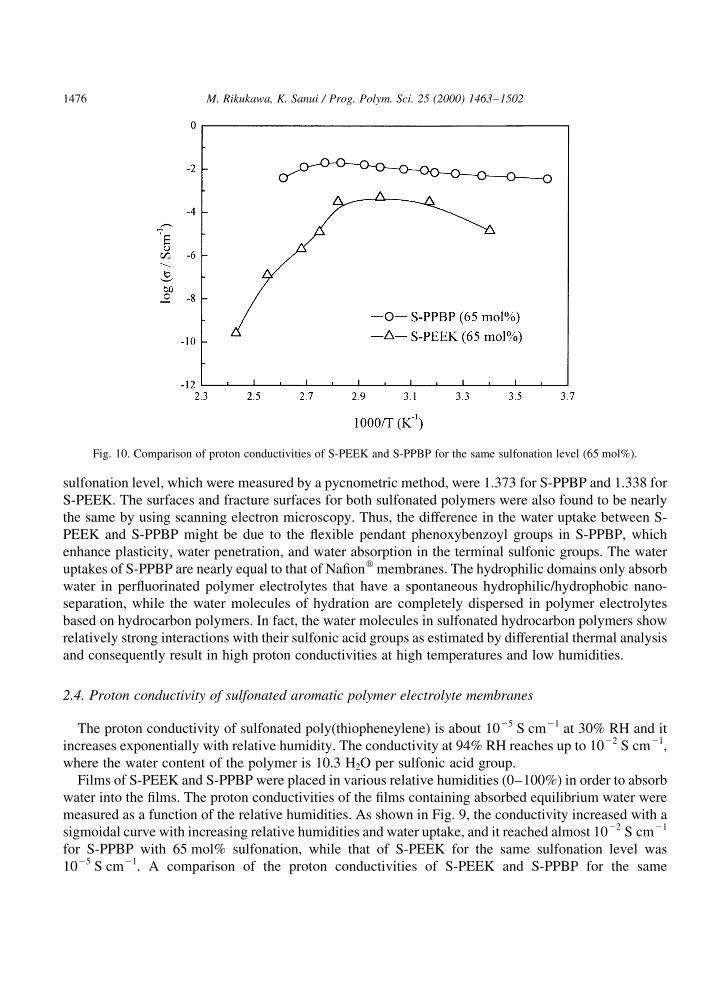
Films of S-PEEK and S-PPBP were placed in various relative humidities (0±100%) in order to absorbwater into the ®lms. The proton conductivities of the ®lms containing absorbed equilibrium water weremeasured as a function of the relative humidities. As shown in Fig. 9, the conductivity increased with asigmoidal curve with increasing relative humidities and water uptake, and it reached almost 1022 S cm21
for S-PPBP with 65 mol% sulfonation, while that of S-PEEK for the same sulfonation level was1025 S cm21. A comparison of the proton conductivities of S-PEEK and S-PPBP for the same
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021476
Fig. 10. Comparison of proton conductivities of S-PEEK and S-PPBP for the same sulfonation level (65 mol%).

sulfonation level (65 mol%) is shown in Fig. 10, where the ®lms were saturated at 100% RH. It is clearthat the proton conductivities and the water absorption of S-PPBP are much higher than those of S-PEEK. Moreover, the proton conductivities of S-PEEK dropped sharply at temperatures above 1008C,while those of S-PPBP exhibited less temperature dependence.
These stable proton conductivities of sulfonated poly(thiophenylene) and S-PPBP at elevatedtemperatures are an interesting feature worthy of further applications such as high temperature andnon-humidifying PEFCs, compared with Na®onw membranes which show a sharp decrease in protonconductivities at temperatures above 1008C due to dehydration. The high water uptake and the strong
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1477
Fig. 11. Schematic view of the cell for galvanostatic four-point-probe electrochemical impedance spectroscopy technique.
Fig. 12. Temperature dependence of proton conductivity for S-PEEK with sulfonation level of 85 mol% under various relative
humidity by using a four-point cell.

interaction between absorbed water and polymer chains might be maintained at elevated temperatures sothat these polymers do not show a measurable reduction in proton conductivity.
The conductivity of S-PPBP was also measured using a galvanostatic four-point-probe electroche-mical impedance spectroscopy technique [35]. A four-point-probe cell with two platinum foil outercurrent-carrying electrodes and two platinum wire inner potential-sensing electrodes was mounted on aTe¯on plate. The schematic view of the cell is illustrated in Fig. 11. Membrane samples were cut intostrips that were approximately 1.0 cm wide, 5 cm long, and 0.01 cm thick prior to mounting in the cell.The cell was placed in a thermo-controlled humidic chamber to measure the temperature and humiditydependence of proton conductivity. With this method, a ®xed AC current is passed between two outerelectrodes, and the conductance of the material is calculated from the ac potential difference observedbetween the two inner electrodes. The method is relatively insensitive to the contact impedance at thecurrent-carrying electrode and is therefore well suited for measuring proton conductances. This open cellis also suited for studying the humidity dependence of conductivity for proton-conducting polymerelectrolytes because of the low interfacial resistance.
In many cases, sealed cells have been used to investigate temperature and water content dependenceof proton conductivity for samples hydrated under certain humidities prior to measurements. Conduc-tivity measurements carried out with the sealed cell are simple and useful for conductivity measurementsabove 1008C and below 08C. However, the water uptake of the hydrated sample in the sealed cellsometimes changes during measurements.
Fig. 12 shows the proton conductivity of S-PPBP with a sulfonation level of 85 mol% under variousrelative humidities as a function of temperature by using a four-point-probe cell. S-PPBP exhibits astrong decrease in conductivity as humidity decreases like Na®onw membranes. This strong dependenceof proton conductivity on humidity re¯ects in part a strong tendency of S-PPBP to absorb water vapor.This behavior in conductivity under various relative humidity is attributed to a liquid-like protonconductivity mechanism where protons are transported as free hydronium ions through water-®lledionic pores in the membrane [36].
Usually per¯uorinated polymer electrolytes exhibit a remarkable decrease in conductivity withincreasing temperature, such that conductivity at 808C is diminished by more than 10 times relativeto that at 608C. Per¯uorinated polymer electrolyte membranes become less conductive under hightemperature conditions because they dry out, causing the channels to collapse and proton transport tobecome more dif®cult. However S-PPBP exhibits little increase in conductivity with increasing tempera-ture under each relative humidity (90±50%). This phenomenon is attributable to the strong interactionbetween the sulfonic acid group and the absorbed water molecules in S-PPBP. Proton-conductingpolymer electrolyte membranes based on hydrocarbon polymers such as S-PPBP and sulfonatedpoly(thiopheneylene) have a tendency to include relatively higher amounts of bound water,resulting in higher conductivities at high temperatures and/or low humidities, as determined bydifferential scanning calorimetry.
3. Proton-conducting polymer electrolyte membranes based on alkylsulfonated aromatic polymers
3.1. Materials
Sulfonation with sulfuric acid, chlorosulfonic acid, sulfur trioxide, and acetylsulfate easily provides
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021478
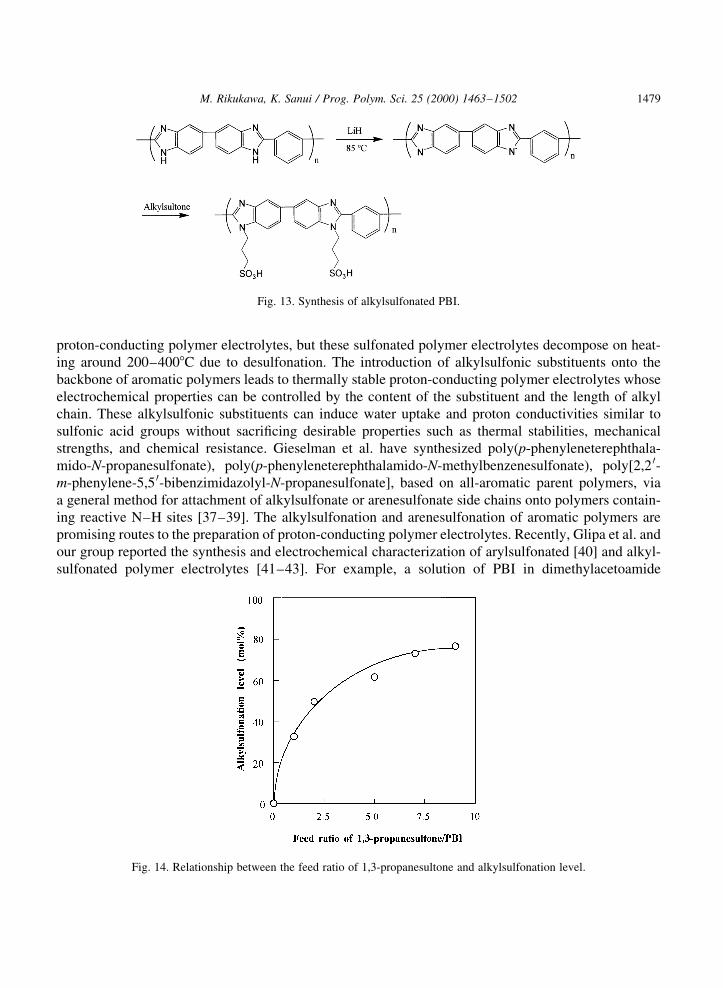
proton-conducting polymer electrolytes, but these sulfonated polymer electrolytes decompose on heat-ing around 200±4008C due to desulfonation. The introduction of alkylsulfonic substituents onto thebackbone of aromatic polymers leads to thermally stable proton-conducting polymer electrolytes whoseelectrochemical properties can be controlled by the content of the substituent and the length of alkylchain. These alkylsulfonic substituents can induce water uptake and proton conductivities similar tosulfonic acid groups without sacri®cing desirable properties such as thermal stabilities, mechanicalstrengths, and chemical resistance. Gieselman et al. have synthesized poly(p-phenyleneterephthala-mido-N-propanesulfonate), poly(p-phenyleneterephthalamido-N-methylbenzenesulfonate), poly[2,2 0-m-phenylene-5,5 0-bibenzimidazolyl-N-propanesulfonate], based on all-aromatic parent polymers, viaa general method for attachment of alkylsulfonate or arenesulfonate side chains onto polymers contain-ing reactive N±H sites [37±39]. The alkylsulfonation and arenesulfonation of aromatic polymers arepromising routes to the preparation of proton-conducting polymer electrolytes. Recently, Glipa et al. andour group reported the synthesis and electrochemical characterization of arylsulfonated [40] and alkyl-sulfonated polymer electrolytes [41±43]. For example, a solution of PBI in dimethylacetoamide
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1479
Fig. 13. Synthesis of alkylsulfonated PBI.
Fig. 14. Relationship between the feed ratio of 1,3-propanesultone and alkylsulfonation level.

(DMAc) is treated LiH or NaH, which deprotonates the nitrogen of the benzimidazole rings in thepolymer backbone, and the addition of propanesulfonate side chains onto PBI was accomplished by thereaction of 1,3-propanesultone and the PBI anion, as shown in Fig. 13. Subsequent ion-exchange withH3PO4 or HCl provides proton-conducting polymer electrolytes based on PBI. The alkylsulfonation ofPBI improved its solubility in organic solvents, where the solubility depended on the alkylsulfonationlevel. Alkysulfonated PBI was more soluble in polar aprotic solvents like methanol, DMAc, and DMSOwhen compared to the parent polymer. The relationship between the feed ratio of 1,3-propanesultone andalkylsulfonation level is shown in Fig. 14. The alkylsulfonation level for the NH groups in PBI wasestimated by elemental analysis and 1H NMR. The alkylsulfonation level was easily controlled byvarying the feed ratio of 1,3-propanesultone and led to almost 60 mol% with the feed ratio of 5.0(1,3-propanesultone/PBI repeat unit) [44].
We also tried to synthesize ethylphosphrylated PBI via the same derivatization procedure for thereactive N±H sites in PBI, as shown in Fig. 15. The substituted polymer was successfully synthesized,but the resulting polymer was insoluble in organic solvents. It can be presumed that the phosphoric acidgroups aggregate with each other during the substitution process. The ethylphosphrylated PBI showed ahigh proton conductivity of 1023 S cm21 even as a compressed pellet. It is found that phosphoric acidgroups are an effective polar group for proton-conducting polymer electrolytes.
3.2. Thermal stability of alkylsulfonated aromatic polymer electrolyte membranes
The arenesulfonated and alkylsulfonated polymers based on all-aromatic backbones maintain highthermal stabilities when compared to the parent materials. The aim of derivatization of the parentpolymers is to improve water absorption and to promote proton conductivity but retain a signi®cantfraction of their high thermal stabilities. The TGA of these polymers were examined in both inert andoxidative atmospheres by Gieselman et al. [38] PBI is extremely thermally stable, as expected for an all-aromatic polymer. In an inert atmosphere, it exhibits an onset of degradation around 6508C and 5 wt%mass loss at 7008C and retains more than 80% of its mass at 8008C. The onset of degradation tempera-tures for the substituted polymers in an inert atmosphere is signi®cantly lower than PBI, as expected forthe addition of pendant groups that are not conjugated with the main chains. The degradation ofpoly[2,2 0-m-phenylene-5,5 0-bibenzimidazolyl-N-methylbenzenesulfonate] at 22% substitution beginsat about 4808C, and poly[2,2 0-m-phenylene-5,5 0-bibezimidazolyl-N-propanesulfonate] at 54% substitu-tion shows mass loss beginning at 4508C. After the removal of the side chains the degradation is gradual
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021480
Fig. 15. Synthesis of phosphoethylated PBI.

with high residual masses (50±60%) at 8008C. The onset of degradation of PBI in an atmosphere of dryair begins at a signi®cantly lower temperature relative to that in an inert atmosphere. PBI begins todegrade at approximately 5208C, a full 1008C below the degradation temperature in an inert atmosphere.The substituted PBI shows little difference in its onset of degradation temperature in changing from aninert to an oxygen-containing atmosphere. The major difference in the behavior in air compared tonitrogen is that the magnitudes of the mass losses are much higher and the percentages of residual char athigh temperatures are much lower in the oxidative atmosphere. This is apparently the result of the lowerthermal stability of PBI in dry air, rather than a consequence of the addition of the pendant side chain.
Poly(p-phenylene terephthalamide) (PPTA) is also thermally stable. The onset of degradation dosenot occur until above 5508C, and a sharp decrease in mass begins at 6008C with a residue of 50% of theoriginal mass. After derivatization with propanesulfonate side chains (66%), the polymer shows reten-tion of thermal integrity until approximately 4008C, followed by a steady degradation to 40 wt% at8008C. The methylbenzensulfonated derivative (66%) is more thermally stable; degradation initiates atabout 4708C. After an initial mass loss, a decrease in mass to a residue of 50% occurs at 8008C. Thermalanalyses of PPTA and its derivatives in an atmosphere of dry air were also carried out by the sameresearch group. The PPTA is much less stable in air with degradation beginning about 708C lower than innitrogen. Once degradation begins, the entire mass of the polymer is volatilized. The TGA traces of themethylbenzensulfonated PPTA (66%) run in air and in N2 indicate a relatively small decrease in theonset of degradation temperature. The major difference in behavior is that the mass decrease is steeperafter the initial mass loss, and the amount of the residue at high temperatures is much lower due tooxidative degradation of the polymer backbone. The addition of pendant groups to aromatic polymersresults in a lowering of thermal stability due to the lower temperature at which the side chain cleaves,regardless of the nature of the atmosphere. This is the expected behavior because the side chain,especially sulfonic acid groups is not stabilized by conjugation to the polymer backbone. It has beenconcluded that the methylbenzenesulfonate side chain is more stable than the propanesulfonate sidechain, regardless of the type of polymer. This result suggests that the point of cleavage of the side chainis not exclusively at the N±C bond. Glipa et al. also investigated the thermal stability of methylben-zensulfonated PBI (substitution 75%) using TGA in air with a heating rate 18C min21 [40]. Theyreported that the addition of the metylbenzenesulfonate group reduced the thermal stability of thepolymer electrolyte. The onset of degradation begins at 3708C with a residue of 60% of the originalmass. They concluded that the mass loss between 370 and 4208C was attributable to the degradation ofthe sulfonic acid groups. The degradation process and mechanism is likely to be more complex. Residualwater, impurities, substitution levels, and measuring conditions signi®cantly affect in the TGA data forthese polymer electrolytes.
As mentioned above, Glipa et al. also reported that arylsulfonated PBI is stable up to 3508C, whileGieselman et al. concluded that the methylbenzensulfonated PBI was stable up to 5008C in air. It isdif®cult to compare these data due to the difference in substitution levels. It can be presumed that highlymethylbenzensulfonated PBI has a lower stability than propanesulfonated PBI due to the weak aryl±Sbond. Actually, the degradation temperature of methylbenzensulfonated PBI is almost the same aspolymer electrolytes that were sulfonated with sulfuric acid, etc.
The thermal stability of anhydrous PBI-PS was also investigated by using TGA at a heating rate of58C min21 under a nitrogen atmosphere by our research group. All samples were vacuum oven-dried at608C for at least 2 days prior to analysis. The PBI-PS is hygroscopic and quickly reabsorbs water aftervacuum drying. Due to the dif®culty encountered in drying and keeping the samples dry, an in situ drying
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1481
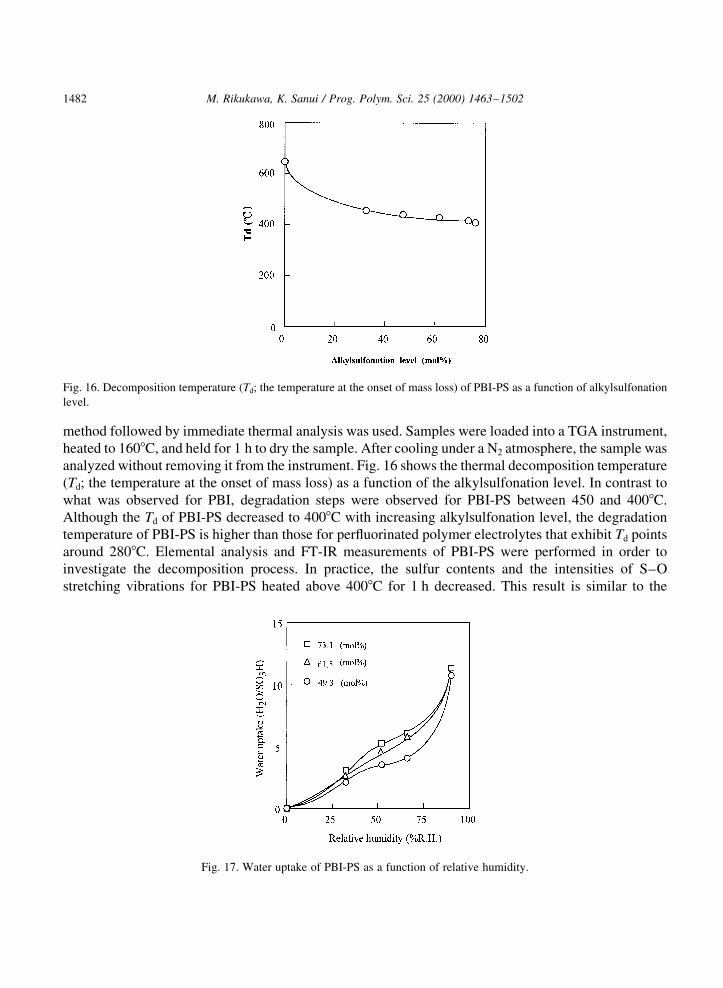
method followed by immediate thermal analysis was used. Samples were loaded into a TGA instrument,heated to 1608C, and held for 1 h to dry the sample. After cooling under a N2 atmosphere, the sample wasanalyzed without removing it from the instrument. Fig. 16 shows the thermal decomposition temperature(Td; the temperature at the onset of mass loss) as a function of the alkylsulfonation level. In contrast towhat was observed for PBI, degradation steps were observed for PBI-PS between 450 and 4008C.Although the Td of PBI-PS decreased to 4008C with increasing alkylsulfonation level, the degradationtemperature of PBI-PS is higher than those for per¯uorinated polymer electrolytes that exhibit Td pointsaround 2808C. Elemental analysis and FT-IR measurements of PBI-PS were performed in order toinvestigate the decomposition process. In practice, the sulfur contents and the intensities of S±Ostretching vibrations for PBI-PS heated above 4008C for 1 h decreased. This result is similar to the
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021482
Fig. 16. Decomposition temperature (Td; the temperature at the onset of mass loss) of PBI-PS as a function of alkylsulfonation
level.
Fig. 17. Water uptake of PBI-PS as a function of relative humidity.

result of Gieselman et al. and indicates that the decomposition of PBI-PS can be attributed to desulfona-tion. Consequently, alkylsulfonated PBI is thermally more stable when compared with sulfonatedaromatic polymer electrolytes whose degradation temperatures are in range of 200±3508C. This thermalstability in alkylsulfonated polymer electrolytes is attributable to the strong chemical bond between thealkyl chain and the sulfonic acid groups. The addition of alkysulfonic acid groups to thermostablepolymers with alkylsultone is one of the most important routes to synthesize thermally stable proton-conducting polymer electrolytes. To design thermostable polymer electrolytes with hydrocarbon poly-mers, the addition of polar groups, as well as the choice of thermostable polymer backbones, areimportant factors.
3.3. Water uptake in alkylsulfonated aromatic polymer electrolyte membranes
The addition of arylsulfonate and alkylsulfonate groups onto aromatic polymers induces waterabsorption and makes the polymer more hygroscopic. The water uptake of PBI-PS was determinedby measuring the change in the mass before and after hydration. The results are shown in Fig. 17 in termsof the moles of water absorbed per mole of sulfonic acid in the polymer. These results show that wateruptake varies with relative humidities. The equilibrium water uptake of PBI-PS ®lms increased withincreasing the relative humidity and alkylsulfonation level. This behavior and the water absorption levelare similar to per¯uorinated polymer electrolyte membranes. Two regions with high and low increases inwater uptake of PBI-PS were observed over the entire range of relative humidity. The water uptake ofPBI-PS with 73.1 mol% alkylsulfonation achieved 11.3 H2O/SO3H at 90% RH and room temperature,while Na®onw 117 showed a water uptake of 11 H2O/SO3H under the same conditions. We alsosynthesized butanesulfonated and methylpropanesulfonated PBI (PBI-BS and PBI-MPS) by the sameprocedure with butanesultone and methylpropanesultone. These polymers showed different features intheir water uptakes compared with PBI-PS. The water uptakes of PBI-BS and PBI-MPS were 19.5 and27.5 H2O/SO3H at 90% RH, respectively. These values for alkylsulfonated PBI were affected by the
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1483
Fig. 18. DSC curve of hydrous PBI-PS (73.1 mol%) ®lm containing 11.3 H2O per sulfonic acid group.

length of alkyl chains and chain branching, and hence the long alkyl chain and chain branching inducedits water uptake. It is thought that this phenomenon is attributed to the greater ¯exibility of long alkylchains and larger water absorbed cavity formed by the branching chains.
Several studies have examined the speci®c role of absorbed water in polymer electrolytes, and thephysical states of absorbed water have been characterized by several techniques such as IR spectroscopy[45] and low temperature 1H NMR relaxation time measurements [46]. We tried to elucidate the physicalstates of the absorbed water in hydrous PBI-PS ®lms by means of DSC. Fig. 18 shows a DSC curve of ahydrous PBI-PS (73.1 mol%) ®lm containing 11.3 H2O per sulfonic acid group. While anhydrous PBI-PS showed no peak, hydrous PBI-PS showed two sharp phase transitions of absorbed water at 236.6 and221.68C, which are attributable to the freezing and melting temperature of the absorbed water, respec-tively. On the other hand, hydrous Na®onw mostly showed a phase transition temperature around 08Cunder the same condition. These results suggest that the absorbed water in PBI-PS is more restricted bysulfonic acid groups on the polymer chains, as compared with Na®onw membranes. Therefore, theabsorbed water tends to be retained in PBI-PS even at elevated temperatures.
3.4. Proton conductivity of alkylsulfonated aromatic polymer electrolyte membranes
The electrical properties of wet PBI-PS ®lms were investigated by both DC measurements and thecomplex impedance method. When direct current was applied to PBI-PS ®lms, the DC conductivitysuddenly decreased. This result demonstrated that there is no electron conduction in PBI-PS, though themain chains are conjugated. In order to clarify the carrier ion in hydrous PBI-PS ®lms, the conductivityof PBI-PS ®lms containing H2O or D2O was measured, and the result is shown in Fig. 19. The conduc-tivity of PBI-PS containing H2O increased with increasing water uptake and was higher than that of PBI-PS containing D2O in the entire temperature range. This result indicates that the carrier ion is a proton(hydronium ion) for hydrous PBI-PS. Although the conventional theory for H/D isotope effects supports
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021484
Fig. 19. Conductivity of PBI-PS (61.5 mol%) ®lms containing H2O or D2O.

that the mobility of a proton is 1.4 times higher than that of deuterium ion, PBI-PS containing H2Oshowed much higher conductivity than that of PBI-PS containing D2O. Further studies regarding thedifferences in conductivity from theoretical values are now in progress.
The proton conductivity of PBI-PS containing equilibrium water was measured as a function oftemperature by the complex impedance method. As shown in Fig. 20, hydrous PBI-PS exhibits ahigh proton conductivity at room temperature. The conductivity of a PBI-PS ®lm containing 3.1H2O/SO3H reached 1025 S cm21 at 808C and decreased slightly at higher temperatures due to a smallloss of water (about 10 wt%). The conductivity of PBI-PS containing more than 5.2 H2O/SO3H increased
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1485
Fig. 20. Temperature dependence of conductivity for PBI-PS. Left: water uptakes of PBI-PS ®lms are almost 48 wt%. Right:
sulfonation level of PBI-PS ®lms is 73.1 mol%.
Fig. 21. A comparison of Arrhenius plots of conductivity between PBI-PS and Na®onw membrane.

with an increase in temperature, and a conductivity of the order of 1023 S cm21 was maintained over1008C. The PBI-PS ®lm containing 11.3 H2O/SO3H showed high proton conductivities on the order of1023 S cm21 and an Arrhenius type dependence of conductivity with the activation energy Ea �9:4 kJ mol21
: A comparison of Arrhenius plots for conductivities between PBI-PS and Na®onw
membranes is shown in Fig. 21. The water uptake of PBI-PS is similar to that of Na®onw membranewhen these ®lms were placed under 90% RH. The proton conductivity of Na®onw membrane reached1023 S cm21 at room temperature but dropped over 1008C due to the loss of absorbed water. On thecontrary, the high proton conductivity of hydrous PBI-PS was retained over 1008C. The higher wateruptake and proton conductivity over 1008C for PBI-PS is attributed to the different water absorptionbehavior and the physical state of the absorbed water within PBI-PS.
On the other hand, Glipa et al. reported the proton conductivities of methylbenzensulfonated PBIunder various relative humidities. The proton conductivity increased with increases in the substitutionlevel. Methylbenzensulfonated PBI with 75% substitution showed a high conductivity of about1022 S cm21 at 408C under 100% RH.
Judging from the results described above, alkylsulfonated aromatic polymer electrolytes possessenough thermal stability for use as an electrolyte in a fuel cells operating near 808C. This is thetemperature where per¯uorinated polymer electrolytes based PEFC are usually operated. The wateruptakes and proton conductivities of these polymers are similar to per¯uorinated polymer electrolytesbelow 808C but are better temperature over 808C. The absorbed water molecules and sulfonic acidgroups in these polymers have relatively strong interactions, compared with per¯uorinated polymerelectrolytes. It is thought that this difference is closely related the water absorption mechanism andphysical state of the absorbed water between these polymers and per¯uorinated polymer electrolytes.Further studies on the proton conduction mechanism in these polymers are required.
4. Proton-conducting polymer electrolyte membranes based on acid±base polymer complexes
4.1. Materials
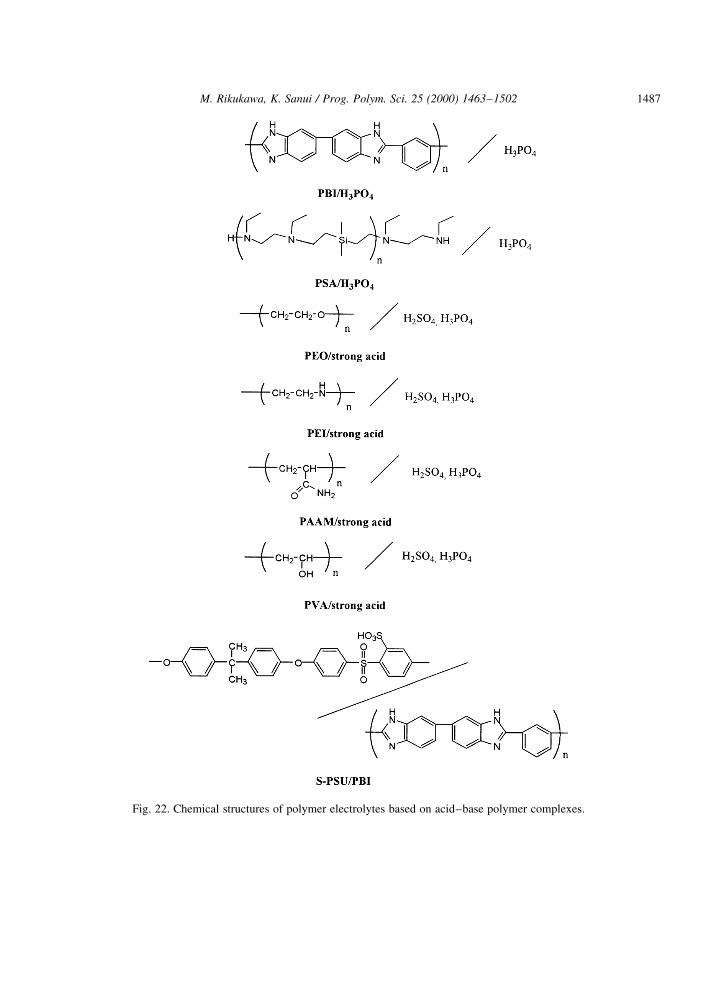
The development of novel proton-conducting polymers for various electrochemical applications isattracting considerable interest. There are still some limitations on the electrochemical properties andwater uptake in proton-conducting polymers, especially at elevated temperatures. Complexes of basicpolymers, such as poly(ethylene oxide) (PEO) [47], poly(ethylene imine) (PEI) [48], poly(acrylamide)(PAAM) [49,50], and poly(vinylalchol) (PVA) [51,52], with strong acids have been shown to possesshigh proton conductivities both in the dehydrated and hydrated states [53±58]. For these systems themechanism of proton conduction has been inferred from conductivity, FT-IR, and nuclear magneticresonance measurements. These complexes are relatively inexpensive and can be easily processed asthin ®lms for applications such as hydrogen sensors [59], electrochromic displays, and PEFC systems.For example, complexes of PVA with orthophosphoric acid in the form of thin ®lms have been preparedby a solution cast technique with different stoichiometric ratios. The ionic transference number ofmobile ions was estimated from Wagner's polarization method and the value was reported to be tion �0:97: At 308C the conductivity of PVA/H3PO4 complexes (molar ratio H1/PVA� 0.77) is about1023 S cm21 [51]. According to Lassegues et al. [58], acid±base polymer complexes ofPAAM and strong acids such as H3PO4 or H2SO3 exhibit a high proton conductivity in the range of
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021486
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1487
Fig. 22. Chemical structures of polymer electrolytes based on acid±base polymer complexes.

1024±1023 S cm21 at ambient temperatures. The proton conductivity increases with temperature toabout 1022 S cm21 at 1008C. However, the mechanical and chemical stability of these complexes isrelatively poor, and chemical degradation is often observed after humidi®cation. Anhydrous acid-basepolymer complexes (PEI/xH2SO4 and PEI/xH3PO4) can also be obtained by protonation of branchedcommercial poly(ethylene imine) with H2SO4 or H3PO4. For partially protonated PEI �x # 0:35�; onlySO4
22 or HPO422 anions are present and the conduction seems to occur by proton exchange between
protonated and unprotonated amine groups. Above x < 0:35; HSO42 or H2PO4
2 anions appear and theconductivity, likely to occur along the SO4
22/HSO42 or HPO4
22/H2PO42 anionic hydrogen-bonded chains,
increases up to values of about 1024 S cm21 for PEI/0.5H2SO4 and about 1025 S cm21 for PEI/0.5H3PO4.Thin transparent ®lms can be produced for x # 0:35; and they lose their mechanical properties andbecome very hygroscopic above this value because of the presence of excess acid. Fig. 22 shows some ofthe chemical structures for these materials.
New proton-conducting polymers based on poly(silamine) (PSA) were synthesized by mixing PSAand H3PO4 by our research group [60]. The reaction of N,N 0-diethylethylenediamine (DEDA) anddimethyldivinylsilane (DVS) in the presence of n-BuLi gave PSA, which was a clear yellow and viscousliquid. The results of 1H NMR analysis for PSA indicated that both ends of the PSA chains carried N-ethylamino groups and that the molecular weight was about 2000. PSA/H3PO4 complexes were obtainedby mixing PSA and H3PO4 in methanol. To a H3PO4 methanol solution, liquid PSA was added dropwise,and then a white precipitate of the resulting polymer complex was obtained. FT-IR measurements werecarried out in order to prove the interaction between PSA and H3PO4 molecules. (Fig. 23) The spectrumof the polymer complex exhibits a broad peak centered at 3000 cm21 and a CN band at 1469 cm21. TheN±H band appears at 3393 cm21 due to the hydrogen bond interaction between the N atoms of PSA andthe phosphoric acid molecules. Because of the presence of the ethylenediamine units in the main chain,PSA shows a two-step protonation process on changing the pH [61]. These results and previous data
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021488
Fig. 23. FT-IR spectra of PSA and PSA/H3PO4 polymer complex.

indicate that PSA is protonated with phosphoric acid to afford a solid complex containing the doublechelating structure. The H3PO4 contents in PSA/H3PO4 polymer complexes were controlled by changingthe feed ratio. The PSA absorbed H3PO4 of 0.8 mol/unit at a feed ratio (H3PO4/PSA unit) of 2. Over aH3PO4 content of 0.8 mol/unit, these PSA/H3PO4 polymer complexes became viscoelastomers. TGAmeasurements for PSA/H3PO4 polymer complexes were carried out to investigate their thermal
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1489
Fig. 24. Thermogravimetric analysis of PSA and PSA/H3PO4 polymer complexes.
Fig. 25. FT-IR spectra of PBI and PBI/strong acid polymer complexes.

stabilities. The results are shown in Fig. 24. The onset of the weight loss for PSA/H3PO4 polymercomplexes began at 2008C, while PSA was stable up to about 3008C. In order to identify the originof weight loss, elemental analysis and FT-IR measurements were carried out before and after the heattreatment of the polymer complexes at 2008C for 1 h. These data indicated that the weight loss at 2008Cresults from the change from orthophosphoric acid to pyrophosphoric acid. Thus, these PSA/H3PO4
complexes were found to be quite stable up to at least 2008C.Recently, new proton-conducting polymer electrolyte membranes based on poly(benzimidazole)
(PBI) have been proposed for use in fuel cells [62±65]. The main advantage compared with per¯uori-nated polymer electrolytes and other acid±base polymer complexes, is that the polymer is conductiveeven when the activity of water is low and has a high thermal stability. These materials are expected tooperate from ambient to high temperatures in humid or dry gas. These PBI complex ®lms are fabricatedby immersing PBI cast ®lms into phosphoric acid solutions.
In our case, the PBI/strong acid polymer complexes were easily prepared by immersing PBI ®lms intoa strong acid/methanol solution [66,67]. The absorption level of strong acid molecule increased withincreasing the concentration of the strong acid, and the highest absorption level of 2.9 mol/unit wasobserved for PBI/H3PO4 polymer complexes. In order to clarify the nature of the interaction between PBIand strong acid molecules, FT-IR measurements were carried out. As shown in Fig. 25, the interactionsbetween strong acids and imidazole groups were observed in the range from 2000 to 3600 cm21. PBIshowed a self-associated NH stretching band at 3139 cm21. Although NH1 stretching bands wereobserved around 2900 cm21 for PBI/H2SO4, CH3SO4, and C2H5SO3H complexes, PBI/H3PO4 did notshow the characteristic absorptions of NH1 groups. These data indicate that the acid molecules, exceptfor H3PO4, protonate the N atom in the imidazole ring. H3PO4 dose not protonate imidazole groups inPBI but interacts with it via strong hydrogen bonding interaction of OH and NH groups. Savinel et al.[62±65] prepared PBI complex ®lms doped with phosphoric acid by immersion in a phosphoric acidaqueous solution for a least 16 h. Equilibration in an 11 M H3PO4 solution yielded a doping level ofroughly ®ve phosphoric acid molecules per polymer repeat unit.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021490
Fig. 26. TG-curves of PBI and PBI/strong acid polymer complexes.

In general, sulfonated aromatic polymers are very brittle in the anhydrous state, which can happen inthe fuel cell application under intermittent conditions. Kerres et al. have discovered new materials,which are not as brittle as anhydrous sulfonated polymers using polymer blend techniques [68]. Thesematerials were synthesized by combining polymeric N-bases, such as ortho-sulfone diamine poly-sulfone, poly(4-vinylpyridine), PBI, and poly(ethyleneimine), and polymeric sulfonic acids such assulfonated poly(etheretherketone) and ortho-sulfone-sulfonated polysulfone, as illustrated in Fig. 22.These blend polymers showed high proton conductivities and moderate swelling values combined withhigh thermal stabilities. The speci®c interaction of the SO3H groups and of the basic N groups wasobserved by FT-IR measurements. The acid±base interaction between both polymers provided suitablemechanical strengths and thermal stabilities. Their thermal and mechanical properties are similar tothose of other acid±base polymer complexes described later.
4.2. Thermal stability of acid±base polymer complexes
The thermal stability of PBI/strong acid polymer complexes was investigated by TG-DTA analysis.Fig. 26 shows TG-curves of PBI and PBI/strong acid polymer complexes. PBI is extremely thermallystable over the entire measuring temperature range. The small mass loss for all samples up to 2008C isattributable to losses of water and solvent, which tend to remain in the membranes. A lowering ofdegradation temperature was expected with the complexation of acid molecules, which easily corrodeand oxidize the polymer backbone. However, the degradation of PBI/H3PO4 polymer complexes in N2
atmosphere was not observed over the entire measuring temperature range, where typical proton-conducting polymer electrolytes show considerable thermal degradation. On the other hand, onsets ofthermal decompositions for PBI/H2SO4, CH3SO4, and C2H5SO3H complexes began at 330, 240, and2208C, respectively. After the thermal degradation of these polymer/strong acid polymer complexesbetween 220 and 4008C, a residue with 50% of the original mass remained. The complexation of H2SO4,CH3SO4, and C2H5SO3H to PBI results in a loss of thermal stability. This decomposition is mainly due tothe elimination of acid molecules from the polymer/strong acid polymer complexes as determined byelemental analysis. Over 4008C, the PBI polymer backbone gradually decomposes due to the presence ofstrong acids and high temperatures.
PBI/H3PO4 complexes, however, were thermally stable up to 5008C and so also PBI. The sulfonationof PBI via acid treatment has been shown to stabilize PBI, and Powers et al. reported that doping PBIwith 27 wt% phosphoric acid also increased thermal stability [69]. They attributed the increased stabilityto the formation of benzimidazonium cations.
Savinell's group also investigated the thermal stability of PBI/H3PO4 polymer complexes in simulatedfuel cell environments [63]. They found that PBI/H3PO4 polymer complexes have promisingproperties for use as polymer electrolytes in hydrogen/air and direct methanol fuel cells. Tosimulate the conditions present in a high temperature fuel cell, PBI/H3PO4 polymer complexeswere loaded with fuel cell grade platinum black, doped with ca. 480 mol% phosphoric acid (4.8H3PO4 molecules per PBI repeat unit) and heated under atmospheres of either nitrogen, 5% hydrogen, orair in a thermogravimetric analyzer. The products of decomposition were taken directly into a massspectrometer for identi®cation. In all cases weight loss below 4008C was found to be due to the loss ofwater. They concluded that the PBI/H3PO4 polymer complexes loaded with fuel cell grade platinumwere quite stable up to 6008C.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1491

4.3. Conductivity of acid±base polymer complex
Savinell's group studied the conductivity of PBI/H3PO4 polymer complexes as a function of watervapor activity, temperature, and acid doping levels [65]. The conductivity of PBI/H3PO4 polymer
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021492
Fig. 27. Temperature dependence of conductivity for anhydrous PBI/strong acid complexes. (X) PBI/H3PO4 (2.0 mol/unit); (O)
PBI/H2SO4 (1.8 mol/unit); (B) MeSO3H (2.0 mol/unit); (V) EtSO3H (1.9 mol/unit).
Fig. 28. Temperature dependence of conductivity for hydrous PBI/strong acid complexes. (X) PBI/H3PO4 (2.0 mol/unit, H2O
13 wt%); (O) PBI/H2SO4 (1.8 mol/unit, H2O 19 wt%); (B) MeSO3H (2.0 mol/unit, H2O 26 wt%); (V) EtSO3H (1.9 mol/unit,
H2O 20 wt%).

complexes at the higher doping level (500 mol%) was roughly twice that for the ®lm doped to 338 mol%under similar conditions of temperature and humidity. For example, the conductivity of PBIdoped with ca. 500 mol% (5 H3PO4 molecules per PBI repeat unit) at 1908C and at watervapor activity of 0.1 was 3:5 £ 1022 S cm21
: The conductivity increased with temperature andwater vapor activity regardless of the doping level of H3PO4. In addition, they found that themethanol crossover with PBI/H3PO4 polymer complexes was one order of magnitude smaller thanthat with per¯uorinated polymer electrolytes. The mechanical strength is three orders of magni-tude greater than that of Na®onw membranes.
We also investigated the proton conductivity of PBI/strong acid polymer complexes prepared by ourmethod with methanol solutions of strong acids. The temperature dependences of conductivity foranhydrous PBI/strong acid polymer complexes at an acid concentration of about 1.9 mol/unit isshown in Fig. 27. All of the anhydrous PBI/strong acid polymer complexes exhibited proton conductionsof 1026±1029 S cm21 at 1008C. The conductivity of PBI/H3PO4 polymer complex reached up to1025 S cm21 at 1608C, while other PBI/strong acid polymer complexes showed a decrease in conduc-tivity over 808C. This result also re¯ects the good thermal stability of PBI/H3PO4 polymer complexes.The ®lms of these PBI/strong acid polymer complexes were placed in a 90% RH vessel for 3 days toprepare hydrous ®lms. The water uptakes of these complexes were estimated to be near 13±26 wt%. Asshown in Fig. 28, the proton conductivities of the hydrous PBI/strong acid polymer complexes were oneorder of magnitude higher than those of anhydrous polymer complexes due to the improvement of carriergeneration by the absorbed water. Especially, remarkable increases in proton conductivities wereobserved at ambient temperatures. Fig. 29 shows the temperature dependence of conductivity foranhydrous PBI/H3PO4 complexes with different acid contents. The conductivity of the PBI/H3PO4
polymer complexes increased with increasing contents of H3PO4. The conductivity of PBI/H3PO4
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1493
Fig. 29. Temperature dependence of conductivity for anhydrous PBI/H3PO4 complexes with different acid contents. H3PO4
contents (mol/unit) (O)2.9; (V) 2.7; (B)2.3; (X)2.0; (W)1.4.

M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021494
Fig. 30. FT-IR spectra of PBI/H3PO4 and H3PO4.
Fig. 31. Temperature dependence of conductivity for anhydrous PSA/H3PO4 complexes with different H3PO4 contents.

complex �x � 1:4� showed a different behavior, having relatively low conductivities over the entiretemperature range. This phenomenon suggests that two H3PO4 molecules interact quantitatively with aPBI unit containing two imidazole groups and consequently an excess of H3PO4 to imidazole groups isnecessary to give suf®cient proton conductivity. In order to clarify the interaction between H3PO4 andPBI, the characteristic absorption of H3PO4 molecules in the PBI/H3PO4 polymer complex was inves-tigated by FT-IR. The results are shown in Fig. 30. Three characteristic absorptions of the HPO4
22 P±OH,and H2PO4
2 groups for PBI/H3PO4 polymer complexes appear at 1090, 1008, and 970 cm21, respectively[49,50,53,70]. The intensity of absorption band of HPO4
22 increases with increase in the concentration ofH3PO4. The presence of HPO4
22 and H2PO42 anions implies that proton conduction may occur according
to the Grotthus mechanism, which involves an exchange of protons between H3PO4 and HPO422 or
H2PO42.
The PSA/H3PO4 polymer complexes also exhibit high proton conductions in the hydrated state as wellas the dehydrated state. Fig. 31 shows the temperature dependence of conductivity in the dehy-drated state for PSA/H3PO4 polymer complexes with different H3PO4 contents. The conductivitiesof the PSA/H3PO4 polymer complexes increased with increasing H3PO4 contents and did notdiminish over 1008C. The PSA/H3PO4 polymer complex possesses a conductivity of1025 S cm21 at 1608C at a concentration of 0.8 H3PO4 per repeat unit. In order to identify thecarrier ion, the conductivities of PSA/H3PO4 and PSA/D3PO4 polymer complexes were measuredunder the same conditions, and the results are shown in Fig. 32. The temperature dependence ofconductivity for the PSA/D3PO4 polymer complex was similar to that of the PSA/H3PO4 polymercomplex, but the conductivity of PSA/H3PO4 complex was higher than the conductivity of thePSA/D3PO4 polymer complex at every temperature. The isotope effect for the conductivity ofthese polymer complexes proves that the carrier ions are protons generated from strong acidmolecules [71].
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1495
Fig. 32. Comparison of temperature dependence of conductivity for PSA/H3PO4 and PSA/D3PO4 complexes.

5. Other proton-conducting polymer electrolyte membranes
As described above, many kinds of proton-conducting polymer electrolyte membranes based onhydrocarbon polymers have been developed not only for PEFC and DMFC applications but also forelectrochromic display applications and sensor devices. There are several reports and patents concerningnew proton-conducting polymer electrolytes having different chemical structures and concepts fromconventional per¯uorinated polymer electrolytes and hydrocarbon materials described above. Thesematerials involve phosphoric acid based polymer electrolytes, silicone based electrolytes, and otherinorganic polymer electrolytes.
Proton-conducting polymer electrolyte membranes for possible use in H2/O2 and direct methanol fuelcells have been fabricated from poly[bis(3-methylphenoxy)phosphazene] by Quo et al. [72]. Thesepolymer electrolytes were sulfonated with SO3 and cast from solution. The ion exchange capacity ofthe membrane was 1.4 mmol/g. The polymer cross-linking was carried out by dissolving a benzophe-none photoinitiator in the casting solution and exposing the resulting cast ®lms to UV light. Thesulfonated and cross-linked polyphosphazene membrane swelled less than Na®on 117w membranesin both water and methanol. The proton conductivity of water equilibrated polymers was similar toNa®onw membranes between 25 and 658C. A Japanese company also claimed gas diffusion electrodesfor fuel cells comprised of polyphosphazene [73].
Gautier-Luneau et al. prepared an organic±inorganic proton-conducting polymer electrolyte to beused as a membrane for DMFC [74]. They synthesized the electrolytes by hydrolysis±co-condensationof three different alkoxy silanes; benzyltriethoxysolane, n-hexyltrimethoxysilane, and triethoxysilane.After co-condensation and solvent evaporation, sulfonation was achieved in dichloromethane by addingchlorosulfonic acid in a stoichiometric amount, with respect to the benzyl groups. The cross-linking wasperformed in THF by hydrosilyation of the silane groups with divinylbenzene using a divinyltetra-methyldisiloxane platinium complex as catalyst. The TGA data showed one broad endothermic peakwith a maximum at 1038C due to the loss of water. They reported that these polymer electrolytes werethermally stable until 2508C and that the cabonization of the organic chains started at 3758C. The protonconductivity of these materials was about 1:6 £ 1022 S cm21
:
Another alkoxy silanes based polymer electrolyte was reported by Honma et al. [75]. Their organic±inorganic hybrid electrolyte membranes have been synthesized by the sol±gel process. Poly(ethylene-glycols) �Mw � 200; 300, 400, 600, 2000) were reacted with 3-isocyanatopropyltriethoxy silane in an N2
atmosphere at 708C to obtain alkoxy end-capped poly(ethyleneoxide) precursors. The hybrid electrolytemembranes were fabricated by hydrolyzing the end-capped precursor with monophenyltriethoxysilane.A composition of the monophenyltriethoxysilane of 20% was found to be the most ¯exible and chemi-cally stable composition for the hybrid electrolyte membranes. The TGA showed that decomposition ofthe hybrid membrane started near 3008C. They reported that the thermal stability of the electrolytemembranes were enhanced enormously compared with poly(ethyleneoxide), which was the base mate-rial of the hybrid membranes due to structural con®nement of the poly(ethyleneoxide) chain between thenano-sized silicate domains. Proton conductivity within the hybrid membranes was provided by incor-porating acid molecules such as monododecylphophate and phosphotungstic acid. The proton conduc-tivity as a function of humidi®er temperature against the constant cell temperature at 808C was measuredfor the electrolyte membrane doped with 20 wt% of monododecylphophate. The proton conductivitywas very small at low humidi®er temperatures and increased with temperature and reached a maximumvalue of 1023 S cm21 above 808C. The hybrid electrolyte membranes needed almost saturated
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021496
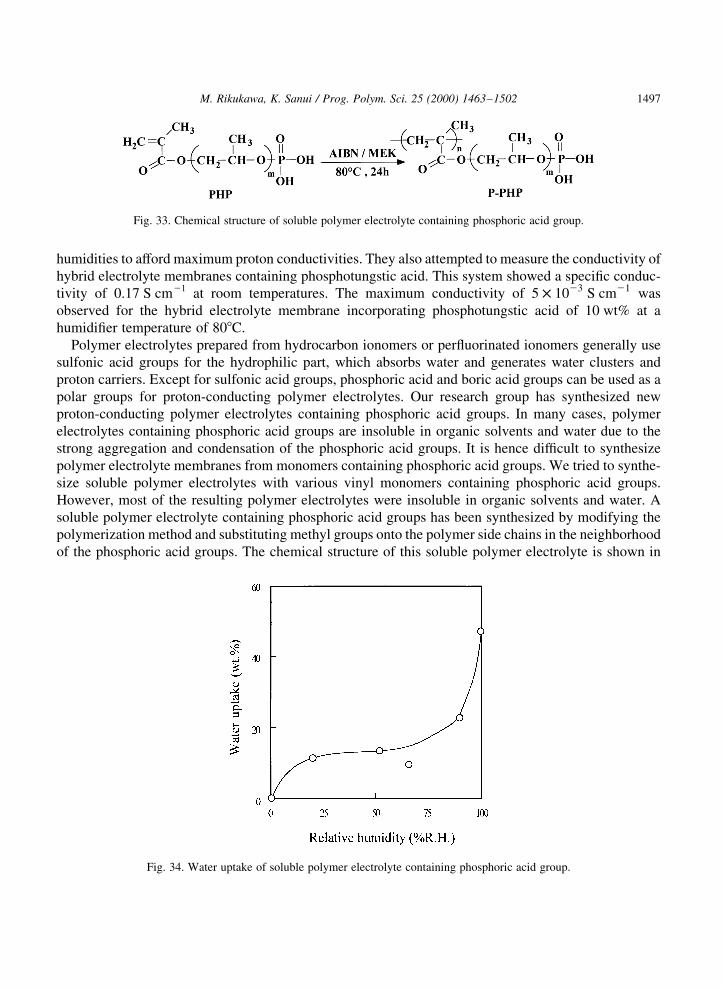
humidities to afford maximum proton conductivities. They also attempted to measure the conductivity ofhybrid electrolyte membranes containing phosphotungstic acid. This system showed a speci®c conduc-tivity of 0.17 S cm21 at room temperatures. The maximum conductivity of 5 £ 1023 S cm21 wasobserved for the hybrid electrolyte membrane incorporating phosphotungstic acid of 10 wt% at ahumidi®er temperature of 808C.
Polymer electrolytes prepared from hydrocarbon ionomers or per¯uorinated ionomers generally usesulfonic acid groups for the hydrophilic part, which absorbs water and generates water clusters andproton carriers. Except for sulfonic acid groups, phosphoric acid and boric acid groups can be used as apolar groups for proton-conducting polymer electrolytes. Our research group has synthesized newproton-conducting polymer electrolytes containing phosphoric acid groups. In many cases, polymerelectrolytes containing phosphoric acid groups are insoluble in organic solvents and water due to thestrong aggregation and condensation of the phosphoric acid groups. It is hence dif®cult to synthesizepolymer electrolyte membranes from monomers containing phosphoric acid groups. We tried to synthe-size soluble polymer electrolytes with various vinyl monomers containing phosphoric acid groups.However, most of the resulting polymer electrolytes were insoluble in organic solvents and water. Asoluble polymer electrolyte containing phosphoric acid groups has been synthesized by modifying thepolymerization method and substituting methyl groups onto the polymer side chains in the neighborhoodof the phosphoric acid groups. The chemical structure of this soluble polymer electrolyte is shown in
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1497
Fig. 33. Chemical structure of soluble polymer electrolyte containing phosphoric acid group.
Fig. 34. Water uptake of soluble polymer electrolyte containing phosphoric acid group.

Fig. 33. Vinyl acrylate phosphoric acid monomer (PHP) and azobisisobulyronitrile (AIBN) were ®rstdissolved in methylethylketone. To methylethylketone heated at 808C, the monomer solution was addeddropwise, and then the mixing solution was maintained at 808C for at least 24 h. The polymer wasobtained and puri®ed by reprecipitation. Copolymers with phenyl mareimide, styrene, or acrylonitrilewere also synthesized by the same polymerization method with AIBN. The mechanical properties,proton conductivities, and solubilities were modi®ed by changing the types of co-monomers and theircontents. The thermal stability of these polymer electrolytes was studied using TGA in nitrogen atmo-sphere. These polymers and copolymers were thermally stable up to 2008C. A ®rst mass loss beganbetween 200 and 3008C in N2 at a heating rate of 108C min21. After the ®rst degradation, 60 wt% of theoriginal mass remained. These results suggest that the dehydration and degradation of P-PHP mainchains occur at that temperature. It can be therefore presumed that these polymer electrolytes havesuf®cient thermal stabilities for most PEFC applications, which are usually operated around 1008C. Theequivalent weight of P-PHP is about 500. Fig. 34 shows the water uptake of P-PHP containing phos-phoric acid. The water absorption behavior of the polymer electrolyte was similar to conventionalper¯uorinated polymer electrolytes, which showed the Brunauer±Emmett±Teller (BET) absorption ofwater. Although P-PHP absorbed 12 H2O molecules per phosphoric acid at 100% RH, less than 4 H2Omolecules per phosphoric acid were absorbed below 60% RH. Its water uptake is very sensitive tohumidity. The proton conductivity of P-PHP showed great dependence on water uptake and temperature.Fig. 35 displays the temperature dependence of conductivity for P-PHP with various water contents. Theconductivity of hydrous P-PHP increased with increasing water content. The P-PHP material containing12 H2O molecules per phosphoric acid group showed 20 times higher conductivity than that of the P-PHP containing only 3.4 H2O molecules per phosphoric acid group. All of these samples showed littledecreases in conductivity up to 1008C. This result indicates that the absorbed water molecules in P-PHP
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021498
Fig. 35. Temperature dependence of conductivity for soluble polymer electrolyte containing phosphoric acid group with various
water contents.

have a strong interaction with the polymer back bones and hence remain in the membranes even atelevated temperatures. Judging from these results, phosphoric acid groups can serve as polar groups withsuf®cient water absorption and proton conductivities for polymer electrolytes in PEFC applications.Since phosphoric acid groups may have different water absorption mechanisms from that of sulfonicacid groups, further studies on the water uptake and physical state of water in these materials are desired.
Doyle et al. [76] demonstrated that per¯uorinated polymer electrolyte membranes such as Na®onw
membrane can be swollen with room temperature molten salts giving composite free-standingmembranes having excellent stabilities and proton conductivities in the high temperature range whileretaining a low volatility of the ionic liquid. Ionic conductivities in excess of 0.1 S cm21 at 1808C havebeen demonstrated using a room temperature molten salt, 1-butyl 3-methyl imidazolium tri¯uoro-methane sulfonate.
On the other hand, a new material, which is permeable to gases and water with ef®cient proton andelectron conductivity, has been developed. Such a material could replace both carbon and Na®onw in thecatalyst layer and should provide enhanced performance on PEFCs. Lefebvre et al. reported the use ofpolypyrrole/poly(styrene-4-sulfonate), poly(3,4-ethylenedioxythiophene)/poly(vinylsulfate), and poly(3,4-ethylenedioxythiophene)/poly(styrene-4-sulfonate) in such a role [10,15,77]. They showed highelectron- and proton conductivities, which facilitated rapid electron chemical reaction rates in thicklayers of catalyst.
6. Fuel cell applications
Two of the acid±base blend polymer electrolytes (type 1: 90 wt% sulfonated PEEK and 10 wt% PBI,type 2: 95 wt% sulfonated poly(ethersulfone) and 5 wt% PBI) were applied in a H2/O2 PEFC. Kerres etal. reported that the current/voltage curves of the acid±base blend polymers in the fuel cell werecomparable with that of Na®on 112w membranes [68]. These fuel cell tests were performed up to 300 h.
Our research group carried out H2/O2 fuel cell tests with sulfonated PPBP membranes. Several cellshave been operated at 808C using E-TEK Pt/C electrodes with a Pt loading of 0.8 mg cm22. The fuelcells were operated at 4 atm, with the reactant gases humidi®ed by bubbling through distilled water.Both O2 and H2 bubblers were typically retained at 808C. The maximum power observed in theseunoptimized cells was 0.3 W cm22 at 800 mA cm22. The electrolyte membrane conductivity determinedusing the current interrupt method was found to be 3 £ 1023 S cm21
: The membrane thickness and areawere 0.01 cm and 3.15 cm2, respectively.
Savinell's group has done H2/O2 and methanol/O2 fuel cell tests with PBI/H3PO4 polymer complexes.For the case of H2/O2 fuel cell tests, several cells were operated at 1508C using E-TEK Pt/C electrodeswith a Pt loading of 0.5 mg cm22. The fuel cells were operated at atmospheric pressure, with the reactantgases humidi®ed by bubbling through distilled water. The bubblers were typically held at 488C (hydro-gen side) and 288C (oxygen side). The maximum power observed in these unoptimized cells was0.25 W cm22 at 700 mA cm22. The electrolyte membrane resistance determined using the currentinterrupt method was found to be 0.4 ohm. The membrane thickness and area were 0.01 cm and1 cm2, respectively, and the doping level was 500 mol%. The measured cell resistance was equivalentto a conductivity of 0.025 S cm21. The cell resistance was found to be essentially independent of gashumidi®cation, indicating that the water produced at the cathode is suf®cient to maintain conductivity inthe electrolyte. A cell of this type was operated continuously at 200 mA cm22 for over 200 h with no
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1499

overall decay in performance. In the case of methanol/O2 fuel cell tests, the fuel cell electrodes were aPr±Ru anode catalyst (4 mg cm22) and a Pt black cathode catalyst (4 mg cm22). A 4:1 water:methanolvapor mixture was fed to the anode. Oxygen for the cathode was humidi®ed at room temperature.Operating at 2008C and atmospheric pressure, the cell produced over 0.1 W cm22 for current densitiesbetween 250 and 500 mA cm22. The conductivities of the membranes in the test cells did not changebetween 30 and 1408C under the same conditions.
7. Summary
This review concerning new proton-conducting polymer electrolytes was devoted to the description ofnumerous synthesis of materials, thermal stabilities, water uptakes, proton conductivities, and fuel cellapplications. The numerous advantages of these materials have been critically reviewed, together withsynthesis methods and characterizations.
Per¯uorinated polymer electrolyte membranes such as Na®onw and Flemionw have been extensivelyused as polymer electrolytes for fuel cells. These polymer electrolytes have suf®cient electrochemicalproperties, mechanical properties, and chemical and thermal stabilities. However, PEFCs constructedwith these per¯uorinated polymer electrolyte membranes tend to be expensive and have severalproblems especially for use in motor vehicles. To overcome these problems and their high costs, thedevelopment of new proton-conducting polymer electrolytes is necessary for extensive applications.Proton-conducting polymer electrolytes based on hydrocarbon polymers or inorganic polymers are oneof the most promising materials for the development of new PEFCs. These polymer materials have greatvariety with regard to chemical structure and can be modi®ed chemically at very low cost. As mentionedin this review, these polymer electrolytes possess high water absorptions and proton conductivities athigh temperatures and low humidities with suf®cient thermal and chemical stabilities. These materialscan be identi®ed as a remarkable family of proton-conducting polymer electrolytes which provide fornew high performance PEFCs that can be operated at high temperatures without humidi®cation. Furtherwork will be required to develop materials with suf®cient long-term stabilities and mechanical strengthsand to further optimize the fuel cell performances.
References
[1] Zawodzinski TA, Derouin C, Radzinski S, Sherman RJ, Smith VT, Springer TE, Gottesfeld S. J Electrochem Soc
1993;140:1041±7.
[2] Watkins DS. In: Blomen LJMJ, Mugerwa MN, editors. Fuel cell systems. New York: Plenum Press, 1993. p. 493.
[3] Higuchi M, Minoura N, Kinoshita T. Chem Lett 1994:227±30.
[4] Gupta B, Buchi FN, Scherer GG. Solid State Ionics 1993;61:213±8.
[5] Flint SD, Slade RCT. Solid State Ionics 1997;97:299±307.
[6] Bredas JL, Chance RR, Silbey R. Phys Rev B 1982;26:5843.
[7] Kobayashi H, Tomita H, Moriyama H. J Am Chem Soc 1994;116:3153±4.
[8] Wang F, Roovers J. Macromolecules 1993;26:5295±302.
[9] Bailly C, Williams DJ, Karasz FE, Macknight WJ. Polymer 1987;28:1009±16.
[10] Qi Z, Pickup PG. J Chem Soc Chem Commun 1998:15.
[11] Wallow TI, Novak BM. J Am Chem Soc 1991;113:7411.
[12] Child AD, Reynolds JR. Macromolecules 1994;27:1975±7.
[13] Kobayashi T, Rikukawa M, Sanui K, Ogata N. Solid State Ionics 1998;106:219±25.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021500

[14] Chalk AJ, Hay AS. J Polym Sci A 1968;7:691.
[15] Qi Z, Lefebvre MC, Pickup PG. J Electroanal Chem 1998;459:9.
[16] Johnson BC, Ylgor I, Iqbal M, Wrightman JP, Lliyd DR, McGrath JE. J Polym Sci: Polym Chem Ed 1984;22:72.
[17] Wei XL, Wang YZ, Long SM, Bobeczko C, Epstein AJ. J Am Chem Soc 1996;118:2545±55.
[18] Miyatake K, Shouji E, Yamamoto K, Tsuchida E. Macromolecules 1997;30:2941±6.
[19] Miyatake K, Iyotani H, Yamamoto K, Tsuchida E. Macromolecules 1996;29:6969±71.
[20] Jin X, Bishop MT, Ellis TS, Karasz FE. Br Polym J 1985;17:4.
[21] Lee J, Marvel CS. J Polym Sci: Polym Chem Ed 1984;22:295.
[22] Noshay A, Robeson LM. J Appl Polym Sci 1976;20:1885.
[23] Litter MI, Marvel CS. J Polym Sci: Polym Chem Ed 1985;23:2205.
[24] Makowsky H, Lundberg RD, Singahl GH. US Patent. US3870841, 1975.
[25] Bishop MT, Karasz FE, Russo PS, Langley KH. Macromolecules 1985;18:86.
[26] Devaux J, Delimoy D, Daoust D, Legras R, Mercier JP, Strazielle C, Neild E. Polymer 1985;26:1994.
[27] Ogawa T, Marvel CS. J Polym Sci: Polym Chem Ed 1985;23:1231.
[28] Iwasaki K, Terahara A, Harada H. Japanese Patent. H11-116679, 1999.
[29] Konishi M, Murata H, Yamamoto F. Japanese Patent. H11-67224, 1999
[30] Chu D, Gervasio D, Razaq M, Yeager EB. J Appl Electrochem 1990;20:157.
[31] Surowiec J, Bogoczek R. J Thermal Anal 1988;33:1097.
[32] Samms SR, Wasmus S, Savinell RF. J Electrochem Soc 1996;143:1498±504.
[33] Gierke TD. J Electrochem Soc 1977;134:319c.
[34] Kreuer KD. Solid State Ionics 1997;97:1±15.
[35] Sumner JJ, Creger SE, Ma JJ, DesMarteau DD. J Electrochem Soc 1998;145:107±10.
[36] Kreuer KD. Chem Mater 1996;8:610.
[37] Gieselman MB, Reynolds JR. Macromolecules 1992;25:4832±4.
[38] Gieselman MB, Reynolds JR. Macromolecules 1993;26:5633±42.
[39] Gieselman MB, Reynolds JR. Macromolecules 1990;23:3188.
[40] Glipa X, Haddad ME, Jones DJ, RozieÁre J. Solid State Ionics 1997;97:323±31.
[41] Tsuruhara K, Hara K, Kawahara M, Rikukawa M, Sanui K, Ogata N. Electrochim Acta 2000;45:1223±6.
[42] Kawahara M, Rikukawa M, Sanui K. Polym Adv Technol 2000;11:1±5.
[43] Kawahara M, Rikukawa M, Sanui K, Ogata N. Proceedings of the Sixth International Symposium on Polymer Electro-
lytes, Extended Abstracts, 1998. p. 98.
[44] Kawahara M, Rikukawa M, Sanui K, Ogata N. Solid State Ionics, in press.
[45] Yoshida H, Hatakeyama T, Hatakeyama H. Polymer 1990;31:693±8.
[46] Folk M. Can J Chem 1980;58:1495.
[47] Donoso P, Gorecki W, Berthier C, Dfendini F, Poinsignon C, Armand MB. Solid State Ionics 1988;28±30:969±74.
[48] Senadeera GKR, Gareem MA, Skaarup S, West K. Solid State Ionics 1996;85:37±42.
[49] Stevens JR, Wieczorek W, Raducha D, Jeffrey KR. Solid State Ionics 1997;97:347±58.
[50] Wieczorek W, Stevens JR. Polymer 1997;38:2057±65.
[51] Gupta PN, Singh KP. Solid State Ionics 1996;86±88:319±23.
[52] Vargas MA, Vargas RA, Mellander B-E. Electrochim Acta 1999;44:4227±32.
[53] Tanaka R, Yamamoto H, Kawamura S, Iwase T. Electrochim Acta 1995;40:2421±4.
[54] Rodriguez D, Jegat C, Trinquet O, Grondin J, LasseÁgues JC. Solid State Ionics 1993;61:195±202.
[55] Weng D, Wainright JS, Landau U, Savinell RF. J Electrochem Soc 1996;143:1260±3.
[56] Lassegues JC, Desbat B, Trinquet O, Cruege F, Poinsignon C. Solid State Ionics 1988;28±30:969.
[57] Petty-Weeks S, Zupancic JJ, Swedo JR. Solid State Ionics 1988;31:117.
[58] Lassegues JC. In: Colomban P, editor. Proton conductors: solids, membranes and gels. Cambridge, UK: Cambridge
University Press, 1992. p. 311±28.
[59] Bouchet R, Siebert E, Vitter G. J Electrochem Soc 1997;144:L95±7.
[60] Tsuruhara K, Rikukawa M, Sanui K, Ogata N, Nagasaki Y, Kato M. Electrochim Acta 2000;45:1391±4.
[61] Nagasaki Y, Kataoka K. Trends Polym Sci 1996;4:59±64.
[62] Wang J-T, Wasmus S, Savinell RF. J Electrochem Soc 1996;143:1233±9.
[63] Samms SR, Wasmus S, Savinell RF. J Electrochem Soc 1996;143:1225±32.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±1502 1501

[64] Savinell RF, Yeager E, Tryk D, Landau U, Wainright JS, Weng D, Lux K, Litt M, Rogers C. J Electrochem Soc
1994;141:L46±8.
[65] Wainright JS, Wang J-T, Weng D, Savinell RF, Lit M. J Electrochem Soc 1995;142:L121±3.
[66] Kawahara M, Morita J, Rikukawa M, Sanui K, Ogata N. Electrochim Acta 2000;45:1395±8.
[67] Rikukawa M, Morita J, Sanui K, Ogata N. Proceedings of the Fifth International Symposium on Polymer Electrolytes,
Abstracts, 1996. p. 32.
[68] Kerres J, Ullrich A, Meier F, Haring T. Solid State Ionics 1999;125:243±9.
[69] Powers ED, Serad GA. High performance polymers: origin and development. New York: Elsevier, 1986. p. 355.
[70] Daniel MF, Destbat B, Cruege F, Trinquet O, Lassegues JC. Solid State Ionics 1988;28±30:637.
[71] Kreuer K-D, Fuchs A, Maier J. Solid State Ionics 1995;77:157.
[72] Quo Q, Pintauro PN, Tang H, O'Connor S. J Membr Sci 1999;154:175±81.
[73] Saito T. Japanese Patent. H11-3715, 1999.
[74] Gautier-Luneau I, Denoyelle A, Sanchez JY, Poinsignon C. Electrochim Acta 1992;37:1615±8.
[75] Honma I, Takeda Y, Bae JM. Solid State Ionics 1999;120:255±64.
[76] Liu J-C, Kunz HR, Cutlip MB, Fenton JM. Proceeding of the 31st Mid-Atlantic Industrial and Hazardous Waste
Conference, 1999. p. 656±62.
[77] Lefebvre MC, Qi Z, Pickup PG. J Electrochem Soc 1999;146:2054±8.
M. Rikukawa, K. Sanui / Prog. Polym. Sci. 25 (2000) 1463±15021502





























