Embed Size (px)
Citation preview

Proteome wide protein production
Hanna Tegel
Royal Institute of Technology
School of Biotechnology
Stockholm 2013

© Hanna Tegel Stockholm 2013 Royal Institute of Technology School of Biotechnology AlbaNova University Center SE-106 91 Stockholm Sweden Printed by Universitetsservice US-AB Drottning Kristinas väg 53B SE-100 44 Stockholm Sweden ISBN 978-91-7501-913-0 TRITA-BIO Report 2013:17 ISSN 1654-2312
Cover illustration by Maria Stenvall

III ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Hanna Tegel (2013): Proteome wide protein production. School of Biotechnology, Royal Institute of Technology (KTH), Stockholm, Sweden
Abstract
Over a decade after the completion of the human genome, researchers around the world are still wondering what information is hidden in the genome. Although the sequences of all human genes are known, it is still almost impossible to determine much more than the primary protein structure from the coding sequence of a gene. As a result of that, the need for recombinantly produced proteins to study protein structure and function is greater than ever. The main objective of this thesis has been to improve protein production, particularly using Escherichia coli. To improve protein production in Escherichia coli there are a number of different parameters to consider. Two very important parameters in the process of protein production are transcription and translation. To study the influence of differences in transcription rate, target proteins with different characteristics were produced under control of three promoters of different strength (lacUV5, trc and T7). Analyzing the total amount of target protein as well as the amount of soluble protein demonstrated the benefits of using a strong promoter such as T7. However, protein production is also highly dependent on translational efficiency, and a drawback associated with the use of Escherichia coli as host strain is that codons rarely used in this host can have a negative effect on the translation. The influence of using a strain supplied with genes for rare codon tRNAs, such as Rosetta(DE3), instead of the standard host strain BL21(DE3), was therefore evaluated. By using Rosetta(DE3) an improved protein yield for many of the poorly produced proteins was achieved, but more importantly the protein purity was significantly increased for a majority of the proteins. For further understanding of the underlying causes of the positive effects of Rosetta(DE3), the improved purity was thoroughly studied. The cause of this improvement was explained by the fact that Rosetta(DE3) has a significantly better read through of the full sequence during translation and thereby less truncated versions of the full-length protein is formed. Moreover, the effect of supplementation of rare tRNAs was shown to be highly dependent on the target gene sequence. Surprisingly, it was not the total number of rare codons that determined the benefit of using Rosetta(DE3), instead it was shown that rare arginine codons and to some extent also rare codon clusters had a much bigger impact on the final outcome. As a result of the increased interest in large-scale studies in the field of proteomics, the need for high-throughput protein production pipelines is greater than ever. For that purpose, a protein production pipeline that allows handling of nearly 300 different proteins per week was set up within the Swedish Human Protein Atlas project. This was achieved by major and minor changes to the original protocol including protein production, purification and analysis. By using this standard setup almost 300 different proteins can be produced weekly, with an overall success rate of 81%. To further improve the success rate it has been shown that by adding an initial screening step, prior high-throughput protein production, unnecessary protein production can be avoided. A plate based micro-scale screening protocol for parallel production and verification of 96 proteins was developed. In that, protein production was performed using the EnBase® cultivation technology followed by purification based on immobilized metal ion affinity chromatography. The protein products were finally verified using matrix-assisted laser desorption ionization time-of-flight MS. By using this method, proteins that will be poorly produced can be sorted out prior high-throughput protein production.
Keywords: protein production, Escherichia coli, transcription, promoter, translation, rare codon, high-throughput, screening
© Hanna Tegel 2013


V ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
“Ta vara på dagen idag, den kommer aldrig åter”


VII ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Till minne av min älskade mor


IX ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
List of publications
This thesis is based upon the five publications listed below. In the text they are referred to by the corresponding Roman number (I-V).
I. Tegel H, Steen J, Konrad A, Nikdin H, Pettersson K, Stenvall M, Tourle S, Wrethagen U, Xu L, Yderland L, Uhlén M, Hober S, Ottosson J. High-throughput protein production--lessons from scaling up from 10 to 288 recombinant proteins per week. Biotechnol J (2009). 4(1):51-57.
II. Tegel H, Tourle S, Ottosson J, Persson A.
Increased levels of recombinant human proteins with the Escherichia coli strain Rosetta(DE3). Protein Expr Purif (2010). 69(2):159-67.
III. Tegel H, Ottosson J, Hober S.
Enhancing the protein production levels in Escherichia coli with a strong promoter. FEBS J (2011). 278(5):729-39.
IV. Tegel H*, Yderland L*, Boström T, Eriksson C, Ukkonen K, Vasala A, Neubauer P,
Ottosson J, Hober S. Parallel production and verification of protein products using a novel high-throughput screening method. Biotechnol J (2011). 6(8):1018-25.
V. Tegel H, Malm K, Halldin A, Älgenäs C, Hober S, Ottosson Takanen J.
In-depth study of the positive effects of Escherichia coli Rosetta(DE3) on recombinant protein production. Manuscript (2013).
*Authors contributed equally Published articles are reproduced with the kind permission of the respective copyright holders.

X ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Related publications Uhlén M, Björling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergström K, Brumer H, Cerjan D, Ekström M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Björklund MG, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, Olsson I, Oster E, Ottosson J, Paavilainen L, Persson A, Rimini R, Rockberg J, Runeson M, Sivertsson A, Sköllermo A, Steen J, Stenvall M, Sterky F, Strömberg S, Sundberg M, Tegel H, Tourle S, Wahlund E, Waldén A, Wan J, Wernérus H, Westberg J, Wester K, Wrethagen U, Xu LL, Hober S, Pontén F. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics (2005). 4(12):1920-32. Fagerberg L, Oksvold P, Skogs M, Älgenäs C, Lundberg E, Pontén F, Sivertsson A, Odeberg J, Klevebring D, Kampf C, Asplund A, Sjöstedt E, Al-Khalili Szigyarto C, Edqvist PH, Olsson I, Rydberg U, Hudson P, Ottosson Takanen J, Berling H, Björling L, Tegel H, Rockberg J, Nilsson P, Navani S, Jirström K, Mulder J, Schwenk JM, Zwahlen M, Hober S, Forsberg M, von Feilitzen K, Uhlén M. (2012). Contribution of Antibody-based Protein Profiling to the Human Chromosome-centric Proteome Project (C-HPP). J Proteome Res (2013). 12(6):2439-48.

XI ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Contents
INTRODUCTION .................................................................................................................................................. 1
1. PROTEINS ................................................................................................................................................................ 1 2. RECOMBINANT PROTEIN PRODUCTION .......................................................................................................................... 3
2.1 Escherichia coli as host for recombinant protein production ....................................................................... 3 2.2 The expression vector ................................................................................................................................... 5 2.3 Replication ................................................................................................................................................... 6
2.3.1 Plasmid copy number ............................................................................................................................................ 6 2.3.2 Plasmid stability and compatibility ........................................................................................................................ 6 2.3.3 Effects of replication on the recombinant protein production .............................................................................. 7
2.4 Regulation on transcriptional level .............................................................................................................. 7 2.4.1 The most common promoters developed from the E. coli genome ...................................................................... 9 2.4.2 The T7 promoter system ...................................................................................................................................... 11 2.4.3 Less common promoters ..................................................................................................................................... 12 2.4.4 Transcriptional terminators ................................................................................................................................. 12 2.4.5 Comparison of different promoters ..................................................................................................................... 13
2.5 Regulation on translational level ............................................................................................................... 14 2.5.1 Translation initiation and termination ................................................................................................................. 14
2.5.1.1 The SD sequence and the spacing ............................................................................................................... 14 2.5.1.2 The initiation codon and its downstream region ........................................................................................ 15
2.5.2 The effect of differences in codon usage ............................................................................................................. 16 2.5.2.1 How to circumvent problems related to codon biases ............................................................................... 17
3. CULTIVATION TECHNIQUES ........................................................................................................................................ 20 3.1 Batch vs. fed‐batch ..................................................................................................................................... 20
3.1.1 The EnBase® technology ...................................................................................................................................... 21 3.2 Cultivation conditions ................................................................................................................................ 21
3.2.1 Culture media ...................................................................................................................................................... 21 3.2.2 Optimal temperature and pH............................................................................................................................... 22 3.2.3 Induction of protein production .......................................................................................................................... 23
3.3 Automated and multi‐parallel systems for high‐throughput protein production ...................................... 24 4. DOWNSTREAM PROCESSING ...................................................................................................................................... 26
4.1 Protein purification by chromatography .................................................................................................... 26 5. PROTEIN ANALYSIS .................................................................................................................................................. 28
5.1 Mass spectrometry ..................................................................................................................................... 28
PRESENT INVESTIGATION ................................................................................................................................. 31
6. THE EFFECTS OF DIFFERENT PROMOTERS ON THE PROTEIN PRODUCTION ............................................................................. 33 7. INCREASED SUCCESS RATE WITH E. COLI ROSETTA(DE3) ................................................................................................. 38 8. COMBINING DIFFERENT PROMOTERS AND E. COLI STRAINS .............................................................................................. 44 9. DEVELOPING A HIGH‐THROUGHPUT PROTEIN PRODUCTION PIPELINE ................................................................................. 47 10. SCREENING TO AVOID UNNECESSARY PROTEIN PRODUCTION .......................................................................................... 50
CONCLUDING REMARKS ................................................................................................................................... 53
POPULÄRVETENSKAPLIG SAMMANFATTNING ................................................................................................... 55
ACKNOWLEDGEMENTS ..................................................................................................................................... 57
REFERENCES ..................................................................................................................................................... 61


XIII ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
Abbreviations
β-IAA β-indoleacrylic acid cAMP cyclic adenosine monophosphate CAP Catabolite gene activator protein cDNA complementary deoxyribonucleic acid DNA Deoxyribonucleic acid E. coli Escherichia coli eGFP enhanced green fluorescent protein EnBase® Enzyme-based-substrate-delivery ESI Electrospray ionization HIC Hydrophobic interaction chromatography His6 Hexahistidine tag HPA Human protein atlas ICR Ion cyclotron resonance IEXC Ion exchange chromatography IMAC Immobilized metal ion affinity chromatography IPTG Isopropyl-β-D-thiogalactopyranoside IT Ion trap LB Luria-Bertani broth LEX Large-scale expression system MALDI Matrix-assisted laser desorption ionization mRNA messenger ribonucleic acid MS Mass spectrometry ori origin of replication PCR Polymerase chain reaction PTM Post-translational modification Q Quadrupole RBS Ribosomal binding site RNA Ribonucleic acid RPC Reversed-phase chromatography SB Super broth SD Shine-Dalgarno SDS-PAGE Sodium dodecyl sulfate - polyacrylamide gel electrophoresis SEC Size exclusion chromatography SOB Super optimal broth SOC Super optimal broth with catabolite repression SPA Staphylococcus aureus protein A TB Terrific broth TOF Time-of-flight tRNA transfer ribonucleic acid TSB Tryptic soy broth


HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1
Introduction
1. Proteins
Proteins are the most abundant molecules in biology after water, and due to their vital functions (e.g.
signaling, transport, enzymatic reactions and immune defense) they are often called the building
blocks of life [1, 2]. When a protein is produced, the genetic code (DNA), which is copied via
replication, is transcribed into mRNA followed by translation of mRNA into a chain of amino acids.
A process called the Central Dogma [3]. However, the resulting amino acid chain, called the primary
protein structure, needs to be folded into an organized three-dimensional structure to become a
functional protein. In a functional protein one generally refers to three different levels of protein
structure (Figure 1). Locally folded structures are termed secondary structures and the two major
secondary structures are called α-helix and β-strand. When secondary structures are packed together
to a domain, the tertiary structure is formed. A protein can also consist of several folded amino acid
chains, called the quaternary structure [2].
Since the sequencing effort of the human genome was completed, the sequence and chromosomal
localization of all human genes are known [4, 5]. However, one remaining question is; what
information is hidden in the genome? By knowing the coding sequence of a gene it is possible to
predict the primary protein structure. But, it is not possible to determine the three-dimensional
structure of the protein due to the complexity of combining 20 different amino acids. In other
words, based on today’s knowledge, the sequence itself says very little about the function. Therefore,
it has become of great importance to systematically study structure and function of proteins,
especially human proteins. This can be done by using different approaches for large-scale
proteomics studies, for example by mass spectrometry (MS)- and antibody-based proteomics as well
as structure determination [6-8]. However, one prerequisite for the latter two approaches is the
availability of the protein of interest and consequently there has been an increased interest in
recombinant protein production. So far, Escherichia coli (E. coli) is the dominating host for that
purpose. If the produced protein is going to be used for structural determination, correctly folded
protein is also a necessity. This could sometimes be a problem when producing recombinant
proteins in bacteria, since the produced proteins easily aggregate and form inclusion bodies. On the

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
2
other hand, if loss of function during inclusion body formation is not a problem or the protein is
easily refolded in vitro, the relatively high purity of the aggregated proteins can instead be highly
beneficial [9]. In summary, the optimal setup for successful recombinant protein production is
highly dependent on the use of the protein.
Figure 1. The levels of protein structure. (Illustration by Maria Stenvall)

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
3
2. Recombinant protein production
Isolation of a protein of interest from its natural source generates in general a very small amount of
protein. One way to circumvent this is by recombinant protein production. Genomic DNA or
cDNA encoding the gene of interest is inserted into a plasmid, a circular molecule of DNA that can
be introduced into host cells. Protein production of the target gene can then be induced, resulting in
large amounts of the target protein. The first successfully produced human protein using this
approach, was the growth hormone somatostatin that was produced in E. coli [10]. Since this first
successful production of a human recombinant protein in 1977, groundbreaking developments have
occurred in the field of recombinant protein production which has enabled a wide range of studies
of proteins. Apart from E. coli there are other bacterial expression systems for protein production as
well as systems based on yeast, insect cells, mammalian cells and also cell-free systems [11-14].
However, E. coli is so far the most commonly used organism for heterologous protein production.
2.1 Escherichia coli as host for recombinant protein production
E. coli was first discovered in 1885 by the German pediatrician Theodor Escherich. Thanks to this
bacterium, the fantastic progress in the field of recombinant protein production during the 20th
century has been possible. E. coli is a rod-shaped, gram-negative, facultative anaerobic bacterium that
is approximately 2 µm long. The benefits of using E. coli for protein production are the large
knowledge available on culture conditions required for optimal growth and that it has an easily
modified genome. This has enabled efficient protein expression that gives high yields at low cost.
On the other hand, disadvantages are that it is difficult to express proteins with disulfide bonds or
post-translational modifications, such as glycosylations, and that inclusion bodies may form when
producing heterologous proteins or proteins at very high concentrations [15]. The inherent
production of endotoxins might also be a problem when producing proteins for medical purposes.
The two most frequently used E. coli strains for routine protein production are BL21 and K-12. To
improve quality and efficiency of producing large amounts of a protein in a foreign host these
strains have been modified in various ways, resulting in several useful derivatives of this important
bacteria [13]. Examples of useful strains are listed in Table 1.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
4
Table 1. Examples of E. coli strains commonly used for recombinant protein production.
Host strain Origin Characteristics
B834 B Protease-deficient, methionine auxotroph
BL21 B834 Deficient in both lon and ompT proteases
Rosetta BL21 Supply tRNAs for codons rarely used in E. coli to enhance the production
of eukaryotic proteins. Deficient in both lon and ompT proteases
BL21 CodonPlus BL21 Supply tRNAs for codons rarely used in E. coli to enhance the production
of eukaryotic proteins. Deficient in both lon and ompT proteases
Origami K-12 trxB and gor mutant, resulting in enhance disulfide bond formation in
the cytoplasm
Origami B BL21 trxB and gor mutant, resulting in enhance disulfide bond formation
in the cytoplasm. Deficient in both lon and ompT proteases
Rosetta gami Origami Combines the features Origami and Rosetta to enhance the production
of eukaryotic proteins as well as disulfide bond formation in cytoplasm
Rosetta gami B Origami B Combines the features Origami B and Rosetta to enhance the production
of eukaryotic proteins as well as disulfide bond formation in cytoplasm.
Deficient in both lon and ompT proteases
BL21 Star BL21 rne mutant, improves the stability of mRNA resulting in increased
protein production. Deficient in both lon and ompT proteases
C41 BL21 Mutated to tolerate production of toxic and membrane proteins.
Deficient in both lon and ompT proteases
C43 C41 Mutated to tolerate production of toxic and membrane proteins.
Deficient in both lon and ompT proteases
To support the T7 promoter system most of these strains are also available as DE3 and pLysS strains. The DE3
lysogen expresses T7 RNA polymerase upon induction while the pLysS plasmid produces T7 lysozyme that reduces the
basal level expression of the gene of interest.
Recombinant protein production in E. coli is influenced by many factors. The processes of
replication, transcription and translation, as well as the stability of the expression vector and mRNA
are all parameters to take into account when choosing an expression system. Furthermore, culture
conditions, proteolytic stability, protein folding and localization need to be considered for optimal
protein production [16, 17]. In other words, to find the ideal conditions for recombinant protein
production in E. coli is not a trivial task, especially if the goal is to find one set of conditions that
works well for large sample sets. However, as a result of the wide range of expression vectors and
strains nowadays available, the chances of designing a suitable protein expression protocol in E. coli
have significantly increased over the last decades [18]. To further improve the production of soluble
protein it is also possible to use fusion tags and chaperones [19-22].

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
5
2.2 The expression vector
An expression vector is a plasmid designed for protein expression of a specific gene in a host cell.
When designing a new expression vector, there are some essential genetic elements needed in the
vector to be able to produce a protein of interest. By careful consideration of the combination of
these elements, a high protein production can often be achieved. In Figure 2 a graphic presentation
of a typical E. coli expression vector is shown. A more detailed description of these elements will be
presented in the following sections.
Figure 2. Schematic presentation of an expression vector and the process of protein production in E. coli. The
origin of replication (ori) is the element that sets the number of plasmids available for transcription. Transcription is
initiated when RNA polymerase binds to the promoter (P). P is a DNA sequence located approximately 10 bp to 100 bp
upstream of the ribosomal binding site (RBS). Promoters from E. coli normally consists of two hexanucleotide
sequences, one located approximately 35 bp upstream (-35) and another located approximately 10 bp upstream (-10) of
the transcription initiation base, TTGACA and TATAAT, respectively [23-25]. These two hexamers are in general
separated by a promoter-spacer with a length of 16 to 18 bp and the optimal promoter-spacer length is most often 17 bp
[24, 26]. The activity of a promoter could be modulated by a regulatory protein. If so, the regulatory gene (R), encoding
for such a regulatory protein, may be present on the vector itself or integrated into the host chromosome. The process
of transcription is finally terminated by a transcription terminator (TT). When translation is initiated the ribosome will
form a complex with the mRNA. The Shine-Dalgarno (SD) sequence will then interact with the 3’-end of the 16S rRNA
of the 30S ribosome [27]. Translation will then start from the initiation codon (IC, +1) and be stopped by the translation
terminator (tt). Features important for an efficient translation initiation is the distance between the SD site and the
translation initiation codon (IC, +1), the spacer, as well as possible secondary structure formations in the mRNA of this
region [28, 29]. Apart from the elements directly involved in the protein production, antibiotic resistance (AR) is used to
ensure that only cells harboring the vector will grow under a selective pressure. (Illustration by Maria Stenvall)

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
6
2.3 Replication
The process of replication is affected by a number of different parameters. Plasmid copy number,
stability and compatibility are all parameters that need to be considered to ensure a successful
protein production.
2.3.1 Plasmid copy number
One parameter affecting replication and thereby the protein production is the plasmid copy number.
The plasmid copy number is determined by the origin of replication (ori), and the plasmid is
preferably replicated in a relaxed fashion, i.e. independently of replication of the host chromosome.
The two most commonly used origins of replication in recombinant protein production are derived
from two naturally occurring plasmids, isolated from E. coli; pMB1 and p15A [30, 31]. Examples of
frequently used expression vectors with pMB1 ori are pBR322 and its derivatives [32, 33]. The pMB1
ori is also used in a modified form in the pUC family of plasmids that is also very often used [34, 35].
Concerning the p15A ori it is used in the pACYC plasmids [36]. The main difference between these
different origins of replication is the copy number. The pACYC, pBR322 and pUC plasmids are
therefore classified as low-copy, medium-copy and high-copy, respectively [37].
The replication machinery of the commonly used ColE1-type plasmids, like pMB1 and p15A, has
been well studied in vitro [38, 39]. What has been concluded is that two RNA molecules encoded by
the plasmid itself, RNAI and RNAII, controls replication of plasmids with these oris, and only
proteins originating from its E. coli host are required for the replication process [40]. A majority of
these plasmids also code for an additional negative control element that stabilizes the interaction
between RNAI and RNAII [41, 42]. By excluding this regulatory region the copy-number of the
ColE1-type plasmids increases [43, 44].
2.3.2 Plasmid stability and compatibility
Apart from the plasmid copy number, structural plasmid stability and segregational plasmid stability
also affect the productivity of a plasmid [45]. A plasmid is called structurally stable when all
generated plasmids have the correct sequence, and segregationally stable when all daughter cells get
at least one plasmid during cell division. Compatibility is also something to take into consideration

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
7
when choosing origins of replication. Theoretically, two different plasmids with the same replication
machinery are incompatible, and therefore they cannot be stably maintained in the same cell [46].
However, lately Velappan and colleagues have shown that under certain conditions such as selective
antibiotic pressure, plasmids containing the same origin of replication are able to be maintained in
the bacteria for a long time, although they belong to the same incompatibility group [47].
2.3.3 Effects of replication on the recombinant protein production
Plasmid copy number and stability, strongly influence the quality and productivity of an expression
system. However, the mechanisms behind these parameters are very complex and so far no general
model has been created. In theory, a plasmid with higher copy number should via a higher number
of mRNA molecules result in a larger amount of recombinantly produced protein. This is supported
by many reports. For example Choi et al report that cells with less than 84 plasmid copies per cell do
not produce detectable levels of bovine growth hormone, but when the plasmid copy number is
increased to about 300 copies per cell, the production increases up to 12.3% of total cell protein
[48]. However, in some cases the plasmid copy number is not the limiting factor but other
parameters affecting protein yield are and then it could actually be beneficial to lower the copy
number. For example, Ramos and colleagues show that the amount of recombinant protein of the
tetanus toxin fragment C does not increase when a higher copy number is used [49].
A drawback of high-copy plasmids is the high metabolic burden they impose on the host. Metabolic
burden is defined as the part of a host cell’s resources, in form of energy and raw material, needed to
maintain and express the foreign DNA in the cell [50]. Production, including replication,
transcription and translation, of a plasmid-encoded protein reduces the growth rate of the host as a
consequence of this additional metabolic burden [51]. Therefore, cells carrying a high-copy plasmid
often display a larger decrease in growth rate after induction compared to cells carrying low-copy
plasmids [52].
2.4 Regulation on transcriptional level
Transcription of a gene of interest is one of the key steps in protein production. This step is initiated
by the binding of RNA polymerase to the promoter sequence. For gene expression in E. coli there

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
8
are many different promoters available (Table 2) and the choice of promoter has great effects on the
protein yield. If a large amount of produced protein per cell is the goal there are some characteristics
to consider. First, a general recommendation regarding the promoter is to use a strong promoter,
allowing accumulation of target protein to approximately 10 to 30% or more of the total cellular
protein [17]. The strength of different promoters is determined by the relative frequency of
transcription initiation, which is mainly affected by the affinity of the RNA polymerase for the
promoter sequence. A tight regulation, resulting in a low level of basal transcription, is another
important parameter to consider when choosing promoter. By choosing a tightly regulated
promoter, the metabolic burden and potential toxic effects on the host cell may be minimized. An
additional key feature of the promoter is inducibility, i.e. the possibility to induce transcription.
Preferably the induction procedure should be simple and inexpensive, and for that reason chemical
and thermal inductions are the two most commonly used methods.
Table 2. A selection of promoters commonly used for recombinant protein production in E. coli.
Promoter Origin Inductiona Reference(s)
lac
E. coli IPTG [53]
lacUV5 E. coli IPTG [54]
trp E. coli Trp starvation/β-IAAb [55]
tac E. coli IPTG [56]
trc E. coli IPTG [57]
araBAD E. coli L-arabinose [58]
T7 Bacteriophage T7 IPTG [59]
T7lac Bacteriophage T7 IPTG [60]
tetA E. coli Anhydrotetracycline [61]
Pm Pseudomonas putida m-toluate [62]
phoA E. coli Phosphate starvation [63-65]
pL Bacteriophage λ Thermal [66]
pR Bacteriophage λ Thermal [67-69]
lac(TS) E. coli Thermal [70]
cspA E. coli Thermal [71]
PSPA Staphylococcus aureus Constitutive [72]
aMost commonly used
bβ-indoleacrylic acid

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
9
2.4.1 The most common promoters developed from the E. coli genome
Promoters originating from native E. coli genes, so called E. coli promoters, are commonly used to
regulate transcription in E. coli. Transcription from these promoters is initiated by the binding of
E. coli RNA polymerase to the promoter region. E. coli RNA polymerase consists of five subunits.
One of the subunits, known as the sigma factor, is involved in the promoter recognition. Since
different E. coli promoters have different sequences in the –10 and –35 regions they are recognized
by different sigma factors and the most commonly used is σ70. Apart from the -10 and -35 region
there are other regions of the promoter, such as the ‘extended –10 element’ [73] and the
‘UP-element’ [74, 75], located immediately upstream of the –35 region, that also can affect the
transcription initiation efficiency through interactions with the RNA polymerase.
The lac operon has for many years served as a paradigm for transcription regulation [76]. Therefore,
many E. coli promoter systems, used for heterologous protein production, are based on its regulatory
elements (e.g. lac, lacUV5, tac and trc). Transcription from these promoters is induced by addition of
the lactose analogue isopropyl-β-D-thiogalactopyranoside (IPTG). The benefit of using IPTG
instead of lactose is that it cannot be metabolized by E. coli and therefore a strong induction is
possible. IPTG-inducible promoters are often used in laboratory-scale. However, since IPTG is
toxic to humans and it is a rather expensive chemical, IPTG-inducible promoters are not ideal for
production of human therapeutic proteins in large culture volumes [17].
The lac promoter and its modified form lacUV5, are examples of two rather weak promoters [53,
54]. Transcription from these IPTG-inducible promoters is regulated by the lac repressor, which
inhibits the binding between the polymerase and the promoter sequence. At induction, IPTG will
bind to the lac repressor and thereby hinder its binding to the operon. This will allow the E. coli
RNA polymerase to initiate transcription. Transcription can be further stimulated by the catabolite
gene activator protein (CAP) [77, 78]. When cyclic AMP (cAMP), whose concentration is high in
absence of glucose, binds to CAP, the CAP-cAMP complex is able to bind to the CAP binding site.
This action will stabilize the binding of E. coli RNA polymerase to the promoter and thereby
increase the transcription rate. The lac and the lacUV5 promoters are very similar. The only
difference is two mutations in the –10 region and one mutation in the gene of the CAP binding site.
As a result of this, the promoter becomes relatively insensitive to cAMP stimulation, and thereby the
promoter strength is increased compared to the wild type lac promoter. However, the lacUV5

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
10
promoter is still rather weak, and therefore, like the lac promoter, rarely used for high-level
production of recombinant protein.
For production in large culture volumes the trp promoter is commonly used [55]. This promoter
originates from the trp operon of E. coli that in contrast to the lac operon is negatively regulated [79,
80]. In other words, transcription will be turned off in the presence of tryptophan and turned on by
tryptophan starvation or by addition of β-indoleacrylic acid (β-IAA). When comparing promoters,
the trp promoter is stronger than the lac promoters and could result in protein accumulation of up to
30% of the total cell protein, but a potential problem of the trp promoter is that it is difficult to
completely down-regulate under non-induced conditions. Therefore, leakage will result in
production of the target protein all through cultivation, also prior to induction. Further
developments of the trp promoter are the two commonly used IPTG-inducible E. coli promoters tac
and trc [56, 57]. These synthetic promoters consist of the –35 region from the trp promoter and the
–10 region from the lacUV5 promoter. The only difference between tac and trc is 1 bp in the length
of the promoter-spacer. The tac and trc promoters are both defined as rather strong promoters and
allow up to 15-30% accumulation of total cell protein [13]. When the tac promoter was compared
with the lacUV5 promoter it was at least five times more efficient [81]. A disadvantage of the tac and
trc promoters is the high basal production level and therefore they are not suitable if the target
protein is toxic to the cell [13]. However, for non-toxic proteins these promoters could be very
useful due to their high-level expression.
Another useful alternative to the IPTG-inducible E. coli promoters is the L-arabinose inducible
araBAD promoter of the arabinose operon [58]. This system is both positively and negatively
regulated by the product of the araC gene. The major benefits of the araBAD promoter are the
inexpensive induction with L-arabinose, the very tight regulation and a linear relationship between
inducer concentration and protein expression. Thanks to the tight regulation and dose-dependent
induction it is possible to achieve a very controlled protein expression. Thereby, this promoter
system is suitable for expression of toxic proteins and proteins that easily form inclusion bodies at
high expression levels [58]. However, it has been shown that to ensure a homogeneous expression in
all cells, a modified strain is needed [82].

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
11
2.4.2 The T7 promoter system
The T7 promoter system, derived from bacteriophage T7, is the most frequently used promoter for
protein production in laboratory-scale today [59]. Transcription from this promoter is, in contrast to
the E. coli promoters, initiated by the T7 RNA polymerase. This polymerase is a single polypeptide
chain with a molecular weight of 99 kDa that requires a specific promoter sequence for efficient
transcription [83, 84]. The gene encoding the T7 RNA polymerase is incorporated into the E. coli
genome and under control of a lacUV5 promoter. Hosts with this chromosomal insert are so called
DE3 lysogens. Addition of IPTG will induce production of T7 RNA polymerase that will be able to
bind to the T7 promoter on the plasmid and initiate transcription of the target gene. As a result of
this two-step process, the T7 promoter system in E. coli is tightly regulated. By adding a lac operator
immediately downstream of the T7 promoter region, a T7lac promoter, that will further tighten the
regulation of the T7 system, is created [60].
Even though the T7 system is a tightly regulated promoter system, there might still be some basal
transcription. By using strains containing the pLysS or pLysE vectors this can be reduced, since
these vectors express T7 lysozyme that degrades T7 RNA polymerase [85]. However, a drawback of
the T7 lysozyme is reduced transcription, also under induced conditions, resulting in lower yields
[86]. Another approach to lower the basal level of T7 RNA polymerase is by addition of 0.5-1.0%
glucose to the medium. This will result in reduced cAMP levels and as a result of that a reduced
activation of the lac operon, and thereby the unwanted production of T7 RNA polymerase before
the induction phase will decrease [87].
When comparing the promoter strength of the E. coli promoters with the T7 promoter, it is clear
that T7 is a very strong promoter. The explanation to this is that T7 RNA polymerase is a very
selective and efficient polymerase, resulting in a high frequency of transcription initiation as well as
efficient elongation. By using the T7 RNA polymerase, a five-fold faster RNA elongation is achieved
compared to what is achieved by the E. coli RNA polymerase [88]. The T7 promoter could therefore
result in an accumulation of the desired protein as high as 40-50% of the total protein content in the
cell [59]. However, this is not always optimal, neither for the host cell nor for the translation
process.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
12
2.4.3 Less common promoters
Apart from the previously described lac, trp and araBAD promoters there are other chemically
induced E. coli promoter systems. One example is tetA that is induced by addition of
anhydrotetracycline at a low concentration [61]. Another chemically induced promoter is Pm [62].
This promoter originates from Pseudomonas putida and it is induced by addition of m-toluate. For
production in large culture volumes, however, it is preferable to avoid addition of extra chemicals.
Therefore, phoA is often used for this purpose since it is induced by phosphate limitation [63-65].
Another alternative, which also avoids addition of extra chemicals, is thermally induced promoters.
These promoters are very simple and cost-effective alternatives to chemically induced promoters.
Examples of well-known thermally inducible promoters are the strong bacteriophage λ promoters pL
[66] and pR [67-69], the thermo sensitive lac promoter lac(TS) [70], and the cspA promoter [71]. The
latter two both originate from E. coli. The promoters pL, pR and lac(TS) are all induced by an
increased temperature, while cspA is induced by a thermal downshift. A drawback of an increased
temperature during protein production is that it could cause formation of inclusion bodies as well as
production of heat-shock proteins, including certain proteases. The phage promoter pL, for example,
is normally induced by increasing the temperature from 30°C to 42°C, a temperature at which the
cI857 repressor is inactive. The cspA promoter, on the other hand, is maximally induced by a
thermal downshift below 25°C [89]. Expression at low temperature could be beneficial for the
soluble expression of proteins prone to form aggregates [90]. Other cultivation conditions used for
induction are, for example, pH and ionic concentrations [91]. Finally, there is a group of constitutive
promoters used for recombinant protein production, such as the Staphylococcus aureus Protein A
(SPA) promoter (PSPA) [72]. This group of promoters is, in contrast to the inducible promoters,
constantly active.
2.4.4 Transcriptional terminators
To ensure an efficient transcription, initiation as well as termination is very important. A
transcription terminator should therefore be placed downstream of the coding sequence. By adding
a transcription terminator, unnecessary transcription can be prevented [17]. There are two types of
bacterial terminators; intrinsic and factor dependent. The intrinsic terminators are the ones used in
recombinant protein production. By using this terminator, a stem-loop structure that will cause
dissociation of the polymerase, is formed in the newly transcribed mRNA. Stem-loops can also

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
13
enhance the mRNA stability in the cases where stem-loop structures are formed at the 3’-end of the
mRNA molecule [92].
2.4.5 Comparison of different promoters
All in all there are several different expression systems using different promoters for recombinant
protein production. The optimal choice of promoter is highly dependent on the target protein and
the final application. If a large total amount of protein is important, a tightly regulated, inducible,
strong promoter is recommended. On the other hand, if the amount of soluble protein is of high
importance, a weaker promoter or a promoter activated by temperature downshift may sometimes
be more useful. However, a number of exceptions to any such generalization made can be found in
the literature. These exceptions stress the importance of experimental data as ground for the
promoter selection. This will be further discussed below.
In a study by Schultz et al, a set of expression vectors with different promoters, inducing agents and
plasmid copy number, was evaluated to try to identify if there is a single parameter controlling
protein solubility [93]. In their study, the highest yield of soluble protein correlates with the lowest
aggregation. What they also observe is that, even though no single vector feature alone appears to be
responsible for the solubility, the total aggregation tends to increase when the combination of the
elements involved in the expression regulation allows a higher expression rate. They also conclude
that an increased aggregation of protein does not automatically result in a low yield of soluble
protein.
In another study, the expression of five different proteins under control of five different promoters
was compared [94]. The promoters studied were T7lac, trc, araBAD, Pm and Pm modified by three
point mutations. It was concluded that if the amount of mRNA is a bottleneck, it is beneficial to use
the T7lac promoter. However, a large fraction of the produced proteins turned out to be insoluble.
Therefore, if it is soluble protein that is desired, the modified Pm could be used instead of T7lac. It
was also concluded that due to high basal transcription in combination with comparatively low
protein production, trc is the least useful promoter. The promoter that had the tightest regulation
was araBAD, which makes this promoter well suited for production of toxic proteins or metabolic

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
14
engineering [95]. In other words, more systematic studies to identify the optimal promoter could be
very useful.
2.5 Regulation on translational level
The process that follows transcription is called translation. During this process, mRNA is translated
into amino acids and a protein is formed. Translation initiation and termination as well as elongation
of the amino acid chain are all important steps in the regulation of protein production.
2.5.1 Translation initiation and termination
Compared to transcription that is controlled by promoters of specific sequences, there is no unique
sequence for efficient translation initiation and still today the mechanism behind translation
initiation is not fully understood [96, 97]. When protein synthesis is initiated, base-paring between
the purine-rich SD sequence of the mRNA and the complementary sequence in the 3’-end of the
16S rRNA (anti-SD sequence) of the 30S ribosome occurs [27]. The efficiency of translation
initiation is influenced by several features, such as the length of the SD sequence, the distance
between SD and the initiation codon, the initiation codon itself, the region downstream of the
initiation codon and mRNA secondary structures formed in the translational initiation region. To
terminate translation of the mRNA in E. coli, a stop codon is needed. Stop codons used in E. coli are
UAA, UGA and UAG. By adding a fourth nucleotide to the commonly used UAA; UAAU, the
translational termination efficiency will improve [98].
2.5.1.1 The SD sequence and the spacing
It has recently been demonstrated that the SD interaction may not be essential for the translation
initiation to occur [99, 100]. To study the relationship between protein production and the presence
of an SD sequence, Ma et al analyzed 30 complete prokaryotic genomes [101]. What they showed
was that a gene predicted to be highly expressed is more likely to have a strong SD sequence. They
also showed that the majority of the highly expressed genes in the E. coli strain K-12 have an SD
sequence that harbor the core motif GGAG or GAGG. However, according to their results the SD
sequence is not mandatory for protein production.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
15
The SD sequence is a sequence in the mRNA consisting of three to nine adjacent bases that can
base-pair to some or all of the bases in the 3’-end of the 16S rRNA (ACCUCCUUA (anti-SD
sequence)). The general opinion is that a short SD sequence, preferably complementary to the anti-
SD sequence CCUCCU, is ideal if high translation efficiency is the goal [102-105]. An explanation
to this is most likely that longer SD sequences will cause ribosome stalling at the initiation site as a
result of too strong interaction between the SD and anti-SD [103]. However, despite the general
opinion that a shorter SD sequence is preferred, Ringquist and co-workers conclude that the longer
SD sequence in their study initiated translation several folds more efficiently and that this is
independent of the length of the spacing between the SD site and the initiation codon [106].
The length of the AU-rich spacer between the SD sequence and the initiation codon is another
feature affecting the translation initiation. To determine the optimal length of the spacer, several
studies have been performed. According to the previously mentioned study by Ringquist et al, each
SD sequence has its own optimal spacer length as well as a minimum spacing required for
translation [106]. In general, the spacing has been defined as the number of nucleotides separating
the SD sequence and the initiation codon. However, the definition “aligned spacing”, which is the
number of nucleotides separating a reference nucleotide in the complete SD sequence from the
initiation codon, is nowadays often used. Chen et al have shown the benefit of using this definition
[29]. By studying the efficiency of two equally long SD sequences corresponding to different
subsequences of the anti-SD, they conclude that the optimal spacing is different, but the optimal
aligned spacing is the same for the two SD sequences. When Ma and colleagues did a comparative
analysis of the distance between the SD sequence and the initiation codon of highly expresses genes
in E. coli K-12, their results agreed with the findings of Chen and coworkers [101]. In other words, if
U in the core anti-SD motif CCUCC is used as reference base, it is recommended to use an aligned
spacing of eight nucleotides.
2.5.1.2 The initiation codon and its downstream region
In E. coli, the most commonly used initiation codon is AUG [107]. As many as 91% of all sequenced
E. coli genes have shown to contain this initiation codon, while 8% contains GUG and only
approximately 1% contains the rarely used UUG [17, 108]. AUG is not only the most frequently
used; it has also been reported to be significantly more efficient than GUG and UUG. However, the
efficiency of the latter two can be increased by changing the codon that follows the initiation codon

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
16
(+2) [106, 109]. Nevertheless, the AUG initiation codon is required for translation of mRNA
sequences lacking a sequence involved in ribosome binding in E. coli [110]. In other words, with
AUG at +1 it is possible to initiate translation in the absence of an SD sequence, although at lower
efficiency.
The region downstream of the initiation codon has, independent of base-pairing between mRNA
and 16S rRNA, shown to also influence the translation initiation. Particularly has the effect of the
codon following the initiation codon (+2) been very well studied. A general conclusion is that this
position is highly important for an efficient translation initiation and in general a high adenine
content of the +2 codon, preferably AAA, results in a high protein synthesis rate [111-114]. Apart
from +2, positions +3 to +7 has also been studied [115, 116]. Depending on the sequence in this
region the protein production could either increase or decrease. For example, AU-rich codons are
positive for the protein production [117] while NGG codons should be avoided [115]. The benefits
of engineering the downstream region has been shown by several groups [118-121].
2.5.2 The effect of differences in codon usage
Apart from initiation and termination of the translation process, protein synthesis is also highly
dependent on elongation of the amino acid chain. One very important parameter for efficient
elongation is codon usage, since different organisms use different codons more or less often. In
other words, a codon frequently used by one organism could be rarely used by another, and this may
cause problems when working with heterologous protein production [122, 123]. Table 3 presents a
comparison of usage frequencies in E. coli B, K-12 and Homo sapiens of codons rarely used in E. coli.
As can be seen in the table, a codon rarely used in E. coli is not necessarily a rare codon in humans.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
17
Table 3. Codon usage in E. coli B, E. coli K-12 and Homo sapiens (data collected from
http://www.kazusa.or.jp/codon/). The codons summarized in this table are compensated for in different commercially
available E. coli strains since they are rarely used in E. coli [124].
A target gene with high frequency or clusters of codons rarely used by the host could sometimes
cause misincorporation of amino acids [125, 126], premature translation termination [127],
translational frameshifting [126, 128] or in-frame translational hop [129]. These actions can prevent
a high protein yield or reduce the quality of the product [123]. It has also been shown that the 5’-end
of a transcript is extra sensitive to the presence of rare codons or codon clusters, especially of the
rare arginine codons AGA and AGG [123, 130-132]. Rare codons in this region may thus affect the
translation efficiency to a great extent.
2.5.2.1 How to circumvent problems related to codon biases
To solve problems related to rare codons as well as differences in codon usage there are two main
strategies. Either the gene sequence could be optimized for its host or, as an alternative; the rare
tRNAs could be co-expressed in the host.
If synthetic genes are used to overcome problems related to differences in codon usage between
organisms, computer based codon optimization is needed [133]. When using this approach the
sequence of the target gene is optimized without altering the amino acid sequence. The synthetic
gene is finally used for recombinant protein production. Today, there are several different codon
usage analysis and optimization tools publically available [134]. The two most commonly used
methods for codon optimization; “one amino acid–one codon” and “codon randomization”, have
Frequency1 Additional tRNA genes in different E. coli strains Codon E. coli B E. coli K-12 Homo sapiens Rosetta Rosetta 2 BL21 CodonPlus-RP BL21 CodonPlus-RIL BL21 CodonPlus-RIPL
AGG 2,1 1,6 12,0 x x x x x
AGA 2,4 1,4 12,2 x x x x x
CCC 2,4 6,4 19,8 x x x x
CUA 3,4 5,3 7,2 x x x x
CGG 5,0 4,1 11,4 x
AUA 5,0 3,7 7,5 x x x x
GGA 8,2 9,2 16,5 x x
1Frequency per thousand.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
18
been compared by Menzella [135]. In the first method, the most abundant codon for a specific
amino acid is used every time that amino acid appears in the target sequence. In the second method
the codons are weighted, based on how frequently the codons are used, and then randomly
distributed in the synthetic gene. The synthetic genes were then produced in E. coli, resulting in
significantly more protein when the codon randomized method was used compared to the native
ones. However, the one amino acid–one codon approach did not show significant improvement. A
likely explanation to this is that the availability of specific tRNAs is limiting the translation when
using the one amino acid-one codon method. Another benefit of using the randomization method is
flexible codon selection, which makes it possible to e.g. avoid repetitive elements, mRNA secondary
structures and restriction sites [136].
The second approach to circumvent problems caused by rare codons is by co-expression of genes
encoding rare tRNAs [137]. For this purpose, either plasmids containing genes for several rare
codon tRNAs or commercially available expression strains overexpressing rare tRNAs (e.g. the
RosettaTM strains (Novagen) and BL21-CodonPlus® strains (Stratagene)) could be used [138], Table
3. Several research groups have shown the benefits of using a host compensating for rare codons
[139-141]. An increased yield is in general the main advantage, but it has also been reported that by
using this approach it is possible to avoid protein truncations and thereby a highly pure homogenous
full-length protein sample is achieved [142].
In a comprehensive study by Maertens and colleagues, the protein production of wild type as well as
codon optimized human genes in E. coli, was compared [143]. The original genes were produced in
E. coli strains which co-express certain rare tRNAs, while the sequence-optimized analogs were
produced in E. coli BL21(DE3) without additional tRNAs. Using codon optimized human genes was
shown to be significantly better than strains compensating for differences in codon usage. The
benefit of using codon optimization instead of a tRNA-supplemented bacterial strain, regarding
protein yield, has also been shown by several other groups [144-146]. Possible explanations to this
are inefficient supply of the rare tRNAs or the additional metabolic burden caused by the extra
plasmid encoding rare tRNAs [146]. However, Burgess-Brown and co-workers conclude that for the
majority of the proteins in their test set, sequence optimization as well as supplementation of rare
tRNAs have a positive effect on heterologous protein expression [139].

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
19
In other words, there are several different parameters affecting recombinant protein production in
E. coli. And to find the optimal design of the expression vector for successful production of a
protein of interest, a combination of theory and experimental data is needed. However, for high-
throughput protein production pipelines it is not possible to identify the optimal design of the
expression vector for each protein. Instead, it is important to design a vector that works for the
majority of all proteins.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
20
3. Cultivation techniques
Depending on what application the target protein will be used for, there are different possible
cultivation strategies. Cell cultivations for industrial production of a protein, for example in the
biopharmaceutical industry, are in general performed in a large reactor, while high-throughput
protein production or screening for research purpose is generally performed in small batches run in
parallel. Cultivation of cells can be performed in different modes and the three main modes of
operation are batch, fed-batch and continuous fermentation [147]. In batch cultivations all nutrients
required for cell growth are added at the start whereas in a fed-batch one or more nutrients are
supplied to the fermenter during the process to control growth [148, 149]. The final product will be
harvested at the end of the run in batch as well as fed-batch fermentation. During the third
commonly used mode of operation, the continuous process, all nutrients are constantly supplied to
the fermenter. In contrast to the two other modes, the culture medium is removed at the same flow
rate as nutrients are added and thereby the culture volume is constant in a continuous bioprocess
[149]. The optimal choice of bioprocess depends on the application the target protein will be used
for as well as the producing organism; bacteria, yeast, insect cells or mammalian cells. However,
most industrial processes are fed-batch processes. In the following sections, laboratory-scale
cultivation with E. coli as the protein-producing organism will be presented.
3.1 Batch vs. fed‐batch
In laboratory-scale, batch cultures in shake flasks are commonly used due to simple handling and
low cost. However, batch cultivation in shake flasks will unavoidably suffer from oxygen limitation,
low pH and overflow metabolism that all will have a negative effect on cell growth and protein yield
[150-152]. Overflow metabolism is a metabolic phenomenon that occurs under aerobic conditions
when there is an excess of the carbon source. The rate of glycolysis will then exceed a critical value,
resulting in production of acetate as a by-product. To solve this problem a fed-batch culture, which
supplies the growth-limiting carbon source in a controlled manner, could be used. This will prevent
overflow metabolism and thereby acetate formation, and as a result of this the cell growth and
product yield will increase [153-155]. The fact that fed-batch cultivation is superior to batch
cultivation in shake flasks has been shown by several groups [156-160]. Restaino and co-workers
actually show as much as a 50-fold increase in production of mammalian 6-O-sulfotransferase in

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
21
E. coli when optimized fed-batch cultivation instead of shake flasks is used [160]. However, it is
important to note that in the previously mentioned studies just a few proteins are handled.
3.1.1 The EnBase® technology
Although fed-batch cultivation is superior it demands expensive equipment. One way to circumvent
this problem, when working in laboratory-scale, is by using an enzyme-based-substrate-delivery
cultivation method called EnBase® developed by Neubauer and co-workers [161]. With this
method, high cell densities as well as productivity can be obtained by glucose-limited fed-batch
cultivation in a shake flask and no bioreactor is needed. The major difference, compared to classical
fed-batch processes, is that no external glucose feed is needed, instead microwell plates or shake
flasks can be used and glucose is released into the growth medium by enzymatic degradation of
starch that is present in the culture from the start [161]. Advantages of using this fed-batch
technology compared to the classical batch format are the possibility to reach high cell densities
without impairing the productivity per cell and an increase in yield of soluble protein [162, 163].
Krause and colleagues report an increased amount of purified soluble protein per volume as high as
13-fold after production in EnBase® compared to a classical batch cultivation system [162]. This
makes it possible to lower the culture volume using EnBase®, which reduces the time and effort
needed for process optimization and downstream processing [162].
3.2 Cultivation conditions
In addition to cultivation technique are cultivation conditions highly important for the success of
recombinant protein production in E. coli. Culture media, temperature, pH, growth phase at
induction, concentration of inducing agent and period of induction are all parameters affecting
protein production. By careful consideration of the different culture conditions it is possible to
maximize recombinant protein production [164].
3.2.1 Culture media
One of the initial decisions to make is what culture medium to use; a nutrient-rich or nutrient-
deficient medium, in other words a complex or chemically defined medium, respectively. Complex

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
22
media that are composed of digests of chemically undefined substances, such as yeast and peptone
(i.e. enzymatic digest of protein), provide all essential nutrients needed to support cell growth and
protein production. However, since the exact composition is unknown and can vary a lot, it is
difficult to control the cultivation process. Therefore, minimal media that contain a defined amount
of carbon source, nitrogen source, minerals and trace elements, are often used in processes where it
is important to be able to fully control the cultivation [165]. Examples of commonly used media are
listed in Table 4. The complex as well as the minimal media can also be modified into an auto-
inducing medium. More details about auto-inducing media are presented in the section about
induction (3.2.3).
Table 4. Commonly used culture media for E. coli.
Nutrient-rich media Nutrient-deficient media
LB - Luria-Bertani broth M9 mimimal salt medium SB - Super Broth M63 minimal salt medium SOB - Super Optimal Broth
SOC - Super Optimal broth with Catabolite repression
TB - Terrific Broth
TSB - Tryptic Soy Broth
2xYT
To obtain the maximum protein yield it is important to identify the optimal medium [166, 167].
Complex media are generally used for recombinant protein production in laboratory-scale. The
benefits of this media on cell density and productivity have been shown by several groups [168,
169]. However, for production of some target proteins, minimal medium has shown to be more
suitable, as in the case of lipoproteins [170]. One way to improve the productivity from a minimal
medium is by supplementing the medium with particular amino acids, based on the amino acid
composition of the target protein, post induction. Babaeipour and colleagues actually show that by
using this approach, protein yields comparable to the yields from a complex culture can be obtained
[169].
3.2.2 Optimal temperature and pH
The optimal growth temperature for E. coli is 37˚C. However, although a high yield of recombinant
protein could be achieved at 37˚C it is often recommended to lower the temperature after induction

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
23
[168, 171]. Lowering the temperature is most important if soluble target protein is needed, since the
process of protein production have a tendency to result in precipitated protein at higher
temperatures [172-177]. Most often 25˚C is the recommended temperature for successful
production of soluble protein. Induction at lower temperature will slow down the protein
production rate and thereby it is more likely that the complex folding process of eukaryotic proteins
in E. coli will be successful. By lowering the temperature it is also possible to reduce synthesis of
stress and heat shock proteins, that can damage the target protein, as well as reduce proteolytic
degradation in E. coli [165].
Optimal pH for growth of E. coli is in the range of 6-7.5, but it varies with the temperature [178].
When using classical shake flasks for cultivation, where all substrate is added at the start, overflow
metabolism resulting in accumulation of acetate is as previously described a common problem. The
increased level of acetate will result in a drop of the pH value that has a negative effect on the
growth of E. coli and thereby the protein yield. One way to reduce this problem is by changing to a
fed-batch system or by using a controlled release of carbon source in shaken cultures [161, 179].
3.2.3 Induction of protein production
During cell cultivation, protein production is initiated at the point of induction. Timing of induction,
concentration of the inducing agent or thermal shift and duration of the induction are all parameters
influencing protein yield as well as solubility. Generally it is recommended to induce protein
production when the cells are in the early or mid-log phase. In a comparative study by the Structural
Proteomics In Europe (SPINE) consortium, induction in the early, mid- and late-log phase as well
as early stationary phase is compared. What they concluded is in accordance with the general
opinion; induction at the early log phase generates the best results [176]. This is also supported in
work by, for example, Muntari and co-workers [180].
It is also of great importance to find the optimal concentration of inducer, since it is important to
find the balance between decreasing cell growth after induction and increasing production of target
protein on a cellular level. In laboratory-scale, IPTG-inducible promoters are commonly used. The
standard recommendation is to use an IPTG concentration in the range of 0.1 to 1.0 mM IPTG
[167, 168, 171]. A good example of how the IPTG concentration in combination with other

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
24
parameters affects protein yield as well as solubility has recently been published by Pranchevicius et
al [172]. They concluded that the combination of incubation at 37˚C, a high shaking speed and 1.0
mM IPTG generated a large amount of protein, but in insoluble form. However, if induction
temperature was decreased to 20˚C, the shaking speed lowered and 0.4 mM IPTG was used for
induction the highest amount of soluble protein was produced. In other words, there are several
parameters influencing solubility of the produced target protein, and one of them is the
concentration of inducing agent.
When working with IPTG-inducible promoters a more convenient way of induction than addition
of IPTG, especially when many cultures are cultured in parallel, is to use an auto-inducing medium
[181]. The medium used for this purpose contains lactose as well as a limited amount of glucose and
the mechanism behind auto-induction is based on that glucose prevents uptake of lactose and
thereby also prevent induction. When the glucose is depleted, lactose can be converted to allolactose
resulting in initiation of transcription. For optimization of the timing of auto-induction, the levels of
glucose and lactose are adjusted. By using an optimized auto-induction protocol, transcription will
always be induced at the optimal time point, without any manual handling. This will most likely have
a positive effect on the average protein yield when handling several parallel cultures.
Another important parameter to consider is for how long protein production should last. The
optimal time post induction is highly dependent on the target protein, but also induction
temperature, timing of induction and concentration of inducing agent. The reported optimal
induction time varies between three and 24 hours [168, 171, 176, 180], but if the temperature is very
low during induction up to 72 hours could be necessary [182].
3.3 Automated and multi‐parallel systems for high‐throughput protein production
As a result of all the sequence information available from different genome projects, the possibility
to study protein function and structure are better than ever. Production of proteins for use in such
experimental studies has dramatically increased and therefore, parallel recombinant protein
production also has become more important. Parallel large-scale production of recombinant

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
25
proteins in a high-throughput manner (i.e. ten or more parallel cultures) is generally performed in
two different formats; shake flasks or bubble columns [183]. The classical way of doing this is by
running several parallel shake flask cultures. Drawbacks of this system are difficulties to control
important variables such as oxygen limitation and that it is labor intensive. However, the benefits of
using shake flasks are simple handling and less infrastructure investments. An alternative to parallel
shake flask cultivations are different parallel bubble columns. Bubble columns have a cylindrical
vessel and gas is sparged into the culture medium via a gas distributor. The benefits of this system
are that it is simple to handle and possible to achieve a high oxygen transfer rate in rather large
culture volumes. The culture volume that is possible using this type of bioreactor makes it suitable
for projects where milligram amounts of soluble protein are needed, for example different structural
genomics projects. Two different parallel bubble column solutions presented in the literature are the
large-scale expression system (LEX, Harbinger Biotech) [184] and the Genomics Institute of the
Novartis Research Foundation (GNF) milliliter systems [185]. The main difference between these
two systems is that a LEX system consists of 24 individual 2L bottles, while the GNF system
consists of 96 round-bottomed 100 mL tubes.
To design a bioprocess, including optimal cultivation conditions for production of a target protein
of a specific purpose, is not a trivial task. However, by careful consideration of the different
parameters it is most likely possible to set up a process for production of the majority of all proteins.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
26
4. Downstream processing
After cultivation, downstream processing is needed to isolate the protein of interest. Initially, the
cells are harvested by separation from the cultivation medium, for example by centrifugation or
filtration. For target proteins that are not secreted to the medium, an action to release the target
protein from the cytoplasm or periplasm is thereafter generally needed. For target proteins produced
intracellularly in E. coli, breakage of the host cells, either mechanically (e.g. by using ultra sound or
pressure) or non-mechanically (e.g. by using physical, chemical or enzymatic lysis), is necessary to
release the proteins [186]. However, if the target protein is exported to the periplasmic space of
E. coli, only disruption of the outer membrane is needed, e.g. by osmotic shock. Once the proteins
are released from the cell, the proteins are separated from the cell debris by centrifugation and or
filtration. Thereafter the target protein can be purified from the host proteins, for example by using
one or more chromatographic methods.
4.1 Protein purification by chromatography
Chromatography is a common term for techniques involving a stationary phase and a mobile phase
for separation of molecules. For protein purification the general setup is based on a stationary phase
that is packed into a column and a mobile phase that will be pumped through the column.
Depending on the choice of stationary phase, different proteins will be separated differently based
on properties such as size, charge, hydrophobicity or specific interactions. Commonly used
chromatographic methods are Size Exclusion Chromatography (SEC), Ion Exchange
Chromatography (IEXC), Reversed-Phase Chromatography (RPC), Hydrophobic Interaction
Chromatography (HIC), Affinity chromatography (AC) and Immobilized Metal Ion Affinity
Chromatography (IMAC) [187-192].
Based on the focus of this thesis, IMAC is the only method that will be presented in detail. IMAC is
a method frequently used for purification of proteins in a laboratory-scale, and it is based on the
affinity of transition metal ions such as Co2+, Ni2+, Cu2+ and Zn2+ to histidine and, although weaker,
also cysteine and tryptophan [191]. To be able to use this affinity for purification purposes, metal
ions, used as affinity ligands, are immobilized on a chromatographic support. The specific residues
in the target protein can then bind to the metal ions. Before elution of the target protein, all

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
27
unbound proteins are washed away. Elution can then be performed in three different ways. i)
Competitive elution with a gradient of increasing concentration of for example imidazole. ii)
Protonation of the histidyl residues by lowering the pH. This will break the interaction between the
ion and the residue. iii) Extraction of the metal ions by addition of a chelating agent such as EDTA
[186].
By producing recombinant proteins with a polyhistidine tag such as His6, the proteins will via its tag
be able to bind to the metal ions with high specificity [193]. The combination of IMAC and His6
makes it possible to purify the target protein from the host proteins in a single step, and although
this setup is generally used for a small number of parallel purifications, it is also possible to use this
method for high-throughput protein purification [194]. Other benefits of using His6 in combination
with IMAC than selective binding are high capacity, mild elution conditions and low cost [195, 196].
Moreover, another advantage is that the interaction between the histidine tag and the metal ion is
structure independent and therefore this purification method is possible to perform under native as
well as denaturing conditions. Also, the histidine tag used to improve the selectivity of IMAC has
low immunogenicity and is relatively small [197, 198]. The benefit of the small size is that it rarely
affects protein folding and function [198]. However, although this method has many advantages it
also has disadvantages. The most prominent drawback is that it is sometimes difficult to eliminate all
contaminants, due to the fact that cysteine- or histidine-rich regions in the host proteins can result in
unwanted binding [196, 197]. One way to control the selectivity and thereby the yield of pure
protein is by including a low concentration of imidazole in the sample and wash buffers [197, 199].

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
28
5. Protein analysis
After purification it is of importance to determine concentration, purity and molecular weight of the
purified protein product. To measure protein concentration Ultraviolet-visible (UV/Vis)
spectroscopy is commonly used. Examples of methods based on UV/Vis spectroscopy are direct
measurement, where the absorbance of the protein solution at 280 nm is used to determine the
protein concentration, and the commercial bicinchoninic acid (BCA) Protein Assay [200]. The purity
of purified protein is routinely determined using Sodium dodecyl sulfate - polyacrylamide gel
electrophoresis (SDS-PAGE). By using this technique all proteins are primarily separated based on
their size [201]. The resulting gel gives information about the purity of the protein but also protein
concentration and molecular weight. If further information about a specific protein is needed,
Western blotting can be used [202]. The proteins are then transferred from the gel to a nitrocellulose
membrane. A primary antibody, directed against the protein of interest, is used for detection. This
interaction is then visualized by a secondary antibody coupled to an enzyme or a fluorescent dye. If
a more precise molecular weight is needed, mass spectrometry (MS) is preferably used. This is a very
powerful technique that will be discussed in the following section.
5.1 Mass spectrometry
Mass spectrometry is an analytical method that is used to determine the mass of a molecule. The
first mass spectrometer was constructed in 1912 by J.J. Thomson [203]. By using this kind of
instrument it is possible to separate molecules based on size (m) and charge (z) and thereby
determine their masses. The three major components of a mass spectrometer are the ion source, the
mass analyzer and the detector. Although this technique has been available for more than hundred
years, it has not been applicable on biomolecules, such as proteins, until 25 years ago due to the lack
of soft ionization techniques.
However, in the late 1980’s two new ionization techniques, enabling analysis of biomolecules, were
developed: Matrix-Assisted Laser Desorption Ionization (MALDI) [204, 205] and Electrospray
Ionization (ESI) [206]. Although both techniques are soft, which allows for mass analysis of intact
macromolecules, the ions are created in different ways. When using MALDI, the sample and a
matrix that absorbs laser energy are co-crystallized on a target. Irradiation of the crystals will then

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
29
result in ionization of the analytes. In contrast to MALDI, ESI produces ions in gas phase from a
sample in liquid phase by applying a high voltage between the spray emitter and the inlet of the mass
analyzer. A benefit of starting from liquid phase, as in the case of ESI, is that it is possible to couple
it to a liquid chromatographic system and thereby include an extra separation step prior ionization.
After ionization the ions will enter the mass analyzer. When analyzing proteins or peptides there are
five commonly used types of mass analyzers: time-of-flight (TOF), quadrupole (Q), ion trap (IT),
orbitrap and ion cyclotron resonance (ICR) [207]. TOF mass analyzers separate ions based on the
time that ions take to move from the source to the detector, while the quadrupole separates ions in
an oscillating electric field according to their mass-to-charge ratio (m/z). Ion traps, which are the
third group of analyzers, use an oscillating electric field to store ions. The ions are then subjected to
an additional electric field that will eject ions of a given mass. The remaining two mass analyzers are
also trapping the ions; the orbitrap in an electric field and the ICR in a high magnetic field. It is also
possible to perform tandem MS (MS/MS). When using TOF or Q two identical or two different
mass analyzers are combined, while MS/MS in time can be performed when using a trapping
instrument (IT, orbitrap or ICR). Tandem MS can for example be used for peptide sequencing.
After separation in the mass analyzer the ions are detected and converted into a usable signal in the
mass analyzer (orbitrap, ICR) or by a separate detector (TOF, Q, IT).
Since the advent of MALDI and ESI, the analysis of biomolecules by MS is frequently done. MS is a
sensitive and fast technique that can be used to analyze primary sequences to identify proteins,
quantification of proteins, post-translational modifications (PTMs) as well as protein-protein
interactions [208]. Initially it was only possible to analyze a small set of samples. However, as a result
of all genomic data available in combination with technical improvement, there are nowadays
instruments and softwares allowing complex samples in large-scale proteomics studies [209]. MS has
therefore become a very important method in proteomics research.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
30

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
31
Present investigation
The main objective of this thesis has been to improve recombinant protein production in E. coli and
increase the throughput of the protein production pipeline within the Swedish Human Protein Atlas
(HPA) project [210]. To study if a strong promoter is always the best choice, if addition of rare
tRNAs to the host cell is beneficial and if it is possible to produce almost 300 different recombinant
proteins in just a week, a number of different projects were initiated. These projects have now
resulted in five papers that this thesis is based upon. The projects can be divided into two groups i)
optimization of the production of recombinant proteins in E. coli (paper II, III, V) and ii)
developments to enhance the throughput in a high-throughput protein production pipeline (paper I,
IV). Altogether, the new findings have resulted in an improved protein production pipeline within
the Swedish Human Protein Atlas project.
To ensure a high yield of recombinant protein, the choice of expression vector and its different
elements are highly important. One of the key steps in protein production is transcription and
therefore the effect of different promoters on the protein yield was evaluated (paper III). When
producing proteins, translation is another very important process. Since the codon usage in humans
and E. coli differs this could affect the translation and thereby the amount of produced target protein
and its quality. For that reason the protein production of human protein fragments in a standard
E. coli strain was compared to the recombinant proteins generated by an E. coli strain that was
supplied with genes for tRNAs that are rare in E. coli (paper II). The positive result of this study
encouraged us to do a thorough analysis to further explain these results (paper V).
Apart from evaluating different promoters and host strains for production of human protein
fragments in E. coli, setting up a high-throughput protein production pipeline has also been a goal
(paper I). The purpose of this project has been to develop a protocol for protein production and
purification to be able to handle parallel production of a large number of antigens. These antigens
are then used for generation of antibodies needed for antibody-based proteomics within the Human
Protein Atlas project. By careful consideration of each step in the process, a robust protocol with a

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
32
reduced number of manual handling steps, enabling high-throughput protein production, has been
developed. Although the overall success rate of this pipeline is high, there are still proteins that are
not successfully produced. For that reason, the idea of developing a screening protocol to use prior
high-throughput protein production came up. A screening protocol has therefore been developed by
mimicking the production workflow in a miniaturized format (paper IV).

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
33
6. The effects of different promoters on the protein
production
Recombinant protein production in E. coli is influenced by many different parameters. The
combined processes of replication, transcription and translation are highly important. However,
culture conditions, the localization of the protein in the cell, how it folds, its proteolytic stability and
how it affects cell growth also have a great impact on the protein production [16, 17, 199]. To
evaluate the influence of transcription on the protein production, sixteen proteins with different
characteristics were produced in E. coli BL21(DE3) under control of three different promoters;
lacUV5, trc and T7 (paper III). All three promoters are IPTG-inducible, but have different promoter
strength. The lacUV5, trc and T7 promoters are classified as rather weak, strong and very strong,
respectively. Hence, T7 has the highest transcription rate and lacUV5 the lowest.
For quantification of the total amount of protein as well as the amount of soluble protein produced
under control of the three different promoters, the cultured cells were disrupted and separated into a
soluble and an insoluble fraction. The two fractions were then analyzed on SDS-PAGE and by
Western blot to detect the His-tagged fusion proteins. As expected, the T7 promoter generated the
largest amount of total protein and the lacUV5 promoter the lowest (Figure 3A). This is in
accordance with the fact that T7 RNA polymerase has a much faster RNA elongation than E. coli
RNA polymerase [88]. The T7 promoter should therefore theoretically result in a higher
accumulation of target protein than for example lacUV5, which was shown here.
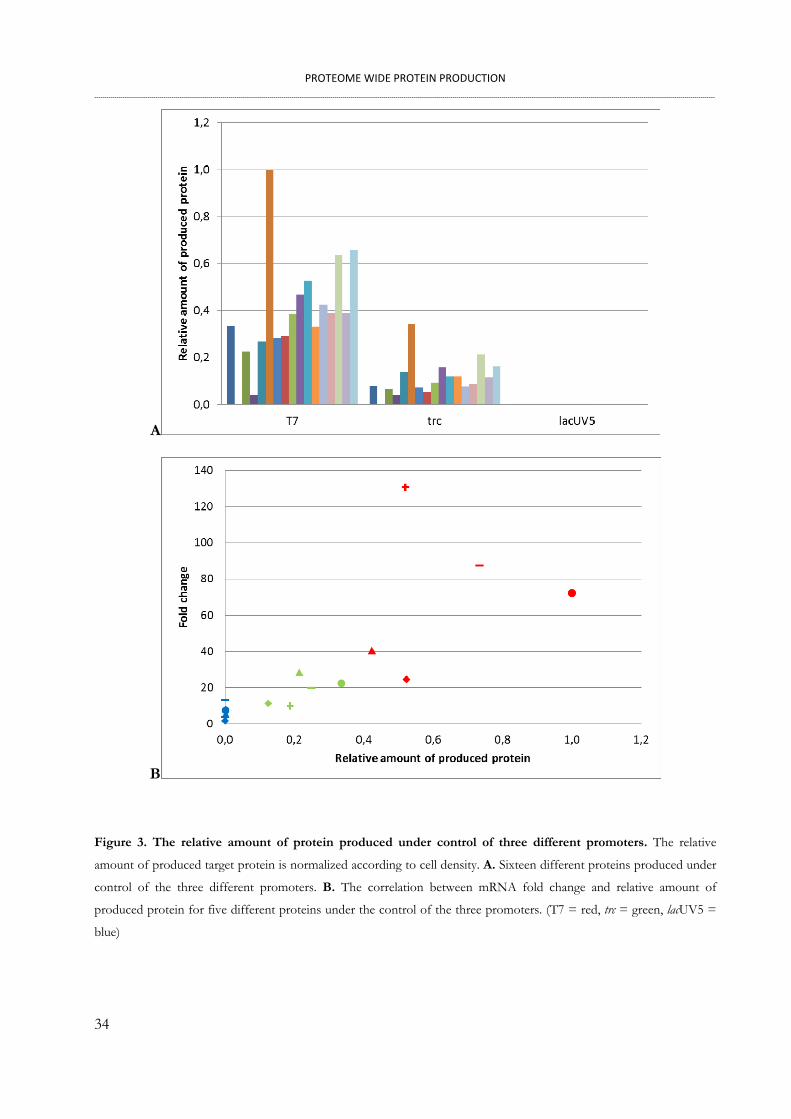
PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
34
A
B
Figure 3. The relative amount of protein produced under control of three different promoters. The relative
amount of produced target protein is normalized according to cell density. A. Sixteen different proteins produced under
control of the three different promoters. B. The correlation between mRNA fold change and relative amount of
produced protein for five different proteins under the control of the three promoters. (T7 = red, trc = green, lacUV5 =
blue)

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
35
To get a deeper understanding of the influence of different mRNA levels on the protein production,
the number of mRNA molecules before and after induction for five of the sixteen proteins was
determined using Real-time PCR. The fold change of mRNA after induction was then compared to
the total amount of produced protein, see Figure 3B. As expected, the promoter with highest
transcription rate (T7) generated the highest fold change of mRNA as well as largest total amount of
protein and vice versa. In other words, there was a correlation between the fold change of mRNA
after induction and the amount of produced target protein. However, the differences within the
group of constructs produced under control of the T7 promoter were larger than expected. Most
likely one explanation to the spread in the amount of produced protein, is the influence of rare
codons on the translation, since a high number of rare codons will slow down the translation
process resulting in lower amount of produced protein and vice versa. Thus, translation is as
important as transcription for successful recombinant protein production.
Furthermore the effect of different promoters on the amount of soluble protein was analyzed. The
general opinion is that a slower protein production is beneficial for production of soluble protein.
When analyzing the results of this study it was clear that the lowest transcription rate (lacUV5)
generated the largest fraction of soluble protein and the highest transcription rate (T7) the lowest
(Figure 4A). However, due to the large total amount of produced protein, the T7 promoter also
generated the most soluble protein and is therefore still preferred for production of soluble protein
(Figure 4B). The fact that a high protein production rate can generate large amounts of soluble
protein is in accordance with a study by Schultz et al [93]. Apart from transcription rate, the amount
of soluble produced protein is also highly dependent on the solubility of the target protein. When
comparing the amount of soluble produced protein and the solubility class, determined by an in vitro
solubility assay [211], there was to some extent a positive relation between the amount of soluble
protein and the in vitro solubility class (Figure 4C). Hence, it is more likely that a protein of a higher
solubility class, i.e. a more soluble protein, will result in a larger amount of soluble protein. This
correlation was shown to be independent of the choice of promoter.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
36
A
B
C
Figure 4. The effect of different promoters on the production of soluble protein. A. The fraction of soluble
protein. B. The relative amount of soluble target protein, normalized according to cell density. C. The relative amount
of soluble target protein compared to the solubility class. (T7 = red, trc = green, lacUV5 = blue)

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
37
The above described comparisons of three different IPTG-inducible promoters have clearly shown
the benefits of using the T7 promoter for protein production in general, but also for production of
soluble proteins. However, transcription is not the only parameter regulating protein production,
translation as well as characteristics of the target protein are also highly important.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
38
7. Increased success rate with E. coli Rosetta(DE3)
To find the optimal conditions for protein production of human proteins in E. coli, there are several
parameters to consider, for instance codon usage. High frequency or clusters of codons rarely used
by E. coli have shown to have a negative influence on translation of the recombinant protein and
thereby the yield of the product [122, 123]. Although sequence optimization, in general, is the best
solution to this problem [143-146], using a strain co-expressing rare tRNAs is a more practical
approach when handling a large number of different target proteins.
For many years the standard host strain used for recombinant protein production in laboratory-scale
has been E. coli BL21(DE3). However, in an attempt to improve the protein production in the
Human Protein Atlas project, the possibility to compensate for codons rarely used in E. coli were
evaluated (paper II). Due to the large number of samples handled, sequence optimization was not
the first choice; instead a strain compensating for rare codons was evaluated. The strain chosen was
E. coli Rosetta(DE3). Rosetta(DE3) is according to the manufacturers very similar to BL21(DE3).
The only difference is an extra plasmid (pRARE) with chloramphenicol resistance, containing genes
for as many as six rare codon tRNAs (AGG, AGA, AUA, CUA, CCC, GGA). A test set of proteins,
previously produced in BL21(DE3), were reproduced in Rosetta(DE3) followed by purification. The
protein yields as well as the purity of the target proteins produced in the two different strains were
evaluated. When comparing the amounts of purified protein, it was obvious that for most of the
proteins that showed a protein yield below one milligram after production in BL21(DE3) followed
by purification, changing to Rosetta(DE3) was very beneficial and significantly improved the protein
production for many of the proteins (Figure 5A). After evaluating the proteins that previously
generated above one milligram of purified protein after production in BL21(DE3), it was concluded
that only a few of the produced proteins showed an improved protein yield in Rosetta(DE3) while
the remaining proteins still produced well above one milligram of purified protein also in
Rosetta(DE3) (Figure 5B). In other words, Rosetta(DE3) is beneficial when trying to produce
proteins that are difficult to produce in sufficient amounts, but in general it is not a disadvantage to
use this strain for easily produced proteins.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
39
A
B
Figure 5. Comparing the amount of purified protein after production in E. coli BL21(DE3) and Rosetta(DE3).
A. Proteins that generated less than one milligram of purified protein after production in BL21(DE3). B. Proteins that
generated more than one milligram of purified protein after production in BL21(DE3). (Blue = BL21(DE3), Red =
Rosetta(DE3))
Furthermore, as the purity is just as important as yield, the purity of the proteins successfully
produced in both strains was also studied. For a majority of the proteins the purity was significantly
increased using Rosetta(DE3). This indicated that by using a strain compensating for rare codons,
both the quantity as well as the quality of the product is increased.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
40
To further evaluate the usage of Rosetta(DE3), a new larger set of 96 different proteins that had
previously failed to produce enough protein in BL21(DE3), was produced in Rosetta(DE3). This
turned out to be so successful that Rosetta(DE3) was implemented as the default strain for
recombinant protein production in the high-throughput production pipeline of the Human Protein
Atlas project. Four years later, it was possible to compare the success rate of 7,080 different proteins
produced in BL21(DE3) and another 13,000 different proteins produced in Rosetta(DE3). All in all,
after comparing data for the first attempt in production of 20,080 different proteins, it was
concluded that the success rate regarding the part of samples with enough protein after purification
was almost the same in the two production strains. Instead, in accordance with the initial test set, the
greatest benefit was a much increased purity. The improved purity had a great impact on the success
rate, resulting in an overall success rate of protein production in Rosetta(DE3) of 76.8% compared
to 64.3% in BL21(DE3).
For better understanding of the underlying causes of the improved success rate in Rosetta(DE3), the
content of codons rarely used in E. coli in the DNA sequences of the 68 different proteins in the
initial test set was analyzed. The result of this analysis was then compared to the total amount of
protein after affinity purification. No clear correlation was found between the occurrence of the six
rare codons that Rosetta(DE3) compensates for and the protein yield in the two strains, though
proteins that produced well in BL21(DE3) most often had few or no rare codons. What could be
seen was that clusters of rare codons, here defined as the number of two rare codons appearing
consecutively or separated by one codon, and a high content of the rare arginine codons AGG and
AGA seemed to have a negative effect on protein production in BL21(DE3). However, for the
purity of the proteins both the total number of rare codons, as well as the number of rare arginine
codons was shown to have a great impact (paper II).
To get a deeper understanding of the improved success rate in Rosetta(DE3), a more thorough
analysis of the differences in protein yield and the cause of the improved purity were done (paper
V). For further evaluation of the improved purity after production in Rosetta(DE3), twenty different
proteins that previously had been produced in BL21(DE3) as well as Rosetta(DE3) with different
success were chosen. These proteins were reproduced in triplicates in BL21(DE3) and
Rosetta(DE3), followed by IMAC purification and analysis on SDS-PAGE and by Western Blot,
using an antibody directed towards the common tag as primary antibody. Comparison of the
impurities on the SDS-PAGE and the proteins that according to the Western Blot analysis

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
41
contained the common tag clearly showed that the remaining impurities in fact were truncated
versions of the target protein (Figure 6). This result is in line with the report of Sorensen et al, who
show that by compensating for rare tRNAs with plasmids like pRARE, truncated forms of the full-
length protein can be avoided [142]. This explains the improved purity after production in
Rosetta(DE3) and stresses that the choice of strain highly affects the possibility of having a pure
protein after purification.
Figure 6. SDS-PAGE and Western Blot to study the effect of using Rosetta(DE3) on the purity. Triplicates of a
representative protein produced in BL21(DE3) (lane 1-3) and Rosetta(DE3) (lane 4-6) were analyzed by SDS-PAGE
(left) and Western Blot (right). The SDS-PAGE shows all proteins remaining after purification, while the Western Blot
identifies all proteins with the common tag.
Another test set of 24 proteins with differences in rare codon content was compiled to gain further
knowledge of the effect of co-expression of rare tRNAs in the host on the recombinant protein
production. These proteins were produced in triplicates in four different strains; BL21(DE3),
Rosetta(DE3) without pRARE, Rosetta(DE3) and BL21(DE3) with pRARE. Also, the proteins
were produced in Rosetta(DE3) without chloramphenicol in the shake flask culture medium to
study if the protein production is affected by the absence of chloramphenicol and if it is possible to
maintain the pRARE plasmid without this antibiotic. The amount of full-length target protein after
purification was quantified from SDS-PAGE gels. Presence of pRARE showed to have a significant
impact on the protein yield for many of the proteins, but for a few it showed no difference at all
(Figure 7). Comparison of the results using the commercial Rosetta(DE3) strain and BL21(DE3)
cells which had been transformed with pRARE as well as BL21(DE3) and Rosetta(DE3) without
pRARE clearly showed that the only difference between the two strains was the pRARE plasmid
(Figure 7).

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
42
Figure 7. SDS-PAGE analysis of two representative proteins to compare the amount of protein produced in
strains with and without pRARE. The proteins were produced in triplicate in BL21(DE3) (lane 1-3), Rosetta(DE3)
without pRARE (lane 4-6), Rosetta(DE3) (lane 7-9), BL21(DE3) with pRARE (lane 10-12) and Rosetta(DE3) without
chloramphenicol in the 100 ml culture (lane 13-15).
However, the effect of adding a pRARE plasmid seemed to be highly target dependent. For that
reason each target gene sequence was analyzed to identify the number of rare codons, clusters of
rare codons as well as rare arginine codons (AGG and AGA). Comparison of the protein yield with
the rare codon content revealed that it was not the number of rare codons that determined the
benefit of using a strain with pRARE. Instead, pRARE appeared to have strongest impact on
proteins rich in rare codon clusters and particularly proteins rich in rare arginine codons. In other
words, if there were a large number of rare arginine codons within the sequence it was shown to be
highly beneficial to use a strain with pRARE. This could be explained by the limited number of
tRNAs for the rare arginine codons AGG and AGA in BL21(DE3), causing a delay in the protein
synthesis and thereby a lower protein yield than in Rosetta(DE3) [137]. In fact, the AGG and AGA
codons for arginine are the least used codons in E. coli, while they are commonly used in human
[122].

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
43
Regarding the effect of chloramphenicol on the protein production in strains with pRARE, the
amount of target protein was shown to be independent of the presence of chloramphenicol in the
culture medium. Also, it was shown that Rosetta(DE3) does not lose pRARE during the timeframe
of protein production despite absence of chloramphenicol. Hence, it is possible to exclude this
harsh antibiotic from the shake flask cultures without interfering with the protein production.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
44
8. Combining different promoters and E. coli strains
As described in the two previous sections, recombinant protein production is affected both by the
choice of promoter and E. coli strain. To evaluate the effect of different combinations of promoter
and strain on protein production, five different proteins were produced under control of three
different promoters (T7, trc and lacUV5) and in two different E. coli strains (BL21(DE3) and
Rosetta(DE3)) (paper III). The total amount of produced protein as well as the amount of soluble
protein was determined using SDS-PAGE and Western Blot to detect the His-tagged fusion
proteins. The results of this study clearly showed that independent of strain, the different promoters
display the expected expression pattern (Figure 8A). However, for all promoters Rosetta(DE3)
generated a larger amount of these five proteins than BL21(DE3). In accordance with the results in
paper II and V, the increased yield in Rosetta(DE3) has most likely a correlation to rare codons.
The fraction of soluble protein produced in the two strains was also compared. What could be seen
was that independent of strain, lacUV5 generated the largest fraction of soluble protein while T7
generated the smallest fraction (Figure 8B). However, in general, it is the amount of soluble protein
that is important and for that purpose the Rosetta(DE3) strain was the preferred one (Figure 8C).
Somewhat surprisingly, the expression pattern of soluble protein changed when using Rosetta(DE3).
What happened was that the combination of Rosetta(DE3) and trc generated a larger amount of
soluble protein than Rosetta(DE3) in combination with T7 in as many as three out of the five cases.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
45
A
B
C
Figure 8. The effect of promoter and host strain on the protein production. A. . The relative amount of produced
target protein, normalized according to cell density. B. The fraction of soluble protein. C. The relative amount of
soluble protein, normalized according to cell density
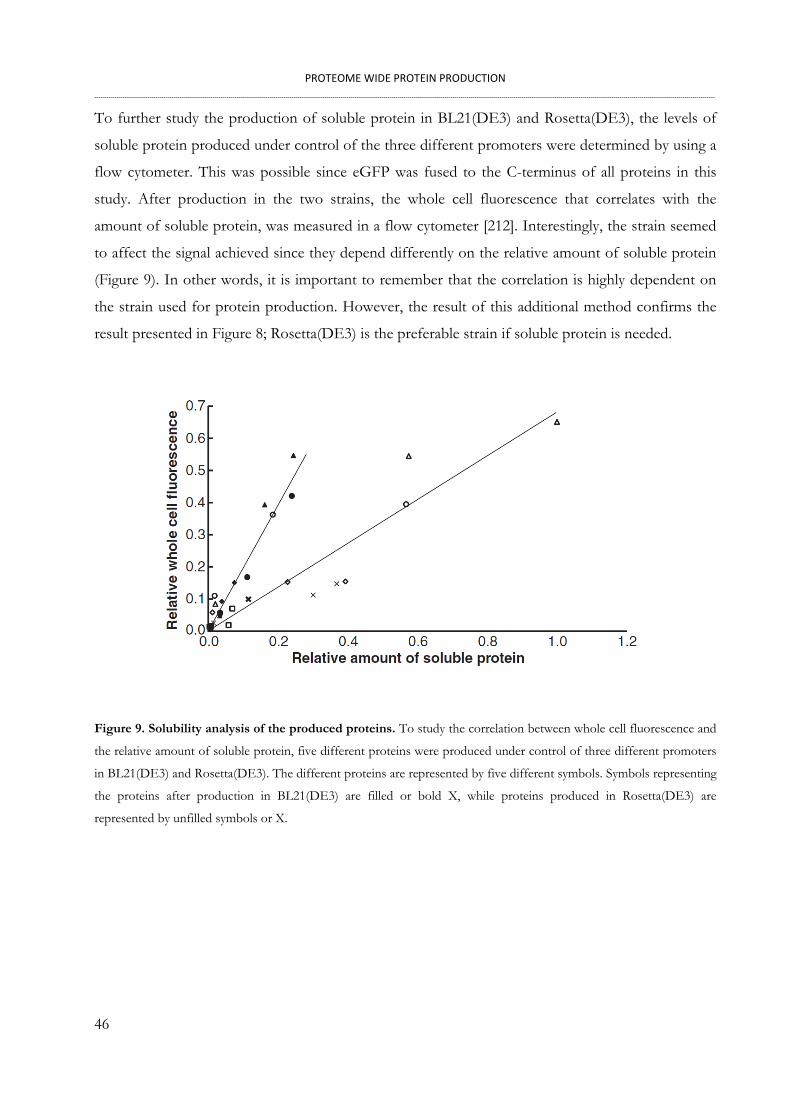
PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
46
To further study the production of soluble protein in BL21(DE3) and Rosetta(DE3), the levels of
soluble protein produced under control of the three different promoters were determined by using a
flow cytometer. This was possible since eGFP was fused to the C-terminus of all proteins in this
study. After production in the two strains, the whole cell fluorescence that correlates with the
amount of soluble protein, was measured in a flow cytometer [212]. Interestingly, the strain seemed
to affect the signal achieved since they depend differently on the relative amount of soluble protein
(Figure 9). In other words, it is important to remember that the correlation is highly dependent on
the strain used for protein production. However, the result of this additional method confirms the
result presented in Figure 8; Rosetta(DE3) is the preferable strain if soluble protein is needed.
Figure 9. Solubility analysis of the produced proteins. To study the correlation between whole cell fluorescence and
the relative amount of soluble protein, five different proteins were produced under control of three different promoters
in BL21(DE3) and Rosetta(DE3). The different proteins are represented by five different symbols. Symbols representing
the proteins after production in BL21(DE3) are filled or bold X, while proteins produced in Rosetta(DE3) are
represented by unfilled symbols or X.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
47
9. Developing a high‐throughput protein production
pipeline
As a result of all the sequence information available from different genome projects, the possibility
to perform large-scale proteomics studies has dramatically increased. Therefore, the need for target
proteins to perform high-throughput experimental studies is greater than ever. To supply the need
of protein in such projects, different high-throughput protein production pipelines have been
developed [184, 185, 213-215]. In the Human Protein Atlas project, milligram amounts of a large
number of different proteins to be used for antibody generation are needed monthly and a high-
throughput protein production pipeline was a prerequisite for success. A high-throughput protein
production pipeline, including protein production, protein purification and protein analysis, has
therefore been set up (Figure 10) (paper I). A major difference between this setup and the ones used
in, for example structural biology projects, is that for antibody generation protein solubility is not an
issue.
Figure 10. A schematic presentation of the developed protein production pipeline. (Illustration by Maria Stenvall)

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
48
In the beginning of this project the production pipeline allowed production of a handful of samples
in parallel. To be able to set a goal of 600 antigens per month, increased capacity as well as success
rate compared to the initial setup was needed. Therefore, it was necessary to evaluate each and every
step in the production pipeline. By major and minor changes of the initial protocol, a standardized
setup that reached the monthly production goals was developed. Examples of major changes that
were essential to be able to increase the throughput as well as the overall success rate were the
change of E. coli strain (paper II) and the implementation of an automated protein purification
system [194]. The minor changes, that did not take great effort in the lab to identify but still had a
large impact on the efficiency was, for example, increased culture volume in the shake flasks and
decreased manual handling by using dispensers and multi-pipettes. When optimizing a protocol for
high-throughput protein production, optimal conditions are not the only thing to consider, it also
has to be practicable. The resulting protocol was therefore a combination of the optimal and the
most practical solutions; some unit operations were optimized, others excluded or automated. By
using this protocol the throughput has increased and the number of days of the entire process
decreased from seven to five. Altogether this has enabled production of up to 288 different proteins
per week, in batches of 72 parallel samples.
The most important improvement of the production pipeline was automation of the purification.
Without this development, handling of 72 parallel samples would not have been possible. In the
Human Protein Atlas project, the purified proteins are used for two purposes; as antigens and as
ligands on affinity columns. The fraction of the protein product used for column coupling needs to
have a high concentration in a rather small volume. For this purpose it was beneficial to identify the
fraction within the elution profile having the highest protein concentration. This could definitely be
a challenge when handling 72 parallel samples. However, since all proteins produced in the Human
Protein Atlas project have the same N-terminal purification tag (His6), the possibility of using a
standardized purification method based on IMAC with a standard elution protocol for all proteins
was evaluated. The elution profile of a large set of different His6-tagged proteins was therefore
analyzed and as can be seen in Figure 11 all His6-tagged proteins display a similar elution profile. The
optimal fraction to be used for column coupling was thereby easily identified; fraction number 2.
This made it possible to set up a standard elution protocol used for all proteins. Implementation of
the automated protein purification including a standardized elution protocol has made it possible to
handle almost 300 different proteins weekly.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
49
Figure 11. Elution profiles of 30 different His6-tagged proteins. The distribution of the eluted proteins within the
five fractions of 500µl is here presented.
All in all the optimized protocol has been a great success for the Protein Factory module in the
Human Protein Atlas project. By using this protocol the throughput as well as the overall success
rate has increased. Today, the number of successfully produced proteins, with purity higher than
80%, results in an overall success rate as high as 81%. However, since the aim of this project differs
from other projects with a high-throughput protein production pipeline, it is unfortunately not
possible to compare this setup with others.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
50
10. Screening to avoid unnecessary protein production
When using the same protocol for production of thousands of proteins, all proteins cannot be
successfully produced. To save time and money, it is therefore common to combine high-
throughput protein production pipelines with an initial small-scale screen. Most available screening
protocols have been developed for screening of soluble protein [184, 213, 214]. As a complement to
already existing screening protocols, a novel high-throughput screening method that enables parallel
production and verification of up to 96 different protein products has been set up (paper IV). By
using this method, proteins that are poorly produced under the chosen conditions can be sorted out
prior large-scale production. The protocol was developed by mimicking the standard workflow used
for protein production in the Human Protein Atlas project. However, in the screening procedure all
steps have been miniaturized and are performed in plate format. For protein production,
purification and verification the chosen solutions are the EnBase® cultivation technology, IMAC
purification in filter plates and MALDI-TOF MS, respectively (Figure 12).
Figure 12. Protein production pipelines. A. The standard workflow for protein production. B. The miniaturized
workflow of the screening setup. (Illustration by Maria Stenvall)

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
51
One very important parameter to evaluate in the beginning of this study was how much it was
possible to reduce the culture volume when using the EnBase® medium and still get sufficient
protein yields. The cultivations were therefore initially performed in deep well plates before
proceeding to micro well plates. Using the EnBase® medium in deep well plates was shown to
generate a substantially larger amount of protein per milliliter culture medium compared to a
standard culture in a shake flask. This result indicated that it was possible to further downscale the
EnBase® cultivations to microliter scale in micro well plates and still get reliable results.
Apart from the culture volume, it was also important to decrease the amount of IMAC matrix
needed for purification to reduce the costs of the screening setup. By replacing the purification
columns with a micro well filter plate, it was possible to downscale the needed volume of IMAC
matrix 80 times compared to the standard protocol. The resulting protocol is thereby based on
miniaturized protein production and purification, and to verify if a protein is produced or not, a
MALDI-TOF MS analysis is used. The agreement was shown to be 91% when the results in the
standard large-scale protein production pipeline and this screening method were compared.
However, by including an extra SDS-PAGE analysis after the MS analysis, the agreement was
further increased to 95%. In other words, a reliable screening method that could be used prior large-
scale production has been developed. By using this method, proteins that will be poorly produced
can be sorted out at an early stage, resulting in reduced costs. The main difference between this
protocol and the most commonly used screening protocols is that the protein product first is
verified by MS instead of SDS-PAGE. As an alternative to SDS-PAGE, dot-blotting or colony
filtration blot could also be used for screening of soluble protein [216]. The benefit of the latter
method is that soluble protein could be screened directly from colonies and thereby it is faster than
the more conventional methods.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
52

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
53
Concluding remarks
As a result of all the new improved sequencing techniques as well as the reduced cost per sample,
there has been a dramatic increase in number of sequenced genomes over the last years. Although
the number of known genomes is steadily increasing, it is still impossible to determine the three-
dimensional structure as well as the function of a protein from the coding sequence of the gene.
Therefore, systematic studies of protein structure and function has become of great importance. For
that purpose, the need for recombinant proteins is greater than ever. To find the optimal setup for
heterologous protein production there are several parameters to consider; for example host cell,
regulation on transcriptional and translational level as well as cultivation conditions.
In the papers that this thesis is based upon, E. coli has been the choice of host for protein
production due to that it is simple to handle and provides high yields at low cost. However, what
have been thoroughly studied are some of the parameters affecting transcription and translation. In
paper III, three commonly used promoters were studied and the results clearly indicate the benefits
of using a strong promoter like T7 compared to the weaker lacUV5 promoter for recombinant
protein production. Although the general opinion often is that a strong promoter is beneficial for
the total amount of produced protein, while a weaker is more suitable for production of soluble
protein, it was clearly shown in paper III that a strong promoter could also be very useful for
production of soluble protein. When focus was shifted from transcription to translation, the effect
of codons rarely used in E. coli on recombinant protein production was studied. What was
concluded was that it is an advantage to use a host strain that compensates for codons rarely used in
the host strain (paper II). Surprisingly the greatest benefit was not an increased yield but, as a result
of less truncated proteins, an increased purity (paper II and V). Also, even though Rosetta(DE3)
compensates for six different rare codons, it was somewhat surprising that it was mainly the number
of rare arginine codons that had an evident effect on the protein production. To study the individual
effect of the six rare codons it would therefore have been interesting to also include codon
optimized constructs, where one codon is optimized at a time. For further understanding of the
complex processes of transcription and translation and the possibility to improve recombinant
protein production, it would in the future be very interesting to set up a large-scale optimization

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
54
protocol for this purpose. By including several different parameters affecting transcription and
translation this may result in a deeper understanding of recombinant protein production as well as
the possibility to identify crucial parameters for successful protein production of proteins that are
not successfully produced in standard protein production pipelines.
Apart from identifying the optimal parameters, such as the design of the expression vector and
cultivation conditions, it is in large-scale projects also important to meet the demand of handling
several parallel samples. For that purpose a standard setup that works for large set of samples is
often needed. However, it is also highly important that the chosen setup can be done in practice. In
paper I, it was shown that by using a relatively simple setup it is possible to produce almost 300
different proteins in just a week. Interestingly, although the proteins have quite different
characteristics, as many as 81% can be successfully produced when using this setup. Hence, an
individually adjusted protocol is not necessary for the majority of all proteins. However, despite the
fact that the number of proteins that cannot be successfully produced is fairly low, by adding an
initial screening step unnecessary attempts of protein production can almost be eliminated (paper
IV). Combining the lessons of paper I-V one may conclude that it is possible to produce a large
number of different proteins by recombinant protein production. So far, as many as 38,000 different
human protein fragments have been successfully produced within the Human Protein Atlas project,
covering 95% of all human genes.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
55
Populärvetenskaplig sammanfattning Till pappa, syrran och alla ni andra som föredrar en kortare summering av vad denna bok egentligen handlar om.
Proteiner är molekyler som är uppbyggda av aminosyror i långa kedjor. Eftersom proteiner är
involverade i en rad olika livsviktiga funktioner, exempelvis som antikroppar i immunförsvaret och
som enzymer i kemiska reaktioner, brukar de ofta kallas för ”livets byggstenar”. Information om hur
ett protein ska se ut finns lagrat i DNAt. Förenklat skulle man kunna säga att DNAt är receptet och
proteinet den färdiga sockerkakan. För att omvandla DNA till protein, med andra ord receptet till en
kaka, krävs en process som kallas ”Det centrala dogmat”. I denna process sker ett informationsflöde
från DNA via mRNA till protein. Det som händer är att informationen i DNAt kopieras till en
budbärarmolekyl (messengerRNA) med hjälp av ett enzym. Den information som finns lagrad i
budbärarmolekylen kan sedan omvandlas till protein genom så kallad proteinsyntes. I detta delsteg
sätter sig en ribosom runt mRNAt. Ytterligare en molekyl, nämligen så kallade transferRNA (tRNA),
transporterar därefter de aminosyror som behövs under proteinsyntesen till ribosomen. Längs denna
kedja av reaktioner ökar komplexiteten dramatiskt när informationen i mRNAt slutligen omvandlas
till ett protein. DNA och RNA är båda uppbyggda av fyra olika kvävebaser, A, T, C och G
respektive A, U, C och G. Proteiner däremot är uppbyggda av 20 olika aminosyror. För att
ribosomen ska kunna översätta mRNAt till protein läser den tre baser i taget, och varje
trebokstavskombination (kodon) motsvarar en aminosyra.
Under de senaste femton åren har ett antal organismers arvsmassa, däribland människans, kartlagts.
Därmed har kunskapen kring varje organisms DNA, som arvsmassan består av, ökat. Att utifrån
baserna i DNAt ta reda på vilka aminosyror som ingår i det resulterande proteinet är inga problem
nuförtiden. Med dagens kunskap är det dock ännu inte möjligt att utifrån DNA- eller
aminosyrasekvensen kunna säga särskilt mycket om ett proteins struktur eller funktion. Behovet av
systematiska studier av proteiners struktur och funktion är därför stort. För att kunna genomföra
dessa studier behövs inte bara några enstaka, utan många olika proteiner. Att utvinna dessa från sin
naturliga miljö är i stort sätt en omöjlighet, men eftersom DNA-sekvensen är känd kan man istället
välja att producera de proteiner som behövs genom så kallad rekombinant proteinproduktion. Detta
innebär att man klistrar in den DNA-sekvens som motsvarar det protein man vill studera i en
vektor. En vektor är en cirkulär DNA-molekyl som innehåller alla komponenter som behövs för att

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
56
reglera den rekombinanta proteinproduktionen. Denna vektor förs sedan in i en värdcell, vanligtvis
en E. coli-bakterie, i vilken proteinet sedan kan produceras. Tack vare vissa komponenter i vektorn
kan proteinproduktionen ske under förhållandevis kontrollerade former.
I de arbeten som denna avhandling baseras på har jag dels studerat de komponenter som reglerar
processen då DNA kopieras till mRNA respektive när mRNA översätts till protein, dels hur man
ska gå till väga för att kunna producera hundratals rekombinanta proteiner per vecka. Vad gäller
själva kopieringen av DNA till mRNA så regleras den av en komponent som kallas promotor. De
promotorer som jag utvärderat kopierar DNA till mRNA olika snabbt. Efter att ha utvärderat
effekten av långsam respektive snabb kopiering kunde jag dra slutsatsen att förutsatt att man vill ha
så mycket protein som möjligt ska man välja en snabbkopierande promotor. En annan process som
studerats är när mRNA översätts till protein. Det har nämligen visat sig att vissa kodon
(trebokstavskombinationer i mRNAt) som är sällsynta i E. coli kan orsaka en del problem när mRNA
ska översättas till protein. Extra problematiskt har det visat sig vara om de sällsynta kodonen
dessutom är vanligt förekommande i den DNA-sekvens som ska produceras. På grund av detta så
har möjligheten att använda en E. coli-stam som försetts med extra tRNA för de kodon som är
sällsynta i E. coli utvärderats. Resultatet av denna studie påvisar klart och tydligt fördelarna med
denna stam. Något oväntat var dock att en ökad renhet, tack vare färre halvfärdiga proteiner, var
den klart största vinsten. Utöver att studera de processer som har en direkt påverkan på
proteinproduktionen, studerades även möjligheten att sätta upp en storskalig proteinproduktion för
nästan 300 olika proteiner per vecka. Detta visade sig, genom ett fåtal större och ett antal mindre
modifieringar av ursprungsprotokollet, vara fullt möjligt. Slutligen så har även ett screening-
protokoll, för att kunna exkludera de proteiner som aldrig kommer att funka i en storskalig
proteinproduktion, utvecklats.
Sammantaget visar studierna i denna avhandling att genom omsorgsfulla val av parametrar som
påverkar proteinproduktion är det full möjligt att producera många olika proteiner i en simpel
tarmbakterie; E. coli. Om man dessutom kan sätta samman ett väl genomtänkt standardprotokoll så
kan ett och samma protokoll användas för att producera tusentals olika protein.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
57
Acknowledgements
Då var det dags att slutligen tacka alla som på ett eller annat sätt funnits med under denna långa men ack så utvecklande och lärorika resa.
Sophia – Denna resa har varit lång och allt annat än rak, men tack vare ditt enorma stöd är målet nära nu. TACK för allt du ger, inte bara på det vetenskapliga planet utan även på det personliga planet. Att arbeta med dig är en ynnest; du är inte bara en framstående forskare och inspirerande handledare, du har dessutom förmågan att se människan bakom alla resultat. Du kan konsten att se helheten! Och trots att du är en väldigt upptagen person med många bollar i luften är du alltid redo att dela såväl framgång som motgång.
Jenny – att det tidsmässigt skulle gå att få ihop en doktorsavhandling var till en början inte självklart, men tack vare din stora förståelse för mitt behov av denna utmaning gick det till slut. Ingen har hejat på mig som du under slutförandet av denna avhandling och utan all avlastning vet jag inte riktigt hur detta skulle ha gått ihop. Känner dessutom stor tacksamhet över att du tror på mig och mina förmågor och alla utmaningar du ställt mig inför. Du anar inte hur utvecklande de har varit.
Att jobba inom ett projekt som HPA är verkligen en förmån. Tack Mathias för att du brinner för detta projekt och att du efter tio år fortfarande orkar vara såväl idéspruta som motor. Och tack Jocke för att du i samband med mitt exjobb öppnade dörren till detta fantastiska projekt.
Alltid närvarande och alltid redo för frågor; Per-Åke jag hoppas verkligen att du förstår vilken tillgång du är för alla doktorander på plan 3. En annan ack så viktig person är vår glada norrlänning, tillika skolchef/städgeneral; Stefan. Tack för den glädje och härliga stämning du sprider. Vill även passa på att tack övriga PIs på plan 3 för att ni bidrar till en trevlig atmosfär och stimulerande forskningsmiljö.
Henrik W, det var under din ledning jag lärde mig att flyga med egna vingar. Tack!
Det sägs att ensam är stark, men utan handledning och engagerade medförfattare undrar jag om resultatet verkligen hade blivit lika bra?! Tack alla PFare som var involverade i utvecklingen av vår storskaliga proteinproduktion; resultatet blev inte bara ett effektivt flöde utan även finfin artikel. Vill även passa på att tacka Anja och Sam för att ni initierade Rosetta-studien, Lotta för att du lärde mig klona, Bahram och MolBio för prima sekvenser, My för att du lärde mig använda flödescytometern, Caroline och Anna S för att ni aldrig tröttnade på alla mina frågor om realtids-PCR, Tove B och Louise för MS-analyser, Cajsa för fantastiska WBs och slutligen Kattis och Anneli för all hjälp med Rosetta2-projektet. I would also like to thank Kaisa, Antti and Peter for all input on the EnBase protocol.
Finaste Johanna, är så himla glad över att jag fått dela denna resa med just dig. Tack för alla samtal om forskning, familjeliv, nuet, framtiden och allt annat som är viktigt i livet. I vått och torrt, i glädje och sorg har vi funnits där för varandra. I dig har jag funnit en vän för livet!
Under mina år som doktorand i Sophias grupp har jag haft förmånen att jobba med ett härligt gäng människor; My, Ronny, Tove A, Cilla, Karin, Johanna, Jenny, Henrik W, Margareta, Anna K, Johan, Tove B, Micke, Sara K, Mattias, Sarah L och alla exjobbare. Tack för forskningsfrukostar, trevliga gruppmiddagar och äventyr till Måla, Funäs, Tårtan och Bredsund.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
58
”HPA-tjejerna”; Anna S, Marica, Caroline, Johanna och Karin, tusen tack för trevliga luncher, fikapauser, biokvällar, förfester, middagar och för att ni är så goa människor. Numer ses vi inte lika ofta, men desto trevligare blir det när vi väl ses.
Min nuvarande skrivrumskamrat tillika världens bästa bollplank: Holger – tack för samtal om vetenskap, ledarskap och allt annat som är viktigt i livet.
Efter över tio år i Proteinfabriken har jag haft förmånen att lära känna en hel drös härliga och hjälpsamma människor. Tack Jenny, Johanna, Ami, Maria S, Anneli K, Robin, Ulla, Asif, Emma, Anna K, Kattis, LanLan, Jens, Lasse, Hero, Louise, Malin, Mia, Martin H, Marie, Maria B, Anna Bä, FeiFan, Sara K, Diana, Alexandra, Adrian, Anneli, Julia, Anton, Elsa, Amanda, Fredrik, Sebastian, Axel, Anna Be, Madeleine, Jenny B, Anne-Sophie, Lisa, Petter, Roxana och all extrapersonal/sommarjobbare för trevliga fikapauser, massa PrESTar och en fantastisk gemenskap!!!
Extra tack till den FoIng-grupp vars kvinnonamn passar väl in i en roman av Marianne Fredriksson. Jag har nog aldrig haft så roliga, långa och produktiva möten som de jag hade tillsammans med er. Anna K att arbeta med dig var ett sant nöje. Du är inte bara omtänksam, kreativ och nyfiken, du har dessutom alltid nära till skratt. Vill även passa på att tacka kloka fina Ulla för allt du ger och att du stod ut med oss ungtuppar! ;)
Övriga HPAare genom åren är givetvis också väl värda ett tack. Extra tack till Pia, Stisse, Mårten, Jenny F och slutligen Martin Z och Åsa för LIMS-stöd och ett oräkneligt antal välbehövliga listor!
Under åren på KTH har jag haft möjligheten att åka på ett antal kurser och konferenser runt om i världen, upplevelserna hade inte varit de samma utan finfint sällskap av kollegor. Extra tack till Tove A, Björn, Hammou och Fredrik E för en galet kul natt i Tokyo under pågående avhandlingsskrivande. Jag vill även passa på att tacka alla som jag delat skrivrum med: HPAarna i rookierummet, Mikaela, Caroline G, Cilla, Margareta, Nina, Johanna, Hovsep, Erik P, Henrik S, Peter, Camilla, Pelin, Cajsa, Helena och Holger.
Utöver alla jobbrelaterade aktiviteter har jag under mina år på AlbaNova även haft förmånen att få sjunga med några av er. Lucia har alltid varit vår höjdpunkt och genom åren har vi inte bara sjungit för kollegor utan även ett antal Nobelpristagare! Jesper, Linda, Cilla, Cajsa, My, Tobbe, Linn, Tove B, Sara K, Anneli, Josefin, Andreas, Micke, Magnus och alla ni andra – tack för alla ljuva toner!!!
Bästa Caroline – sen en augustidag i K2 har vi följts åt. Oavsett avstånd mellan oss kan du konsten att finnas där för mig när jag behöver det som mest. Är dig och Micke evigt tacksam för att ni lockade med mig på Esquadern, utan den seglatsen undrar jag hur mitt liv sett ut idag! ;)
Maria och Otto, underbara vänner, tack för att ni är ni, för härliga fjällsemestrar och supermysiga helger i Falun med tid för umgänge och glada skratt men även för eftertanke. Vill även passa på att tack dig Maria för att du piggat upp den färglösa textmassan i denna avhandling med fantastiska illustrationer!
Louise, Cecilia och Harriet, vad vore livet utan fina vänner? Vår gemensamma resa är lång och antalet minnesvärda stunder oräkneliga. Vi har inte bara delat glädje med varandra utan även livets svåra stunder. Att veta att ni finns är en trygghet.
Sara R – det var i sandådan vi lärde känna varandra. Under barndomen visionerade vi ofta om framtiden; du skulle bli författare och tilldelas Nobelpriset i litteratur, själv skulle jag satsa på vetenskap och samma år ta hem fysikpriset. Några pris lär del väl inte bli, men drömmarna har blivit verklighet.
Uffe och Iris, jag ser fram emot att snart åter få tid för ett rafflande parti Settlers!

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
59
Har sen barnsben haft förmånen att ha två vuxna medvandrare. I helt olika sammanhang lärde jag känna er, men ni har båda gjort ett tydligt avtryck i mig och mitt ledarskap. Monne – tack för samtal, läger, ledarskap, fjällvandring och många glada skratt. Håkan M – tack för fredagsmys i mittcirkeln, minnesvärda framgångar med F80, ständiga möjligheter till extrajobb och en målmedvetenhet som smittar av sig.
Kjelle och Lisa – tack för mysiga middagar, tips om trädgårdsskötsel och världens bästa Sebastian. Christoffer och Millan, tyvärr hinner vi inte ses alltför ofta, men vi har det desto trevligare när vi väl ses!
Ö-vikssläkten med moster Ann-Marie, morbror Håkan och Lisbet och givetvis alla kusiner – tack för oförglömliga sommarlov och härliga vinteräventyr. Det är alltid lika kul att åka upp!
Finaste morbror Nicke och Inger, jag är så himla glad att jag har er. Det är en trygghet att omges av två så kloka, omtänksamma och stöttande människor som ni.
Sara, Tero och de tre godingarna Svea, Walter och Ester – det är en ynnest att ha er ett stenkast bort. Tack för hjälp med barnen när tiden inte riktigt räcker till!
Världens bästa Mamma och Pappa – tack för gränslös kärlek och trygghet! Ser idag tillbaka på min uppväxt med glädje och önskar att jag kan ge mina barn lika mycket som ni har givit mig. Mamma – kloka, underbara, omtänksamma lilla mor, ingen känner mig så väl som du gjorde. Du visste alltid när jag behövde ditt stöd som mest. Pappa – ”Liten” har blivit stor och ser med tacksamhet tillbaka på en uppväxt där jag tilläts vara den pojkflicka jag är. Minns med glädje alla idrottsupplevelser som vi delat genom åren.
Underbara älskade Sebastian, är glad, stolt och tacksam över att få dela mitt liv med dig! Du gör mig hel! Tack för att du har stått ut med mig under denna intensiva slutfas och för att du snabbt knackar ihop ett program när du tycker att mina manuella manövrar känns tidsineffektiva. Astrid och Alvar, älskade mirakel, vad vore livet utan er?! Tack för att ni konstant påminner mig om vad som är viktigt i livet!
This work has been financially supported by grants from the Knut and Alice Wallenberg Foundation.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
60

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
61
References 1. Dobson, C.M., Experimental investigation of protein folding and misfolding. Methods, 2004. 34(1): p. 4-14. 2. Brandén, C. and J. Tooze, Introduction to Protein Structure, 2nd edition. 1999. 3. Crick, F., Central dogma of molecular biology. Nature, 1970. 227(5258): p. 561-3. 4. Lander, E.S., et al., Initial sequencing and analysis of the human genome. Nature, 2001. 409(6822): p. 860-921. 5. Venter, J.C., et al., The sequence of the human genome. Science, 2001. 291(5507): p. 1304-51. 6. Legrain, P., et al., The human proteome project: current state and future direction. Mol Cell Proteomics, 2011. 10(7): p.
M111 009993. 7. Gileadi, O., et al., The scientific impact of the Structural Genomics Consortium: a protein family and ligand-centered approach
to medically-relevant human proteins. J Struct Funct Genomics, 2007. 8(2-3): p. 107-19. 8. Protein Structure Initiative (PSI). Available from: http://www.nigms.nih.gov/Research/FeaturedPrograms/PSI/. 9. Villaverde, A. and M.M. Carrio, Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol
Lett, 2003. 25(17): p. 1385-95. 10. Itakura, K., et al., Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science, 1977.
198(4321): p. 1056-63. 11. Andersen, D.C. and L. Krummen, Recombinant protein expression for therapeutic applications. Curr Opin Biotechnol,
2002. 13(2): p. 117-23. 12. Assenberg, R., et al., Advances in recombinant protein expression for use in pharmaceutical research. Curr Opin Struct Biol,
2013. 23(3): p. 393-402. 13. Terpe, K., Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical
fundamentals to commercial systems. Appl Microbiol Biotechnol, 2006. 72(2): p. 211-22. 14. Mattanovich, D., et al., Recombinant protein production in yeasts. Methods Mol Biol, 2012. 824: p. 329-58. 15. Demain, A.L. and P. Vaishnav, Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv,
2009. 27(3): p. 297-306. 16. Baneyx, F., Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol, 1999. 10(5): p. 411-21. 17. Makrides, S.C., Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev, 1996. 60(3): p.
512-38. 18. Hunt, I., From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Protein Expr
Purif, 2005. 40(1): p. 1-22. 19. Esposito, D. and D.K. Chatterjee, Enhancement of soluble protein expression through the use of fusion tags. Curr Opin
Biotechnol, 2006. 17(4): p. 353-8. 20. Walls, D. and S.T. Loughran, Tagging recombinant proteins to enhance solubility and aid purification. Methods Mol Biol,
2011. 681: p. 151-75. 21. Baneyx, F. and M. Mujacic, Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol, 2004.
22(11): p. 1399-408. 22. Georgiou, G. and P. Valax, Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol, 1996. 7(2):
p. 190-7. 23. Harley, C.B. and R.P. Reynolds, Analysis of E. coli promoter sequences. Nucleic Acids Res, 1987. 15(5): p. 2343-61. 24. Hawley, D.K. and W.R. McClure, Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids
Res, 1983. 11(8): p. 2237-55. 25. Rosenberg, M. and D. Court, Regulatory sequences involved in the promotion and termination of RNA transcription. Annu
Rev Genet, 1979. 13: p. 319-53. 26. Mulligan, M.E., J. Brosius, and W.R. McClure, Characterization in vitro of the effect of spacer length on the activity of
Escherichia coli RNA polymerase at the TAC promoter. J Biol Chem, 1985. 260(6): p. 3529-38. 27. Shine, J. and L. Dalgarno, The 3'-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense
triplets and ribosome binding sites. Proc Natl Acad Sci U S A, 1974. 71(4): p. 1342-6. 28. Hall, M.N., et al., A role for mRNA secondary structure in the control of translation initiation. Nature, 1982. 295(5850): p.
616-8. 29. Chen, H., et al., Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation
initiation codon of Escherichia coli mRNAs. Nucleic Acids Res, 1994. 22(23): p. 4953-7. 30. Betlach, M., et al., A restriction endonuclease analysis of the bacterial plasmid controlling the ecoRI restriction and modification
of DNA. Fed Proc, 1976. 35(9): p. 2037-43. 31. Cozzarelli, N.R., R.B. Kelly, and A. Kornberg, A minute circular DNA from Escherichia coli 15. Proc Natl Acad Sci
U S A, 1968. 60(3): p. 992-9.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
62
32. Bolivar, F., et al., Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene, 1977. 2(2): p. 95-113.
33. Balbas, P. and F. Bolivar, Back to basics: pBR322 and protein expression systems in E. coli. Methods Mol Biol, 2004. 267: p. 77-90.
34. Lin-Chao, S., W.T. Chen, and T.T. Wong, High copy number of the pUC plasmid results from a Rom/Rop-suppressible point mutation in RNA II. Mol Microbiol, 1992. 6(22): p. 3385-93.
35. Vieira, J. and J. Messing, The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene, 1982. 19(3): p. 259-68.
36. Chang, A.C. and S.N. Cohen, Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol, 1978. 134(3): p. 1141-56.
37. Mayer, M.P., A new set of useful cloning and expression vectors derived from pBlueScript. Gene, 1995. 163(1): p. 41-6. 38. Tomizawa, J., Control of ColE1 plasmid replication: the process of binding of RNA I to the primer transcript. Cell, 1984.
38(3): p. 861-70. 39. Tomizawa, J., Control of ColE1 plasmid replication: initial interaction of RNA I and the primer transcript is reversible. Cell,
1985. 40(3): p. 527-35. 40. Donoghue, D.J. and P.A. Sharp, Replication of colicin E1 plasmid DNA in vivo requires no plasmid-encoded proteins. J
Bacteriol, 1978. 133(3): p. 1287-94. 41. Tomizawa, J. and T. Som, Control of ColE1 plasmid replication: enhancement of binding of RNA I to the primer transcript
by the Rom protein. Cell, 1984. 38(3): p. 871-8. 42. Cesareni, G., M.A. Muesing, and B. Polisky, Control of ColE1 DNA replication: the rop gene product negatively affects
transcription from the replication primer promoter. Proc Natl Acad Sci U S A, 1982. 79(20): p. 6313-7. 43. Som, T. and J. Tomizawa, Regulatory regions of ColE1 that are involved in determination of plasmid copy number. Proc
Natl Acad Sci U S A, 1983. 80(11): p. 3232-6. 44. Twigg, A.J. and D. Sherratt, Trans-complementable copy-number mutants of plasmid ColE1. Nature, 1980. 283(5743): p.
216-8. 45. Friehs, K., Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol, 2004. 86: p. 47-82. 46. Davison, J., Mechanism of control of DNA replication and incompatibility in ColE1-type plasmids--a review. Gene, 1984.
28(1): p. 1-15. 47. Velappan, N., et al., Plasmid incompatibility: more compatible than previously thought? Protein Eng Des Sel, 2007. 20(7):
p. 309-13. 48. Choi, J.W., K.S. Ra, and S.L. Lee, Enhancement of bovine growth hormone gene expression by increasing the plasmid copy
number. Biotechnology Letters, 1999. 21: p. 1-5. 49. Ramos, C.R., et al., A high-copy T7 Escherichia coli expression vector for the production of recombinant proteins with a
minimal N-terminal His-tagged fusion peptide. Braz J Med Biol Res, 2004. 37(8): p. 1103-9. 50. Glick, B.R., Metabolic load and heterologous gene expression. Biotechnol Adv, 1995. 13(2): p. 247-61. 51. Bentley, W.E., et al., Plasmid-encoded protein: the principal factor in the "metabolic burden" associated with recombinant
bacteria. Biotechnol Bioeng, 1990. 35(7): p. 668-81. 52. Jones, K.L. and J.D. Keasling, Construction and characterization of F plasmid-based expression vectors. Biotechnol
Bioeng, 1998. 59(6): p. 659-65. 53. Gronenborn, B., Overproduction of phage lambda repressor under control of the lac promotor of Escherichia coli. Mol Gen
Genet, 1976. 148(3): p. 243-50. 54. Wanner, B.L., R. Kodaira, and F.C. Neidhardt, Physiological regulation of a decontrolled lac operon. J Bacteriol, 1977.
130(1): p. 212-22. 55. Yansura, D.G. and D.J. Henner, Use of Escherichia coli trp promoter for direct expression of proteins. Methods Enzymol,
1990. 185: p. 54-60. 56. de Boer, H.A., L.J. Comstock, and M. Vasser, The tac promoter: a functional hybrid derived from the trp and lac promoters.
Proc Natl Acad Sci U S A, 1983. 80(1): p. 21-5. 57. Brosius, J., M. Erfle, and J. Storella, Spacing of the -10 and -35 regions in the tac promoter. Effect on its in vivo activity. J
Biol Chem, 1985. 260(6): p. 3539-41. 58. Guzman, L.M., et al., Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD
promoter. J Bacteriol, 1995. 177(14): p. 4121-30. 59. Studier, F.W. and B.A. Moffatt, Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned
genes. J Mol Biol, 1986. 189(1): p. 113-30. 60. Dubendorff, J.W. and F.W. Studier, Controlling basal expression in an inducible T7 expression system by blocking the target
T7 promoter with lac repressor. J Mol Biol, 1991. 219(1): p. 45-59. 61. Skerra, A., Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli.
Gene, 1994. 151(1-2): p. 131-5. 62. Mermod, N., et al., Vector for regulated expression of cloned genes in a wide range of gram-negative bacteria. J Bacteriol,
1986. 167(2): p. 447-54.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
63
63. Kikuchi, Y., et al., The nucleotide sequence of the promoter and the amino-terminal region of alkaline phosphatase structural gene (phoA) of Escherichia coli. Nucleic Acids Res, 1981. 9(21): p. 5671-8.
64. Miyake, T., et al., Secretion of human interferon-alpha induced by using secretion vectors containing a promoter and signal sequence of alkaline phosphatase gene of Escherichia coli. J Biochem, 1985. 97(5): p. 1429-36.
65. Oka, T., et al., Synthesis and secretion of human epidermal growth factor by Escherichia coli. Proc Natl Acad Sci U S A, 1985. 82(21): p. 7212-6.
66. Bernard, H.U., et al., Construction of plasmid cloning vehicles that promote gene expression from the bacteriophage lambda pL promoter. Gene, 1979. 5(1): p. 59-76.
67. Meyer, B.J., R. Maurer, and M. Ptashne, Gene regulation at the right operator (OR) of bacteriophage lambda. II. OR1, OR2, and OR3: their roles in mediating the effects of repressor and cro. J Mol Biol, 1980. 139(2): p. 163-94.
68. Ptashne, M., et al., How the lambda repressor and cro work. Cell, 1980. 19(1): p. 1-11. 69. Zabeau, M. and K.K. Stanley, Enhanced expression of cro-beta-galactosidase fusion proteins under the control of the PR
promoter of bacteriophage lambda. EMBO J, 1982. 1(10): p. 1217-24. 70. Bukrinsky, M.I., E.V. Barsov, and A.A. Shilov, Multicopy expression vector based on temperature-regulated lac repressor:
expression of human immunodeficiency virus env gene in Escherichia coli. Gene, 1988. 70(2): p. 415-7. 71. Tanabe, H., et al., Identification of the promoter region of the Escherichia coli major cold shock gene, cspA. J Bacteriol, 1992.
174(12): p. 3867-73. 72. Lofdahl, S., et al., Gene for staphylococcal protein A. Proc Natl Acad Sci U S A, 1983. 80(3): p. 697-701. 73. Keilty, S. and M. Rosenberg, Constitutive function of a positively regulated promoter reveals new sequences essential for activity.
J Biol Chem, 1987. 262(13): p. 6389-95. 74. Estrem, S.T., et al., Identification of an UP element consensus sequence for bacterial promoters. Proc Natl Acad Sci U S A,
1998. 95(17): p. 9761-6. 75. Ross, W., et al., A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase.
Science, 1993. 262(5138): p. 1407-13. 76. Jacob, F. and J. Monod, Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol, 1961. 3: p. 318-56. 77. Busby, S. and R.H. Ebright, Transcription activation by catabolite activator protein (CAP). J Mol Biol, 1999. 293(2): p.
199-213. 78. Reznikoff, W.S., Catabolite gene activator protein activation of lac transcription. J Bacteriol, 1992. 174(3): p. 655-8. 79. Monod, J. and G. Cohen-Bazire, [The effect of specific inhibition in biosynthesis of tryptophan-desmase by Aerobacter
aerogenes]. C R Hebd Seances Acad Sci, 1953. 236(5): p. 530-2. 80. Yanofsky, C., et al., The complete nucleotide sequence of the tryptophan operon of Escherichia coli. Nucleic Acids Res, 1981.
9(24): p. 6647-68. 81. Amann, E., J. Brosius, and M. Ptashne, Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned
genes in Escherichia coli. Gene, 1983. 25(2-3): p. 167-78. 82. Morgan-Kiss, R.M., C. Wadler, and J.E. Cronan, Jr., Long-term and homogeneous regulation of the Escherichia coli
araBAD promoter by use of a lactose transporter of relaxed specificity. Proc Natl Acad Sci U S A, 2002. 99(11): p. 7373-7. 83. Chamberlin, M. and J. Ring, Characterization of T7-specific ribonucleic acid polymerase. 1. General properties of the
enzymatic reaction and the template specificity of the enzyme. J Biol Chem, 1973. 248(6): p. 2235-44. 84. Moffatt, B.A., J.J. Dunn, and F.W. Studier, Nucleotide sequence of the gene for bacteriophage T7 RNA polymerase. J Mol
Biol, 1984. 173(2): p. 265-9. 85. Moffatt, B.A. and F.W. Studier, T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell, 1987. 49(2): p. 221-7. 86. Studier, F.W., Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol, 1991. 219(1): p.
37-44. 87. Grossman, T.H., et al., Spontaneous cAMP-dependent derepression of gene expression in stationary phase plays a role in
recombinant expression instability. Gene, 1998. 209(1-2): p. 95-103. 88. Golomb, M. and M. Chamberlin, Characterization of T7-specific ribonucleic acid polymerase. IV. Resolution of the major in
vitro transcripts by gel electrophoresis. J Biol Chem, 1974. 249(9): p. 2858-63. 89. Vasina, J.A. and F. Baneyx, Recombinant protein expression at low temperatures under the transcriptional control of the major
Escherichia coli cold shock promoter cspA. Appl Environ Microbiol, 1996. 62(4): p. 1444-7. 90. Vasina, J.A. and F. Baneyx, Expression of aggregation-prone recombinant proteins at low temperatures: a comparative study of
the Escherichia coli cspA and tac promoter systems. Protein Expr Purif, 1997. 9(2): p. 211-8. 91. Goldstein, M.A. and R.H. Doi, Prokaryotic promoters in biotechnology. Biotechnol Annu Rev, 1995. 1: p. 105-28. 92. Newbury, S.F., et al., Stabilization of translationally active mRNA by prokaryotic REP sequences. Cell, 1987. 48(2): p.
297-310. 93. Schultz, T., L. Martinez, and A. de Marco, The evaluation of the factors that cause aggregation during recombinant
expression in E. coli is simplified by the employment of an aggregation-sensitive reporter. Microb Cell Fact, 2006. 5: p. 28. 94. Balzer, S., et al., A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene
expression in Escherichia coli. Microb Cell Fact, 2013. 12(1): p. 26. 95. Keasling, J.D., Gene-expression tools for the metabolic engineering of bacteria. Trends Biotechnol, 1999. 17(11): p. 452-
60.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
64
96. Malys, N. and J.E. McCarthy, Translation initiation: variations in the mechanism can be anticipated. Cell Mol Life Sci, 2011. 68(6): p. 991-1003.
97. Scherer, G.F., et al., The ribosome binding sites recognized by E. coli ribosomes have regions with signal character in both the leader and protein coding segments. Nucleic Acids Res, 1980. 8(17): p. 3895-907.
98. Poole, E.S., C.M. Brown, and W.P. Tate, The identity of the base following the stop codon determines the efficiency of in vivo translational termination in Escherichia coli. EMBO J, 1995. 14(1): p. 151-8.
99. Fargo, D.C., et al., Shine-Dalgarno-like sequences are not required for translation of chloroplast mRNAs in Chlamydomonas reinhardtii chloroplasts or in Escherichia coli. Mol Gen Genet, 1998. 257(3): p. 271-82.
100. Nakamoto, T., A unified view of the initiation of protein synthesis. Biochem Biophys Res Commun, 2006. 341(3): p. 675-8.
101. Ma, J., A. Campbell, and S. Karlin, Correlations between Shine-Dalgarno sequences and gene features such as predicted expression levels and operon structures. J Bacteriol, 2002. 184(20): p. 5733-45.
102. De Boer, H.A., et al., A hybrid promoter and portable Shine-Dalgarno regions of Escherichia coli. Biochem Soc Symp, 1983. 48: p. 233-44.
103. Komarova, A.V., et al., Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA, 2002. 8(9): p. 1137-47.
104. Schurr, T., E. Nadir, and H. Margalit, Identification and characterization of E.coli ribosomal binding sites by free energy computation. Nucleic Acids Res, 1993. 21(17): p. 4019-23.
105. Vimberg, V., et al., Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol, 2007. 8: p. 100. 106. Ringquist, S., et al., Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol,
1992. 6(9): p. 1219-29. 107. Stormo, G.D., T.D. Schneider, and L.M. Gold, Characterization of translational initiation sites in E. coli. Nucleic
Acids Res, 1982. 10(9): p. 2971-96. 108. Schneider, T.D., et al., Information content of binding sites on nucleotide sequences. J Mol Biol, 1986. 188(3): p. 415-31. 109. Stenstrom, C.M., E. Holmgren, and L.A. Isaksson, Cooperative effects by the initiation codon and its flanking regions on
translation initiation. Gene, 2001. 273(2): p. 259-65. 110. Van Etten, W.J. and G.R. Janssen, An AUG initiation codon, not codon-anticodon complementarity, is required for the
translation of unleadered mRNA in Escherichia coli. Mol Microbiol, 1998. 27(5): p. 987-1001. 111. Looman, A.C., et al., Influence of the codon following the AUG initiation codon on the expression of a modified lacZ gene in
Escherichia coli. EMBO J, 1987. 6(8): p. 2489-92. 112. Sato, T., et al., Codon and base biases after the initiation codon of the open reading frames in the Escherichia coli genome and
their influence on the translation efficiency. J Biochem, 2001. 129(6): p. 851-60. 113. Stenstrom, C.M., et al., Codon bias at the 3'-side of the initiation codon is correlated with translation initiation efficiency in
Escherichia coli. Gene, 2001. 263(1-2): p. 273-84. 114. Zamora-Romo, E., et al., Efficient expression of gene variants that harbour AGA codons next to the initiation codon.
Nucleic Acids Res, 2007. 35(17): p. 5966-74. 115. Gonzalez de Valdivia, E.I. and L.A. Isaksson, A codon window in mRNA downstream of the initiation codon where
NGG codons give strongly reduced gene expression in Escherichia coli. Nucleic Acids Res, 2004. 32(17): p. 5198-205. 116. Stenstrom, C.M. and L.A. Isaksson, Influences on translation initiation and early elongation by the messenger RNA region
flanking the initiation codon at the 3' side. Gene, 2002. 288(1-2): p. 1-8. 117. Krishna Rao, D.V., et al., Optimization of the AT-content of codons immediately downstream of the initiation codon and
evaluation of culture conditions for high-level expression of recombinant human G-CSF in Escherichia coli. Mol Biotechnol, 2008. 38(3): p. 221-32.
118. Ahn, J.H., J.W. Keum, and D.M. Kim, High-throughput, combinatorial engineering of initial codons for tunable expression of recombinant proteins. J Proteome Res, 2008. 7(5): p. 2107-13.
119. Bandmann, N. and P.A. Nygren, Combinatorial expression vector engineering for tuning of recombinant protein production in Escherichia coli. Nucleic Acids Res, 2007. 35(5): p. e32.
120. Makino, T., et al., Comprehensive engineering of Escherichia coli for enhanced expression of IgG antibodies. Metab Eng, 2011. 13(2): p. 241-51.
121. Simmons, L.C. and D.G. Yansura, Translational level is a critical factor for the secretion of heterologous proteins in Escherichia coli. Nat Biotechnol, 1996. 14(5): p. 629-34.
122. Gustafsson, C., S. Govindarajan, and J. Minshull, Codon bias and heterologous protein expression. Trends Biotechnol, 2004. 22(7): p. 346-53.
123. Kane, J.F., Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol, 1995. 6(5): p. 494-500.
124. Chen, D. and D.E. Texada, Low-usage codons and rare codons of Escherichia coli. Gene Ther Mol Biol, 2006. 10: p. 1-12.
125. Calderone, T.L., R.D. Stevens, and T.G. Oas, High-level misincorporation of lysine for arginine at AGA codons in a fusion protein expressed in Escherichia coli. J Mol Biol, 1996. 262(4): p. 407-12.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
65
126. McNulty, D.E., et al., Mistranslational errors associated with the rare arginine codon CGG in Escherichia coli. Protein Expr Purif, 2003. 27(2): p. 365-74.
127. Gerchman, S.E., V. Graziano, and V. Ramakrishnan, Expression of chicken linker histones in E. coli: sources of problems and methods for overcoming some of the difficulties. Protein Expr Purif, 1994. 5(3): p. 242-51.
128. Spanjaard, R.A. and J. van Duin, Translation of the sequence AGG-AGG yields 50% ribosomal frameshift. Proc Natl Acad Sci U S A, 1988. 85(21): p. 7967-71.
129. Kane, J.F., et al., Novel in-frame two codon translational hop during synthesis of bovine placental lactogen in a recombinant strain of Escherichia coli. Nucleic Acids Res, 1992. 20(24): p. 6707-12.
130. Goldman, E., et al., Consecutive low-usage leucine codons block translation only when near the 5' end of a message in Escherichia coli. J Mol Biol, 1995. 245(5): p. 467-73.
131. Kim, S. and S.B. Lee, Rare codon clusters at 5'-end influence heterologous expression of archaeal gene in Escherichia coli. Protein Expr Purif, 2006. 50(1): p. 49-57.
132. Rosenberg, A.H., et al., Effects of consecutive AGG codons on translation in Escherichia coli, demonstrated with a versatile codon test system. J Bacteriol, 1993. 175(3): p. 716-22.
133. Hatfield, G.W. and D.A. Roth, Optimizing scaleup yield for protein production: Computationally Optimized DNA Assembly (CODA) and Translation Engineering. Biotechnol Annu Rev, 2007. 13: p. 27-42.
134. Angov, E., Codon usage: nature's roadmap to expression and folding of proteins. Biotechnol J, 2011. 6(6): p. 650-9. 135. Menzella, H.G., Comparison of two codon optimization strategies to enhance recombinant protein production in Escherichia coli.
Microb Cell Fact, 2011. 10: p. 15. 136. Villalobos, A., et al., Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC
Bioinformatics, 2006. 7: p. 285. 137. Brinkmann, U., R.E. Mattes, and P. Buckel, High-level expression of recombinant genes in Escherichia coli is dependent on
the availability of the dnaY gene product. Gene, 1989. 85(1): p. 109-14. 138. Baca, A.M. and W.G. Hol, Overcoming codon bias: a method for high-level overexpression of Plasmodium and other AT-rich
parasite genes in Escherichia coli. Int J Parasitol, 2000. 30(2): p. 113-8. 139. Burgess-Brown, N.A., et al., Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene
study. Protein Expr Purif, 2008. 59(1): p. 94-102. 140. Fu, W., J. Lin, and P. Cen, 5-Aminolevulinate production with recombinant Escherichia coli using a rare codon optimizer host
strain. Appl Microbiol Biotechnol, 2007. 75(4): p. 777-82. 141. Kleber-Janke, T. and W.M. Becker, Use of modified BL21(DE3) Escherichia coli cells for high-level expression of
recombinant peanut allergens affected by poor codon usage. Protein Expr Purif, 2000. 19(3): p. 419-24. 142. Sorensen, H.P., H.U. Sperling-Petersen, and K.K. Mortensen, Production of recombinant thermostable proteins
expressed in Escherichia coli: completion of protein synthesis is the bottleneck. J Chromatogr B Analyt Technol Biomed Life Sci, 2003. 786(1-2): p. 207-14.
143. Maertens, B., et al., Gene optimization mechanisms: a multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Sci, 2010. 19(7): p. 1312-26.
144. Han, J.H., et al., Codon optimization enhances protein expression of human peptide deformylase in E. coli. Protein Expr Purif, 2010. 70(2): p. 224-30.
145. Li, A., et al., Optimized gene synthesis and high expression of human interleukin-18. Protein Expr Purif, 2003. 32(1): p. 110-8.
146. Zhou, Z., et al., Enhanced expression of a recombinant malaria candidate vaccine in Escherichia coli by codon optimization. Protein Expr Purif, 2004. 34(1): p. 87-94.
147. Enfors, S.O. and L. Häggström, Bioprocess Technology - Fundamentals and Applications. 2000. 148. Lee, J., et al., Control of fed-batch fermentations. Biotechnol Adv, 1999. 17(1): p. 29-48. 149. Yamane, T. and S. Shimizu, Fed-batch Techniques in Microbial Processes. 1984. 150. Buchs, J., Introduction to advantages and problems of shaken cultures. Biochem Eng J, 2001. 7(2): p. 91-98. 151. Losen, M., et al., Effect of oxygen limitation and medium composition on Escherichia coli fermentation in shake-flask cultures.
Biotechnol Prog, 2004. 20(4): p. 1062-8. 152. Vasala, A., et al., A new wireless system for decentralised measurement of physiological parameters from shake flasks. Microb
Cell Fact, 2006. 5: p. 8. 153. Lee, S.Y., High cell-density culture of Escherichia coli. Trends Biotechnol, 1996. 14(3): p. 98-105. 154. Shiloach, J. and R. Fass, Growing E. coli to high cell density--a historical perspective on method development. Biotechnol
Adv, 2005. 23(5): p. 345-57. 155. Strandberg, L. and S.O. Enfors, Batch and fed batch cultivations for the temperature induced production of a recombinant
protein in Escherichia coli. Biotechnology Letters, 1991. 13(8): p. 609-614. 156. Lutomski, D., et al., Purification of human galectin-1 produced in high-cell density cultures of recombinant Escherichia coli: a
comparison with classic shake flask cultivation. J Chromatogr B Analyt Technol Biomed Life Sci, 2004. 808(1): p. 105-9.
157. Goyal, D., G. Sahni, and D.K. Sahoo, Enhanced production of recombinant streptokinase in Escherichia coli using fed-batch culture. Bioresour Technol, 2009. 100(19): p. 4468-74.

PROTEOME WIDE PROTEIN PRODUCTION
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
66
158. Ren, Q., et al., High level production of tyrosinase in recombinant Escherichia coli. BMC Biotechnol, 2013. 13: p. 18. 159. Tripathi, N.K., et al., Development of a simple fed-batch process for the high-yield production of recombinant Japanese
encephalitis virus protein. Appl Microbiol Biotechnol, 2010. 86(6): p. 1795-803. 160. Restaino, O.F., et al., High cell density cultivation of a recombinant E. coli strain expressing a key enzyme in bioengineered
heparin production. Appl Microbiol Biotechnol, 2013. 97(9): p. 3893-900. 161. Panula-Perala, J., et al., Enzyme controlled glucose auto-delivery for high cell density cultivations in microplates and shake
flasks. Microb Cell Fact, 2008. 7: p. 31. 162. Krause, M., et al., A novel fed-batch based cultivation method provides high cell-density and improves yield of soluble
recombinant proteins in shaken cultures. Microb Cell Fact, 2010. 9: p. 11. 163. Mahboudi, F., et al., A fed-batch based cultivation mode in Escherichia coli results in improved specific activity of a novel
chimeric-truncated form of tissue plasminogen activator. J Appl Microbiol, 2013. 114(2): p. 364-72. 164. Ryan, W., S.J. Parulekar, and B.C. Stark, Expression of beta-lactamase by recombinant Escherichia coli strains containing
plasmids of different sizes - effects of pH, phosphate, and dissolved oxygen. Biotechnology and Bioengineering, 1989. 34: p. 309-319.
165. Donovan, R.S., C.W. Robinson, and B.R. Glick, Review: optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter. J Ind Microbiol, 1996. 16(3): p. 145-54.
166. Hortsch, R. and D. Weuster-Botz, Growth and recombinant protein expression with Escherichia coli in different batch cultivation media. Appl Microbiol Biotechnol, 2011. 90(1): p. 69-76.
167. Athmaram, T.N., et al., Optimization of Dengue-3 recombinant NS1 protein expression in E. coli and in vitro refolding for diagnostic applications. Virus Genes, 2013. 46(2): p. 219-30.
168. Collins, T., et al., Batch production of a silk-elastin-like protein in E. coli BL21(DE3): key parameters for optimisation. Microb Cell Fact, 2013. 12: p. 21.
169. Babaeipour, V., et al., Enhancement of human granulocyte-colony stimulating factor production in recombinant E. coli using batch cultivation. Bioprocess Biosyst Eng, 2010. 33(5): p. 591-8.
170. Tseng, C.L. and C.H. Leng, Influence of medium components on the expression of recombinant lipoproteins in Escherichia coli. Appl Microbiol Biotechnol, 2012. 93(4): p. 1539-52.
171. Liu, J.F., et al., Significantly enhanced production of recombinant nitrilase by optimization of culture conditions and glycerol feeding. Appl Microbiol Biotechnol, 2011. 89(3): p. 665-72.
172. Pranchevicius, M.C., et al., Characterization and optimization of ArtinM lectin expression in Escherichia coli. BMC Biotechnol, 2012. 12: p. 44.
173. Pacheco, B., et al., A screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr Purif, 2012. 81(1): p. 33-41.
174. Wang, X., et al., Evaluation of different culture conditions for high-level soluble expression of human cyclin A2 with pET vector in BL21 (DE3) and spectroscopic characterization of its inclusion body structure. Protein Expr Purif, 2007. 56(1): p. 27-34.
175. Correa, A. and P. Oppezzo, Tuning different expression parameters to achieve soluble recombinant proteins in E. coli: advantages of high-throughput screening. Biotechnol J, 2011. 6(6): p. 715-30.
176. Berrow, N.S., et al., Recombinant protein expression and solubility screening in Escherichia coli: a comparative study. Acta Crystallogr D Biol Crystallogr, 2006. 62(Pt 10): p. 1218-26.
177. Vincentelli, R., et al., High-throughput protein expression screening and purification in Escherichia coli. Methods, 2011. 55(1): p. 65-72.
178. Davey, K.R., Modelling the combined effect of temperature and pH on the rate coefficient for bacterial growth. Int J Food Microbiol, 1994. 23(3-4): p. 295-303.
179. Scheidle, M., et al., Controlling pH in shake flasks using polymer-based controlled-release discs with pre-determined release kinetics. BMC Biotechnol, 2011. 11: p. 25.
180. Muntari, B., et al., Recombinant bromelain production in Escherichia coli: process optimization in shake flask culture by response surface methodology. AMB Express, 2012. 2: p. 12.
181. Studier, F.W., Protein production by auto-induction in high density shaking cultures. Protein Expr Purif, 2005. 41(1): p. 207-34.
182. San-Miguel, T., P. Perez-Bermudez, and I. Gavidia, Production of soluble eukaryotic recombinant proteins in is favoured in early log-phase cultures induced at low temperature. Springerplus, 2013. 2(1): p. 89.
183. Weuster-Botz, D., Parallel reactor systems for bioprocess development. Adv Biochem Eng Biotechnol, 2005. 92: p. 125-43.
184. Graslund, S., et al., The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif, 2008. 58(2): p. 210-21.
185. Lesley, S.A., High-throughput proteomics: protein expression and purification in the postgenomic world. Protein Expr Purif, 2001. 22(2): p. 159-64.
186. Janson, J.-C., Protein Purification: Principles, High Resolution Methods, and Applications, 3rd edition. 2011. 187. Porath, J. and P. Flodin, Gel filtration: a method for desalting and group separation. Nature, 1959. 183(4676): p. 1657-9. 188. Sober, H.A. and E.A. Peterson, Protein chromatography on ion exchange cellulose. Fed Proc, 1958. 17(4): p. 1116-26.

HANNA TEGEL
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
67
189. Howard, G.A. and A.J. Martin, The separation of the C12-C18 fatty acids by reversed-phase partition chromatography. Biochem J, 1950. 46(5): p. 532-8.
190. Yon, R.J., Chromatography of lipophilic proteins on adsorbents containing mixed hydrophobic and ionic groups. Biochem J, 1972. 126(3): p. 765-7.
191. Porath, J., et al., Metal chelate affinity chromatography, a new approach to protein fractionation. Nature, 1975. 258(5536): p. 598-9.
192. Cuatrecasas, P., M. Wilchek, and C.B. Anfinsen, Selective enzyme purification by affinity chromatography. Proc Natl Acad Sci U S A, 1968. 61(2): p. 636-43.
193. Hochuli, E., et al., Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent. Nature Biotechnology, 1988. 6: p. 1321 - 1325
194. Steen, J., et al., High-throughput protein purification using an automated set-up for high-yield affinity chromatography. Protein Expr Purif, 2006. 46(2): p. 173-8.
195. Chaga, G.S., Twenty-five years of immobilized metal ion affinity chromatography: past, present and future. J Biochem Biophys Methods, 2001. 49(1-3): p. 313-34.
196. Lichty, J.J., et al., Comparison of affinity tags for protein purification. Protein Expr Purif, 2005. 41(1): p. 98-105. 197. Young, C.L., Z.T. Britton, and A.S. Robinson, Recombinant protein expression and purification: a comprehensive review of
affinity tags and microbial applications. Biotechnol J, 2012. 7(5): p. 620-34. 198. Xu, X., et al., The tandem affinity purification method: an efficient system for protein complex purification and protein interaction
identification. Protein Expr Purif, 2010. 72(2): p. 149-56. 199. Graslund, S., et al., Protein production and purification. Nat Methods, 2008. 5(2): p. 135-46. 200. Smith, P.K., et al., Measurement of Protein Using Bicinchoninic Acid. Anal Biochem., 1985. 150(1): p. 76-85. 201. Laemmeli, U.K., Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 1970. 227: p.
680-685. 202. Towbin, H., T. Staehelin, and J. Gordon, Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets:
procedure and some applications. Proc. Natl. Acad. Sci., 1979. 76(9): p. 4350-4354. 203. Thomson, J.J. Rays of positive electricity. in Proc. R. Soc. Lond. A. 1913. 204. Tanaka, K., et al., Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid
communications in mass spectrometry, 1988. 2(8): p. 151-153. 205. Karas, M. and F. Hillenkamp, Laser desorption ionization of proteins with molecular masses exceeding 10 000 Dalton. Anal.
Chem., 1988. 60: p. 2299-2301. 206. Fenn, J.B., et al., Electrospray ionization for mass spectrometry of large biomolecules. Science, 1989. 246(4926): p. 64-71. 207. Holcapek, M., R. Jirasko, and M. Lisa, Recent developments in liquid chromatography-mass spectrometry and related
techniques. J Chromatogr A, 2012. 1259: p. 3-15. 208. Aebersold, R. and M. Mann, Mass spectrometry-based proteomics. Nature, 2003. 422(6928): p. 198-207. 209. Roepstorff, P., Mass spectrometry based proteomics, background, status and future needs. Protein Cell, 2012. 3(9): p. 641-
7. 210. Uhlen, M., et al., A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics,
2005. 4(12): p. 1920-32. 211. Stenvall, M., et al., High-throughput solubility assay for purified recombinant protein immunogens. Biochim Biophys Acta,
2005. 1752(1): p. 6-10. 212. Hedhammar, M., et al., A novel flow cytometry-based method for analysis of expression levels in Escherichia coli, giving
information about precipitated and soluble protein. J Biotechnol, 2005. 119(2): p. 133-46. 213. Elsliger, M.A., et al., The JCSG high-throughput structural biology pipeline. Acta Crystallogr Sect F Struct Biol Cryst
Commun, 2010. 66(Pt 10): p. 1137-42. 214. Xiao, R., et al., The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J
Struct Biol, 2010. 172(1): p. 21-33. 215. Savitsky, P., et al., High-throughput production of human proteins for crystallization: the SGC experience. J Struct Biol,
2010. 172(1): p. 3-13. 216. Cornvik, T., et al., Colony filtration blot: a new screening method for soluble protein expression in Escherichia coli. Nat
Methods, 2005. 2(7): p. 507-9.





























