Embed Size (px)
Citation preview

Protein Folding in the 2D HP Model
Alexandros Skaliotis – King’s College London
Joint work with:•Andreas Albrecht (University of Hertfordshire)•Kathleen Steinhöfel (King’s College London)

Overview
1. Proteins
2. Protein Folding
3. 2D HP Model
4. Simple Example
5. Local Search for Protein Folding
6. Set of Moves
7. Logarithmic Cooling Schedule
8. Selected Benchmarks
9. Experiment

1. Proteins
• A protein is a sequence of amino acids encoded by a gene in a genome.
• There are 20 different amino acids.• The length of the sequence can range from about 20 to
3500.• The function of a protein is determined by its three-
dimensional structure.• Predicting this structure is quite daunting and very
expensive.

2. Protein Folding
• Protein Folding is the process by which a sequence of amino acids conforms to a three-dimensional shape.
• Anfinsen’s hypothesis suggests that proteins fold to a minimum energy state.
• So, our goal is to find a conformation with minimum energy.
• We want to investigate algorithmic aspects of simulating the folding process.
• We need to simplify it.

3.1 2D HP Model [Dill et al. 1985]
1. Classify each amino acid as hydrophobic (H) or hydrophilic (P).
2. Confine consecutive amino acids to adjacent nodes in a lattice (Treat search space as a grid).
3. Flatten the search on a 2D lattice.
• Function HHc: Number of new HH contacts
• Parameter ξ < 0: Influence ratio of the new HH
contacts (usually ξ = -1)
• Objective Function = HHc * ξ = -HHc

3.2 2D HP Model [Dill et al. 1985]
• Protein Folding in the 2D HP Model is NP-Hard for a variety of lattice structures [Paterson/Przytycka 1996; Hart/Istrail 1997; Berger/Leighton 1998; Atkins/Hart 1999].
• Constant factor approximations in linear time but not helpful for predictions of real protein sequences [Hart/Istrail 1997].
• Exact methods work only for sequences up to double digits length.

4. Simple Example
• Normally the energy is a positive number• But we have a minimisation problem, so we talk about
negative energies
Energy = 0
Energy = -3
H = RED
P = PINK

5. Local Search for Protein Folding
• A wide range of heuristics have been applied to find optimal HP structures, especially evolutionary algorithms.
• Lesh et. Al (2003) and Blazewicz et al. (2005) applied tabu search to the problem.
• We apply Logarithmic Simulated Annealing.• To move in the search space we employ a complete
and reversible set of moves proposed by Lesh et al. in 2003 and Blazewicz et al. in 2005.

6. Set of Moves
L
L
C
L
3
2
3
1
4 5
6
4 5
6
LL
4
2
4 5
6
33
11

7. Logarithmic Cooling Schedule
• Following Hajek’s theorem (1988), we are guaranteed to find the optimal solution after an infinite number of steps if and only if .
• is the maximum value of the minimal escape heights from local minima.
• Albrecht et al. show that after transitions, the probability to be in a minimum energy conformation is at least , where n is the maximum size of the neighbourhood of sequences.
• Cooling Function:
,...1,0,)2ln(
)(
kk
gkc
g
)()/( gOn
1

8. Selected Benchmarks
• S36: 3P 2H 2P 2H 5P 7H 2P 2H 4P 2H 2P 1H 2P• S60: 2P 3H 1P 8H 3P 10H 1P 1H 3P 12H 4P 6H
1P 2H 1P 1H 1P• S64: 12H 1P 1H 1P 1H 2P 2H 2P 2H 2P 1H 2P
2H 2P 2H 2P 1H 2P 2H 2P 2H 2P 1H 1P 1H 1P 12H
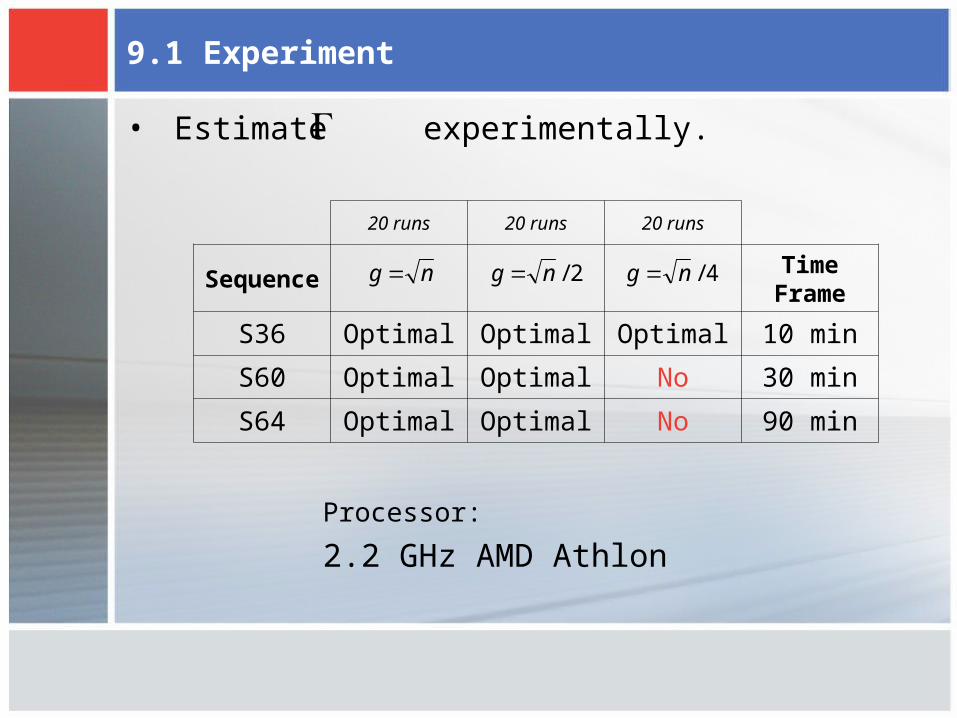
9.1 Experiment
• Estimate experimentally.
20 runs 20 runs 20 runs
SequenceTime
Frame
S36 Optimal Optimal Optimal 10 min
S60 Optimal Optimal No 30 min
S64 Optimal Optimal No 90 min
Processor:
2.2 GHz AMD Athlon
ng 2/ng 4/ng

9.2 Experiment
• We found that is a good estimated upper bound for .
• We checked this against S85 and got the best known results in 10 / 10 runs.
• Of course we need more benchmarks.• But this can be a good starting point in trying to
develop a formal proof for the value of .
2/n





























