Embed Size (px)
Citation preview

98 TIBTECH - APRIL 1990 [Vol. 8]
14 Privalov, P. L. (1979) Adv. Protein Chem. 33, 167-241
15 Becktel, W. J. and Schellman, J. A. (1987) Biopolymers 26, 1859-1877
16 Chen, B. and Schellman, J. A. (1989) Biochemistry 28, 685-691
17 Privalov, P. L., Gricko, Y., Venyaminov, S. Y. and Kutyshenko (1986) J. Mol. Biol. 190, 487-498
18 Baldwin, R. L. (1986) Proc. NatlAcad. Sci. USA 83, 8069-8072
19 Privalov, P. L. and Gill, S. J. (1988) Adv. Protein Chem. 39, 191-234
20 Alber, T. (1989) Annu. Rev. Biochem. 58, 765-798
21 Shirley, B. A., Stanssens, P., Steyaert, J. and Pace, C. N. (1989) J. Biol. Chem. 264, 11621-11625
22 States, D. J., Creighton, T. E., Dobson, C. M. and Karplus, M. (1987) J. Mol. Biol. 195, 731-739
23 Johnson, R. E., Adams, P. and Rnpley, J. A. (1978) Biochemistry 17, 1479-1484
24 Lin, S. H., Konishi, Y., Denton, M. E. and Scheraga, H. A. (1984) Biochemistry 23, 5504-5512
25 Goto, Y., Tsunenaga, M., Kawata, Y. and Hamaguchi, K. (1987)J. Biochem. 101,319-329
26 Pace, C. N., Grimsley, G. R., Thomson, J. A. and Barnett, B. J. (1988) J. Biol. Chem. 263, 11820-11825
27 Matsumura, M., Signor, G. and Matthews, B. W. (1989) Nature 342, 291-293
28 Pace, C. N. and Grimsley, G. R. (1988) Biochemistry 27, 3242-3246
29 Mitani, M., Harushima, Y., Kuwajima, K., Ikeguchi, M. and Sngai, S. (1986)J, Biol. Chem. 261, 8824-8829
30 Pakula, A. A. and Sauer, R. T. (1989) Proteins Struct. Funct. Genet. 5, 202-210
31 Des, G., Hickey, D. R., McLendon, D., McLendon, G. and Sherman, F. (1989) Proc. Nat] Acad. Sci. USA 86,
496-499 32 Kellis, J. T., Nyberg, K. and Fersht,
A. R. (1989) Biochemistry 28, 4914-4922
33 Yutani, K., Ogasahara, K., Tsujita, T. and Sugino, Y. (1987) Proc. Natl Acad. Sci. USA 84, 4441-4444
34 Matsumura, M., Becktel, W. J., Levitt, M. and Matthews, B. W. (1989) Proc. Natl Acad. Sci. USA 86, 6562-6566
35 Pace, C. N. and McGrath, T. (1980) J. Biol. Chem. 255, 3862-3865
36 Kostrewa, D., Choe, H-W., Heine- mann, U. and Saenger, W. (1989) Biochemistry 28, 7592-7599
37 Acharya, K. R., Stuart, D. I., Walker, N. P. C., Lewis, M. and Phillips, D. C. (1989) J. Mol. Biol. 208, 99-127
38 Pantoliano, M. W. et el. (1988) Biochemistry 27, 8311-8317
39 Kuroki, R., Taniyama, Y., Nakmura, H., Kikuchi, M. and Ikehara, M. (1989) Proc. Natl Acad. Sci. USA 86, 6903-6907
[ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ] [ ]
Prospects for genetic diagnosis of inherited
predisposition to cancer Bruce A. J. Ponder
The best long-term prospects for the reduction in the number of deaths due to cancer lie with screening and prevention rather than with therapy of the advanced disease. Screening eff iciency wil l be improved by the identification of individuals at inherited high risk of cancer. Understanding the mechanism of predisposition may provide the basis for rational attempts at prevention, as wel l as for new
approaches to therapy.
Cancer is a genetic disease at the level of the somat ic cell. Several sequent ia l events are required to turn a no rma l cell into a cancer cell, and mos t or all of these involve muta t ions - for example , the ac t iva t ion of oncogenes or the loss of suppres so r genes. The idea of cancer as a genetic disease at the level of the germl ine is less well- es tabl ished, but it fits easi ly into the same scheme. Ind iv idua l s wi th in-
B. A. J. Ponder is at the CRC Human Cancer Genetics Research Group, Depart- ment of Pathology, University of Cam- bridge, Tennis Court Road, Cambridge CB2 1QP, UK.
her i ted p red i spos i t i on to cancer m a y have inheri ted:
• one of the muta t iona l events a l ready c o m p l e t e d in the germline; • a trait w h i c h makes one of these muta t iona l s teps more l ikely to h a p p e n (e.g. a p o l y m o r p h i s m in me tabo l i sm of an exogenous chemica l w h i c h increases its con- vers ion to a carcinogen); or • a trait w h i c h makes the conse- quences of one of the s teps in carc inogenesis more severe in te rms of progress ion to cancer (e.g. a p o l y m o r p h i s m in endocr ine metab- o l i sm wh ich favours the prol i fera t ion of an al tered pre -cancerous cell).
© 1990, Elsevier Science Publishers Ltd (UK) 0167 - 9430/90/$2.00
The great difference in invest i - gating cancer c o m p a r e d with, for example , inher i ted p r ed i sp o s i t i o n to ca rd iovascu la r d isease (where the genes i nvo lved in l i pop ro t e in metab- o l i sm were an obvious target), is that there are a lmos t no clues as to wha t any of the p red i spos ing genes for cancer migh t be.
In the cancers where inher i t ed risk is sufficient to cause obv ious famil ia l clustering, it is poss ib le to explo i t the c lus ter ing to make an empi r i ca l search for the genes, us ing the t echn iques of genetic l inkage 1. Each of a series of DNA marke r s is tes ted for co- inher i tance of one al lele of a res t r ic t ion f ragment length p o ly mo r - p h i s m wi th the cancer in several m e m b e r s of a family. If a marke r is found whose inher i t ance co inc ides wi th that of the cancer, the ma rk e r locus m u s t lie close to the cancer- de te rmin ing gene on the same c h r o m o s o m e , o therwise they w o u l d p robab ly have been separa ted by genetic r e combina t i o n in thei r passage th rough the family. Famil ies wi th several affected ind iv idua l s are needed for this t echn ique to work. A signif icant c o m p o n e n t of inher i ted p red i spos i t i on to cancer may , how- ever, occur outs ide obv ious ' cancer famil ies ' (see below) 2. Here, genet ic l inkage cannot be appl ied . All that can be done is to guess the genes that

TIBTECH - APRIL 1990 [Vol. 8] 99
~ T a b l e I
Classi f icat ion o f genet ic pred ispos i t ion to cancer by strength o f fami l ia l c luster ing
Class Examples
Inherited cancer syndromes
Familial clusters of cancer
Genetic predisposition without evident familial clustering (still hypothetical)
Familial polyposis; Multiple endocrine neoplasia types 1 and 2; Von HippeI-Lindau syndrome.
Breast cancer; Ovarian cancer; Non- polyposis colorectal cancer.
Metabolic polymorphisms determining response to exogenous or endogenous carcinogens.
may be involved, or their pheno- types, and test these guesses empiri- cally in case-control studies of individuals with or without cancer.
Finding the predisposing genes and recognition of inherited predis- position depends, therefore, upon the degree of familial clustering. From this practical viewpoint, cancers can be classified into three groups (Table 1). The features of each group, and their implications for genetic diagnosis, will be discussed in turn.
Inherited cancer syndromes In these syndromes, which account
for perhaps 2% of all cancer inci- dence, there is strong predisposition consistent with the effect of a single autosomal dominant gene (Fig. 1; for review, see Refs 3, 4). Family members who have inherited the gene have a high probability (usually at least 50% lifetime risk) of develop- ing one or more specific cancers which are characteristic of each syndrome. A child of an affected parent is at 50% risk of inheriting the
'cancer gene'. Therefore, the most recent generations of such a family are a high risk group on whom, provided there is effective treatment, screening should be concentrated.
In most of these syndromes, devel- opment of the cancer is preceded by a characteristic phenotypic abnormal- ity in one or a few tissues (generally the tissues in which the cancer will develop): for example, C-cell hyper- plasia 5 or colonic polyps (Fig. 2; Ref. 6). These phenotypes are important for two reasons. First, they signal clearly which cancers have a familial basis and which do not: in common cancers such as breast and ovarian where such marker phenotypes have not been found, the distinction between family clusters which are due to chance and those which represent true predisposition can be very difficult (see below). Second, they indicate, before the cancers develop, which family members are gene carriers and therefore at risk, and which are not. Such marker phenotypes therefore provide the basis for screening tests in the
- -F ig . 1
,. 5 5 5 c
iv [
~42
ca ca 40 38 28
22
.47
1965
20 22 20 15 14
~30 ~ d . 4 0
d.35 [ ~ ,
,I 23
Pedigree of family with familial polyposis of the colon. Affected individuals are indicated by black (cancer) or hatched (polyps only) symbols. The family was recognized when the individual in generation III (arrowed) presented with colonic cancer and was found to have multiple colonic polyps. Subsequently, other members of the family (indicated by asterisk) have been screened by examination of the bowel through a colonoscope. Three were found already to have cancer; five siblings and a cousin in generation IV had polyps only and were treated by prophylactic colectomy. A line through the symbol indicates the individual died. (d.37 = died age 37; Ca 28 = cancer diagnosed age 28.)

100 TIBTECH - APRIL 1990 [Vol. 8]
management of high-risk families 7,8. The mapping of the genetic loci for
six of the inherited cancer syndromes by genetic linkage, in the space of just over a year (Table 2; Ref. 9), testifies to the pace of research once the necessary technical development - in this case, an outline human genome map - has been achieved. For the most part, the mapping is still based on simple two-allele poly- morphisms and these markers are still a few recombination units away from the disease gene locus. Never- theless, there is already the possi- bility of using the linked genetic markers for genetic diagnosis of gene carriers in families.
The further development of genetic diagnosis using these markers in- volves both technical and biological problems. The technical require- ments are straightforward: closer markers, markers which lie either side of the gene, and multi-allele polymorphisms will improve the accuracy of prediction and the proportion of families in which prediction is possible (e.g. Ref. 10). Ultimately, probes which can detect the mutation in the predisposing gene itself will be required. Probes are already available for retino- blastoma ~. Such probes have several advantages:
• they provide the greatest accuracy of diagnosis;
• if different mutations are associ- ated with different behaviour of the disease, probes which recognize each mutation will provide additional prognostic information;
• they do not have the limitation, inherent in the use of linked markers, that two or more affected family members must be available to pro- vide the linkage 'phase' on which to base prediction.
The biological problems in the application of genetic diagnosis are more subtle. Figure 3 shows the use of a linked probe (14.34), to predict the inheritance of the predisposing gene for multiple endocrine neo- plasia type 2 (MEN 2) in a family. There are 3 types of MEN 2: MEN 2A, MEN 2B and MTC-only, each with specific features, probably due to different mutations at the MEN2 locus. (Nomenclature: MEN 2A, disease; MEN2A, gene.) People carry-
--Fig. 2
,e
F
i
S
(a) Polyps in colon of indiv idual with famil ia l polyposis. Two large polyps and a number of smal ler ones are shown arising from the surface of the colonic epithelium. Bar = 5 mm. (Photograph provided by the ICRF Colorectal Cancer Unit, St. Mark's Hospital.) (b) Focus of C-cell hyperplasia in the thyroid of a patient with MEN 2A. A 41~m paraffin section of thyroid stained immunochemica l ly for calcitonin. A hyperplastic focus of C-cells (arrowed) is seen in the centre of the photograph, lying between, and extending into adjacent thyroid follicles. F indicates the lumen of the foll icle on the right. Bar = 100 Hm.
ing the MEN2A gene develop C-cell hyperplasia as a prelude to develop- ing medullary thyroid carcinoma. (C-cells are located in the thyroid and produce calcitonin - hyperplasia results in elevated production of calcitonin.) The 7-year-old boy is predicted to be a gene carrier; the prediction was confirmed by a grossly abnormal plasma calcitonin level,, reflecting advanced C-cell hyperplasia 5 (Fig. 2) or early tumour; he underwent thyroidectomy with removal of a 6 mm tumour. The
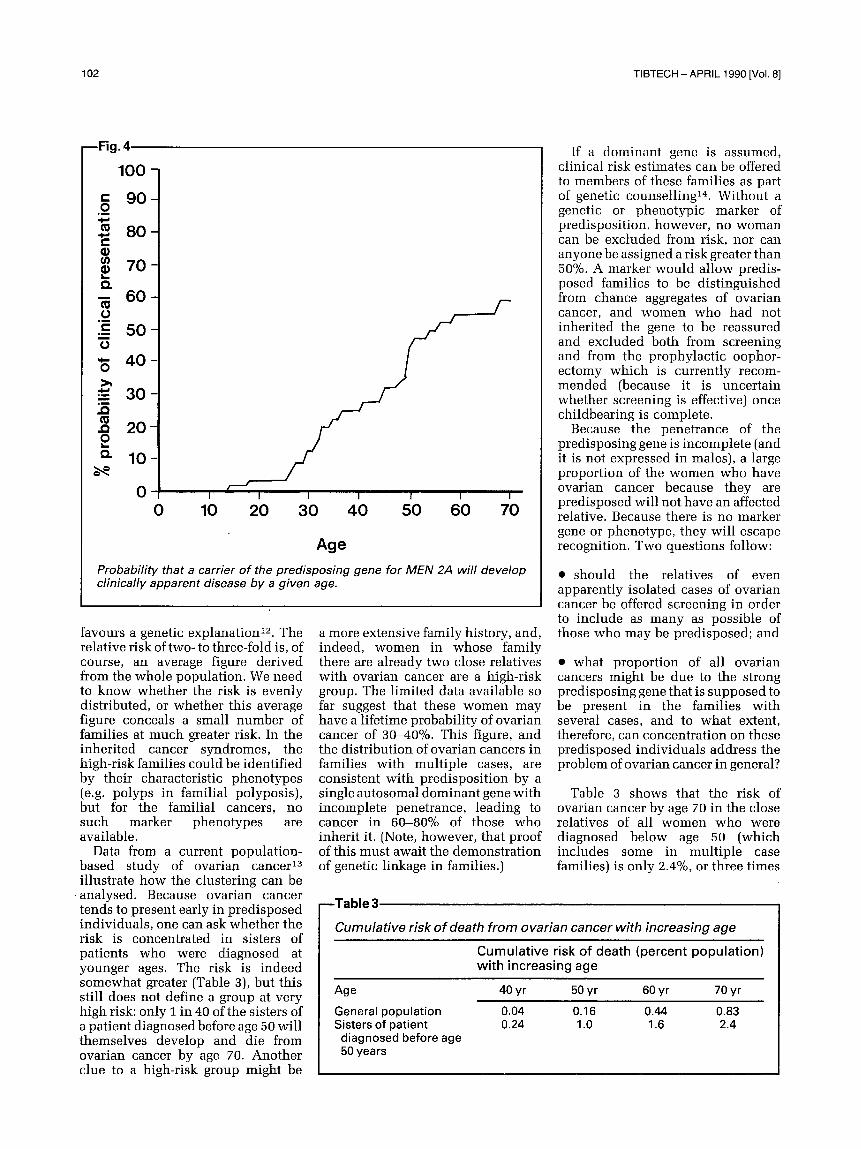
decision for surgery was easy because the raised calcitonin indicated the development of the tumour: but what if no such marker phenotype had been available? Figure 4 shows the age-related probability that a carrier of the MEN2A gene will develop disease sufficient to take him to a doctor 8. Hardly anyone has sympto- matic disease before the age of 20 years: 40% of gene carriers are still clinically unaffected at the age of 70 years. Without the confirmatory biochemical evidence that a tumour

TIBTECH-APRIL 1990 [Vol. 8] 101
~ T a b l e 2
Mapping o f loci o f predisposit ion to inherited cancer syndromes
S y n d r o m e C h r o m o s o m e
Retinoblastoma Wilms tumour
•Familial polyposis /Mul t ip le endocrine neoplasia type 1
All mapped ]Mul t ip le endocrine neoplasia type 2 between | V o n HippeI-Lindau syndrome 1987 and 1988|Neurof ibromatosis type 1
~, Neurofibromatosis type 2
13q 11p a
5q 11q 10 3p
17q 22
lead to more intensive screening rather than directly to treatment.
Famil ia l clusters o f cancer This group includes cancers which
have a tendency to cluster in families, but where the genetic basis of the clustering is not at present so clearly defined as in the inherited cancer syndromes. The degree of clustering
Familial melanoma lp (unconfirmed)
aThe locus for the familial form of Wilms tumour maps to another chromosomal region, still not identified.
was developing; a decision for thy- roidectomy in this 7-year-old based on genetic diagnosis alone would have been difficult. On the other hand, the prediction from Fig. 3 that the boy's three sisters aged 15, 13 and 3 years are not carriers of the gene is very helpful. Because of the variation in pace of development of the tumour in different individuals 8, a negative result from the current biochemical screening test in childhood is only weak evidence against carriage of the predisposing gene. The biochemical testing (which is rather uncomfort- able and provokes a good deal of anxiety) must therefore be repeated every year from the age of 5 until the mid-20s, and at longer intervals until the age of 40 years, before one can be confident that the individual is not a gene carrier. A negative prediction using DNA markers, however, can already reduce the risk to around 2% in the best situation (and will reduce it below 1% as markers improve), on the basis of a single blood sample in early childhood.
The most straightforward use of DNA prediction in the inherited cancer syndromes at present is therefore to exclude family members from risk, and so (at a level of risk which will vary from case to case) from the necessity of screening. On the other hand, because of the variation in expression of predis- posing genes within and between families, genetic diagnosis of the presence of the predisposing gene may still leave considerable uncer- tainty about the probability and timing of the development of cancer. Clinical decisions must balance this uncertainty against the severity of the treatment. So long as prophylactic treatment for those at risk in the inherited cancer syndromes remains
largely surgical, a genetic diagnosis of the inherited gene by itself, without the implication of certain and imminent disease, is likely to
- -Fig. 3,
is measured by the relative risk (RR) of a specific cancer occurring in siblings of a patient, compared with the risk in the general population. For most cancers, including breast and ovarian, the RR is around two- to three-fold higher. This clustering could be due to either environmental or genetic effects; but analysis of the patterns of cancer among relatives
~i 7i:~i:~i ::: ~
Genetic prediction in multiple endocrine neoplasia type 2 (MEN 2A) using a linked DNA marker. An autoradiograph of a Southern blot of DNA from family members, hybridized with a cosmid DNA probe (14.34) closely linked to the MEN2A locus on chromosome 10. The alleles recognized by the probe are A and B. The bands immediately below B are non-polymorphic DNA fragments which also hybridize with the probe. The affected grandfather (see pedigree at top of gel) is homozygous BB. One of his B alleles, which must be linked to the M E N 2A gene, is inherited by his affected daughter (genotype AB). As her husband is AA, each of their children must have received an A from the father: the inheritance of A or B from their mother predicts the inheritance of predisposition to the disease.
: :ii:iiii~ii!ii~i ::: : :ii:i~:~i/ili::i::ii:: :::ii::ii:~ :: ̧̧
i
102 TIBTECH- APRIL 1990 [Vol. 8]
~ F i g . 4
100
9O
8O
60
"E 50
"5 40
• =>' 30
, ' , 2 0 0 L _
~- 10
0 I I
0 10 20
/ /
I I
30 40 50
/ -
I I
60 70
Age
Probability that a carrier of the predisposing gene for MEN 2A will develop clinically apparent disease by a given age.
favours a genetic explanation t2. The relative risk of two- to three-fold is, of course, an average figure derived from the whole population. We need to know whether the risk is evenly distributed, or whether this average figure conceals a small number of families at much greater risk. In the inherited cancer syndromes, the high-risk families could be identified by their characteristic phenotypes (e.g. polyps in familial polyposis), but for the familial cancers, no such marker phenotypes are available.
Data from a current population- based study of ovarian cancer 13 illustrate how the clustering can be
analysed . Because ovarian cancer tends to present early in predisposed individuals, one can ask whether the risk is concentrated in sisters of patients who were diagnosed at younger ages. The risk is indeed somewhat greater (Table 3), but this still does not define a group at very high risk: only i in 40 of the sisters of a patient diagnosed before age 50 will themselves develop and die from ovarian cancer by age 70. Another clue to a high-risk group might be
a more extensive family history, and, indeed, women in whose family there are already two close relatives with ovarian cancer are a high-risk group. The limited data available so far suggest that these women may have a lifetime probability of ovarian cancer of 30-40%. This figure, and the distribution of ovarian cancers in families with multiple cases, are consistent with predisposition by a single autosomal dominant gene with incomplete penetrance, leading to cancer in 60-80% of those who inherit it. (Note, however, that proof of this must await the demonstration of genetic linkage in families.)
If a dominant gene is assumed, clinical risk estimates can be offered to members of these families as part of genetic counselling 14. Without a genetic or phenotypic marker of predisposition, however, no woman can be excluded from risk, nor can anyone be assigned a risk greater than 50%. A marker would allow predis- posed families to be distinguished from chance aggregates of ovarian cancer, and women who had not inherited the gene to be reassured and excluded both from screening and from the prophylactic oophor- ectomy which is currently recom- mended (because it is uncertain whether screening is effective) once childbearing is complete.
Because the penetrance of the predisposing gene is incomplete (and it is not expressed in males), a large proportion of the women who have ovarian cancer because they are predisposed will not have an affected relative. Because there is no marker gene or phenotype, they will escape recognition. Two questions follow:
• should the relatives of even apparently isolated cases of ovarian cancer be offered screening in order to include as many as possible of those who may be predisposed; and
• what proportion of all ovarian cancers might be due to the strong predisposing gene that is supposed to be present in the families with several cases, and to what extent, therefore, can concentration on these predisposed individuals address the problem of ovarian cancer in general?
Table 3 shows that the risk of ovarian cancer by age 70 in the close relatives of all women who were diagnosed below age 50 (which includes some in multiple case families) is only 2.4%, or three times
- -Table 3.
Cumulative risk o f death from ovarian cancer with increasing age
Cumulat ive risk of death (percent populat ion) wi th increasing age
Age 40 yr 50 yr 60 yr 70 yr
General population 0.04 0.16 0.44 0.83 Sisters of patient 0.24 1.0 1.6 2.4
diagnosed before age 50 years

TIBTECH - APRIL 1990 [Vol. 8] 103
- - T a b l e 4,
Possible effects of hypothetical cancer-predisposing genes causing the concentration of cancer incidence in a minor i ty of the population a
Gene Propor t ion of R isk to Percentage RR to f r equency popu la t i on c a r r i e r v s of cancer sibl ing
w h o are non-carr ier w h i c h carr iers occurs in
carr iers b
Dominant 0.1 0.19 100x 95 2.89
Recessive 0.5 0.25 1 0 0 × b 97 2.07
aThese major effects may not necessarily give rise to obvious familial clustering, as shown bythe small relative risks (RR) in siblings. (Adapted from Ref. 2.) b In homozygote.
the general-population risk. Concen- trating screening on these relatives does, therefore, offer a small gain in efficiency over screening the whole population, but still only about 1 in 1000 women in this group will develop ovarian cancer each year. Table 3 also shows that the excess risk of ovarian cancer by age 70 in the close relatives of patients, compared with the risk in the general popu- lation, is about 1.6%. If all of this excess were due to the inheritance by these relatives of the single putative predisposing gene, it would imply that about 4% of the affected index cases had the gene (since it is dominant, 2% of their relatives would have it, and 80% penetrance in them would give an incidence of 1.6%). This means that only about 1 in 25 of ovarian cancers diagnosed below age 50 can be attributed to the predisposing gene. (If the gene is less completely penetrant, the proportion may be a little higher, but almost certainly less than 10%.) Thus, while the ovarian cancer families are clearly a very high risk group that require special consideration, screening targeted by family history does not appear to be a solution to the public health problem of ovarian cancer. It follows that a genetic marker for the predisposing gene, while it would undoubtedly be valuable in the management of the high risk group, would have a limited impact on the disease as a whole.
Inherited predisposition without evident family clustering
So far, only the possible role of a rare single gene of incomplete but fairly high penetrance has been considered. Part of the increased relative risk in siblings could, alter-
natively, be explained by a relatively common predisposing gene or genes with much lower penetrance. If individuals with the gene had only a 1 in 10 chance of developing cancer, they would still be at substantially increased risk, but extensive family clusters would be rare and the predisposition would be hard to recognize. Such a gene could never- theless result in most of the incidence of a particular cancer being concen- trated in a minority of the population, with important implications for screening and prevention. The theor- etical arguments have been set out by Peto 2. These show clearly that a common predisposing gene for a common cancer can result in quite striking concentrations of cancer incidence in a minority of the population, without drawing atten- tion to itself by causing extensive familial aggregation.
Examples of the effects that could be seen for dominant or recessive predisposing genes are given in Table 4. A study 15 in which colonic polyps were tested as a marker phenotype for a common gene predisposing to colonic cancer (separate from the inherited cancer syndrome polyposis coli) provides support for these calculations. Colonic cancer tends to cluster in families but, like breast and ovarian cancer, only a small minority of cases have a family history which clearly signals predisposition. When the relatives of isolated cases of colonic cancer are systematically examined for colonic polyps, how- ever, they are found to have a significantly increased number com- pared to controls. The data can be interpreted to show that the majority of colon cancer incidence may be concentrated in a minority of the
population who are predisposed and who, in this case, can be recognized by the phenotype of colonic polyps. If these conclusions can be confirmed, they have obvious and very import- ant implications for the reduction of cancer deaths in a much larger proportion of the population than are included in the recognizable familial syndromes. First, screening might be concentrated on a minority of the population which contains most of the risk. Second, elucidation of the mechanism of predisposition may in some cases provide opportunities for prevention. For example, the well- known examples of polymorphism in metabolism of drugs suggest that at least some instances of genetic predisposition to cancer will be due to comparable polymorphisms in metabolism of exogenous or en- dogenous carcinogens (for examples and discussion, see Ref. 16). If so, elucidation of the mechanisms may lead to better identification of the carcinogens - about which infor- mation is still surprisingly scanty - and thence to methods of prevention by avoiding them or altering their metabolism.
Exciting though this prospect is, it remains largely hypothetical. Ex- amples of metabolic polymorphism which appear to be associated with increased cancer risk have been described 17,18, but they are still few and awaiting confirmation. Progress is slow because, in the absence of familial clusters, the empirical methods of genetic linkage cannot be used to find the genes. Candidate genes or phenotypes must be guessed at, and tested in case-control studies. With a hundred hypotheses to be tested and no strong clue where to start, this is a laborious business.
Conclusion and prospects In most of the inherited cancer
syndromes, genetic diagnosis using linked markers is already feasible; and it will probably soon become so in at least some of the common familial cancers such as breast cancer. Together, these may account for about 5% of cancer incidence. In these cases, the practical value of the diagnosis at present is to identify those not at risk. Further progress is possible in several directions:
• better definition of marker pheno- types will improve the recognition of

104 T IBTECH- APRIL 1990 [Vol. 8]
those at inher i ted risk and make easier genetic l inkage studies to find the genes;
• better def ini t ion of preneoplas t ic lesions may improve screening and the power to predic t wh ich predis- posed individuals are at immedia te risk;
• c loning of the predispos ing genes will a l low direct assay for the mutan t gene in individuals at risk and so genetic diagnosis wi thout the need for linkage in family members ;
• it may be possible to ident i fy other genes which modi fy the express ion of a predispos ing gene, and so to predic t whe the r a p red i sposed in- d ividual will be severely or mi ld ly affected; and
• if the mechan i sm of predis- pos i t ion is unders tood it may be possible to use this knowledge to design strategies to retard or p reven t the deve lopmen t of cancer, as an al ternat ive to the prophylac t ic sur- gery which will mos t ly on ly be acceptable to those at ve ry high risk.
Fur ther ahead, but potent ia l ly more impor tan t in terms of the numbers affected, it may be possible to ident i fy individuals genet ical ly p red i sposed at a lower level, who comprise a substantial f ract ion of the cancer inc idence in the popula t ion . Once again, candidate pheno types - whe the r preneoplas t ic lesions such as polyps, or metabol ic po lymor-
phisms in ho rmone or carc inogen metabol ism - are l ikely to be vital in providing clues to the recogni t ion of these groups. Wi thout clear family clusters, a genetic l inkage approach will be difficult, a l though if h ighly po lymorph ic DNA markers can be def ined at regular intervals along the h u m a n gene map, an empir ical approach using pairs of affected siblings might just succeed. (It may also be wor th invest igat ing the predispos ing loci ident i f ied in the inher i ted cancer syndromes , in case 'weak' alleles at these loci are responsible for low-level predis- posi t ion in the populat ion.) Because the lifetime risk of cancer in these individuals is still low, and their numbers will be large, invasive or expens ive measures such as surgery will not be acceptable. In cancers where screening is effective, this can be made more efficient. The long- term goal, however , mus t be the same as for the familial clusters of cancer: to use genetic p red ispos i t ion to discover the env i ronmenta l or en- dogenous factors wh ich interact wi th the genotype to cause cancer, and use these as a means to prevent ion .
References 1 White, R. (1985) Trends Genet. 1,
177-180 2 Peto, J. (1980) Cancer Incidence in
Defined Populations, Banbury Report, 4 (Cairns, J., Lyon, J. L. and Skolnick, M., eds), pp. 203-213, Cold Spring Harbor Laboratory
3 Harnden, D., Morten, J. and Feather-
stone, T. (1984) Adv. Cancer Res. 41, 185-255
4 Mulvihill, ]. J., Miller, R. W. and Franmeni, J. F. (eds) (1977) Progress in Cancer Research and Therapy Vo]. 3, Raven Press
5 Wolfe, H. J. and DeLellis, R. A. (1981) Clin. Endocrino]. Metab. 10, 351-365
6 Watne, A. L. (1987) Semin. Surgical Oncol. 3, 71-76
7 Murday, V. and Slack, J. (1989) Cancer Surv. 8, 139-157
8 Ponder, B. A. J., Ponder, M. A., Coffey, R., Pembrey, M. E., Gage], R. F., Telenius-Berg, M., Semple, P. and Easton, D. F. (1988) Lancet i, 397-400
9 Ponder, B. A. J. (1988) Nature 335, 400-402
10 Telenius, H., Mathew, C. G. P., Nakamura, Y., Easton, D. F., Clark, J., Neumann, H. P. H., Ziegler, W. H., Schinzel, A. and Ponder, B. A. J. Ear. J. Surg. Onco]. (in press)
11 Yandell, D. W. and Dryja, T. P. (1989) Am. J. Hum. Genet. 45, 547-555
12 Williams, W. R. and Anderson, D. E. (1984) Genet. Epidemio]. 1, 7-20
13 Ponder, B. A. J., Easton, D. F. and Peto, J. (1989) in Ovarian Cancer (Sharp, F., Mason, W. P. and Leake, R. E., eds), pp. 3-6, Chapman and Hall
14 Harper, P. S. (1988) Practical Genetic Counselling (3rd edn), Wright
15 Cannon-Albright, L. A., Skolnick, M. H., Bishop. D. T., Lee, R. G. and Burt, R. W. (1988) New Engl. J. Med. 319,533-537
16 Omenn, G. S. and Gelboin, H. V. (eds) (1984) Genetic Variability in Responses to Chemical Exposure, Ranbury Report, 16, Cold Spring Harbor Laboratory
17 Wolf, C. R. (1986) Trends Genet. 2, 209-214
18 Hein, D. W. (1988) BioEssays 9, 200-204
i: . T R E N D S J O U R N A L S : :I
P T Voiumes, No~21 ~ I : I /I:::I:I':::. I:~ :: : : I I:I:~I:~: TIG~: Latin Amer ican w o r k s h o p for the eva iuat ion Of DnA:probes: : i: :::: UnStable l iaisonS: the u ~ e o f t ~ a n s p o ~ s : in p i~nt -~e6et i~ : fo r le ishmanias is , .: : ~ i : . . . . i ~-:~ ~ii: .~:i: engineer ing; : ~ :;:::~ ;::~; : : : ~ : i : :
: by L: A. Labrada and D: S. smi th :i :: ':: : / : ::i:i:: : : : : : / b y A n g e l o S p e h a : : ! :t: : t:: " i. T ransgen ic mosqu i toes : A f u t u r e v e c t d r c o - n t 6 Strdtecjy~i : : : : :::<:i: - : : : ::: :: - ; :::: : : : : :
byJ. Crampton, A. Morris, G; Lycett, A: Warren and ::::: :::: :* ~ : V o l u m d ) t~ N01:2 ::./i:: : : ::::: :: :::: i: : ; : : P. Eggleston : :: - : : : ..... : : : : : % : : ; : Immunoog ica lad juvants ! ;~ : io ie :o i i : : : : :S :m:s : ; : : : : : ::;; ::::::: :
... . . . . . . ~ . . , : :: :.. [ ::::i : ::':: : : : ; : ,:::' :5 : : : : :: : : : : . by Gregory Gregor iadis; : : :::: ...... : : : : :
A n o v e l p a t h w a y f o r s e c r e t o r y p r O t e n s ? ::: : : t : : .... : :: . . . . . :: : : : : : : - . . . . . . : : : - : : : - : : : : : : : : : : - : : : : ::.::t:; : - ' A M es h - ' " ; . . . . " : : ; :-%::~:::: : : : :T IPS : ; Votu~e::Z[ No:~:: :t
oy , u c . ~. t~ar~mann ~. Honoe :/4~ Huoarrem : : : : : : : : M a s s s e c t r o m e t ' a : '~ ' ; : : : : : : ; : : : : : : : : : :%' : : :: -': ::~ ::: : o o:.:~__.~-~ ^ ~, . . . . . . . " - ' ":1: :::::::: : p ry t n gh mass V r m e s a n c l v i c e s o t s o m e : . : r l . , . . ~ / u ~ d / S U I . / 4 . m a f l J l J U l J U f t : - : : : : : : [ : > , i . . . . . . : : : : : I
n e w approacnes, ' The predic t ion of t r a n s m e m b r a n e : - , ~ . - . . ~ ~/::[::: i, ::::::. : % : t ::: : : : :% :: ::::% ] Dy btmon J (sasKefl t he i r con fo rma t i on : an evaluat ion, -:-:: . . . . . . : ' : : : ::::: :::i::: :: : :: % t : : : : : ]
byGera ld D. Fasman and Wifliam AI G i l b e r t : ::: : : : : : : : : : : : :::: : - : : : : i :::: :: : : : : :: : : ::; i : :





























