Embed Size (px)
Citation preview

Probabilistic Roadmaps: A Tool for Computing
Ensemble Properties of Molecular Motions
Serkan Apaydin, Doug Brutlag1
Carlos Guestrin, David Hsu2
Jean-Claude Latombe, Chris Varma
Computer Science DepartmentStanford University
1 Department of Biochemistry, Stanford University2 Computer Science Department, University of North Carolina

Goal of our ResearchGoal of our Research
Develop efficient computational representations and algorithms to study molecular pathways for protein folding and ligand-protein binding
Protein folding RECOMB ’02Ligand-protein binding ECCB ‘02

AcknowledgementsAcknowledgements
People: Leo Guibas Michael Levitt, Structural BiologyItay Lotan Vijay Pande, ChemistryFabian Schwarzer Amit SinghRohit Singh
Funding: NSF-ITR ACI-0086013Stanford’s Bio-X and Graduate Fellowship
programs
Analogy with RoboticsAnalogy with Robotics

Configuration SpaceConfiguration Space
Approximate the free space by random sampling
Probabilistic Roadmaps

Probabilistic RoadmapProbabilistic Roadmap
free space
[Kavraki, Svetska, Latombe,Overmars, 95][Kavraki, Svetska, Latombe,Overmars, 95]

Probabilistic CompletenessProbabilistic Completeness
The probability that a roadmap fails to correctly capture the connectivity of the
free space goes to 0 exponentially in the number of milestones (~ running time).
Random sampling is convenient incremental
scheme for approximating the free space

Computed ExamplesComputed Examples

Biology Biology Robotics Robotics
Energy field, instead of joint controlContinuous energy field, instead of binary free and in-collision spacesMultiple pathways, instead of single collision-free pathPotentially many more degrees of freedomRelation to real world is more complex

Initial WorkInitial Work[Singh, Latombe, Brutlag, 99][Singh, Latombe, Brutlag, 99]
Study of ligand-protein bindingProbabilistic roadmaps with edges weighted by energetic plausibilitySearch of most plausible paths

Initial WorkInitial Work[Singh, Latombe, Brutlag, 99][Singh, Latombe, Brutlag, 99]
Study of ligand-protein bindingProbabilistic roadmaps with edges weighted by energetic plausibilitySearch of most plausible pathsStudy of energy profiles along such paths
CatalyticSite
energy

Initial WorkInitial Work[Singh, Latombe, Brutlag, 99][Singh, Latombe, Brutlag, 99]
Study of ligand-protein bindingProbabilistic roadmaps with edges weighted by energetic plausibilitySearch of most plausible pathsStudy of energy profiles along such pathsExtensions to protein folding[Song and Amato, 01] [Apaydin et al., 01]
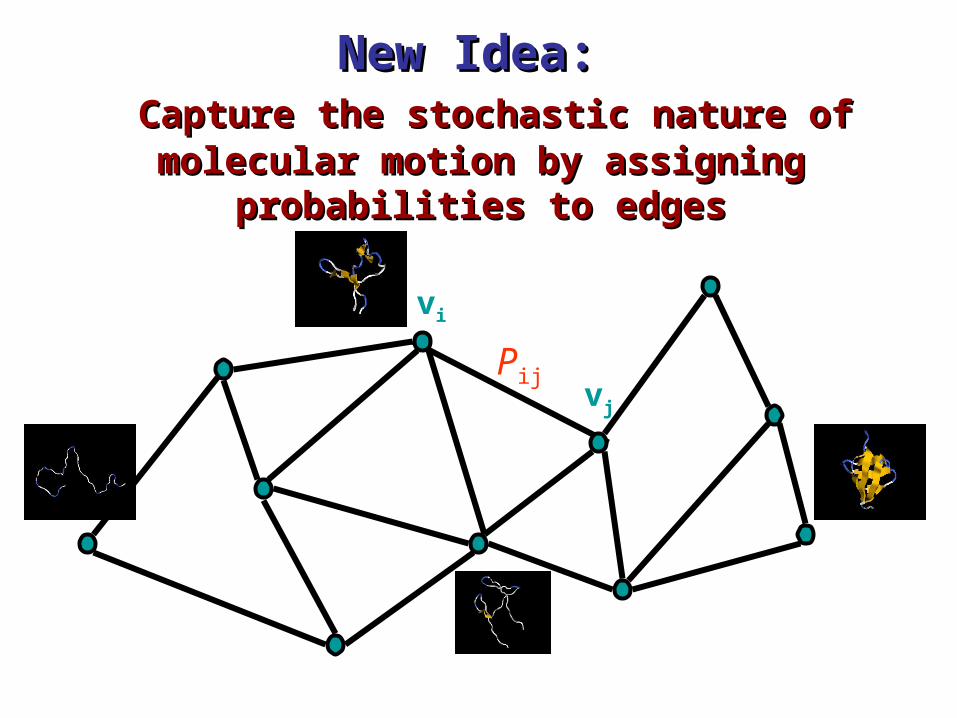
New Idea: New Idea: Capture the stochastic nature of molecular Capture the stochastic nature of molecular motion by assigning probabilities to edgesmotion by assigning probabilities to edges
vi
vj
Pij

Why is this a good idea?Why is this a good idea?
1) We can approximate Monte Carlo simulation as closely as we wish
2) Unlike with MC simulation, we avoid the local-minima problem
3) We can consider all pathways in the roadmap at once to compute ensemble properties

Edge probabilitiesEdge probabilities
Follow Metropolis criteria:
otherwise. ,
1
;0 if ,)/exp(
i
iji
Bij
ij
N
EN
TkE
P
Self-transition probability:
ijijii PP 1
vj
vi
Pij
Pii

Stochastic simulation on roadmap and Monte Carlo simulation converge to same Boltzmann distribution
S
Stochastic Roadmap SimulationStochastic Roadmap Simulation
Pij

Problems with Problems with Monte Carlo SimulationMonte Carlo Simulation
Much time is wasted in local minima Each run generates a single pathway

SolutionSolution
Pij
Treat roadmap as a Markov chain and use the First-Step Analysis tool

Example #1: Example #1:
Probability of Folding pProbability of Folding pfoldfold
Unfolded set Folded set
pfold1- pfold
“We stress that we do not suggest using pfold as a transition coordinate for practical purposes as it is
very computationally intensive.” Du, Pande, Grosberg, Tanaka, and Shakhnovich “On the Transition
Coordinate for Protein Folding” Journal of Chemical Physics (1998).
HIV integrase[Du et al. ‘98]

Pii
F: Folded setU: Unfolded set
First-Step AnalysisFirst-Step Analysis
Pij
i
k
j
l
m
Pik Pil
Pim
Let fi = pfold(i)After one step: fi = Pii fi + Pij fj + Pik fk + Pil fl + Pim fm
=1 =1
One linear equation per node Solution gives pfold for all nodes
No explicit simulation run All pathways are taken into account Sparse linear system

In Contrast …In Contrast …
Computing pfold with MC simulation requires:
Performing many MC simulation runs
Counting the number of times F is attained firstfor every conformation of interest:

Computational TestsComputational Tests• 1ROP (repressor of
primer)• 2 helices• 6 DOF
• 1HDD (Engrailed homeodomain)
• 3 helices• 12 DOF
H-P energy model with steric clash exclusion [Sun et al., 95]

1ROP
Correlation with MC ApproachCorrelation with MC Approach

Correlation with MC ApproachCorrelation with MC Approach
1HDD

Computation Times (1ROP)Computation Times (1ROP)
Monte Carlo:
49 conformations Over 11 days ofcomputer time
Over 106 energy
computations
Roadmap:
5000 conformations1 - 1.5 hours ofcomputer time
~15,000 energycomputations
~4 orders of magnitude speedup!

Example #2: Example #2: Ligand-Protein InteractionLigand-Protein Interaction
Computation of escape time from funnels of attraction around potential binding sites(funnel = ball of 10A rmsd)

Computing Escape Time with Computing Escape Time with RoadmapRoadmap
Funnel of Attraction
ij
kl
m
Pii
Pim
PilPikPij
i = 1 + Pii i + Pij j+ Pik k + Pil l + Pim m
(escape time is measured as number of stepsof stochastic simulation)
= 0

Similar Computation Similar Computation Through Simulation Through Simulation [Sept,
Elcock and McCammon `99]
10K to 30K independent simulations

ApplicationsApplications
1) Distinguishing catalytic site: Given several potential binding sites, which one is the catalytic site?

Complexes StudiedComplexes Studied
ligand protein # random nodes
# DOFs
oxamate 1ldm 8000 7
Streptavidin 1stp 8000 11
Hydroxylamine 4ts1 8000 9
COT 1cjw 8000 21
THK 1aid 8000 14
IPM 1ao5 8000 10
PTI 3tpi 8000 13

Distinction Based on Distinction Based on EnergyEnergy
Protein Bound state
Best potential binding site
1stp -15.1 -14.6
4ts1 -19.4 -14.6
3tpi -25.2 -16.0
1ldm -11.8 -13.6
1cjw -11.7 -18.0
1aid -11.2 -22.2
1ao5 -7.5 -13.1 (kcal/mol)
Able to distinguish
catalytic site
Not able
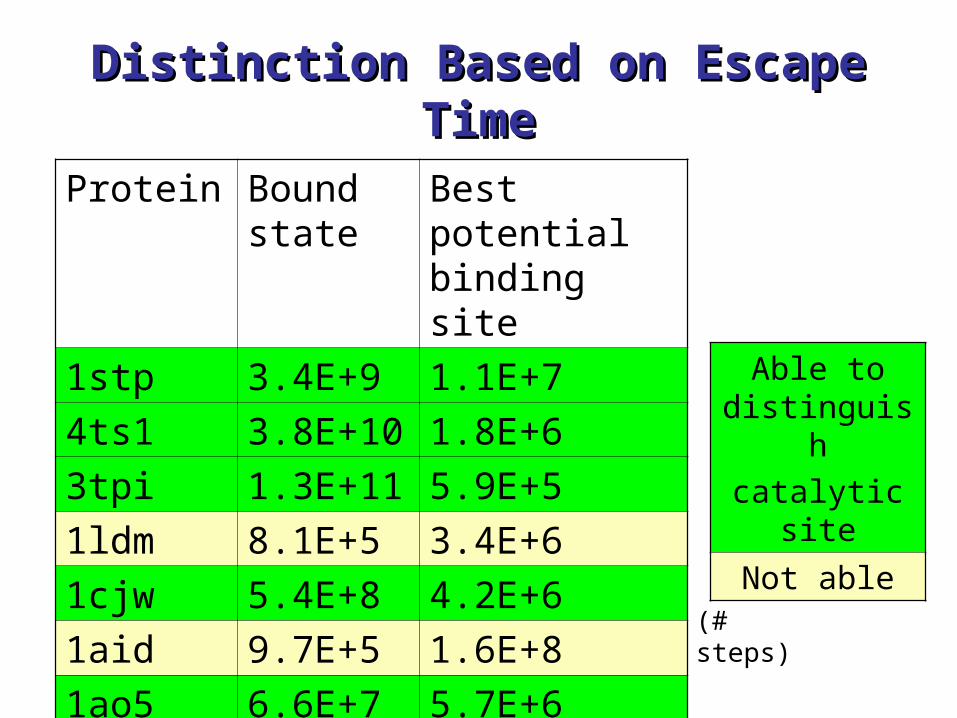
Distinction Based on Escape Distinction Based on Escape TimeTime
Protein Bound state
Best potential binding site
1stp 3.4E+9 1.1E+7
4ts1 3.8E+10 1.8E+6
3tpi 1.3E+11 5.9E+5
1ldm 8.1E+5 3.4E+6
1cjw 5.4E+8 4.2E+6
1aid 9.7E+5 1.6E+8
1ao5 6.6E+7 5.7E+6(# steps)
Able to distinguishcatalytic
site
Not able

ApplicationsApplications
1) Distinguishing catalytic site2) Computational mutagenesis
C
C
OO
O
GLN-101
ARG-106
ASP-195HIS-193
ASP-166
ARG-169
NADH
+
+
+
Loop
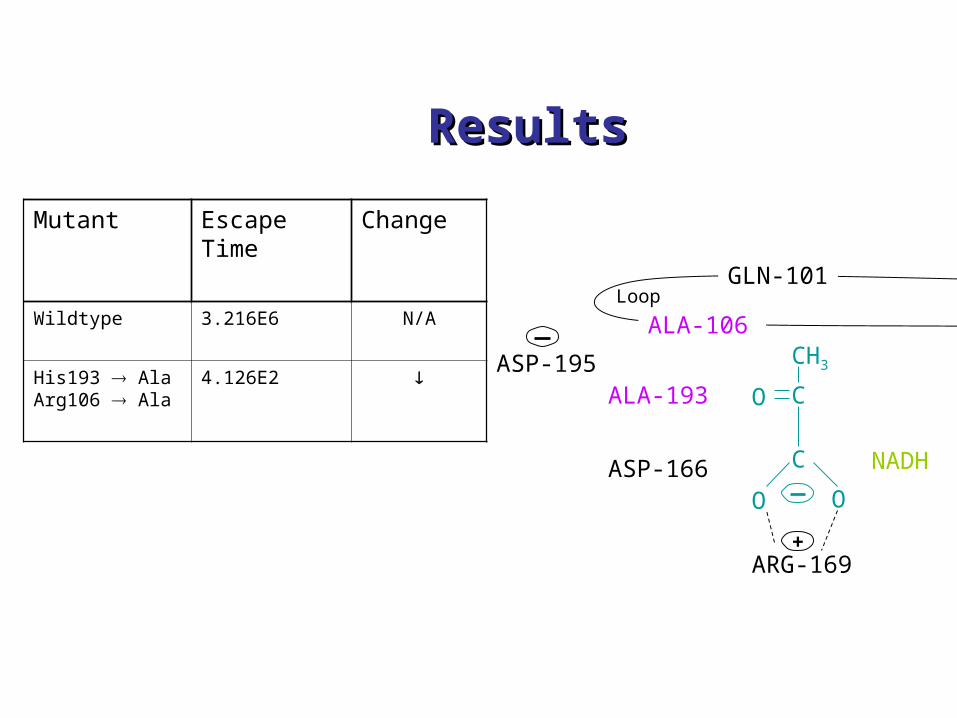
Chemical environment of LDH-NADH-substrate complex (pyruvate) (catalyzes conversion of pyruvate to lactate in the presence of NADH
CH3
Some amino acids aredeleted entirely, replaced by other amino acids, or sidechains altered
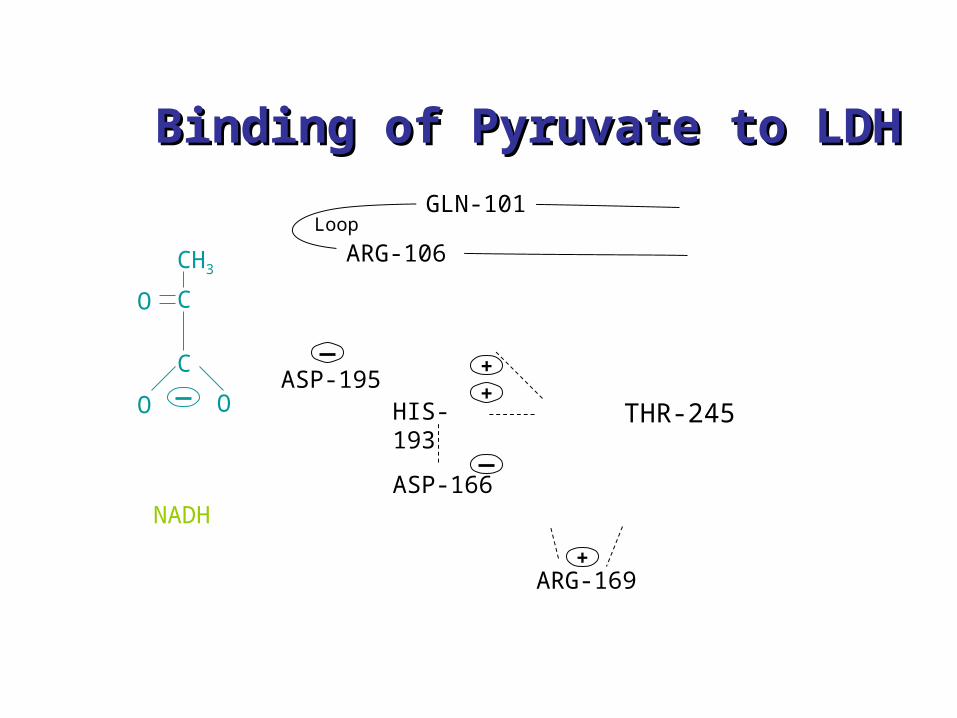
Binding of Pyruvate to LDHBinding of Pyruvate to LDH
ASP-195HIS-193
ASP-166
ARG-169
+
+
+
THR-245
C
C
OO
O
CH3
NADH
GLN-101
ARG-106Loop

ResultsResults
C
C
OO
O
GLN-101
ARG-106
ASP-195HIS-193
ASP-166
ARG-169
NADH
+
+
+
Loop
CH3
THR-245
Mutant Escape Time
Change
Wildtype 3.216E6 N/A
ResultsResults
C
C
OO
O
GLN-101
ALA-106
ASP-195ALA-193
ASP-166
ARG-169
NADH
+
Loop
CH3
Mutant Escape Time
Change
Wildtype 3.216E6 N/A
His193 AlaArg106 Ala
4.126E2

ResultsResults
Mutant Escape Time Change
Wildtype 3.216E6 N/A
His193 AlaArg106 Ala
4.126E2
His193 Ala 3.381E3
Arg106 Ala 2.550E2
Asp195 Asn 5.221E7
Gln101 Arg 1.669E6 No change
Thr245 Gly 4.607E5
C
C
OO
O
GLN-101
ARG-106
ASP-195HIS-193
ASP-166
ARG-169
NADH
+
+
+
Loop
CH3
GLY-245

ConclusionConclusion
Probabilistic roadmaps are a promising computational tool for studying ensemble properties of molecular pathwaysCurrent and future work: Better kinetic/energetic models Experimentally verifiable tests Non-uniform sampling strategies Encoding MD simulation

Stochastic simulation on a roadmap and MC simulation converge to the same distribution (Boltzman):For any set S, >0, >0,>0, there exists N such that a roadmap with N milestones has error bounded by:
with probability at least 1- )1)(()(ˆ)1)(( SSS
vs
vg
S
Stochastic Roadmap SimulationStochastic Roadmap Simulation
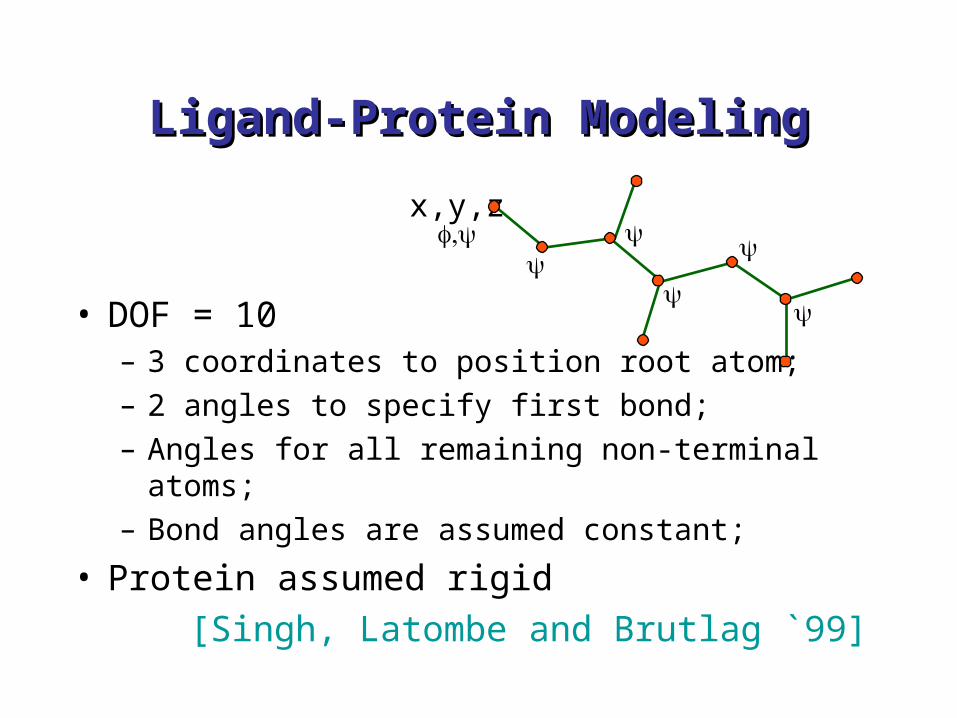
Ligand-Protein ModelingLigand-Protein Modeling
• DOF = 10 – 3 coordinates to position root atom;– 2 angles to specify first bond;– Angles for all remaining non-terminal atoms;– Bond angles are assumed constant;
• Protein assumed rigid[Singh, Latombe and Brutlag `99]
x,y,z

Energy of InteractionEnergy of Interaction
Ev
Rij
Ec
Rij
Ev = 0.2[(R0/Rij)12 - 2(R0/Rij)
6 ]Ec = 332 QiQj/(Rij)
Energy = van der Waals interaction (Ev)
+ electrostatic interaction (Ec)

Solvent Effects
• Is only valid for an infinite medium of uniform dielectric;• Dielectric discontinuities result in induced surface
charges;
Solution: Poisson-Boltzman equation
Ec = 332 QiQj/(Rij)
Use Delphi [Rocchia et al `01] Finite Difference solution is based on discretizing
the workspace into a uniform grid.
[(r) . (r)] - (r)k(r)2sinh([(r)] + 4rf(r)/kT = 0





























