Embed Size (px)
Citation preview

THE JOURXAL OP BIOLOGICAL CHEMISTRY Vol.248, No.22, Issue of November25, pp.7729-7741. 1073
Printed in U.S.A.
Simultaneous Purification of Bovine Prothrombin and Factor X
ACTIVATIO?? OF PROTHROMBIN BY TRYPSIN-ACTIVATED FACTOR X”
(Received for publication, April 2: 1973)
SATYA P. BAJAJ AND KENNETH G. R/IANN$
From the Department of Biochemistry, University of Minnesota, St. Paul, Minnesota55101, and the Hematology Research Section, Mayo Clinic, Rochester, Minnesota 55901
SUMMARY
A simple reproducible procedure has been developed for the large scale isolation of bovine factor X and prothrom- bin in highly purified states. Both proteins isolated by this method show a single component in four different elec- trophoretic systems. The specific activity of prothrombin obtained by this procedure is 1300 f 100 NIH thrombin units per mg of prothrombin, whereas the specific activity of factor X is 90 + 10 units of activated factor X (factor Xa) per mg of factor X, when assayed by the standard tech- nique (BACHMANN, F., DUCKERT, F., AND KOLLER, F. (1958) Thromb. Diath. Haemorrh. 2, 24). The over-all yields of bovine prothrombin and factor X are 70 mg and 4.5 mg, respectively, per liter of starting plasma. The isolation procedure involves adsorption of factor X and prothrombin on to barium citrate, their elution from the barium citrate complex with EDTA, extensive dialysis against 0.02 M tri- sodium citrate and 0.9% sodium chloride, ammonium sul- fate fractionation, diisopropylfluorophosphate treatment, and DEAE-cellulose chromatography. DEAE-cellulose chromatography in (sodium) citrate buffer at pH 6.0 effec- tively and completely separates prothrombin from factor X. Factor X is then further purified by DEAE-Sephadex chro- matography. Both proteins are completely stable in 50% glycerol-HsO (v/v) at -20’ for at least 18 months.
The molecular weights of bovine prothrombin and factor X as determined by sodium dodecyl sulfate polyacrylamide electrophoresis are 72,000 f 4,000 and 56,000 + 2,000, respectively. The molecular weight of native factor X, determined by sedimentation equilibrium, is 59,000 f 2,000. Factor X protein is composed of two chains of 44,000 and 15,500 daltons as determined by gel filtration of the reduced and S-carboxymethylated protein in 6 M guanidinium chloride.
Activation of factor X by trypsin insolubilized on poly- acrylamide produced a trypsin-free factor Xa(factor Xa(trygsin)) with a maximum yield of 750 + 50 units of factor
* This research was supported by Grant HL-15381 from the National Heart-Lung Institute, United States Public Health Service, and by the Mayo Foundation. An account of this work was presented at t,he Fifteenth Annual West Central States Bio- chemistry Conference (1972), the University of Iowa, Iowa City, Iowa; and has also appeared in American Society of Hematology abstracts (197‘.‘.) p. 118.
1 Recipient of a Camille and Henry Drefus Foundation Teacher- Scholar grant ; to w-horn correspondence should be addressed.
Xa per mg of factor X, which is 8 times higher than the yield obtainable by the action of Russell’s viper venom (RW) on factor X.
Activation of prothrombin by factor Xa(trypsin), has been carried out under conditions in which the possibility of throm- bin contamination has been eliminated. The rate of the factor Xa(trypsin) catalyzed activation of prothrombin is accelerated 50-fold in the presence of Ca2+ and go-fold in the presence of both Ca2+ and phospholipid. However, under all conditions studied, and all types of factor Xa (fac- tor Xahyp8in, RVV , citrate)) used, the products of prothrom- bin activation are the same and electrophoretically equiv- alent to those described previously by this laboratory (MANN,
K. G., HELDEBRANT, C. M., AND FASS, D. N. (1971) J. Biol.
Chem. 246, 6106).
Research in this laboratory has been aimed at elucidating the mechanism of prothrombin activation by different physiological and nonphysiological clotting agents (l-3).’ In order to in- vestigate the molecular mechanism of prothrombin activation mediated by the circulating plasma coagulation factors, rela- tively large quantities of highly purified factors X, II, and V2 are required. Although procedures for the isolation of bovine factor X have been published (5-8)) major problems have existed with respect to lack of reproducibility, low yield, and contamina- tion of factor X with other clotting factors. It was felt, there- fore, that it was necessary either to extensively modify these procedures or to develop an entirely new procedure for the isola- tion of bovine factor X. Various attempts to modify existing procedures were unsuccessful. The second approach has proved to bc more valuable in that not only has bovine factor X been isolated, but also very high quality bovine prothrombin is ob- tained in excellent yield as a by-product of this procedure.
Before factor X can participate in the conversion of pro-
1 Mr. 1% J. Butkowski has activated bovine, canine, and human prothrombin with tiger snake venom. His studies show that intermediates formed are the same as in citrate-Xa activation and in all three species are approximately of the same molecular weight.
2 The nomenclature for various coagulation factors conforms to the recommendations of the International Nomenclature Com- mittee on blood clotting factors (4).
7729
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7730
thrombin to thrombin, it must be converted to its active en- zymatic form (9-11). Activated factor X (factor Xa) can be obtained from factor X by the action of either: (a) a complex of factors IXa, VIII, calcium, and phospholipid (12-14); (b) plasma factor VII and tissue factor (15-19) ; (c) a specific protein from Russell’s viper venom (5, 6, 20); or (d) trypsin (6, 21, 22). Although the factor Xa3 obtained by these various activating agents can participate in the coagulation process, there is no information available to suggest that these differently derived species of factor Xa are structurally identical or that they each participate in the prothrombin activation process in the same manner.
This communication describes the complete procedure for the simultaneous purification of bovine prothrombin and factor X. Some of the physical properties of the factor X obtained are re- ported. The method for the conversion of factor X to its catalytically active form by the action of insoluble trypsin is described. Finally, the intermediates produced during the ac- tivation of prothrombin with trypsin activated factor X (factor Xa(tryr,sin)) will be discussed with respect to the intermediates produced by other activators of prothrombin.
EXPERIMENTAL PROCEDURE
Materials
Bovine blood was obtained from G. Bartusch Packing Com- pany, St. Paul, Minn., and from Hormel, Inc., Austin, Minn. NIH standard thrombin, lot B-3, was obtained from the National Institutes of Health. Gum acacia was obtained from Scharr and Company, Chicago, Ill., and was purified by the technique described in the NIH clotting procedure (23). Prothrombin two-stage reagent and AC-globulin were from Difco, Detroit, Mich. Factor X-deficient plasma, rabbit brain cephalin, and fibrinogen (82.7 y0 clottable) were obtained from Sigma Chemical Company, St. Louis, MO. Russell’s viper venom (stypven preparation) was from Burroughs Wellcome, New York, N.Y. Human factor X, factor VII, and factor IX deficient plasmas were kindly provided by Dr. E. J. W. Bowie of the Mayo Clinic. Rabbit brain thromboplastin was made by the procedure out- lined in the “Mayo Clinic Laboratory Manual of Hemostasis”
(24). Acrylamide, N, N’-methylenebisacrylamide, 2-mercaptoeth-
anol, basic fuchsin, and N , N , N’ , N’-tetramethylenediamine were from Eastman, Rochester, N. Y. Acrylamide and N ,N’- methylenebisacrylamide were recrystallized from chloroform and 1-propanol, respectively. Guanidinium chloride (ultrapure) was from Heico Chemical Company, Delaware Watergap, Pa. So- dium dodecyl sulfate (DodSOS was from Schwarz-Mann, Orangeburg, N. Y. DEAE-cellulose was purchased from Schleicher and Schuell, Keene, N. H., and was purified by the method of Sophianopolous and Vestling (25). DEAE-Sepha- dex A-50 and Sepharose 6B were from Pharmacia Fine Chemi- cals, Piscataway, N. J. Iodoacetic acid was obtained from Matheson, Coleman and Bell Manufacturing Chemists, Nor- wood, Ohio, and was recrystallized from n-hexane and stored in the dark at -20”. Periodic acid was from Baker Chemical Company, Phillipsburg, N. J. Tosyl-r-arginine methyl ester (TosArgOMe) was obtained from the Cycle Chemical Corpora-
3 The abbreviat,ions used are: Xa, activated factor X; DFP, diisopropylfluorophosphate; DodS04, sodium dodecyl sulfate; TosArgOMe, N-a-tosyl-L-arginine methyl ester; Xa@n,, Xa (trypsin), Xac,itratc), activated form of factor X obtained by the action of Russell’s viper venom, trypsin, and 25% trisodium citrate, respectively.
tion, Los Angeles, Calif. Trypsin and insoluble polyacrylamide trypsin were obt,ained from Sigma. Diisopropylfluorophosphate (DFP) was from Sigma and was used as 1 hl solution in anhy- drous isopropanol. Bovine thrombin (topical thrombin) was obtained from Parke-Davis, Detroit, Mich., and was purified by the method of Lundblad (26). All other reagents were of the best available commercial grade.
Methods
Assays
Prothrombin was determined by the modification of the two stage method of Ware and Seegers (27) described previously (2). Thrombin assays were performed essentially according to the standard NIH procedure (23) as modified by lMann et al. (1). Factor IX was assayed according to the method described in the “Mayo Clinic Laboratory Manual of Hemostasis” (24).
Factor X was assayed by the technique of Bachmann et al. (28) using asbestos adsorbed bovine plasma (obtained from Sigma) as a factor X-deficient substrate and with a congenitally deficient human substrate. The assay is conducted as follows. To 0.1 ml of either substrate plasma, 0.1 ml of test solution is added; Russell’s viper venom in cephalin (0.1 ml) is then added and the mixture is incubated for 30 s at 37”. A O.l-ml volume of prewarmed (37”) 0.025 M CaClz solution is added quickly with a lOO+l syringe to this incubation mixture, and the clotting time is recorded from the point of addition of CaC&. Various dilu- tions of the test solution are made with 0.0168 M imidazole, 0.145 M NaCl, pH 7.4 (imidazole-saline, pH 7.4).” A standard curve is constructed daily by using various dilutions between 1: 10 and 1: 100 of pooled titrated bovine plasma. A linear cali- bration curve is obtained when the log of the plasma dilutions are plotted versus the log of the clotting times. Since the assay is based on the conversion of factor X to its activated form, one unit of factor Xa is arbitrarily defined as that amount of activity which would evolve from 1 ml of undiluted titrated bovine plasma when factor X is activated in the standard Bachmann assay (28) described above. The Bachmann assay is essentially a two-stage measurement, involving first conversion of factor X to factor Xa and then subsequent measurement of the degree of shortening of the clotting time of the factor X-deficient plasma by the factor Xa generated. These two stages may be conducted separately. Purified factor X was incubated with Russell’s viper venom and the factor Xa thus produced was assayed with the standard Bachman assay reagents (omitting the Russell’s viper venom). The activity of factor Xa obtained in this manner was the same as when purified factor X was assayed in the conven- tional manner. These results indicate that the step involving the activation of factor X to factor Xa is not dependent upon the presence of the factor X-deficient plasma in the Bachmann assay, and it is of no significance whether factor Xa is generated in the assay system or extraneously. It appears more appro- priate to consider the Bachmann assay to be one which provides primary data related directly to the factor Xa activity (second stage). We therefore, will express activities in units as those of factor Xa defined by the Bachmann (Russell’s viper venom) assay; 1 mg of factor X will, however, produce different amounts of factor Xa activity under different activating conditions.
Factor VII was assayed by a modification of the procedure de- scribed in “Mayo Clinic Laboratory Manual of Hemostasis” (24). Factor VII-deficient human plasma, 0.1 ml, is mixed with 0.1 ml
4 We shall refer to this buffer throughout this communication as imidazole-saline, pH 7.4.
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

of test solution to which 0.1 ml of rabbit brain thromboplastin is added, and the mixture is incubated for a few seconds at 37”. To this, 0.1 ml of prewarmed 0.025 M CaClz is added and the timer is started. The mixture is agitated in a 37” water bath for 10 S, then the tube is tipped rapidly, and clot formation is timed.
During the early stages of purification of prothrombin and factor X, protein concentration was estimated spectrophoto- metrically assuming Ei$ of 10. The concentration of purified thrombin, prothrombin and factor X were determined spectro- photometrically using the Ei% of 19.5 (29), 14.4 (30), and 12.4 (8), respectively. The activity of factor Xa towards TosArgOMe as a substrate was measured spectrophotometrically by the procedure of Hummel (31). Analyses were conducted at 25”
using a Beckman ACTA CZI spectrophotometer equipped with
a Heath EV-205-11 chart recorder, Esterase activities are ex- pressed as A Ap47 per min.
Electrophoresis
DodS04 polyacrylamide gel electrophoresis was performed by a modification of the method of Weber and Osborn (32) as de- scribed by Mann et al. (1). Protein samples to be analyzed by DodSOl electrophoresis were dialyzed against 0.2 M acetic acid and lyophilized. The lyophilized samples were then dissolved to a concentration of 1 mg per ml in DodSOc sample preparation buffer (170 DodSO+ 0.025 M sodium phosphate, pH 7.1). For disulfide bond reduction, 2-mercaptoethanol was added to each sample (10% by volume, final concentration). Samples were incubated overnight at 37” before application to the gels. Ten to 40 ~1 (30 to 50 pg) of the samples were applied to each gel. In all the determinations of apparent molecular weight by this technique, we have used reduced and S-carboxymethylated standards. When reduced samples were run, the standard was composed of reduced and S-carboxymethylated proteins, and in- cluded 10 y0 (v/v) 2-mercaptoethanol. The protein standards used were human transferrin (77,000), bovine serum albumin (69,000)) ovalbumin (43,000)) carbonic anhydrase (29,500) and cytochrome c (12,400).
Urea-acetic acid gel electrophoresis was performed according to the method of Panyim and Chalkley (33). Disc gel electro- phoresis was performed according to the method of Davis (34) at p1-I 8.9 and Williams and Reisfeld (35) at pH 7.5. DodSOd gels were stained with Coomassie blue. All other acrylamide gels were stained with Amido black. Gels were scanned as described previously (1)
Molecular Weight Determination
In addition to the DodSOc electrophoretic technique, molecu- lar weights were determined using the techniques of sedimenta- tion equilibrium for the native protein and gel filtration in 6 M
guanidinium chloride for the component chains. Sedimentation equilibrium studies were conducted using the short column, high speed technique of Yphantis (36). Protein stored in 50% glyc- erol-water (v/v) was diluted in 0.025 M (sodium) citrate-O.1 M
NaCl (pH 6) to a final concentration of 0.02% and dialyzed against the same solvent for 30 hours. A column height of ap- proximately 3 mm and a temperature of 22” was used. The partial specific volume (u) of factor X has been reported as 0.717 ml per g (37) and as 0.707 ml per g (38) ; both of these values of u were used in calculations of molecular weight.
Gel Filtration
The molecular weights of the constituent chains of the reduced, S-r.a.rhoxvmethvlated. and fluorescein-labeled protein were de-
7731
termined by gel filtration in 6 JI guanidinium chloride. Reduc- tion and S-carboxymethylation were carried out using the pro- cedure of Crestfield et al. (39). Fluorescein labeling of the protein and gel filtration in 6 M guanidinium chloride have been described elsewhere (40, 41).
Isolation of Bovine Prothrombin and Factor X
We routinely carry out the isolation procedure for prothrombin and factor X with 10 to 20 liters of plasma. The specific purifica- tion illustrated was carried out with 14 liters of plasma. All steps are performed at 4” unless specified. Fig. 1 shows the flow diagram of the steps involved in the purification.
1 Collection of Blood-The technology employed in the first two steps is that of Moore et al. (42). Slaughterhouse blood is collected in 2.85% trisodium citrate (blood, 8 parts; antico- agulant, 1 part) and the cells and plasma separated by the means of a continuous flow separator (De Lava1 No. 518). This step is performed at room temperature.
2. Barium Citrate Adsorption and Elution-The vitamin K-de- pendent factors are initially adsorbed onto barium citrate. The plasma is gently stirred and 1 M BaClg (80 ml per liter of plasma, or 1120 ml of BaC12/14 liters of plasma) is added dropwise. Stirring is continued for 10 min after all the BaCl* has been added. The suspension then is centrifuged for 30 min at 3600 x g. We have used a Lourdes continuous flow centrifuge (BETA-FUGE, model-A-2, rotor 1010) for large preparations with good results. The supernatant from the barium citrate adsorption may be used as a source of factors V and VIII. The barium citrate precipitate obtained is suspended in a 1: 9 dilution of stock citrate-saline (9% T\‘aCl and 0.2 M trisodium citrate, diluted 1:9 with distilled HzO) by means of a Waring Blendor at a low speed. The total volume of diluted citrate-saline used is one-third of the volume of starting plasma (4670 ml for 14 liters of plasma). The resuspended protein is reprecipitated by addi- tion of the same volume of 1 RI BaCls used in the initial precipita- tion step. Stirring is continued for 10 min following the addition
Cltmted blood Cel15--~~l:~~~sel~“‘“tor
j (1) Add B&l2
supernatant- I (21 Centrlfu!ze (source 3f factors v & VIII) c
Barium citrate precipitate , j(l) Dissolve in citrate-saline
TWIG? ’
: (2) Add B&l2
Supernatant- 1’3’ Centrlh~e
(discard) Barium citrate precipitate (1) Dialyze Into EDTA (2) Dialyze into citrate-sakne (3) Add saturated ammonium sulfate
to 407 saturation Precipitate--~ (4) Centrifqe
(discard) supernatant
/ Add saturated ainmonium sulfate Supernatant - to 60% saturation
(discard) Preclpltate
(1) Dissolve in 0.025M citrate,
1
pH 6, 10-3M DFP (2) dialyze Into 0.025M citrate,
pH 6 (3) DEAE-cellulose chromatovaphy
O.lM ckloride eluate 0.3M chloride eluate (Factor II) (Factor X, -95% pU,U”Z)
DEAE -Sephadex
--I chromnto$raphy
contaminants Factor X
FIG. 1. Purification scheme for bovine prothrombin and factor x.
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7732
of B&l2 and the suspension is covered with parafilm and allowed to stand for 1 hour. The suspension may be allowed to stand overnight, if desired. The suspension is then centrifuged for 30 min at 3600 x g and the supernatant is discarded. The pre- cipitate is resuspended in diluted citrate-saline as before and the precipitation is repeated with same volume of 1 M BaC&. The barium citrate precipitate obtained at this stage may be stored indefinitely in the frozen state ( - 20’).
The barium citrate precipitate is suspended in cold 0.2 M
EDTA, pH 7.4 (120 ml per liter of plasma). A Waring Blendor at low speed is used to obtain a homogenous suspension. The suspension is dialyzed for 40 min versus 10 volumes of a mixture of 0.2 RI EDTA, pH 7.4, stock citrate-saline (0.2 M trisodium citrate-0.97, KaCl), and distilled water (I:1 :8). Dialysis is continuous versus a similar volume of citrate-saline (stock citrate- saline-distilled water, 1:9) for an additional 3 hours with the dialysate changed about every 30 min and then continued over- night. With each dialysate change, bags are tilted gently to ensure complete mixing.
3. Fractionation with Ammonium sulfate-The viscous sus- pension (EDTA eluate) obtained in Step 2 is brought to 40% saturation with respect to ammonium sulfate. A saturated solution of ammonium sulfate (pH 7.0, adjusted with con- centrated SHdOH) is added dropwise to the slowly stirring EDTA eluate. Stirring is continued for 15 min after all the ammonium sulfate solution has been added. The opaque sus- pension is centrifuged at 3500 x g for 30 min and the precipitate is discarded. The supernatant is then brought to 60% satura- tion with respect to ammonium sulfate by the dropwise addition of saturated ammonium sulfate. Stirring is continued for an additional 10 min. Then the suspension is allowed to stand for 20 min after which it is centrifuged at 3500 x g for 30 min. The supernatant is discarded.
4. DEAE-cellulose Chromatography-The precipitate obtained in Step 3 is dissolved in a minimum volume of 0.025 M (sodium) citrate buffer, pH 6. The protein solution is made 1 mM in DFP by the addition of 1 M DFP in isopropanol. The DFP- treated protein is dialyzed versus 2 liters of 0.025 M (sodium) citrate buffer, pH 6. The dialysis is continued overnight with two changes.
Invariably after dialysis the solution has a small amount of insoluble protein; this is removed by centrifugation at 3500 x g for 20 min. The supernatant is then applied to a DEAE-cellu- lose column (45 x 4 cm) equilibrated with the same buffer. The column is then washed with 0.025 M (sodium) citrate buffer, pH 6.0, until the absorbance of the effluent at 280 nm is less than 0.02. The buffer is then changed to 0.025 M (sodium) citrate- 0.1 M KaCl, pH 6. Prothrombin is eluted with this buffer. Following elution of prothrombin, the column is washed with the same buffer until the effluent has an absorbance of less than 0.01. _4t this point a linear gradient of sodium chloride is applied to the column to clute the factor X. The gradient is formed by the presence of 1400 ml of 0.025 M (sodium) citrate-O.5 M Tu’aCX,
pH 6, in the reservoir and 1400 ml of 0.025 M (sodium) citrate- 0.1 al ?;aCl, pH 6, in the mixing chamber. The flow rate of the column is usually 80 ml per hour.
5. DEAE-Sephadex Chromatography-Factor X obtained by DEAE-cellulose chromatography may be further purified by DEAE-Sephades chromatography if desired. Fractions from the DEAE-cellulose column containing factor X activity are pooled and protein is precipitated by the addition of solid am- monium sulfate (to 80% saturation). The pellet is dissolved in 0.025 M (sodium) citrate-O.1 M NaCl, pH 6, and the protein solu-
tion is made 1 mM in DFP. The solution then is dialyzed versus 0.025 M (sodium) citrate-O.1 M NaCI, pH 6. The dialyzed pro- tein is applied to the DEAE-Sephadex column (2.5 x 40 cm) equilibrated with 0.025 M (sodium) citrate, 0.1 M NaCl, pH 6. The column is washed with the same buffer until the effluent has an absorbance of less than 0.01. Factor X activity is eluted by a linear gradient in sodium chloride formed by the presence of 600 ml of 0.025 M (sodium) citrate-O.1 M NaCl, pH 6, in the mixing chamber and 600 ml of 0.025 M (sodium) citrate-O.7 M NaCl, pH 6, in the reservoir. A flow rate of 40 to 50 ml per hour is maintained.
RESULTS
Purification of Bovine Prothrombin and Factor X-The adsorp- tion of prothrombin to barium citrate was first demonstrated by Lewis and Ware (43). Elution of prothrombin from such a complex with trisodium citrate solutions was not very effective and Lanchantin suggested that a stronger complexing agent, EDTA, be used (44). Remarkably good results were obtained with bovine plasma (42). However, no attempts to determine the factor X activity were reported. In the procedure described here, factor X, as well as prothrombin is eluted effectively and without denaturation from the barium citrate complex. Selec- tive ammonium sulfate fractionation of the EDTA eluatc of the barium citrate complex yields about SO7o of the prothrombin activity and 60% of the factor X activity present in the starting plasma. This step occasionally results in the recovery of more activity for both prothrombin and factor X than the preceding step. This could be due to the presence of trace amounts of inhibitory Ba2+ ions in the EDTA eluate. Ammonium sulfate fractionation might also result in the loss of certain inhibitors which could interfere with the prothrombin two-stage assay and the Bachmann assay for factor X. The most effective step in the purification procedure is DEAE-cellulose chromatography in (sodium) citrate buffers, pH 6. This pH was chosen in order to minimize the proteolytic losses of factor X which result when DEAE-cellulose chromatography is carried out at the higher pH (pH 8.0) used by other workers (8). Use of this chromato- graphic procedure not only results in an increase of the specific activities of bovine prothrombin and factor X but also achieves the complete separation of these factors from each other. A typical separation of bovine prothrombin and factor X effected by the DEAE-cellulose column is shown in Fig. 2. Following application of the prothrombin-factor X-containing sample to the column, washing the column with 0.025 M (sodium) citrate buffer, pH 6, results in the elution of Peak A. Neither prothrom- bin nor factor X activity was present in this peak. DodSOe gel electrophoresis of the nonreduced and reduced samples (gels not shown) from this peak revealed a major protein component (&5%, by visual examination) composed of a single chain and having an apparent molecular weight of 55,000 i 5,000 (reduced sample). The ionic strength of the eluting buffer was then in- creased by the addition of iYaC1 to 0.1 M and two partially re- solved peaks (Peaks B and C) were eluted. Both peaks contained prothrombin of identical specific activities (1300 f 100 NIH thrombin units per mg of protein). This elution pattern was characteristic of every preparation. However, occasionally, the prothrombin in Peak C gave lower specific activity (=lOOO KJH thrombin units per mg of protein). These two prothrombin peaks (Peaks B and C) did not contain detectable levels of thrombin or factors VII, IX, or X. The electrophoretic behavior of the protein(s) in Peaks B and C is described later in this sec- tion. Two additional peaks (Peaks D and E) were eluted from
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7733
o.iM NaCl O.I-0.5M NaCl
12OJ c B B
t
3.0 11
190 270 350 430 490
Fraction number
FIG. 2. Elution profile for DEAE-cellulose column (see “Meth- ods”). Absorbance (O), factor X activity per ml (X), and factor X specific activity (0) are plotted versus fraction number. The dashed line represents the increasing concentration of NaCl in the gradient. Approximately 2200 mg of protein in 120 ml was ap- plied to the column and 7-ml fractions were collected. The col- umn was initially developed with 0.025 M (sodium) citrate, pH 6.0. Peak A was eluted in this buffer and contained neither prothrom- bin nor factor X activity. Peaks B and C were eluted with 0.025 M (sodium) citrate-O.1 M NaCl, pH 6. Both these peaks correspond to nrothrombin. FactorX (Peak E) was eluted in the gradient at a concentration of approximately 0.3 M NaCl. -
the column by the application of the sodium chloride gradient. Peak D was devoid of activity when assayed for the presence of prothrombin and factor X. Peak E contained factor X. Al- though this peak is very symmetrical and the factor X specific activity is constant throughout, electrophoretic analysis of the protein components of this peak in different systems (Fig. 3) revealed a contaminant (-5% by visual examination). The contaminating protein exhibited a lower mobility when compared to the mobility of factor X in all the analytical electrophoretic systems used. The fractions containing prothrombin (Peaks B and C) were pooled separately, as were the fractions containing factor X (Peak E) . Solid ammonium sulfate was added to 80% saturation, the suspensions were centrifuged and the resulting precipitates were taken up in cold 5Ooj, glycerol-Hz0 (v/v) and stored at -20”. Both proteins when stored in this manner are completely stable for at least 18 months. Neither the specific activity nor the electrophoretic behavior of the proteins change with storage.
The factor X may be further purified by DEAE-Sephadex chromatography. An elution profile of a column of this type is shown in Fig. 4. Non-factor X impurities are eluted prior to the elution of factor X, which is eluted in the gradient at approxi- mately 0.5 M NaCI concentration. This step also resulted in a slight increase in the specific activity of the factor X. The specific activity of the factor X obtained by this step is 90 zt 10 units of factor Xa per mg of protein when assayed by the con- ventional Bachmann technique. Prothrombin, thrombin, factor Xa, factor VII, and factor IX were not present in detectable levels when assayed at a concentration of 1 mg per ml of fact,or X. Factor X-containing fractions were pooled and the protein was precipitated by the addition of solid ammonium sulfate to 80% saturation; the resulting precipitate was dissolved in 50% glycerol-Hz0 (v/v) and stored at -2O”, conditions under which it is stable for at least 1 year.
Electrophoretic analysis of the prothrombin from the DEAE- cellulose column (Peaks B and C of Fig. 2) and the factor X from
FIG. 3. A photograph of polyacrylamide gel electrophoreto- grams of factor X obtained from a DEAE-cellulose column (Fig. 2. Fraction numbers 410 to 470). 1, DodSOd (sample unreduckd)u; S; DodSOc (sample reduced); S, urea-acetic acid (pH 3.2); 4, disc (Davis method pH 8.9).
0.0 [
120
Fraction number
FIG. 4. Elution profile for DEAE-Sephadex column (see “Meth- ods”). Absorbance at 280 nm (0 ), factor X activity per ml (X), and factor X specific activity (0) are plotted versus fraction number. The dashed line represents the increasing concentration of NaCl in the gradient. Approximately 80 mg of protein in 20 ml was applied to the column and 10.5-ml fractions were collected.
the DEAE-Sephadex column (Fig. 4) in four different systems is shown in Fig. 5. The mobility of the prothrombin from Peaks B and C is identical in all these systems. Both the prothrombin (from the DEAE-cellulose column, Fig. 2) and the factor X
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from
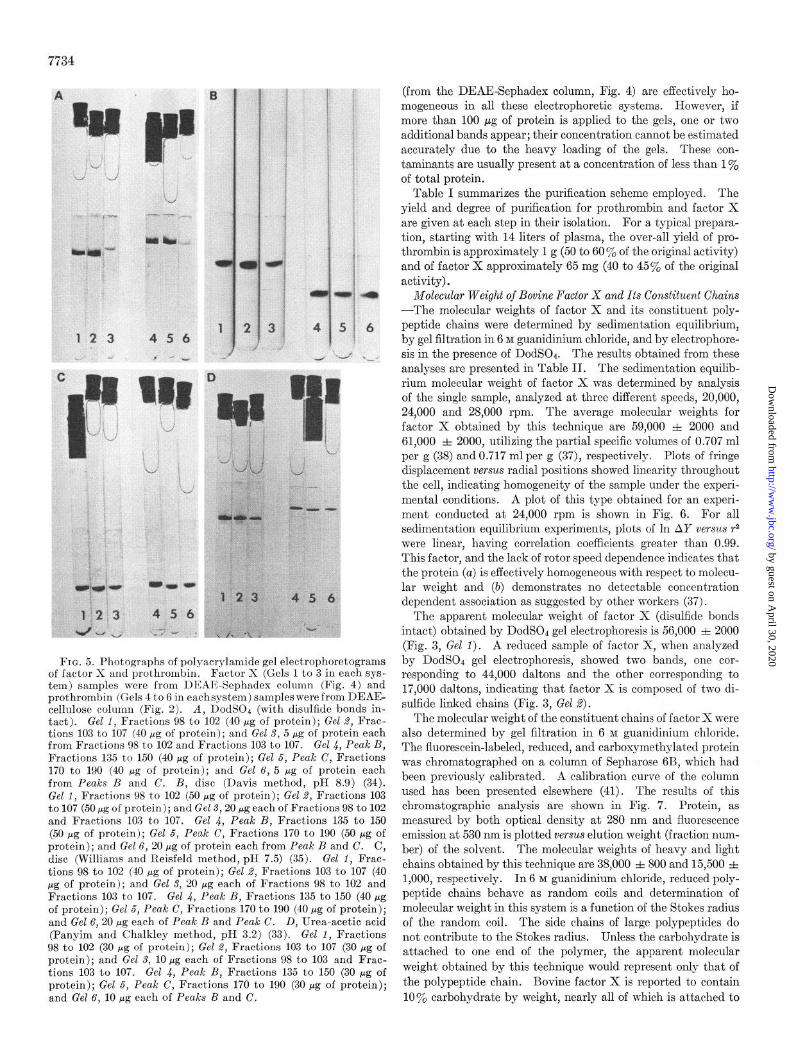
FIG. 5. Photographs of polyacrylamide gel electrophoretograms of factor X and prothrombin. Factor X (Gels 1 to 3 in each sys- tem) samples were from DEAE-Sephadex column (Fig. 4) and prothrombin (Gels 4 to 6 in eachsystem) samples were from DEAE- cellulose column (Fig. 2). A, DodSOc (with disulfide bonds in- tact). Gel 1, Fractions 98 to 102 (40 pg of protein); Gel 2, Frac- tions 103 to 107 (40 pg of protein); and Gel 3, 5 pg of protein each from Fractions 98 to 102 and Fractions 103 to 107. Gel 4, Peak B, Fractions 135 to 150 (40 pg of protein); Gel 5, Peak C, Fractions 170 to 190 (40 pg of protein); and Gel 6, 5 pg of protein each from Peaks B and C. B, disc (Davis method, pH 8.9) (34). Gel 1, Fractions 98 to 102 (50 fig of protein); Gel 2, Fractions 103 to 107 (50 pg of protein); and Gel 3,20 fig each of Fractions 98 to 102 and Fractions 103 to 107. Gel 4, Peak B, Fractions 135 to 150 (50 pg of protein); Gel 5, Peak C, Fractions 170 to 190 (50 rg of protein); and Gel 6, 20 pg of protein each from Peak B and C. C, disc (Williams and Reisfeld method, pH 7.5) (35). Gel 1, Frac- tions 98 to 102 (40 pg of protein); Gel 2, Fractions 103 to 107 (40 pg of protein); and Gel 3, 20 pg each of Fractions 98 to 102 and Fractions 103 to 107. Gel 4, Peak B, Fractions 135 to 150 (40 pg of protein); Gel 5, Peak C, Fractions 170 to 190 (40 Mg of protein); and Gel 6,20 pg each of Peak B and Peak C. D, Urea-acetic acid (Panyim and Chalkley method, pH 3.2) (33). Gel 1, Fractions 98 to 102 (30 pg of protein); Gel 2, Fractions 103 to 107 (30 pg of protein); and Gel 3, 10 fig each of Fractions 98 to 103 and Frac- tions 103 to 107. Gel 4, Peak B, Fractions 135 to 150 (30 pg of protein); Gel 5, Peak C, Fractions 170 to 190 (30 pg of protein); and Gel 6, 10 pg each of Peaks B and C.
(from the DEAE-Sephadex column, Fig. 4) are effectively ho- mogeneous in all these electrophoretic systems. However, if more than 100 E.cg of protein is applied to the gels, one or two additional bands appear; their concentration cannot be estimated accurately due to the heavy loading of the gels. These con- taminants are usually present at a concentration of less than 1% of total protein.
Table I summarizes the purification scheme employed. The yield and degree of purification for prothrombin and factor X are given at each step in their isolation. For a typical prepara- tion, starting with 14 liters of plasma, the over-all yield of pro- thrombin is approximately 1 g (50 to 60 ‘$& of the original activity) and of factor X approximately 65 mg (40 to 45% of the original activity).
Molecular Weight of Bovine Factor X and Its Constituent Chains -The molecular weights of factor X and its constituent poly- peptide chains were determined by sedimentation equilibrium, by gel filtration in 6 M guanidinium chloride, and by electrophore- sis in the presence of DodSOr. The results obtained from these analyses are presented in Table II. The sedimentation equilib- rium molecular weight of factor X was determined by analysis of the single sample, analyzed at three different speeds, 20,000, 24,000 and 28,000 rpm. The average molecular weights for factor X obtained by this technique are 59,000 & 2000 and 61,000 f 2000, utilizing the partial specific volumes of 0.707 ml per g (38) and 0.717 ml per g (37), respectively. Plots of fringe displacement versus radial positions showed linearity throughout the cell, indicating homogeneity of the sample under the experi- mental conditions. A plot of this type obtained for an experi- ment conducted at 24,000 rpm is shown in Fig. 6. For all sedimentation equilibrium experiments, plots of In AY versus r2 were linear, having correlation coefficients greater than 0.99. This factor, and the lack of rotor speed dependence indicates that the protein (a) is effectively homogeneous with respect to molecu- lar weight and (b) demonstrates no detectable concentration dependent association as suggested by other workers (37).
The apparent molecular weight of factor X (disulfide bonds intact) obtained by DodSOI gel electrophoresis is 56,000 =t 2000 (Fig. 3, Gel 1). A reduced sample of factor X, when analyzed by DodSOd gel electrophoresis, showed two bands, one cor- responding to 44,000 daltons and the other corresponding to 17,000 daltons, indicating that factor X is composed of two di- sulfide linked chains (Fig. 3, Gel 2).
The molecular weight of the constituent chains of factor X were also determined by gel filtration in 6 M guanidinium chloride. The fluorescein-labeled, reduced, and carboxymethylated protein was chromatographed on a column of Sepharose 6B, which had been previously calibrated. A calibration curve of the column used has been presented elsewhere (41). The results of this chromatographic analysis are shown in Fig. 7. Protein, as measured by both optical density at 280 nm and fluorescence emission at 530 nm is plotted versus elution weight (fraction num- ber) of the solvent. The molecular weights of heavy and light chains obtained by this technique are 38,000 f 800 and 15,500 f 1,000, respectively. In 6 M guanidinium chloride, reduced poly- peptide chains behave as random coils and determination of molecular weight in this system is a function of the Stokes radius of the random coil. The side chains of large polypeptides do not contribute to the Stokes radius. Unless the carbohydrate is attached to one end of the polymer, the apparent molecular weight obtained by this technique would represent only that of the polypeptide chain. Bovine factor X is reported to contain 10% carbohydrate by weight, nearly all of which is attached to
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7735
TABLE I
Purification of bovine factors II and X
Prothrombin (factor II) Factor X
Volume
Purification stage I I ,, ,^ Units/
mg
7
Total units %.age yield Purifi. catior
Purifi- cation I
I- __--~ ml
1.4 x 104 1 80” 0.0125 1.4 X lo* 100
2580 3.6 9288 66
128 62 23.5d 2.63 7936 57
ml
Plasma............. 1.4 X lo4 195 80” EDTA eluate from
barium citrate.. 2580 853 Ammonium sulfate
fractionation. 128 1.8 X lo4 23.5d DEAE-cellulose
chromatography. 800 1.7 x 103 1.31 DEAE-Sephadex
chromatography.
2.44
750
1300
1
310
535
210
6400
7200
2.73 X lo6 100
2.2 X lo6 80
2.3 X lo6 84
1.36 X 10” 50
(L ?jational Institutes of Health (NIH) units (23). c Long (45). b One unit of factor Xa activity is the amount which would d Determined by the absorbance assuming E$ = 10.
evolve from 1 ml of pooled titrated bovine plasma when factor X 6 Spectrophotometrically estimated using Ei$, of 14.4 (30). is activated in the standard Bachmann assay (28). / Obtained spectrophotometrically using I$,“6 of 12.4 (8).
Molecular weights of bovine factor X ancl its constituent chainsa
Technique Molecular weight of factor X I
Molecular weights of constituent chains
1. Sedimentation equilibriumb (unreduced sample
in aqueous solvent) B = 0.717 ml/g (37) 0 = 0.707 ml/g (38)
2. Gel filtration in 0 M guanidinium chloridec 3. Electrophoresis
DodSOc (unreduced sample)e DodSOa (reduced sample)f
61,000 f 2,000 59,000 It 2,000
44,000 rt 800;d 15,500 f 1,000
56,000 f 2,000 44,000 xt 2,000; 17000 * 2,000
a Error in molecular weights is expressed as the maximum devi- ation from the mean value.
* Average of three runs. c Average of two runs.
d Includes the contribution which would have been made by the
carbohydrate content attached to this chain (see text for explana- tion).
e Average of 10 runs. f Average of 5 runs.
,b’
I
the heavy chain (37). Based on the molecular weight of factor X as 60,000, the mass of the carbohydrate attached would be 6000, hence the molecular weight of the heavy chain, 44,000 i 800 (38,000-dalton polypeptide + 6000-dalton carbohydrate).
Production of Factor Xa by Russell’s Viper Venom and by
Trypsin-Factor XacRvv, was produced by incubating factor X at 37” in imidazole-saline, pH 7.4, with Russell’s viper venom. Samples were assayed at different times for factor Xa and Tos- ArgOMe esterase activity. In a typical experiment, factor X (11 pg per ml) was incubated with (a) 0.1 pg per ml and (b)
0.05 pg per ml of Russell’s viper venom. Aliquots of the samples were drawn, diluted lo-fold, and assayed for factor Xa activity. In each experiment, the units of factor Xa generated were identi- cal (e90 units of factor Xa per mg of factor X) and the activation reached a plateau at 50 min. Although the final concentrations of Russell’s viper venom in the assay mixtures in these experi- ments were 2.5 ng per ml in (a) and 1.25 ng per ml in (b), the factor Xa activity generated was equivalent to that obtained in the conventional Uachmann assay which provides a final Russell’s viper venom concentration of 1.25 pg per ml. This suggests that the presence of Russell’s viper venom in the assay mixture has
70
66
62
38Eu -I-
49.8 500 502 50.4 50.6 50.8 51.0 51 2
r’ (cm’)
FIG. 6. Sedimentation equilibrium centrifugation of bovine factor X obtained from DEAE-Sephadex chromatography. The natural logarithm of the fringe displacement (corrected for water blank) in microns (In A Y) is presented as a function of the square of the radial distance (+). Speed, 24,000 rpm; temperature, 20.35”; buffer, 0.025 M (sodium) citrate, 0.1 M NaCl, pH 6; +,’ represents the ceU bottom.
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7736
Fraction number
FIG. 7. Elution profile of the gel filtration of the fluorescein- labeled, reduced, and S-carboxymethylated factor X in 6 1~ guanidinium chloride on Sepharose 6B. Absorbance at 630 nm, blue dextran (cl); 280 nm, protein (X ); 360 nm, dinitrophenyl- alanine (A); and relative fluorescence (see text) (0) are plotted versus fraction number.
little effect upon the assay and that it is of no significance whether the factor Xa activity is generated extraneously or in the assay system. To further test this hypothesis, 100 pg of factor X per ml (final concentration) was incubated with 1 fig per ml of Russell’s viper venom (final concentration). Aliquots of the mixture were removed throughout the course of the ex- periment, diluted 100 times, and assayed for factor Xa activity. The activation was complete within less than 20 min, and the maximum level of factor Xa activity (factor Xa units) gen- erated was the same (92 units of factor Xa per mg of factor X) as in the previous experiments, although the concentration of Russell’s viper venom was only 2.5 ng per ml in the final assay mixture.
We conclude from these observations that: (a) although the rate of factor X activation depends upon the venom concentra- tion, the final yield of factor Xa activity generated does not and (b) within the range tested (1.25 ng per ml to 1.25 pg per ml) the clotting behavior of the second stage of the assay system is in- dependent of the Russell’s viper concentration. Additional studies reported below further indicate that the second stage functions normally in the absence of Russell’s viper venom. The maximum level of TosArgOMe esterase activity of factor Xa, obtained by the action of Russell’s viper venom was 0.115 AA per min per mg of factor X. Russell’s viper venom, at a con- centration timesgreater than that present in the factor Xa(nvv) samples did not possess any detectable TosArgOMe esterase activity, nor did factor X itself.
Studies of trypsin activation of factor X were conducted using a short column of trypsin insolubilized on polyacrylamide. The resin (obtained from Sigma) was equilibrated with imidazole- saline, pH 7.4. Resin containing approximately 15 units5 of trypsin activity was packed into a polyethylene column (1.3 X 8 cm) equipped with a fritted polyethylene disc. Two to 3 mg of factor X in 4 ml of imidazole saline, pH 7.4 at 25”, was applied to the column. Fractions were collected at intervals and assayed for factor Xa and TosArgOMe esterase activity. Results of a typical experiment are shown in Fig. 8. There is a coincident rise in the TosArgOMe esterase and factor Xa clotting activity. Following the attainment of maximum factor Xa activity, there is a slight plateau and then a rapid decline in the factor Xa clot- ting activity, whereas the TosArgOMe esterase activity declines very slowly. The maximum factor Xa activity generated in this system was 750 =t 50 units per mg of factor X. This activity is
5 One unit of trypsin will hydrolyze 1.0 pmole of benzoyl-n- arginine ethyl ester per min at pH 8.0 at 30”.
Incubation time, minutes
FIG. 8. Activation pattern of factor X with insoluble trypsin. Percentage of maximum clotting activity (Xactrrpain) clotting) and TosAraOMe (I’AME) esterase activity is plotted as a func- tion of time of activation. Actual specific activities of factor Xa(trypsin) obtained are also shown. The experimental con- ditions for activation are described under “Results.” Factor X applied to the trypsin-acrylamide column represents the zero time sample.
measured in the second stage of the standard Bachmann assay and the values obtained in the total absence of Russell’s viper venom were identical to those obtained if Russell’s viper venom was present in the cephalin reagent used for factor X assays. The maximum TosArgOMe esterase activity of factor Xa ob- tained from the trypsin-acrylamide column was 0.890 AA per min per mg of factor X.
The maximum yield of factor Xa obtained from 1 mg of factor X by the action of Russell’s viper venom and by the action of insoluble-trypsin under the conditions used are substantially different. The yield of factor Xa obtained by the action of trypsin on factor X is approximately 8 times higher than that obtained by the action of Russell’s viper venom on factor X, both in terms of clotting and TosArgOMe esterase activity. Factor Xa(tr,,sin) is stable in 50% glycerol-Hz0 (v/v) at -20” for at least 5 months. During storage at 4” in imidazole-saline, pH 7.4, factor Xa(+,rypsin) retained 90% of its original activity after 1 week. Examination of Fig. 8 reveals that the factor Xa pro- duced by trypsin is rapidly degraded in the presence of the activator. Following approximately 35 min of treatment at 25”, essentially all activity is destroyed. The trypsin sensitivity of the factor Xa(trypsin) and the storage data indicate that no trypsin is present as a contaminant in the factor Xa removed from the column. Moreover, trypsin (at a concentration 10 times greater than the factor Xa(trypsin) used) did not shorten the clotting time of the factor X-deficient plasma in the Bach- mann assay system. The stability of the factor Xa(trypsin) prepared by this technique makes it possible to use the enzyme as a laboratory reagent.
Prothrombin contamination of factor X would result in high apparent yield of factor Xa following trypsin activation. If factor X is contaminated with prothrombin, the action of trypsin on the factor X preparation would also convert prothrombin to thrombin, or the factor Xa generated would act on prothrombin to yield thrombin; either event, therefore, could cause a high apparent yield of factor Xa obtained by the action of trypsin. To probe this possibility, the differential rate of inhibition of factor Xa, thrombin and trypsin by 1 mM DFP proved to be useful. Thrombin, purified by the Lundblad procedure (26) and trypsin were used at concentrations which gave rates of
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7737
TosArgOMe hydrolysis equivalent to that of the typical factor Xa(trypsin) solution. Sufficient 1 M DFP was then added to each tube to provide a final concentration of 1 InM. Since the Tos- ArgOMe esterase assay itself is not affected by the presence of DFP in the assay mixture, aliquots of these samples were taken at different times and analyzed for their ability to hydrolyze TosArgOMe. Within 5 min, thrombin and trypsin were both inactivated, whereas there was no detectable change in the rate of hydrolysis of TosArgOMe by factor Xa(trypain). This factor Xa<trypsin) then was dialyzed versus imidazole-saline, pH 7.4, and assayed for its clotting activity. More than 90% of the clotting activity was recovered (680 factor Xa units per mg of factor X) . These results indicate that the higher yield of factor Xa obtained by the action of trypsin is not due to contamination of the factor Xa with trypsin or thrombin and that a DFP treatment of controlled and short duration provides a convenient means of the selective inactivation of thrombin in a factor Xa preparation.
During the activation of factor X by insoluble trypsin, aliquots were withdrawn and analyzed by DodSOe gel electrophoresis. In all of our experiments dealing with the production of factor XactryBsin) we have observed that the production of factor Xa coincides with the formation of an electrophoretic component with an apparent molecular weight in DodSOa gels of 52,000 f 2000. Conversely, disappearance of the component correspond- ing to 52,000 dalt,ons parallels the loss of the factor Xa clotting activity. Fig. 9 shows a photograph of DodSOd gel electro- phoretograms of factor X activation samples (disulfide bonds intact) removed at different times. The zero time gel is the factor X sample (DodSOa apparent mol wt, 56,000) prior to ap- plication to the trypsin column. The 52,000-dalton component is apparent in the 5- and lo-min activation samples. Residual factor X can still be seen after 5 min of activation. Five addi- tional components of lower molecular weight are also evident on the gels. The apparent molecular weights of these components obtained by this technique are also shown in Fig. 9. A further comparison of Figs. 8 and 9 indicates that a substantial propor- tion (at least 40%) of the TosArgOMe esterase activity is asso- ciated with components of molecular weights lower than 52,000 possessing essentially no factor Xa clotting activity. This ob- servation in conjunction with the association of the factor Xa ac- tivity with the 52,000-dalton component suggests that the action of trypsin on factor X initially produces factor Xa, which has a DodSOc apparent molecular weight (with disulfide bonds intact) of 52,000 =t 2000; and low molecular weight products which do not possess appreciable clotting activity but retain their ability to hydrolyze TosArgOMe are the result of either additional cleav- ages of factor Xa or nonproductive cleavages of factor X by trypsin.
A similar DodSOl gel electrophoretic analysis of the factor X activation samples in the reduced state was conducted. Fig. 10 shows the scans of the DodSOl gel electrophoretograms of the unreduced (lower curve) and reduced (upper curve) 5-min activa- tion sample. This activation sample was chosen because it contained a representative distribution of all the components detected in reduced gels. The 44,000-dalton component in the reduced gel was only present if the 56,000-dalton component (residual factor X) was present in the unreduced sample and represents the heavy chain of factor X (see molecular weight de- termination section). Similarly in the reduced samples the 40,000-dalton component was associated with the presence of the 52,000.dalton component in the unreduced samples and it was present only when the 52,000-dalton component was present in
FIG. 9. A photograph of DodSOl gel electrophoretograms of the nonreduced samples during the time activation of factor X with insoluble trypsin. The vertical column of numbers on the left represents the apparent DodSOb molecular weights X 10-a (disulfide bonds intact) of the stained bands. The numbers on the gel tubes correspond to activation time in minutes. The zero time sample corresponds to the factor X applied to trypsin- acrylamide column.
I3
Positlon In gel, cm
FIG. 10. Absorbance scans of DodSOd electrophoretograms of trypsin activated factor X (5 min incubation time, Fig. 9). Absorbance at 560 nm is plotted 2rersus gel position (centimeters). A, unreduced sample; B, reduced sample. In each case 30 pg of protein were applied to the gel. The numbers associated with each peak represent the DodSOa apparent molecular weights.
the unreduced state. Since the clotting activity of factor Xa coincides with the presence of the 52,000-dalton component in the unreduced samples and the 40,000-dalton component in the reduced samples, the 40,000-dalton component in the reduced samples represents the heavy chain of factor Xa. Since the difference between the apparent molecular weights of factor X (56,000) and factor Xa(trypsinj (52,000) is approximately 4,000 and the same difference (4000) is observed between the heavy chain (44,000) of factor X and the heavy chain of fact.or Xa(trYpsin), these observations suggest that during the conversion of factor X to factor Xa(trypsin) a small peptide of molecular
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from
7738
Incubation time, hours
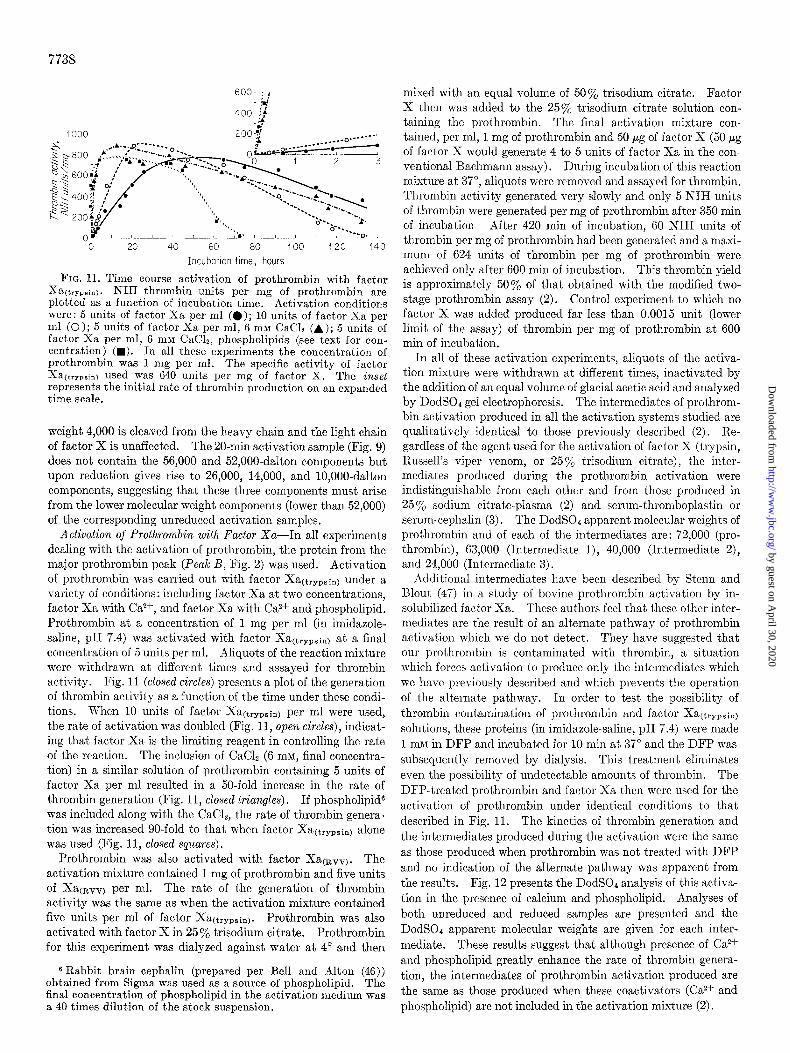
FIG. 11. Time course activation of prothrombin with factor Y i a(tryph). NIH thrombin units per mg of prothrombin are plotted as a function of incubation time. Activation conditions were: 5 units of factor Xa per ml (0); 10 units of factor Xa ner ml (0); 5 units of factor Xa per ml, 6 mM CaCh (A); 5 units of factor Xa per ml, 6 mM CaCL. uhosnholioids (see text for con- centration) (w). ‘In all these experiments the concentration of prothrombin was 1 mg per ml. The specific activity of factor Xa(trypain) used was 640 units per mg of factor X. The inset represents the initial rate of thrombin production on an expanded time scale.
weight 4,000 is cleaved from the heavy chain and the light chain of factor X is unaffected. The 20.min activation sample (Fig. 9) does not contain the 56,000 and 52,000-dalton components but upon reduction gives rise to 26,000, 14,000, and lO,OOO-dalton components, suggesting that these three components must arise from the lower molecular weight components (lower than 52,000) of the corresponding unreduced activation samples.
Activation of Prothronzbin witlz Factor Xa-In all experiments dealing with the activation of prothrombin, the protein from the major prothrombin peak (Peak B, Fig. 2) was used. Activation of prothrombin was carried out with factor Xa(trypsin) under a variety of conditions: including factor Xa at two concentrations, factor Xa with Ca*f, and factor Xa with Ca*+ and phospholipid. Prothrombin at a concentration of 1 mg per ml (in imidazole- saline, pH 7.4) was activated with factor Xa(trypsin) at a final concentration of 5 units per ml. Aliquots of the reaction mixture were withdrawn at different times and assayed for thrombin activity. Fig. 11 (closed circles) presents a plot of the generation of thrombin activity as a function of the time under these condi- tions. When 10 units of factor Xa(trypsin) per ml were used, the rate of activation was doubled (Fig. 11, open circles), indicat- ing that factor Xa is the limiting reagent in controlling the rate of the reaction. The inclusion of CaCl2 (6 mM, final concentra- tion) in a similar solution of prothrombin containing 5 units of factor Xa per ml resulted in a 50-fold increase in the rate of thrombin generation (Fig. 11, closed triangles). I f phospholipid6 was included along with the CaC12, the rate of thrombin genera- tion was increased go-fold to that when factor Xa(trypsin) alone was used (Fig. 11, closed squares).
Prothrombin was also activated with factor Xa(nvv). The activation mixture contained 1 mg of prothrombin and five units of Xa(avv) per ml. The rate of the generation of thrombin activity was the same as when the activation mixture contained five units per ml of factor Xa(trypsin). Prothrombin was also activated with factor X in 25% t.risodium citrate. Prothrombin for this experiment was dialyzed against water at 4” and then
6 Rabbit brain cephalin (prepared per Bell and Alton (46)) obtained from Sigma was used as a source of phospholipid. The final concentration of phospholipid in the activation medium was a 40 times dilution of the stock suspension.
mixed with an equal volume of 5Ooj, trisodium citrate. Factor X then was added to the 25% trisodium citrate solution con- taining the prothrombin. The final activation mixture con- tained, per ml, 1 mg of prothrombin and 50 pg of factor X (50 pg of factor X would generate 4 to 5 units of factor Xa in the con- ventional Bachmann assay). During incubation of this reaction mixture at 37”, aliquots were removed and assayed for thrombin. Thrombin activity generated very slowly and only 5 NIH units of thrombin were generated per mg of prothrombin after 350 min of incubation After 420 min of incubation, 60 IC’IH units of thrombin per mg of prothrombin had been generated and a maxi- mum of 624 units of thrombin per mg of prothrombin were achieved only after 600 min of incubation. This thrombin yield is approximately 50% of that obtained with the modified two- stage prothrombin assay (2). Control experiment to which no factor X was added produced far less than 0.0015 unit (lower limit of the assay) of thrombin per mg of prothrombin at 600 min of incubation.
In all of these activation experiments, aliquots of the activa- tion mixture were withdrawn at different times, inactivated by the addition of an equal volume of glacial acetic acid and analyzed by DodSOe gel electrophoresis. The intermediates of prothrom- bin activation produced in all the activation systems studied are qualitatively identical to those previously described (2). Re- gardless of the agent used for the activation of factor X (trypsin, Russell’s viper venom, or 25% trisodium citrate), the inter- mediates produced during the prothrombin activation were indistinguishable from each other and from those produced in 25% sodium citrate-plasma (2) and serum-thromboplastin or serum-cephalin (3). The DodSOa apparent molecular weights of prothrombin and of each of the intermediates are: 72,000 (pro- thrombin), 63,000 (Intermediate 1)) 40,000 (Intermediate 2), and 24,000 (Intermediate 3).
Additional intermediates have been described by Stem and Blout (47) in a study of bovine prothrombin activation by in- solubilized factor Xa. These authors feel that these other inter- mediates are the result of an alternate pathway of prothrombin activation which we do not detect. They have suggested that our prothrombin is contaminated with thrombin, a situation which forces activation to produce only the intermediates which we have previously described and which prevents the operation of the alternate pathway. In order to test the possibility of thrombin contamination of prothrombin and factor Xa(trypsin) solutions, these proteins (in imidazole-saline, pH 7.4) were made 1 m&l in DFP and incubated for 10 min at 37” and the DFP was subsequently removed by dialysis. This treatment eliminates even the possibility of undetectable amounts of thrombin. The DFP-treated prothrombin and factor Xa then were used for the activation of prothrombin under identical conditions to that described in Fig. 11. The kinetics of thrombin generation and the intermediates produced during the activation were the same as those produced when prothrombin was not. treated with DFP and no indication of the alternate pathway was apparent from the results. Fig. 12 presents the DodSOd analysis of this activa- tion in the presence of calcium and phospholipid. Analyses of both unreduced and reduced samples are presented and the DodSOd apparent molecular weights are given for each inter- mediate. These results suggest that although presence of Cazf and phospholipid greatly enhance the rate of thrombin genera- tion, the intermediates of prothrombin activation produced are the same as those produced when these coactivators (Ca2+ and phospholipid) are not included in the activation mixture (2).
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

FIG. 12. A photograph of DodSOd gel electrophoretograms of nonreduced (left) and reduced (right) samples taken during the activation of prothrombin with factor Xaorypein) in the presence of Ca* and phospholipids. A representative sample of the total
DISCUSSION
The yield of factor X obtained by the isolation procedure described is about 5 mg per liter of the starting plasma. This is 5 times higher than that obtained by the procedure of Jackson et al. (8). The procedure is relatively simple and in addition offers simultaneous isolation of highly purified prothrombin in good yield. By carrying out the chromatographic steps of the purification at pH 6, the possibility of proteolysis of factor X and prothrombin by plasma proteases is diminished. We have fur- ther reduced this possibility by DFP treatment as initially sug- gested by Jackson et al. (8). However, because of the relatively low pH in our procedure, we have not found it necessary to include DFP in every buffer. The recent isolation procedure of Fujikawa et al. (37) makes use of several protease inhibitors (soybean trypsin inhibitor, benzamidine-HCl, Heparin) during collection of blood. The complete stability of our factor X in 50% glycerol-H,0 at -20” for at least 18 months and the fact that it is a single component in four different electrophoretic systems lead us to believe that it is not necessary to use these inhibitors. The recovery of factor X reported by these workers is 40 to 50% of the starting plasma which is quite comparable to the yield (45%) obtained by our procedure. The specific ac- tivity of their purified product range from 90 to 130 units per mg of protein which is comparable to the specific activity (90 & 10 units of factor Xa per mg of factor X) obtained by our procedure. Jackson (38) and Fujikawa et al. (37) have also observed resolu- tion of bovine factor X into two components (denoted factor X1 and factor X2) upon DEAE-Sephadex chromatography. Jack- son (38) has reported that factor X2 contains 2 hexose and 1 to 2 more sialic acid residues than factor Xi. However, Fujikawa et al. (37) found no significant difference in the carbohydrate content of factor X1 and X2. Under the conditions of our DEAE- Sephadex chromatography (pH 6), we have not observed any resolution of the two factor X proteins. However, when chro- matography was carried out at pH 8 as described by Jackson and Hanahan (48), factor X protein(s) partially resolved into two peaks. At the present time we are not certain whether these two species of factor X arise as a result of the modification of one of these factor X species at pH 8 by a plasma enzyme present at a level too low to detect or whether there actually exist two differ-
number of gels is depicted. The numbers on the vertical axes represent the DodS04 apparent molecular weights for the stained bands in each set of gels. The numbers on the gels correspond to the activation times.
ent forms of factor X in plasma. It is interesting to note that observations of Fujikawa et al. (37) show that factor Xp (56,000) is of slightly higher molecular weight than factor X1 (53,000). It is quite feasible that a time-dependent reaction occurs when factor X is exposed to a pH of 8. Does the ratio of factor X1 to factor X2 change after long exposure to pH 8? We have not yet explored this possibility.
The yield of prothrombin obtained by this procedure is approxi- mately 70 mg per liter of the starting plasma, which is comparable to the best isolation procedures reported so far (42, 49). The quality of the product is evident from it.s homogeneity in four different electrophoretic systems. The prothrombin is partially resolved into two components on the DEAE-cellulose column. Although the minor prothrombin peak (Peak C, Fig. 2) occa- sionally was of slightly lower specific activity, it is not a degrada- tion product of the major prothrombin peak, as its electrophoretic behavior in a variety of systems is identical to that of the major prothrombin peak. (Both prothrombins co-migrate in all the systems used.) For four preparations of prothrombin, the major prothrombin peak amounted to 85 f 3y0 of the total prothrom- bin eluted. Both of the prothrombin components (disulfide bonds reduced) have an apparent molecular weight of 72,000 I 4,000 in the DodSOc gel dectrophoretic system. This value is in good agreement with the values of 68,000 to 74,000 reported previously (49, 50).
The DodSOd apparent molecular weight of factor X (56,000) with disulfide bonds intact is slightly lower than the sum of the apparent molecular weights (44,000 + 17,000) of the constituent chains obtained by the same technique. The protein-DodS04 complex is not a random coil but has a high degree of ordered structure and the conformation of the protein with disulfide bonds intact is different from that of the protein in the reduced state (51). Pitt-Rivers and Impiombato (52) have shown that following the reductive cleavage of disulfide bonds, proteins may bind 50% more DodSOd. Their data also support the view that diminished binding of DodSOd by proteins with disulfide bonds intact is due to their restricted configuration. As a consequence of this restriction, the presence of disulfide bonds may result in low values for the apparent molecular weight of nonreduced proteins (1).
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7740
The sedimentation equilibrium molecular weight of native factor X reported by Jackson (38) and Fujikawa et al. (37) is approximately 55,000. When we use the values of v reported by these authors, our data provide a slightly higher molecular weight (59,000 for a 1s of 0.707 ml per g (38) and 61,000 for a v of 0.717 ml per g (37)). We have not observed any heterogeneity or con- centration dependence of our factor X preparations as seen by Fujikawa et al. (37). These authors attribute the heterogeneity they observed to limited association of factor Xi and factor Xa protein to form higher molecular weight species.
The molecular weight of factor X obtained by sedimentation equilibrium in 6 M guanidinium chloride with disulfide bonds intact has been reported by Furie et al. (53) as 59,000. However, these authors did not report the value of v used in their studies. In the present study, the molecular weight of the heavy chain of factor X obtained by gel filtration in 6 M guanidinium chloride is 44,000. This value is slightly higher than that reported by Jackson (38) (41,000 for factor X2) and Fujikawa et al. (37) (36,000 for factor X1 and 41,000 for factor X2). The molecular weight of the light chain of factor X obtained by gel filtration in 6 M guanidinium chloride in the present study is 15,500. This value is in close agreement with the values obtained by Jackson (38) (15,000) and Fujikawa et al. (37) (16,000 to 18,000).
Our preliminary data suggest that factor Xa(trypsin) has an apparent molecular weight of 52,000 in the unreduced state as judged by DodS04 gel electrophoretic technique. The molecular weight of factor Xa(nvv) obtained by sedimentation equilibrium in different laboratories is 44,000 (27,000-s-s-17,000) (54), 48,000 (30,000-s-s-20,000) (55), and 57,800 (53). The apparent DodSOc molecular weight of factor Xac nvv) (unreduced sample) obtained by Furie et al. (53) is 56,000 (35,000-s-s-19,000). From these data it is hard to view whether different agents which can acti- vate factor X result in the formation of the same or different activated species of the factor X zymogen. However, irrespec- tive of the agent used to activate factor X, the action of factor Xa (produced by Russell’s viper venom, by trypsin, or by citrate) on prothrombin produces the same intermediates of prothrombin activation which are seen in citrate-plasma (2) and serum throm- boplastin (3) activation of prothrombin.
Roth Russell’s viper venom and trypsin represent nonphysio- logical activators of factor X. In both cases, multiple products are produced upon the activation of factor X. The higher yield of factor Xa obtained by the action of insolubilized trypsin as compared to that obtained by the action of Russell’s viper venom cannot be explained with the data presently in hand. Three alternatives exist, which may account for the difference in the specific activity of factor Xa(nvv) and factor Xa(tryps+. These alternatives are as follows. (a) The different activators produce different products, with different specific activities. (5) Of the multiple products produced by each activator, only one repre- sents an active factor Xa molecule, the others being degradation products or side reactions of the activator with factor X. If the relative rates of the side reactions which lead to nonactive prod- ucts or the rates of destruction of factor Xa by the activation are different in the case of Russell’s viper venom and trypsin, then the observed specific activity of the final products would be different. (c) If either of the activators produces, in addition to the active factor Xa, side products which are inhibitory to factor Xa, a specific activity difference would also be observed. At the present time, we do not have adequate data to decide which of the above three alternatives is indeed the cause of the difference in the specific activity of factor Xa(nvv) and factor Xa (trypsin). This is, however, currently under investigation.
Under the conditions used to activate prothrombin, we have observed one unique pathway of activation which results in the formation of same intermediates as in citrate-plasma (2). How- ever, further studies in our laboratory (56) have shown that the first step of the activation (which can be caused by factor Xa or thrombin) results in the formation of Intermediate 1 (COOH- terminal region of prothrombin) and Intermediate 3 (NH2- terminal region of prothrombin) . Intermediate 1 is then cleaved to produce Intermediate 2 (COOH-terminal region of Inter- mediate 1) and Intermediate 4 (NHz-terminal region of Inter- mediate 1). This step is the result of action of factor Xa on Intermediate 1. Thrombin is ineffective in carrying out this reaction. Factor Xa further acts on Intermediate 2 to produce a-thrombin. Thrombin again is ineffective in carrying out this reaction. With the exception of Intermediate 1 which provides an erroneously high molecular weight (63,000) on DodSOa gel electrophoresis, the DodSOc molecular weights of the other inter- mediates are consistent with values determined by sedimentation equilibrium. The sedimentation equilibrium molecular weights of Intermediates 1,2, and 3 are 51,000,40,000, and 23,000 respec- tively (56). The molecular weight of intermediate 4 as deter- mined by gel filtration in 6 M guanidinium chloride is 13,000 (56). All these intermediates of prothrombin activation were seen in the present study irrespective of the origin of factor Xa. The exact location of Intermediate4 in the DodS04 gels in the present experiments could not be visualized because of the presence of degradation products of a-thrombin. An alternative hypothesis for the mechanism of activation of prothrombin proposed by Stenn and Blout (47) has not been observed in the present in- vestigation. At the present time, however, we cannot rule out the possibility suggested by these authors that the action of factor Xa on prothrombin produces Intermediate 2 and a frag- ment composed of Intermediates 3 and 4. The prothrombin and factor Xa used in our studies, however, do not contain thrombin as suggested by these authors (47). The important question which still remains to be answered is whether prothrombinase complex (factor Xa, factor V, Ca2+, and phospholipid) produces these very intermediates observed in the present study or not. However, the inclusion of CaZ+ and phospholipid in the prothrom- bin activation reaction mixture produced electrophoretically indistinguishable intermediates to those produced in the absence of these accelerators. Only the rate of activation was enhanced.
Acknowledgments-The authors are greatly indebted to Dr. D. N. Fass for his constant encouragement, advice, and critical discussions throughout the course of these investigations. The authors are grateful to Drs. E. J. W. Uowie and Charles A. Owen for their constant encouragement, support, and advice. The authors thank Ms. Mary Miskimen for her excellent technical assistance. The authors also express their appreciation to Ms. Therese L. Cole for her help in performing factors VII and IX assays.
REFERENCES
1. MANN, K. G., HJSLDEBRAST, C. AI., AND Fass, D. N. (1971) J. Biol. Chem. 246, 5994
2. MANN, K. ct., HELDEHRANT, C. RI., AND FASS, D. N. (1971) J. Biol. Chem. 246, 6106
3. FASS, D. N., AND MANN, K. G. (1973) J. Biol. Chem. 248, 3280
4. WRIGHT, I. (1959) J. Amer. Med. Ass. 170, 325 5. ESNOUF, M. P., AND WILLIAMS, W. J. (1962) Biochem. J. 84,
62 6. PAPAHADJOPOULOS, D., YIN, E. T., AND HANAHAN, D. J.
(1964) Biochemistry 3, 1931
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

7741
7. LECHNER, K., AND DEUTSCH, E. (1965) Thromb. Diath. Haemorrh. 13, 314
8. JACKSON, C. M., JOHNSON, T. F., HANAHAN, D. J. (1968) Bio- chemistry 7, 4492
9. DAVIE, E. W., AND RATNOFF, 0. D. (1964) Science 146, 1310 10. MACFARLANE, R. G. (1964) AJature 202, 498 11. DAVIE, 1:. W., HOUGIIC, C., AND LUNDBLAD, R. L. (1969) in
Recent Advances in Blood Coagulation (POLLAR, L., ed), p. 13, Little, Brown and Company, Boston
12. BERGGAGEL, 1~. E., AND HOGIE, C. (1956) Brit. J. Haematol. 2, 113
13. HOUGIE, C., DENSON, K. W. E., END BIGGS, R. (1967) Thromb. Diath. Haemorrh. Suppl. 18, 211
14. OSTERUD, B., AWD RAPAPORT, S. I. (1970) Biochemistry 9, 1854
15. FLYXN, J. 15., AND COON, R. W. (1953) Amer. J. Physiol. 176, 289
16. HOUGIE, C. (1959) Proc. Sot. Exp. Biol. Med. 101, 132 17. NEMERSON, Y., AND SPAET, T. H. (1964) Blood 23, 657 18. NEMERSON, Y. (1966) Biochemistry 6, 601 19. WILLIAMS, W. J., AND NORRIS, D. G. (1966) J. BioZ. Chem.
241, 1847 20. MACFARLANE, R. G. (1961) Brit. J. Haematol. 7, 496 21. FERGUSON, J. H., WILSON, E. G., IATRIDIS, S. G., RIERSON,
H. A., AND JOHNSTON, B. R. (1960) J. CZin. Invest. 39, 1942 22. PECHET, L., AND ALEXANDER, B. (1960) Fed. Proc. 19, 64 23. Minimum Requirements for Dried l’hrombin (1946) Second
Revision, National Institutes of Health, Bethesda, Maryland 24. BOWIE, E. J. W., THOMPSON, J. H. JR., DIDISHEIM, P., AND
OWEN, C. A., JR. (1971) Mayo Clinic Laboratory Manual of Haemostasis, W. B. Saunders Company, Philadelphia, Pa.
25. SOPHIAXOPOLOUS, A. J., AND VESTLING, C. 8. (1962) Biochem. Prep. 9, 102
26. LTJXDBLAD, R. L. (1971) Biochemistry 10, 2501 27. WARE, A. G., AND SEEGERS, W. H. (1949) Amer. J. CZin. Pathol.
19, 471 28. BACHMANN, F., DUCI<I’:RT, F., AND HOLLER, F. (1958) Thromb.
Diath. Haemorrh. 2, 24’ 29. WINZOR. D. J.. AND SCHERAGA. H. A. 11964) J. Phus. Chem.
68, 338 ’ \ I
30. Cox, A. C., AND HANAHAN, D. J. (1970) Biochim. Biophys. Acta 207, 49
31. HUMMEL, B. C. W. (1959) Can. J. Biochem. Physiol. 37, 1393 32. WEBISR, K., AXD OSBORN, M. (1969) J. BioZ. Chem.
244, 440G
33. PANYIM, S., AND CH~LKLEY, R. (1969) Arch. Biochem. Bio- phys. 130, 337
34. DAVIS, B. J. (1964) Ann. N. Y. Acad. Sci. 121, 404 35. WILLIAMS, D. E., AND REISFELD, R. A. (1964) Ann. N. Y.
Acad. Sci. 121, 373 36. YPHANTIS, D. A. (1964) Biochemistry 3, 297 37. FUJII~AWA, K., LEGAZ, M. E., AND D~VIE, E. W. (1972) Bio-
chemistry 11, 4882 38. JACKSON, C. M. (1972) Biochemistry 11, 4873 39. CRESTFIELD, A. M., MOORE, S., AND STEIN, W. 1~. (1963) J.
Biol. Chem. 233, 622 40. MANN, K. G., FASS, D. N., AND FISH, W. W. (1973) in Advances
in Chemistry (GOULD, 1~. F., ed), American Chemical Soci- ety, Washington, D. C.
41. MANX, K. G., .4ND FISH, W. W. (1972) Melhods Enzymol. 26, 28
42. MOORE, H. C., Lux, S. E., MALHOTRA, 0. P., BAIUZMIN, S., AND CARTER. J. R. (1965) Biochem. Bionhus. Acta 111, 174 ’
~ I ‘ y
43. LEWIS, M. J., AND WARE, A. G. (1953) Proc. Sot. Exp. Biol. Med. 34, 636
44. LANCHANTIN, G. F. (1958) Amer. J. Physiol. 194, 7 45. LONG, C. (1961) Biochemist’s Handbook; Princeton, N. J. 46. BELL. W. N.. AND ALTON. H. G.. (1954) Nature 174. 880 47. STEN&, K. ‘S., AND BLOUT, k: R.’ (1972) Biochemistry
11, 4502 48. JACI~SON, C. M., AND HANAHAN, D. J. (1968) Biochemistry
7, 4506 49. INGWALL, J. S., AND SCHERAGA, H. A. (1969) Biochemistry
6, 1860 50. ARONSON, D. L., AND M~~NAcH~, D. (1966) Biochemistry 6,
2635 51. FISH, W. W., REYNOLDS, J. A., AND TANFORD, C. (1970) J.
BioZ. Chem. 246, 5166 52. PITT-RIVERS, R., AND IMPIOMBATO, F. S. A. (1968) Biochem.
J. 109, 825 53. FURIE, B. C., FURIE, B., GOTTLIWB, A. J., AND WILLIAMS,
W. J. (1972) Fifteenth Annual Meeting, American Society of Hematology, p. 121
54. FUJIKAWA, K., LEGAZ, M. E., AND DAVIE, E. W. (1972) Bio- chemistry 11; 4892
55. RADCLIFFE. R. D.. AND BARTON. P. G. (1972) J. Biol. Chem. 247, 7735’ ’
\ ,
56. HELDEBRANT, C. M., BUTKOWSKI, R. J., BAJAJ, S. P., AND MANN, K. G. (1973) J. BioZ. Chem. 248, 7149
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Satya P. Bajaj and Kenneth G. MannOF PROTHROMBIN BY TRYPSIN-ACTIVATED FACTOR X
Simultaneous Purification of Bovine Prothrombin and Factor X: ACTIVATION
1973, 248:7729-7741.J. Biol. Chem.
http://www.jbc.org/content/248/22/7729Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/248/22/7729.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 30, 2020
http://ww
w.jbc.org/
Dow
nloaded from





























