Embed Size (px)
Citation preview

1
Name student: R.G.M. Lammerts
Student number: S1836005
Faculty supervisor: H.G.D. Leuvenink
External supervisor: Prof. R.J. Ploeg
Daily supervisor: Z.M. Akhtar
Preventing brain death induced kidney injury through
exploiting the hypoxia inducible factor pathway
Oxford Transplant Centre, Department of Surgical Sciences. University of Oxford

2
Abstract in English
Background and aims: Brain death-derived kidney allografts have inferior short and
long-term outcomes when compared to the living donated allograft, even when HLA
mismatches and cold ischemia times are taken into consideration. Brain death leads to a
physiologically abnormal state resulting in dramatic hemodynamic, hormonal, coagulation
and inflammatory disturbances culminating in kidney injury prior to organ procurement.
Two promising therapies evaluated in the brain death setting include heme-oxygenase 1
(HO-1) and erythropoietin (EPO) administration. These are both downstream effectors of the
hypoxia inducible factor (HIF) pathway. HIF is the universal cellular oxygen sensing
mechanism and mediates over 100 hypoxia responsive genes. The HIF pathway is
responsible for the early and delayed response of ischaemic preconditioning, a pathway in
what exposure of an organ to a short period of ischemia protects the organ against exposure
to subsequent periods of ischemia. We hypothesized that up regulation of HIF in brain death
donors could protect against brain death induced kidney injury and improve the outcomes of
kidney transplantation. To begin to evaluate this hypothesis I characterized the effects of
brain death on HIF1α expression in comparison to other models of hypoxic/ischemic injury.
Methods: the expression of HIF1α was characterized in four models of kidney
ischemic injury. Adult male Fischer rats were used in the described animal model.
(n=8, 230-350g). Brain death (DBD) was induced by a gradual inflation of a subdurally
placed balloon catheter. In addition an ischemia reperfusion model(n=2) and a deceased after
circulatory death model (n=2) was developed. Cell culture NRK-49 cells were grown in
DMEM medium. Western blotting was performed to characterize the differential expression
of HIF and HO-1, a downstream effector of HIF.
Results: A brain death animal model was developed. Induction of brain death showed
a drop in blood pressure after approximately 40 min. It was not possible to increase the blood
pressure to 80 mmHg after brain death induction. Western blotting showed HIF1α expression
in the 4 models of kidney injury. HO-1 expression was seen in 3 models.
Conclusions: The BD model needs optimization to maintain the animals stable for a
longer period of time. These results suggest that HIF1α is activated in the 4 different models
of kidney injury. Western blotting for kidney samples showed more HIF1α and HO-1
expression in the IRI experimental group than in the control group. In the DCD model HIF1α
is less expressed in the experimental group than in the control group, whereas HO-1 is more
expressed in the DCD experimental group. As for the DBD group, both HIF1α and HO-1 are
more expressed in the experimental group than in the control group. Western blotting for
liver samples showed more HIF1α expression in the IRI control group than in the IRI
experimental group. In the liver DCD model HIF1α is more expressed in the control group
than in the DCD experimental group, whereas in the DBD model both HIF1α and HO-1 are
more expressed in the experimental group than in the control group. However, to confirm this
further research needs to be performed.
Expression of the prolyl hydroxylases 1,2 and 3 and expression of HIF2 α and HIF 3α are
questions yet to be answered before administration of a prolyl hydroxylase inhibitor and
translation to the clinical setting.
Keywords:HIF, hypoxia inducible factor, DBD, DCD, brain death, deceased after
circulatory death, transplantation, ischemia reperfusion, IRI, animals, heme oxygenase-1,
HO-1, EPO,1, erythropoietin

3
Samenvatting in het Nederlands
Achtergrond en doelstellingen: Nieren verkregen van hersendode orgaan donoren
hebben een slechtere korte en lange termijn overleving wanneer dit wordt vergeleken met de
levende donor, zelfs wanneer HLA mismatches en koude ischemie tijden in beschouwing
worden genomen. Hersendood leidt tot een fysiologische abnormale staat die resulteert in
dramatische hemodynamische-, hormonale-, coagulatie-, en ontstekings- verstoringen die
samenklonteren in de nier voordat orgaan donatie begint. Twee veel belovende therapieën in
de hersendood setting zijn de toediening van heme-oxygenase-1 en erythropoietin (EPO). Dit
zijn beide eindproducten van de hypoxia inducible factor(HIF) pathway. HIF is een cellulair
zuurstof gevoelig mechanisme en medieert meer dan 100 genen die reageren op hypoxie. HIF
is verantwoordelijk voor de vroege en de late fase van preconditie, een pathway waarin de
blootstelling van een orgaan aan een korte periode van ischemie, het orgaan beschermd tegen
de volgende langere periode van ischemie. De hypothese is dat de activatie van HIF in
hersendode orgaandonoren bescherming biedt aan nierschade door hersendood en hiermee de
uitkomsten van niertransplantatie verbeteren. Om te beginnen met het testen van deze
hypothese, werden de effecten van hersendood op HIF1α expressie gekarakteriseerd in
vergelijking met andere modellen van hypoxie/ischemie schade.
Methoden: De expressie van HIF1α werd gekarakteriseerd in vier modellen van nier
ischemie schade. Volwassen mannelijke Fischer ratten werden gebruikt in het beschreven
diermodel. (n=8, 230-350g). Hersendood (DBD) werd geïnduceerd door een langzaam
opgeblazen dubduraal geplaatste ballon katheter. Ook werd ischemie reperfusie model (IRI)
(n=2) en een overleden na circulatie model (DCD) (n=2) ontwikkeld. Het was niet mogelijk
om de bloeddruk weer naar 80 mmHg te laten stijgen na hersendood inductie. NRK-49 cellen
werden gekweekt in DMEM medium. Western blotting werd gebruikt om de verschillen in
expressie van HIFα en HO-1, een eindproduct van HIF1α, te karakteriseren.
Resultaten: Een hersendood dier model werd ontwikkeld. Inductie van hersendood liet
een daling in bloeddruk zien na ongeveer 40 minuten. Western blotting liet HIF1α expressie
zien in de 4 modellen van nier schade. HO-1 expressie werd gezien in 3 modellen.
Conclusie: Het hersendood model heeft optimalisatie nodig om de dieren stabiel te houden
voor een langere tijd. Deze resultaten suggereren dat HIF1α is geactiveerd in de 4
verschillende modellen van nier schade. Western blotting voor de nier liet zien dat HIF1α en
HO-1 meer tot expressive komen in de experimentele IRI groep dan in de IRI controle groep.
In de DCD setting HIF1α komt minder tot expressie in de experimentele groep dan in de
controle groep, terwijl HO-1 meer tot expressie komt in de experimentele groep. In de DBD
groep, komen beide HIF1α en HO-1 meer tot expressie in de DCD experimentele groep dan
in de controle groep. Western blotting voor de lever liet zien dat HIF1α meer tot expressie
komt in de controle groep dan in de experimentele groep. In het lever DCD model komt
HIF1α meer tot expressie in de controle groep dan in de DCD experimentele groep terwijl in
het DBD model beide HIF1α en HO-1 meer tot expressie komen in de experimentele groep
dan in de controle groep. Echter, om dit te bevestigen is er meer onderzoek nodig. Expressie
van de prolyl hydroxylases 1,2 en 3 en expressie van HIF2α en HIF3α zijn vragen die nog
beantwoord dienen te worden voordat administratie van een prolyl hydroxylase inhibitor en
vertaling naar de klinische setting kan plaatsvinden.
Sleutel woorden: HIF, hypoxia inducible factor, DBD, DCD, IRI, hersendood, overladen
naar circulatie stop, transplantatie, ischemie reperfusie, dieren, heme oxygenase-1, HO-1,
EPO, erythropoietin

4
Preface
This is the final report of the scientific clerkship for the Master in Medicine. The emphasis of
this project lies on 3 major aims;
- The development of a slow induction brain death animal model in rodents;
- The exploitation of the hypoxia inducible factor pathway;
- The establishment of the QUality in Organ Donation (QUOD) bio-bank.
Once this clerkship is finished, research and collaboration between the University of Oxford
and the University of Groningen will continue. Experience gained from previous research
projects plus this clerkship in Oxford encourages my ambition to keep on working within this
research field.
The main goal of the scientific internship was to improve different skills: performing animal
experiments, performing laboratory work, scientific writing, scientific thinking, to take part
in a management team and of course the experience of living abroad. This research is based
on developing a brain death animal model, deceased after circulatory death model and
ischemia reperfusion model. While analyzing the samples creative scientific thinking was
required to develop and proceed to the next step.
To develop a parallel translational strategy the Quality in Organ Donation (QUOD) initiative
was established. The key of QUOD is to increase the number and quality of organs procured
from deceased donors for transplantation by evaluating pathways of injury and repair in
organ donors and establishing a platform for the investigation of interventional strategies in
organ donors. In fact comparable to the animal work but QUOD is the translation to the
human setting. During my stay in Oxford a management team was constructed for the
establishment of the QUOD initiative. The management team was constructed to establish the
collection of biological materials and clinical data from organ donors.
It is important for optimizing donor organ quality to bring together all the participants in
organ retrieval, including clinical and scientific experts. The QUOD initiative is a beginning
project providing opportunities for clinicians and researchers to aid and perform research in
the optimization of donation and transplantation.
Working in the QUOD management team gave me the chance to gain experience in writing
protocols, understanding the UK transplant system and work with people from different
fields.
Whilst living abroad during my scientific internship and working in the QUOD national
management team I gained a lot of life and-, research experience and developed my
communication skills.
Rosa Lammerts

5
Table of contents
Abstract in English .................................................................................................................................. 2
Samenvatting in het Nederlands ............................................................................................................. 3
Preface .................................................................................................................................................... 4
Table of contents ..................................................................................................................................... 5
Introduction ............................................................................................................................................. 6
Internship in Oxford ............................................................................................................................ 6
Organ Donation and transplantation .................................................................................................. 7
Donation after brain death (DBD) ....................................................................................................... 7
Donation after circulatory death (DCD) .............................................................................................. 8
Phases of organ injury during donation .............................................................................................. 8
Ischemia/reperfusion injury (IRI) ........................................................................................................ 9
Hypoxia inducible factor ................................................................................................................... 12
Rationale ............................................................................................................................................... 14
Methods ................................................................................................................................................ 15
Results ................................................................................................................................................... 20
Conclusion and Discussion ................................................................................................................... 28
Translational strategy; relevancy for the human setting .................................................................. 32
Conflict of interest ................................................................................................................................ 35
Acknowledgements ............................................................................................................................... 35
References ............................................................................................................................................. 36
Supplementary protocols ...................................................................................................................... 40

6
Introduction
Internship in Oxford
Following participation in the Transplantation summer school I was inspired to carry out my
scientific internship at the Oxford Transplant Centre, University of Oxford under the
guidance of Prof Rutger J Ploeg. Prof Ploeg had recently been appointed as Professor of
Transplant Biology and Honorary Consultant Transplant Surgeon in Oxford and was
establishing a number of exciting initiatives.
I planned to join Prof Ploeg and his team in Oxford for 6 months to work on two of the
major initiatives:
- To work with a DPhil student, Zeeshan Akhtar, to establish a rodent model of brain
death in Oxford. This model had been developed in Groningen by the Surgical
Research Laboratory and has been used as a basic science model to investigate the
pathways responsible for organ injury following brain death and the effects of
therapeutic interventions. (1)We aimed to collaborate on a project looking at novel
interventions in the brain dead organ donor. In preparation for this aspect of the
project I spent several weeks during the summer becoming familiar with the model in
Groningen. I learned the western blotting laboratory technique in Oxford, which was
used to characterize the hypoxia inducible factor pathway.
- To aid in the establishment of a national organ donor bio-repository to identify
biomarkers in the donor that can be used to predict the outcomes of transplantation. A
project termed Quality in Organ Donation (QUOD) and sponsored by NHS Blood and
Transplant.
Both of these initiatives concern addressing the most important issue in transplantation; the
lack of good quality organs. It is estimated in the UK that one in three patients a day will
either die or become too unwell to receive a transplant whilst on the waiting list. Thus there is
an urgent need to protect, preserve and repair donor organs whilst also identifying markers to
predict the outcomes of transplantation. It is the translational prospect of these projects and
their clinical relevance that appeared scientifically and clinically interesting to me. My
previous experience as President of the Prometheus kidney team would aid me in assisting in
the establishment of the QUOD bio-repository. I participated as part of this team collecting
biopsies during my Bachelor and as a President responsible for the operational aspects of the
team.
One of the trends observed across Europe in the last decade has been that increasingly
“marginal donors” are being used to address the organ deficit. These are organs obtained
from older donors with additional co-morbities, termed extended criteria donors (ECDs) and
even donors after cardiocirculatory arrest (DCDs). The vast majority of organs obtained from
deceased individuals still come from brain dead organ donors (DBDs), although record
numbers of donors were achieved from DCD donors last year in the UK accounting for 40%
of all deceased donation.
The application I wrote 7 months ago concerned the effect of antithymocyte globulin (ATG)
on renal injury in brain stem dead rats. Since submission of the application a different
approach to addressing kidney injury following brain death has been pursued, by attempting
to exploit the hypoxia inducible factor (HIF) pathway. The rationale for this change was
because of an increasing body of literature supporting the exploitation of the HIF pathway as
an ischemic pre-conditioning and post-conditioning strategy. In addition work performed by
another group in Argentina and presented at the American Transplant Congress demonstrated
7
only a modest improvement in terms of organ injury when ATG was administered to brain
dead rats(2).
Organ Donation and transplantation
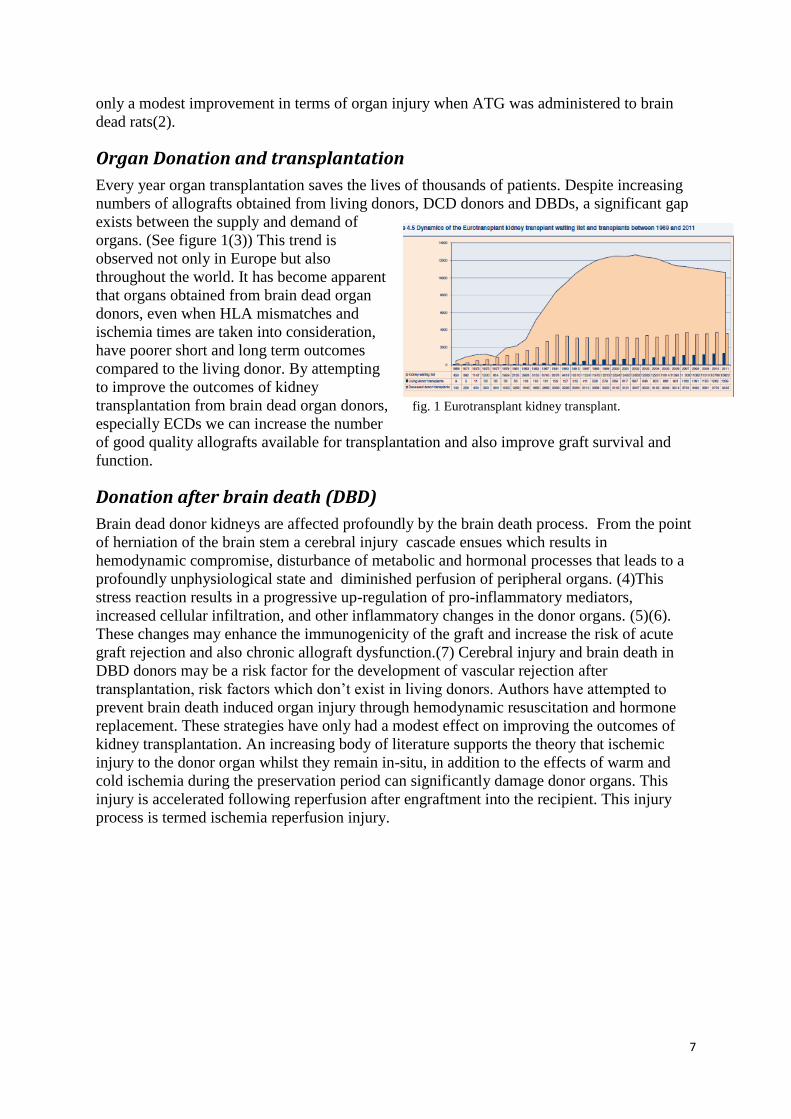
Every year organ transplantation saves the lives of thousands of patients. Despite increasing
numbers of allografts obtained from living donors, DCD donors and DBDs, a significant gap
exists between the supply and demand of
organs. (See figure 1(3)) This trend is
observed not only in Europe but also
throughout the world. It has become apparent
that organs obtained from brain dead organ
donors, even when HLA mismatches and
ischemia times are taken into consideration,
have poorer short and long term outcomes
compared to the living donor. By attempting
to improve the outcomes of kidney
transplantation from brain dead organ donors, fig. 1 Eurotransplant kidney transplant.
especially ECDs we can increase the number
of good quality allografts available for transplantation and also improve graft survival and
function.
Donation after brain death (DBD)
Brain dead donor kidneys are affected profoundly by the brain death process. From the point
of herniation of the brain stem a cerebral injury cascade ensues which results in
hemodynamic compromise, disturbance of metabolic and hormonal processes that leads to a
profoundly unphysiological state and diminished perfusion of peripheral organs. (4)This
stress reaction results in a progressive up-regulation of pro-inflammatory mediators,
increased cellular infiltration, and other inflammatory changes in the donor organs. (5)(6).
These changes may enhance the immunogenicity of the graft and increase the risk of acute
graft rejection and also chronic allograft dysfunction.(7) Cerebral injury and brain death in
DBD donors may be a risk factor for the development of vascular rejection after
transplantation, risk factors which don’t exist in living donors. Authors have attempted to
prevent brain death induced organ injury through hemodynamic resuscitation and hormone
replacement. These strategies have only had a modest effect on improving the outcomes of
kidney transplantation. An increasing body of literature supports the theory that ischemic
injury to the donor organ whilst they remain in-situ, in addition to the effects of warm and
cold ischemia during the preservation period can significantly damage donor organs. This
injury is accelerated following reperfusion after engraftment into the recipient. This injury
process is termed ischemia reperfusion injury.
8
Donation after circulatory death (DCD)
In order to enlarge the donor pool, many centres are revisiting donation after circulatory
death (DCD). In DCD donors the organs are subjected to a substantial period of oxygen
deprivation and ischaemic injury prior to retrieval. In addition, in this donor pool the organs
suffer from warm and cold ischemia. The amount of injury differs for the various donor
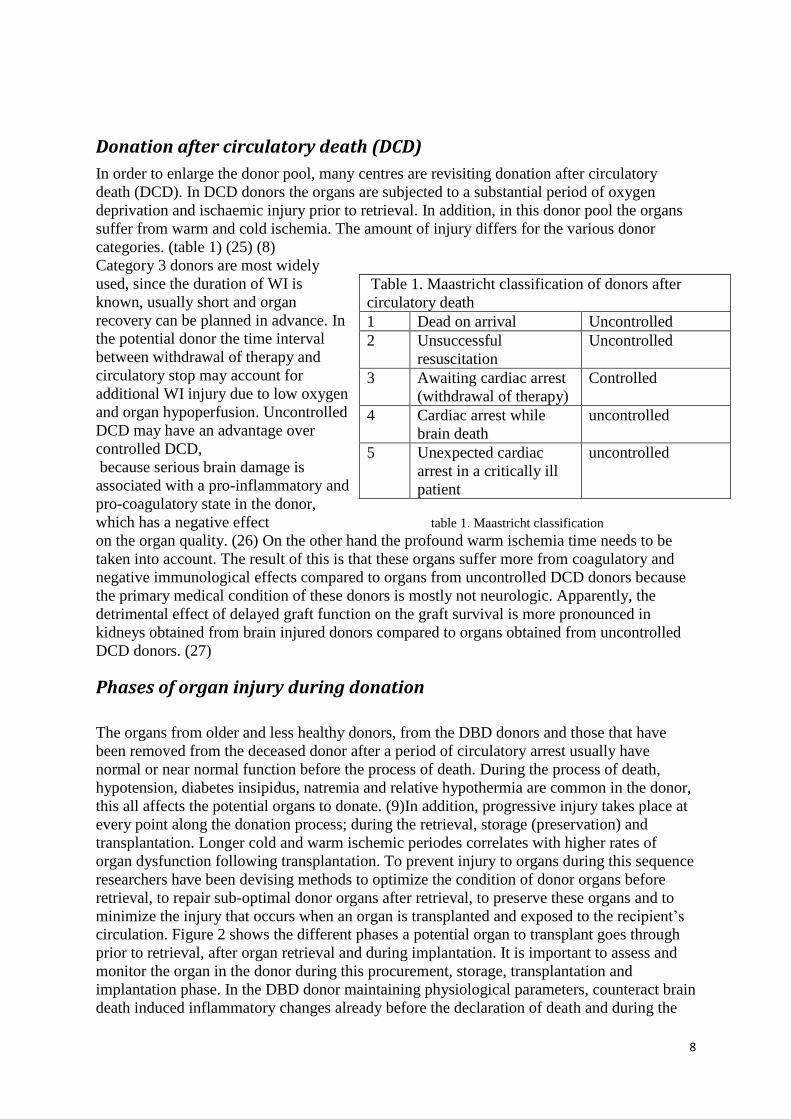
categories. (table 1) (25) (8)
Category 3 donors are most widely
used, since the duration of WI is
known, usually short and organ
recovery can be planned in advance. In
the potential donor the time interval
between withdrawal of therapy and
circulatory stop may account for
additional WI injury due to low oxygen
and organ hypoperfusion. Uncontrolled
DCD may have an advantage over
controlled DCD,
because serious brain damage is
associated with a pro-inflammatory and
pro-coagulatory state in the donor,
which has a negative effect table 1. Maastricht classification
on the organ quality. (26) On the other hand the profound warm ischemia time needs to be
taken into account. The result of this is that these organs suffer more from coagulatory and
negative immunological effects compared to organs from uncontrolled DCD donors because
the primary medical condition of these donors is mostly not neurologic. Apparently, the
detrimental effect of delayed graft function on the graft survival is more pronounced in
kidneys obtained from brain injured donors compared to organs obtained from uncontrolled
DCD donors. (27)
Phases of organ injury during donation
The organs from older and less healthy donors, from the DBD donors and those that have
been removed from the deceased donor after a period of circulatory arrest usually have
normal or near normal function before the process of death. During the process of death,
hypotension, diabetes insipidus, natremia and relative hypothermia are common in the donor,
this all affects the potential organs to donate. (9)In addition, progressive injury takes place at
every point along the donation process; during the retrieval, storage (preservation) and
transplantation. Longer cold and warm ischemic periodes correlates with higher rates of
organ dysfunction following transplantation. To prevent injury to organs during this sequence
researchers have been devising methods to optimize the condition of donor organs before
retrieval, to repair sub-optimal donor organs after retrieval, to preserve these organs and to
minimize the injury that occurs when an organ is transplanted and exposed to the recipient’s
circulation. Figure 2 shows the different phases a potential organ to transplant goes through
prior to retrieval, after organ retrieval and during implantation. It is important to assess and
monitor the organ in the donor during this procurement, storage, transplantation and
implantation phase. In the DBD donor maintaining physiological parameters, counteract brain
death induced inflammatory changes already before the declaration of death and during the
Table 1. Maastricht classification of donors after
circulatory death
1 Dead on arrival Uncontrolled
2 Unsuccessful
resuscitation
Uncontrolled
3 Awaiting cardiac arrest
(withdrawal of therapy)
Controlled
4 Cardiac arrest while
brain death
uncontrolled
5 Unexpected cardiac
arrest in a critically ill
patient
uncontrolled

9
agonal phase is of a great value, whereas in the DCD donor it is important to reduce the warm
ischemia time in this period. In both the DCD and DBD donors it is important to reduce
damage to the organ during organ procurement, ex-vivo storage and transportation. By
cooling the procured organs as soon as possible warm ischemia injury could be prevented. In
addition, preserving the organs through in-vivo and ex-vivo perfusion with nutrients, oxygen
and other additives is an intervention which provides benefit in reducing the injury that
occurs in donor organs. In the donation and transplantation process ischaemia reperfusion
injury is a central mechanism resulting in organ injury. It is important to begin to address this
injury mechanism in the donor, through understanding the pathways of injury.
Figure 2
Ischemia/reperfusion injury (IRI)
Ischemia
The definition of ischemic injury is cessation of arterial blood flow with immediate oxygen
deprivation of cells (ie, hypoxia with accumulation of metabolic products.)
Ischemia/reperfusion injury (IRI) is unfortunately unavoidable in transplantation and it can be
a serious complication of kidney transplantation.
Different organs have different degrees of tolerance to ischemic injury. The brain for example
suffers permanent loss of function when exposed to a few minutes of ischemia as illustrated
in rodent models of ischemia reperfusion injury. (10)In kidney transplantation allograft
damage is reversible if the warm ischemia time of the kidney is less than 30 minutes,
prolonging the warm ischemia time results in delayed graft function (DGF) and diminished
allograft survival. (11)
Ischemia involves oxygen and nutrient deprivation, switch to anaerobic metabolism and the
accumulation of catabolites with the capacity to induce cell death and attract erythrocytes,
leukocytes and platelets. The renal cortex has an especially high requirement for oxygen.
Under normal physiological conditions in normoxia, intracellular potassium and magnesium

10
levels are kept relatively high, whereas sodium and calcium concentrations are kept low. This
physiological state is maintained due to the activity of the ATP dependent sodium-potassium
pump and calcium-magnesium pump. In an hypoxic/ischemic cell, ATP is no longer
produced and thus these pumps are no longer able to maintain the concentration gradients.
Sodium, chloride and water diffuse into the cell and potassium and magnesium diffuse out.
Cell swelling ensues and cell lysis pathways are activated.(10).
During organ donation an organ is damaged during recovery, preservation and transplantation
and this occurs primarily as a result of ischemia. Techniques for organ preservation, like
cooling of the organs to decrease cell metabolism, serve to minimize this damage. In a brain
death donor the brain death process itself leads to a physiologically abnormal state resulting
in dramatic hemodynamic, hormonal, coagulatory and inflammatory disturbances
culminating in kidney injury prior to organ procurement. This injury is exacerbated during
the preservation phases and worsened on reperfusion in the repecient..(12)
Repefusion injury is irreversible damage after ischemia. Figure 3 illustrates the effects of
hypoxia and ischemia on renal cell metabolism. During warm ischemia, adenosine tri-
phosphate is degraded to adenosine monophosphate. Because of calcium influx the cytosolic
proteases are activated. They produce xanthin with the formation of superoxide free radical
and hydrogen peroxide.
Also, after ischemia the cell no longer has its usual reserve of free radical scavengers. This
increases the vulnerability of cellular and subcellular membranes to injury by lipid
peroxidation. The subcellular and cellular membranes are damaged, so after reperfusion the
capacity of restoration of homeostasis in the cell is decreased. Erythrocytes, leukocytes and
platelets adhere to damaged vasculature, because production of oxygen free radicals also
initiates production of prostaglandins, including leukotriene B4, which is an chemoattractant.
These neutrophils can cause secondary hypoxia in the kidney, followed by further
preservation injury. (10)
Figure 3. Results of hypoxia and warm ischemia on renal cell metabolism.

11
Protection of the kidney from ischemia injury
In recent years authors have investigated strategies to tackle ischemia reperfusion injury.
Examples of these treatments include; calcium channel blockers, allopurinol, free radical
scavengers, steroids, vasoactive drugs energy replenishment therapies and several others. But
despite the plethora of therapies the efficacy of these agents to reduce ischemic damage is
limited.
Two promising therapies have emerged showing an ability to ameliorate brain death induced
kidney injury in an experimental setting. This has been the application of heme-oxygenase 1
(HO1) upregulators and carbamylated erythtopoietin (cEPO). (13)These are both downstream
effectors of the hypoxia inducible factor (HIF) pathway, and are cytoprotective molecules
that are thought to protect against ischemic injury. They decrease the expression of several
proinflammatory genes, including vascular adhesion molecule-1, an adhesion molecule that
regulates leukocyte migration from the blood into tissues in addition to p-selectin and e-
selectin. These are both mediating the attachment of leukocytes to endothelial cells and are
early adhesion molecules. cEPO also impairs IL-1 and IL-6 expression, which are important
proinflammatory cytokines. HO-1 and EPO also decrease the polymorphonuclear (PMN) cell
infiltration (leukocyte migration) in the kidney. (14)
In addition, HO-1 is responsible for the breakdown of heme proteins, resulting in the
generation of biliverdin, carbon monoxide (CO) and iron. These products play a critical role
in the normal function of the kidney as well as protecting the kidney from ischemic insults
and exposure to nephrotoxins, so they have an important anti-oxidant, anti-apoptotic,
cytoprotective and anti-inflammatory property. (15)
EPO has, in addition to its ability to decrease PMN cell infiltration more complex actions.
EPO rescues cells from apoptosis (programmed cell death) to increase their survival. Other
effects include stimulation of angiogenesis, stimulation of endothelial and vascular smooth
muscle cell proliferation, increasing endothelin production, up-regulation of tissue renin and
change in vascular tissue prostaglandins production. (16)
In the recent years authors have shown that activation of HIF and its downstream effectors
prior to renal injury protects the kidney from ischemia reperfusion injury. (17)This is called
ischemic preconditioning (IPC) and it is a therapeutic intervention which aims at protecting
against subsequent exposure to ischemic injury. This was first described by Murry et al. (18).
They showed that brief periods of coronary occlusion followed by a short period of
reperfusion reduced the infarct size caused by an ischemic insult in a canine model. After this
discovery it has been found to be a near-universal phenomenon in all organs. IPC is a
biphasic phenomenon. It has a short lasting phase of about 2 hours of protection within
minutes of the initial ischaemic insult and a delayed phase of protection which becomes
apparent around 24 hours later and lasts for about 3 days.(19) IPC is a potent renoprotective
strategy which has not yet been translated successfully into clinical practice, despite
impressive preclinical results. However, it remains interesting to induce genes that are
normally activated during the early phase of an ischemic insult. Small molecule inhibition of
the oxygen-sensing HIF-prolyl hydroxylases have been identified. This offers the possibility
to mimic the hypoxic response, by pharmacological stabilization of HIF in order to achieve
organ protection. Therefore, a promising therapeutic strategy for the prevention of organ
failure and organ injury is oxygen-independent activation of HIF.

12
Hypoxia inducible factor
Hypoxia inducible factor (HIF) is a DNA-binding transcription factor that associates with
specific nuclear cofactors under hypoxic conditions. HIF consists of two subunits subunit
HIFα and an abundantly expressed β-subunit, HIFβ. Both subunits are part of the basic Helix-
Loop-Helix PER-ARNT-SIM (bHLH-PAS) family of transcription factors. (20) HIFα is
constitutively transcribed and translated in cells, but under normoxia it has a short half-life of
less than 5 min. There are three HIFα isoforms; HIF1α, HIF2α and HIF3α, these isoforms
are all oxygen dependent.
The isoforms consist of an oxygen
dependent degradation domain (ODD)
and an N-terminal transactivation
domain (NAD), HIF1α and HIF2α also
have a C-terminal transactivation
domain (CAD). The N-terminal
transactivation domains are essential
for targeting gene specificity, CAD on
the other hand contributes to the
regulation of most HIF target genes.
(21)HIF1α and HIF2α have a similar
architecture and are regulated in the
same
manner, whereas HIF3α is less closely
related and its regulation is less well
understood. The HIF1α and HIF2α oxygen dependent degradation domains are located in
the central region of the molecule. HIF1α is the subject to a further control that involves a
nuclear localization, figure 4. HIF and its downstream effectors
which is mediated by the active exclusion of
HIF1α from the nucleus in the presence of oxygen. (22) To date, far more than 100 HIF target
genes have been identified, including the previous described EPO and HO-1, further
downstream effectors are VEGF and glucose transporters. These downstream effectors have
all the potential to protect the kidney under different conditions. (23) (See figure 4)

13
HIFα regulation by prolyl hydroxylation and factor inhibiting HIF
HIFα protein levels are regulated by several mechanisms. The most important one is the
degradation pathway. Under aerobic conditions, HIFα undergoes proteasomal degradation.
This happens via the ubiquitin-dependent pathway and involves hydroxylation of specific
proline residues within the ODD domain. This degradation is done by prolyl hydroxylases
(PHDs), non-heme, oxygen-, Fe(II)- and 2-oxoglutarate-dependent dioxygenases. The PHD
proteins act as oxygen sensors and so far three of them have been identified: PHD1, 2 and 3.
(24) Hydroxylated HIFα is bound by the von Hippel-Lindau protein (pVHL). This protein
targets HIFα for degradation by the 26S proteasome. Binding of pVHL to HIFα is prevented
when PHDs are inactive, this is during hypoxia or in case of lack of the cofactors Fe(II) or 2-
oxoglutarate. In this case HIFα is able to escape ubiquitination and proteasomal degradation
and is transported to the nucleus where it binds with HIF β. After recruitment of co-activators
they bind together to the hypoxia responsive element (HRE) at the target gene loci. (figure 5)
Figure 5. HIF1α regulation by prolyl hydroxylation

14
Rationale
Ischemia related injury plays a central role in both the pathophysiology of organ injury from
both brain dead and deceased circulatory death donors. Following brain dead dramatic
physiological disturbances occur, resulting in non-function of the central nervous system,
hemodynamic instability, systemic hormonal changes and diminished perfusion of peripheral
organs. This is worsened by exposure to periods of ischemia during the preservation period
followed by reperfusion in the recipient, this activates toxic reactive oxygen species
production and cell death via apoptosis. These changes may enhance the immunogenicity of
the grafts and increase the risk of acute graft rejection.
In the DCD setting the organs are subjected to a substantial period of oxygen deprivation and
ischemic injury, prior to the retrieval. Concluded from the data from the previous described in
the DCD chapter, is reasonable to suggest that in the controlled DCD group warm ischemia
plus profound cerebral injury could account for a different form of delayed graft function
than observed in uncontrolled DCD group that only have suffered warm ischemia. It would
be interesting to see whether and on what level the hypoxia inducible factor is up-regulated in
a rat model which mimics uncontrolled DCD.
After promising studies in the experimental brain dead setting of application of HO-1, one of
the most important downstream effectors of HIF, and HIF being negatively regulated by
PHD1,2 and 3, we reasoned that the induction of protective mechanisms before the initiation
of the acute injury associated with transplantation of BD and DCD kidneys might be an
effective way to improve early and possibly late graft function. Our hypothesis is that the
induction of genes that have become inducible by hypoxia could confer such protection. For
testing this hypothesis we first aimed to characterize the expression of HIF and its
downstream effector HO-1 following both brain death and also deceased after circulatory
death.
Main objectives
- To develop a gradual onset brain death model in rats.
- To determine the effect of 4 methods of renal ischemia on HIF1α and HO-1
expression. The models include an ischemia reperfusion model (IRI),donated after
circulatory death (DCD), donated after brain death (DBD) and NRK-49 cell culture.
- To aid in the development of a national transplant donor bio-bank.

15
Methods Animal justification
This animal research was carried out in accordance with the Home Office guidance on
welfare for animals and the experiments were approved by the animal committee.
We have employed the principles of reduction, replacement and refinement on the animal
study. The study has been reviewed by the Oxford veterinary services. Experiments were
performed in the Animal facility in the South Parks road, Oxford. The number of animals for
the experiments have been based on the following question: What are the effects of BD and
DCD on HIF and HIF downstream effector production in the donor kidney?
For the establishment of the BD model 30 animals are going to be used because the model
needs to be set up and optimized. 8 animals were used during my stay in Oxford for the pilot
experiments to begin to characterize the HIF pathway in 4 different types of kidney injury.
Two animals were used per group.
.
Animals
Adult male Fischer rats where used weighing 230- 350g were used. Animals were housed in
cages at 22˚C with a light-dark cycle of 12/12h and were allowed free access to food and
water. Acclimatization period was of at least 1 week before starting experiments.
Experimental group brain death model
To establish the brain death animal model one group were used.(n=8)
Experimental groups DBD, DCD, IRI and NRK-49 cells
To study the amount of HIF1α upregulation and its downstream effector HO-1 in different
models of kidney injury, 3 groups were used.(Figure 7) The samples from group 2 were
historical samples taken from previous BD experiments done in the University medical centre
Groningen. Rat kidney cells exposed to 24 hours of hypoxia were used as a positive control.
Group 1: deceased brain death group. (n=2)
Group 2: donor after circulatory death. (n=2)
Group 3: ischaemia reperfusion group. (n=2)
Group 4: rat kidney cells 24 hours hypoxia.
Fig. 7 Experimental protocol for the DBD, DCD and IRI experimental groups.

16
Brain death model
Experimental preparation
Protocols for the animal experiments were written and worksheets were designed for
recording the physiological parameters e.g. temperature, mean arterial pressure, heart rate and
oxygen saturations. (See supplementary protocols and worksheets.)
To set up the brain death model we had to monitor several parameters, and investigate which
set up and anesthesia settings were optimal. This is done by monitoring parameters including
peak pressures in the lungs, the tidal volume (the volume of gas inhaled and exhaled during
one respiratory cycle), the oxygen flow and % isoflurane administered. We calibrated the
optimal settings for rodents of different weights. The normal lung pressure of a male rat is
10-12 mmHg.
Anaesthesia and surgical preparation
Rats were anesthetised using 3-5% isoflurane with 100% oxygen (1L/minute) and during the
procedure maintained at 2% isoflurane on average. The rats were weighed and placed in
supine position. After shaving and cleaning the operation field with 70% alcohol, the
operation was commenced.
Brain death animal model
For the brain death rat experiments we used a gradual onset brain death model. This model
has been described previously (1) (figure 8) The aim was to simulate cerebral haemorrhage
by slowly increasing the intracranial pressure. Anaesthesia and preparation was performed
like described previously. Via a tracheostomy the animals were intubated and ventilated
throughout the experiment with a respiratory rate of 50-70 per minute. In this model we
mimicked intracranial hemorrhage, which finally led to brain death, with increasing a
subdurally placed no.4 Fogarty catheter (Edwards Lifesciences Co., Irvine, CA). The catheter
was slowly inflated (16 μl/min) with saline using a syringe pump. During the brain death
induction the percentage of isoflurane was reduced to 2 %. To monitor the blood pressure a
polyethylene catheter was inserted into a femoral artery. After brain death establishment the
anesthesia was turned off and the rats were ventilated with oxygen (1L/min), this was
continued during the whole experiment.
Fig. 8. Brain death model

17
Brain death procedure (DBD)
Using a microdrill a hole ( 2mm) was drilled through the skull frontolateral to the bregma.
Into the extradural space a balloon catheter was inserted. By gradually increasing the
intracranial pressure by inflating the balloon with 16μL saline per min, induction of brain
death was started. Stopping Inflation of the balloon and withdrawal of anaesthesia was done
after a hypotensive period followed by a short peak and a subsequent drop in blood pressure
and after this returning of the blood pressure to its basal level again. Until the end of the
experiment the balloon was kept inflated. Confirmation of brain death was done 30 -40 min
after the onset of brain death, by the absence of corneal reflexes and a positive apnoea test.
Donor management
A mean arterial pressure (MAP) above 80 mmHg was considered to be normotensive during
brain death. Blood pressure was recorded by the Labview program nerve Chart version 1.02
on a device provided by the Evans group. When blood pressure decreased below 80 mmHg,
venous return was increased by repositioning the rodent. If this failed to increase the MAP
colloid infusion were administered in boluses of 0.2 ml (Voluven). By using a heating pad
and a rewarming lamp we attempted to maintain the body temperature at 37°C.
Collecting tissues
A midline laparotomy was performed 10 minutes before the end of the experiment. Urine was
collected just before the organs were flushed with 40 mL ice-cold saline via aortic infusion.
The kidneys were collected after complete blood flush out. The tissue samples were fixated in
4% paraformaldehyde and frozen on dry ice (+/- ethanol).
DBD samples, DCD model, IRI model and NRK-49 cells
Deceased brain death samples (DBD)
Historical kidney and liver samples were taken from previous BD experiments done in the
University of Groningen. There the BD procedure described by Kolkert et al. was performed.
(1)
Donation after circulatory death model (DCD)
Anaesthesia and preparation was performed in a similar manner to that described previously.
The left and right kidney were randomized by flipping a coin. The control kidney was taken
out at time point 0 by ligating the renal pedicle and dissection of the kidney. After tying the
kidney was retrieved, dissected longitudinally and frozen on dry ice (+/- ethanol) or fixated in
4% paraformaldehyde. The rat was kept anesthetized and the wound was closed.
Subsequently cervical dislocation was performed. Cessation of cardiac death was monitored
by palpation. After 30 minutes following cervical dislocation the abdomen was reopened and
the second kidney retrieved, dissected longitudinally and frozen on dry ice (+/- ethanol) or
fixated in 4% paraformaldehyde.
Ischaemia reperfusion model (IRI)
Anaesthesia and preparation was performed in a similar manner to that described previously.
The left and right kidney were randomized to IRI vs control and the renal artery and vein
were dissected. After dissecting a non crushing bulldog arterial clamp was applied for a
period of 45 min. 10 U / ml heparin was administered subcutaneously 10min prior to
removal of the clamp. After 45 min the abdomen was reopened again and the clamp removed.
Bupivicaine was administered as pain relief (10 mg/ml) and the wound closed. The rat was
recovered using 100% O2 and placed in a heating room for slowly waking up. After 4 hours

18
the experiment was terminated. Anestheasia was induced with 3-5% isoflurane and the
abdomen opened again. The aorta was cannulated and flushed with normal saline. Both
kidneys were retrieved, dissected longitudinally into halfs and frozen on dry ice (+/- ethanol)
or fixated in 4% paraformaldehyde. Following the first anesthesia period the animals woke
up and were alert after approximately 30 min, while being kept in a heating room.
Preparation of slices
Laboratory experiments were performed in the welcome Wellcome Trust Centre for Human
Genetics.
Precision-cut kidney slices of 50 mg were prepared on dry ice. For the homogenisation 1 ml
of urea /SDS lysis buffer was used for 100 mg of tissue. The ultraturrax T25 homogenizer
was used and samples were homogenized for one minute. After homogenizing the tissue
homogenate were kept on urea SDS buffer for proper lysising and vortexed every minute.
The cells were centrifuged twice at 13k rpm for 10 minutes for separation with the
supernatant. The supernatant was piptetted into fresh Eppindorfs. After a last spin and an
additional equal volume of lysis buffer the samples were stored in a -20°C freezer.
Cell culture
As a positive control, NRK-49 cells were exposed to 1% of hypoxia for 24 hours in a hypoxia
station.
Initial culture
For the initial culture NRK-49 cells were the cells of interest. The cells were defrosted by
placing them in a 37 °C bath until defrosted. The cells were prepared in the culture hood. 5
mL of DMEM culture medium, 5 mL of a 5% glutamine dilution, 5 mL of a 5% Pen and
Strep dilution and 50 mL of a 10% Bovine serum dilution was added. The supernatant was
removed and the pellet was resuspend in 18 mL of culture medium. After assessment of cell
roundness, fullness and quality the box with cells was placed into the incubator at 37 °C with
5% CO2.
Splitting cells
Cells were daily checked under the microscope. Confluency of the cells was an indicator that
splitting needed to take place. The medium was removed from the cells and disposed.
Phosphate buffered saline (PBS) was added to the non cell side of the flask to wash the cells
and the PBS was removed after washing. Then 1 mL of trypsin is added to make the cells
detach from the bottom of the plate. Trypsin works best in a warm surrounding, so the plate is
incubated for 1-2 minutes in 37 °C. The cells are tapped to free them up and examined under
the microscope to see whether they move or not. In order to stop the reaction and resuspend
the cells 10 mL of medium is added. To make up the right dilution (1:10) 9 mL of the
medium was discard. To make up to the volume started with 17 mL of medium was added.
Seeding cells
For splitting the cells into the right concentration that will be the most useful in the
experiment the cells were seeded. The confluences needed were 60% and 50% to allow them
to be approximately 80% confluent after the 24 hypoxia period.
The cells were washed in the culture bottle. First the DMEM medium was removed and the
cells were washed using 10 mL phosphate buffered saline (PBS). After washing the PBS was
removed. 1 mL of trypsin was added and incubated for 1 minute at 37 °C with 5% CO2. The
cells were rescued by adding 14 mL of the DMEM medium. To make a confluence of 60%,
355 μL was added in 9 separate wells. The wells were further filled with DMEM until a

19
volume of 1mL in each well. After seeding the cells were stored in the incubator at 37 °C
with 5% CO2.
Exposing to hypoxia
To expose the cells to hypoxia, the wells with a 50% and 60% confluence were placed in the
hypoxia chamber for 24 hours. The oxygen tension maintained was 1%.
Harvesting cells
To harvest the cells, the control cells (normoxia cells) were harvested first. 8 μL of 4x SDS
sample buffer (2.0 mL 1M tris-HCl pH6.8, 0.8 g SDS, 4.0 mL 100% glycerol, 0.4 mL 14.7 M
β-mercaptoeethanol, 1.0 mL 0.5 M EDTA, 8mg bromophenol Blue) was added to prelabeled
eppindorfs. The DMEM medium was removed from the wells containing the samples. 34 μL
USDS lysis medium was added to the wells. The samples were aspirated and placed into the
prelabeled eppindorfs containing the 4x SDS sample buffer.The eppindorfs were centrifuged
for one minute and vortexed. The same steps were followed for the positive control hypoxia
cells. Once all samples were obtained the were heated at 95°C for 5 minutes and virgorously
vortexed for 15 sec per sample. After centrifugation the cells were checked for siffucient
liquification and stored at -20 °C.
Western blotting
We evaluated whether exposing cells to 24 hours of hypoxia could induce HIF1α response
using Western blotting. For this equal amounts of protein were loaded on to SDS/PAGE gels.
(stacking solution; Tris/HCL/SDS pH 8.8, 10% acrylamide, dH20, TEMED, 10%APS.
Resolving solution; Tris/HCL/SDS pH6.8, acrylamide, dH20, TEMED, 10%APS) The first
well was filled by a page marker and the gels were run at 180V and 60-80 mA for 1 hour.
The proteins were transferred on to nitrocellulose membranes by running at 100V for 1 hour
and incubated with Novus Biologicals NB100-479 antibody for 1 hour, an antibody used in
the laboratory which shows cross-reactivity with rat HIF1α. The blots were subsequently
incubated with a rabbit anti- IgG antibody a as an secondary antibody for 1 hour. Β-actin was
used as a loading control and was detected with conjugated antibody. Visualization was
peformed by incubation of the membranes with SSWD/SSWS solutions (super signal west
dura peroxide buffer/super signal west dura luminal/enhancer solution) and exposing films on
the membrane to infrared light. Detected signal was quantified an normalized. After a
positive result, confirmation of hypoxic cells, detected by the Novus biological antibody we
compared 3 different types of antibodies on the hypoxic and normoxic cells, to see which
antibody gives the strongest signal. After detection of the best antibody (Novus Biological
NB100-479 in a 1:1000 solution) we tested whether this antibody was cross-reactive with
our tissue samples.
Statistical analyses
Using the photography software program image J for scientific image processing the western
blots were converted raw data.
The statistical program Instat 3 was used to perform the statistical analyses.The distribution
of data was assessed with Q-Q plots and the Kolmogorov-Smirnov test for normality.
Normally distributed data are going to be expressed as mean ± standard error of the mean.
Non-normally distributed data are going to be tested with the Mann-Witney U test.

20
Results
Brain death experiments
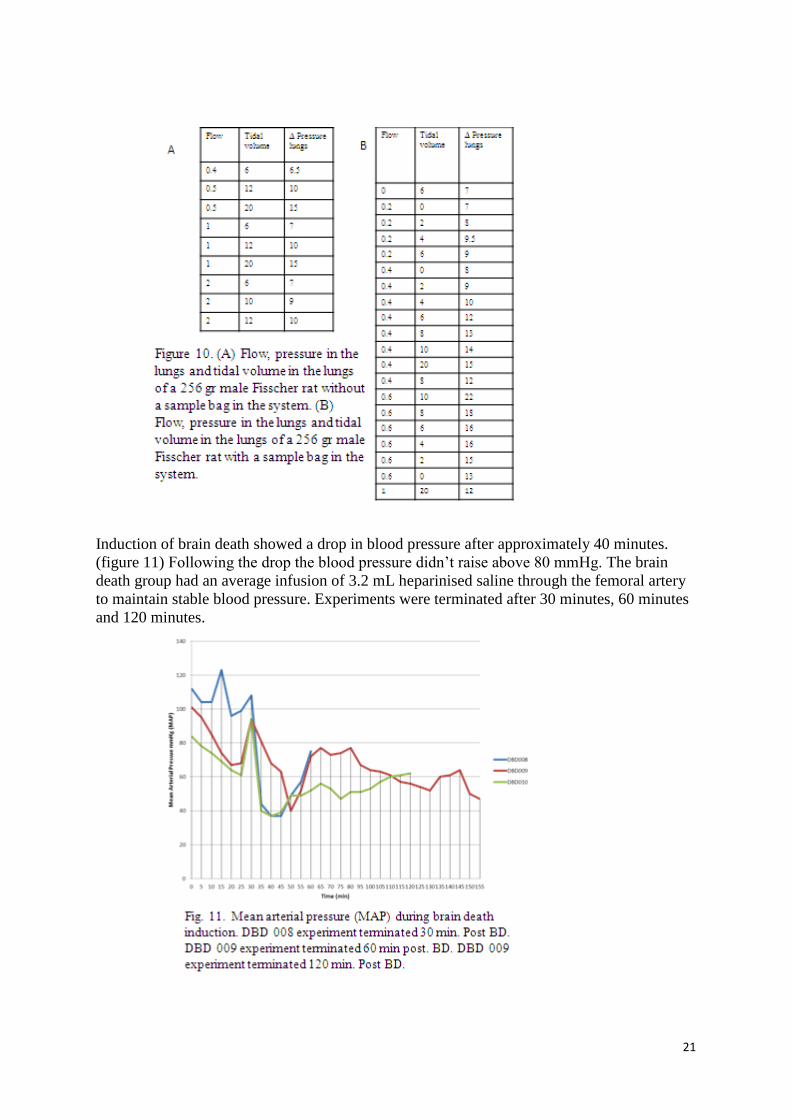
To optimize the ventilation settings and to set a start point for each following experiment, the
pressure in the lungs, tidal volume and flow was measured. (figure 9 and 10) The tidal
volume could be adjusted by turning a button on the ventilator. A sample bag, from what the
ventilator could sample oxygenated air mixed with isoflurane was used in the brain death
model from the University of Groningen. In our brain death model set up a comparison was
made whether optimal anaesthesia was with or without the sample bag. The normal pressure
in the lungs of a rat is 10 -12 mmHg, so without the sample bag, a tidal volume of 8 turns and
a flow of 0.4 L/min the optimal starting settings are achieved.
21
Induction of brain death showed a drop in blood pressure after approximately 40 minutes.
(figure 11) Following the drop the blood pressure didn’t raise above 80 mmHg. The brain
death group had an average infusion of 3.2 mL heparinised saline through the femoral artery
to maintain stable blood pressure. Experiments were terminated after 30 minutes, 60 minutes
and 120 minutes.

22
Deceased after circulatory death experiments
2 DCD experiments were performed. Cervical dislocation resulted in immediate cessation of
breathing and cessation of cardiac contractility at 5 min after cervical dislocation. After 30
minutes of warm ischemia the second kidney was retrieved.
Ischemia reperfusion injury experiments
The renal artery of the randomized kidney was clamped to induce ischemia, which was
verified by the change in renal color. During ischemia the abdomen was closed. Time
between start of the experiment and a clamp on the randomized kidney was the same in all
experiments and took 25 minutes. After 45 minutes of WI the abdomen was reopened, the
clamp was removed and reperfusion started. The pain relief bupivicaine was administered
before closure of the wound. After 4 hours of reperfusion the kidneys were retrieved.
Cell culture
The cells were harvested at a confluence of 50% and 60%. (figure 12)

23
Detection of HIF1α in the normoxia(Nx) and hypoxia(Hx) cells.
Western blotting was used to detect HIF1α in the grown cells with a confluence of 50% and
60%. (figure 13) The antibody Novus Biologicals NB100-479, known to be cross reactive
with kidney epithelial cells, was used to incubate and detect the proteins with. Β-actin was
used as a control. Figure 14 shows a significant upregulation of HIF1α in the hypoxic cells.
Figure 13. Detection of HIF-1α in normoxic (Nx) vs hypoxic (Hx, 1% O2 for 24 hours) NRK-49 cells using
anti-rat HIF1a antibody (Novus Biologicals NB100-479) comparing cells harvested from 50% and 60%
confluence at seeding. Β- actin as a control.
Figure 14. Detection of HIF 1 α expression in Nx and Hx cell culture. 24 hours of 1% hypoxia significantly
induced HIF 1 α in the hypoxic cells.
Antibody determination
To find the antibody which detects HIF1α the best in the samples of interest using western
blots, two different antibodies were tested one of them in a 1:1000 and a 1:3000 dilution,
using the Nx and Hx cells. The first one was anti-rat HIF1α Novus Biological (NB100-479),
raised in rabbit, in a 1:1000 dilution and in a 1:3000 dilution. The second one was anti-rat
HIF1α Transduction Laboratories (610959) raised in mouse, in a 1:1000 dilution. As shown
in figure 15, the anti-rat HIF1α Novus Biological (NB100-479), raised in rabbit, in a 1:1000
dilution is the best antibody to use.
Figure 15. Comparison of
antibodies detecting HIF1α using
normoxic (Nx) and hypoxic (Hx)
NRK-49 cells, B-actin as control.

24
Comparison of HIF1a expression in 4 models of kidney ischemic injury
After finding and a detection of the HIF1α by the antibody Novus Biological (NB100-479),
raised in rabbit, in a 1:1000 dilution, 4 models of kidney ischemic injury were compared. As
shown in fig. 16 and figure 19 HIF1α is more expressed after 45 min of WI in the IRI group
than in the IRI control group.
30 min post cervical dislocation there is less HIF1α expression than in the DCD control
group. Also, after 4 hours of brain death, the HIF1α expression is less HIF1α upregulation
than in the control group.
Figure 16. Comparison of HIF1α
expression in 4 models of kidney
ischemic injury: cell hypoxia (NRK-
49 cells 1%O2 for 24 hours), IRI
model (45 min WI 4 hours
reperfusion), DCD model (30min
WI following cervical dislocation),
DBD model (4 hours of BD). Β-
actin as a control.
25
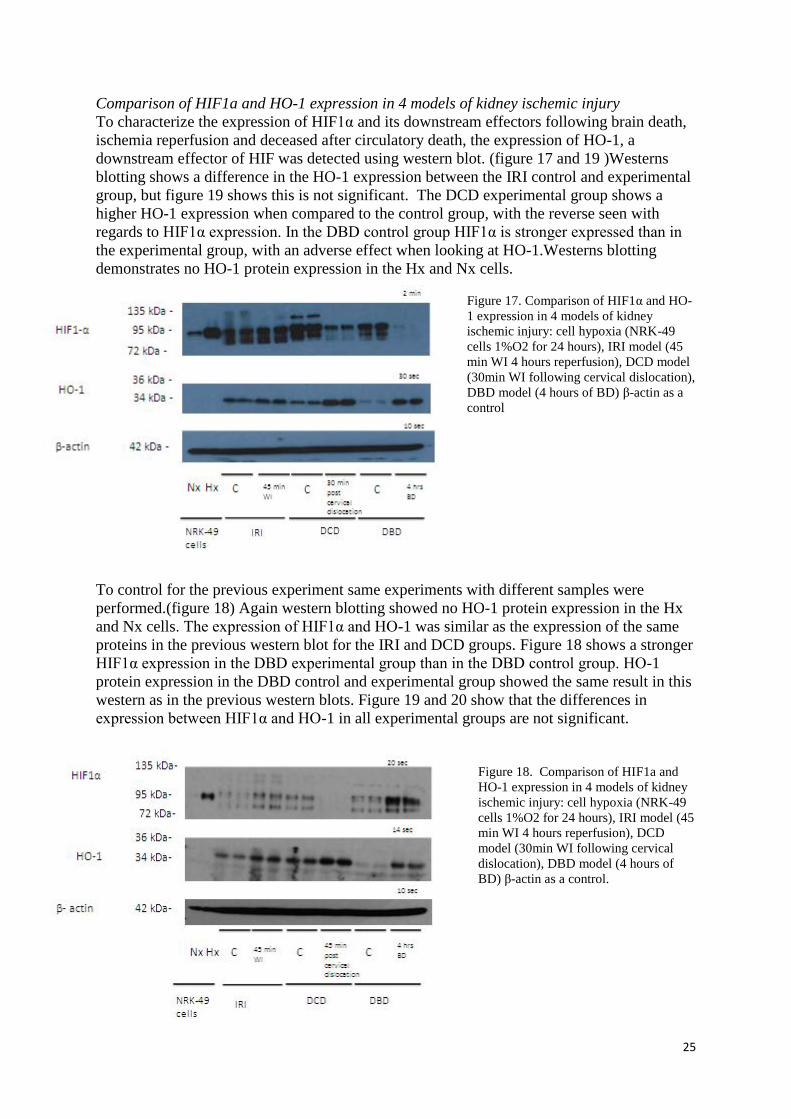
Comparison of HIF1a and HO-1 expression in 4 models of kidney ischemic injury
To characterize the expression of HIF1α and its downstream effectors following brain death,
ischemia reperfusion and deceased after circulatory death, the expression of HO-1, a
downstream effector of HIF was detected using western blot. (figure 17 and 19 )Westerns
blotting shows a difference in the HO-1 expression between the IRI control and experimental
group, but figure 19 shows this is not significant. The DCD experimental group shows a
higher HO-1 expression when compared to the control group, with the reverse seen with
regards to HIF1α expression. In the DBD control group HIF1α is stronger expressed than in
the experimental group, with an adverse effect when looking at HO-1.Westerns blotting
demonstrates no HO-1 protein expression in the Hx and Nx cells.
Figure 17. Comparison of HIF1α and HO-
1 expression in 4 models of kidney
ischemic injury: cell hypoxia (NRK-49
cells 1%O2 for 24 hours), IRI model (45
min WI 4 hours reperfusion), DCD model
(30min WI following cervical dislocation),
DBD model (4 hours of BD) β-actin as a
control
To control for the previous experiment same experiments with different samples were
performed.(figure 18) Again western blotting showed no HO-1 protein expression in the Hx
and Nx cells. The expression of HIF1α and HO-1 was similar as the expression of the same
proteins in the previous western blot for the IRI and DCD groups. Figure 18 shows a stronger
HIF1α expression in the DBD experimental group than in the DBD control group. HO-1
protein expression in the DBD control and experimental group showed the same result in this
western as in the previous western blots. Figure 19 and 20 show that the differences in
expression between HIF1α and HO-1 in all experimental groups are not significant.
Figure 18. Comparison of HIF1a and
HO-1 expression in 4 models of kidney
ischemic injury: cell hypoxia (NRK-49
cells 1%O2 for 24 hours), IRI model (45
min WI 4 hours reperfusion), DCD
model (30min WI following cervical
dislocation), DBD model (4 hours of
BD) β-actin as a control.

26
27
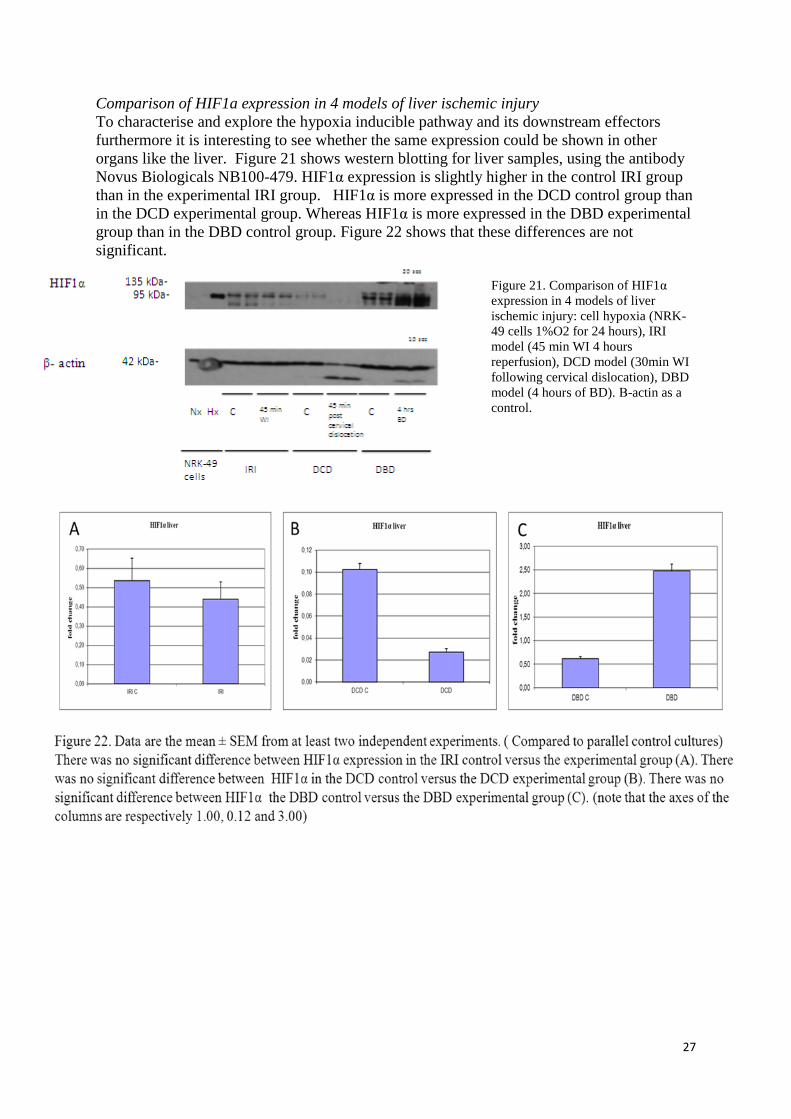
Comparison of HIF1a expression in 4 models of liver ischemic injury
To characterise and explore the hypoxia inducible pathway and its downstream effectors
furthermore it is interesting to see whether the same expression could be shown in other
organs like the liver. Figure 21 shows western blotting for liver samples, using the antibody
Novus Biologicals NB100-479. HIF1α expression is slightly higher in the control IRI group
than in the experimental IRI group. HIF1α is more expressed in the DCD control group than
in the DCD experimental group. Whereas HIF1α is more expressed in the DBD experimental
group than in the DBD control group. Figure 22 shows that these differences are not
significant.
Figure 21. Comparison of HIF1α
expression in 4 models of liver
ischemic injury: cell hypoxia (NRK-
49 cells 1%O2 for 24 hours), IRI
model (45 min WI 4 hours
reperfusion), DCD model (30min WI
following cervical dislocation), DBD
model (4 hours of BD). Β-actin as a
control.

28
Comparison of HIF1a expression in 2 models of liver ischemic injury
To explore the HIF1α pathway in the DBD group western blotting was performed on liver
samples and HIF1α and HO-1 expression was detected. Figure 23 shows a stronger
expression of HO-1 in the DBD experimental group than in the control group. The same
conclusion can be made regarding to HIF1α expression. Figure 24 shows a significant
difference in expression of HO-1 in de DBD experimental group versus the DBD control
group. Figure 24 shows no significant difference between the HIF1α expression in the DBD
control and DBD experimental group.
Figure 23. Comparison of HIF1α
and HO-1 expression in 2 models of
kidney ischemic injury: cell hypoxia
(NRK-49 cells 1 % O2 for 24 hours)
and the DBD model (4 hours of BD)
β-actin as a control.

29
Conclusion and Discussion
This study was designed to develop a brain death model which was the same as described
before(1), but then on a different location. This preliminary study aimed to characterise the
effect of brain death on HIF1α expression and HO-1 in comparison to other models of kidney
ischemic injury.
The importance of this study lies in the fact that the donor and pre-donation management and
state of the organs determines post-transplant function and graft survival.
Brain death model
A brain death animal model was established. Figure 11 shows that the results in this model
are extreme variable. There is a large variation in the parameters within the same group. This
variation could be due to technical problems and/or physiological problems. During the
performance of the experiments we faced several technical problems with the ventilator and
blood pressure measurements. On the other hand, the technical failures reflect an ‘unstable’
BD organ donor on the ITU and therefore are representative for the clinical setting.
Concluding from the fact that the lack of reflexes, a positive apnea test and the pressure
change pattern are similar to that of previous experiments,(1) it can be stated that the animals
were brain death after the brain death induction.
We observed after the initial decrease in blood pressure, a gradual increase in blood pressure
just before a peak was observed. This is due to the physiological response to increasing
intracranial pressure and is followed by a decrease in cerebral perfusion pressure. Normally
this event leads to ischemia in the brainstem, which results in peripheral vasoconstriction due
to excitation of different neurons and a subsequent increase in arterial pressure.(25) The
arterial pressure exceeds the intracranial pressure and blood flow to the brain stem is ensured.
But in our experiments the MAP of the peak wasn’t as high as described before by Kolkert et
al. (1)This effect could be because of hemodynamic effects due to brain death and this was
not corrected properly enough. Also, it could be due to reduced release in catecholamines and
pulmonary changes. In most studies using a sudden inflammation animal brain death model
(a model which simulates acute and significant cerebral trauma by increasing the intracranial
pressure within 30-60 sec.) a hypotensive period after the catecholamine-induced peak has
been described.(26) (27) Our results show a decrease in blood pressure after the peak
occurred, which is similar to the observations described in the sudden onset brain death
model. Kolkert et al. described the gradual onset brain death model and they showed a
plateau at levels of 100 mmHg after the peak in blood pressure. In our model optimization
and clarification needs to be performed to make sure we induce brain death on a gradual
onset instead of a sudden onset, which is what the blood pressure curve suggests.
In our brain dead model we didn't have a venous cannula. So blood pressure couldn’t be kept
stable by administration of HAES or noradrenalin, as described before. (1)A next step would
be to insert a venous cannula.
In our setting we used mechanical ventilation. In the past researchers looked at how to apply
mechanical ventilation and it has become evident that a number of ventilator strategies can
produce or worsen lung injury. The use of large tidal volumes (VT)(28), high inspiratory
flows,(29) high peak airway pressures, (30) and high respiratory rates (RR)(31)play a role in
the pathogenesis of ventilator induced injury. The epithelial and endothelial barrier is
damaged and compartments of alveolar cytokines can be lost into the vascular system during
acute lung injury. A process that can initiate a systemic response. (32). Tremblay et al. have
shown that ventilation with zero positive end-expiratory pressure (PEEP) and/or excessive

30
end-inspiratory lung volume increased the concentration of lung cytokines and ‘leak’ into the
systemic circulation. (33) During the optimization process we started doing the animal
experiments with a respiration frequency of the animal around 80 breaths per minute and with
a PEEP of zero. We corrected for this ventilation settings during the optimization of the brain
death model.
The occurrence of ischemia was one potential important primary trigger which was analyzed
in relation to the brain death process. Like described before, (34)periods in the slow-
induction model of BD and the IRI model are associated with a blood pressure drop followed
by a period of hypotension. The vasoconstriction and increase in peripheral vascular
resistance due to catecholamines release after the sharp increase in blood pressure can result
in a period of hypotension and a decrease in peripheral flow to the abdominal organs. This is
something which results in the occurrence of ischemic episodes and the occurrence of HIF1α
activation due to ischemia followed by hypotension instead of hypoxia.
Eckhardt. Et al. described an ischemia reperfusion model in which the control kidney was
removed before the ischemia/reperfusion of the contralateral kidney, to obtain information
about the efficacy of precondition.(19) in our experimental setting the control kidney of the
DCD is not the same as the control kidney of the IRI group. This is due to a different
procedure and therefore the 2 control groups are not comparable. The IRI control kidney was
removed after two periods of anaesthesia, opening the abdomen and placing a clamp on the
ischemia/reperfusion kidney. After this procedure western blotting showed a different
accumulation of HIF 1α in the DCD versus the IRI model.(figure 16, 17, 18, 21, 23)
Benedikt et al. have shown that anaesthetics induce early and late preconditioning in several
organs by activation of the transcription factor HIF 1 α and its downstream effectors. (35)
This might be an explanation of the difference between the HIF 1 α accumulation in our DCD
and IRI model.
Collection, preparation and analysing of slices
HIF1α and HIF2α have been shown to be differentially expressed in the kidney following
systemic hypoxia. HIF1 α is predominantly found in the tubular cells and in particular in the
medullary collecting ducts. HIF2 α is expressed in the glomerular cells, peritubular
endothelial cells and interstitial fibroblasts. (36) In our models we dissected the explanted
kidneys longitudinally into halfs and snap froze the pieces. 50 mg of these were prepared on
ice for analysis. Since we were first looking at HIF1 α we tried to cut the 50 mg pieces
longitudinally, so that we got the region were HIF1 α was expressed and we could detect it. It
was possible to control the way the tissues were collected and prepared in the experiments
performed by ourselves , however, the possibility we only received the cortex of the organs
taken from the BD experiments done in the Medical Centre in Groningen and therefore have
less tissue with a HIF1 α response is not excluded.
Interestingly, figure 18 shows a stronger HIF1α expression in the DBD experimental group
than in the DBD control group. This is in contrast with the western blot shown in 17 and 16.
We can’t explain this observation other than only having prepared the cortex instead of the
medulla where HIF1α is strongly expressed. Wrong loading during western blotting is not a
possible reason, because HO-1 is in both figure 17and 18 stongly expressed in the DBD
control group. For further research it is essential to develop a strategy to prepare the slices so
that the localization of HIF1α is not missed. For example through homogenezing whole
kidneys.

31
Figures 16, 17 and 18 of the kidney samples show a low HIF1α protein expression in the
experimental DCD group. Testing for HO-1 in figure 17 and 18 shows the reverse effect.
This phenomenon could be explained because of the short half-life of HIF1 α (37) and the
longer half-life of HO-1. HIF1α could already be disappeared when left on the bench for 30
minutes in the DCD group, whereas HO-1 is still detectable. So we could ‘miss’ seeing
HIF1α and we need to look at other time points during the experiments.
Figure 21, the western blot of liver samples, shows a low HO-1 protein expression in the
DCD experimental group when compared to the control group. As seen in the β-actin control
this effect could be due to technical reasons and another western blot needs to be performed
before making any conclusions.
Another explanation of the high expression of HO-1 where there is low expression of HIF1α
could be the fact that HO-1 is a heat shock protein, because of its responsiveness to thermal
stress.(38) Thermal stress is stress caused by temperature change. Because the thermometer
used during the animal experiments was not accurate there might be a possibility HO-1 is
highly expressed because of the temperature change instead of being a downstream effect of
HIF1α.
HO-1 could also be activated by a pathway independent from HIF1α. HO-1 is an enzyme that
has a transcription which is regulated by a large variety of stimuli. These include its substrate,
heme, signaling proteins nerve growth factor, TNFα, IL1β and interferon γ.(39) These are all
enzymes expressed as an adaptive response that increases cell resistance to oxidative injury.
However, HO-1 still maintains one of the major downstream effectors of HIF (40) so further
research needs to be performed to determine whether HO-1 is upregulated by HIF1α in these
models of kidney injury.
As shown in the results, in none of the groups the difference in HIF1α is significantly
different expressed. This is probably due to the size of the n and/or the constructed models.
More experiments with different samples need to be performed to reduce large variations in
the outcomes within the same groups and to make conclusions which could be significant.
The assumption that HIF activation protects tissues against hypoxic damage so far mainly has
been indirect, all on the basis of the characeristics of HIF and its downstream effectors. For
example it has been shown that EPO expression is predominantly HIF 2 α and not HIF 1 α
dependent. (41) So for the next step in our approach on the HIF pathway it is interesting to
clarify what happens with HIF downstream effectors like HO-1, EPO and VEGF.
Furthermore, a clarification of the prolyl hydroxylase 1,2 and 3 and of HIF 1 α and HIF2 α
expression in the different models would be of a great value. In addition a time course effect
of the HIF accumulation at 0.5, 1, 2, and 4 hours is interesting to see.

32
Recommendations for further research
After a complete clarification of the hypoxia inducible factor pathway it is interesting to
investigate whether the pharmacological induction of HIF and its protective downstream
effectors target ischaemia related injury in the brain death setting and deceased after
circulatory death setting. In the clinical context of organ donation, the opportunity to
‘pretreat’ organs is limited. In an IRI study performed by Bernhardt et al. animals were
treated with prolyl hydroxylase inhibitors prior to ischaemia.
In normoxia the prolyl hydroxylases are continuously inactivating HIFα. In this way they
prevent the dimerisation with HIFβ and consequently preventing the transcription of hypoxia
responsive genes. In addition to oxygen, prolyl hydroxylases also require Fe2+, 2-
oxoglutarate, and ascorbate. Knowing this, there is a possibility to pharmacologically activate
PHDs in the BD and DCD setting by for example the administration of DMOG
(dimethyloxalylglycine) a 2-oxoglutrate analogue, or Fe2+ and cobaltous ions (42).
Oxyglutarate analogues act as competitive inhibitors of PHDs and other HIF stabilizers. (17)
No data exists on whether treatment can be administered after ischaemic kidney injury. (43)
However, a rodent study found that administration of the prolyl hydroxylase inhibitor
dimenthyloxaylglycine (DMOG) 30-60 min following middle cerebral artery occlusion
reduced ischaemic injury (44) and in cardiac postconditioning HIF plays a critical role (45).
So after completing to assess the effect BD and DCD have on HIF and HIF target genes in
the kidney, investigating whether DMOG can activate HIF following deceased after
circulatory death and brain death is of great value. Activating HIF following an ischaemic
period by DMOG administration might reduce the pro inflammatory state of the donor and
subsequent effects of IRI, this improves the outcome of kidney transplantation.
Figure 16, 17, 18 and 21 showed HIF1α expression in an model which mimics uncontrolled
DCD. After further exploitation and confirmation of the HIF1α expression in an uncontrolled
DCD model it will be interesting to see whether and on what level the hypoxia inducible
factor is up-regulated in a rat model which mimics controlled DCD. At a later state it would
be interesting to investigate whether it is possible to use the hypoxia inducible factor to
precondition DCD donors.
Translational strategy; relevancy for the human setting
Quality in Organ donation
In order to attempt the issue that fewer optimal organ donors will be available, because of
reasons like changes in neurosurgical practice as well as improvements in the
management of cardiovascular disease and a fall in road accident deaths, the
transplantation community has been turning to organs preciously considered
unsuitable for donation. These are organs from older donors following brain
death, the extended criteria donors (ECD) and donors following circulatory
arrest (DCD). The abnormal physiological state in these donors results in
significant organ damage even prior to procurement. Investigating these injury
mechanisms in the donor, by understanding the pathways of injury and
applying therapeutics, we think that organ injury can be prevented and short and long-term
organ survival as well as the function of the graft can be improved. We propose that it helps
transplant teams in deciding the suitability of specific organs for individual patients if we are
able to identify markers in the donor to predict the short and long-term outcome of
transplantation. This would also help to improve or alter the post-operative management in
the specific recipient.

33
Success in transplantation starts in the organ donor. There is a number of points at which
there is a possibility to intervene to prevent organ damage, from the moment of the initial life
threatening event on. (See figure) These potential points include interventions during the
ICU, management of the brain dead and dying patient, strategies to prevent ischemia
reperfusion injury and optimizing retrieval and organ preservation techniques.
The key aims of the QUality in Organ Donation (QUOD) project are:
To increase the number and quality of organs procured from DBD and DCD donors for
transplantation by optimizing donor management and resuscitating and preserving marginal
organs.
- To make previously unusable organs transplantable and increase the ‘donor pool’.
- To identify pathways of injury and apply targeted interventions to repair donor organ
injury.
- To translate validated experimental methods and technologies into clinical use and
best practice protocols.
- To identify bio-markers and functional parameters that predict outcome following
transplantation.
- To streamline collaboration and dissemination between scientific and clinical experts
in academic institutions across the United Kingdom.
The team which worked on this project consisted of Prof RJ Ploeg, who was the co-
ordinating principle investigator, Zeeshan Akhtar and Maria Kaisar, the research students,
Luke Marsden and Christian Brailsford the expert advisors, Sandrine Rendel, the national
operational coordinator and myself, also a research student with the capacity to aid in
establishing the national donor bio-bank because of my previous experience as President of
the Prometheus kidney team in the University medical Centre in Groningen. Together we are
the QUOD National management team and we propose to create a national Consortium on
donor organ quality. We want to bring together all the participants in the National Organ

34
Retrieval System (NORS), these include clinical and scientific experts from the ICUs and
transplant centres in the United Kingdom. The centres to start the project include Cambridge,
Edingburgh, Newcastle-upon-Tyne, Kings College from London and Oxford. The idea is to
collect demographic and clinical information from organ donors following informed consent.
Also, blood, urine and tissue biopsies will be collected at certain timepoints during
procurement. Tissue biopsies include biopsies from the left and right kidney, from the liver,
the ureter and the spleen. In addition, information about ex-vivo preservation techniques and
procurement related injury will be recorded. We will use the samples to analyse pathways
involved in organ injury and repair and to identify novel bio-markers. This will be correlated
with the organ outcomes in the recipient. The samples are going to be stored in a central
biobank and this biobank will be accessible to researchers.
The outcome measures are;
Assessment of donor organ injury using clinical (donor profile and organ function) and
biological parameters.
Clinical functional assessment in the recipient (primary non-function, delayed graft function,
immediate function) and expression of relevant biological parameters for injury and/or repair.
Identification of donor risk factors
Evaluation of specific post-post transplant related complications related to donor organ
quality.
My role in the QUOD national management team was to aid in the establishment of this
organ donor database and bio-bank using my expertise, gained as being part and president of
the Prometheus Kidney team in the University medical centre in Groningen. I used this
expertise to establish, implement, check and control laboratory and procedure protocols. I
wrote protocols for the specialized nurses in organ donation (SNODs) which we provided to
NHSBT. To check protocols before implementation into the QUOD standards I went with the
retrieval surgeons on organ retrieval and followed and revised the protocols. Furthermore I
wrote the worksheets on what the SNODs and perfusionists have to record the time, places,
facts and data of the collection of biological materials and clinical data.
I communicated between the members of the management team and encouraged
collaboration of the different participants of organ retrieval; clinical and scientific experts
from all over England and Scotland as well. I wrote patient leaflets and worked on ethics
documents which had to be submitted and approved before starting the collection of samples
and biopsies. All together this became the QUOD booklet to which participants of the QUOD
team are able to refer to and adjust documents when required.

35
Conflict of interest
The author declares that she has no conflict of interest.
Acknowledgements
First of all I want to thank prof. Rutger Ploeg. Thank you for giving me the opportunity to
come to Oxford and work with you and your team. Secondly Henri Leuvenink, thank you for
all your visits and the support in hard times. Zeeshan Akhtar, Zee, thank you for all the hours
we worked together and for becoming such a good friend. Petra Ottens, thank you for coming
over to Oxford and helping us with the brain death model. Furthermore I want to thank
Michiel Voskuil, Sandrine Rendel, Maria Kaisar and Beverly Burcham. It has been a
pleasure to work with you!

36
References
(1) Kolkert JL, t Hart NA, van Dijk A, Ottens PJ, Ploeg RJ, Leuvenink HG. The gradual
onset brain death model: a relevant model to study organ donation and its consequences on
the outcome after transplantation. Laboratory animals 2007 Jul;41(3):363-71.
(2) Cicora F, Stringa P, Guerrieri D, Roberti J, Ambrosi N, Toniolo F, et al. Amelioration of
renal damage by administration of anti-thymocyte globulin to potential donors in a brain
death rat model. Clinical and experimental immunology 2012 Sep;169(3):330-7.
(3) Eurotransplant. Eurostransplant international foundation annual report 2011.
(4) Bos EM, Leuvenink HG, van Goor H, Ploeg RJ. Kidney grafts from brain dead donors:
Inferior quality or opportunity for improvement? Kidney international 2007 Oct;72(7):797-
805.
(5) de Vries DK, Lindeman JH, Ringers J, Reinders ME, Rabelink TJ, Schaapherder AF.
Donor brain death predisposes human kidney grafts to a proinflammatory reaction after
transplantation. American journal of transplantation : official journal of the American Society
of Transplantation and the American Society of Transplant Surgeons 2011 May;11(5):1064-
70.
(6) Kusaka M, Pratschke J, Wilhelm MJ, Ziai F, Zandi-Nejad K, Mackenzie HS, et al.
Activation of inflammatory mediators in rat renal isografts by donor brain death.
Transplantation 2000 Feb 15;69(3):405-10.
(7) Sanchez-Fructuoso AI, Prats D, Marques M, Blanco J, Torrente J, Conesa J, et al. Does
donor brain death influence acute vascular rejection in the kidney transplant? Transplantation
2004 Jul 15;78(1):142-6.
(8) Ridley S, Bonner S, Bray K, Falvey S, Mackay J, Manara A, et al. UK guidance for non-
heart-beating donation. Br J Anaesth 2005 Nov;95(5):592-595.
(9) Mascia L, Mastromauro I, Viberti S, Vincenzi M, Zanello M. Management to optimize
organ procurement in brain dead donors. Minerva Anestesiol 2009 Mar;75(3):125-133.
(10) Morris P. Kidney Transplantation; principles and practice. 2001.
(11) Florack G, Sutherland DE, Ascherl R, Heil J, Erhardt W, Najarian JS. Definition of
normothermic ischemia limits for kidney and pancreas grafts. The Journal of surgical
research 1986 Jun;40(6):550-63.
(12) Belzer FO, Southard JH. Principles of solid-organ preservation by cold storage.
Transplantation 1988 Apr;45(4):673-6.
(13) Westendorp WH, Leuvenink HG, Ploeg RJ. Brain death induced renal injury. Current
opinion in organ transplantation 2011 Apr;16(2):151-6.

37
(14) Nijboer WN, Ottens PJ, van Dijk A, van Goor H, Ploeg RJ, Leuvenink HG. Donor
pretreatment with carbamylated erythropoietin in a brain death model reduces inflammation
more effectively than erythropoietin while preserving renal function. Critical care medicine
2010 Apr;38(4):1155-61.
(15) Jarmi T, Agarwal A. Heme oxygenase and renal disease. Curr Hypertens Rep 2009
Feb;11(1):56-62.
(16) Fisher JW. Erythropoietin: physiology and pharmacology update. Exp Biol Med
(Maywood) 2003 Jan;228(1):1-14.
(17) Bernhardt WM, Gottmann U, Doyon F, Buchholz B, Campean V, Schodel J, et al. Donor
treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in
an allogenic kidney transplant model. Proceedings of the National Academy of Sciences of
the United States of America 2009 Dec 15;106(50):21276-81.
(18) Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal
cell injury in ischemic myocardium. Circulation 1986 Nov;74(5):1124-36.
(19) Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, et al.
Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal
failure. Journal of the American Society of Nephrology : JASN 2006 Jul;17(7):1970-8.
(20) Greer S. The updated biology of hypoxia-inducible factor. The EMBO Journal
2012;Apr31(11):12-2448.
(21) Loboda A. HIF-1 and HIF-2 transcription factors-similar but not identical. molecules
and cells 2010;28(5):7-435.
(22) Schofield J. Oxygen sensing by HIF hydroxylases. Molecular cell Biology
2004;5(May5):11-343.
(23) Bernhardt WM, Warnecke C, Willam C, Tanaka T, Wiesener MS, Eckardt KU. Organ
protection by hypoxia and hypoxia-inducible factors. Methods in enzymology 2007;435:221-
45.
(24) Epstein A. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases
that regulate HIF by prolyl hydroxylation. Cell 2001;107:11-43.
(25) Grady PA, Blaumanis OR. Physiologic parameters of the Cushing reflex. Surg Neurol
1988 Jun;29(6):454-461.
(26) van der Hoeven JA, Ploeg RJ, Postema F, Molema I, de Vos P, Girbes AR, et al.
Induction of organ dysfunction and up-regulation of inflammatory markers in the liver and
kidneys of hypotensive brain dead rats: a model to study marginal organ donors.
Transplantation 1999 Dec 27;68(12):1884-1890.
(27) van Loon J, Shivalkar B, Plets C, Goffin J, Tjandra-Maga TB, Flameng W.
Catecholamine response to a gradual increase of intracranial pressure. J Neurosurg 1993
Nov;79(5):705-709.

38
(28) Parker JC, Hernandez LA, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit
Care Med 1993 Jan;21(1):131-143.
(29) Corbridge TC, Wood LD, Crawford GP, Chudoba MJ, Yanos J, Sznajder JI. Adverse
effects of large tidal volume and low PEEP in canine acid aspiration. Am Rev Respir Dis
1990 Aug;142(2):311-315.
(30) Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema.
Respective effects of high airway pressure, high tidal volume, and positive end-expiratory
pressure. Am Rev Respir Dis 1988 May;137(5):1159-1164.
(31) Peevy KJ, Hernandez LA, Moise AA, Parker JC. Barotrauma and microvascular injury
in lungs of nonadult rabbits: effect of ventilation pattern. Crit Care Med 1990 Jun;18(6):634-
637.
(32) Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic
cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care
Med 1999 Jul;160(1):109-116.
(33) Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies
increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest
1997 Mar 1;99(5):944-952.
(34) van der Hoeven JA, Ploeg RJ, Postema F, Molema I, de Vos P, Girbes AR, et al.
Induction of organ dysfunction and up-regulation of inflammatory markers in the liver and
kidneys of hypotensive brain dead rats: a model to study marginal organ donors.
Transplantation 1999 Dec 27;68(12):1884-1890.
(35) Hieber S, Huhn R, Hollmann MW, Weber NC, Preckel B. Hypoxia-inducible factor 1
and related gene products in anaesthetic-induced preconditioning. Eur J Anaesthesiol 2009
Mar;26(3):201-206.
(36) Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, et al.
Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat
kidneys. J Am Soc Nephrol 2002 Jul;13(7):1721-1732.
(37) Qutub AA, Popel AS. A computational model of intracellular oxygen sensing by
hypoxia-inducible factor HIF1 alpha. J Cell Sci 2006 Aug 15;119(Pt 16):3467-3480.
(38) Morse D, Choi AM. Heme oxygenase-1: from bench to bedside. Am J Respir Crit Care
Med 2005 Sep 15;172(6):660-670.
(39) Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, et al. Regulation of heme
oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2
transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem 2004
Mar 5;279(10):8919-8929.
(40) Westendorp W. Brain death induced renal injury. cur opin organ transplantation
2011;Apr;16(2):6-151.

39
(41) Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, et al.
Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha
(EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in
Hep3B and Kelly cells. FASEB J 2004 Sep;18(12):1462-1464.
(42) Hsieh MM, Linde NS, Wynter A, Metzger M, Wong C, Langsetmo I, et al. HIF prolyl
hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and
modest fetal hemoglobin expression in rhesus macaques. Blood 2007 Sep 15;110(6):2140-
2147.
(43) Bernhardt WM, Gottmann U, Doyon F, Buchholz B, Campean V, Schodel J, et al. Donor
treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in
an allogenic kidney transplant model. Proc Natl Acad Sci U S A 2009 Dec
15;106(50):21276-21281.
(44) Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of prolyl hydroxylases by
dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia
inducible factor-1alpha. Neurobiol Dis 2012 Feb;45(2):733-742.
(45) Zhao HX, Wang XL, Wang YH, Wu Y, Li XY, Lv XP, et al. Attenuation of myocardial
injury by postconditioning: role of hypoxia inducible factor-1alpha. Basic Res Cardiol 2010
Jan;105(1):109-118.

40
Supplementary protocols
Brain Death
1.0 Brain death induction
In most brain death models, an increase in intracranial pressure is generated by an
expanding intracranial balloon, finally leading to brain death. Our model simulates
cerebral hemorrhage by slowly increasing the intracranial pressure, resulting in less
hemodynamic instability and maintenance of normotension during brain death for several
hours.
2.0 Equipment
O2/isoflurane anesthesia
Compressed air
Ventilator: Harvard apparatus, model 683
Trachea canula/”tube”: shortened 16G i.v. cathether ( 2cm) (BD insyte)
Equipment for bloodpressure measurements (Meetcase, Organ Assist)
A. femoralis canula: PE canula 0.40x0,80 mm, 6 cm long with a blunted end 26G
needle.
V. femoralis canule: silicone canule (0.5x1.0 mm, 8 cm long)with a blunt ended 25G
Ligatures 6/0 (Braun: 11340119) and 3/0 (Braun: 01134043)
Suture 4/0 mersilene (R-683H, Ethicon)
Heparin 10 U / ml in saline. (Pharmacy, 5000 IU / ml)
Arterial embolectomy catheter 3F (UMCG, inkoop)
Shaver
10% HAES (37oC). (Pharmacy)
70% alcohol.
papaverine sulphate (50 mg / ml), lidocaine (10 mg / ml), nor-epinephrine (1 mg/ml)
(Pharmacy)
Syringe Pump (Terufusion Syringe Pump, model STC-521)
3.0 Procedure
1. Start surgery:
Anestethise the rat using O2/isoflurane 5%,
Anesthesia is maintained with O2/isoflurane 2%
Turn on the heating mat (stand 2)
Weigh the rat
Shaving: head, throat and one of the legs.
Place the rat in supine position
Clean the operation areas with 70% alcohol.
2. Femoral artery en vene canulation

41
Start with a medial incision in the leg of the rat.
The femoral artery and vein are directly visible under the skin.
Dissection of the femoral artery and vein.
Put two ligatures (6/0) around the femoral vein.
Ligate the distal ligature.
Cut a small hole in the vein and insert the vene canula into the vein.
Fixate the canula with the rostral ligature. Infuse some saline with heparin into the
femoral vein.
Place a drop of papaverine on the artery.
Put two ligatures (6/0) around the femoral artery
Ligate the distal ligature.
Bring a blunt curved forceps rostral under the artery to lift and close the artery.
Cut a hole in the artery and insert the canula.
Fixate the canula with the rostral ligature. Infuse some heparinised saline into the canula
Suture the wound (4/0).
3. Intubation of the rat
Prepare a medial incision through the skin of the throat.
Pull the salivary glands laterally from each other.
Watch out for the blood vessels at the rostral side of the salivary glands that may not be
damaged.
Separate the m. sternohyoideus (muscles)
Dissect the blood vessels from the trachea
Place two ligatures around the trachea (3/0 suture)
Just before intubation, the ventilator has to be switched on.
Slightly lift the trachea with the rostral trachea ligature, make a little incision between 2
cartilage rings below the thyroid glands and the tube can be inserted inside the trachea.
Don’t insert the tube more than 1 cm, because otherwise one of the two lungs will be
ventilated.
Connect the tube to the ventilator and fixate the tube with the caudal ligature.
Ventilator:
Breathing frequency: 50 breaths/min, and a peak of 13-15 (acceptable until 100
breaths/min)
Tidal volume: 3-4 ml/stroke
The EtCO2 will be measured.
If the EtCO2 > 22 mmHg increase the breathing frequency
If the EtCO2 < 20 mmHg decrease the breathing frequency
Suture the wound (4/0 suture) and fixate the tube.
4. Turn rat to prone position
Disconnect the blood pressure measurements.
Disconnect the ventilator and change the rat form supine position to prone position.
Connect the ventilator and the blood pressure measurements.
Infuse some heparinised saline through the a. femoralis to avoid blood clotting.
Place a temperature probe into the rectum of the rat. Control the temperature by turning
on the lamp.
Connect the pulse oximeter.

42
5. Craniotomie + insertion of the catheter
Prepare an incision in the skin above the skull.
Place some drops of lidocaïne / marcaïne on the skull. Wait for several minutes.
Drill a hole through the skull frontolateral to the bregma, using a microdrill. Don’t hit the
dura.
Drill also a line rostral from the hole to gently infuse the catheter.
Using a blunt forceps, prepare some extradural space between the dura and the skull.
Check whether the catheter is intact (with some fluid)
Fill the balloon catheter with 1 ml demi water without air bubbles in the syringe or catheter.
Place the catheter with syringe in an infusion pump and set the infusion rate to 16 ml/h
(equal to 0.5 ml fluid in the catheter during 30 minutes)
Gently insert the balloon catheter until the end of the balloon is just underneath the skull.
Wait till the blood pressure is about 80-100 mmHg
Start the brain death induction.
During balloon inflation, a hypotensive period occurred followed by a short peak and a
subsequent drop in blood pressure.
When the blood pressure returned to it’s basal level during an increasing peak, inflation
of the balloon is stopped and anesthesia is withdrawn.
The first 30 minutes of brain death the rat will be ventilated with 100% O2 (1000 ml/min).
Confirm the state of brain death 30 minutes after onset of brain death:
The absence of corneal reflexes
A positive apnoea test
Disconnect the ventilator for approximately 1 minute.
if the rat isn’t brain death, he will start breathing, otherwise the test will be
positive.
6. Donor managment:
After 30 minutes of brain death, the ventilator will be switched to O2 / air (1:1).
Blood pressure:
A mean arterial pressure (MAP) above 80 mmHg is considered to be normotensive.
o When MAP decreased below 80 mmHg, a drip with norepinephrine
(0.01 mg/ml) is connected to the femoral vene canula.
Start slowly with the infusion untill the MAP will be 100 mmHg. If the blood
pressure is stable at 100 mmHg, slowly decrease the infusion rate of the drip
to zero.
o With the use of a norepinephrine drip we can manage the MAP till 80-100
mmHg
Temperature:
o The temperature of the rat will be controlled with a lamp in combination with
a heating mat
Ventilation:
The EtCO2 will be maintained to 20-22 mmHg.
o If the EtCO2 > 22 mmHg increase the breathing frequency
o If the EtCO2 < 20 mmHg decrease the breathing frequency
7. 4 hrs of Brain Death

43
15 min before 4 hrs of BD, administration of Esmeron (0.6 mg/kg)
o 0,2 ml/100 gram i.a (333 ul stock + 9667 saline)
10 min before 4 hrs of BD, turn the rat in suprine position and open the abdomen
5 min before 4 hrs of BD, administration of heparin i.a
At 4 hrs of BD, collect blood, urine and organs.

44
DCD model
1.0 the model
the model is relevant to non-recovery of rodents
2.0 Equipment
O2/isoflurane anesthesia
Compressed air
Ligatures 6/0 (Braun: 11340119) and 3/0 (Braun: 01134043)
Suture 4/0 mersilene (R-683H, Ethicon)
Heparin 10 U / ml in saline. (Pharmacy, 5000 IU / ml)
Shaver
70% alcohol.
papaverine sulphate (50 mg / ml), lidocaine (10 mg / ml), nor-epinephrine (1
mg/ml) (Pharmacy)
Scale
3.0 Procedure
1. Start surgery
Anesthetise the rat using O2/isoflurane 5%,
Anesthesia is maintained with O2/isoflurane 2%
Turn on the heating mat (stand 2)
Weigh the rat
Place the rat in supine position
Shave the operation field
Clean the operation areas with 70% alcohol.
2. Retrieving first kidney
Midline abdominal incision
Randomize the kidneys
Tie the renal artery
Retrieve first kidney
Dissect longitudinally into halfs- ½ snap freeze, ½ in formalin
Close wound
3. Cervical dislocation
Keep the rat anesthetised
Dislocate the cervix of the rat, using a suffle
Check the heart rate and breathing
Wait for the determined time period
4. Retrieving second kidney
Open abdomen
Retrieve the second kidney
Retrieve other organs as controls
Dissect longitudinally into halfs- ½ snap freeze, ½ in formalin
Close wound

45
Renal ischemia reperfusion injury model
1.0 The model
This model is relevant to non-recovery of rodents.
2.0 Equipment
O2/isoflurane anesthesia
Compressed air
Equipment for blood-pressure measurements (Meetcase, Organ Assist)
A. femoralis canula: PE canula 0.40x0,80 mm, 6 cm long with a blunted end 26G
needle.
Ligatures 6/0 (Braun: 11340119) and 3/0 (Braun: 01134043)
Suture 4/0 mersilene (R-683H, Ethicon)
Heparin 10 U / ml in saline. (Pharmacy, 5000 IU / ml)
Shaver
70% alcohol.
papaverine sulphate (50 mg / ml), lidocaine (10 mg / ml), nor-epinephrine (1 mg/ml)
(Pharmacy) #
scale
3.0 Procedure
1. Start surgery:
Anesthetise the rat using O2/isoflurane 5%,
Anesthesia is maintained with O2/isoflurane 2%
Turn on the heating mat (stand 2)
Weigh the rat
Place the rat in supine position
Shaving: operation field
Clean the operation areas with 70% alcohol.
3. Clamping of the renal artery
Midline abdominal incision
Randomize the kidneys to IRI vs control
Dissect renal artery and vein and apply non crushing clamp for period e.g. 30-45min
Administer heparin through femoral arterial access (check if required)
Close wound
Reopen following ischemic period and remove the clamp
Administer pain relief
Close wound
4. Terminating experiment following ischemic period
Open abdomen
Cannulate aorta
Flush with normal saline
Explant kidneys
Dissect longitudinally into halfs- ½ snap freeze, ½ in formalin

46
DBD experiment
Date:
Ref e.g. DBD001
Type rat:
Male/female:
Weight:
Time start operation:
Time arterial cannulation:
Time intubation:
End of experiment:
Event time temp Heart rate %SpO2 MAP ETCO2
Brain death induction
Time:
5’
10’
15’
20’
25’
30’
45’
1hr
1hr 15’
1hr 30’
1hr 45’
2hrs
2hrs 15’
2hrs 30’
2hrs 45’
3 hrs
3hrs 15’
3hrs 30’
3hrs 45’
4hrs
Time perfusion of abdominal organs:
Volume of urine:
Volume of blood:
Number Eppindorfs kidney left:
Number Eppindorfs kidney right:
Number Eppindorfs pancreas:
Number Eppindorfs liver:

47
DCD experiment
Date:
Ref e.g. DCD001
Type rat:
Male/female:
Weight:
Time of anaesthesia:
Cervical dislocation
Time to cessation of cardiac contractility
Time start operation:
Administration of heparin:
Length of warm ischemia kidney left:
Length of warm ischemia kidney right:
Time perfusion of abdominal organs
End of experiment:
Time perfusion of abdominal organs:
Volume of urine
Volume of blood
Number Eppindorfs kidney left:
Number Eppindorfs kidney right:
Number Eppindorfs pancreas:
Number Eppindorfs liver:

48
Renal IRI experiment
Date:
Ref e.g. RIS001
Type rat:
Male/female:
Weight:
Time of anaesthesia:
Time start operation:
Time re-open the abdomen after 4 hours:
Clamp on left/ right :
Clamp off (reperfusion):
Time ischemic period:
Administration of heparin time & amount:
Time arterial cannulation & flush:
End of experiment:
Length of warm ischemia:
Event time temp Heart rate %SpO2 MAP ETCO2
Application of clamp
Time:
05’
10’
15’
20’
25’
30’
35’
40’
45’
1hr
1hr 15’
Time administration pain relief:
Abdomen closed:
Amount administration normal saline:
Event time temp Heart rate %SpO2 MAP ETCO2
Reperfusion:
15’
30’
Time animal in oxygen/ recovery cabin:
Time waking up:
Time normal behaviour:
Time perfusion of abdominal organs:
Volume of urine:
Volume of blood:
Number Eppindorfs kidney left:
Number Eppindorfs right:
Number Eppindorfs pancreas:
Number Eppindorfs liver:





























