Embed Size (px)
Citation preview

doi.org/10.26434/chemrxiv.12331025.v1
Prediction of Drug Metabolites Using Neural Machine TranslationEleni Litsa, Payel Das, Lydia Kavraki
Submitted date: 19/05/2020 • Posted date: 20/05/2020Licence: CC BY-NC-ND 4.0Citation information: Litsa, Eleni; Das, Payel; Kavraki, Lydia (2020): Prediction of Drug Metabolites UsingNeural Machine Translation. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.12331025.v1
We present an end-to-end learning-based method for predicting possible human metabolites of smallmolecules including drugs. The metabolite prediction task is approached as a sequence translation problemwith chemical compounds represented using the SMILES notation. We perform transfer leaning on aSeq2Seq Transformer model originally trained on chemical reaction data to predict the outcome of humanmetabolic reactions. We further build an ensemble model to account for multiple and diverse metabolites.Extensive evaluation reveals that the proposed method generalizes well to different enzyme families, as it cancorrectly predict metabolites for phase I and phase II drug metabolism reactions as well as for other enzymes.
File list (1)
download fileview on ChemRxivMetabolitesPrediction.pdf (823.85 KiB)

PREDICTION OF DRUG METABOLITES USING NEURALMACHINE TRANSLATION
A PREPRINT
Eleni E. Litsa1, Payel Das2,3 ∗, and Lydia E. Kavraki1†
1Department of Computer Science, Rice University, Houston, TX2IBM Research AI, IBM Thomas J. Watson Research Center, Yorktown Heights, NY
3Applied Physics and Applied Mathematics, Columbia University, New York, NY
ABSTRACT
Metabolic processes in the human body can alter the structure of a drug affecting its efficacy andsafety. As a result, the investigation of the metabolic fate of a candidate drug is an essential part ofdrug design studies. Computational approaches have been developed for the prediction of possibledrug metabolites in an effort to assist the traditional and resource-demanding experimental route.Current methodologies are based upon expert-derived metabolic transformation rules, and thereforelack scalability and generalization. We present a rule-free, end-to-end learning-based method forpredicting possible human metabolites of small molecules including drugs. The metabolite predictiontask is approached as a sequence translation problem with chemical compounds represented using theSMILES notation. We perform transfer leaning on a Seq2Seq Transformer model originally trainedon chemical reaction data to predict the outcome of human metabolic reactions. We further build anensemble model to account for multiple and diverse metabolites. Extensive evaluation reveals that theproposed method generalizes well to different enzyme families, as it can correctly predict metabolitesfor phase I and phase II drug metabolism reactions as well as for other enzymes. For CYP450,the major enzyme family in drug metabolism, our method is able to retrieve a similar number ofmetabolites, when compared to existing tools specifically developed for this enzyme family. Ourresults indicate that an end-to-end learning based approach for metabolite prediction can replace theneed for expert-derived transformation rules, while improving model generalization and scalability.
Keywords Drug Metabolism · Metabolite Prediction · Neural Machine Translation · Transformer
1 Introduction
Metabolic processes involve chemical reactions that are mediated by enzymes and take place to sustain life, either byproviding energy and building blocks to the cells or by eliminating potentially harmful compounds. Certain enzymefamilies are responsible for the elimination of xenobiotics, that is compounds that do not naturally occur in the humanbody, such as, drugs, pesticides and pollutants. Metabolism of xenobiotics commonly takes place in the liver in twophases [1]. Phase I includes oxidation reactions, mediated mainly by the cytochrome P450 (CYP450) enzyme family,which prepare the molecule to undergo a conjugation reaction in phase II. The conjugation reactions, which are mediatedby transferases, serve two purposes: first they deactivate possibly toxic compounds and second, they increase theirpolarity to aid excretion from the body.
Although these processes constitute protection mechanisms for the elimination of xenobiotics from the body, in thecase of drugs they can lead to reduced efficacy and raise safety issues. Phase I reactions, and less commonly phase II,can lead to the formation of toxic metabolites posing threats for liver toxicity. Indeed, a number of drugs have beenwithdrawn from the market due to hepatotoxicity with the leading cause being the formation of active metabolites [2].In addition, metabolism can affect the bioavailability of the drug and can be the cause of drug-drug interactions. Asa result, drug metabolism studies constitute an essential part of drug development. They can provide insights on the∗[email protected]†[email protected]

suitability of a compound as a drug or indicate possible chemical modifications that will improve the metabolic profileof a lead compound. Traditionally drug metabolism is studied experimentally using analytical techniques, such as massspectrometry, through a resource-demanding and time-consuming process.
Multiple efforts have been made for developing computational tools for drug metabolism prediction [3, 4] to assist theresource-demanding experimental evaluation and also facilitate the incorporation of metabolic studies at the early stagesof drug development [5]. Most of the existing tools are specifically designed for predicting metabolism through CYP450enzymes that are responsible for metabolising about 70-80% of existing drugs. Methods that have gained popularity,both from a computational and a practical standpoint, are the ones that aim at identifying the atoms within the moleculeinvolved in the metabolic transformation, called sites of metabolism [3, 4]. In practice, if the sites of metabolism areknown, the structure of a lead compound can be modified in order to manipulate its metabolism. However, the sites ofmetabolism per se do not give insights on the structure of metabolites that may cause toxicity or other complications.
The metabolite prediction problem has been studied to a much less extent and the existing tools have not been widelyadopted. Current approaches are rule-based, as they utilise expert-derived transformation rules for generating possiblemetabolites. The problems with the current rule-based approach are multiple: First, the derivation of the transformationrules in most cases requires expert knowledge which limits scalability. As a result, those methods cover for only certainenzyme families, with a focus on the CYP450 family, and therefore fail to provide a comprehensive view of drugmetabolism. Another problem is that the application of the transformation rules can lead to a very large number ofpossible metabolites, resulting in a low precision performance, which averts pharmacologists from adopting such tools[6]. There have been some noteworthy efforts for addressing these problems, which mainly attempt to reduce the numberof false positives. Some approaches apply statistical analysis or heuristics to rank the generated metabolites [7, 8].Others apply machine learning techniques in order, either to identify the sites of metabolism prior to the applicationof rules [8], or to predict substrate specificity excluding unlikely reaction types [9]. There have been also efforts forobtaining greater coverage of the metabolite space by developing multiple models, each one intended for a differentenzyme family [9]. An additional problem inherent to the rule-based methods is that they fail to generalize for a varietyof substrates, as a rule is applied only when there is an exact match between the substrate and the rule pattern.
The metabolite prediction problem relates closely to that of reaction outcome prediction, which has attracted greatinterest and has seen significant advancements the last few years. Similar to the metabolite prediction, most approaches,and especially the early ones, are rule-based [10, 11, 12]. The adoption of deep learning methodologies though,along with the availability of massive datasets of chemical reactions, such as the Lowe’s dataset [13], have led tosignificant improvements in terms of accuracy [14]. In an effort to deal with the lack of generalization capabilitiesof rule-based methods, the application of end-to-end learning has also been explored aiming at using neural networkbased architectures for directly converting the reactant molecules into the product molecules bypassing the need ofexplicitly encoding transformations rules. More specifically, the reaction prediction problem has been formulated as asequence translation problem where the reactants are translated into the products, relying on a sequence representationof molecules, similar to natural language translation [15]. One of the first approaches was developed upon a sequencetranslation model which relies on recurrent neural networks for capturing dependencies within the sequence [16]. Amore recent model, called Molecular Transformer [17], further improved upon its predecessor by adopting a newerarchitecture for neural machine translation, called Transformer [18], which relies solely on attention layers for capturing
(a) Chemical reaction(b) Drug Metabolism
Figure 1: Drug metabolites prediction as opposed to reaction outcome prediction: In drug metabolism (i) multipleproducts are possible, and, (ii) transferred structures (highlighted in red) are not known in advance.
2

inter-dependencies in sequences. Very recently, the Molecular Transformer proved to be a good starting point forderiving a model that is specialised on predicting outcomes for a specific reaction class through transfer learning [19].
The lack of data seems to be the most important factor impeding the application of an end-to-end learning based methodfor the task of predicting drug metabolites. In addition to that, the metabolite prediction problem exhibits a number ofadditional intrinsic difficulties when compared to that of reaction outcome prediction, as it is illustrated in Figure 1: Amolecule may be metabolized in different ways through multiple enzymes and the various metabolites may be quitediverse in terms of structure. Oxidative enzymes for example, which include the CYP450 family, cause small localchanges. Transferases increase the size of the molecule attaching a new structure to it, while hydrolases, may breakdown the molecule into smaller structures. Therefore, in the context of the reaction prediction problem, the predictionof drug metabolites can be seen as predicting incomplete reactions in which multiple outcomes are possible.
Herein, we present Metabolite Translator: a rule-free, end-to-end learning-based method for predicting human metabo-lites of small molecules. We approach the metabolite prediction problem as a sequence translation problem based onthe SMILES representation of molecules. We construct a training dataset, relying on human metabolism data frompublicly available databases, that we are making available in order to encourage further development. Due to the limitedavailability of metabolic data, we apporach the problem of metabolite prediction by transfer learning from a MolecularTransformer [17], pre-trained on general chemical reactions, to a model that is specifically tuned on human metabolicreactions. We further build an ensemble model to account for multiple and diverse metabolites. We evaluate our methodspecifically on predicting metabolites for drugs and compare it against three existing drug metabolite prediction tools(SyGMa [7], Glory [8], BioTransformer [9]) on CYP450 metabolism.
2 Methodology
Our approach consists of pre-training a Transformer model on a set of chemical reactions and subsequently fine-tuningit on a dataset of human metabolic transformations as shown in Figure 2. The ensemble model is constructed bycombining the output of multiple fine-tuned models. In the following, we discuss the three main components of thiswork: i) the datasets, including the data used for pre-training, fine-tuning and testing the model, ii) the hyperparametersof the training process, including pre-training and fine-tuning, and, iii) the ensemble model.
Pre-training Fine-Tuning
ENSEMBLE
MetaboliteTranslator
TRAIN
ING
Input Molecule
Predicted Metabolites
PREDICTIO
N
MolecularTransformer
[17]
Transformer[18]
MetabolicTransformations
ChemicalReactions
[13]
model
data
MetaboliteTranslator 2
MetaboliteTranslator 3
MetaboliteTranslator 4
MetaboliteTranslator 5
MetaboliteTranslator 1
Figure 2: The Metabolite Translator is derived through fine-tuning the Molecular Transformer on metabolic reactions. Duringinference, the ensemble model outputs the metabolites predicted by 5 Metabolite Translator models.
3
2.1 Data
2.1.1 Pre-training Data
The dataset for pre-training the Transformer model [18] is a subset of the Lowe’s dataset of chemical reactions [13],consisting of about 900,000 single-product reactions. The input sequence to the translation model consists of thereactants and reagents separated with a special token while the output sequence is the product molecule. Molecules arerepresented using the canonical SMILES notation [20].
2.1.2 Fine-tuning Data
The dataset for fine-tuning the pre-trained model consists of pairs of parent molecules and human metabolites inSMILES notation. The metabolites are derived through one-step enzymatic reactions. The dataset contains metabolitesfor both, xenobiotics and endogenous compounds, in an effort to obtain a comprehensive human metabolism datasetthat is not restricted to a specific enzyme family. It should be noted that although the outcome of a metabolic reactiondepends on the metabolising enzyme, information on the enzyme and its action is not encoded in the dataset. Doing socould possibly limit scalability and generalization of the method similar to the rule-based approaches. On top of that,the metabolising enzyme is not known in advance when predicting metabolites for new molecules, and therefore thiscould further limit applicability.
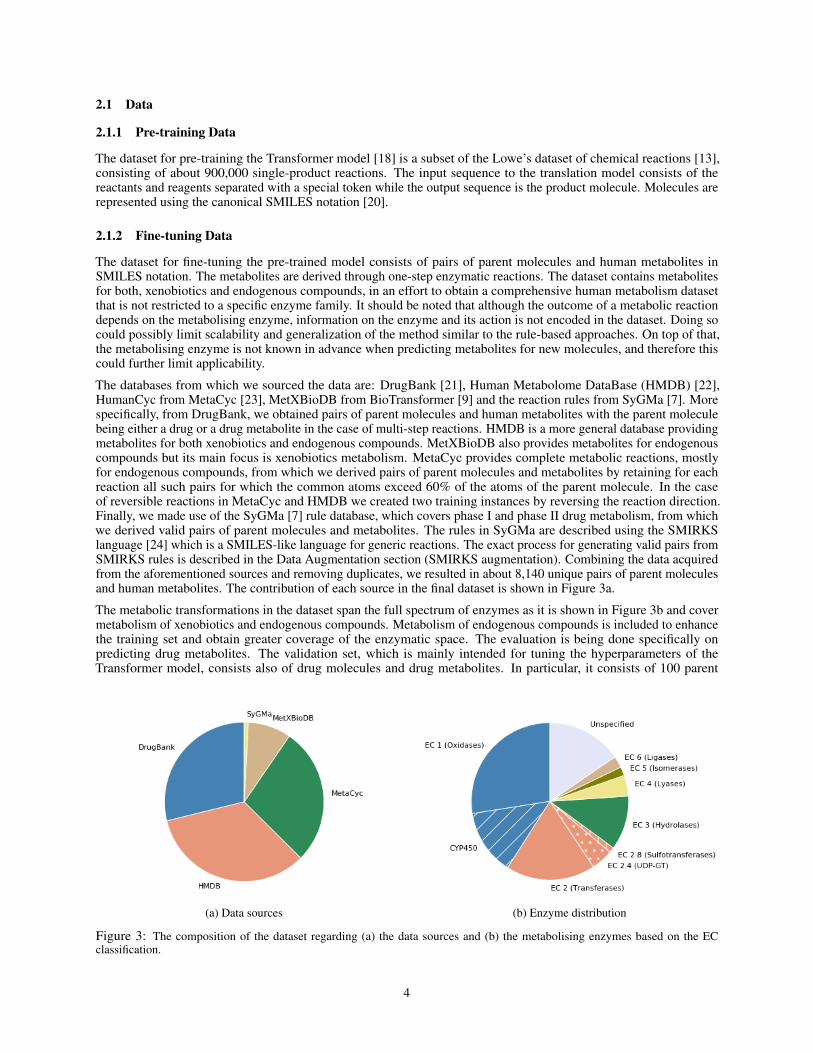
The databases from which we sourced the data are: DrugBank [21], Human Metabolome DataBase (HMDB) [22],HumanCyc from MetaCyc [23], MetXBioDB from BioTransformer [9] and the reaction rules from SyGMa [7]. Morespecifically, from DrugBank, we obtained pairs of parent molecules and human metabolites with the parent moleculebeing either a drug or a drug metabolite in the case of multi-step reactions. HMDB is a more general database providingmetabolites for both xenobiotics and endogenous compounds. MetXBioDB also provides metabolites for endogenouscompounds but its main focus is xenobiotics metabolism. MetaCyc provides complete metabolic reactions, mostlyfor endogenous compounds, from which we derived pairs of parent molecules and metabolites by retaining for eachreaction all such pairs for which the common atoms exceed 60% of the atoms of the parent molecule. In the caseof reversible reactions in MetaCyc and HMDB we created two training instances by reversing the reaction direction.Finally, we made use of the SyGMa [7] rule database, which covers phase I and phase II drug metabolism, from whichwe derived valid pairs of parent molecules and metabolites. The rules in SyGMa are described using the SMIRKSlanguage [24] which is a SMILES-like language for generic reactions. The exact process for generating valid pairs fromSMIRKS rules is described in the Data Augmentation section (SMIRKS augmentation). Combining the data acquiredfrom the aforementioned sources and removing duplicates, we resulted in about 8,140 unique pairs of parent moleculesand human metabolites. The contribution of each source in the final dataset is shown in Figure 3a.
The metabolic transformations in the dataset span the full spectrum of enzymes as it is shown in Figure 3b and covermetabolism of xenobiotics and endogenous compounds. Metabolism of endogenous compounds is included to enhancethe training set and obtain greater coverage of the enzymatic space. The evaluation is being done specifically onpredicting drug metabolites. The validation set, which is mainly intended for tuning the hyperparameters of theTransformer model, consists also of drug molecules and drug metabolites. In particular, it consists of 100 parent
(a) Data sources (b) Enzyme distribution
Figure 3: The composition of the dataset regarding (a) the data sources and (b) the metabolising enzymes based on the ECclassification.
4

molecules that we randomly sampled from the molecules derived from DrugBank with the constraint that it includesmolecules that are metabolized by other than CYP450 enzymes in addition to the dominant CYP450 cases. Finally, weshould note that since each molecule may yield multiple metabolites, the dataset includes cases that share the sameparent molecule but differ in the resulting metabolites. However, we ensure that instances that share the same parentmolecule are always in the same data partition (training, validation, test).
2.1.3 Test Set
The parent molecules in the test set are mainly drugs and, to a smaller extent, drug metabolites. We constructed thetest set as follows: We first sourced 29 drugs from a manually curated dataset that was recently made available by thedevelopers of the GLORY method for predicting CYP450 metabolites [8]. The dataset includes in total 81 metabolitesspecifically through CYP450 enzymes, sourced from the scientific literature. We expanded the GLORY test set with16 additional metabolites, through mainly other than CYP450 enzymes, that we sourced from DrugBank. We alsoadded 19 more drugs and 6 drug metabolites as parent molecules. The drug metabolites were introduced as parentmolecules first because we are testing the model for predicting one-step reactions, and second, because transferasereactions commonly occur on drug metabolites. The added drug molecules were manually chosen from DrugBank in aneffort to create a more diverse test set in terms of metabolizing enzymes. We paid attention on including drugs that aremetabolized by phase II enzymes, hydrolases, and also other enzymes less commonly involved in drug metabolism. Wealso included drugs that are metabolized by multiple different enzymes and therefore can result in structurally differentmetabolites since these are particularly challenging cases. The resulting test set consists of 54 molecules of which48 are drugs and 6 are drug metabolites. Within this set, 35 compounds are metabolized by CYP450 enzymes (E.C.1.14.14.- and 1.14.13.-), 11 compounds are metabolized by glucuronosyltransferases, also known as UDP-GT, (E.C.2.4.1.17), 3 are metabolized by sulfotransferases (E.C. 2.8.2.-), 1 molecule is metabolized by an acetyltransferase (E.C.2.3.1.5), 1 by an amine oxidase (E.C. 1.11.-), 4 molecules are metabolized by hydrolases (E.C. 3.-), and for 17 reactionsthe enzyme was not specified in DrugBank.
2.1.4 Data Augmentation
Data augmentation is a common practice for enhancing the training set, especially in cases with limited data, by creatingnew valid training instances from the existing ones. We used two techniques to augment the dataset of metabolicreactions: i) SMILES augmentation, and ii) SMIRKS augmentation, with the first one accounting for the biggestpart of the augmented data. SMILES augmentation refers to generating randomized SMILES representations fromthe canonical SMILES by randomizing the order of the atoms in the molecular graph. This augmentation techniquehas been found to be beneficial when training neural network-based architectures and generative models in particular[25, 26]. With SMIRKS augmentation we refer to generating multiple valid pairs of parent molecules and metabolitesthat satisfy a SMIRKS pattern [24]. The SMIRKS language is used for describing generic reactions in which thesubstrates and products may contain generic atoms. In order to generate pairs of parent molecules and metabolites, wesubstitute the generic atoms with one of the common atoms in organic chemistry (C, O, S, H, N) and subsequentlycheck the generated SMILES for validity. The SMIRKS augmentation was applied on the transformation rules fromSyGMa and the entries from MetaCyc that include generic atoms. The SMILES augmentation was applied on the entiredataset.
2.2 Training
2.2.1 Pre-Training on General Chemical Reactions
We pre-trained the Transformer model on single-product chemical reactions from the Lowe’s dataset [13] accordingto the specifications of the Molecular Transformer model [17]. We additionally experimented with reducing the sizeof the Transformer model and also augmenting the dataset using SMILES augmentation. The final selection of theseparameters was based on the validation accuracy of the fine-tuned model.
2.2.2 Fine-Tuning on Human Metabolic Reactions
The Metabolite Translator model is obtained by fine-tuning the pre-trained Transformer model on the set of metabolicreactions. The training specifications for fine-tuning the model were chosen based on the accuracy on the validationset. We experimented with various parameters including the data augmentation method, the learning rate schedule andstrength and warmup steps, and the batch size. Subsequently, we created an ensemble model, combining models trainedusing different hyperparameters and data-augmentation, in order to increase the diversity of the output as we discussnext.
5

2.3 Ensembling
An important challenge introduced when moving from reaction outcome prediction to metabolite prediction is thepossibly multiple and diverse metabolites that can be formed through different enzymes. To account for such diverseoutcomes, on top of the standard beam search algorithm for generating multiple sequences, we are constructing anensemble Metabolite Translator model.
The beam search algorithm, instead of expanding the generated sequence with a greedy approach choosing the mostlikely character, explores all possible characters and keeps the k most likely sequences. We tried beam sizes of 5, 10, 15and 20. With a smaller beam size we expect to get the most likely metabolites while increasing the beam size we canobtain larger coverage of the metabolite space.
The ensemble model is created by combining the outputs of multiple Metabolite Translator models trained underdifferent specifications. More specifically, we fine-tuned multiple models varying the model hyperparameters and thedata augmentation process, we selected the ones that obtained higher accuracy on the validation set and subsequentlyselected a small number of models to form an ensemble model. The selection process of the individual models wasbased on finding a trade-off between maximizing the number of correctly identified metabolites and keeping the outputsize low. The individual models are combined in a bagging fashion where the final prediction is the union of the setsof predicted metabolites from each individual model. The final ensemble model consists of 5 Metabolite Translatormodels.
2.4 Post-processing
The model output is being filtered in order to discard invalid SMILES and unlikely metabolites. The second caseincludes: i) metabolites that have gained atoms whose species are not among the parent molecule atoms or amongthe common atoms in organic molecules, ii) metabolites whose size, in terms of atoms, significantly deviates fromthe parent molecule size, and iii) metabolites which exhibit low fingerprint similarity when compared to to the parentmolecule. The cut-offs for the molecular size and fingerprint similarity are determined on the validation set. As a sidenote, for the individual models the output size with a beam size of k will eventually be at most k since some predictionsmay be filtered-out. For the ensemble model though, the output size will be larger than k, since the output is the unionof the 5 individual models.
3 Method Evaluation
The evaluation of the method mainly lies on the number of correctly identified metabolites as well as the outputsize that is an indication of the number of false positives. In order to assess the ability of the algorithm to properlyrank the generated metabolites we present results for beam sizes of 5, 10, 15 and 20. We additionally compare theperformance of the ensemble model with the averaged performance of the 5 individual models in order to assess thebenefit of ensembling. Finally, we compare our method against 3 existing drug metabolite prediction tools specificallyon predicting CYP450 metabolites and we further examine the capability of the model to predict metabolites throughother than CYP450 enzymes.
The evaluation is based on comparing the SMILES representations between the predicted and the actual metabolites.Certain cases in the test set were verified after manual inspection and for ambiguous cases after consulting the literature.In general, for the comparison between the predicted and the actual metabolites, discrepancies in stereochemistry andatom charges are disregarded. Additionally, for the case of CYP450 metabolites, aldehydes are considered equivalent tocarboxylic acids since they commonly constitute intermediate compounds that are further being oxidised to carboxylicacids without leaving the active center of the enzyme [27, 8].
3.1 Baseline Model
As a reference point, we tested the performance of the original Transformer trained on chemical reactions on themetabolite test set without any fine-tuning. With a beam size of 20, the model identified 21 metabolites in total outof 138 with an output size of 781 which corresponds to a specificity value of about 2% and recall 15%. Interestingly,the model was able to retrieve only CYP450 metabolites, that is metabolites that result from oxidation reactions. Inother words, the model was not able to handle cases where there is a more noticeable change in the size of the inputmolecule such as transferase and hydrolase reactions. Oxidation reactions commonly involve bond changes and possiblyattachment of oxygen atoms and usually do not change the size of the molecule. This result is expected considering thenature of the dataset used for pre-training which includes complete chemical reactions.
6

Table 1: Assessment of the ranking capability of the ensemble model in terms of the number of drugs for which at least one, at leasthalf and all known metabolites have been identified as well as the total number of identified metabolites. The table also shows thetotal output size for all 54 test cases as well as the average output size per input
Beam At least one At least half All Total identified Total Averagesize metabolite metabolites metabolites metabolites out. size out. size Precision Recall
5 45 37 22 74 486 9.0 0.15 0.5410 49 42 27 89 951 17.61 0.09 0.6415 49 45 28 97 1425 26.39 0.07 0.7020 49 46 29 101 1928 35.70 0.05 0.73
Table 2: Averaged prediction performance and standard deviation of the individual models that comprise the ensemble model fordifferent beam sizes
Beam At least one At least half All Total identified Total Averagesize metabolite metabolites metabolites metabolites out. size out. size Precision Recall
5 33.8±2.2 20.8±1.3 8.8±1.9 46.4±3.0 196.6±6.8 3.6±0.1 0.24±0.01 0.34±0.0210 40±2.5 30.2±2.8 13.8±1.5 63.8±3.8 388.4±7.2 7.2±0.1 0.16±0.01 0.46±0.0315 42.6±1.8 33.2±2.4 15.6±1.1 70.6±3.4 572.6±21.7 10.6±0.4 0.12±0.01 0.51±0.0220 43±1.6 35.8±2.6 17.8±1.8 76.2±3.0 758.2±31.4 14.02±0.6 0.1±0.01 0.55±0.02
3.2 Prediction Accuracy
Next, we evaluate the prediction accuracy of the ensemble Metabolite Translator. We report the results with varyingchoices of beam size, as shown in Table 1. The results show that with a beam size of 5, which corresponds to about 9predictions per input molecule, the ensemble model identifies at least one metabolite for about 83% of the drugs (45 outof 54) while it successfully retrieves more than half of the verified metabolites (recall 0.54). With a beam size of 15, themodel finds at least one metabolite for about 90% of the drugs, it correctly identifies all known metabolites for morethan half of the drugs while in total it successfully retrieves 70% of the known metabolites. Further increase of the beamsize does not seem beneficial, since the average output size increases considerably without significantly improvement inthe recall rate.
Next we assess the averaged prediction performance and standard deviation of the individual models that comprisethe ensemble, as shown in Table 2. According to the results, on average the individual models with a beam size of 20achieve comparable performance with the ensemble model with beam size 5 in terms of precision and recall. However,overall the ensemble model seems to retrieve a larger number of metabolites proving that the ensembling techniqueindeed increases the output diversity.
It is worth mentioning that although we assess the model for predicting metabolites through one-step reactions, wewere able to identify metabolites that are formed through multi-step reactions among the predicted molecules. Morespecifically, in DrugBank we found metabolites that are the end products of multi-step reactions for 6 out of the 54compounds. The model correctly predicted in total 5 such metabolites for 4 of the 6 compounds.
3.3 Performace on CYP450 Metabolism and Comparison with Existing Tools
Next, we assess the capability of the ensemble model to identify metabolites specifically through CYP450 enzymesusing as reference three existing drug metabolite prediction tools: GLORY [8], BioTransformer [9] and SyGMa [7].The CYP450 enzyme family is of particular interest since it accounts for the metabolism of the big majority of drugs.Most of the existing computational tools for assisting drug metabolism studies are specifically focused on this enzymefamily. GLORY is one of the tools that focuses solely on identifying CYP450 metabolites and has two operationmodes: maxEfficiency and maxCoverage. SyGMa relies on a rule database that covers phase I and phase II reactions.BioTransformer has even greater coverage and consists of 5 different modules, each one intended for a specific setof enzymes, among them one module that is specific to CYP450 metabolism. The research group that developed theGLORY method published a comparative evaluation between their method, the SyGMa tool using only the rules forphase I reactions and the CYP450 module of the BioTransformer for identifying CYP450 metabolites on the test set of29 drugs and 81 metabolites that they have released [8].
We applied the ensemble Metabolite Translator on the same dataset with different beam sizes. The results of thiscomparison are shown in Table 3. SyGMa, BioTransformer and the maxEfficiency mode of GLORY identified 60,
7

Table 3: Comparison with three existing tools for predicting CYP450 metabolites
SyGMa BioTransformer GLORY Metabolite TranslatormaxCov maxEffic beam=5 beam=10 beam=15 beam=20
Identified Metabolites 60 58 67 52 46 54 60 62Output Size 406 344 793 327 270 522 787 1069
58 and 52, respectively, metabolites with a comparable output size ranging from 327 to 406. The larger output sizecorresponds to SyGMa which is the only method among the 3 existing tools that covers phase I reactions insteadof focusing solely on CYP450. With a beam size of 10 and a comparable output size of 522 our ensemble modelidentifies 54 metabolites. A larger output size is expected since our method is not restricted to CYP450 metabolism andcovers human metabolism in general. The maxCoverage mode of GLORY identified 67 metabolites but at a cost ofa significant increase of the number of false positives which is not tolerable considering that GLORY predicts onlyCYP450 metabolites. With a beam size of 15, the ensemble model identified 60 CYP450 metabolites. Interestinglythough, we were able to verify an additional number of mainly non-CYP450 metabolites. More specifically, we foundadditional metabolites from DrugBank for 6 out of the 29 test cases and eventually verified 7 additional metabolitesthrough UDP-GT, myeloperoxidases, CYP450 and other non-specified enzymes. Hence, it is safe to argue that thelarger output size from our method is explained by the fact that the model indeed covers a much larger range of enzymescomparing to existing tools.
In general, our comparison shows that the Metabolite Transformer retrieved a comparable number of CYP450 metabo-lites with existing tools at the expense of a larger output size which is justified from the larger enzyme coverage. It isimportant to recall here that our method has been trained on a dataset of general metabolic reactions and is not specificto drug metabolism. On the contrary the methods to which we are comparing against have been specifically developedfor predicting drug metabolites. The fact that the Metabolite Translator retrieves a comparable number of metaboliteswith methods that have been optimised for the specific enzyme family confirms the capability of our model to identifyrelevant metabolites without requiring enzyme specific information. On top of that, the end-to-end learning basedapproach has eliminated the need for expert-derived rules and in general has minimised the need for expert intervention.Without special care of each enzyme family, we have developed a single model that can predict human metabolitesfor major enzyme families and also enzymes that are not commonly involved in drug metabolism. Having a singlemodel can potentially decrease the overall number of false positives. The current approach for covering a larger rangeof enzymes is to develop multiple models, however, applying multiple models on the query molecule translates to anaccumulation of false positives across all models. Finally, we should note that our method predicted metabolites forall test cases even for molecules in the upper limit of molecular size. On the contrary the GLORY method, whichinvolves machine learning for identifying the sites of metabolism, could not be applied in one test case because thetest compound included a boron atom which was not present in their training set. The metabolite Translator, whichwas pre-trained on a large set of chemical reactions and therefore has seen molecules with rarely occurring atoms, wasapplied successfully on that case.
3.4 Analysis per Enzyme Family
Next, we are assessing the prediction accuracy of the ensemble model as well as the relevance of the predicted metabolitesfor different enzyme families. The test set includes metabolites from the CYP450 enzyme family, glucurosyltransferases(UDP-GT) and sulfotransferases, hydrolases, a smaller number of enzymes that are not commonly involved in drugmetabolism and finally metabolites from unspecified enzymes. The prediction accuracy for each enzyme class is shownin Table 4 where we see that the recall rate varies noticeably between different enzyme families. However, we shouldnote that not all enzymes are equally represented in the test set. For example, although the model appears to have aperfect recall for sulfotransferase and hydrolase reactions, the number of test cases for these two enzyme families isvery low as they are significantly under-represented in the entire dataset (Figure 3b). The same holds also for the case ofglucurosyltransferases. However, despite the fact that these enzymes are significantly under-represented in the trainingset, our results indicate that the model is still able to predict relevant reactions and identify correct metabolites despitethe fact that the enzyme action is not directly encoded in the data.
More specifically, we manually inspected the predicted metabolites in order to get insights on how relevant thepredictions are for each enzyme family. Some interesting cases are depicted in Figure 4 where we see the structureof the drug, the actual metabolite and the closest predicted metabolite in the 3 columns, respectively. To start with,for 6 out of 9 drugs that have metabolites through more than one enzymes, the model correctly identified metabolitesfrom more than one enzyme. Looking into specific enzyme families, the case of glucuronidation reactions appearsto be an especially challenging study case possibly because a large and complex structure is transferred to the parent
8

Figure 4: Drug structure, actual metabolite and closest prediction for a small number of test cases.
9

Table 4: Accuracy per enzyme family.
Total number IdentifiedEnzyme Family of metabolites Metabolites Recall
CYP450 95 66 0.69UDP-GT 12 6 0.5
Sulfotransferases 3 3 1Hydrolases 4 4 1
Other 23 18 0.78
molecule. The Metabolite Translator correctly identified 6 out of 12 glucuronidation metabolites. For additional 3parent molecules the model was able to correctly identify the reaction type, that is conjugation with a glucuronic acid,but the predicted structure was not entirely correct (such as in the case of Tamezapam and Lamotrigine of Figure 4).
The case of sulfotransferase reactions, on the other side, does not seem to be challenging for the model despite thefact that this enzyme family is even more under-represented in the training set. Contrary to the glucuronic acid, thesulfate structure is much smaller and simpler. Interestingly, the model has learnt that the positions that are likely to beconjugated with a glucuronic acid can also be conjugated with a sulfate structure. As we noticed, in many cases sulfationand glucuronidation reactions are predicted for the same molecules and in most cases these molecules indeed undergophase II metabolism. Raloxifene (third case in Figure 4) is a very interesting case which has two major metabolites thatresult from conjugations with the glucuronic acid in two different positions which the model has correctly identified.An additional metabolite though, which is the end-product of a double-conjugation with a glucuronic acid, can also beformed. The Metabolite Translator has predicted the very rare event of a double-conjugation but with a sulfate structureinstead of a glucuronic acid. The case of hydrolase reactions, where the molecule is being truncated, does not seem tobe challenging either.
3.5 Invalid Predictions and Post-processing
Besides the prediction accuracy of the model, we finally evaluate two additional aspects: first, how susceptible themodel is for generating invalid SMILES, and, second what is the effect of the post-processing filtering. Our analysisshows that the model generated at least one valid SMILES for all drugs in both, validation and test sets, for all beamsizes. The size of the molecules in these sets ranged from 6 to 65 atoms. The average number of invalid predictions perinput molecule ranged from 0.33 for a beam size of 5, to 2.26 for a beam size of 20. These findings indicate that theissue of generating non-valid SMILES is insignificant for small molecules. Regarding the effect of the post-processingfiltering, our analysis showed that a very small percentage of the predicted molecules is filtered out proving that thebig majority of the predictions are actually relevant. More specifically, for a beam size of 15, only 42 predictions arefiltered out in total which corresponds to less than 3% of the total predictions.
4 Conclusions
We have presented an end-to-end learning-based method for predicting human metabolites of small molecules. Ourmodel has been trained by using transfer learning on a dataset of human metabolic reactions without restricting to certainenzyme families. We evaluate the performance of our method on predicting drug metabolites. Results show that ourmethod is able to correctly predict metabolites from enzymes of phase I and phase II drug metabolism and also enzymesthat are less commonly involved in drug metabolism. Regarding the CYP450 enzyme family, which is responsible forthe metabolism of the big majority of drugs, we show that our method can identify a comparable number of metaboliteswith existing tools that have been specifically developed for this enzyme family. The proposed approach addresses theproblems of limited scalability and lack of generalization of the existing rule-based methods. It additionally provides amore comprehensive study of drug metabolism since it is trained on data that cover human metabolism in its entirety asopposed to the enzyme-specific existing methods. As more data become available, especially on drug metabolism, theperformance of this approach can be further improved encouraging the adoption of such tools in drug discovery foraccelerating and enhancing the safety studies.
References
[1] Edward Croom. Chapter three - metabolism of xenobiotics of human environments. In Ernest Hodgson, editor,Toxicology and Human Environments, volume 112 of Progress in Molecular Biology and Translational Science,
10

pages 31 – 88. Academic Press, 2012.[2] F. Peter Guengerich. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab
Pharmacokinet., (1), 2011.[3] Jonathan D. Tyzack and Johannes Kirchmair. Computational methods and tools to predict cytochrome p450
metabolism for drug discovery. Chem Biol Drug Des., (4):377–386, 2019.[4] Sayada Reemsha Kazmi, Ren Jun, Myeong-Sang Yu, Chanjin Jung, and Dokyun Na. In silico approaches and
tools for the prediction of drug metabolism and fate: A review. Computers in Biology and Medicine, 106:54 – 64,2019.
[5] Naiem T. Issa, Henri Wathieu, Abiola Ojo, Stephen W. Byers, and Sivanesan Dakshanamurthy. Drug metabolismin preclinical drug development: A survey of the discovery process, toxicology, and computational tools. CurrentDrug Metabolism, 18(6):556–565, 2016.
[6] Johannes Kirchmair, Andreas H. Göller, Dieter Lang, Jens Kunze, Bernard Testa, Ian D. Wilson, Robert C. Glen,and Gisbert Schneider. Predicting drug metabolism: experiment and/or computation? Nat Rev Drug Discov,(6):387–404, 2015.
[7] Lars Ridder and Markus Wagener. Sygma: combining expert knowledge and empirical scoring in the predictionof metabolites. ChemMedChem, 3(5):821–832, 2008.
[8] Christina de Bruyn Kops, Conrad Stork, Martin Sicho, Nikolay Kochev, Daniel Svozil, Nina Jeliazkova, andJohannes Kirchmair. Glory: Generator of the structures of likely cytochrome p450 metabolites based on predictedsites of metabolism. Frontiers in Chemistry, 7:402, 2019.
[9] Yannick Djoumbou-Feunang, Alberto Fiamoncini, Jarlei andGil-de-la-Fuente, Russell Greiner, Claudine Manach,and David S. Wishart. Biotransformer: a comprehensive computational tool for small molecule metabolismprediction and metabolite identification. Journal of Cheminformatics, 11(2), 2019.
[10] Timothy D. Salatin and William L. Jorgensen. Computer-assisted mechanistic evaluation of organic reactions. 1.overview. The Journal of Organic Chemistry, 45(11):2043–2051, 1980.
[11] Hiroko Satoh and Kimito Funatsu. Sophia, a knowledge base-guided reaction prediction system - utilization ofa knowledge base derived from a reaction database. Journal of Chemical Information and Computer Sciences,35(1):34–44, 1995.
[12] Peter Röse and Johann Gasteiger. Automated derivation of reaction rules for the eros 6.0 system for reactionprediction. Analytica Chimica Acta, 235:163 – 168, 1990.
[13] Daniel Lowe. Chemical reactions from us patents (1976-sep2016). 2017.[14] Connor W. Coley, Wengong Jin, Luke Rogers, Timothy F. Jamison, Tommi S. Jaakkola, William H. Green,
Regina Barzilay, and Klavs F. Jensen. A graph-convolutional neural network model for the prediction of chemicalreactivity. Chem. Sci., 10:370–377, 2019.
[15] Juno Nam and Jurae Kim. Linking the neural machine translation and the prediction of organic chemistry reactions,2016.
[16] Philippe Schwaller, Théophile Gaudin, Dávid Lányi, Costas Bekas, and Teodoro Laino. “found in translation”:predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem.Sci., 9:6091–6098, 2018.
[17] Philippe Schwaller, Teodoro Laino, Théophile Gaudin, Peter Bolgar, Christopher A. Hunter, Costas Bekas, andAlpha A. Lee. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACSCentral Science, 5(9):1572–1583, 2019.
[18] Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Ł ukasz Kaiser, andIllia Polosukhin. Attention is all you need. pages 5998–6008, 2017.
[19] Giorgio Pesciullesi, Philippe Schwaller, Teodoro Laino, and Jean-Louis Reymond. Carbohydrate transformer:Predicting regio- and stereoselective reactions using transfer learning, Mar 2020.
[20] David Weininger, Arthur Weininger, and Joseph L. Weininger. Smiles. 2. algorithm for generation of uniquesmiles notation. J. Chem. In. Comput. Sci., 29(2):97–101, 1989.
[21] David S Wishart, Yannick D Feunang, An C Guo, Elvis J Lo, Ana Marcu, Jason R Grant, Tanvir Sajed, DanielJohnson, Carin Li, Zinat Sayeeda, Nazanin Assempour, Ithayavani Iynkkaran, Yifeng Liu, Adam Maciejewski,Nicola Gale, Alex Wilson, Lucy Chin, Ryan Cummings, Diana Le, Allison Pon, Craig Knox, and Michael Wilson.DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research, 46(D1):D1074–D1082,11 2017.
11

[22] David S Wishart, Yannick Djoumbou Feunang, Ana Marcu, An Chi Guo, Kevin Liang, Rosa Vázquez-Fresno,Tanvir Sajed, Daniel Johnson, Carin Li, Naama Karu, Zinat Sayeeda, Elvis Lo, Nazanin Assempour, MarkBerjanskii, Sandeep Singhal, David Arndt, Yonjie Liang, Hasan Badran, Jason Grant, Arnau Serra-Cayuela,Yifeng Liu, Rupa Mandal, Vanessa Neveu, Allison Pon, Craig Knox, Michael Wilson, Claudine Manach, andAugustin Scalbert. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Research, 46(D1):D608–D617, 11 2017.
[23] Ron Caspi, Richard Billington, Carol A Fulcher, Ingrid M Keseler, Anamika Kothari, Markus Krummenacker,Mario Latendresse, Peter E Midford, Quang Ong, Wai Kit Ong, Suzanne Paley, Pallavi Subhraveti, and Peter DKarp. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Research, 46(D1):D633–D639,10 2017.
[24] Smirks. a reaction transform language. https://daylight.com/dayhtml/doc/theory/theory.smirks.html.
[25] Esben Jannik Bjerrum. SMILES enumeration as data augmentation for neural network modeling of molecules.CoRR, abs/1703.07076, 2017.
[26] Josep Arús-Pous, Simon Johansson, Oleksii Prykhodko, Esben Jannik Bjerrum, Christian Tyrchan, Jean-LouisReymond, Hongming Chen, and Ola Engkvist. Randomized SMILES strings improve the quality of moleculargenerative models. J. Cheminformatics, 11(1):71:1–71:13, 2019.
[27] F. Peter Guengerich, Christal D. Sohl, and Goutam Chowdhury. Multi-step oxidations catalyzed by cytochromep450 enzymes: Processive vs. distributive kinetics and the issue of carbonyl oxidation in chemical mechanisms.Archives of Biochemistry and Biophysics, 507(1):126 – 134, 2011. P450 Catalysis Mechanisms.
12

download fileview on ChemRxivMetabolitesPrediction.pdf (823.85 KiB)





























