Embed Size (px)
Citation preview

1
U N I V E R S I T Y O F C O P E N H A G E N
F A C U L T Y O F S C I E N C E
D E P A R T M E N T O F B I O L O G Y
This thesis has been submitted to the PhD School of The Faculty of Science, University of
Copenhagen
PhD Thesis Michael Christiaan Greeff
Suppressing autoimmunity in Arabidopsis
thaliana with dominant negative immune
receptors.
Academic advisor: Morten Petersen
Submitted 30/6/2014

2
Table of contents
ABSTRACT ......................................................................................................................... 4
ABSTRAKT ......................................................................................................................... 5
PREFACE AND ACKNOWLEDGEMENTS ......................................................................... 6
Abbreviations .................................................................................................................................................................... 7
INTRODUCTION ................................................................................................................. 9 Plant Disease Resistance ................................................................................................................................................ 9 Structural plant features in defense .............................................................................................................................. 10 Pathogen associated molecular patterns (PAMPS) ...................................................................................................... 11 Effectors ....................................................................................................................................................................... 13 R proteins ..................................................................................................................................................................... 14 NBS Domain ................................................................................................................................................................ 15 The TIR domain ........................................................................................................................................................... 16 The CC domain ............................................................................................................................................................ 17 The LRR domain.......................................................................................................................................................... 18 Model of R Protein function ........................................................................................................................................ 18 General factors required for R protein signaling .......................................................................................................... 19 Models of effector recognition ..................................................................................................................................... 21 R Protein pairs ............................................................................................................................................................. 22 NB-LRR Interaction with transcription factors ............................................................................................................ 23 The Hypersensitive Response ...................................................................................................................................... 25 Hormone signaling in plant defense ............................................................................................................................. 26 Autoimmune mutants ................................................................................................................................................... 29 The CAMTA3 autoimmune mutant ............................................................................................................................. 32 Aim of our work........................................................................................................................................................... 34
RESULTS .......................................................................................................................... 36
Chapter 1. acd11 suppressors ........................................................................................................................................ 36 A CC-NBS-LRR contributes to acd11 autoimmune phenotype .................................................................................. 36
Chapter 2. Validating P-loop mutants dominant negative effects .............................................................................. 40 RPM1 P-loop mutants are specific and dominant negative. ........................................................................................ 40 P-loop dominant negative specificity ........................................................................................................................... 43 P-loop DN effects partially extends to R gene complexes. .......................................................................................... 46 PAT1 and SUMM2-D .................................................................................................................................................. 47
Chapter 3. Autoimmune mutant library and P-loop screens ...................................................................................... 49 P-loop library construction and cloning optimization. ................................................................................................. 49 Concluded transformations and screening of individual autoimmune mutants............................................................ 55
Chapter 4. Suppression of camta3-1 phenotypes .......................................................................................................... 57 Suppressor mutants from screen and visual phenotypes. ............................................................................................. 57 Genotyping of DSC-D lines ......................................................................................................................................... 59 Expression levels of DSC mRNA ................................................................................................................................ 60 Cell death and ROS phenotype .................................................................................................................................... 61 Defense marker gene PR1 ............................................................................................................................................ 63

3
Enhanced Disease resistance ........................................................................................................................................ 65 Necrotrophic infections ................................................................................................................................................ 67 Ethylene Signalling ...................................................................................................................................................... 68 EDS1 gene regulation .................................................................................................................................................. 70 Recessive DSC mutants do not supress camta3-1 ....................................................................................................... 72
Preliminary data ............................................................................................................................................................. 74 Cold responsive genes .................................................................................................................................................. 74 The role of CAMTA3 in SAR ...................................................................................................................................... 76 Noco2 infection and resistance .................................................................................................................................... 78
Chapter 5. CAMTA3 complexes .................................................................................................................................... 81 Localisation Studies ..................................................................................................................................................... 81 FRET interaction study ................................................................................................................................................ 84 BiFC study ................................................................................................................................................................... 85
CONCLUSION ................................................................................................................... 88
PERSPECTIVES ............................................................................................................... 90
MATERIALS AND METHODS .......................................................................................... 91 Statistical analysis ........................................................................................................................................................ 91 Plants growth conditions .............................................................................................................................................. 91 Maintenance of pathogenic isolates ............................................................................................................................. 91 Ion leakage assays ........................................................................................................................................................ 91 Cloning and generation of transgenic plants ................................................................................................................ 91 Trypan blue staining .................................................................................................................................................... 93 DAB staining ............................................................................................................................................................... 93 Bacterial growth assays ................................................................................................................................................ 93 Botrytis cinerea infection ............................................................................................................................................. 93 Hyaloperonospora arabidopsidis infection ................................................................................................................. 93 Confocal microscopy ................................................................................................................................................... 93 RT-PCR ....................................................................................................................................................................... 94 SAR assay .................................................................................................................................................................... 94 Triple Response ........................................................................................................................................................... 94 Cold Shock ................................................................................................................................................................... 94 Cloning PCR ................................................................................................................................................................ 95 PCR purification cleanup ............................................................................................................................................. 95 Primers ......................................................................................................................................................................... 95
REFERENCES .................................................................................................................. 97
MANUSCRIPT ................................................................................................................. 121
PUBLISHED REVIEW ..................................................................................................... 162

4
Abstract
A small set of Resistance proteins (R-proteins), guards plants against a large set of pathogen effector proteins
that can suppress or subvert plant defense responses. The guard model attempts to solve this discrepancy by
proposing that a major function of R proteins is to monitor host effector targets. In response to effector mediated
changes on targets, R proteins trigger the hypersensitive response.
We have proposed that a corollary to this 'guard model' is that forms of plant autoimmunity are due to
inappropriate R protein activation. For example, we showed that a knockout of Accelerated Cell Death
11 (acd11) leads to inappropriate activation of hypersensitive cell death. We have previously performed a
large-scale survival screen for suppressors of acd11 and found that cell death in acd11 is suppressed by
mutations in a gene encoding an R protein. We have thus proposed that loss of ACD11 results in HR cell death
because LAZ5 directly or indirectly guards it. The LAZ5 alleles we first found were dominant negative (laz5-
DN). The laz-DN allele mutation was found in a conserved functionally important ATP binding region, the P-
loop. Site-directed DN mutant alleles can be made for other R genes, as we have recently found that
transgenics with similarly mutated rpm1-DN alleles lose resistance to Pseudomonas syringae expressing
the AvrRpm1 effector. Accordingly, we have constructed a collection of 100 R-DN alleles and transformed
them into other autoimmune mutants including camta3. CAMTA3 was previously shown to be a negative
regulator of plant defense by inhibiting transcription of EDS1 and NDR1, important downstream signaling
components of R-protein signaling. We found that two dominant negative alleles, DSC-D and DSC2-D, can
suppress all tested camta3-1 phenotypes. We hypothesize that like acd11 and other autoimmune mutants, the
increased levels of defense genes like EDS1 in camta3-1 might be a consequence of R protein activation and
not merely as a result of negative regulation of plant defense responses as was previously proposed. DSC
and CAMTA3 are part of a nuclear localized complex supporting the possibility that DSC is directly guarding
CAMTA3.

5
Abstrakt
Et lille antal af resistensproteiner (R proteiner), bevogter planter mod et stort antal af patogene
effektorproteiner, der kan undertrykke eller undergrave planters forsvarsmekanismer. ”Guard”-modellen
forsøger at løse denne numeriske uoverensstemmelse ved at foreslå, at en stor funktion af R proteiner er at
overvåge værtens proteiner, som er effektorene er målrettet mod. Som svar på effektor-medierede ændringer
udløser R proteinerne en allergisk reaktion.
Vi har foreslået, at en naturlig følge denne "vagt model" er, at nogle former for autoimmunitet i planter skyldes
unaturlig R protein aktivering. For eksempel viste vi, at en knockout af Accelerated Cell Death 11 (acd11) fører
til uhensigtsmæssig aktivering af hypersensitivt response (HR), celledød. Vi har tidligere udført et storstilet
overlevelses-screen for suppressorer af acd11 og fandt, at celledød i acd11 undertrykkes af mutationer i et
gen, der koder for et R-protein. Vi har således foreslået, at tab af ACD11 resulterer i HR celledød, fordi LAZ5
direkte eller indirekte bevogter det. De LAZ5 alleler vi først fandt var dominerende negative (laz5-DN). Den
laz-DN allelmutation blev fundet i en konserveret funktionelt vigtigt ATP-bindene region, P-sløjfen. Site-
directed DN mutantalleler kan laves for andre R-gener, som vi for nylig har fundet, at transgene planter med
tilsvarende muterede RPM1-DN alleler mister modstand mod Pseudomonas syringae, som udtrykker
AvrRpm1 effektoren. Derfor har vi konstrueret en samling af 100 R-DN alleler og transformeret dem i andre
autoimmune mutanter herunder camta3. CAMTA3 blev tidligere foreslået at være en negativ regulator af plante
forsvar ved at hæmme transkription af EDS1 og NDR1, vigtige downstream signaleringskomponenter af R-
protein-signalering. Vi fandt, at to dominerende negative alleler, DSC-D og DSC2-D, kan undertrykke alle
testede camta3-1 fænotyper. Vi foreslår, at ligesom acd11 og andre autoimmune mutanter, kan det øgede
niveauer af forsvarsrelaterede gener som EDS1 i camta3-1 være en konsekvens af R proteinaktivering og ikke
blot som følge af negativ regulering af planters forsvarsrespons, som tidligere foreslået. DSC og CAMTA3 er
en del af et kernelokaliseret kompleks, hvilket understøtter muligheden for, at DSC direkte bevogter CAMTA3.

6
Preface and acknowledgements
This thesis concludes my PhD work at the department of Biology, University of Copenhagen. The
project resulted in manuscript submitted to PLoS Pathogens and a review published in Frontiers in
Plant Science “Receptor-like kinase complexes in plant innate immunity.”
Klaus Petersen started this work and cloned together with Simon Bressendorff, TIR-NBS-LRR P-
loop mutants and transformed Col-0, camta3, and mpk4 with all cloned constructs. This laid the
foundation for most of my work in this thesis. I was tasked with cloning CC-NBS-LRR P-loop
mutants and Eleazar Rodriguez took over the CC-NBS-LRR project and proceeded with
transforming candidate autoimmune mutants. Signe Lolle made BiFC constructs and performed
BiFC and FRET assays under my supervision as a very independent and capable Masters student.
Initial work with Michael Krogh Jensen on a potential suppressor P-loop was done but the project
ended. Michael also included my suppressor in a Yeast two hybrid screen for interactors not
included in this manuscript. Unfortunately putative interactions were shown not to be real
interactions. Some of this work was performed by a Bachelors student Signe Steinhoff.
I would like to thank my main supervisor Morten Petersen and co-supervisor John Mundy for their
support and guidance during my research. Thank you for the tremendous opportunity you gave me
to work in your lab. I would like to thank Suksawad Vongvisuttikun for his technical assistance.
Thank you to all the current and previous lab members that provided an interesting and supportive
working environment. Your suggestion and assistance were invaluable. They are Eleazar, Rachel,
David, Chandra , Frederikke, Simon, Magnus, Milena , Kenneth, Peter ,Kris , Azra, Helena,
Thank you to my family and friends for your continued support.
Lastly and most importantly thank you to my wife Milena for all her love and tremendous support
throughout my studies.
Michael Christiaan Greeff

7
Abbreviations
ACD11 accelerated cell death 11
A.th. Arabidopsis thaliana
ABA abscisic acid
ABA abscisic acid
ACD1 accelerated cell death 1
ACD5 Accelerated cell death 5
ADP Adenosine diphosphate
ADR1 activated disease resistance
ADR1-L2 ADR1 like-2
ADR1-L2-D ADR1 like-2 ploop mutant
AGO Argonaute
APAF-1 Apoptotic protease activating factor 1
ATP Adenosine triphosphate
Avr avrirulence
B. Cinerea Botrytis cinerea
BAK1 Brassinosteroid Insensitive1-associated Kinase1
BiFC Bimolecular fluorescence complementation
BIK1 Botrytis induced Kinase 1
C. Elegans Caenorhabditis elegans
CaM calmodulin
CAMTA calmodulin binding transcription activators
CAT3 Catalase3
CBF1 C-repeat/DRE binding factor 1
CBF2 C-repeat/DRE binding factor 1
CC coiled coil
ccdB Cytotoxic protein
cDNA complementary DNA
CED-4 Cell death protein 4
CERK1 chitin elicitor receptor kinase 1
CFP cyan fluorescent protien
ChIP chromatin immunoprecipitation
CMPG1 Cys, Met, Pro, and Gly protein 1
CNL CC-NBS-LRR
COI1 Coranitine insensitive 1
Col-0 Columbia
COR cold-regulated
CP viral coat protein
CUL3 Cullin 3
Cyt[Ca²⁺] cytoplasmic Ca²⁺ levels
DAB 3,3′-diaminobenzidine
DCL dicer-like proteins
DN Dominant negative
DNA Deoxyribonucleic acid
DND1 defense, no death 1
DND2 defense, no death 2
dNTP deoxynucleotide triphosphates
DSC Dominant supressor of camta3
DSC-D DSC p-loop mutant
EDS1 Enhanced disease susceptibility 1
EFR EF-Tu receptor
EF-Tu Bacterial elongation factor Tu
EILs EIN3-like proteins
EIN ethylene insensitive
EIN2 ethylene insensitive 2
ERS ethylene response sensor
ET Ethylene
ETI Effector triggered immunity
ETR ET receptor
ETS Effector Triggered susceptibility
Flg22 Flagellin derived peptide 22
FliC Flagellin
FLS2 Flagellin Sensing 2
FRET Förster resonance energy transfer
GFP green fluorescent protein
H2O2 Hydrogen peroxide
H3K36 Histone H3 lysine 36 methylation
H3K4 Histone H3 lysine 4 methylation
Hpa Hyaloperonospora parasitica
HR hyper sensitive response
HSP90 heat shock protein 90
ICS1 Isochorismate Synthase 1
IL-1 Interleukin 1
INF1 Phytophthora cactorum Infestin 1
IP immunoprecipitation
JA jasmonic acid
JAI3 jasmonate-insensitive 3
JAZ jasmonate ZIM-domain
Kan Kanamycin

8
LAZ5 Lazarus 5
LRR Leucine rich repeat
LSD1 lesion stimulation disease 1
LUC Luciferase
MAPK Mitogen activated protein kinases
MC1 Metacaspase1
MEKK MAP kinase kinase kinase
MIP MPK4 interacting protien
miRNA microRNA
MKK Map kinase kinase
MPK4 MAP kinase 4
mRNA messanger RNA
MYB myeloblastosis
NahG Pseudomonas salicylate hydroxylase gene
NASC Nottingham Arabidopsis Stock Centre
NB-ARC nucleotide-binding adaptor shared by APAF-1, R proteins, and CED-4
NB-LRR Nucleotide binding-Leucine rich repeat
NBS nucleotide binding site domain
Nd-1 Niederzenz
NDR1 non-race-specific disease resistance
NPR1 non-expresser of PR genes 1
NRG1 N requirement gene 1
NRIP1
Ori Origen of replication
p50 tobacco mosaic virus protien
PAD4 Phytoalexin deficient 4
PAMPs pathogen associated molecular patterns
PCD programmed cell death
PCR polymerase chain reaction
PDF1.2 plant defensin 1.2
PGPR plant growth-promoting rhizobacteria
phasiRNA phased siRNAs
PIE1 photoperiod-independent early flowering1
PR pathogen related
PRR Pattern recognition receptors
Pst Pseudomonas syringae
PTI PAMP triggered immunity
PVX Potato virus X
QPCR Quantitive PCR
R resistance genes
RanGAP2 Ran GTPase Activating Protein 2
RAR1 required for Mla12 resistance 1
R-DN Ploop R gene
RIN4 RPM1 interacting protein 4
RNA Ribonucleic acid
ROS Reactive oxygen species
RPM1 resistance to p. syringae pv maculicola 1
RPM1-D RMP1 p-loop mutant
RPP recognition of peronospora parasitica
RPS resistant to p. Syringae
RRS resistance to Ralstonia solanacearum
RT-PCR Realtime PCR
SA Salicylic acid
SAG101 Senescence-associated gene 101
SAR systemic acquired resistance
SBP squamosa promoter binding protein
SD Short Days
SDG8 Set Domain Group 8
SGT1 suppressor of G2 allele of skp1
SIZ1 SUMO E3 ligase
SKP1 S-phase-kinase-associated protein-1
SLH1 sensitive to Low Humidity 1
SNC1 suppressor of npr1-1, constitutive 1
SPL6 SBP-domain transcription factor 6
SR1IP1 AtSR1-interaction-protein 1
SSI SA insensitivity
STAND signal transduction ATPases with numerous domains
SUMM2 SUPPRESSOR OF MKK1 MKK2 2
SUMM2-D SUMM2 p-loop mutant
SUMO Small ubiquitin-like modifier
TAIR The Arabidopsis Information Resource
TAO1 Target of AvrB Operation
TIG transcription factor immunoglobulin
TIR Toll/interleukin-1 IL-1 receptor
TLR Toll like receptors
TMV Tobacco mosaic virus
TNL TIR-NSB-LRR
TPR1 Topless-related 1
TTSS type III secretion systems
UBQ10 Ubiquitin 10
USER Uracil-Specific Excision Reagent
VAD1 vascular associated death
YFP Yellow florecent protien

Introduction
Plant Disease Resistance
Plants are primary producers and therefore a source of nutrients to many organisms
(Lindeman 1991). Root exudates for example provide nutrients for several species of soil-
borne organisms (Lundberg et al. 2012). Many of these organisms can be beneficial to plant
growth, like plant growth-promoting rhizobacteria (PGPR) (Gopal, 2013). However many
organisms have a detrimental effect on plant growth and long-term survival (Dangl and
Jones 2001). Logically if plants cannot defend their resources from being depleted by
pathogens and herbivores they cannot thrive. Availability of energy resources is directly
correlated to reproduction and thus to evolutionary fitness (Janczur 2009). The cost of the
defense should be less than the loss of resources to pathogens or herbivores to be effective
as a survival mechanism (Coley, Bryant, and Chapin 1985). Over-investing in defense in
the absence of pathogens can be just as detrimental to survival as disease.
Plant defense is energetically very demanding, so strict modulation of defense activation is
required (Freeman 2008). This modulation occurs in a spatio-temporal way and requires in
most cases recognition of pathogens directly or indirectly. For instance, plants can directly
detect bacterial flagella (Chinchilla et al. 2006) or they can indirectly detect tissue damage
from pathogens by responding to the endogenous peptide product, AtPEP1 (Krol et al.
2010).
Much of what we know today is through studies on the plant model organism Arabidopsis
thaliana (A.th.). (Wienkoop, Baginsky, and Weckwerth 2010) initially chosen for this
purpose because it has a small genome, a short life cycle, produces lots of seed, and is
easy to transform and self-pollinates making genetic work much easier. In the genomics
era this model is still very useful to work with, due to established protocols and annotated
databases and ever increasing molecular tools (Wigge and Weigel 2001). In the last 20
years this model plant has been used, among other topics, to study how plants defend
themselves against pathogens, since A.th. like other plants has a plethora of defense
mechanisms.

10
Structural plant features in defense
Plants have defense barriers to protect themselves from pathogens. This physical barrier
that separates plants from the outside world is called the cuticle. The cuticle consists of
polymeric lipids, soluble waxes and polymeric cutin. It protects the epidermal cell wall in
arial part of plants from pathogens, water loss and abiotic stresses (Jeffree 2007). It
discourages attachment of microorganisms and insects and helps keep surfaces clean
ensuring optimal light harvesting for photosynthesis. This current dogma however might be
an oversimplification. Some data show that a defective cuticle leads to increased resistance
to Botrytis cinerea. The exact mechanism is still unknown but there likely exists a complex
dynamic process that includes cuticle structures that control plant diseases (Chassot,
Nawrath, and Metraux 2008).
The leaf surface is covered by the cuticle and one cannot fail to also notice that the leaf
surface is punctuated with structures called trichomes. These thorny projections serve as
a physical and chemical deterrent against herbivores and also pathogen vectors (Traw and
Bergelson 2003).
Other important structures in the plant-microbe milieu are the stomata. These openings are
formed by specialized plant cells called guard cells (Assmann and Shimazaki 1999). The
word comes from “stoma” Greek for mouth. These structures are openings in plant leaves
that allow for moisture and gaseous exchange between the environment and deep plant
tissue. While the cuticle excludes pathogens, the stomata together with wounds are prime
entry points for plant pathogens (Underwood, Melotto, and He 2007). Stomatal aperture is
controlled by turgor pressure in the guard cells that form the “mouth”. This aperture is very
tightly controlled by several stress hormones most notably abscisic acid (ABA) (Pillitteri and
Dong 2013). Hydathodes and nectaries are specialized stomata. Hydathodes cannot
control their aperture so they particularly serve as prime pathogen entry points (Carlton,
Braun, and Gleason 1998). Stomata respond to the presence of pathogenic bacteria and
fungi and close rapidly (Gudesblat, Torres, and Vojnov 2009). This closure depends on the
recognition of pathogen associated molecular patterns (PAMPs) by cell surface pattern
recognition receptors (PRRs). For example when Pseudomonas syringae (Pst) gains
access to the apoplast via stomata, a receptor like kinase called FLAGELLIN SENSING 2

11
(FLS2) (Gómez-Gómez and Boller 2002) recognizes bacterial flagellin and leads to closure
of stomata (Melotto et al. 2006).
If pathogens can pass the cuticle or enter through stomata they have to face another
formidable obstacle, the cell wall. The structure of the cell wall is a complex dynamic mixture
of lignin, cellulose, hemicellulose, pectic substances, proteins, enzymes and water
(Heredia, Jiménez, and Guillén 1995). This mixture allows it to serve as the plant
exoskeleton providing mechanical support but it also serves as a physical barrier, important
for resistance to pathogens. An example of the role the cell wall plays in resistance comes
from certain types of fungi or oomycetes that form specialized cell-penetrating feeding
structures that first need to pierce the cell wall to access nutrients (Szabo and Bushnell
2001). When attempted penetration, i.e. cell wall damage, is detected the cell wall is
remodeled and reinforced at the penetration site by formation of cell wall-associated
structures like papillae (Aist 1976). This reaction prevents infection of individual cells and
stunts pathogen growth. Other classes of pathogens can enzymatically degrade the cell
wall to better access the nutrients within (Arnab Kapat 1998). Cell wall degradation
products, also called damage associated molecular patterns (DAMPs), are known trigger
defense responses that include cell wall reinforcement (D’Ovidio et al. 2004). Furthermore,
several species of bacteria colonize the spaces between cells (apoplast), and interaction
with the cell wall is required to overcome defense responses and achieve full pathogenicity
(Hauck, Thilmony, and He 2003). Even though these structures have in the past been
regarded as “passive”, recent research has shown that they are very dynamic and intricately
connected to the “active defenses” (Traw and Bergelson 2003).
Pathogen associated molecular patterns (PAMPS)
PAMP triggered immunity (PTI) represents the first line of active defense in the zig-zag
model of plant defense shown in figure I (Jones and Dangl 2006).

12
Figure I 1
Pathogens contain pathogen associated molecular patterns (PAMPs) that can be detected by plant cells
leading to a defense response. Pathogens can overcome PAMP triggered immunity (PTI) by secreting effector
proteins that can disable defenses called effector triggered susceptibility (ETS). Plants employ cytosolic
receptors that detect effectors and launch a stronger response called effector triggered immunity (ETI).
(Jones and Dangl 2006)
The perception of PAMPS has been reviewed by many labs including our own (Greeff et
al. 2012). There are three PAMPs that have been well studied in the last few years. They
are flagellin, bacterial elongation factor-Tu (EF-Tu), and fungal chitin. Flagella are bacterial
organelles required for propulsion. A major component of this organelle is the protein
flagellin (FliC) (Macnab 2004). Flagellin is secreted by Pst and is perceived by flagellin
sensing 2 (FLS2), a leucine-rich repeat (LRR)-receptor kinase located in the plasma
membrane (Gómez-Gómez and Boller 2002). Upon perception of flagellin or a derived
peptide called flg22, FLS2 dimerizes (Sun et al. 2012) and hetero-dimerizes with a co-
receptor, called BAK1 (BRI1-associated kinase 1). BAK1 is a plasma membrane-bound
receptor-like kinase that is required for flg22 PAMP signaling (Chinchilla et al. 2007). BAK1
phosphorylates FLS2 as well as Botrytis-induced kinase 1 (BIK1) (Lu 2012). Upon BIK1
phosphorylation, its association with BAK1 is diminished and it is released to affect

13
downstream components. Flg22 perception leads to PTI consisting of rapid reactive oxygen
species (ROS) production, alkalinization of the apoplast, genetic reprogramming, callose
deposition and enhanced disease resistance (Nicaise, Roux, and Zipfel 2009) . There is
also an increase in cytosolic calcium (Ca²⁺)(Garcia-Brugger et al. 2006) and nuclear Ca²⁺
(Lecourieux, Ranjeva, and Pugin 2006) after PAMP perception that is an important
messenger for PTI responses. Similarly to flg22 and its receptor FLS2, another PAM, EF-
Tu is perceived by the EF-Tu receptor (EFR), which is also a BAK1-dependent pattern
recognition receptor (PRR) (Zipfel et al. 2006) (Roux et al. 2011). The final PAMP, fungal
chitin, is recognized by chitin elicitor receptor kinase 1 (CERK1) (Miya et al. 2007)(Wan et
al. 2008) and is BAK1-independent.
Effectors
If pathogens cannot overcome this first recognition or subsequent defensive mechanisms
they cannot effectively reproduce. Selective pressure is therefore placed upon pathogens
to overcome PTI (Jones and Dangl 2006). Successful pathogens have evolved effector
proteins that can disable PTI. For example, Pseudomonas syringae (Pst), Xanthomonas
and Ralstonia all have type III secretion systems (TTSS) used to inject effectors into host
cells. The importance of these effectors is illustrated by the observation that mutants of the
TTSS machinery often cannot overcome PTI and successfully infect plants (Hauck,
Thilmony, and He 2003). These effectors have many functions and can target specific
organelles to change host metabolism or disable defense responses. Examples of effector
activities are ubiquitin E3 ligase-like AvrPtoB (Duplan and Rivas 2014), transcription factors
like TAL effectors (Boch et al. 2009), phosphatases like HopAO1 (Underwood, Zhang, and
He 2007), proteases like AvrPphB (Qi et al. 2014, 1) or they can merely be PAMP
scavengers (Dean 2011).
Pst DC3000 injects some 30 effectors through the TTSS including AvrRpt2 and AvrRpm1,
which target and modify the host protein, RPM1 interacting protein 4 (RIN4). AvrRpt2
causes cleavage of RIN4 (Mackey et al. 2003) and AvrRpm1 leads to phosphorylation of
RIN4 (Mackey et al. 2002).

14
Other plant pathogens like oomycete Phytophthora and Hyaloperonospora have hundreds
of RXLR effectors. These oomycetes infect plants by forming specialized feeding structures
called haustoria (Voegele and Mendgen 2003). Unlike Type III effectors, it is thought that
these RXLR effectors can gain entry into host cells even in absence of pathogens (Tyler et
al. 2013). This suggests that host processes are required for the import of these cytoplasmic
effectors (Morgan and Kamoun 2007).
One specific example of this class of effector is the well-studied Avr3a. Avr3a is recognized
by potato resistance gene product R3a. Silencing Avr3a leads to decreased infectivity of
Phytophthora infestans, highlighting its importance in virulence. Avr3a interacts with and
stabilizes host ubiquitin E3 ligase CMPG1 leading to decreases degradation of CMPG1 and
its substrates. This results in increased susceptibility to Phytophthora infestans (Bos et al.
2010). This increased susceptibility is due to suppression of defense induced by another
Phytophthora infestans protein called INF1 (Wawra et al. 2012).
R proteins
Unperturbed effector action on plant cells would be devastating thus not surprisingly in an
evolutionary arms-race, plants have evolved to guard against disarming of pathogen
recognition and consequent defense responses. Even though the details of this secondary
defense layer remained obscure, it was long known that plant-pathogen interactions
function on a gene-for-gene basis (Flor 1971). Breeders used this knowledge to breed
disease resistance crops without knowing the real biochemical basis behind this resistance.
The presence of a gene in both pathogen and in plants is required for full immunity, an
avirulence (Avr) gene in the pathogen and a resistance (R) gene in plants. This was a
striking paradox that remained unresolved for a long time. Why would certain pathogen
strains carry a gene that makes them less effective in infecting plants? When Jeff Ellis
cloned the flax L6 resistance gene, the biochemical interpretation became much clearer.
This gene contained a nucleotide binding motif and a LRR domain and these two motifs
characterize almost all known resistance (R) genes known today and for this reason
referred to synonymously as NB-LRRs. Avr gene products are often effectors that are
recognized in plants by R proteins and this causes disease resistance that is also referred

15
to as effector triggered immunity (ETI) (Jones and Dangl 2006)(Ellis, Dodds, and Lawrence
2007). The pathogen thus retains the Avr gene product in certain isolates because it is
required to overcome PTI in plants lacking the appropriate NB-LRR.
Plants employ R proteins to guard against effectors. The A. thaliana Columbia (Col-0)
accession has around 150 R genes present in its genome (Meyers et al. 2003). The
encoded R proteins typically have a Toll/interleukin-1 (IL-1) receptor (TIR) or a coiled coil
(CC) N-terminal domain, a central nucleotide binding site domain (NBS) and a C-terminal
LRR domain (McHale et al. 2006). These structures are shown in Figure I2. The domains
of R proteins, not surprisingly, have unique roles in NB-LRR function as I will discuss below.
Figure I2 CC, TIR LRR and NB-ARC structures that are the building blocks of R proteins. (Frank L W Takken
and Goverse 2012)
NBS Domain
The NBS domain that is also called ‘nucleotide-binding adaptor shared by APAF-1, R
proteins, and CED-4’ (NB-ARC) is highly conserved in many species. APAF1 is a protein
that controls apoptosis in mammalian development and CED-4 is the C. elegans
counterpart (Cecconi et al. 1998). It contains several defined motifs typical of the ‘signal
transduction ATPases with numerous domains’ (STAND) family of ATPases (McHale et al.
2006). These motifs have various roles in R protein function. Mutations in the MHD domain
cause auto-activation, indicating a possible role in negative regulation of R protein function
(Bendahmane et al. 2002). The Walker B motif binds a water molecule facilitating
adenosine triphosphate (ATP) hydrolysis and the Walker-A or P-loop motif co-ordinates the
nucleotide β- and γ-phosphates and a Mg²⁺ ion (Bell 2005). Not surprisingly then the I2 and

16
Mi-1 NBS domains have been confirmed to be functional ATP-binding proteins with ATPase
activity (Tameling et al. 2002).
NBS domains are generally thought to act as molecular switches. In the case of NBS-LRR
proteins, ATP-bound is considered the ‘on’ state and adenosine diphosphate (ADP)-bound
the ‘off’ state. This is thought to be conserved throughout the STAND family (Danot et al.
2009). Hydrolysis of ATP returns the protein to the off state and is considered to be another
level of negative regulation (F L W Takken and Tameling 2009). This model is supported
by the fact that auto-active mutations in the MHD motif result in a higher affinity for ATP
(Williams et al. 2011) and P-loop mutants that are generally non-functional are known to
bind ATP with less affinity. Besides ATP hydrolysis, the LRR domain of NB-LRRs plays a
role in keeping the NBS domain in an inactive state (Moffett et al. 2002).
The TIR domain
The TIR domain found in both plants and animals consists of about 200 residues with three
conserved motifs (Slack et al. 2000). Most TIR-NSB-LRRs (TNLs) share a requirement for
enhanced disease susceptibility 1 (EDS1) for downstream signaling. This implies that the
TIR domain is involved in regulating downstream signaling interactions (Nicole Aarts et al.
1998, 1). Interestingly, expressing the TIR domains of R proteins, like L6 and RPS4, causes
cell death. This adds to speculation that the TIR domain confers downstream signaling
(Frank L W Takken and Goverse 2012). The L6 TIR domain is known to have homotypic
interaction and this self-association seems to be required for immune signaling (Bernoux et
al. 2011). Mutational analysis of the TMV resistance protein N revealed that its TIR domain
also engages in homotypic interactions (Mestre and Baulcombe 2006). This is apparently
a conserved feature since this is also true for animal TIR domain-containing Toll-like
receptors (TLRs) like TLR4 that recognizes lipopolysaccharides (Tapping 2009). The TIR
domain seems likely to function in downstream signaling, however some data have
suggested that it is also involved in determining ligand specificity. In L6 the TIR domain is
important for recognition specificity to different rust strains but this is not due to recognition
of its associated effectors but rather depends on a dominant rust inhibitor protein that
targets the TIR domain (Ellis, Dodds, and Lawrence 2007). The TIR domain of N however
was shown to be important for recognition of its ligand p50, a tobacco mosaic virus protein.

17
This is an illustration that TIRs can also have a function in determining binding specificity
(Burch-Smith et al. 2007a).
The CC domain
R proteins with CC N-terminal require the non-race-specific disease resistance (NDR1)
gene for downstream signaling (Nicole Aarts et al. 1998, 1). Studies with the CC-NBS-LRR
(CNL) resistance protein Rx showed that the CC domain interacts with the NBS-LRR
domain and this interaction is disrupted upon activation of Rx (Moffett et al. 2002). Complete
activation of Rx also requires the loss of interaction between the NBS-LRR domains and
the CC domain. P-loop mutants of the Rx NBS domain abolish the CC / NBS-LRR
interaction although this does not activate Rx and therefore further nucleotide exchange is
likely needed for activation. The CC domain appears to be a recognition domain since many
CNLs have been shown to interact with several different proteins through this domain. CC
domain interactions include RPM1 interaction with RIN4, PBS1 with RPS5 and Pto with Prf
(Rairdan et al. 2008). Recent crystal structure determination of Rx and its binding partner
RanGAP2 confirmed that the interaction is also through CC domain. However substituting
the CC domain of RPS5 with the RPS2 CC domain does not change the binding specificity
to PBS1, instead a full LRR was required, arguing against CC being mainly a recognition
domain (Qi, DeYoung, and Innes 2012). Expressing CC domains of certain CNLs, like
activated disease resistance 1 (ADR1) and N requirement gene 1 (NRG1), on their own
also causes a hyper sensitive response (HR) which favors the signaling function of these
domains (Collier, Hamel, and Moffett 2011). However these CNLs have ‘helper’ roles in that
they are required for signaling of other R proteins and cannot be considered an indication
of general function (Bonardi et al. 2011). The MLA10 CC domains form homotypic
interactions even in the inactive state. This interaction is required for MLA10 signaling
function. Expression of the MLA10 CC domain alone causes cell death in plants (Maekawa
et al. 2011). This again indicates a signaling function.
Defining general functions to TIR or CC domains is difficult due to conflicting information,
however they surely play a role in downstream signaling because they dictate downstream
signaling requirements. At this stage there is no solid general model for the recognition
behavior of all TNLs and CNLs and generalization should be considered with caution.

18
CC/TIR and LRR domains likely both contribute to recognition as will be clarified later with
the bait and switch model of NB-LRR function.
The LRR domain
LRR domains are versatile binding motifs found in thousands of proteins in diverse species
from viruses to eukaryotes (Bella et al. 2008). They play a key part in plant immunity since
they are found in PRRs like FLS2 (Dunning et al. 2007) and most R proteins (McHale et al.
2006). Solvent-exposed residues in the LRR domains of R proteins are hypervariable and
under positive selection. This indicates that this domain is a main platform for ligand
recognition. Positive selection of this domain is often found in host pathogen interactions
(Mondragón-Palomino et al. 2002)(Meyers et al. 1998). The domain is characterized by
LxxLxLxxN/CxL (where x can be any amino acid and L positions can also be occupied by
valine, isoleucine and phenylalanine). This motif typically results in horseshoe shapes or
more accurately a super helix (Kobe and Kajava 2001). The LRR domain is thought to have
a dual role in R protein function. The N-terminal part is thought to act in negative regulation
of the protein since mutantion in the N-terminal LRR domain of Rx causes slight auto-
activation in the absence of viral coat protein (CP) (Bendahmane et al. 2002)(Lukasik and
Takken 2009). The C-terminal part of the LRR domain is generally thought to facilitate target
recognition, based on domain-swapping experiments of highly similar R proteins (B. Zhou
et al. 2006)(Ellis, Dodds, and Lawrence 2007)(Shen et al. 2003).
Model of R Protein function
Although there are probably exceptions, the current working model of NBS-LRR ‘nibbler’
function is shown in Figure I3. Briefly, interactions with effectors cause changes in LRR
conformation, thereby releasing the NBS domain for nucleotide exchange. This exchange
causes further release of the LRR domain, and subsequent release of TIR/CC domain for
downstream signaling (F L W Takken and Tameling 2009). Further ATP cycling causes

19
the formation of an active signaling complex (Bonardi 2012)(Tameling et al. 2002)(F. L.
Takken, Albrecht, and Tameling 2006).
(F L W Takken and Tameling 2009)
Figure I3. General model of R protein signaling
General factors required for R protein signaling
As briefly mentioned above, EDS1 and NDR1 are required for TIR and CC signaling
respectively. The EDS1 signaling complex also includes Phytoalexin deficient 4 (PAD4)
and Senescence-associated gene 101 (SAG101). PAD4, EDS1 and SAG101 bear some
resemblance to lipases but so far, an enzymatic function has not been established but might
play a role in signaling. These three components form two different complexes in which
PAD4 and SAG101 show partially redundant functions (Wiermer, Feys, and Parker
2005)(Wagner et al. 2013). PAD4/EDS1/SAG101 complexes have been confirmed and
shown to be preferentially nuclear-localized, where the EDS1 nuclear localization is critical
for the signaling function. Cytoplasmic PAD4/EDS1/SAG101 complexes are not seen, only
PAD4/EDS1 complexes have been detected. Due to the spatial distribution of these distinct
complexes it appears that PAD4 and SAG101 might control EDS1 localization and this in
turn affects defense signaling activity. Overexpressing SAG101 results in predominant

20
nuclear EDS1 localization where PAD4 can disrupt this effect and retain some EDS1 in the
cytosol. It would therefore appear that changes in abundance of these three proteins during
a defense response are possibly responsible for regulating the EDS1 related signal. Much
of the functioning of this downstream signaling is unfortunately still unknown (S. Zhu et al.
2011). Some data show that some R proteins, like RPS4, directly interact with EDS1
(Heidrich et al. 2011) and the dynamics of this interaction may be required to produce a
mobile R protein signal required to activate defense (Bhattacharjee et al. 2011).
CNL signaling however is independent of EDS1 and instead requires non-race resistance
1 (NDR1), a membrane bound integrin-like protein. Compared to EDS1 no progress has
been made to elucidate how NDR1 effects downstream CNL signaling (Century, Holub, and
Staskawicz 1995)(Knepper, Savory, and Day 2011).
Intriguingly some R genes like RPP13 (Bittner-Eddy and Beynon 2001), RPP7 and RPP8
(McDowell et al. 2000) do not require either EDS1 or NDR1 for signaling. We see this clearly
in eds1 ndr1 double mutants that still show normal resistance in response to RPP13. These
R proteins also act independently from the salicylic acid pathway. This suggests that a third
signaling pathway exists in addition to EDS1 and NDR1.
Others factors are also important for the stability and accumulation of R proteins. Presence
of the guardee is often required for R protein accumulation, as in the case of RIN4 and
RPM1. In the absence of RIN4, RPM1 does not accumulate (Belkhadir et al. 2004).
Producing signaling competent NB-LRRs further requires additional factors. A suppressor
of G2 allele of skp1 (SGT1)/ Heat shock protein 90 (HSP90)/ RAR1 complex containing
highly conserved eukaryotic proteins is also required for accumulation and function of NB-
LRRs (Takahashi et al. 2003). SGT1 binds HSP90 and RAR1 (Azevedo et al. 2006) and
together these proteins form a chaperone complex that maintains the signal-competent
state of R proteins (Ken Shirasu 2009).
Controlling steady state levels of NB-LRRs is also important for avoiding inappropriately
activated responses. NB-LRRs have also been shown to be regulated at a post-
transcriptional level by phased siRNAs (phasiRNA). MicroRNA (miRNA) is generated from
hairpin secondary structures mostly from antisense transcripts by dicer-like proteins (DCL)
(Voinnet 2009). These miRNAs, in a complex with argonaute (AGO), lead to sequence-
specific cleavage of mRNA (Baumberger and Baulcombe 2005). The resulting cleaved

21
RNA is processed into small interfering RNA (Vazquez et al. 2004). Some of these products
are called phasiRNA and are trans-acting silencers of target transcripts levels (Fei, Xia, and
Meyers 2013). NB-LRR proteins are extensively targeted by these phasiRNAs and probably
serve as a means of fine control of NB-LRR protein levels (Zhai et al. 2011). The exact role
for these phasiRNAs is yet to be determined.
Models of effector recognition
Direct detection of effectors by R proteins was long considered as the default scenario and
in support of this, direct interaction between RRS1 and AvrPopP2 was reported by Laurent
and co-workers (Laurent Deslandes et al. 2003). However, it also became evident that
many R proteins did not interact with their cognate effectors, like RPM1 and AvrRPM1
(Mackey et al. 2002).
In light of this, a model was proposed that could explain why interaction between the Avr
gene product and R proteins is not more common. The guard model proposes that there is
an indirect detection of effectors by monitoring for a change in effector targets caused by
effectors (E. Van der Biezen and Jones 1998)(Dangl and Jones 2001). This would also
explain why structurally diverse effectors can be detected by a single R protein, like the
example of AvrB and AvrRpm1 that are detected by RPM1 (T. K. Eitas, Nimchuk, and Dangl
2008). A prime example of the guard hypothesis is RIN4, briefly mentioned above, which is
monitored by at least two R proteins, RPM1 and RPS2. RPM1 and RPS2 guard against
RIN4 modification brought about by AvrRPM1 and AvrRpt2 respectively. Multiple R proteins
can also guard against single effectors. As example, AvrB can target RIN4, leading to
activation of both RPM1 and TAO1 (T. K. Eitas, Nimchuk, and Dangl 2008, 1).
The guard model asserts that the target being guarded is important in terms of disease
resistance. This is however not always true, as for the kinase Pto guarded by the resistance
protein Prf. AvrPto is an effector that binds to FLS2 and EFR, thereby inhibiting their
function. AvrPto also targets Pto that is guarded by Prf and in the absence of Pto AvrPto
still contributes to virulence. This implies that Pto is a decoy for FLS2 and EFR rather than
an important immune component that is targeted (Xiang et al. 2008). Why would decoys be
beneficial? Interaction of an R protein with a defense signaling component may induce an

22
evolutionarily unstable situation since there would be two opposing selection forces. In the
absence of an R protein, the effector target would mutate to bind the effector with less
affinity. When an R protein is present, increased binding and detection of the effector would
be favored. It has been suggested that gene duplication might produce dedicated effector
decoys and a more stable situation. These decoys can mutate freely to better detect
effectors without the constraint of maintaining their function and interactions. The decoy
itself would possibly lose its original function in plant defense (Van Der Hoorn and Kamoun
2008).
The key feature to consider when discriminating between the above-mentioned decoy or
guard models is whether effector action on its target leads to an advantage for the
pathogen. When RIN4 is targeted by HopF2 it leads to increased disease susceptibility,
therefore RIN4 does not fit with this definition of the decoy model (Wilton et al. 2010).
Studies aimed to identify the recognition domain of NB-LRR proteins often showed
conflicting results or indicated that TIR and LRR domains can determine specificity. Since
both domains are sometimes required for Avr recognition, the bait-and-switch model was
proposed. Here, the CC/TIR domain interacts with the ‘bait’ target and the LRR of the NB-
LRR protein interacts with the effector (Collier and Moffett 2009). Illustration of this model
can be found for the tobacco resistance protein N. N-receptor-interacting protein (NRIP)
can bind to recognized versions of N inducing viral protein, p50. N also interacts with
recognized p50 as well as NRIP1 (Ueda, Yamaguchi, and Sano 2006)(Caplan et al. 2008).
In this way, N binds to NRIP1 with its CC domain and subsequent binding of p50 to NRIP
is detected through the interaction with the LRR domain. This second interaction would
trigger downstream signaling.
R Protein pairs
A very interesting development in recent years is the finding that some R proteins work as
pairs in the perception of effectors (T. Eitas and Dangl 2010). The first of such pairs were
RPP2A and RPP2B. Mutations in either one causes a loss of resistance to
Hyaloperonospora arabidopsidis (Hpa) Cala2 (Sinapidou et al. 2004). In rice, resistance
pairs RGA4/RGA5 are required for direct recognition of Avr1-CO39 (Okuyama et al. 2011).
Further, both the TNL N and the CNL NRG1 are required for TMV resistance (Peart et al.

23
2005). ADR1 together with NRG1 is required for resistance conferred by RPS2 and RPP4
(Bonardi et al. 2011).
Another example is the R protein pair RPS4 and RRS1 from the Arabidopsis Ws-0
accession. These R proteins have heterotypic interactions and both are required for the
recognition of AvrRps4 (Narusaka et al. 2009) and the Ralstonia effector PopP2 (Narusaka
et al. 2013a). RRS1 has also been shown to directly interact with PopP2. Not surprisingly,
these R-pairs are also genetically linked. RPS4 and RRS1 are in a head-to-head genomic
conformation and generally this feature indicates co-regulated genes (Y.-Y. Li et al. 2006).
Several R proteins have been shown to have a similar head-to-head conformation and may
also represent R gene pairs (Narusaka et al. 2009).
RRS1 is an unusual R protein in that it contains an additional N-terminal WRKY domain
(Heidrich et al. 2013, 1). WRKY domains are DNA-binding motifs found in the plant-specific
WRKY family of transcription factors involved in plant disease resistance (Fiil and Petersen
2011). A WRKY domain mutation of SLH1, an R protein identical to RRS1 from Arabidopsis
accession Nd-1, causes spontaneous lesions. This indicates that the WRKY domain might
be involved in negative regulation of defense activation. Since WRKY transcription factors
have been shown to associate with NB-LRRs, SLH1 might also be a decoy, mimicking
another unknown WRKY/NB-LRR complex (Noutoshi et al. 2005).
The most unusual potential R protein pair is At4g12010 and WRKY19. WRKY19 contains
in addition to TIR, NBS and LRR domains, an additional WRKY domain and also a mitogen-
activated protein kinase kinase kinase (MEKK) domain (Narusaka et al. 2009, 4).
Unfortunately nothing more is known about this gene pair, but WRKY19 might play a similar
decoy role to SLH1 and RRS1.
NB-LRR Interaction with transcription factors
Several families of transcription factors have been shown to be involved in defense gene
regulation. Two prominent well-studied families include the WRKY transcription factors
mentioned above and MYB transcription factors.
WRKY transcription factors are so named due to an almost invariant WRKY amino acid
sequence at their N-terminus (Eulgem and Somssich 2007). The first WRKY discovered

24
was SPF1 in sweet potato shown to be a novel sequence-specific DNA-binding protein
(Ishiguro and Nakamura 1994, 1). Since then, many WRKY genes have been identified and
74 WRKY genes are expressed in Arabidopsis, almost all of which bind the W-Box DNA
sequence (Chi et al. 2013). Among these, several have been shown to be important in
disease resistance. Loss of WRKY70 for example, makes plants more susceptible to Pst
as well as Botrytis cinerea (Knoth et al. 2007). WRKY33 has been shown to have a role in
resistance to B. cinerea since loss of WRKY33 leads to increased susceptibility to this
necrotrophic pathogen (Zheng et al. 2006). Other WRKY transcription factors have partially
redundant functions in regulating disease resistance. Single mutants of WRKY18, WRKY40
and WRKY60 show little change in resistance to Pst and B. cinerea but double mutants
wrky18 wrky40 and wrky18 wrky60 are significantly more susceptible to B. cinerea and
resistant to Pst. These WRKYs also exist in protein complexes in planta and their
interactions affect their binding affinity to DNA (X. Xu et al. 2006). Apart from interactions
among different WRKYs, several reports have also documented interaction of WRKYs with
signaling molecules like calmodulin, MAP kinases and 14-3-3 proteins and this gives an
idea of the complexity involved in these signaling networks but this will not be discussed in
detail here. Nevertheless, WRKYs are major players in regulating responses to biotrophic
or necrotrophic pathogens (Chi et al. 2013).
MYB transcription factors are characterized by varying numbers of MYB DNA-binding
motifs in the N-terminal region. 94 MYB transcription factors found in Arabidopsis (Stracke,
Werber, and Weisshaar 2001). These transcription factors have numerous roles in plant
signaling and development but are also involved in signaling connected to plant hormones
important for stress and defense responses namely salicylic acid (SA), jasmonic acid (JA)
and abscisic acid (ABA) (Lorenzo et al. 2004)(Raffaele, Rivas, and Roby 2006)(Ambawat
et al. 2013). MYB30 is one of the best-studied examples related to defense. This
transcription factor is a target of Xanthomonas effector XopD that disarms MYB30. MYB30
is thought to be a key player in establishment of HR since its overexpression leads to
increased HR and knockdown to a reduced HR response (Raffaele and Rivas 2013).
Mechanisms of R protein signaling downstream of EDS1 and NDR1 remain obscure but a
direct link to transcription factors has been shown for some NB-LRRs that points to a clear
path between R protein activation and defense gene induction. MLA10 binds two WRKY

25
transcription factors WRKY1 and WRKY2. WRKY1/2 are repressors of basal defense
(Shen, Saijo, Mauch, Biskup, Bieri, Keller, Seki, Ulker, et al. 2007). Barley MLA10 also
interacts with transcription factor MYB6 and MLA10 binding is needed to release MYB6
function from being antagonized by WRKY1 (Cheng Chang et al. 2013). In addition, the R
protein ‘suppressor of npr1-1, constitutive 1’ (SNC1) has also been shown to interact with
the transcription co-repressor Topless-related 1 (TPR1) (Z. Zhu et al. 2010) and loss of
TPR1 compromises SNC1-mediated immunity indicating that SNC1 functions through
TPR1. Lastly, N interaction with squamosa promoter-binding protein (SBP)-domain
transcription factor, SPL6, during an active immune response has been confirmed. P-loop
mutations in N that abolished N function also inhibit the SPL6 interaction (Padmanabhan
et al. 2013).
The Hypersensitive Response
HR is a rapid localized programmed cell death following infection and is characterized
mostly by morphological features (Mur et al. 2008). The presence of effector and cognate
R protein (M. Grant et al. 2000)(Peart et al. 2005), overexpression of R proteins (Stokes,
Kunkel, and Richards 2002) and expression of auto-active mutants all lead to formation of
the HR (Gao, Gao, et al. 2011). This illustrates that HR is a consequence of NB-LRR
activation. The consequences of HR include increased levels of several defense genes.
These transcriptional changes are distinctly different from developmental programmed cell
death (PCD) indicating that they are distinct processes (M. Kim et al. 2006). HR also
typically shows changes in location and production of signaling molecules like Ca²⁺ ion
fluxes (Yucel, Xiao, and Hutcheson 1989) and reactive oxygen species (ROS) production
(Zurbriggen, Carrillo, and Hajirezaei 2010). HR’s major consequences are cell death and
resistance to specifically recognized effector-carrying strains of pathogens.
Resistance and cell death however can be uncoupled. HR-related cell death appears to be
dispensable in gene-for-gene disease resistance. This is shown by certain mutants like
defense no death 1 (DND1) that can still exhibit Avr effector-dependent resistance but do
not display HR (Clough et al. 2000). Resistance conferred by Rx against potato virus X
(PVX) is also not dependent on HR (Bendahmane, Kanyuka, and Baulcombe 1999). These
results make the exact role of HR-related cell death in plant disease resistance unclear.

26
The prevailing hypothesis however is that localized cell death results in restricted moisture
and nutrients for pathogen growth (Lam, Kato, and Lawton 2001).
Many of the animal PCD components are not well conserved in plant cells so consequently
few parallels can be drawn (Q. Xu and Zhang 2009). In animal systems, several caspases
are important for cell death (Cardone et al. 1998) but in Arabidopsis no homologous
caspases are found. Instead metacaspases have been found to control HR cell death.
Metacaspase 1 (MC1) is a positive regulator of HR and metacaspase 2 is a negative
regulator of HR. Mc1 mutants do not show cell death but still show gene-for-gene resistance
(Coll et al. 2010). This is yet another example of uncoupling of HR cell death and resistance
responses.
Lastly, certain types of HR have been shown to be dependent on autophagy, a conserved
biological process that involves engulfment of cytoplasmic constituents into
autophagosomes that are then degraded. In brief, Hofius and co-workers have shown that
atg7 and atg9 mutants, that do not have functional autophagy, do not display a full HR in
response to Pst DC3000 (AvrRPS4) (Hofius et al. 2009). This indicates that autophagy is
required for the HR.
Hormone signaling in plant defense
Several plant hormones play key roles in regulating plant defense responses including the
well-studied SA which acts as a signal to activate several plant defense responses both
locally and systemically (Durner, Shah, and Klessig 1997). Some of the first experiments
with SA proved that application of SA to tobacco induces pathogen related (PR) gene
expression and enhances resistance to pathogens (Malamy et al. 1990). In line with this,
Arabidopsis mutants with defects in SA biosynthesis genes like isochorismate synthase 1
(ics1) display reduced PR1 gene expression upon infection and are more susceptible to
certain pathogens (Wildermuth et al. 2001)(Garcion et al. 2008)(Spoel, Johnson, and Dong
2007). Similarly, expression of the Pseudomonas putida NahG gene, encoding an SA
hydroxylase, which degrades SA, results in increased disease susceptibility, and abolished
PR1 gene expression that indicates a role for SA in resistance (Delaney et al. 1994).

27
SA responses additionally require the non-expresser of PR genes 1 (NPR1) transcription
co-factor for defense gene activation and NPR1 was shown to be a SA receptor as it binds
to SA (Y. Wu et al. 2012). Redox changes in the cell due to increased SA result in formation
of monomers of NPR1. This in turn leads to NPR1 localization to the nucleus where NPR1
interacts with other transcription factors effecting changes in defense gene expression
(Lindermayr et al. 2010). Recently NPR3 and 4, Cullin 3 (CUL3) E3 ligase adapters, have
been shown to bind SA and this in turn inhibits degradation of NPR1. This mechanism is
thought to control spontaneous defense activation (Fu et al. 2012).
SA’s role in defense seems to be to potentiate defense responses since application of SA
at physiological levels had little effect other than defense gene induction. However
subsequent challenges by pathogens induced much stronger responses in the presence of
SA (K. Shirasu et al. 1997). SA is also required for the development of systemic acquired
resistance (SAR), a systemic long-lived increase in resistance in distal tissue (Durrant and
Dong 2004).
SA-dependent pathways play a major role in defense against biotrophic pathogens like Pst
(Oirdi et al. 2011). Conversely, alterations that inhibit SA-dependent responses such as
mutations in NPR1 or expression of NahG often have little effect on resistance to
necrotrophic pathogens like B. cinerea (Glazebrook 2005)(Ferrari et al. 2003). Infection with
B. cinerea causes increased expression of PDF1.2 (Manners et al. 1998), which encodes
an antifungal defensin-like peptide used as a defense marker for necrotrophic infections,
akin to PR1. Mutations like ethylene insensitive 2 (ein2) and coronatine-insensitive protein
1 (coi1) which affect ethylene (ET) and jasmonic acid (JA) signaling pathways respectively,
result in increased susceptibility to Botrytis and failure to induce PDF1.2 (Manners et al.
1998)(Thomma et al. 1999a) (Thomma et al. 1998). It is generally thought that resistance
to biotrophs requires SA-dependent pathways and resistance to necrotrophs require JA/ET-
dependent pathways (Glazebrook 2005). In line with this general evaluation, JA also
antagonizes SA-mediated responses and vice versa (Oirdi et al. 2011). This is illustrated
by Pst DC3000 secreting the phytotoxin coronatine, that mimics action of JA and
suppresses SA-mediated defenses, thereby allowing it to infect host plants (Youfu Zhao et
al. 2003). These two hormones therefore play a role in modulating appropriate defense
responses depending on the class of pathogen (Thaler, Humphrey, and Whiteman 2012).

28
Knowledge of JA signaling has expanded rapidly in recent years. COI1 is an F-box protein,
a class of proteins that are receptors enabling the recruitment of regulatory proteins as
substrates for ubiquitin-mediated destruction in the proteasome (Xie et al. 1998). The E3
ubiquitin ligase known as the SCF complex consisting of S-phase-kinase-associated
protein-1 (SKP1), cullin and RING-finger protein (Rbx) interacts with the COI1 F box protein
(C. Bai et al. 1996). All these components forms a SCFCOI1 complex that acts as the JA
receptor and control protein degradation in response to JA (Devoto et al. 2002). The targets
of these complexes were found to be jasmonate ZIM-domain (JAZ) proteins. One of these
JAZ proteins is JASMONATE-INSENSITIVE 3 (JAI3). JAI3 inhibits the function of MYC2 a
key transcriptional activator of JA responsive genes. When JAI3 is degraded, MYC2 can
function unhindered (Chini et al. 2007).
ET signaling has also been well studied. Many ET receptors are present in the endoplasmic
reticulum membrane. They are ET receptor 1 (ETR1), ethylene response sensor 1 (ERS1),
ETR2, ERS2, and ethylene insensitive 4 (EIN4) (Jian Hua et al. 1998, 2)(J Hua et al.
1995)(Merchante, Alonso, and Stepanova 2013). The CTR1 protein kinase phosphorylation
of EIN2 is inhibited by ET perception and causes the C-terminal end of EIN2 to translocate
to the nucleus (Ju et al. 2012, 1). The EIN2 C-terminus stabilizes EIN3/ EIN3-like
proteins(EILs) in the nucleus, resulting in transcriptional responses to ethylene (Qiao et al.
2012).
ET fine-tunes appropriate defense responses by inhibiting JA-mediated defense
suppression by SA (Leon-Reyes et al. 2010) adding another level of control. Ein2 mutants
are not compromised in resistance to Pst DC3000 (AvrRpm1/AvrRpt2/AvrB) (Bent et al.
1992) although they are slightly more resistant to Pst DC3000 (Boutrot et al. 2010) .
However, ein2 is more susceptible to B. cinerea (Thomma et al. 1999a). Furthermore, EIN2
is also required for flagellin perception by FLS2, indicating ethylene plays a role in PTI
(Boutrot et al. 2010).
This is a simplistic interpretation of available data but interplay between SA, JA and ET is
undoubtedly far more complex especially considering other plant hormones, like ABA, also
seem to be involved in modulating immune responses (Adie et al. 2007).

29
Autoimmune mutants
Since erroneous activation of HR is detrimental to the host it makes sense that there should
be tight regulation of HR. We already presented many genetic features and processes
involved in maintaining a signal-competent state of NB-LRRs while avoiding auto-
activation. A collection of mutants have been uncovered that show spontaneous HR in the
absence of pathogens, or display runaway cell death upon HR induction. The simplest
explanation of these mutants is that they represent negative HR regulators and many are
described as such (Galon et al. 2008)(J T Greenberg, Silverman, and Liang 2000)(S. Yang
et al. 2006). However this interpretation could in some cases be misdirected. For instance,
some mutations may also cause perturbations resembling effector action thereby triggering
NB-LRR activation and leading to the observed spontaneous HR.
Autoimmune mutants typically show common phenotypes. These include elevated defense
gene expression, such as PR1 and PR2, increased salicylic acid (SA) levels, increased
ROS levels, dependence on either EDS1/PAD4 or NDR1, stunted growth and necrotic or
chlorotic lesions (Lorrain et al. 2003)(Moeder and Yoshioka 2008).
We know the underlying causes for several autoimmune mutants. These include
uncontrolled ROS production, imbalances in ceramide metabolism, disrupted Ca²⁺
signaling and disrupted MAPK signaling that can all lead to autoimmunity. These examples
will be discussed separately below.
The first autoimmune mutant is the well-characterized lesion stimulation disease 1 (lsd1)
(Dietrich et al. 1994). It is classified as a propagation lesion mimic mutant. This means that
lsd1 plants develop normally but application of SA or pathogen challenge leads to a
runaway cell death. It is known that lsd1 runaway cell death requires a functional ADR1 (a
helper R gene) and metacaspase 1 (MC1) but these factors act downstream of other
autoimmune mutants and do not explain the phenotype. LSD1 is known to interact with
CATALASE3 (CAT3) that can catalyze the conversion of hydrogen peroxide (H2O2) to water
(Mhamdi et al. 2010). Additionally it is known that H2O2 is an important signaling molecule
for HR, since application of H2O2 induces HR (Levine et al. 1994). The LSD1-CAT3
interaction suggests that uncontrolled ROS production resulting in H2O2 is the underlying
reason for the runaway cell death in lsd1 mutants. Therefore, LSD1 and CAT3 are important

30
in preventing the spread of HR by reducing levels of H2O2 after defense induction (Yansha
Li et al. 2013, 3).
Autoimmune mutants also illustrate that sphingolipid metabolism is an important aspect in
establishment of HR. Accelerated cell death 5 (ACD5) is a ceramide kinase and loss of
ACD5 causes typical autoimmune phenotypes (J T Greenberg, Silverman, and Liang 2000,
5). C2 ceramides are known to cause HR and in their phosphorylated form partially block
HR. In acd5, the sphingolipid profile is altered towards un-phosphorylated C2 ceramides,
explaining the resulting autoimmunity (Liang et al. 2003). Further illustration of the
importance of ceramides during HR is that the fumonisin toxin is known to induce PCD by
disruption of sphingolipid metabolism (Asai et al. 2000).
Another sphingolipid-related autoimmune mutant is Accelerated cell death 11 (acd11),
mutated in a gene that encodes a sphingolipid transfer protein. In acd11 severe
autoimmunity can be seen (Brodersen et al. 2002). In contrast to acd5 this mutant has been
shown to rely on the LAZ5 TNL for runaway HR and related phenotypes. Dominant P-loop
mutants of LAZ5 completely suppress all acd11 phenotypes where laz5 T-DNA insertion
mutants show only partial suppression. This suggests that activation of other R proteins
might contribute to the acd11 phenotype. Complete suppression of acd11 is also obtained
in acd11 sdg8 double mutants, since LAZ5 expression is epigenetically controlled by the
SET domain group 8 (SDG8) histone H3 methyltransferase (Berr et al. 2010a). The
mechanism by which the loss of ACD11 triggers LAZ5 is not known, but cellular
perturbations caused by the loss of ACD11 may resemble those caused by pathogen attack
that is monitored by LAZ5 (Palma et al. 2010).
Autoimmunity can also be caused by defects in calcium signaling, known to be important
for HR. Two spikes in calcium occur during HR following inoculation with effector-secreting
Pst strains, prior to cell collapse. An initial burst at around ten minutes and a second
sustained elevated level of cyt[Ca²⁺] (M. Grant et al. 2000). It is also known that calcium
channel blockers such as lanthanum ions can inhibit HR (Atkinson et al. 1990). In line with
this, Defense no death (DND) 1 and DND2 encode nucleotide-gated calcium ion channels
whose absence results in autoimmunity (Clough et al. 2000). DND1 and DND2 play a role
in regulating cytosolic calcium influx (Leng et al. 1999). Therefore its been suggested that
the inability of dnd mutants to change the level of cyt[Ca²⁺] leads to a defective HR.

31
Intriguingly, dnd mutants produce no HR but retain gene-for-gene resistance (Jurkowski et
al. 2004) indicating that calcium signaling is dispensable for gene-for-gene resistance.
The next autoimmune mutant shows that disrupting guarded immune components can
trigger HR. An important immunity-related mitogen-activated protein (MAP) kinase cascade
consists of the upstream MAP kinase kinase kinase (MEKK) MEKK1 that activates the MAP
kinase kinase (MKK) MKK1/2, that leads to activation of the MAP kinase MPK4. The
activation of this cascade by PAMPs leads to ROS production and SA accumulation
(Pitzschke et al. 2009). Loss of MEKK1 (Ichimura et al. 2006) and MPK4 (Petersen et al.
2000) also results in autoimmunity. Autoimmunity of mekk1, mkk1/2 and mpk4 can be
suppressed by loss of MEKK2, also called suppressor of mkk1/2 (SUMM1). Autoimmunity
from overexpression of MEKK2 in turn can be suppressed by mutation in SUMM2, a TNL
resistance protein. Disruption of this kinase cascade therefore ultimately leads to HR by
triggering SUMM2 (Kong et al. 2012). Further, the effector HopAI targets MPK4 and
activates SUMM2. Here we have a striking example of the guard model since it is clear that
SUMM2 is guarding this kinase cascade against HopAI. This is not surprising since this
cascade is important for PTI (Z. Zhang et al. 2012, 2). The autoimmune phenotype of
mutants in this MAP kinase pathway illustrates that autoimmunity resulting from loss of key
signaling components could indicate that these are guarded by the immune system.
In addition to uncovering NB-LRRs as the underlying cause of some autoimmune mutants,
some autoimmune mutants are due to perturbations in NB-LRRs themselves. For example,
snc1-1 is a gain of function mutation that constitutively activates the resistance protein
SNC1 leading to HR (Yuelin Zhang et al. 2003). Similarly, the autoimmune mutant,
suppressor of salicylic acid insensitive 4 (ssi4) is a gain of function autoimmune mutant in
the NBS domain of the TNL SSI4 protein rendering it auto-active (Shirano et al. 2002).
Accumulation of R proteins can also cause autoimmunity. The F-box protein CRP1
regulates levels of SNC1 and when CRP1 is lost, elevated SNC1 levels cause
autoimmunity (Gou et al. 2012). These findings are hardly surprising since autoimmunity
and NB-LRR activation are mostly indistinguishable.

32
The CAMTA3 autoimmune mutant
Intracellular calcium levels are thousands of times lower than apoplastic levels. This
differential is exploited by cells as a means of signal transduction (Lecourieux, Ranjeva,
and Pugin 2006). Cytoplasmic Ca²⁺ concentration (Cyt[Ca²⁺]) influx can be triggered by
many factors including pathogen challenge. Pst DC3000 (AvrRpm1) elicits an initial spike
around 10 minutes after infiltration, followed by a sustained rise in cyt[Ca²⁺] levels. This rise
is important for development of HR since lanthanum, a calcium channel blocker, causes
diminished H2O2 accumulation and loss of HR (M. Grant et al. 2000). Expressing effectors
inside plant cells through inducible promoters also results in a similar cyt[Ca²⁺ ] increases
(Ranf et al. 2011).
This increase in cyt[Ca²⁺ ] is likely to be decoded by calcium binding proteins. One such
protein is the messenger calcium binding protein, calmodulin (CaM) (Toutenhoofd and
Strehler 2000). The key structural feature of this calcium binding protein is the EF-hand
motif that is responsible for calcium binding (Lewit-Bentley and Réty 2000). Upon binding
of calcium to CaM, a conformational change occurs that changes its interaction with other
proteins thereby modulating their activity (Snedden and Fromm 1998).
CaM interacts with a novel family of CaM-binding transcription activators (CAMTAs)
identified in multicellular organisms. This finding is exciting since it provides a direct line
between Ca²⁺ signaling and resulting transcriptional changes. The CAMTA family in
Arabidopsis consists of 6 members, CAMTA1-6 (Figure I4). Members of this family have a
unique CG-1 DNA binding domain (T. Yang and Poovaiah 2002), varying numbers of IQ
calmodulin binding domains, ankyrin repeats and a transcription factor immunoglobulin
(TIG) domain (Bouché et al. 2002).
Figure I4 Phylogenic Tree of the CAMTA family in Arabidopsis.
CAMTA3 mutants show spontaneous lesions at 4-weeks of growth and have elevated SA
levels and defense related gene expression (Galon et al. 2008). CAMTA3 is therefore

33
described as a negative regulator of defense gene expression.Camta1/3 and camta2/3
mutants have even higher SA levels and growth inhibition. Microarray data lead the same
authors to conclude that CAMTA1, CAMTA2 and CAMTA3 act largely redundantly to control
gene expression (Y. Kim et al. 2013). In some instances however CAMTA3 has been shown
to act as a positive regulator. CAMTAs have been shown to be involved in cold responses.
Increased cyt[Ca²⁺ ] is an early response to cold treatment, ultimately leading to increased
expression of cold-related genes causing cold resistance and freezing tolerance in
Arabidopsis (Knight, Trewavas, and Knight 1996). CAMTA1 and CAMTA3 have been
implicated in regulating cold expression genes and freezing tolerance, as well as positive
regulators of a cold responsive gene, ‘DREB subfamily A-1 of ERF/AP2 transcription factor’
(CBF2) (Doherty et al. 2009).
Apart from cold signaling and defense, CAMTA3 is also involved in ethylene signaling.
Firstly CAMTA3 expression is up-regulated by ethylene treatment (Laluk et al. 2012). The
link to CAMTA3 is not that surprising, as ethylene signaling additionally requires intact
calcium signaling. In line with this, blocking calcium signaling results in loss of ethylene-
induced genetic changes (Raz and Fluhr 1992). CAMTA3 has also been shown to directly
bind to the EIN3 promoter in a chromatin immunoprecipitation (ChIP) assay (Nie et al.
2012). Due to these findings, it seems that CAMTA3 might play a role in controlling genetic
changes in response to ethylene perception.
EDS1 gene regulation by CAMTA3 has been particularly well-studied. CAMTA3 has been
shown to bind the EDS1 promoter and that mutation of its CG-1 box causes loss of EDS1
promoter binding (Du et al. 2009). Furthermore, chromatin immunoprecipitation (ChIP) with
CAMTA3 also showed enrichment for EDS1 (Nie et al. 2012, 1). Also in line with this data,
in camta3, the EDS1 promoter-luciferase (LUC) construct shows enhanced LUC
expression. The results above therefore suggest a role for CAMTA3 in the negative
transcriptional regulation of EDS1. Additional support for negative regulatory function is
given by the following. CaM binding to CAMTA3 is Ca²⁺-dependent (Nie et al. 2012, 1) and
therefore CAMTA3 binding to CG-1 promoter element is also dependent on Ca²⁺/CaM (Du
et al. 2009). Further it was shown that Ca²⁺/CaM binding to CAMTA3 is required for its

34
transcription inhibition function. Increased cyt[Ca²⁺ ] and nuclear[Ca²⁺] during a defense
and consequent suppression of defense activation can only be explained as a mechanism
of negative feedback of defense responses. This seems to be the case, since a recent
report indicated that pathogen challenge by Pst DC3000 (avrRPS4) leads to degradation
of CAMTA3 through cullin 3-based E3 ubiquitin ligase with SR1IP1 as a substrate adapter.
Thus, CAMTA3 removal is required for subsequent ETI (L. Zhang et al. 2014).
The remaining support for the negative regulation is that overexpression of CAMTA3 and a
dominant mutation camta3-3D causes increased disease susceptibility (Jing et al. 2011).
All this provides quite a strong case for the negative regulatory role of CAMTA3 in plant
immunity.
However, camta3 autoimmunity can be suppressed by high temperature and pad4. This
implicates R protein involvement in the phenotype of this mutant since PAD4 is a known
downstream signaling component of TNLs (Du et al. 2009) and it was recently shown that
high temperature suppresses ETI-related responses (Cheng et al., 2013). If NB-LRRs are
indeed activated by the loss of CAMTA3, many of the reported phenotypes could be
attributed to this activation and not the negative regulatory function of CAMTA3. To
discriminate between camta3-1 phenotypes that are related to negative regulation by
CAMTA3 and those that follow with NB-LRR activation, we would first need to determine
whether NB-LRRs are activated, and identify them.
Aim of our work
The guard model leads to the prediction that certain mutations might mimic the action of
effectors, leading to autoimmunity. For example loss of RIN4 is lethal because it mimics
effector action of AvrRpt2. Rin4 rps2 mutants are viable because the R protein detecting
the faux effector action is absent (Belkhadir et al., 2004). A major aim of this work was to
conduct a novel, directed, dominant-negative screen to determine if certain autoimmune
phenotypes are indeed caused by the activation of an NB-LRR. Autoimmune mutants that
share dependencies of NB-LRR signaling like requirements for EDS1 or NDR1 and
temperature sensitivity are prime candidates for this screen. Since autoimmune phenotypes
could be due to activation of R proteins, triggered by disruption of a guarded defense

35
component, suppressors would allow us to evaluate the function of the guardee in the
absence of induced autoimmunity.
In a larger scope, transferring R proteins between different plant families often leads to
necrosis or non-functionality due to incompatibility of R protein and guardees or missing
signaling partners. This is referred to as restricted taxonomic functionality (Narusaka et al.
2013b). Through connecting R proteins to guardees we could in future facilitate transfer of
R proteins between families by also transferring appropriate decoys, guardees or signaling
partners (Narusaka et al. 2013a). A thorough knowledge of the function and interactions of
specific R proteins is therefore required.

36
Results
Chapter 1. acd11 suppressors
A CC-NBS-LRR contributes to acd11 autoimmune phenotype
In 2010, our lab reported multiple dominant alleles of an TIR-NBS-LRR gene, LAZ5, that
was capable of suppressing acd11 autoimmunity (Palma et al. 2010). The strongest
suppressor contained a mutation in the P-loop in the NBS domain and was named laz5-
D2. Interestingly, laz5-D2 suppresses acd11 cell death much more sufficiently than laz5
null-mutants. The phenotypic rescue can be seen in Figure 1A in plants grown under short-
day (SD) light conditions (Figure 1a). It is not unusual for dominant-negative (DN) mutants
like laz5-D2 to have superior inhibitory function over knockout lines. Factors that are known
to require dimerization for activity are rendered inactive by forming complexes with inactive
dominant negative proteins (Veitia 2007). Indeed, several R proteins are known to form
homotypic interactions, such as N, RPS5, MLA, L6 and Prf (Gutierrez et al. 2010)(Mestre
and Baulcombe 2006)(Bernoux et al. 2011)(Maekawa et al. 2011)(Ade et al. 2007). It is
also possible that a DN NB-LRR and other NB-LRRs could share a binding site on a
gaurdee that is mutually exclusive, DN binding thereby preventing signaling competence or
activation of a second R protein. Given the the dominant nature of laz5-D2 and partial
suppression of acd11 by the laz5 null-mutant, NB-LRRs other than LAZ5 could contribute
to acd11 phenotypes. This is not unusual since many defense components can be guarded
by multiple R genes. RIN4 for example is guarded by RPM1, RPS2 and TAO1 (Belkhadir
et al. 2004). acd11 or a closely associated protein may be similarly guarded by multiple R
proteins.

37
Figure 1A. Partial suppression of acd11 phenotype in laz5-1 knockout mutants. (Palma et al. 2010)
NDR1 is a critical signalling component downstream of CNL R proteins (N Aarts et al. 1998,
1). To test whether any CNLs might also contribute to acd11 cell death, we crossed acd11
laz5-1 to ndr1 and saw complete suppression of the acd11 laz5-1 phenotype (Figure 1B).
This therefore strongly suggests that a CNL could be responsible for the remaining cell
death and autoimmunity observed in acd11 laz5-1 double mutants.
Figure 1B. 4-week-old, short-day, soil-grown plants. acd11 laz5-1 (far right) retains conditional cell death
but had a less severe phenotype compared to acd11. acd11 laz5-1 ndr1 triple mutants (second from right)
however show complete suppression of the acd11 phenotype, similar to WT Col-0 (far left).
We could narrow down the possible CNLs by employing another suppressor of acd11
uncovered in the same suppressor screen, LAZ2 (Palma et al. 2010). LAZ2 was mapped
to Set Domain Group 8 (SDG8). Generally histone H3 lysine 4 (H3K4-) and H3K36-
methylation is associated with active transcription, where H3K27-methylation is associated
with silenced genes. Establishment of these marks depends in part on the SET domain-
containing proteins. Sdg8 knock-out plants show decreased H3K36 methylation in turn

38
associated with decreased transcription (Berr et al. 2010b). Since acd11 sdg8-2 (figure1B)
and acd11 laz5-1 ndr1 (figure 1A) both showed complete acd11 suppression, we
hypothesized that an SDG8 -dependant CNL (Table1a) might be the additional R protein
contributing to the acd11 phenotype.
Figure 1C. sdg8 completely reverts the acd11 autoimmune phenotype (Palma et al. 2010).
Locus Domains Name
At5g44870 TIR-NBS-LRR LAZ5
At1g56510 TIR-NBS-LRR
At1g57650 TIR-NBS-LRR
At5g46270 TIR-NBS-LRR
At5g45000 TIR-TIR-NBS-LRR
At1g72980 TIR-NBS
At2g32140 TIR-X
At3g07040 CC-NBS-LRR
At1g12220 CC-NBS-LRR RPM1
At4g14610 CC-NBS-LRR RPS5
At3g14470 NBS-LRR
Table 1A. TIR-NBS-LRR genes under transcriptional control of SDG8.
Since mutations in the P-loops of resistance proteins are documented to be dominant
negative like laz5-D2 (Palma et al. 2010) and N P-loop mutants (Dinesh-Kumar, Tham, and
Baker 2000) we could use these mutants to test the involvement of specific CNLs in acd11
autoimmunity. Dominant-negative mutants are polypeptides that can disrupt the function of
wild-type (WT) genes when overexpressed (Herskowitz 1987). We therefore cloned
dominant-negative P-loop versions of 4 CC-NBS-LRRs known to be under the control of

39
SDG8. USER FUSION was used to generate GXXXIGKTT to GXXX(N)GKTT P-loop
mutations in At3g07040, RPM1, RPS5 and At3g14470. We subsequently transformed
acd11/ACD11 as well as acd11 laz5-1 with over-expression constructs and selected for
transformed plants for resistance to the herbicide BASTA. Since the segregation of
acd11/ACD11 would yield WT plants with no experimental value to us, resistant plants were
first genotyped to confirm the presence of the acd11 T-DNA insertion. We then evaluated
if any one of these CNLs could be the additional CNL involved in the acd11 phenotype and
therefore be able to supress acd11.
Four independent acd11/ACD11/35S:CNL-D T2 lines were grown in short day conditions
to see if similar suppression to acd11 laz5-1 ndr1 could be observed. None of the lines
screened showed any degree of suppression. We saw normal 1:4 segregation ratios for the
acd11 phenotype as expected for offspring from a heterozygous acd11 parent. Any
deviation from this segregation ratio could have indicated suppression.
Since there is a chance that these dominant P-loop mutants might not be able to overcome
LAZ5 activation, we also transformed acd11 laz5 with the same 4 P-loop mutant constructs.
None of these transformants gave suppression of the acd11 laz5 phenotype.
Since the 4 selected candidates P-loop mutants did not show any suppression, they are
unlikely to be activated by loss of acd11. To uncover the activated CNL, acd11 laz5 will be
included in a larger P-loop screen that is explained in the next chapter. There might be an
additional CNL under control of SDG8 that is unknown to us that contributes to acd11
phenotypes.

40
Chapter 2. Validating P-loop mutants dominant negative effects
RPM1 P-loop mutants are specific and dominant negative.
The central domain of plant resistance genes, the NB-ARC or NBS domain, is involved in
ATP binding and hydrolysis (E. A. van der Biezen and Jones 1998). This ATP binding and
hydrolysis is critical for functioning of this domain as a molecular switch (Tameling et al.
2002).
The P-loop motif, GXXXXGKT(T/S) (Walker et al. 1982), in the NBS domain of resistance
proteins is well conserved in all signal transduction ATPases with numerous domains
(STAND) family members and important in coordinating binding of ATP beta, gamma
phosphates and the catalytic Mg²⁺ ion (Yan et al. 2005). Mutations in this motif, especially
the GK residues, greatly reduce ATP-binding capacity and always lead to loss of function
in this family (Tameling et al. 2002).
For instance, N (Dinesh-Kumar, Tham, and Baker 2000), M (Williams et al. 2011), Rx
(Bendahmane et al. 2002), NRG1 (Peart et al. 2005) and L6 (Dodds et al. 2006) R proteins
require intact P-loops for their function. The functional significance of P-loops also extends
to other members of the STAND family. CED4 requires an intact P-loop for its function
(Kanuka et al. 1999) as does APAF1 (Hu et al. 1999). The remarkable functional and
structural conservation of this motif highlights its importance in the functioning of these
ATPases as molecular switches.
In addition, mutations of the P-loop domain of N (Dinesh-Kumar, Tham, and Baker 2000),
LAZ5 (Palma et al. 2010) and ADR1 (Roberts et al. 2013) are dominant negative (DN).
Indeed remarkable conservation of NBS structure and function is apparent when
considering the examples of Saccharomyces cerevisiae Ras P-loop mutants (Modzelewska
et al. 2007), Gα subunit P-loop mutants (Bosch et al. 2012) as well as mutS P-loop mutants
from Escherichia coli (T. H. Wu and Marinus 1994) that are also dominant negative. This is
an important feature and the central concept in this work. If DN effects of P-loop mutations
extend to most STAND family members, it could be a useful tool for dominant-negative
screens by selectively inhibiting WT R protein function with P-loop constructs.
Given common downstream signalling components of NB-LRRs and conservation of the P-
loop in the NBS domain of NBS-LRR resistance proteins, we are confident that our

41
extrapolation of the DN effect of P-loops to a large collection of NB-LRR R proteins is
reasonable.
To test the utility of P-loop mutants we conducted a pilot study on the well-studied R protein
RPM1. RPM1 is known to monitor RIN4 for changes in phosphorylation brought about by
effectors including AvrRpm1 (Belkhadir et al. 2004, 1). We created P-loop constructs of
RPM1 by USER Fusion into pLIFE a binary 35S plant expression vector. After
transformation of Col-0 by floral dip, BASTA-resistant lines were selected. To obtain a
homogenous test population we identified T3 lines homozygous for the mutant construct by
selecting lines that showed no segregation of BASTA herbicide resistance.
We first examined whether Pst DC3000 (AvrRpm1), when syringe infiltrated, could trigger
HR in RPM1-D lines by measuring ion leakage. Increased conductivity correlates well with
progression of HR since membrane integrity is lost during ETI and conductivity can be
measured in water with the infiltrated leaf disks (Goodman 1972)(Mackey et al. 2003).
When infiltrating Arabidopsis Col-0 leaf discs with Pst DC3000 (AvrRpm1), AvrRpm1
triggered a rapid rise in ion leakage that plateaued after 6 - 7 hours in Col-0, as expected
(Gao, et al. 2011). However the rpm1-3 and two independent RPM1-D lines did not show
this increase and exhibited similarly levels of low conductance (Figure 1A). AvrRpm1 failed
to induce HR in the RPM1-D lines even though WT RPM1 NB-LRR is still present,
confirming that RPM1-D indeed functions as dominant negative.

42
Figure 2A. Ion leakage profiles of Col-0, rpm1-3, RPM1-D1 and RPM1-D2 lines showing that RPM1
function in RPM1-D lines is compromised to rpm1-3 knockout levels. ANOVA Col-0 vs rpm1, RPM1-D1 and
RPM1-D2 (p<0.01)
HR is clearly compromised in RPM1-D lines in response to AvrRpm1. However, resistance
to pathogens is not always directly related to HR (Coll et al. 2010) (Clough et al. 2000, 1).
Therefore we further wanted to assess whether gene-for-gene resistance conferred by
RPM1 is compromised in the RPM1-D lines. Two this end, two independent lines with 35S
RPM1-D were assayed for resistance to Pst DC3000 (avrRpm1) infection. Plants lacking
RPM1 typically have 2 log fold higher bacterial growth of this specific strain of DC3000 than
wild-type Col-0 (Belkhadir et al. 2004). Bacterial growth assays of Pst DC3000 (AvrRpm1)
were performed on Col-0, rpm1-3, RPM1-D1 and RPM1-D2. While Col-0 only supported
minor growth of the bacteria, RPM1-D lines had the same level of high bacterial growth
seen in rpm1-3 (Figure 2B). This confirms that the RPM1-D constructs are dominant-
negative, inhibiting WT RPM1 gene-for-gene resistance.
0
20
40
60
80
100
120
1 2 3 4 5 6 7
µS
/cm
⁻¹
H.P.I.
Pst DC3000(avrRpm1) induced Ion Leakage
Col-0
rpm1-3
RPM1-D1
RPM1-D2

43
Figure 2B. Bacterial growth at 0 and 3 days after infection of Col-0, rpm1-3, RPM1-D1 and RPM1-D2 with
Pst DC3000 (AvrRpm1). In RPM1-D lines, resistance to Pst DC3000 (avrRpm1) is compromised. ANOVA of
Col vs rpm1-3, Col vs RPM1-D1 and Col vs RPM1-D2 shows p<0.01.
P-loop dominant negative specificity
We find that P-loop mutants are dominant-negative and, to our knowledge, there are
currently no documented exceptions. Thus, we expect that P-loop mutants can be
employed in a screen to uncover specific R-protein associated processes. Very importantly
for this purpose, the P-loop mutant must inhibit the WT NB-LRR with reasonable specificity.
To establish if this is true, we tested additional strains of Pst, secreting different effectors
that rely on NB-LRRs other than RPM1. Two strains were selected for this purpose. The
first Pst DC3000 (AvrRpt2) strain relies on the CNL R protein, RPS2 for full resistance (Bent
et al. 1994, 2). This would give an indication whether the function of the CC class NB-LRR
proteins is still intact. We therefore inoculated RPM1-D lines and Col-0 with Pst DC3000
(AvrRpt2). Since resistance to this strain relies on NDR1 (N Aarts et al. 1998, 1), an
important molecule for CNL signalling, we saw compromised resistance in ndr1. Resistance
to this strain was not affected in rpm1-3, Col-0 and, importantly, RPM1-D did not show
increased levels of bacterial growth as was previously seen for Pst DC3000 (AvrRpm1)
(Figure 2C). However, whereas RPM1-D2 did not exhibit any statistically significant
0
1
2
3
4
5
6
7
8
Col-0 rpm1-3 RPM1-D1 RPM1-D2
C.F
.U./cm
²
Pst DC3000 (avrRpm1)
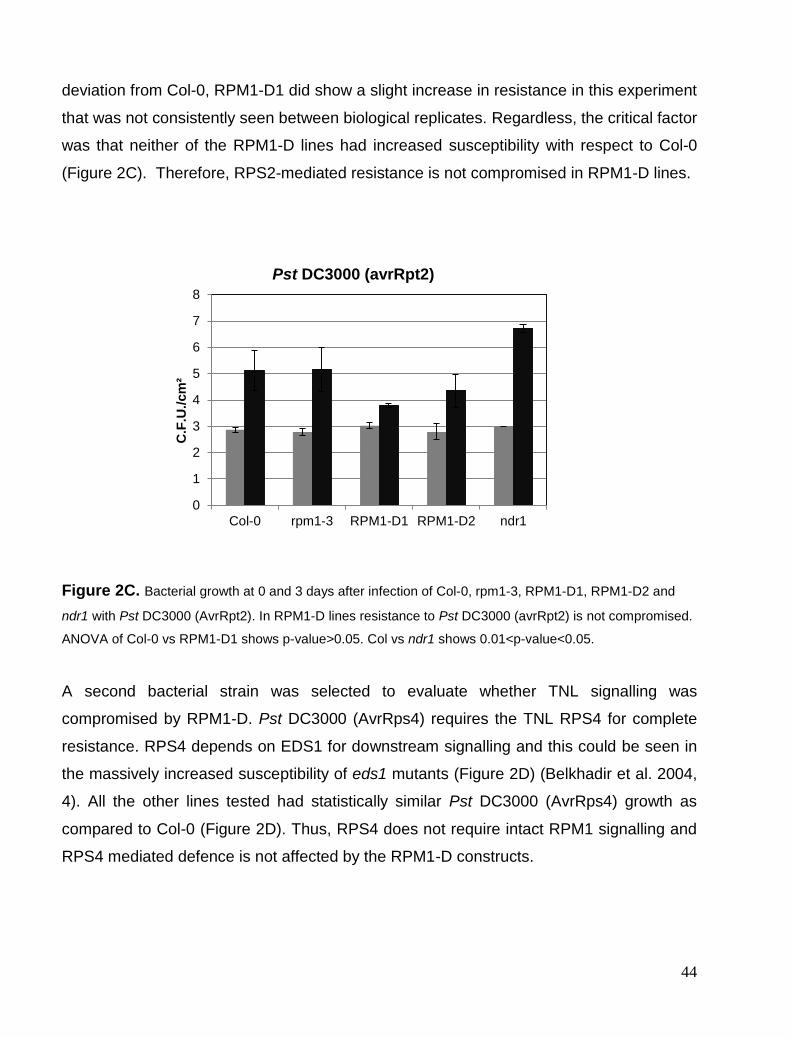
44
deviation from Col-0, RPM1-D1 did show a slight increase in resistance in this experiment
that was not consistently seen between biological replicates. Regardless, the critical factor
was that neither of the RPM1-D lines had increased susceptibility with respect to Col-0
(Figure 2C). Therefore, RPS2-mediated resistance is not compromised in RPM1-D lines.
Figure 2C. Bacterial growth at 0 and 3 days after infection of Col-0, rpm1-3, RPM1-D1, RPM1-D2 and
ndr1 with Pst DC3000 (AvrRpt2). In RPM1-D lines resistance to Pst DC3000 (avrRpt2) is not compromised.
ANOVA of Col-0 vs RPM1-D1 shows p-value>0.05. Col vs ndr1 shows 0.01<p-value<0.05.
A second bacterial strain was selected to evaluate whether TNL signalling was
compromised by RPM1-D. Pst DC3000 (AvrRps4) requires the TNL RPS4 for complete
resistance. RPS4 depends on EDS1 for downstream signalling and this could be seen in
the massively increased susceptibility of eds1 mutants (Figure 2D) (Belkhadir et al. 2004,
4). All the other lines tested had statistically similar Pst DC3000 (AvrRps4) growth as
compared to Col-0 (Figure 2D). Thus, RPS4 does not require intact RPM1 signalling and
RPS4 mediated defence is not affected by the RPM1-D constructs.
0
1
2
3
4
5
6
7
8
Col-0 rpm1-3 RPM1-D1 RPM1-D2 ndr1
C.F
.U./cm
²
Pst DC3000 (avrRpt2)

45
Figure 2D Bacterial growth at 0 and 3 days after infection of Col-0, rpm1-3, RPM1-D1, RPM1-D2 and
ndr1 with Pst DC3000 (AvrRps4). In RPM1-D lines resistance to Pst DC3000 (avrRps4) is not
compromised. ANOVA of Col-0 vs eds1 shows p-value<0.01.
Since RPM1-D mutants behave functionally equivalent to RPM1 knockout mutants, P-loop
mutants do not generally affect other R genes and downstream signalling key members of
two major classes of R proteins, CNLs and TNLs. Thus, P-loop mutants exhibit specificity
and function similarly to null-mutants.
How exactly these DN P-loop mutants function is of course interesting, but unfortunately
we do not know the exact mechanism behind this. However, one explanation is that P-loop
mutants possibly interfere with ATP cycling and homotypic interactions with WT proteins
could lead to inactive aggregates (Bratton and Salvesen 2010). MLA10, Prf and RPS5 have
been shown to form homotypic interactions in the inactive state and this interaction in
required for their function. It is not known whether P-loop mutations affect this interaction
(S. Bai et al. 2012). The formation of inactive R gene complexes consisting of P-loop and
WT R proteins provides one plausible explanation for P-loop function.
Upon activation, N forms homotypic interaction important for its signalling function.
However P-loop mutations of N lead to loss of this homotypic interaction (Mestre and
Baulcombe 2006). This argues against the formation of inactive complexes and in the case
of N, competitive binding to NRIP might be an alternative explanation of the DN behaviour.
0
1
2
3
4
5
6
7
8
9
Col-0 rpm1-3 RPM1-D1 RPM1-D2 eds1
C.F
.U./cm
²
Pst DC3000 (avrRps4)

46
Further research on the mechanism of P-loop DN behaviour should be performed and could
provide interesting information on mechanisms of R protein signalling.
P-loop DN effects partially extends to R gene complexes.
AvrRpm1 activates a single R protein, RPM1. However, AvrB targets two known R proteins.
These are the CNL RPM1 and the TNL TAO1 (T. K. Eitas, Nimchuk, and Dangl 2008).
Since RPM1 and TAO1 target the same guardee and respond to the same effector, we
wanted to see what the effect of RPM1-D would have on ion leakage profile of Pst DC3000
(AvrB). We chose to use ion leakage assay because TAO’s contribution to AvrB resistance
is less than RPM1’s and the bacterial growth inhibition assay may not be sensitive enough
to detect subtle differences.
Figure 2E Ion leakage of Col-0, rpm1-3, ndr1, RPM1-D1 and RPM1-D2 in response to 0.2OD Pst DC3000
(avrB). ANOVA Col-0 vs rpm1,ndr1 p <0.01. rpm1,ndr1 vs RPM1-D1,RPM1-D2 (p<0.01).
0
50
100
150
200
250
0 2 4 6 8 10 12 14
uS/
cm-¹
H.P.I.
Pst DC3000 (avrB) induced Ion leakage
Col-0
rpm1
RPM1-D1
RPM1-D2
ndr1

47
To this end, we infiltrated Pst DC3000 (AvrB) into leaves of Col-0, rpm1-3, ndr1, RPM1-D1
and RPM1-D2 plants. Rpm1-3 and ndr1 had similar ion leakage profiles but significantly
lower than Col-0 (Figure 2E). This is expected because TAO1 is a TNL and ndr1 does not
affect TAO1 signalling, only RPM1. Similarly, the remaining cell death observed in rpm1
likewise showed the contribution of TAO1 to AvrB-triggered cell death since RPM1 is non-
functional in rpm1 and ndr1 (T. K. Eitas, Nimchuk, and Dangl 2008). However, RPM1-D1
and RPM1-D2 had the lowest statistically significant ion leakage compared to Col-0, ndr1
and rpm1-3 at 12 hrs post-infiltration (H.P.I.). This indicates that RPM1-D1 and RPM1-D2s
inhibit WT signalling of both RPM1 and TAO1.
This is a similar situation as has been described for LAZ5-D (Figure 1A). Thus the P-loop
appears to have a stronger suppression compared to KO mutants. One explanation for this
is it inhibits multiple R proteins that function together in a complex, for example guarding
the same guardee.
P-loop mutations can possibly poison R protein complexes and could, for this reason, be
superior in uncovering R proteins involved in autoimmunity since R gene redundancy is
overcome. Importantly, RPS2, that also guards RIN4, is not affected by RPM1-D. It may be
that RPM1 and TAO1 compete for the same binding site whereas RPS2 binding is
independent of RPM1 and TAO1.
These results give us a good framework to further study P-loop mutant function. We will in
future assess whether RPM1-D leads to loss of accumulation of WT RPM1 and TAO1.
Certain TAO1 mutants have been shown to affect RPM1 accumulation (T. K. Eitas,
Nimchuk, and Dangl 2008, 1) and loss of RIN4 leads to loss of RPM1 accumulation
(Belkhadir et al. 2004). RPM1-D might lead to loss of WT RPM1 and TAO1 protein
accumulation due to its interaction with RIN4 (Figure 2E).
PAT1 and SUMM2-D
In addition to evaluating the utility of p-loop mutants of RPM1 we also performed a small
pilot study with another system, autoimmunity in Atpat1 null-alleles. In yeast, PAT1
(topoisomerase II-associated protein 1) is a decapping activator, translational repressor
and also a facilitator of processing body formation (Coller and Parker 2005). In
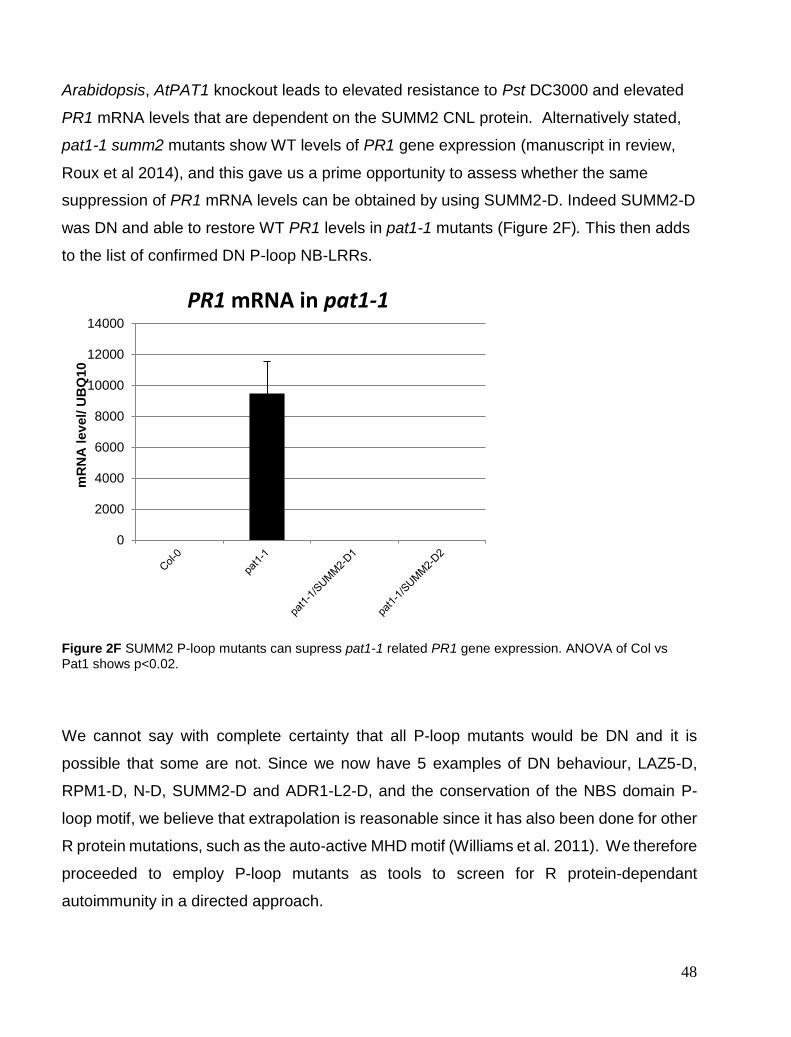
48
Arabidopsis, AtPAT1 knockout leads to elevated resistance to Pst DC3000 and elevated
PR1 mRNA levels that are dependent on the SUMM2 CNL protein. Alternatively stated,
pat1-1 summ2 mutants show WT levels of PR1 gene expression (manuscript in review,
Roux et al 2014), and this gave us a prime opportunity to assess whether the same
suppression of PR1 mRNA levels can be obtained by using SUMM2-D. Indeed SUMM2-D
was DN and able to restore WT PR1 levels in pat1-1 mutants (Figure 2F). This then adds
to the list of confirmed DN P-loop NB-LRRs.
Figure 2F SUMM2 P-loop mutants can supress pat1-1 related PR1 gene expression. ANOVA of Col vs Pat1 shows p<0.02.
We cannot say with complete certainty that all P-loop mutants would be DN and it is
possible that some are not. Since we now have 5 examples of DN behaviour, LAZ5-D,
RPM1-D, N-D, SUMM2-D and ADR1-L2-D, and the conservation of the NBS domain P-
loop motif, we believe that extrapolation is reasonable since it has also been done for other
R protein mutations, such as the auto-active MHD motif (Williams et al. 2011). We therefore
proceeded to employ P-loop mutants as tools to screen for R protein-dependant
autoimmunity in a directed approach.
0
2000
4000
6000
8000
10000
12000
14000
mR
NA
lev
el/ U
BQ
10
PR1 mRNA in pat1-1

49
Chapter 3. Autoimmune mutant library and P-loop screens
P-loop library construction and cloning optimization.
Analysis of the Arabidopsis genome revealed that the Col-0 ecotype contains around 150
R proteins encoded in the genome divided into some 100 TIR-NBS-LRRs and 50 CC-NBS-
LRRs (Meyers et al. 2003). A phylogenetic tree of TNLs and CNLs based on the NBS
domain can be seen in Figure 3A.

50
Figure3A CNL (A) and TNL(B) R genes found in Arabidopsis Col-0 accession (Meyers et al. 2003).

51
Since we have observed that specific mutations in the P-loop of R proteins cause specific
DN effects and autoimmunity can be caused by inappropriate R protein activation (Palma
et al. 2010; Dinesh-Kumar, Tham, and Baker 2000; Roberts et al. 2013) we decided to
introduce P-loop mutations in a large collection of R genes found in the Arabidopsis genome
and subsequently transform them into a collection of autoimmune mutants. To this end, we
generated 35S promoter-fusion P-loop constructs for over-expression of as many NB-LRRs
as possible. Initial cloning of P-loop constructs was performed by overlap-extension PCR.
Two PCR fragments were generated using primers containing a mismatch correlating to
the GXXXXGKT(T/S) to GXXXXAAT(T/S) mutation in the P-loop motif of the R proteins.
These two fragments were then amplified using flanking PCR primers and the
complementary ends served as internal primers to obtain a full-length mutated clones as
illustrated in Figure 3B.
Figure 3B. Overlap PCR for mutant generation (Ho et al. 1989). Two PCR fragments with overlapping
sequence are generated and used to self-prime a larger fragment. The larger fragment is then amplified by
flanking primers.
Full-length mutant constructs were subsequently amplified with Uracil-containing primers
to facilitate USER cloning into the pLIFE vector. The procedure is illustrated in in Figure
3C.

52
Figure 3C. USER cloning (Nour-Eldin, Geu-Flores, and Halkier 2010). Digestion of a USER cassette by
restriction enzymes PacI and Nt.BbvCi and treating Uracil containing PCR fragments with USER enzyme
creates 8bp complementary overhangs that require no ligation prior to E. coli transformation.
Using this approach, 73 TIR-NBS-LRRs were cloned by Klaus Petersen, Simon
Bressendorf and Azra Omerovic (Table 4A). The CC cloning was initially approached with
the same protocol as for TNLs. However for the CNL P-loops, the procedure proved
unreliable and only resulted in amplification of WT genes due to original DNA template
contamination. Optimisation of the initial PCR and overlap PCR reactions did not improve
my success rate.
Given these drawbacks, the CNL P-loop mutations were generated instead using USER
fusion with mismatched internal primers. This proved to be far more reliable and eliminated
the problem of screening out WT clones. We employed a touchdown PCR approach to
amplify gene fragments since it gave increased specificity and far less optimisation was
required. We also used Pfu X7 polymerase (Nørholm 2010) which gave much higher fidelity
of amplification. The USER fusion approach is shown in Figure 3D.

53
Figure 3D. User fusion cloning used to generate P-loop mutants. Uracil containing primers can be
employed to generate compatible sticky ends with treatment by USER enzyme. Multiple PCR products can
be assembled seamlessly. If the uracil-containing primers contain a deliberate mismatch, mutations can be
introduced at will.
Our last challenge was that empty background colonies obtained due to incomplete vector
digestion slowed the cloning progress due to the increased amount of colony screening
needed. To eliminate empty vector background we cloned the ccdB toxin cassette from the
pGWB series vectors (Nakagawa et al. 2007) by USER cloning into the pLIFE vector. This
created a vector that gave zero background after 1 hr of digestion with PacI and Nt.BBvCi
that required only heat-inactivation of restriction enzymes for subsequent use. A similar
vector was created for USER cloning into the pENTR-D-TOPO vector (Invitrogen). First, a
USER cassette was cloned into pENTR-D-TOPO followed by USER cloning of the ccdB
toxin cassette (Figure 3E). We then tested the vector by cloning GFP into this new vector
(pENTRU-ccdB). Following LR cloning into a destination expression vector, strong GFP
expression was obtained in Nicotiana benthamiana.

54
This vector will save time and money in the future since we can now re-use USER cloning
primers to clone any P-loop candidate into multiple destination vectors for subsequent work.
Figure 3E. Constructed USER cloning-compatible Gateway entry vector. PacI and BbvCi sites form 8bp
overhangs and cut out ccdB toxin cassette. Kanamycin (Kan) gene and Origin of replication (Ori) are also
indicated. The AttL1 and AttL2 facilitated LR cloning of in-frame cloned gene into compatible destination
vectors.
Initially, I tried to clone and mutate some 50 CC P-loops; 35 gave sequence-confirmed P-
loop constructs that have been transformed into Agrobacterium tumefaciens for further
work (Table 3A). The remaining genes did not yield either one or both required fragments
for USER Fusion after multiple rounds of optimisation. With the 15 remaining genes, a
nested PCR approach also did not improve the success rate. Nevertheless, since we cloned
around 70% of the known R genes form Col-0 we could increase our chances of uncovering
suppressors by transforming multiple autoimmune mutants with the generated TIR P-loops
and CC P-loops.
The 73 TIR-NBS-LRR P-loop constructs were transformed into Col-0, mpk4-/+, camta3-1,
pie1-3 and siz1-3 since these were the first autoimmune mutants available to us that fit the
criteria for possible TIR-dependent phenotypes. The criteria chosen were that mutants
showed a temperature- and EDS1- or PAD4-dependent phenotype. Ten individual T2 lines
that survived BASTA selection were collected for suppression screens. Autoimmune
mutants were screened for suppression of their phenotype and any recovered lines were
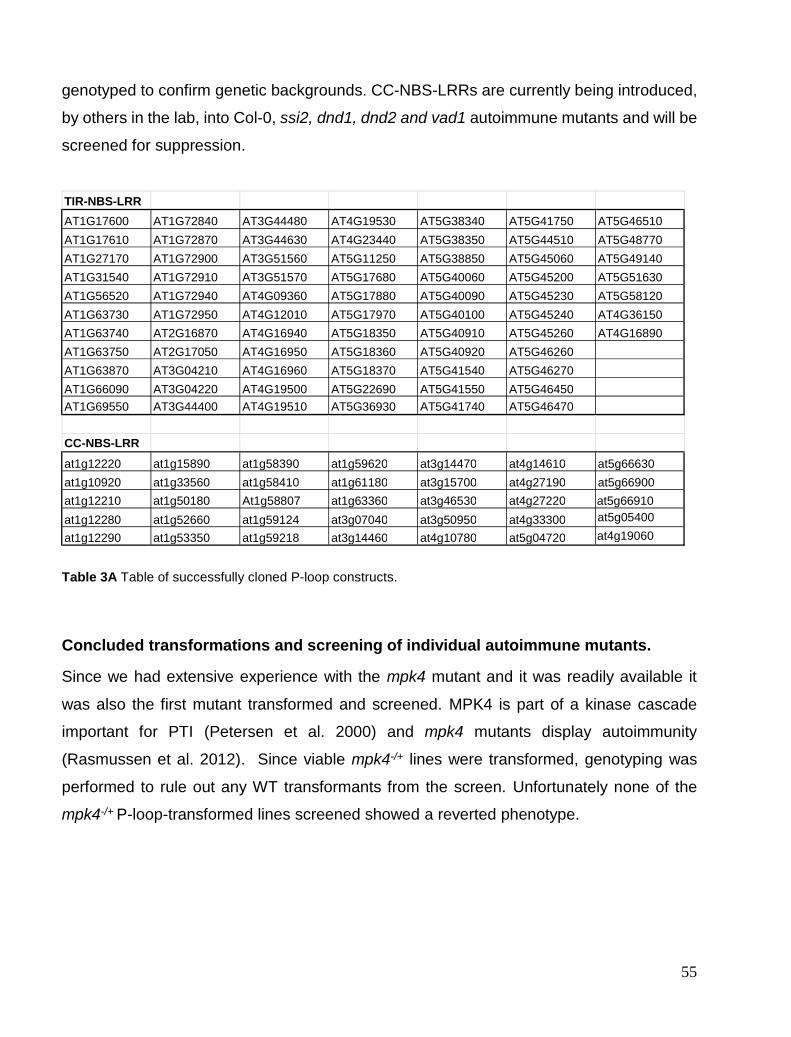
55
genotyped to confirm genetic backgrounds. CC-NBS-LRRs are currently being introduced,
by others in the lab, into Col-0, ssi2, dnd1, dnd2 and vad1 autoimmune mutants and will be
screened for suppression.
Table 3A Table of successfully cloned P-loop constructs.
Concluded transformations and screening of individual autoimmune mutants.
Since we had extensive experience with the mpk4 mutant and it was readily available it
was also the first mutant transformed and screened. MPK4 is part of a kinase cascade
important for PTI (Petersen et al. 2000) and mpk4 mutants display autoimmunity
(Rasmussen et al. 2012). Since viable mpk4-/+ lines were transformed, genotyping was
performed to rule out any WT transformants from the screen. Unfortunately none of the
mpk4-/+ P-loop-transformed lines screened showed a reverted phenotype.
TIR-NBS-LRR
AT1G17600 AT1G72840 AT3G44480 AT4G19530 AT5G38340 AT5G41750 AT5G46510
AT1G17610 AT1G72870 AT3G44630 AT4G23440 AT5G38350 AT5G44510 AT5G48770
AT1G27170 AT1G72900 AT3G51560 AT5G11250 AT5G38850 AT5G45060 AT5G49140
AT1G31540 AT1G72910 AT3G51570 AT5G17680 AT5G40060 AT5G45200 AT5G51630
AT1G56520 AT1G72940 AT4G09360 AT5G17880 AT5G40090 AT5G45230 AT5G58120
AT1G63730 AT1G72950 AT4G12010 AT5G17970 AT5G40100 AT5G45240 AT4G36150
AT1G63740 AT2G16870 AT4G16940 AT5G18350 AT5G40910 AT5G45260 AT4G16890
AT1G63750 AT2G17050 AT4G16950 AT5G18360 AT5G40920 AT5G46260
AT1G63870 AT3G04210 AT4G16960 AT5G18370 AT5G41540 AT5G46270
AT1G66090 AT3G04220 AT4G19500 AT5G22690 AT5G41550 AT5G46450 AT1G69550 AT3G44400 AT4G19510 AT5G36930 AT5G41740 AT5G46470
CC-NBS-LRR
at1g12220 at1g15890 at1g58390 at1g59620 at3g14470 at4g14610 at5g66630
at1g10920 at1g33560 at1g58410 at1g61180 at3g15700 at4g27190 at5g66900
at1g12210 at1g50180 At1g58807 at1g63360 at3g46530 at4g27220 at5g05400 at1g12280 at1g52660 at1g59124 at3g07040 at3g50950 at4g33300
at1g12290 at1g53350 at1g59218 at3g14460 at4g10780 at5g04720
at5g66910
at4g19060

56
Pie1 represents another autoimmune mutant we selected for our screen. PIE1 is a
chromatin remodelling protein of the ISWI family whose deletion also results in enhanced
PR1 gene expression, stunted growth and early flowering (Alvarez, Nota, and Cambiagno
2010). However, after transformation and screening pie1-3, we did not uncover any P-loop
constructs capable of supressing pie1-3 growth and early flowering phenotypes.
Siz1-3 was yet another candidate that was selected for transformation and screening. Small
ubiquitin-like modifier (SUMO) proteins are involved in many processes. Siz1-3 is an E3
SUMO protein ligase involved in plant immunity suggested to negatively regulate SA-
mediated defences. siz1-3 is a typical autoimmune mutant showing spontaneous PAD4-
dependent cell death and thus is an excellent candidate to transform with our P-loop
mutated repertoire (Jin et al. 2008). Attempts at transforming siz1-3 yielded few or no
transformants after several rounds of transformation. Very low viable seed yield and
enhanced defences made plants highly sensitive to either Agrobacterium or silwett-L77
surfactant used in the floral dip transformation and resulted in immediate death of the
majority of plants after dipping with Agrobacterium. Rinsing dipped plants with distilled
water after 24hrs did not improve success. We recommend that future transformation be
done in NahG backgrounds as was done for the acd11 suppressor screen (Palma et al.
2010). Healthier plants that yield more seeds and are less prone to stress after
transformation would greatly speed up screening. BTH, a salicylic analogue, sprayed on
these transformants afterward would reveal any suppressors.
In addition to these autoimmune mutant screens we also assayed all P-loop-transformed
Col-0 lines for loss of PAMP perception. Since TTSS can sometimes inject flagellin into the
cytosol and this flagellin can be perceived in mammals by TLRC4 (Yue Zhao et al. 2011)
we wanted to see if any NB-LRR can contribute to flagellin sensing. All Col-0 P-loop lines
were screened for loss of flg22 and elf18 PAMP perception but none displayed altered
seedling growth inhibition. A recent report also confirmed that in Nicotiana benthamiana,
no cytosolic receptors are present (Wei et al. 2012).

57
Chapter 4. Suppression of camta3-1 phenotypes
Suppressor mutants from screen and visual phenotypes.
As discussed previously, camta3-1 is an autoimmune mutant that shows spontaneous
lesions and stunted growth at the 4-week stage when grown at 22°C under normal lighting
conditions. Camta3-1 shows dependence on phytoalexin deficient 4 (PAD4) and low
temperature for its phenotype (Du et al. 2009). This implicates activated R proteins in the
phenotype and therefore camta3-1 was added to the TNL P-loop screen as a candidate.
We successfully transformed camta3-1 with 73 TIR-NBS-LRR P-loop mutants. Sixteen
plants, from eight independent T2 lines for each P-loop construct were grown under short
day (SD) conditions for 6 weeks in order to identify transformants with WT growth
phenotypes. Two P-loop constructs were uncovered that were capable of completely
reverting camta3-1 growth and lesion phenotypes. These were the At4g12010 P-loop and
At5g18370 P-loop. Six-week-old camta3-1/At5g19370-D is shown on the far right in Figure
4A. In the T2 generation, 2/8 lines expressing At5g18370-D showed growth and lesion
phenotypic rescue where 6/8 T2 lines expressing At4g12010-D had complete reversal of
camta3-1 phenotype (not shown).
Figure 4A. 4 week-old short-day grown Arabidopsis plants. Col-0 left, camta3-1 middle, camta3-1/at5g18370
right.
Since 6/8 T2 lines showed suppression and to simplify further experiments, At4g12010 was
chosen for further comprehensive analysis. At4g12010 was renamed Dominant Suppressor

58
of Camta3-1 (DSC) with –D indicating the P-loop allele and a subsequent number
identifying individual lines. At4g12010 is a typical TIR-NBS-LRR R gene that is found in the
genome in a head-to-head conformation with At4g12020 (WRKY19), a MEKK-TIR-NBS-
LRR-WRKY gene (Figure 4B). It is possible that the DSC R protein together with WRKY19
forms a so called R-gene pair (Narusaka et al. 2009, 4). No published data currently exist
for at4g12010 other than the R-pair like conformation.
Figure 4B. DSC and WRKY19 in a head-to-head conformation similarly to RPS4 and RRS1 (TAIR
www.arabisopsis.org ).
We firstly obtained T3 seeds from the T2 plants that survived BASTA treatment and
analysed segregation of BASTA resistance to obtain lines homozygous for 35S R protein
DSC P-loop for further characterisation. These lines were renamed camta3-1/DSC-D1,
camta3-1/DSC-D2, DSC-D3 and DSC-D4, the latter two serving as controls of DSC-D
constructs in the Col-0 background.
Figure 4C. Suppression of the camta3-1 phenotype in 6-week-old SD-grown plants. Only D1 and D3 are
shown but D2 and D4 have identical appearance. The size of the camta3 plants harbouring the DSC-D
construct is identical to WT Col-0. Chlorosis and cell death of older leaves are also absent from camta3-
1/DSC-D1 and camta3-1/DSC-D2. Black bars represent 1 cm.

59
Camta3-1 growth is stunted after 4 weeks of growth, especially under SD growth conditions
(Galon et al. 2008). The phenotype becomes progressively worse with age, and by 6 weeks
the camta3-1 phenotype was very pronounced (Figure 4C). The spontaneous lesions and
chlorosis seen in older rosette leaves of camta3-1 were completely absent from the camta3-
1/DSC-D1 and camta3-1/DSC-D2 lines. Expression of DSC-D in Col-0 had no effect on
plant growth and induced no visible cell death (Figure 4C). This is often not true for
overexpression of functional WT R proteins, that often trigger cell death (Yi and Richards
2009)(Yan Zhang et al. 2004). Indeed, overexpression of WT DSC in Arabidopsis caused
camta3-like phenotypes. Pictures are not shown due to failure of these lines to produce
viable seed.
Genotyping of DSC-D lines
Due to the remarkable reversal of the camta3 phenotypes by the dominant P-loop R protein
we first confirmed that the lines were indeed null for CAMTA3 mRNA (Figure 4D, Left).
Camta3 and camta3/DSC-D1/2 had critically reduced levels of CAMTA3 mRNA confirming
they were true CAMTA3 knockout lines.
Figure 4D. CAMTA3 and genotyping confirmation of DSC lines. qPCR analysis of CAMTA3 (left), genotyping
PCR (right).
0
0.2
0.4
0.6
0.8
1
1.2
mR
NA
lev
el/U
BQ
10
CAMTA3 mRNA

60
Genotyping was also performed on these lines to confirm that all test lines contained the
transgenic DNA of the P-loop construct. Using two sets of vector/gene specific primers, we
could see inserts present in DSC-D lines but not Col-0 (Figure 4D, Right). Sizes
corresponded to expected products. Therefore we are certain that suppression of camta3-
1 by DSC-D is not a result of seed contamination or outcrossing of camta3-1 to WT.
Expression levels of DSC mRNA
Figure 4E. qPCR analysis of mRNA levels of DSC + DSC-D in Col-0, camta3-1, camta3-1/DSC-D1/2 and
DSC-D3/4.
Up-regulation of DSC expression levels in camta3-1 could be an explanation for the
resulting phenotype, since R gene overexpression in known to cause HR (Yi and Richards
2009). For example SNC1 is elevated by SFR1 mutations and the resulting increased SNC1
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
mR
NA
lev
el/U
BQ
10
DSC mRNA

61
expression leads to autoimmunity (Yingzhong Li et al. 2010). DSC does not contain any
CG-1 CAMTA3 binding sites in its promoter region (TAIR 10) and is therefore unlikely to be
directly regulated by CAMTA3. In line with this, DSC mRNA levels in camta3-1 were not
elevated compared to Col-0 and negative regulation of DSC is therefore unlikely to be the
cause of DSC activation in camta3-1 (Figure 4E).
Additional information gained from these results was that the combined expression of WT
DSC plus DSC-D gene showed that DSC + DSC-D mRNA levels were similar to WT DSC
levels in camta3-1/DSC-D1/2 and DSC-D3/4. Since different degrees of camta3-1 rescue
were not seen, we were not concerned with DSC-D expression on its own. If DSC-D dosage
effects were in play, different levels of suppression would have been observed. The DSC
+ DSC-D mRNA levels observed were not statistically different from those detected in Col-
0 (Figure 4E). For genes under the control of the 35S promoter, one should expect 2 orders
of magnitude increase in the levels of the transgene expression (Yoo et al. 2005). Therefore
some level of post transcriptional regulation could be involved in regulation DSC and DSC-
D mRNA levels as has been showed for the RPP5 and SNC1 NB-LRRs (Yi and Richards
2007).
Endogenous gene silencing is another plausible explanation for DN suppression of P-loop
mutants. The DSC-D construct did not result in silencing of DSC and DSC-D in these lines,
since decreases in both endogenous and transgene transcripts would have resulted. P-
loop mutants may therefore rather exert their dominant-negative effects at the protein level.
Cell death and ROS phenotype
Macroscopic lesions in camta3-1 appeared to be absent but whether smaller microscopic
lesions are still found in the camta3-1/DSC-D lines was investigated. Closer analysis of cell
death by trypan blue staining confirmed that spontaneous lesions of camta3-1 were
completely absent from DSC-D-containing lines (Figure 4F). Expression of DSC-D leads to
complete loss of HR cell death in camta3-1 and does not induce cell death by itself.
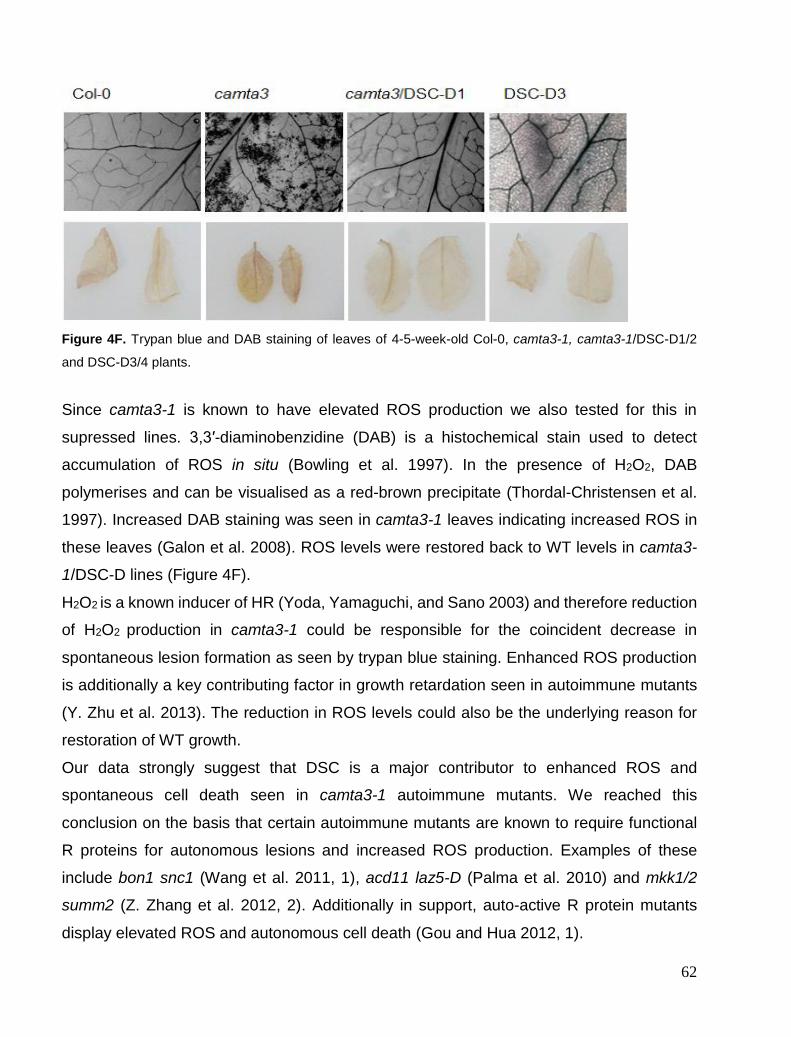
62
Figure 4F. Trypan blue and DAB staining of leaves of 4-5-week-old Col-0, camta3-1, camta3-1/DSC-D1/2
and DSC-D3/4 plants.
Since camta3-1 is known to have elevated ROS production we also tested for this in
supressed lines. 3,3′-diaminobenzidine (DAB) is a histochemical stain used to detect
accumulation of ROS in situ (Bowling et al. 1997). In the presence of H2O2, DAB
polymerises and can be visualised as a red-brown precipitate (Thordal-Christensen et al.
1997). Increased DAB staining was seen in camta3-1 leaves indicating increased ROS in
these leaves (Galon et al. 2008). ROS levels were restored back to WT levels in camta3-
1/DSC-D lines (Figure 4F).
H2O2 is a known inducer of HR (Yoda, Yamaguchi, and Sano 2003) and therefore reduction
of H2O2 production in camta3-1 could be responsible for the coincident decrease in
spontaneous lesion formation as seen by trypan blue staining. Enhanced ROS production
is additionally a key contributing factor in growth retardation seen in autoimmune mutants
(Y. Zhu et al. 2013). The reduction in ROS levels could also be the underlying reason for
restoration of WT growth.
Our data strongly suggest that DSC is a major contributor to enhanced ROS and
spontaneous cell death seen in camta3-1 autoimmune mutants. We reached this
conclusion on the basis that certain autoimmune mutants are known to require functional
R proteins for autonomous lesions and increased ROS production. Examples of these
include bon1 snc1 (Wang et al. 2011, 1), acd11 laz5-D (Palma et al. 2010) and mkk1/2
summ2 (Z. Zhang et al. 2012, 2). Additionally in support, auto-active R protein mutants
display elevated ROS and autonomous cell death (Gou and Hua 2012, 1).

63
Defense marker gene PR1
PR1 was reported to be one of the genes negatively regulated by CAMTA3 (Galon et al.
2008). Pathogenesis-related protein 1 (PR1) gene expression is often elevated in
autoimmune mutants and serves as a key marker for defence induction and SAR (Lorrain
et al. 2003). Since camta3-1 displays elevated SA levels and increased PR1 levels (Du et
al. 2009), the increased PR1 is possibly a consequence of increased SA levels. Support of
this comes from data that shows that by expressing NahG, a bacterial protein that
hydrolyses SA, in camta3-1 supresses the enhanced defence phenotype and PR1
expression (Du et al. 2009). Additionally, PR1 expression is enhanced by application of SA
and SA analogues known to induce plant defence (Vlot, Dempsey, and Klessig 2009).
Therefore SA accumulation is upstream of PR1 induction. Absence of PR1 expression
therefore also implies reduced SA levels. We tested the PR1 mRNA levels in Col-0,
camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 to evaluate whether the loss
of a visual phenotype is reflected in PR1 expression levels. It is possible that some PR1
gene expression remains when other visual phenotypes are absent, indicating partial
suppression.

64
Figure 4G. qPCR analysis of mRNA levels of PR1, a well-known defence marker gene. Camta3-1 displays
highly elevated levels of PR1. DSC-D1 and DSC-D2 lines in camta3 background results in WT levels of PR1.
ANOVA for Col-0 vs camta3-1 shows p<0.01. ANOVA of Col-0 vs camta3-1/DSC-D1, Col-0 vs camta3-
1/DSC-D2, Col-0 vs DSC-D3 and Col-0 vs DSC-D4 showed no significant deviations.
Camta3-1 showed typically elevated PR1 mRNA expression levels (Galon et al. 2008) while
suppressed camta3-1/DSC-D1 and camta3-1/DSC-D2 lines displayed PR1 mRNA levels
restored to those of Col-0 WT (Figure 4G). Since PR1 is a known defence marker gene,
this suggests that the enhanced disease resistance phenotype could also be suppressed
in DSC-D lines. WT levels of PR1 in DSC-D lines suggest that the loss of CAMTA3 does
not have a regulatory effect on this gene in the absence of DSC. The PR1 gene promoter
region does not contain a CG-1 CAMTA3 binding box (TAIR) and therefore CAMTA3-
mediated direct negative regulation is not expected. Elevated PR1 expression is thus likely
0
200
400
600
800
1000
1200
1400
mR
NA
lev
el/U
BQ
10
PR1 mRNA

65
due to DSC activation in camta3-1. This is plausible because PR1 gene expression is a
known consequence of R protein activation (Tsuda et al. 2013)(Belkhadir et al. 2004).
Figure4H. 4-week-old plants were spray-inoculated with Pst DC3000 and Pst DC3000 (AvrRps4). Bacterial
growth was assayed after 3 days. ANOVA of Col vs camta3-1 bacterial growth to both strains shows p<0.01.
ANOVA of Col vs eds1 and camta3-1 vs eds1 shows p<0.01.
Enhanced Disease resistance
As seen above, the PR1 levels of camta3-1 are reduced to WT levels by DSC-D. This
suggests that the reported increased disease resistance to pathogens seen in camta3-1
knockouts (Du et al. 2009) would also be restored back to WT levels. In agreement with
previous reports camta3-1 showed increased resistance to both Pst DC3000 and Pst
DC3000 (AvrRps4) in our study (Figure 4H). However the DSC-D transgene was capable
of reversing this enhanced resistance phenotype since camta3-1/DSC-D1 and camta3-
1/DSC-D2 showed WT levels of bacterial growth to both isolates (Figure 4H). Since WT
levels of bacterial growth were seen in camta3-1/DSC-D, it’s unlikely that CAMTA3
functions as a negative regulator in resistance and that increased resistance of camta3-1
is probably a consequence of DSC activation. If CAMTA3 did play a role in regulating
defence we would have expected a difference between camta3-1/DSC-D and Col-0.
0
1
2
3
4
5
6
7
8
log
10 c
fu/c
m²
Pst DC3000
0
1
2
3
4
5
6
7
log
10
cfu
/cm
²
Pst DC3000 (avrRPS4)

66
Additional information that can be gained from the Pst DC3000 (AvrRps4) data is that DSC-
D does not compromise RPS4 signalling since this would also have resulted in eds1-like
bacterial growth. This is important since it indicates that suppression by DSC-D has the
expected specificity as we have seen in Results chapter 1 (Figure 4H, right).
An example of NB-LRR-dependant reversal of the enhanced defence phenotype is seen in
the autoimmune mutant mkk1 mkk2. Lacking redundant MAPKKKs MKK1 and MKK2, this
double mutant has elevated PR1 mRNA levels and likely is also more resistant to
pathogens similarly to camta3-1 (Kong et al. 2012, 2). This resistance cannot be directly
tested because of severe autoimmune phenotypes of this double mutant (Kong et al. 2012,
2). However the enhanced PR1 expression is restored to WT levels in mkk1 mkk2 summ2
triple mutants. Additionally in mkk1 mkk2 summ2 we do not see WT levels of pathogen
growth, but instead far enhanced growth (Z. Zhang et al. 2012, 2). This indicates that MKK1
and MKK2 play a role in disease resistance but these effects are masked by NB-LRR
activation.
Although loss of CAMTA3 did not affect resistance, it is too early to rule out any function in
defence at this stage. Since there are 6 members in the CAMTA family (Figure I4) there is
a chance that redundancy masks the defence contribution of CAMTA3. Camta1 camta3
and camta2 camta3 show much more severe growth retardation and PR1 overexpression
phenotypes than camta3 alone (Nie et al. 2012) Camta1 and camta2 do not show
autoimmunity and neither does camta1 camta2. It therefore appears that only CAMTA3 is
guarded by DSC and that CAMTA1 and CAMTA2 partially suppress autoimmunity resulting
from loss of CAMTA3 (Y. Kim et al. 2013). CAMTA1 and CAMTA2 could possibly negatively
regulate defence gene expression independently from CAMTA3. If this is true, then the loss
of all 3 CAMTAs in a DSC-D containing line could show altered resistance compared to
camta3-1/DSC-D.
In future work we aim to evaluate the effects of DSC-D on these double and triple camta
mutants in terms of altered disease susceptibility. Since the enhanced defence phenotype
is far more severe in the double and triple camta mutants and seed yield is very low

67
compared to WT, several attempts to transform these mutants have so far failed. Crosses
of camta1 camta3 and camta2 camta3 to camta3-1/DSC-D are being performed instead.
Necrotrophic infections
Although we were unable to detect any difference in growth of Pst in camta3-1/DSC-D and
WT plants, CAMTA3 may be important for defences aimed at combating other types of
pathogens. Therefore, to extend our analysis we also challenged camta3-1/DSC-D lines
with the necrotrophic pathogen Botrytis cinerea. We inoculated Col-0, camta3-1, camta3-
1/DSC-D1, camta3-1/DSC-D2, DSC-D3, DSC-D4 and susceptible control wrky33 (Zheng
et al. 2006) mutant with 5 µL of 5x10⁵ spores/ml B. cinerea. After 5 days necrotic lesions
were examined.
Figure 4I B. cinerea infection of camta3 mutants. A 5 µL drop of 5x10⁵ spores/ml was placed on leaves of 4-
week-old plants and disease progression recorded after 5 days.
Similar to what others have also reported, we saw slight increases in chlorosis surrounding
B. cinerea infection of camta3-1, but overall lesion size remained the same in most lines
tested (Figure 4I). WRKY33 is known to be important in B. cinerea resistance (Zheng et al.
2006) and in line with this, wrky33 showed drastically increased lesion size and in some
cases resulted in complete maceration of infected tissue (Figure 4I). Because B. cinerea is
known to produce ethylene during infection (Cristescu et al. 2002), the chlorosis in camta3-

68
1 surrounding B. cinerea infection might be due to increased sensitivity of camta3-1 to this
B. cinerea-produced ethylene (Nie et al. 2012). This was not however seen in camta3-
1/DSC-D lines, which suggests that increased ethylene sensitivity is restored in these lines
and might be a pleiotropic effect due to defence activation. Why B. cinerea produces
ethylene during infection is puzzling since ein2, an ethylene insensitive mutant, is far more
susceptible to B. cinerea (Thomma et al. 1999b). This indicates that ethylene is important
for resistance to the fungus. Regardless of this contradiction, loss of CAMTA3 in supressed
DSC-D lines had no clear effect on B. cinerea resistance.
Ethylene Signalling
As mentioned above camta3-1 has been shown to display increased ethylene-induced
senescence. Furthermore, camta3 ein3 mutants indicated that ethylene signalling is
required for the spontaneous cell death seen in camta3-1 (Nie et al. 2012). These results
therefore suggest that camta3-1 could display increased ethylene sensitivity or even
spontaneous ethylene production. To study the response to ethylene we employed the
triple response. The triple response is characterized by thickening and shortening of the
hypocotyl with a distinct apical hook seen in seedlings grown in the dark in the presence of
ethylene or ethylene precursors like 1-aminocyclopropane-1-carboxylic acid (ACC).
Defects in one or more of these traits are typical of ethylene insensitive mutants (Guzmán
and Ecker 1990) while mutants with continuous ethylene signalling or spontaneous
ethylene production exhibit the triple response in the absence of ethylene (C Chang and
Meyerowitz 1995).
The ethylene precursor ACC was used to trigger the seedling triple response in Col-0,
camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 (Figure 4K, right)
and compared to untreated seedlings (Figure 4K, left). No significant differences were seen
with regard to triple response in untreated seedlings in terms of hypocotyl growth (Figure
4K, left), where spontaneous ethylene production would have resulted in shortened,
engorged hypocotyls, similar to the response seen in Figure 4K (right). Camta3-1 showed
shorter hypocotyl length compared to Col-0 (Figure 4K right) and slightly more pronounced
apical hooking (Figure 4L) which indicates increased sensitivity to ethylene. This shortening
and increased apical hooking was restored in camta3-1/DSC-D therefore the increased

69
ethylene sensitivity is not a consequence of loss of CAMTA3. In contrast to our results,
camta3 has been shown by others to be unaffected in triple response (Nie et al. 2012).
However these authors used 5 µM ACC treatments, ten times more than in our assays, and
may have saturated the response of their lines. We will in future work also evaluate
ethylene-induced senescence in Col-0, camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2,
DSC-D3 and DSC-D4 as confirmation of results seen in the B. cinerea infection and to
support the triple response assays. Both the camta3-1 (Nie et al. 2012) and acd1 (Jean T.
Greenberg and Ausubel 1993) autoimmune mutants are known to be more sensitive to
ethylene-induced senescence. Since CAMTA3 and ACD1 functions are unrelated the
increased ethylene sensitivity could be due to activated defence responses and could
therefore appear normal in camta3-1/DSC-D.
Figure 4K Triple response to ACC in Col-0, camta3-1, camta3-1/DSC-D1/2 and DSC-D3/4 seedlings.
Figure 4L. Apical hook in ACC-treated dark-grown seedlings shows normal ACC apical hooking in Col-0,
camta3-1/DSC-D1/2 and DSC-D3/4 and increased hooking in camta3-1.
camta3-1/
DSC-D1
camta3-1/
DSC-D2 Col-0 camta3-1 DSC-D3 DSC-D4

70
EDS1 gene regulation
Camta3-1/DSC-D lines behaved similarly to Col-0 in all experiments thus far, suggesting
that CAMTA3 does not play a role in any of these processes. To find a niche for CAMTA3
in plant immunity, we decided to investigate whether EDS1 gene regulation is affected by
loss of CAMTA3. Since CAMTA3 can bind a CGCG box (vCGCGb) in the EDS1 promoter
region in a calmodulin and Ca2⁺-dependent manner and EDS1 mRNA accumulate in
camta3 mutants it has been concluded that CAMTA3 negatively regulates EDS1
expression (Du et al. 2009) Galon et al. 2008). This would imply that EDS1 should still
accumulate in camta3-1/DSC-D lines as it does in camta3-1 and we decided to test this
hypothesis. However when we investigated the EDS1 mRNA levels we found that EDS1
expression in camta3-1/DSC-D was restored to WT levels (Figure 4M). Thus, it is unlikely
that CAMTA3 functions in the negative regulation of EDS1 expression.
Figure 4M qPCR of EDS1 mRNA in 6-week-old soil grown Col-0, camta3-1, camta3-1/DSC-D1/2 and DSC-
D3/4. ANOVA of Col vs camta3-1 shows p<0.01.
0
1
2
3
4
5
6
7
8
mR
NA
lev
el/U
BQ
10
EDS1 mRNA

71
It is a possibility that overexpression of DSC-D could lead to suppression of EDS1 levels
seen in camta3-1/DSC-D lines (Figure 4M). Since it is known that Pst DC3000 (AvrRps4)
infiltration leads to increased EDS1 expression (García et al. 2010) we could use this
knowledge to test for normal EDS1 regulation and exclude this possibility. If DSC-D
supressed EDS1 transcription, we could expect to see differences in AvrRps4-induced
EDS1 levels in WT and DSC-D lines. We consistently saw EDS1 levels of camta3-1/DSC-
D1/2 and DSC-D3/4 induced to a similar extent as WT levels after Pst DC3000 (AvrRps4)
challenge (Figure 4N). Therefore we do not suspect that DSC-D causes aberrant regulation
of EDS1 mRNA levels.
Further support that EDS1 up-regulation is related to autoimmunity is seen in the fact that
at 4-weeks of growth when camta3-1 phenotypes are not yet apparent, no significant
difference in EDS1 mRNA expression could be seen between Col-0 and camta3-1 (Figure
4N, Black bars). We saw similar results in initial studies of EDS1 expression in 2-week-old
seedlings. This shows us that up-regulated EDS1 mRNA levels correlate to autoimmune
phenotypes and therefore could be a consequence of this autoimmunity. It would be very
informative to compare microarray data of two-week-old plants with six-week-old plants.
We plan to perform this experiment after having evaluated whether DSC-D is capable of
completely suppressing camta1 camta3, camta2 camta3 and camta1 camta2 camta3
phenotypes.

72
Figure 4N qPCR analysis of Induction of EDS1 mRNA by 0.05 OD600 Pst DC3000 (AvrRps4). No
statistically significant changes after induction (Grey bars) were seen.
The failure of CAMTA3 to negatively regulate EDS1 may not be so surprising. Its well known
that EDS1 transcripts accumulate in many autoimmune mutants and similar to what is
observed for camta3 mutants, mutations in PAD4 suppress this autoimmunity indicating
that NB-LRR triggering leads to up-regulated EDS1 (Brodersen et al. 2006) (Brodersen et
al. 2002)(F. Zhou et al. 2008). Equally important, overexpression of EDS1 alone does not
cause HR in Arabidopsis (Xing and Chen 2006) and thus accumulation of EDS1 seems to
be symptomatic to autoimmunity in many cases.
The loss of CAMTA3 cannot lead to elevation of EDS1 mRNA expression levels without
activation of R proteins. Our data indicates that DSC likely contributes to this up-regulation
in uninfected camta3-1 mutants.
Recessive DSC mutants do not supress camta3-1
Up to this point all work has been performed on the DSC-D suppressor lines. Since DSC-
D suppression strongly points to DSC activation as an underlying cause of camta3-1
0
2
4
6
8
10
12
14
16
mR
NA
lev
el/U
BQ
10
EDS1 induction by Pst DC3000 (avrRPS4)

73
phenotypes, we needed to investigate if DSC T-DNA insertion lines could similarly suppress
camta3-1 phenotypes. We therefore crossed camta3-1 to two independent DSC T-DNA
lines and visually assessed the rosette size and chlorosis phenotypes. We did not however
see similar suppression in camta3-1 dsc (Figure 4O) as with camta3-1/DSC-D. The lack of
suppression in these lines suggests that other R proteins may contribute to camta3-1
autoimmunity. We have reason to suspect that DSC-D can suppress more than one R
protein since we have seen that RPM1-D inhibits both RPM1 and TAO1, which guard
against the same Avr gene and are part of the same immune complex. Despite this, RPM1-
D retains some specificity since it does not affect the function of the closely associated R
protein RPS2. Further, we also showed that laz5-DN completely suppresses the acd11
phenotype but acd11 laz5 only shows partial rescue (Figure 1A). Crossing acd11 laz5 to
ndr1 gave us complete suppression (Results chapter 1) clearly implicating a second CNL
R protein in its phenotype. It is therefore not unreasonable to think that DSC-D could show
similar behaviour.
This behaviour could be explained by the fact that dominant mutants are known to be able
to poison complexes (Sheppard 1994). The dominant nature of the P-loop construct is
capable of resulting in a far greater effect than that of simple knockouts of single R genes
and thus might also be a great advantage in our dominant screens.
In order to obtain full suppression of camta3-1 by recessive R gene mutants we would
require knowledge of all R proteins activated by loss of CAMTA3. We also uncovered a
second dominant suppressor, DSC2 that may contribute to the camta3-1 phenotype in the
absence of DSC. A T-DNA insertion of the second dominant suppressor gene
At5g18370/DSC2 (Figure 4A) also failed to suppress the camta3-1 phenotype (not shown)
likely because DSC is still functional in these backgrounds. Triple camta3-1 dsc dsc2
mutants may show some degree of phenotypic rescue and are currently being generated
to investigate if these two NB-LRRs are redundant.

74
Figure 4O 6-week-old plants showing phenotypes of two camta3-1 dsc T-DNA knockout lines.
Preliminary data
This section represents data of preliminary experiments and several of the assays should
be repeated. However it gives us useful initial information and further attempts to obtain
consistent replicates with our experimental setup will be done.
Cold responsive genes
CAMTA3 has been shown to bind the CBF1 promoter and is reported to be a positive
regulator of CBF1 gene expression (Doherty et al. 2009). CBF1 is a transcriptional activator
that binds to the CRT/DRE sequence - a cold-responsive sequence - and causes increased
cold regulated (COR) gene expression. CBF1 is therefore a regulator of the cold response
(Jaglo-Ottosen et al. 1998). This means that exposure to non-freezing temperatures causes
CAMTA3-dependent expression of COR genes leading to freezing tolerance in
Arabidopsis.
Separately, cold treatment also leads to a rise in cyt[Ca²⁺] that is a key signalling event
needed for freezing tolerance (Knight, Trewavas, and Knight 1996). The link between these
two responses is still unclear but CAMTA3 is a prime candidate in linking the early cyt[Ca²⁺]
rises to transcriptional changes. CBF1 expression in camta3-1 is reduced upon cold
treatment relative to Col-0. This suggests a positive regulatory function for camta3-1 that
can be tested in DSC-D-expressing lines. In this case, we would expect that the camta3-
1/DSC-D lines would behave similarly to camta3-1.

75
Figure 4P shows induction of the CBF1 gene after 4 hours of cold treatment at 4°C. Black
bars show untreated CBF1 mRNA levels and grey bars cold-treated CBF1 mRNA levels.
All samples showed up-regulation of CBF1 (Doherty et al. 2009). Camta3-1 showed
reduced levels of CBF1 up-regulation as previously reported (Figure 4P). Camta3-1/DSC-
D1 and camta3-1/DSC-D2 did not appear to be affected in CBF1 up-regulation since mRNA
levels accumulated to WT levels (Figure 4P). If true, the reduction in CBF1 mRNA
expression in camta3-1 might also be pleiotropic effect of defence induction. Support for
this comes from another autoimmune mutant siz1-3 that shows a similar reduction in CBF1
expression during cold treatment (Miura et al. 2007). It is also possible that redundancy
masks the effect of losing CAMTA3, since CBF1 expression in camta1 camta2, camta2
camta3 and camta1 camta3 is far more severely affected after cold treatment than in
camta3 alone. This indicates that CAMTA1/2/3 function together to induce CBF1
expression (Y. Kim et al. 2013). No solid conclusions can be made due to unexpected
differences in Col-0 and DSC3/4 CBF1 mRNA levels.
Unfortunately data were not reproducible over several experiments. All genotypes
displayed CBF1 up-regulation following cold treatment, but the degree of up-regulation
varied between biological replicates. Col-0 and camta3-1 always showed a similar trend.
We attempted to solve this variability by using seedlings grown in agar instead of soil since
large volume of soil might have been causing differences. This however did not solve the
inconsistencies. We will in future do a kinetic study with points at 30 min, 1 hr, 2 hrs and 4
hrs to find more consistent cold treatment conditions.

76
Figure 4P qPCR anlaysis of cold-induced CBF1 mRNA after 4 hr at 4°C.
The role of CAMTA3 in SAR
Due to reports that a dominant allele of CAMTA3, camta3-D3 completely inhibited SAR, we
wanted to evaluate CAMTA3’s role in SAR establishment. Induction of SAR causes distal
PR1 mRNA up-regulation and is a widely used marker for SAR induction (Ward et al. 1991).
We therefore infiltrated proximal leaves with Pst DC3000 (avrRPM1) and after two days
evaluated distal PR1 levels. Due to already elevated PR1 levels in camta3-1, PR1 levels of
other genotypes are shown on separate plot (Figure 4Q top). Distal PR1 levels were further
elevated in camta3-1 upon infiltration of 3 proximal leaves with Pst DC3000 (AvrRpm1)
(Figure 4P, top).
0
0.2
0.4
0.6
0.8
1
1.2
CB
F1 r
ela
tiv
e t
o U
BQ
10
CBF1 mRNA levels
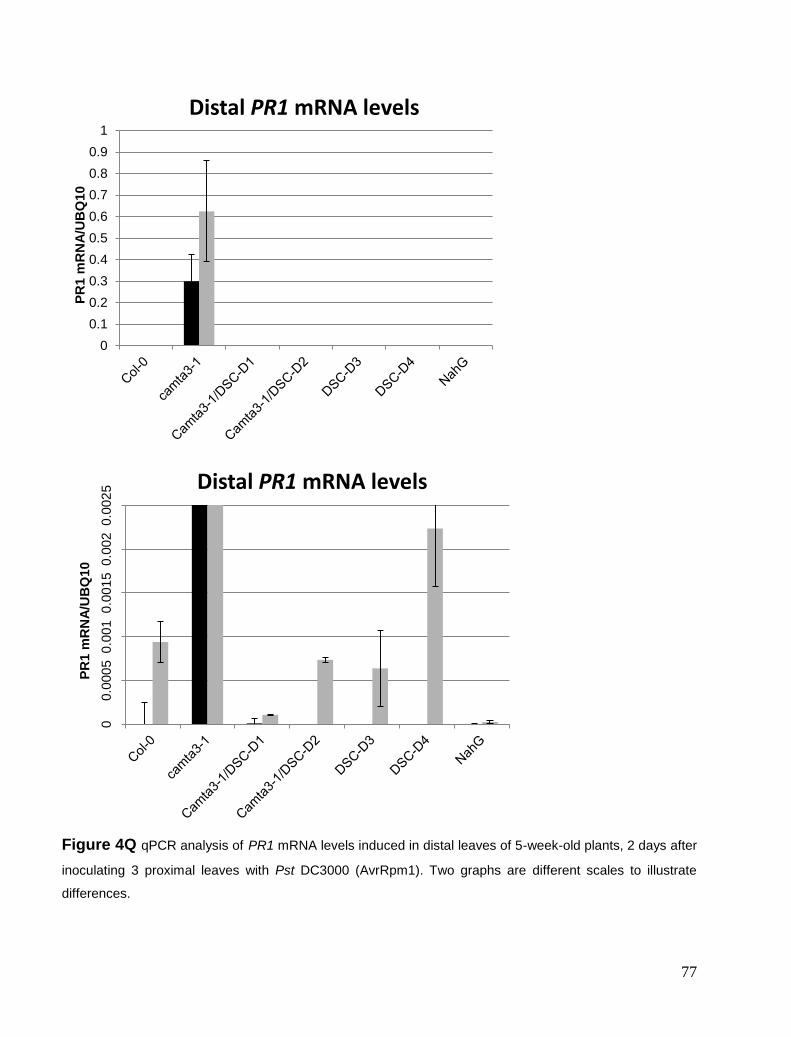
77
Figure 4Q qPCR analysis of PR1 mRNA levels induced in distal leaves of 5-week-old plants, 2 days after
inoculating 3 proximal leaves with Pst DC3000 (AvrRpm1). Two graphs are different scales to illustrate
differences.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
PR
1 m
RN
A/U
BQ
10
Distal PR1 mRNA levels0
0.0
00
50.0
01
0.0
01
50.0
02
0.0
02
5
PR
1 m
RN
A/U
BQ
10
Distal PR1 mRNA levels

78
This in itself indicates that SAR is still induced in camta3-1 mutants and loss of CAMTA3
does not compromise SAR. The camta3-1/DSC-D2 line had similar levels of PR1 gene
expression as WT, indicating that SAR induction in these lines was normal. NahG
transgenic plants, that are SAR defective, displayed no PR1 induction as expected
(Nawrath and Métraux 1999). CAMTA3 OE and dominant alleles of CAMTA3 are reported
to have a compromised SAR (Jing et al. 2011) and one would therefore imagine a higher
PR1 mRNA in camta3-1/DSC-D1 and camta3-1/DSC-D2 in distal tissue after bacterial
challenge similarly to what has been shown for another negative regulator of SAR, RFC3
(Xia et al. 2009). Neither of the camta3-1/DSC-D lines had higher levels of distal PR1
mRNA induction than Col-0 (Figure 4Q). Therefore, CAMTA3 does not appear to affect
normal induction of SAR. Specifically the camta3-1/DSC-D2 and DSC-D3 lines showed
Col-0-like SAR induction, suggesting that SAR is not compromised by loss of camta3-1.
We will gather more data to support this finding.
Noco2 infection and resistance
Downy mildew, Hyaloperonospora arabidopsidis (Hpa) isolate Noco2 is a biotrophic
oomycete pathogen that can infect Col-0 (Parker et al. 1997). We spray-inoculated 2-week-
old soil-grown seedlings with Noco2 spores and assessed infection after 5 days.
Col-0, camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 all showed
formation of sporangiophores 5 days after infection (Figure 4R). We harvested spores from
2 seedlings in three replicates and counted spores/ml using a haemocytometer. This gave
us a quantifiable indication of the level of infection.

79
Figure 4R Conidiophore formation on Col-0, camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3
and DSC-D4, 5 days after inoculation with 5 x 10⁵ Noco2 spores.
The initial test indicated that camta3-1/DSC-D1 and camta3-1/DSC-D2 might be
compromised in resistance to Hpa Noco2 (Figure 4S) since these lines supported increased
spore production correlating to increased pathogen growth. Interestingly, DSC-D3 and
DSC-D4 also showed increased spore production compared to Col-0 and camta3-1,
indicating that compromising DSC itself could have an effect on Hpa Noco2 growth. It would
be very useful if Hpa Noco2 secretes an effector that somehow targets CAMTA3 and that
is guarded for by DSC. This effector could be used to link DSC to CAMTA3 if it targets
CAMTA3 and relies on DSC for HR. Subsequent tests unfortunately failed to show any
growth of Hpa Noco2 in all lines tested including Col-0 controls. We suspected subsequent
contamination of Hpa Noco2 strain occurred and frozen stocks are currently being revived
to retest this result. If this result can be reproduced, CAMTA3 might be shown play an
important role in resistance to Hpa infection.
Col-0 Camta3-1 Camta3-1/DSC-D1 Camta3-1/DSC-D2
Eds1 DSC-D3 DSC-D4

80
Figure 4S. Quantification of Hpa Noco2 infection by spore counting on infected 2-week-old plants (n=6).
0
10000
20000
30000
40000
50000
60000
70000
Sp
ore
s/m
l
Hpa Noco2 spore count

81
Chapter 5. CAMTA3 complexes
Localisation Studies
R protein localisation is abundantly variable. Many R proteins like Rx (Tameling et al.
2010), N (Burch-Smith et al. 2007b), RPS4 (Wirthmueller et al. 2007) and Mla10 (Shen,
Saijo, Mauch, Biskup, Bieri, Keller, Seki, Ulker, et al. 2007) display nucleocytoplasmic
localization. Mla10 changes localisation upon activation and excluding Mla10 from the
nucleus inhibits its signalling function (Shen, Saijo, Mauch, Biskup, Bieri, Keller, Seki, Ülker,
et al. 2007). RPS4 also requires nuclear localization for signaling, however significant
changes in localization do not occur after activation by AvrRps4 (Wirthmueller et al. 2007).
N function also requires nuclear localization, but exclusive cytoplasmic expression of its
ligand p50 does not hinder signaling (Burch-Smith et al. 2007b). RRS1 is only detectable
in the nucleus when its effector PopP2 is also present, no signal could be observed in the
absence of PopP2 (L Deslandes et al. 1998). Rx has been shown to require both
cytoplasmic and nuclear localization for its function. It is believed that the Rx ligand CP is
recognized in the cytoplasm after which Rx is relocated to the nucleus where signaling
proceeds (Slootweg et al. 2010). Nuclear localization is however not critical for the function
of all R proteins. RPM1 is a plasma-membrane-localized protein that does not require
nuclear localization for its signaling function. Exactly how RPM1 signaling occurs is still
uncertain (Gao, Chung, et al. 2011, 1). Nevertheless, these examples illustrate that nuclear
localization is critical for the signaling of certain NB-LRRs. Several NB-LRR also directly
interact with transcription factors and nuclear localization is required to modulate these
transcription factors (Padmanabhan et al. 2013) (S. Bai et al. 2012).
We saw in the previous section that DSC-D is capable of suppressing most camta3-1-
related phenotypes. If indeed CAMTA3 is guarded by DSC, we could expect physical
interaction of CAMTA3 and DSC since NB-LRRs often co-localise with their cognate
effectors or guardees (Frank L W Takken and Goverse 2012). First we evaluated whether
CAMTA3 and DSC have similar subcellular localisations. To this end, we cloned genomic
DSC and CAMTA3, through a Gateway cloning strategy, into 35S tagged expression
vectors and analysed the expression patterns in by agroinfiltration of N. benthamiana.
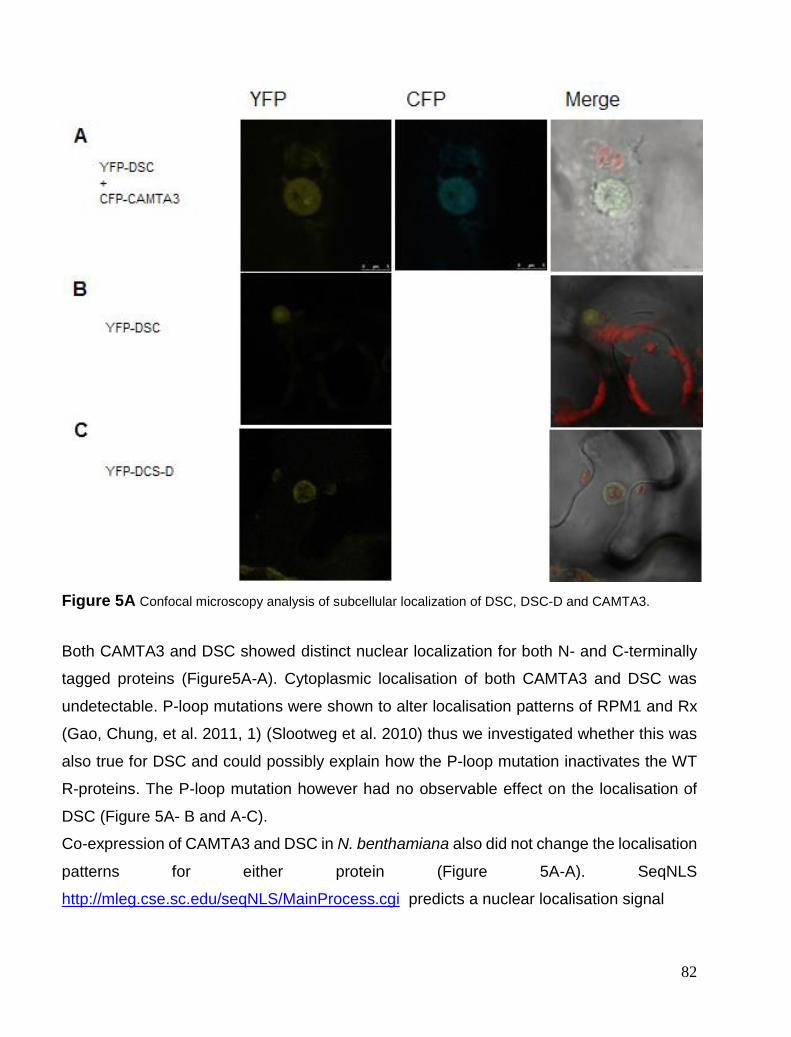
82
Figure 5A Confocal microscopy analysis of subcellular localization of DSC, DSC-D and CAMTA3.
Both CAMTA3 and DSC showed distinct nuclear localization for both N- and C-terminally
tagged proteins (Figure5A-A). Cytoplasmic localisation of both CAMTA3 and DSC was
undetectable. P-loop mutations were shown to alter localisation patterns of RPM1 and Rx
(Gao, Chung, et al. 2011, 1) (Slootweg et al. 2010) thus we investigated whether this was
also true for DSC and could possibly explain how the P-loop mutation inactivates the WT
R-proteins. The P-loop mutation however had no observable effect on the localisation of
DSC (Figure 5A- B and A-C).
Co-expression of CAMTA3 and DSC in N. benthamiana also did not change the localisation
patterns for either protein (Figure 5A-A). SeqNLS
http://mleg.cse.sc.edu/seqNLS/MainProcess.cgi predicts a nuclear localisation signal

83
Figure 5B. Confocal images of N. benthamiana leaves expressing N-terminal YFP-DSC and N-terminal CFP-
CAMTA3. The first panel shows YFP-DSC localization before bleach, second panel shows CFP-CAMTA3
pre-YFP bleach. Third panel is CFP-CAMTA3 intensity post-YFP bleach. Last panel is heat map of change
in CFP florescence indicating FRET.
B, Table summarizing averages for multiple FRET analyses and average FRET for controls.
(NLS) for CAMTA3 (RKVLRYFRKDGHNWRKK), while DSC does not encode a NLS. It
appears that DSC nuclear localisation is controlled by other factors, perhaps even by its
association with CAMTA3. N and MLA also lack a NLS but nuclear accumulation is seen
and required for signalling (Slootweg et al. 2010). Once we have identified all the R proteins
activated in camta3-1 we can study the effect of nuclear exclusion on DSC with regard to
camta3 phenotypes and determine if nuclear localisation is also required for its signalling
function. At this stage, it is likely that the simultaneous activation of a second R protein
could mask the contribution of DSC. Localisation data of DSC suggest that both recognition
and signalling by DSC occurs in the nucleus.

84
FRET interaction study
Since YFP and CFP localisation confirmed that both proteins are nuclear localised we could
proceed to conduct Förster resonance energy transfer (FRET) analysis to see if the two
proteins are in close proximity and possibly be part of the same nuclear complex.
FRET occurs when a fluorophore (donor) in an excited state transfers its energy to a
neighboring molecule (acceptor) (Berney and Danuser 2003), when proteins are less than
6 nm apart (Jares-Erijman and Jovin 2003). In plants, interaction assays can then be
performed using a biological fluorescent FRET pair like CFP/YFP. The emission spectrum
of the donor needs to overlap with the excitation spectrum of the acceptor. When the donor
is excited, FRET signal from the acceptor can be recorded as FRET. Since there can be
some overlap in the excitation spectra of these molecules, false FRET signal can be
detected and interpretation of results can be complicated. Instead we used photo-bleaching
of the acceptor molecule that leads to increased donor fluorescence due to removal of
acceptor quenching. This is a far more reliable approach (Piston and Kremers 2007).
High FRET efficiency could be seen for CAMTA3 and DSC but not CAMTA3 and a second
CAMTA3-unrelated, nuclear localized R protein, SUMM2. Increased intensity of the CFP
signal after acceptor bleaching can clearly be seen (Figure 5B-A third panel). It is interesting
to note that FRET of N-terminally tagged CFP-CAMTA3 shows higher FRET efficiency that
C-terminally tagged CAMTA3-CFP (Figure 5B-B). This implies a specific orientation of the
interaction.
Interestingly, the highest signal intensity of FRET could be seen in the nuclear membrane.
There are strong indications that nucleocytoplasmic trafficking is important for ETI. This can
be seen in the mos7 mutant that lacks part of the nuclear pore complex involved in nuclear
cytoplasmic trafficking. Loss of this component leads to compromised signaling by SNC1
and RPS5 (Cheng et al. 2009). Furthermore, RanGAP2 is important for regulating the small
GTPase Ran known to be important for nucleocytoplasmic trafficking of macromolecules
through the nuclear pores (Tameling and Baulcombe 2007). RanGAP2 is required for Rx-
mediated resistance responses and Rx is known to interact with RanGAP2 (Tameling et
al. 2010). Thus, it is possible that DSC might interact and be retained by a nuclear envelope
protein to control its localization.

85
Figure 5C A, BiFC signal form N. benthamiana expressing N-terminal tagged, C-terminal-YFP-DSC and N-
terminal tagged, N-terminal-YFP-CAMTA3. Chlorophyll, DIC channels and merged image follow in order. B,
BiFC signal from N. benthamiana expressing N-terminal tagged, C-terminal-YFP-DSC-D and N-terminal
tagged N-terminal-YFP-CAMTA3. Chlorophyll, DIC channels and merged image follow in order.
BiFC study
We additionally used a second approach to confirm the FRET signal seen between DSC
and CAMTA3. Bimolecular fluorescence complementation (BiFC) employs two non-
fluorescent segments of YFP on separate proteins. When the proteins come into close
proximity, YFP is reconstituted and a fluorescence signal can be observed. Typically
interactions need to be within 15 nm to observe the reconstituted signal. Reconstitution of
the fluorophore is highly stable and can detect weaker transient interactions. Due to this
signal stability, negative controls are important to rule out false positive results (Kodama
and Hu 2012). YFP signal was observed for N-terminal-nYFP(NnYFP)-CAMTA and N-
terminal-cYFP(NcYFP)-DSC (Figure 5C-A). YFP signal was not observed for CAMTA3-C-
terminal-nYFP(CnYFP) and NcYFP-DCS. The orientation of the interaction is known to be
important in BiFC assays (Kodama and Hu 2012). N-terminally-tagged DSC and CAMTA3
also displayed higher FRET efficiencies, thus both assays indicated that N-terminal tags
are in closer proximity than the N/C terminal combination. This could indicate that

86
interaction occurs via the DSC TIR domain as was shown for Rx and N with their respective
ligands (Rairdan et al. 2008) (Burch-Smith et al. 2007b).
No BIFC could be seen for negative controls with nuclear localized proteins (Figure 5D)
Similar signal intensity to that detected for CAMTA3 and DSC was seen between two known
interactors, MPK4 and PAT1 (Figure 5D). The interaction between MPK4 and the PAT1
has been previously confirmed through immunoprecipitation (IP) (Roux et al., in revision).
IP experiments between CAMTA3 and DSC were attempted by others in the lab, but
detection of both CAMTA3 and DSC tagged constructs was problematic. We will proceed
with smaller HA and MYC tags instead in an attempt to obtain detectable expression in a
western blot. We also attempted to generate Arabidopsis transgenic lines over-expressing
tagged DCS/CAMTA3 for IP. WT R proteins are known to cause cell death and stable
transformed DSC lines displayed spontaneous lesions causing the loss of the first batch of
transformants. We are attempting to generate these lines in Col-0 eds1 background
instead.
Generation of CAMTA3-specific antibodies for use in IP experiments and ChIP assays was
attempted through an external company. Conserved CAMTA3 epitopes
(QNKFRGYKGRKDYLI and SASVNGFHSPELEDAE) were used for immunization but the
resulting serum failed to detect any difference between WT and camta3-1 protein extracts
after extensive attempts at western blot optimization. Lastly we are currently also
conducting Y2H interaction studies to further support our interaction data derived from BiFC
experiments.
It is likely that DSC also interacts with WRKY19. This is based on the R protein pair RRS1
and RPS4 whose interaction shows a similar head-to-head conformation. This predicts a
CAMTA3/WRKY19/DSC complex. The WRKY19 TNL has two additional atypical domains,
a WRKY domain and MEKK domain. The Rosetta stone principal dictates that if a chimera
like WRKY19 exists, similar complexes can likely be seen for separate proteins (Enright
and Ouzounis 2001). WRKY53 and MEKK1 are known to interact in Arabidopsis (Miao et
al. 2007, 53) so it is possible that these WRKY19 domains could also interact. We
hypothesize a CAMTA3/WRKY19/DSC complex where the loss of CAMTA3 causes
changes in MEKK1 and WRKY interaction triggering WRKY19 and DSC. WRKY19 could
87
be a decoy for effectors that target similar R-protein/transcription factor complexes. We will
attempt to investigate this in future studies.
Figure 5D A, Negative BiFC from N. benthamiana expressing N-terminally tagged, C-terminal-YFP-SUMM2
and N-terminally tagged, N-terminal-YFP-CAMTA3. B, Negative BiFC from N. benthamiana expressing N-
terminally tagged, C-terminal-YFP-DSC and N-terminally tagged N-terminal-YFP-MPK4.. C, Positive control
BiFC signal from N. benthamiana expressing N-terminally tagged, C-terminal-YFP-PAT1 and N-terminally
tagged N-terminal-YFP-MPK4. Chlorophyll, DIC channels and merged images follow in order.

88
Conclusion
Camta3-1 phenotypes are completely rescued by DSC-D but not dsc T-DNA lines. This
suggests that DSC and other R proteins act redundantly to guard against the loss of
CAMTA3 or associated disrupted processes. Many of the reported phenotypes of camta3-
1 might therefore also be pleiotropic effects of DSC activation and not related to loss of
CAMTA3. No major perturbation between WT and camta3-1/DSC-D was found and we
suspect CAMTA1 and CAMTA2 could act redundantly with CAMTA3.
Recessive dsc mutants did not share DSC-D camta3-1 supressing capabilities. That does
not mean that DSC has no role in the camta3-1 phenotypes since we have seen that LAZ5-
D and RPM1-D also inhibit at least two R proteins other than LAZ5 and RPM1 respectively.
In the case of RPM1, both these R proteins, RPM1 and TAO1, are known to be associated
with RIN4. Therefore dominant negative P-loop mutants could be capable of inhibiting
autoimmunity by poisoning R protein complexes. In addition, DSC2-D, another suppressor
of camta3-1, has been uncovered. It could be that DSC and DSC2 both guard CAMTA3.
Triple camta3-1 dsc dsc2 mutants may show rescue of camta3-1 phenotypes and confirm
this hypothesis.
FRET and BiFC indicated that DSC is closely associated with CAMTA3 in the nucleus. This
suggests that DSC is directly guarding CAMTA3 for effector-induced changes and this fits
well with our data.
The serendipitous discovery that the laz5 P-loop mutants are dominant negative alleles that
can supress acd11 lead to the idea that P-loop mutants could be a good diagnostic tool to
study autoimmunity caused by NB-LRRs. We have shown that even with a partial collection
of P-loop mutants we could uncover suppressors of autoimmunity and with an even larger
collection our success rate would likely also increase. This method is a good complement
to other screening methods especially since this strategy could overcome some
redundancy issues in R protein signalling. This fact might reveal autoimmunity triggered by
NB-LRRs that has been overlooked by classical methods. Autoimmunity is often attributed
to negative regulation of HR or plant defence and indeed in many cases this should be true.
However, since around 150 NB-LRRs survey the integrity of defence signalling networks is
only logical that there could be several mutational triggers that could result in autoimmunity

89
even though the underlying factors might not necessarily be involved in negative regulation.
This is ultimately important for assigning the correct functions to loci that trigger
autoimmunity and we hope to find many more partners in pathogen perception.

90
Perspectives
We will repeat CBF1 gene expression, SAR and Hpa Noco2 experiments to obtain reliable
data. Triple camta3-1 dsc dsc2 mutants will be analysed for suppression of camta3-1
phenotype.
Since we did not find any major changes in camta3-1/DSC-D disease resistance, we will
also evaluate if camta1 camta3, camta2 camta3 and camta1 camta2 camta3 can be
suppressed by DSC-D. If this is the case, these lines might show differences in pathogen
infection and gene expression to a much greater extent than camta3-1 /DSC-D alone.
Microarray analysis of these lines could possibly yield interesting data on the roles of
CAMTA1, CAMTA2 and CAMTA3 in transcriptional responses that are not skewed by
autoimmunity. We will conduct IP and yeast two-hybrid assays between DSC and CAMTA3
to further confirm the interactions seen in BiFC and FRET analysis.
We will further work on WRKY19, the R gene pair partner of DSC. We will test whether
WRKY domain mutants in WRKY19 cause autoimmunity, as for SLH, and if this resulting
autoimmunity can be suppressed by DSC-D or dsc. Mutations in the kinase domain of
WRKY19 will also similarly be evaluated.
Possible interactions between DSC and WRKY19 will be studied. We will also generate
WRKY19-D and evaluate if it can also suppress the camta3-1 phenotypes. Since the WRKY
domain of WRKY19 and MEKK domain resemble the interaction of MEKK1 and WRKY53,
we will test if the up-regulated WRKY53 levels in camta3-1 play any role in camta3-1
phenotypes by crossing to wrky53 KO. Interestingly WRKY53 OE lines are phenotypically
similar to camta3-1 and this OE might be a trigger for DSC and possibly WRKY19
activation. We will also test if WRKY53 OE lines can be supressed by DSC-D if indeed we
see rescue in camta3-1 wrky53 double mutants. Interaction between CAMTA3 and WKRY
transcription factors, specifically WRKY53, is also of interest. Since WRKY53 can also bind
CaM these players might function together to affect transcriptional changes.
We will attempt to further expand our library of P-loop mutants and screen more
autoimmune mutants for suppression. Effectors could also be tested in Col-0 P-loop lines
to connect effectors to their cognate R proteins, since this seems feasible given the loss of
Pst DC3000 (AvrRpm1) resistance seen in RPM1-D.

91
Materials and methods
Statistical analysis
Multiple ANOVA analysis was performed, aided by Graphpad Prism software. Significant
results were considered to have p values < 0.05.
Plants growth conditions
Arabidopsis thaliana Col-0 accession was used as wild-type in all experiments. Salk lines
were obtained from the Nottingham Arabidopsis Seed Centre (NASC) and primers
designed using the iSect tool (http://signal.salk.edu/isectprimers.html) were used to
genotype lines for homozygosity. Lines obtained from NASC include SALK_001152
(camta3-1), SALK_009668 (dsc1-1) and SAIL_49_C05 (dsc1-2). The mutants ndr1-1
(Century, Holub, and Staskawicz 1995, 1), eds1-2 (N Aarts et al. 1998), rpm1-3 (M. Grant
et al. 2000), wrky33 (Zheng et al. 2006, 33) have been described previously.
Maintenance of pathogenic isolates
Pseudomonas syringae pv. tomato DC3000 containing the avirulence genes AvrRps4
(Hinsch and Staskawicz 1996), AvrRpm1 (M. R. Grant et al. 1995), AvrRpt2 (Bent et al.
1994), in the broad host range vector pVSP61 or DC3000 containing empty pVSP61 were
cultured at 28 °C on NYG plates or NYG media (5 g Bactopeptone, 3 g Yeast Extract ,22,99
g Glycerol (87%), 1L H2O) with appropriate antibiotics.
Ion leakage assays
Conductivity assays were conducted as previously described (Aviv et al. 2002) on 6 x 9mm
diameter Arabidopsis leaf disk in 4 replicates, after inoculation of 0.2 OD600 Pst DC3000
AvrRpm1.
Cloning and generation of transgenic plants
P-loop TIR-NBS-LRRs were created from genomic DNA by USER mutagenesis as
described in (Nour-Eldin et al. 2006) and cloned into modified pCAMBIA-3300, using a
uracil-excision based cloning technique (USER, New England Biolabs). Cloning primers
were tagged with 5′-ggcttaaU-X3′ for the forward primer and 5′-ggtttaaUX-3′ for the reverse

92
primer. Mutant primers were made containing the mutation GKTT to AATT or IGKTT to
NGKTT of the P-loop motif and appropriate uracil incorporated to give seamless overlap of
two fragments (Geu-Flores et al. 2007). Uracil-containing primers used for generation of
all P-loop mutants can be found in appendix I. We used USER Forward and USER mutants
Rev for 5’ fragment and USER reverse and USER mutant forward for 3’ fragment cloning.
Sequencing primers were designed by using Primer3plus online tool
(http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) which gave excellent
results. Sequencing primers will not be included in the manuscript and can be generated
from this tool if desired using a selection window of 800 bp.
WT DSC, DSC-D1 mutant and CAMTA3 were amplified from genomic DNA (Col-0) or
plasmid template, cloned into pENTR/D-TOPO (Invitrogen) and transferred to Gateway-
compatible constitutive expression vectors by LR recombination reactions (Invitrogen).
Destination vectors used were pGWB644 (35S pro C-Terminal CFP), pGWB645 (35S pro
N-Terminal CFP)(Nakamura et al. 2010), pGWB541(35S pro C-Terminal YFP), pGWB542
(35S pro N-Terminal YFP)(Nakagawa et al. 2007). BiFC vectors used were nEYFP/pUGW2
(35S pro, C-nEYFP), cEYFP/pUGW2(35S pro, C-cEYFP), nEYFP/pUGW0(35S pro, N-
nEYFP) and cEYFP/pUGW0(35S pro, N-cEYFP)(Hino et al. 2011). GFP Antibody (B-2):
sc-9996 (Santa Cruz) was used to detect expression of both YFP fragments.
The final constructs were verified by sequencing (MWG, Germany). Primers were designed
using Primer3Plus online (http://www.bioinformatics.nl/cgi-
bin/primer3plus/primer3plus.cgi/). Sequence-confirmed plasmids were electroporated into
Agrobacterium tumefaciens strain GV3101 and used to transform camta3-1 or wild-type
Col-0 plants by the floral dip method (Clough and Bent 1998). Transgenic plants were
selected on soil by spraying seedlings with the herbicide BASTA (100μg/mL).
Plasmid extraction
Alkaline plasmid miniprep was performed as described in (Engebrecht, Heilig, and Brent
2001).

93
Trypan blue staining
Staining for cell death was done as described in (Koch and Slusarenko 1990)
DAB staining
Staining was performed on 5 proximal 6-week-old leaves as described in (Thordal-
Christensen et al. 1997)
Bacterial growth assays
Performed as described in (Katagiri, Thilmony, and He 2002) by syringe infiltration of
bacteria resuspended in 10 mM MgCl2 to 0.001 OD600 or by spray inoculation of 0.2 OD600.
Samples were harvested at day 0 to ensure that no statistical difference was present at day
0 and that day 3 showed bacterial growth.
Botrytis cinerea infection
Fungus was grown on 1xPDA and spores harvested in 1/4xPDA and filtered through
Miracloth. Harvested spores were counted on a hemocytometer and concentration adjusted
to 5 x 10⁵ spores/ml. 4-week-old soil grown Arabidopsis plants were inoculated by placing
5 µL drops on 2 leaves from 5 different plants. Plants were kept humid for 1 day and
infection recorded after 7 days.
Hyaloperonospora arabidopsidis infection
Hpa Noco2 isolates were propagated at 17°C, 10 h light period, in +75% humidity.
2 week old seedlings were sprayed with freshly isolated Hpa Noco2 spores at 5 x 104
spores/ml. Infection was scored after 5 days by shaking 2 seedlings, in 3 replicates, in 5
ml water. Counting of spores/ml was done using a hemocytometer.
Confocal microscopy
Agrobacterium strains containing plant expression vectors were grown in LB media
overnight at 28°C. Relevant cultures were centrifuged and cells resuspended in 10 mM
MgCl2/10 mM MES pH 5.6. After two days, leaves were infiltrated with water and leaf discs
sampled. Confocal microscopy and FRET-AB were performed on a Leica SP5-X confocal

94
microscope equipped with a HCX lambda blue PL APO 320 objective (0.7 numerical
aperture). Excitation of fluorophores was done at the respective absorption maxima. FRET
analysis was performed using Leica FRET-AB wizard software.
RT-PCR
Total RNA was extracted with Trizol reagent (Sigma) or RNA extraction kit (Machery-Nagel)
and integrity confirmed by agarose gel electrophoreses. Turbo DNase treatment (Ambion)
was performed. cDNA synthesis was done using oligodT primers and RevertAid First
Strand cDNA Synthesis Kit (Thermo Scientific). qPCR was performed with Luminaris Color
HiGreen qPCR Kit (Thermo Scientific). Primers for EDS1, PR1, DSC, DSC2, CAMTA3 and
UBQ10 quantification follow below in “Primers”. qPCR temperature cycles were as follows:
95°C :5’, (94°C:20’`;60°C:30’’;72°C:60’’) x 40 cycles followed by a melting curve. Melting
curves were inspected to confirm single product amplification.
SAR assay
Three proximal leaves of 4-week-old plants were infiltrated with 0.2 OD600 Pst DC3000
(AvrRpm1) or 10 mM MgCl2. After 2 days distal tissue was collected and frozen in N2(l).
qPCR for PR1 was performed as described above.
Triple Response
Seed were stratified for 3 days at 4°C. Seedlings were incubated upright in dark for 3 days
on 0 µM and 0.5 µM ACC containing ½ Murashige and Skoog (Duchefa) media agar plates
and triple response recorded.
Cold Shock
Four-week-old soil-grown plants were incubated at 4°C for 4 hrs and tissue harvested and
frozen in N2(l). Room temperature control plants were harvested at the same time. qPCR
analysis of CBF1 expression was performed.

95
Cloning PCR
PCR master mix containing 1U PfuX7, 2.5mM MgCl2, 200 µM each dNTPs, HF phusion
buffer and 0.2 µM forward and reverse primers. Touchdown PCR program was as follows.
98 °C:2’||(95°C:1’|65 °C -2°C per cycle:1’|74°C:30sec/KB )X5 ||
(95°C:1’|55°C:1’|74°C:30’’/KB)X 35 || 74°C:5’. If this program failed a gradient PCR was
used instead 98°C:2’||(95°C:1’|50-65°C:1’|74°C:30sec/KB )X40 || 74°C:5’.
PCR purification cleanup
PCR fragments were cut from 1% ethidium bromide-containing agarose gels under UV light
or in white light using 1 % agarose crystal violet (100 ul/ 100 ml) gel. Gel fragments were
crudely purified by centrifuging gel slices on top of nylon wool at 4000 x g for 2 min or by
using the commercial NucleoSpin® Gel and PCR Clean-up Kit (Machery-Nagel) as per
manufacturer’s protocols. Both methods yielded similar results.
Primers
QPCR
DSC-F: CAC ACG AAT GCG AGT CAC TTG AAA C DSC-R:TGG CAG AAC CAA CTC GGT ATT TCA G DSC2-F:GGG GCT GTT TCA GGC CAT TAG AGA A DSC2-R:CCT CGA CGG CGT ATA GCT CAA GAC EDS1-F:CGA AGG GGA CAT AGA TTG GA EDS1-R:ATG TAC GGC CCT GTG TCT TC PR1-F:GTA GGT GCT CTT GTT CTT CCC PR1-R:CAC ATA ATT CCC ACG AGG ATC UBq10-F:GGC CTT GTA TAA TCC CTG ATG AAT AAG UBq10-R:AAA GAG ATA ACA GGA ACG GAA ACA TAG T
Genotyping Primers
Camta3-1_FW1:CCG AGT CTC AAG TTT TTG ATA GAG AAG Camat3-1_RV1:GAG CTA TGA GTG AAC CAA TTA TCC TCT SALK_099668LP:TTA AGC GGA AAC AAC ATC GAG SALK_099668RP:GGT TCT TCA CCA CCA ACA AAC SAIL_49_C05LP:GAG CTT TTC TCC CAT GGA ATC SAIL_49_C05LP:GCC TTT GAT CGT CTC AAT CAG lb1.3:ATT TTG CCG ATT TCG GAA C lb4:CGT GTG CCA GGT GCC CAC GGA ATA GT acd11F: TAT ACA GGC CAA ATA TTG CCG

96
acd11R: CAC TGA GAA AAA TCT CAG CCG ndr1F: GGA GAA CGA AAA CGT GTG AAG GTC ndr1R: CCT TTC TTC TGA GCT TTA ACT CCA TC Laz5-1:TCA GTA CCA AGC CAC AAT TCC Laz5-1: GCT TGT GAA GCA AGT TCC TTG Lb1.3 ATTTTGCCGATTTCGGAAC LB4 : CGTGTGCCAGGTGCCCACGGAATAGT

97
References
Aarts, N, M Metz, E Holub, B Staskawicz, M Daniels, and J Parker. 1998. “Different
Requirements for EDS1 and NDR1 by Disease Resistance Genes Define at Least Two R
Gene-Mediated Signaling Pathways in Arabidopsis.” Proceedings of the National Academy
of Sciences of the United States of America 95 (17): 10306–11. doi:10.2307/45649.
Aarts, Nicole, Matthew Metz, Eric Holub, Brian J. Staskawicz, Michael J. Daniels, and Jane E.
Parker. 1998. “Different Requirements for EDS1 and NDR1 by Disease Resistance Genes
Define at Least Two R Gene-Mediated Signaling Pathways in Arabidopsis.” Proceedings
of the National Academy of Sciences 95 (17): 10306–11. doi:10.1073/pnas.95.17.10306.
Ade, Jules, Brody J DeYoung, Catherine Golstein, and Roger W Innes. 2007. “Indirect Activation
of a Plant Nucleotide Binding Site-Leucine-Rich Repeat Protein by a Bacterial Protease.”
Proceedings of the National Academy of Sciences of the United States of America 104 (7):
2531–36. doi:10.1073/pnas.0608779104.
Adie, Bruce A. T., Julián Pérez-Pérez, Manuel M. Pérez-Pérez, Marta Godoy, José-J. Sánchez-
Serrano, Eric A. Schmelz, and Roberto Solano. 2007. “ABA Is an Essential Signal for Plant
Resistance to Pathogens Affecting JA Biosynthesis and the Activation of Defenses in
Arabidopsis.” The Plant Cell Online 19 (5): 1665–81. doi:10.1105/tpc.106.048041.
Aist, J R. 1976. “Papillae and Related Wound Plugs of Plant Cells.” Annual Review of
Phytopathology 14 (1): 145–63. doi:10.1146/annurev.py.14.090176.001045.
Alvarez, María E, Florencia Nota, and Damián A Cambiagno. 2010. “Epigenetic Control of Plant
Immunity.” Molecular Plant Pathology 11 (4): 563–76. doi:10.1111/j.1364-
3703.2010.00621.x.
Ambawat, Supriya, Poonam Sharma, Neelam R. Yadav, and Ram C. Yadav. 2013. “MYB
Transcription Factor Genes as Regulators for Plant Responses: An Overview.” Physiology
and Molecular Biology of Plants 19 (3): 307–21. doi:10.1007/s12298-013-0179-1.
Arnab Kapat, Gilly Zimand. 1998. “Biosynthesis of Pathogenicity Hydrolytic Enzymes by Botrytis
Cinerea during Infection of Bean Leaves and in Vitro.” Mycological Research 102 (8):
1017–24. doi:10.1017/S0953756297006023.
Asai, T, J M Stone, J E Heard, Y Kovtun, P Yorgey, J Sheen, and F M Ausubel. 2000. “Fumonisin
B1-Induced Cell Death in Arabidopsis Protoplasts Requires Jasmonate-, Ethylene-, and
Salicylate-Dependent Signaling Pathways.” The Plant Cell 12 (10): 1823–36.
Assmann, Sarah M., and Ken-ichiro Shimazaki. 1999. “The Multisensory Guard Cell. Stomatal
Responses to Blue Light and Abscisic Acid.” Plant Physiology 119 (3): 809–16.
doi:10.1104/pp.119.3.809.
Atkinson, Merelee M., L. Dale Keppler, Elizabeth W. Orlandi, C. Jacyn Baker, and Charles F.
Mischke. 1990. “Involvement of Plasma Membrane Calcium Influx in Bacterial Induction
of the K+/H+ and Hypersensitive Responses in Tobacco 1.” Plant Physiology 92 (1): 215–
21.
Aviv, Daniel H., Christine Rustérucci, Ben F. Holt Iii, Robert A. Dietrich, Jane E. Parker, and
Jeffery L. Dangl. 2002. “Runaway Cell Death, but Not Basal Disease Resistance, in lsd1 Is
SA- and NIM1/NPR1-Dependent.” The Plant Journal 29 (3): 381–91. doi:10.1046/j.0960-
7412.2001.01225.x.
Azevedo, Cristina, Shigeyuki Betsuyaku, Jack Peart, Akira Takahashi, Laurent Noël, Ari
Sadanandom, Catarina Casais, Jane Parker, and Ken Shirasu. 2006. “Role of SGT1 in

98
Resistance Protein Accumulation in Plant Immunity.” The EMBO Journal 25 (9): 2007–16.
doi:10.1038/sj.emboj.7601084.
Bai, Chang, Partha Sen, Kay Hofmann, Lei Ma, Mark Goebl, J. Wade Harper, and Stephen J.
Elledge. 1996. “SKP1 Connects Cell Cycle Regulators to the Ubiquitin Proteolysis
Machinery through a Novel Motif, the F-Box.” Cell 86 (2): 263–74. doi:10.1016/S0092-
8674(00)80098-7.
Bai, Shiwei, Jie Liu, Cheng Chang, Ling Zhang, Takaki Maekawa, Qiuyun Wang, Wenkai Xiao, et
al. 2012. “Structure-Function Analysis of Barley NLR Immune Receptor MLA10 Reveals
Its Cell Compartment Specific Activity in Cell Death and Disease Resistance.” PLoS
Pathogens 8 (6). doi:10.1371/journal.ppat.1002752.
http://dx.doi.org/10.1371/journal.ppat.1002752.
Baumberger, N, and D C Baulcombe. 2005. “Arabidopsis ARGONAUTE1 Is an RNA Slicer That
Selectively Recruits microRNAs and Short Interfering RNAs.” Proceedings of the National
Academy of Sciences of the United States of America 102 (33): 11928–33.
doi:10.1073/pnas.0505461102.
Belkhadir, Youssef, Zachary Nimchuk, David A. Hubert, David Mackey, and Jeffery L. Dangl.
2004. “Arabidopsis RIN4 Negatively Regulates Disease Resistance Mediated by RPS2 and
RPM1 Downstream or Independent of the NDR1 Signal Modulator and Is Not Required for
the Virulence Functions of Bacterial Type III Effectors AvrRpt2 or AvrRpm1.” The Plant
Cell Online 16 (10): 2822–35. doi:10.1105/tpc.104.024117.
Bell, Charles E. 2005. “Structure and Mechanism of Escherichia Coli RecA ATPase.” Molecular
Microbiology 58 (2): 358–66. doi:10.1111/j.1365-2958.2005.04876.x.
Bella, J., K. L. Hindle, P. A. McEwan, and S. C. Lovell. 2008. “The Leucine-Rich Repeat
Structure.” Cellular and Molecular Life Sciences 65 (15): 2307–33. doi:10.1007/s00018-
008-8019-0.
Bendahmane, Abdelhafid, Garry Farnham, Peter Moffett, and David C Baulcombe. 2002.
“Constitutive Gain-of-Function Mutants in a Nucleotide Binding Site-Leucine Rich Repeat
Protein Encoded at the Rx Locus of Potato.” The Plant Journal: For Cell and Molecular
Biology 32 (2): 195–204.
Bendahmane, Abdelhafid, Konstantin Kanyuka, and David C. Baulcombe. 1999. “The Rx Gene
from Potato Controls Separate Virus Resistance and Cell Death Responses.” The Plant Cell
Online 11 (5): 781–91. doi:10.1105/tpc.11.5.781.
Bent, A F, R W Innes, J R Ecker, and B J Staskawicz. 1992. “Disease Development in Ethylene-
Insensitive Arabidopsis Thaliana Infected with Virulent and Avirulent Pseudomonas and
Xanthomonas Pathogens.” Molecular Plant-Microbe Interactions: MPMI 5 (5): 372–78.
Bent, A F, B N Kunkel, D Dahlbeck, K L Brown, R Schmidt, J Giraudat, J Leung, and B J
Staskawicz. 1994. “RPS2 of Arabidopsis Thaliana: A Leucine-Rich Repeat Class of Plant
Disease Resistance Genes.” Science (New York, N.Y.) 265 (5180): 1856–60.
Berney, Claude, and Gaudenz Danuser. 2003. “FRET or No FRET: A Quantitative Comparison.”
Biophysical Journal 84 (6): 3992–4010. doi:10.1016/S0006-3495(03)75126-1.
Bernoux, Maud, Thomas Ve, Simon Williams, Christopher Warren, Danny Hatters, Eugene
Valkov, Xiaoxiao Zhang, Jeffrey G Ellis, Bostjan Kobe, and Peter N Dodds. 2011.
“Structural and Functional Analysis of a Plant Resistance Protein TIR Domain Reveals
Interfaces for Self-Association, Signaling, and Autoregulation.” Cell Host & Microbe 9 (3):
200–211. doi:10.1016/j.chom.2011.02.009.
Berr, Alexandre, Emily J. McCallum, Abdelmalek Alioua, Dimitri Heintz, Thierry Heitz, and
Wen-Hui Shen. 2010a. “Arabidopsis Histone Methyltransferase SDG8 Mediates Induction

99
of the Jasmonate/Ethylene-Pathway Genes in Plant Defense Response to Necrotrophic
Fungi.” Plant Physiology, September, pp.110.161497. doi:10.1104/pp.110.161497.
———. 2010b. “Arabidopsis Histone Methyltransferase SDG8 Mediates Induction of the
Jasmonate/Ethylene-Pathway Genes in Plant Defense Response to Necrotrophic Fungi.”
Plant Physiology, September, pp.110.161497. doi:10.1104/pp.110.161497.
Bhattacharjee, Saikat, Morgan Halane, Sang Kim, and Walter Gassmann. 2011. “Pathogen
Effectors Target Arabidopsis EDS1 and Alter Its Interactions with Immune Regulators.”
Science (New York, N.Y.) 334 (6061): 1405–8. doi:10.1126/science.1211592.
Bittner-Eddy, P D, and J L Beynon. 2001. “The Arabidopsis Downy Mildew Resistance Gene,
RPP13-Nd, Functions Independently of NDR1 and EDS1 and Does Not Require the
Accumulation of Salicylic Acid.” Molecular Plant-Microbe Interactions: MPMI 14 (3):
416–21. doi:10.1094/MPMI.2001.14.3.416.
Boch, Jens, Heidi Scholze, Sebastian Schornack, Angelika Landgraf, Simone Hahn, Sabine Kay,
Thomas Lahaye, Anja Nickstadt, and Ulla Bonas. 2009. “Breaking the Code of DNA
Binding Specificity of TAL-Type III Effectors.” Science 326 (5959): 1509–12.
doi:10.1126/science.1178811.
Bonardi, Vera. 2012. “How Complex Are Intracellular Immune Receptor Signaling Complexes?”
Frontiers in Plant Proteomics 3: 237. doi:10.3389/fpls.2012.00237.
Bonardi, Vera, Saijun Tang, Anna Stallmann, Melinda Roberts, Karen Cherkis, and Jeffery L.
Dangl. 2011. “Expanded Functions for a Family of Plant Intracellular Immune Receptors
beyond Specific Recognition of Pathogen Effectors.” Proceedings of the National Academy
of Sciences 108 (39): 16463–68. doi:10.1073/pnas.1113726108.
Bos, Jorunn I. B., Miles R. Armstrong, Eleanor M. Gilroy, Petra C. Boevink, Ingo Hein, Rosalind
M. Taylor, Tian Zhendong, et al. 2010. “Phytophthora Infestans Effector AVR3a Is
Essential for Virulence and Manipulates Plant Immunity by Stabilizing Host E3 Ligase
CMPG1.” Proceedings of the National Academy of Sciences 107 (21): 9909–14.
doi:10.1073/pnas.0914408107.
Bosch, Dustin E., Francis S. Willard, Ravikrishna Ramanujam, Adam J. Kimple, Melinda D.
Willard, Naweed I. Naqvi, and David P. Siderovski. 2012. “A P-Loop Mutation in Gα
Subunits Prevents Transition to the Active State: Implications for G-Protein Signaling in
Fungal Pathogenesis.” PLoS Pathog 8 (2): e1002553. doi:10.1371/journal.ppat.1002553.
Bouché, Nicolas, Ariel Scharlat, Wayne Snedden, David Bouchez, and Hillel Fromm. 2002. “A
Novel Family of Calmodulin-Binding Transcription Activators in Multicellular
Organisms.” The Journal of Biological Chemistry 277 (24): 21851–61.
doi:10.1074/jbc.M200268200.
Boutrot, Freddy, Cécile Segonzac, Katherine N Chang, Hong Qiao, Joseph R Ecker, Cyril Zipfel,
and John P Rathjen. 2010. “Direct Transcriptional Control of the Arabidopsis Immune
Receptor FLS2 by the Ethylene-Dependent Transcription Factors EIN3 and EIL1.”
Proceedings of the National Academy of Sciences of the United States of America 107 (32):
14502–7. doi:10.1073/pnas.1003347107.
Bowling, S A, J D Clarke, Y Liu, D F Klessig, and X Dong. 1997. “The cpr5 Mutant of
Arabidopsis Expresses Both NPR1-Dependent and NPR1-Independent Resistance.” The
Plant Cell 9 (9): 1573–84.
Bratton, Shawn B, and Guy S Salvesen. 2010. “Regulation of the Apaf-1-Caspase-9 Apoptosome.”
Journal of Cell Science 123 (Pt 19): 3209–14. doi:10.1242/jcs.073643.
Brodersen, Peter, Morten Petersen, Henrik Bjørn Nielsen, Shijiang Zhu, Mari-Anne Newman,
Kevan M. Shokat, Steffen Rietz, Jane Parker, and John Mundy. 2006. “Arabidopsis MAP

100
Kinase 4 Regulates Salicylic Acid- and Jasmonic Acid/ethylene-Dependent Responses via
EDS1 and PAD4.” The Plant Journal 47 (4): 532–46. doi:10.1111/j.1365-
313X.2006.02806.x.
Brodersen, Peter, Morten Petersen, Helen M. Pike, Brian Olszak, Søren Skov, Niels Ødum, Lise
Bolt Jørgensen, Rhoderick E. Brown, and John Mundy. 2002. “Knockout of Arabidopsis
ACCELERATED-CELL-DEATH11 Encoding a Sphingosine Transfer Protein Causes
Activation of Programmed Cell Death and Defense.” Genes & Development 16 (4): 490–
502. doi:10.1101/gad.218202.
Burch-Smith, Tessa M, Michael Schiff, Jeffrey L Caplan, Jeffrey Tsao, Kirk Czymmek, and
Savithramma P Dinesh-Kumar. 2007a. “A Novel Role for the TIR Domain in Association
with Pathogen-Derived Elicitors.” PLoS Biol 5 (3): e68. doi:10.1371/journal.pbio.0050068.
———. 2007b. “A Novel Role for the TIR Domain in Association with Pathogen-Derived
Elicitors.” PLoS Biol 5 (3): e68. doi:10.1371/journal.pbio.0050068.
Caplan, J.L., P. Mamillapalli, T.M. Burch-Smith, K. Czymmek, and S.P. Dinesh-Kumar. 2008.
“Chloroplastic Protein NRIP1 Mediates Innate Immune Receptor Recognition of a Viral
Effector.” Cell 132 (3): 449–62.
Cardone, Michael H., Natalie Roy, Henning R. Stennicke, Guy S. Salvesen, Thomas F. Franke,
Eric Stanbridge, Steven Frisch, and John C. Reed. 1998. “Regulation of Cell Death
Protease Caspase-9 by Phosphorylation.” Science 282 (5392): 1318–21.
doi:10.1126/science.282.5392.1318.
Carlton, W. M., E. J. Braun, and M. L. Gleason. 1998. “Ingress of Clavibacter Michiganensis
Subsp. michiganensis into Tomato Leaves Through Hydathodes.” Phytopathology 88 (6):
525–29. doi:10.1094/PHYTO.1998.88.6.525.
Cecconi, Francesco, Gonzalo Alvarez-Bolado, Barbara I Meyer, Kevin A Roth, and Peter Gruss.
1998. “Apaf1 (CED-4 Homolog) Regulates Programmed Cell Death in Mammalian
Development.” Cell 94 (6): 727–37. doi:10.1016/S0092-8674(00)81732-8.
Century, K. S., E. B. Holub, and B. J. Staskawicz. 1995. “NDR1, a Locus of Arabidopsis Thaliana
That Is Required for Disease Resistance to Both a Bacterial and a Fungal Pathogen.”
Proceedings of the National Academy of Sciences 92 (14): 6597–6601.
Chang, C, and E M Meyerowitz. 1995. “The Ethylene Hormone Response in Arabidopsis: A
Eukaryotic Two-Component Signaling System.” Proceedings of the National Academy of
Sciences of the United States of America 92 (10): 4129–33.
Chang, Cheng, Deshui Yu, Jian Jiao, Shaojuan Jing, Paul Schulze-Lefert, and Qian-Hua Shen.
2013. “Barley MLA Immune Receptors Directly Interfere with Antagonistically Acting
Transcription Factors to Initiate Disease Resistance Signaling.” The Plant Cell Online,
March, tpc.113.109942. doi:10.1105/tpc.113.109942.
Chassot, Celine, Christiane Nawrath, and Jean-Pierre Metraux. 2008. “The Cuticle: Not Only a
Barrier for Plant Defence.” Plant Signaling & Behavior 3 (2): 142–44.
Cheng, Yu Ti, Hugo Germain, Marcel Wiermer, Dongling Bi, Fang Xu, Ana V. García, Lennart
Wirthmueller, et al. 2009. “Nuclear Pore Complex Component MOS7/Nup88 Is Required
for Innate Immunity and Nuclear Accumulation of Defense Regulators in Arabidopsis.”
The Plant Cell Online 21 (8): 2503–16. doi:10.1105/tpc.108.064519.
Chi, Yingjun, Yan Yang, Yuan Zhou, Jie Zhou, Baofang Fan, Jing-Quan Yu, and Zhixiang Chen.
2013. “Protein–Protein Interactions in the Regulation of WRKY Transcription Factors.”
Molecular Plant, March, sst026. doi:10.1093/mp/sst026.

101
Chinchilla, Delphine, Zsuzsa Bauer, Martin Regenass, Thomas Boller, and Georg Felix. 2006.
“The Arabidopsis Receptor Kinase FLS2 Binds Flg22 and Determines the Specificity of
Flagellin Perception.” The Plant Cell Online 18 (2): 465–76. doi:10.1105/tpc.105.036574.
Chinchilla, Delphine, Cyril Zipfel, Silke Robatzek, Birgit Kemmerling, Thorsten
N|[uuml]|rnberger, Jonathan D. G. Jones, Georg Felix, and Thomas Boller. 2007. “A
Flagellin-Induced Complex of the Receptor FLS2 and BAK1 Initiates Plant Defence.”
Nature 448 (7152): 497–500. doi:10.1038/nature05999.
Chini, A., S. Fonseca, G. Fernández, B. Adie, J. M. Chico, O. Lorenzo, G. García-Casado, et al.
2007. “The JAZ Family of Repressors Is the Missing Link in Jasmonate Signalling.”
Nature 448 (7154): 666–71. doi:10.1038/nature06006.
Clough, Steven J., and Andrew F. Bent. 1998. “Floral Dip: A Simplified Method
forAgrobacterium-Mediated Transformation ofArabidopsis Thaliana.” The Plant Journal
16 (6): 735–43. doi:10.1046/j.1365-313x.1998.00343.x.
Clough, Steven J., Kevin A. Fengler, I-ching Yu, Bernadette Lippok, Roger K. Smith, and Andrew
F. Bent. 2000. “The Arabidopsis dnd1 ‘Defense, No Death’ Gene Encodes a Mutated
Cyclic Nucleotide-Gated Ion Channel.” Proceedings of the National Academy of Sciences
of the United States of America 97 (16): 9323–28.
Coley, Phyllis D, John P Bryant, and F. Stuart Chapin. 1985. “Resource Availability and Plant
Antiherbivore Defense.” Science 230 (4728): 895–99. doi:10.1126/science.230.4728.895.
Coll, Nuria S, Dominique Vercammen, Andrea Smidler, Charles Clover, Frank Van Breusegem,
Jeffery L Dangl, and Petra Epple. 2010. “Arabidopsis Type I Metacaspases Control Cell
Death.” Science 330 (6009): 1393–97. doi:10.1126/science.1194980.
Coller, Jeff, and Roy Parker. 2005. “General Translational Repression by Activators of mRNA
Decapping.” Cell 122 (6): 875–86. doi:10.1016/j.cell.2005.07.012.
Collier, Sarah M, Louis-Philippe Hamel, and Peter Moffett. 2011. “Cell Death Mediated by the N-
Terminal Domains of a Unique and Highly Conserved Class of NB-LRR Protein.”
Molecular Plant-Microbe Interactions: MPMI 24 (8): 918–31. doi:10.1094/MPMI-03-11-
0050.
Collier, Sarah M, and Peter Moffett. 2009. “NB-LRRs Work a ‘Bait and Switch’ on Pathogens.”
Trends in Plant Science 14 (10): 521–29. doi:10.1016/j.tplants.2009.08.001.
Cristescu, Simona M, Domenico De Martinis, Sacco Te Lintel Hekkert, David H Parker, and Frans
J M Harren. 2002. “Ethylene Production by Botrytis Cinerea in Vitro and in Tomatoes.”
Applied and Environmental Microbiology 68 (11): 5342–50.
Dangl, Jeffery L., and Jonathan D. G. Jones. 2001. “Plant Pathogens and Integrated Defence
Responses to Infection.” Nature 411 (6839): 826–33. doi:10.1038/35081161.
Danot, Olivier, Emélie Marquenet, Dominique Vidal-Ingigliardi, and Evelyne Richet. 2009.
“Wheel of Life, Wheel of Death: A Mechanistic Insight into Signaling by STAND
Proteins.” Structure 17 (2): 172–82. doi:10.1016/j.str.2009.01.001.
Dean, Paul. 2011. “Functional Domains and Motifs of Bacterial Type III Effector Proteins and
Their Roles in Infection.” FEMS Microbiology Reviews 35 (6): 1100–1125.
doi:10.1111/j.1574-6976.2011.00271.x.
Delaney, Terrence P., Scott Uknes, Bernard Vernooij, Leslie Friedrich, Kris Weymann, David
Negrotto, Thomas Gaffney, et al. 1994. “A Central Role of Salicylic Acid in Plant Disease
Resistance.” Science 266 (5188): 1247–50. doi:10.1126/science.266.5188.1247.
Deslandes, L, F Pileur, L Liaubet, S Camut, C Can, K Williams, E Holub, J Beynon, M Arlat, and
Y Marco. 1998. “Genetic Characterization of RRS1, a Recessive Locus in Arabidopsis
Thaliana That Confers Resistance to the Bacterial Soilborne Pathogen Ralstonia

102
Solanacearum.” Molecular Plant-Microbe Interactions : MPMI 11 (7): 659–67.
doi:10.1094/MPMI.1998.11.7.659.
Deslandes, Laurent, Jocelyne Olivier, Nemo Peeters, Dong Xin Feng, Manirath Khounlotham,
Christian Boucher, Imre Somssich, Stephane Genin, and Yves Marco. 2003. “Physical
Interaction between RRS1-R, a Protein Conferring Resistance to Bacterial Wilt, and PopP2,
a Type III Effector Targeted to the Plant Nucleus.” Proceedings of the National Academy of
Sciences of the United States of America 100 (13): 8024–29.
doi:10.1073/pnas.1230660100.
Devoto, Alessandra, Manuela Nieto-Rostro, Daoxin Xie, Christine Ellis, Rebecca Harmston,
Elaine Patrick, Jackie Davis, Leigh Sherratt, Mark Coleman, and John G. Turner. 2002.
“COI1 Links Jasmonate Signalling and Fertility to the SCF Ubiquitin–ligase Complex in
Arabidopsis.” The Plant Journal 32 (4): 457–66. doi:10.1046/j.1365-313X.2002.01432.x.
Dietrich, Robert A., Terrence P. Delaney, Scott J. Uknes, Eric R. Ward, John A. Ryals, and Jeffery
L. Dangl. 1994. “Arabidopsis Mutants Simulating Disease Resistance Response.” Cell 77
(4): 565–77. doi:10.1016/0092-8674(94)90218-6.
Dinesh-Kumar, S P, W H Tham, and B J Baker. 2000. “Structure-Function Analysis of the
Tobacco Mosaic Virus Resistance Gene N.” Proceedings of the National Academy of
Sciences of the United States of America 97 (26): 14789–94.
doi:10.1073/pnas.97.26.14789.
Dodds, Peter N., Gregory J. Lawrence, Ann-Maree Catanzariti, Trazel Teh, Ching-I. A. Wang,
Michael A. Ayliffe, Bostjan Kobe, and Jeffrey G. Ellis. 2006. “Direct Protein Interaction
Underlies Gene-for-Gene Specificity and Coevolution of the Flax Resistance Genes and
Flax Rust Avirulence Genes.” Proceedings of the National Academy of Sciences 103 (23):
8888–93. doi:10.1073/pnas.0602577103.
Doherty, Colleen, Heather Van Buskirk, Susan Myers, and Michael Thomashow. 2009. “Roles for
Arabidopsis CAMTA Transcription Factors in Cold-Regulated Gene Expression and
Freezing Tolerance.” The Plant Cell 21 (3): 972–84. doi:10.1105/tpc.108.063958.
Du, Liqun, Gul S. Ali, Kayla A. Simons, Jingguo Hou, Tianbao Yang, A. S. N. Reddy, and B. W.
Poovaiah. 2009. “Ca2+/calmodulin Regulates Salicylic-Acid-Mediated Plant Immunity.”
Nature 457 (7233): 1154–58. doi:10.1038/nature07612.
Dunning, F. Mark, Wenxian Sun, Kristin L. Jansen, Laura Helft, and Andrew F. Bent. 2007.
“Identification and Mutational Analysis of Arabidopsis FLS2 Leucine-Rich Repeat Domain
Residues That Contribute to Flagellin Perception.” The Plant Cell Online 19 (10): 3297–
3313. doi:10.1105/tpc.106.048801.
Duplan, Vincent, and Susana Rivas. 2014. “E3 Ubiquitin-Ligases and Their Target Proteins during
the Regulation of Plant Innate Immunity.” Frontiers in Plant Science 5: 42.
doi:10.3389/fpls.2014.00042.
Durner, Jörg, Jyoti Shah, and Daniel F. Klessig. 1997. “Salicylic Acid and Disease Resistance in
Plants.” Trends in Plant Science 2 (7): 266–74. doi:10.1016/S1360-1385(97)86349-2.
Durrant, W E, and X Dong. 2004. “Systemic Acquired Resistance.” Annual Review of
Phytopathology 42: 185–209. doi:10.1146/annurev.phyto.42.040803.140421.
Eitas, Timothy, and Jeffery Dangl. 2010. “NB-LRR Proteins: Pairs, Pieces, Perception, Partners,
and Pathways.” Current Opinion in Plant Biology 13 (4): 472–77.
doi:10.1016/j.pbi.2010.04.007.
Eitas, Timothy K, Zachary L Nimchuk, and Jeffery L Dangl. 2008. “Arabidopsis TAO1 Is a TIR-
NB-LRR Protein That Contributes to Disease Resistance Induced by the Pseudomonas

103
Syringae Effector AvrB.” Proceedings of the National Academy of Sciences of the United
States of America 105 (17): 6475–80. doi:10.1073/pnas.0802157105.
Ellis, Jeffrey G, Peter N Dodds, and Gregory J Lawrence. 2007. “Flax Rust Resistance Gene
Specificity Is Based on Direct Resistance-Avirulence Protein Interactions.” Annual Review
of Phytopathology 45: 289–306. doi:10.1146/annurev.phyto.45.062806.094331.
Engebrecht, J, J S Heilig, and R Brent. 2001. “Preparation of Bacterial Plasmid DNA.” Current
Protocols in Neuroscience / Editorial Board, Jacqueline N. Crawley ... [et Al.] Appendix 1
(May): Appendix 1J. doi:10.1002/0471142301.nsa01js10.
Enright, A J, and C A Ouzounis. 2001. “Functional Associations of Proteins in Entire Genomes by
Means of Exhaustive Detection of Gene Fusions.” Genome Biology 2 (9):
RESEARCH0034.
Eulgem, Thomas, and Imre E Somssich. 2007. “Networks of WRKY Transcription Factors in
Defense Signaling.” Current Opinion in Plant Biology 10 (4): 366–71.
doi:10.1016/j.pbi.2007.04.020.
Fei, Qili, Rui Xia, and Blake C. Meyers. 2013. “Phased, Secondary, Small Interfering RNAs in
Posttranscriptional Regulatory Networks.” The Plant Cell Online 25 (7): 2400–2415.
doi:10.1105/tpc.113.114652.
Ferrari, Simone, Julia M Plotnikova, Giulia De Lorenzo, and Frederick M Ausubel. 2003.
“Arabidopsis Local Resistance to Botrytis Cinerea Involves Salicylic Acid and Camalexin
and Requires EDS4 and PAD2, but Not SID2, EDS5 or PAD4.” The Plant Journal: For
Cell and Molecular Biology 35 (2): 193–205.
Fiil, Berthe Katrine, and Morten Petersen. 2011. “Constitutive Expression of MKS1 Confers
Susceptibility to Botrytis Cinerea Infection Independent of PAD3 Expression.” Plant
Signaling & Behavior 6 (10): 1425–27. doi:10.4161/psb.6.10.16759.
Flor, H H. 1971. “Current Status of the Gene-For-Gene Concept.” Annual Review of
Phytopathology 9 (1): 275–96. doi:10.1146/annurev.py.09.090171.001423.
Freeman. 2008. “An Overview of Plant Defenses against Pathogens and Herbivores.” The Plant
Health Instructor. doi:10.1094/PHI-I-2008-0226-01.
http://www.apsnet.org/edcenter/intropp/topics/Pages/OverviewOfPlantDiseases.aspx.
Fu, Zheng Qing, Shunping Yan, Abdelaty Saleh, Wei Wang, James Ruble, Nodoka Oka,
Rajinikanth Mohan, et al. 2012. “NPR3 and NPR4 Are Receptors for the Immune Signal
Salicylic Acid in Plants.” Nature 486 (7402): 228–32. doi:10.1038/nature11162.
Galon, Yael, Roy Nave, Joy Boyce, Dikla Nachmias, Marc Knight, and Hillel Fromm. 2008.
“Calmodulin-Binding Transcription Activator (CAMTA) 3 Mediates Biotic Defense
Responses in Arabidopsis.” FEBS Letters 582 (6): 943–48.
doi:10.1016/j.febslet.2008.02.037.
Gao, Zhiyong, Eui-Hwan Chung, Timothy K. Eitas, and Jeffery L. Dangl. 2011. “Plant
Intracellular Innate Immune Receptor Resistance to Pseudomonas Syringae Pv. Maculicola
1 (RPM1) Is Activated At, and Functions On, the Plasma Membrane.” Proceedings of the
National Academy of Sciences 108 (18): 7619–24. doi:10.1073/pnas.1104410108.
Gao, Zhiyong, Zhiyoug Gao, Eui-Hwan Chung, Timothy K Eitas, and Jeffery L Dangl. 2011.
“Plant Intracellular Innate Immune Receptor Resistance to Pseudomonas Syringae Pv.
Maculicola 1 (RPM1) Is Activated At, and Functions On, the Plasma Membrane.”
Proceedings of the National Academy of Sciences of the United States of America 108 (18):
7619–24. doi:10.1073/pnas.1104410108.
García, Ana V., Servane Blanvillain-Baufumé, Robin P. Huibers, Marcel Wiermer, Guangyong Li,
Enrico Gobbato, Steffen Rietz, and Jane E. Parker. 2010. “Balanced Nuclear and

104
Cytoplasmic Activities of EDS1 Are Required for a Complete Plant Innate Immune
Response.” PLoS Pathog 6 (7): e1000970. doi:10.1371/journal.ppat.1000970.
Garcia-Brugger, Angela, Olivier Lamotte, Elodie Vandelle, Stéphane Bourque, David Lecourieux,
Benoit Poinssot, David Wendehenne, and Alain Pugin. 2006. “Early Signaling Events
Induced by Elicitors of Plant Defenses.” Molecular Plant-Microbe Interactions: MPMI 19
(7): 711–24. doi:10.1094/MPMI-19-0711.
Garcion, Christophe, Antje Lohmann, Elisabeth Lamodière, Jérémy Catinot, Antony Buchala,
Peter Doermann, and Jean-Pierre Métraux. 2008. “Characterization and Biological
Function of the ISOCHORISMATE SYNTHASE2 Gene of Arabidopsis.” Plant Physiology
147 (3): 1279–87. doi:10.1104/pp.108.119420.
Geu-Flores, Fernando, Hussam H. Nour-Eldin, Morten T. Nielsen, and Barbara A. Halkier. 2007.
“USER Fusion: A Rapid and Efficient Method for Simultaneous Fusion and Cloning of
Multiple PCR Products.” Nucleic Acids Research 35 (7): e55. doi:10.1093/nar/gkm106.
Glazebrook, Jane. 2005. “Contrasting Mechanisms of Defense Against Biotrophic and
Necrotrophic Pathogens.” Annual Review of Phytopathology 43 (1): 205–27.
doi:10.1146/annurev.phyto.43.040204.135923.
Gómez-Gómez, Lourdes, and Thomas Boller. 2002. “Flagellin Perception: A Paradigm for Innate
Immunity.” Trends in Plant Science 7 (6): 251–56.
Goodman, R. N. 1972. “Electrolyte Leakage and Membrane Damage in Relation to Bacterial
Population, pH, and Ammonia Production in Tobacco Leaf Tissue Inoculated with
Pseudomonas Pisi.” Phytopathology 62 (11): 1327. doi:10.1094/Phyto-62-1327.
Gou, Mingyue, and Jian Hua. 2012. “Complex Regulation of an R Gene SNC1 Revealed by Auto-
Immune Mutants.” Plant Signaling & Behavior 7 (2): 213–16. doi:10.4161/psb.18884.
Gou, Mingyue, Zhenying Shi, Ying Zhu, Zhilong Bao, Guoying Wang, and Jian Hua. 2012. “The
F-Box Protein CPR1/CPR30 Negatively Regulates R Protein SNC1 Accumulation.” The
Plant Journal 69 (3): 411–20. doi:10.1111/j.1365-313X.2011.04799.x.
Grant, M R, L Godiard, E Straube, T Ashfield, J Lewald, A Sattler, R W Innes, and J L Dangl.
1995. “Structure of the Arabidopsis RPM1 Gene Enabling Dual Specificity Disease
Resistance.” Science (New York, N.Y.) 269 (5225): 843–46.
Grant, Murray, Ian Brown, Sally Adams, Marc Knight, Alastair Ainslie, and John Mansfield. 2000.
“The RPM1 Plant Disease Resistance Gene Facilitates a Rapid and Sustained Increase in
Cytosolic Calcium That Is Necessary for the Oxidative Burst and Hypersensitive Cell
Death.” The Plant Journal 23 (4): 441–50. doi:10.1046/j.1365-313x.2000.00804.x.
Greeff, Christiaan, Milena Roux, John Mundy, and Morten Petersen. 2012. “Receptor-like Kinase
Complexes in Plant Innate Immunity.” Frontiers in Plant Proteomics 3: 209.
doi:10.3389/fpls.2012.00209.
Greenberg, J T, F P Silverman, and H Liang. 2000. “Uncoupling Salicylic Acid-Dependent Cell
Death and Defense-Related Responses from Disease Resistance in the Arabidopsis Mutant
acd5.” Genetics 156 (1): 341–50.
Greenberg, Jean T., and Frederick M. Ausubel. 1993. “Arabidopsis Mutants Compromised for the
Control of Cellular Damage during Pathogenesis and Aging.” The Plant Journal 4 (2):
327–41. doi:10.1046/j.1365-313X.1993.04020327.x.
Gudesblat, Gustavo E, Pablo S Torres, and Adrian A Vojnov. 2009. “Stomata and Pathogens:
Warfare at the Gates.” Plant Signaling & Behavior 4 (12): 1114–16.
Gutierrez, Jose R, Alexi L Balmuth, Vardis Ntoukakis, Tatiana S Mucyn, Selena Gimenez-Ibanez,
Alexandra M E Jones, and John P Rathjen. 2010. “Prf Immune Complexes of Tomato Are
Oligomeric and Contain Multiple Pto-like Kinases That Diversify Effector Recognition.”

105
The Plant Journal: For Cell and Molecular Biology 61 (3): 507–18. doi:10.1111/j.1365-
313X.2009.04078.x.
Guzmán, P, and J Ecker. 1990. “Exploiting the Triple Response of Arabidopsis to Identify
Ethylene-Related Mutants.” The Plant Cell 2 (6): 513–23. doi:10.1105/tpc.2.6.513.
Hauck, Paula, Roger Thilmony, and Sheng Yang He. 2003. “A Pseudomonas Syringae Type III
Effector Suppresses Cell Wall-Based Extracellular Defense in Susceptible Arabidopsis
Plants.” Proceedings of the National Academy of Sciences of the United States of America
100 (14): 8577–82. doi:10.1073/pnas.1431173100.
Heidrich, Katharina, Kenichi Tsuda, Servane Blanvillain-Baufumé, Lennart Wirthmueller,
Jaqueline Bautor, and Jane E Parker. 2013. “Arabidopsis TNL-WRKY Domain Receptor
RRS1 Contributes to Temperature-Conditioned RPS4 Auto-Immunity.” Frontiers in Plant
Science 4: 403. doi:10.3389/fpls.2013.00403.
Heidrich, Katharina, Lennart Wirthmueller, Céline Tasset, Cécile Pouzet, Laurent Deslandes, and
Jane Parker. 2011. “Arabidopsis EDS1 Connects Pathogen Effector Recognition to Cell
Compartment-Specific Immune Responses.” Science (New York, N.Y.) 334 (6061): 1401–4.
doi:10.1126/science.1211641.
Heredia, A, A Jiménez, and R Guillén. 1995. “Composition of Plant Cell Walls.” Zeitschrift Für
Lebensmittel-Untersuchung Und -Forschung 200 (1): 24–31.
Herskowitz, Ira. 1987. “Functional Inactivation of Genes by Dominant Negative Mutations.”
Nature 329 (6136): 219–22. doi:10.1038/329219a0.
Hino, Takeshi, Yuji Tanaka, Makoto Kawamukai, Kohji Nishimura, Shoji Mano, and Tsuyoshi
Nakagawa. 2011. “Two Sec13p Homologs, AtSec13A and AtSec13B, Redundantly
Contribute to the Formation of COPII Transport Vesicles in <I>Arabidopsis thaliana</I>.”
Bioscience, Biotechnology, and Biochemistry 75 (9): 1848–52.
Hinsch, M, and B Staskawicz. 1996. “Identification of a New Arabidopsis Disease Resistance
Locus, RPs4, and Cloning of the Corresponding Avirulence Gene, avrRps4, from
Pseudomonas Syringae Pv. Pisi.” Molecular Plant-Microbe Interactions: MPMI 9 (1): 55–
61.
Ho, Steffan N., Henry D. Hunt, Robert M. Horton, Jeffrey K. Pullen, and Larry R. Pease. 1989.
“Site-Directed Mutagenesis by Overlap Extension Using the Polymerase Chain Reaction.”
Gene 77 (1): 51–59. doi:10.1016/0378-1119(89)90358-2.
Hofius, Daniel, Torsten Schultz-Larsen, Jan Joensen, Dimitrios I. Tsitsigiannis, Nikolaj H. T.
Petersen, Ole Mattsson, Lise Bolt Jørgensen, Jonathan D. G. Jones, John Mundy, and
Morten Petersen. 2009. “Autophagic Components Contribute to Hypersensitive Cell Death
in Arabidopsis.” Cell 137 (4): 773–83. doi:10.1016/j.cell.2009.02.036.
Hu, Y, M A Benedict, L Ding, and G Núñez. 1999. “Role of Cytochrome c and dATP/ATP
Hydrolysis in Apaf-1-Mediated Caspase-9 Activation and Apoptosis.” The EMBO Journal
18 (13): 3586–95. doi:10.1093/emboj/18.13.3586.
Hua, J, C Chang, Q Sun, and E M Meyerowitz. 1995. “Ethylene Insensitivity Conferred by
Arabidopsis ERS Gene.” Science (New York, N.Y.) 269 (5231): 1712–14.
Hua, Jian, Hajime Sakai, Saeid Nourizadeh, Qianhong G. Chen, Anthony B. Bleecker, Joseph R.
Ecker, and Elliot M. Meyerowitz. 1998. “EIN4 and ERS2 Are Members of the Putative
Ethylene Receptor Gene Family in Arabidopsis.” The Plant Cell Online 10 (8): 1321–32.
doi:10.1105/tpc.10.8.1321.
Ichimura, Kazuya, Catarina Casais, Scott C Peck, Kazuo Shinozaki, and Ken Shirasu. 2006.
“MEKK1 Is Required for MPK4 Activation and Regulates Tissue-Specific and

106
Temperature-Dependent Cell Death in Arabidopsis.” The Journal of Biological Chemistry
281 (48): 36969–76. doi:10.1074/jbc.M605319200.
Ishiguro, S, and K Nakamura. 1994. “Characterization of a cDNA Encoding a Novel DNA-
Binding Protein, SPF1, That Recognizes SP8 Sequences in the 5’ Upstream Regions of
Genes Coding for Sporamin and Beta-Amylase from Sweet Potato.” Molecular & General
Genetics: MGG 244 (6): 563–71.
Jaglo-Ottosen, K R, S J Gilmour, D G Zarka, O Schabenberger, and M F Thomashow. 1998.
“Arabidopsis CBF1 Overexpression Induces COR Genes and Enhances Freezing
Tolerance.” Science (New York, N.Y.) 280 (5360): 104–6.
Janczur, M. K. 2009. “Optimal Energy Allocation to Growth, Reproduction, and Production of
Defensive Substances in Plants: A Model.” Evolutionary Ecology Research 11 (3): 447–70.
CABDirect2.
Jares-Erijman, Elizabeth A., and Thomas M. Jovin. 2003. “FRET Imaging.” Nature Biotechnology
21 (11): 1387–95. doi:10.1038/nbt896.
Jeffree, Christopher E. 2007. “The Fine Structure of the Plant Cuticle.” In Annual Plant Reviews
Volume 23: Biology of the Plant Cuticle, edited by rkus Riederer and Caroline Müller, 11–
125. Blackwell Publishing Ltd.
http://onlinelibrary.wiley.com/doi/10.1002/9780470988718.ch2/summary.
Jin, Jing Bo, Yin Hua Jin, Jiyoung Lee, Kenji Miura, Chan Yul Yoo, Woe-Yeon Kim, Michael
Van Oosten, et al. 2008. “The SUMO E3 Ligase, AtSIZ1, Regulates Flowering by
Controlling a Salicylic Acid-Mediated Floral Promotion Pathway and through Affects on
FLC Chromatin Structure.” The Plant Journal 53 (3): 530–40. doi:10.1111/j.1365-
313X.2007.03359.x.
Jing, Beibei, Shaohua Xu, Mo Xu, Yan Li, Shuxin Li, Jinmei Ding, and Yuelin Zhang. 2011.
“Brush and Spray: A High-Throughput Systemic Acquired Resistance Assay Suitable for
Large-Scale Genetic Screening.” Plant Physiology 157 (3): 973–80.
doi:10.1104/pp.111.182089.
Jones, Jonathan D. G., and Jeffery L. Dangl. 2006. “The Plant Immune System.” Nature 444
(7117): 323–29. doi:10.1038/nature05286.
Ju, Chuanli, Gyeong Mee Yoon, Jennifer Marie Shemansky, David Y Lin, Z Irene Ying, Jianhong
Chang, Wesley M Garrett, et al. 2012. “CTR1 Phosphorylates the Central Regulator EIN2
to Control Ethylene Hormone Signaling from the ER Membrane to the Nucleus in
Arabidopsis.” Proceedings of the National Academy of Sciences of the United States of
America 109 (47): 19486–91. doi:10.1073/pnas.1214848109.
Jurkowski, Grace I., Roger K. Smith, I-ching Yu, Jong Hyun Ham, Shashi B. Sharma, Daniel F.
Klessig, Kevin A. Fengler, and Andrew F. Bent. 2004. “Arabidopsis DND2 , a Second
Cyclic Nucleotide-Gated Ion Channel Gene for Which Mutation Causes the ‘ Defense, No
Death ’ Phenotype.” Molecular Plant-Microbe Interactions 17 (5): 511–20.
doi:10.1094/MPMI.2004.17.5.511.
Kanuka, Hirotaka, Shin Hisahara, Kazunobu Sawamoto, Shin-ichi Shoji, Hideyuki Okano, and
Masayuki Miura. 1999. “Proapoptotic Activity of Caenorhabditis Elegans CED-4 Protein in
Drosophila: Implicated Mechanisms for Caspase Activation.” Proceedings of the National
Academy of Sciences 96 (1): 145–50. doi:10.1073/pnas.96.1.145.
Katagiri, Fumiaki, Roger Thilmony, and Sheng Yang He. 2002. “The Arabidopsis Thaliana-
Pseudomonas Syringae Interaction.” The Arabidopsis Book / American Society of Plant
Biologists 1 (March). doi:10.1199/tab.0039.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3243347/.

107
Kim, Moonil, Sanghyeob Lee, Kyoungsook Park, Eun-Ju Jeong, Choong-Min Ryu, Doil Choi, and
Hyun-Sook Pai. 2006. “Comparative Microarray Analysis of Programmed Cell Death
Induced by Proteasome Malfunction and Hypersensitive Response in Plants.” Biochemical
and Biophysical Research Communications 342 (2): 514–21.
doi:10.1016/j.bbrc.2006.01.176.
Kim, Yongsig, Sunchung Park, Sarah Gilmour, and Michael Thomashow. 2013. “Roles of
CAMTA Transcription Factors and Salicylic Acid in Configuring the Low Temperature
Transcriptome and Freezing Tolerance of Arabidopsis.” The Plant Journal : For Cell and
Molecular Biology. doi:10.1111/tpj.12205. http://dx.doi.org/10.1111/tpj.12205.
Knepper, Caleb, Elizabeth A Savory, and Brad Day. 2011. “Arabidopsis NDR1 Is an Integrin-like
Protein with a Role in Fluid Loss and Plasma Membrane-Cell Wall Adhesion.” Plant
Physiology 156 (1): 286–300. doi:10.1104/pp.110.169656.
Knight, H, A J Trewavas, and M R Knight. 1996. “Cold Calcium Signaling in Arabidopsis
Involves Two Cellular Pools and a Change in Calcium Signature after Acclimation.” The
Plant Cell 8 (3): 489–503. doi:10.1105/tpc.8.3.489.
Knoth, Colleen, Jon Ringler, Jeffery L Dangl, and Thomas Eulgem. 2007. “Arabidopsis WRKY70
Is Required for Full RPP4-Mediated Disease Resistance and Basal Defense against
Hyaloperonospora Parasitica.” Molecular Plant-Microbe Interactions: MPMI 20 (2): 120–
28. doi:10.1094/MPMI-20-2-0120.
Kobe, Bostjan, and Andrey V Kajava. 2001. “The Leucine-Rich Repeat as a Protein Recognition
Motif.” Current Opinion in Structural Biology 11 (6): 725–32. doi:10.1016/S0959-
440X(01)00266-4.
Koch, E., and A. Slusarenko. 1990. “Arabidopsis Is Susceptible to Infection by a Downy Mildew
Fungus.” The Plant Cell Online 2 (5): 437–45. doi:10.1105/tpc.2.5.437.
Kodama, Yutaka, and Chang-Deng Hu. 2012. “Bimolecular Fluorescence Complementation
(BiFC): A 5-Year Update and Future Perspectives.” BioTechniques 53 (5): 285–98.
doi:10.2144/000113943.
Kong, Qing, Na Qu, Minghui Gao, Zhibin Zhang, Xiaojun Ding, Fan Yang, Yingzhong Li, et al.
2012. “The MEKK1-MKK1/MKK2-MPK4 Kinase Cascade Negatively Regulates
Immunity Mediated by a Mitogen-Activated Protein Kinase Kinase Kinase in Arabidopsis.”
The Plant Cell Online, May. doi:10.1105/tpc.112.097253.
http://www.plantcell.org/content/early/2012/05/27/tpc.112.097253.
Krol, Elzbieta, Tobias Mentzel, Delphine Chinchilla, Thomas Boller, Georg Felix, Birgit
Kemmerling, Sandra Postel, et al. 2010. “Perception of the Arabidopsis Danger Signal
Peptide 1 Involves the Pattern Recognition Receptor AtPEPR1 and Its Close Homologue
AtPEPR2.” The Journal of Biological Chemistry 285 (18): 13471–79.
doi:10.1074/jbc.M109.097394.
Laluk, K, K Prasad, T Savchenko, H Celesnik, K Dehesh, M Levy, T Mitchell-Olds, and A Reddy.
2012. “The Calmodulin-Binding Transcription Factor SIGNAL RESPONSIVE1 Is a Novel
Regulator of Glucosinolate Metabolism and Herbivory Tolerance in Arabidopsis.” Plant &
Cell Physiology 53 (12): 2008–15. doi:10.1093/pcp/pcs143.
Lam, Eric, Naohiro Kato, and Michael Lawton. 2001. “Programmed Cell Death, Mitochondria and
the Plant Hypersensitive Response.” Nature 411 (6839): 848–53. doi:10.1038/35081184.
Lecourieux, David, Raoul Ranjeva, and Alain Pugin. 2006. “Calcium in Plant Defence-Signalling
Pathways.” The New Phytologist 171 (2): 249–69. doi:10.1111/j.1469-8137.2006.01777.x.

108
Leng, Qiang, Richard W. Mercier, Weizhe Yao, and Gerald A. Berkowitz. 1999. “Cloning and
First Functional Characterization of a Plant Cyclic Nucleotide-Gated Cation Channel.”
Plant Physiology 121 (3): 753–61.
Leon-Reyes, Antonio, Yujuan Du, Annemart Koornneef, Silvia Proietti, Ana P Körbes, Johan
Memelink, Corné M J Pieterse, and Tita Ritsema. 2010. “Ethylene Signaling Renders the
Jasmonate Response of Arabidopsis Insensitive to Future Suppression by Salicylic Acid.”
Molecular Plant-Microbe Interactions: MPMI 23 (2): 187–97. doi:10.1094/MPMI-23-2-
0187.
Levine, Alex, Raimund Tenhaken, Richard Dixon, and Chris Lamb. 1994. “H2O2 from the
Oxidative Burst Orchestrates the Plant Hypersensitive Disease Resistance Response.” Cell
79 (4): 583–93. doi:10.1016/0092-8674(94)90544-4.
Lewit-Bentley, Anita, and Stéphane Réty. 2000. “EF-Hand Calcium-Binding Proteins.” Current
Opinion in Structural Biology 10 (6): 637–43. doi:10.1016/S0959-440X(00)00142-1.
Li, Yansha, Lichao Chen, Jinye Mu, and Jianru Zuo. 2013. “LESION SIMULATING DISEASE1
Interacts with Catalases to Regulate Hypersensitive Cell Death in Arabidopsis.” Plant
Physiology 163 (2): 1059–70. doi:10.1104/pp.113.225805.
Li, Yingzhong, Shuxin Li, Dongling Bi, Yu Ti Cheng, Xin Li, and Yuelin Zhang. 2010. “SRFR1
Negatively Regulates Plant NB-LRR Resistance Protein Accumulation to Prevent
Autoimmunity.” PLoS Pathog 6 (9): e1001111. doi:10.1371/journal.ppat.1001111.
Li, Yuan-Yuan, Hui Yu, Zong-Ming Guo, Ting-Qing Guo, Kang Tu, and Yi-Xue Li. 2006.
“Systematic Analysis of Head-to-Head Gene Organization: Evolutionary Conservation and
Potential Biological Relevance.” PLoS Comput Biol 2 (7): e74.
doi:10.1371/journal.pcbi.0020074.
Liang, Hua, Nan Yao, Jong Tae Song, Song Luo, Hua Lu, and Jean T Greenberg. 2003.
“Ceramides Modulate Programmed Cell Death in Plants.” Genes & Development 17 (21):
2636–41. doi:10.1101/gad.1140503.
Lindeman, Raymond L. 1991. “The Trophic-Dynamic Aspect of Ecology.” Bulletin of
Mathematical Biology 53 (1-2): 167–91. doi:10.1007/BF02464428.
Lindermayr, Christian, Simone Sell, Bernd Müller, Dario Leister, and Jörg Durner. 2010. “Redox
Regulation of the NPR1-TGA1 System of Arabidopsis Thaliana by Nitric Oxide.” The
Plant Cell Online 22 (8): 2894–2907. doi:10.1105/tpc.109.066464.
Lorenzo, Oscar, Jose M. Chico, Jose J. Sánchez-Serrano, and Roberto Solano. 2004.
“JASMONATE-INSENSITIVE1 Encodes a MYC Transcription Factor Essential to
Discriminate between Different Jasmonate-Regulated Defense Responses in Arabidopsis.”
The Plant Cell Online 16 (7): 1938–50. doi:10.1105/tpc.022319.
Lorrain, Séverine, Fabienne Vailleau, Claudine Balagué, and Dominique Roby. 2003. “Lesion
Mimic Mutants: Keys for Deciphering Cell Death and Defense Pathways in Plants?”
Trends in Plant Science 8 (6): 263–71. doi:10.1016/S1360-1385(03)00108-0.
Lu, Shunwen. 2012. “Use of the Yeast Two-Hybrid System to Identify Targets of Fungal
Effectors.” In Plant Fungal Pathogens, edited by Melvin D. Bolton and Bart P.H.J.
Thomma, 835:165–89. Totowa, NJ: Humana Press.
http://www.springerlink.com/content/j55qrt3h16m70p0l/#section=1006223&page=1.
Lukasik, Ewa, and Frank LW Takken. 2009. “STANDing Strong, Resistance Proteins Instigators
of Plant Defence.” Current Opinion in Plant Biology 12 (4): 427–36.
doi:10.1016/j.pbi.2009.03.001.

109
Lundberg, Derek S, Sarah L Lebeis, Sur Herrera Paredes, Scott Yourstone, Jase Gehring,
Stephanie Malfatti, Julien Tremblay, et al. 2012. “Defining the Core Arabidopsis Thaliana
Root Microbiome.” Nature 488 (7409): 86–90. doi:10.1038/nature11237.
Mackey, David, Youssef Belkhadir, Jose M Alonso, Joseph R Ecker, and Jeffery L Dangl. 2003.
“Arabidopsis RIN4 Is a Target of the Type III Virulence Effector AvrRpt2 and Modulates
RPS2-Mediated Resistance.” Cell 112 (3): 379–89.
Mackey, David, Ben F Holt 3rd, Aaron Wiig, and Jeffery L Dangl. 2002. “RIN4 Interacts with
Pseudomonas Syringae Type III Effector Molecules and Is Required for RPM1-Mediated
Resistance in Arabidopsis.” Cell 108 (6): 743–54.
Macnab, Robert M. 2004. “Type III Flagellar Protein Export and Flagellar Assembly.” Biochimica
et Biophysica Acta (BBA) - Molecular Cell Research 1694 (1–3). Protein Export/Secretion
in Bacteria: 207–17. doi:10.1016/j.bbamcr.2004.04.005.
Maekawa, Takaki, Wei Cheng, Laurentiu N Spiridon, Armin Töller, Ewa Lukasik, Yusuke Saijo,
Peiyuan Liu, et al. 2011. “Coiled-Coil Domain-Dependent Homodimerization of
Intracellular Barley Immune Receptors Defines a Minimal Functional Module for
Triggering Cell Death.” Cell Host & Microbe 9 (3): 187–99.
doi:10.1016/j.chom.2011.02.008.
Malamy, Jocelyn, John P. Carr, Daniel F. Klessig, and Ilya Raskin. 1990. “Salicylic Acid: A
Likely Endogenous Signal in the Resistance Response of Tobacco to Viral Infection.”
Science 250 (4983): 1002–4. doi:10.1126/science.250.4983.1002.
Manners, John M., Iris A. M. A. Penninckx, Katrien Vermaere, Kemal Kazan, Rebecca L. Brown,
Andrew Morgan, Donald J. Maclean, Mark D. Curtis, Bruno P. A. Cammue, and Willem F.
Broekaert. 1998. “The Promoter of the Plant Defensin Gene PDF1.2 from Arabidopsis Is
Systemically Activated by Fungal Pathogens and Responds to Methyl Jasmonate but Not to
Salicylic Acid.” Plant Molecular Biology 38 (6): 1071–80. doi:10.1023/A:1006070413843.
McDowell, J M, A Cuzick, C Can, J Beynon, J L Dangl, and E B Holub. 2000. “Downy Mildew
(Peronospora Parasitica) Resistance Genes in Arabidopsis Vary in Functional Requirements
for NDR1, EDS1, NPR1 and Salicylic Acid Accumulation.” The Plant Journal: For Cell
and Molecular Biology 22 (6): 523–29.
McHale, Leah, Xiaoping Tan, Patrice Koehl, and Richard W Michelmore. 2006. “Plant NBS-LRR
Proteins: Adaptable Guards.” Genome Biology 7 (4): 212. doi:10.1186/gb-2006-7-4-212.
Melotto, M., W. Underwood, J. Koczan, K. Nomura, and S.Y. He. 2006. “Plant Stomata Function
in Innate Immunity against Bacterial Invasion.” Cell 126 (5): 969–80.
Merchante, Catharina, Jose M Alonso, and Anna N Stepanova. 2013. “Ethylene Signaling: Simple
Ligand, Complex Regulation.” Current Opinion in Plant Biology 16 (5). Cell Signalling
and Gene Regulation: 554–60. doi:10.1016/j.pbi.2013.08.001.
Mestre, Pere, and David Baulcombe. 2006. “Elicitor-Mediated Oligomerization of the Tobacco N
Disease Resistance Protein.” The Plant Cell 18 (2): 491–501. doi:10.1105/tpc.105.037234.
Meyers, Blake C., Alexander Kozik, Alyssa Griego, Hanhui Kuang, and Richard W. Michelmore.
2003. “Genome-Wide Analysis of NBS-LRR–Encoding Genes in Arabidopsis.” The Plant
Cell Online 15 (4): 809–34. doi:10.1105/tpc.009308.
Meyers, Blake C., Katherine A. Shen, Pejman Rohani, Brandon S. Gaut, and Richard W.
Michelmore. 1998. “Receptor-like Genes in the Major Resistance Locus of Lettuce Are
Subject to Divergent Selection.” The Plant Cell Online 10 (11): 1833–46.
doi:10.1105/tpc.10.11.1833.
Mhamdi, Amna, Guillaume Queval, Sejir Chaouch, Sandy Vanderauwera, Frank Van Breusegem,
and Graham Noctor. 2010. “Catalase Function in Plants: A Focus on Arabidopsis Mutants

110
as Stress-Mimic Models.” Journal of Experimental Botany 61 (15): 4197–4220.
doi:10.1093/jxb/erq282.
Miao, Ying, Thomas M. Laun, Anja Smykowski, and Ulrike Zentgraf. 2007. “Arabidopsis
MEKK1 Can Take a Short Cut: It Can Directly Interact with Senescence-Related WRKY53
Transcription Factor on the Protein Level and Can Bind to Its Promoter.” Plant Molecular
Biology 65 (1-2): 63–76. doi:10.1007/s11103-007-9198-z.
Miura, Kenji, Jing Bo Jin, Jiyoung Lee, Chan Yul Yoo, Vicki Stirm, Tomoko Miura, Edward N.
Ashworth, Ray A. Bressan, Dae-Jin Yun, and Paul M. Hasegawa. 2007. “SIZ1-Mediated
Sumoylation of ICE1 Controls CBF3/DREB1A Expression and Freezing Tolerance in
Arabidopsis.” The Plant Cell Online 19 (4): 1403–14. doi:10.1105/tpc.106.048397.
Miya, Ayako, Premkumar Albert, Tomonori Shinya, Yoshitake Desaki, Kazuya Ichimura, Ken
Shirasu, Yoshihiro Narusaka, Naoto Kawakami, Hanae Kaku, and Naoto Shibuya. 2007.
“CERK1, a LysM Receptor Kinase, Is Essential for Chitin Elicitor Signaling in
Arabidopsis.” Proceedings of the National Academy of Sciences 104 (49): 19613–18.
doi:10.1073/pnas.0705147104.
Modzelewska, Katarzyna, Marc G. Elgort, Jingyu Huang, Gregg Jongeward, Amara Lauritzen,
Charles H. Yoon, Paul W. Sternberg, and Nadeem Moghal. 2007. “An Activating Mutation
in Sos-1 Identifies Its Dbl Domain as a Critical Inhibitor of the Epidermal Growth Factor
Receptor Pathway during Caenorhabditis Elegans Vulval Development.” Molecular and
Cellular Biology 27 (10): 3695–3707. doi:10.1128/MCB.01630-06.
Moeder, Wolfgang, and Keiko Yoshioka. 2008. “Lesion Mimic Mutants.” Plant Signaling &
Behavior 3 (10): 764–67.
Moffett, Peter, Garry Farnham, Jack Peart, and David C. Baulcombe. 2002. “Interaction between
Domains of a Plant NBS-LRR Protein in Disease Resistance-Related Cell Death.” The
EMBO Journal 21 (17): 4511–19. doi:10.1093/emboj/cdf453.
Mondragón-Palomino, Mariana, Blake C. Meyers, Richard W. Michelmore, and Brandon S. Gaut.
2002. “Patterns of Positive Selection in the Complete NBS-LRR Gene Family of
Arabidopsis Thaliana.” Genome Research 12 (9): 1305–15. doi:10.1101/gr.159402.
Morgan, William, and Sophien Kamoun. 2007. “RXLR Effectors of Plant Pathogenic Oomycetes.”
Current Opinion in Microbiology 10 (4): 332–38. doi:10.1016/j.mib.2007.04.005.
Mur, Luis A J, Paul Kenton, Amanda J Lloyd, Helen Ougham, and Elena Prats. 2008. “The
Hypersensitive Response; the Centenary Is upon Us but How Much Do We Know?”
Journal of Experimental Botany 59 (3): 501–20. doi:10.1093/jxb/erm239.
Nakagawa, Tsuyoshi, Takamasa Suzuki, Satoko Murata, Shinya Nakamura, Takeshi Hino,
Kenichiro Maeo, Ryo Tabata, et al. 2007. “Improved Gateway Binary Vectors: High-
Performance Vectors for Creation of Fusion Constructs in Transgenic Analysis of Plants.”
Bioscience, Biotechnology, and Biochemistry 71 (8): 2095–2100.
Nakamura, Shinya, Shoji Mano, Yuji Tanaka, Masato Ohnishi, Chihiro Nakamori, Masami Araki,
Tomoko Niwa, et al. 2010. “Gateway Binary Vectors with the Bialaphos Resistance Gene,
<I>bar</I>, as a Selection Marker for Plant Transformation.” Bioscience, Biotechnology,
and Biochemistry 74 (6): 1315–19.
Narusaka, Mari, Yasuyuki Kubo, Katsunori Hatakeyama, Jun Imamura, Hiroshi Ezura, Yoshihiko
Nanasato, Yutaka Tabei, Yoshitaka Takano, Ken Shirasu, and Yoshihiro Narusaka. 2013a.
“Interfamily Transfer of Dual NB-LRR Genes Confers Resistance to Multiple Pathogens.”
PLoS ONE 8 (2): e55954. doi:10.1371/journal.pone.0055954.
———. 2013b. “Breaking Restricted Taxonomic Functionality by Dual Resistance Genes.” Plant
Signaling & Behavior 8 (6): e24244. doi:10.4161/psb.24244.

111
Narusaka, Mari, Ken Shirasu, Yoshiteru Noutoshi, Yasuyuki Kubo, Tomonori Shiraishi, Masaki
Iwabuchi, and Yoshihiro Narusaka. 2009. “RRS1 and RPS4 Provide a Dual Resistance-
Gene System against Fungal and Bacterial Pathogens.” The Plant Journal 60 (2): 218–26.
doi:10.1111/j.1365-313X.2009.03949.x.
Nawrath, Christiane, and Jean-Pierre Métraux. 1999. “Salicylic Acid Induction–Deficient Mutants
of Arabidopsis Express PR-2 and PR-5 and Accumulate High Levels of Camalexin after
Pathogen Inoculation.” The Plant Cell Online 11 (8): 1393–1404.
doi:10.1105/tpc.11.8.1393.
Nicaise, Valerie, Milena Roux, and Cyril Zipfel. 2009. “Recent Advances in PAMP-Triggered
Immunity against Bacteria: Pattern Recognition Receptors Watch over and Raise the
Alarm.” Plant Physiology 150 (4): 1638–47. doi:10.1104/pp.109.139709.
Nie, Haozhen, Chunzhao Zhao, Guangheng Wu, Yingying Wu, Yongfang Chen, and Dingzhong
Tang. 2012. “SR1, a Calmodulin-Binding Transcription Factor, Modulates Plant Defense
and Ethylene-Induced Senescence by Directly Regulating NDR1 and EIN3.” Plant
Physiology 158 (4): 1847–59. doi:10.1104/pp.111.192310.
Nørholm, Morten HH. 2010. “A Mutant Pfu DNA Polymerase Designed for Advanced Uracil-
Excision DNA Engineering.” BMC Biotechnology 10 (March): 21. doi:10.1186/1472-6750-
10-21.
Nour-Eldin, Hussam H, Fernando Geu-Flores, and Barbara A Halkier. 2010. “USER Cloning and
USER Fusion: The Ideal Cloning Techniques for Small and Big Laboratories.” Methods in
Molecular Biology (Clifton, N.J.) 643: 185–200. doi:10.1007/978-1-60761-723-5_13.
Nour-Eldin, Hussam H., Bjarne G. Hansen, Morten H. H. Nørholm, Jacob K. Jensen, and Barbara
A. Halkier. 2006. “Advancing Uracil-Excision Based Cloning towards an Ideal Technique
for Cloning PCR Fragments.” Nucleic Acids Research 34 (18): e122–e122.
doi:10.1093/nar/gkl635.
Noutoshi, Yoshiteru, Takuya Ito, Motoaki Seki, Hideo Nakashita, Shigeo Yoshida, Yves Marco,
Ken Shirasu, and Kazuo Shinozaki. 2005. “A Single Amino Acid Insertion in the WRKY
Domain of the Arabidopsis TIR-NBS-LRR-WRKY-Type Disease Resistance Protein SLH1
(sensitive to Low Humidity 1) Causes Activation of Defense Responses and Hypersensitive
Cell Death.” The Plant Journal: For Cell and Molecular Biology 43 (6): 873–88.
doi:10.1111/j.1365-313X.2005.02500.x.
Oirdi, Mohamed El, Taha Abd El Rahman, Luciano Rigano, Abdelbasset El Hadrami, María
Cecilia Rodriguez, Fouad Daayf, Adrian Vojnov, and Kamal Bouarab. 2011. “Botrytis
Cinerea Manipulates the Antagonistic Effects between Immune Pathways to Promote
Disease Development in Tomato.” The Plant Cell Online 23 (6): 2405–21.
doi:10.1105/tpc.111.083394.
Okuyama, Yudai, Hiroyuki Kanzaki, Akira Abe, Kentaro Yoshida, Muluneh Tamiru, Hiromasa
Saitoh, Takahiro Fujibe, et al. 2011. “A Multifaceted Genomics Approach Allows the
Isolation of the Rice Pia-Blast Resistance Gene Consisting of Two Adjacent NBS-LRR
Protein Genes.” The Plant Journal: For Cell and Molecular Biology 66 (3): 467–79.
doi:10.1111/j.1365-313X.2011.04502.x.
Padmanabhan, Meenu S., Shisong Ma, Tessa M. Burch-Smith, Kirk Czymmek, Peter Huijser, and
Savithramma P. Dinesh-Kumar. 2013. “Novel Positive Regulatory Role for the SPL6
Transcription Factor in the N TIR-NB-LRR Receptor-Mediated Plant Innate Immunity.”
PLoS Pathog 9 (3): e1003235. doi:10.1371/journal.ppat.1003235.
Palma, Kristoffer, Stephan Thorgrimsen, Frederikke Gro Malinovsky, Berthe Katrine Fiil, H. Bjørn
Nielsen, Peter Brodersen, Daniel Hofius, Morten Petersen, and John Mundy. 2010.

112
“Autoimmunity in Arabidopsis acd11 Is Mediated by Epigenetic Regulation of an Immune
Receptor.” PLoS Pathog 6 (10): e1001137. doi:10.1371/journal.ppat.1001137.
Parker, J. E., M. J. Coleman, V. Szabò, L. N. Frost, R. Schmidt, E. A. van der Biezen, T. Moores,
C. Dean, M. J. Daniels, and J. D. Jones. 1997. “The Arabidopsis Downy Mildew
Resistance Gene RPP5 Shares Similarity to the Toll and Interleukin-1 Receptors with N
and L6.” The Plant Cell Online 9 (6): 879–94. doi:10.1105/tpc.9.6.879.
Peart, Jack, Pere Mestre, Rui Lu, Isabelle Malcuit, and David Baulcombe. 2005. “NRG1, a CC-
NB-LRR Protein, Together with N, a TIR-NB-LRR Protein, Mediates Resistance against
Tobacco Mosaic Virus.” Current Biology : CB 15 (10): 968–73.
doi:10.1016/j.cub.2005.04.053.
Petersen, M, P Brodersen, H Naested, E Andreasson, U Lindhart, B Johansen, H B Nielsen, et al.
2000. “Arabidopsis Map Kinase 4 Negatively Regulates Systemic Acquired Resistance.”
Cell 103 (7): 1111–20.
Pillitteri, Lynn Jo, and Juan Dong. 2013. “Stomatal Development in Arabidopsis.” The
Arabidopsis Book / American Society of Plant Biologists 11 (June). doi:10.1199/tab.0162.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3711358/.
Piston, David W., and Gert-Jan Kremers. 2007. “Fluorescent Protein FRET: The Good, the Bad
and the Ugly.” Trends in Biochemical Sciences 32 (9): 407–14.
doi:10.1016/j.tibs.2007.08.003.
Pitzschke, Andrea, Armin Djamei, Frederique Bitton, and Heribert Hirt. 2009. “A Major Role of
the MEKK1-MKK1/2-MPK4 Pathway in ROS Signalling.” Molecular Plant 2 (1): 120–37.
doi:10.1093/mp/ssn079.
Qi, Dong, Brody J. DeYoung, and Roger W. Innes. 2012. “Structure-Function Analysis of the
Coiled-Coil and Leucine-Rich Repeat Domains of the RPS5 Disease Resistance
Protein1[W][OA].” Plant Physiology 158 (4): 1819–32. doi:10.1104/pp.112.194035.
Qi, Dong, Ullrich Dubiella, Sang Hee Kim, D Isaiah Sloss, Robert H Dowen, Jack E Dixon, and
Roger W Innes. 2014. “Recognition of the Protein Kinase AVRPPHB SUSCEPTIBLE1 by
the Disease Resistance Protein RESISTANCE TO PSEUDOMONAS SYRINGAE5 Is
Dependent on S-Acylation and an Exposed Loop in AVRPPHB SUSCEPTIBLE1.” Plant
Physiology 164 (1): 340–51. doi:10.1104/pp.113.227686.
Qiao, Hong, Zhouxin Shen, Shao-shan Carol Huang, Robert J Schmitz, Mark A Urich, Steven P
Briggs, and Joseph R Ecker. 2012. “Processing and Subcellular Trafficking of ER-Tethered
EIN2 Control Response to Ethylene Gas.” Science (New York, N.Y.) 338 (6105): 390–93.
doi:10.1126/science.1225974.
Raffaele, Sylvain, and Susana Rivas. 2013. “Regulate and Be Regulated: Integration of Defense
and Other Signals by the AtMYB30 Transcription Factor.” Plant-Microbe Interaction 4:
98. doi:10.3389/fpls.2013.00098.
Raffaele, Sylvain, Susana Rivas, and Dominique Roby. 2006. “An Essential Role for Salicylic
Acid in AtMYB30-Mediated Control of the Hypersensitive Cell Death Program in
Arabidopsis.” FEBS Letters 580 (14): 3498–3504. doi:10.1016/j.febslet.2006.05.027.
Rairdan, Gregory J., Sarah M. Collier, Melanie A. Sacco, Thomas T. Baldwin, Teresa Boettrich,
and Peter Moffett. 2008. “The Coiled-Coil and Nucleotide Binding Domains of the Potato
Rx Disease Resistance Protein Function in Pathogen Recognition and Signaling.” The Plant
Cell Online 20 (3): 739–51. doi:10.1105/tpc.107.056036.
Ranf, Stefanie, Lennart Eschen-Lippold, Pascal Pecher, Justin Lee, and Dierk Scheel. 2011.
“Interplay between Calcium Signalling and Early Signalling Elements during Defence

113
Responses to Microbe- or Damage-Associated Molecular Patterns.” The Plant Journal :
For Cell and Molecular Biology 68 (1): 100–113. doi:10.1111/j.1365-313X.2011.04671.x.
Rasmussen, Magnus W, Milena Roux, Morten Petersen, and John Mundy. 2012. “MAP Kinase
Cascades in Arabidopsis Innate Immunity.” Frontiers in Plant Science 3: 169.
doi:10.3389/fpls.2012.00169.
Raz, V., and R. Fluhr. 1992. “Calcium Requirement for Ethylene-Dependent Responses.” The
Plant Cell Online 4 (9): 1123–30. doi:10.1105/tpc.4.9.1123.
Roberts, Melinda, Saijun Tang, Anna Stallmann, Jeffery L Dangl, and Vera Bonardi. 2013.
“Genetic Requirements for Signaling from an Autoactive Plant NB-LRR Intracellular
Innate Immune Receptor.” PLoS Genetics 9 (4): e1003465.
doi:10.1371/journal.pgen.1003465.
Roux, Milena, Benjamin Schwessinger, Catherine Albrecht, Delphine Chinchilla, Alexandra Jones,
Nick Holton, Frederikke Gro Malinovsky, Mahmut Tör, Sacco de Vries, and Cyril Zipfel.
2011. “The Arabidopsis Leucine-Rich Repeat Receptor–Like Kinases BAK1/SERK3 and
BKK1/SERK4 Are Required for Innate Immunity to Hemibiotrophic and Biotrophic
Pathogens.” The Plant Cell Online 23 (6): 2440–55. doi:10.1105/tpc.111.084301.
Shen, Qian-Hua, Yusuke Saijo, Stefan Mauch, Christoph Biskup, Stéphane Bieri, Beat Keller,
Hikaru Seki, Bekir Ulker, Imre E Somssich, and Paul Schulze-Lefert. 2007. “Nuclear
Activity of MLA Immune Receptors Links Isolate-Specific and Basal Disease-Resistance
Responses.” Science (New York, N.Y.) 315 (5815): 1098–1103.
doi:10.1126/science.1136372.
Shen, Qian-Hua, Yusuke Saijo, Stefan Mauch, Christoph Biskup, Stéphane Bieri, Beat Keller,
Hikaru Seki, Bekir Ülker, Imre E. Somssich, and Paul Schulze-Lefert. 2007. “Nuclear
Activity of MLA Immune Receptors Links Isolate-Specific and Basal Disease-Resistance
Responses.” Science 315 (5815): 1098–1103. doi:10.1126/science.1136372.
Shen, Qian-Hua, Fasong Zhou, Stephane Bieri, Thomas Haizel, Ken Shirasu, and Paul Schulze-
Lefert. 2003. “Recognition Specificity and RAR1/SGT1 Dependence in Barley Mla
Disease Resistance Genes to the Powdery Mildew Fungus.” The Plant Cell Online 15 (3):
732–44. doi:10.1105/tpc.009258.
Sheppard, D. 1994. “Dominant Negative Mutants: Tools for the Study of Protein Function in Vitro
and in Vivo.” American Journal of Respiratory Cell and Molecular Biology 11 (1): 1–6.
doi:10.1165/ajrcmb.11.1.8018332.
Shirano, Yumiko, Pradeep Kachroo, Jyoti Shah, and Daniel F. Klessig. 2002. “A Gain-of-Function
Mutation in an Arabidopsis Toll Interleukin1 Receptor–Nucleotide Binding Site–Leucine-
Rich Repeat Type R Gene Triggers Defense Responses and Results in Enhanced Disease
Resistance.” The Plant Cell Online 14 (12): 3149–62. doi:10.1105/tpc.005348.
Shirasu, K., H. Nakajima, V. K. Rajasekhar, R. A. Dixon, and C. Lamb. 1997. “Salicylic Acid
Potentiates an Agonist-Dependent Gain Control That Amplifies Pathogen Signals in the
Activation of Defense Mechanisms.” The Plant Cell Online 9 (2): 261–70.
doi:10.1105/tpc.9.2.261.
Shirasu, Ken. 2009. “The HSP90-SGT1 Chaperone Complex for NLR Immune Sensors.” Annual
Review of Plant Biology 60 (1): 139–64. doi:10.1146/annurev.arplant.59.032607.092906.
Sinapidou, Eva, Kevin Williams, Lucy Nott, Saleha Bahkt, Mahmut Tör, Ian Crute, Peter Bittner-
Eddy, and Jim Beynon. 2004. “Two TIR:NB:LRR Genes Are Required to Specify
Resistance to Peronospora Parasitica Isolate Cala2 in Arabidopsis.” The Plant Journal: For
Cell and Molecular Biology 38 (6): 898–909. doi:10.1111/j.1365-313X.2004.02099.x.

114
Slack, Jennifer L., K. Schooley, Timothy P. Bonnert, Jennifer L. Mitcham, Eva E. Qwarnstrom,
John E. Sims, and Steven K. Dower. 2000. “Identification of Two Major Sites in the Type I
Interleukin-1 Receptor Cytoplasmic Region Responsible for Coupling to Pro-Inflammatory
Signaling Pathways.” Journal of Biological Chemistry 275 (7): 4670–78.
doi:10.1074/jbc.275.7.4670.
Slootweg, Erik, Jan Roosien, Laurentiu Spiridon, Andrei-Jose Petrescu, Wladimir Tameling,
Matthieu Joosten, Rikus Pomp, et al. 2010. “Nucleocytoplasmic Distribution Is Required
for Activation of Resistance by the Potato NB-LRR Receptor Rx1 and Is Balanced by Its
Functional Domains.” The Plant Cell 22 (12): 4195–4215. doi:10.1105/tpc.110.077537.
Snedden, Wayne A, and Hillel Fromm. 1998. “Calmodulin, Calmodulin-Related Proteins and Plant
Responses to the Environment.” Trends in Plant Science 3 (8): 299–304.
doi:10.1016/S1360-1385(98)01284-9.
Spoel, Steven H., Jessica S. Johnson, and Xinnian Dong. 2007. “Regulation of Tradeoffs between
Plant Defenses against Pathogens with Different Lifestyles.” Proceedings of the National
Academy of Sciences 104 (47): 18842–47. doi:10.1073/pnas.0708139104.
Stokes, Trevor L., Barbara N. Kunkel, and Eric J. Richards. 2002. “Epigenetic Variation in
Arabidopsis Disease Resistance.” Genes & Development 16 (2): 171–82.
doi:10.1101/gad.952102.
Stracke, Ralf, Martin Werber, and Bernd Weisshaar. 2001. “The R2R3-MYB Gene Family in
Arabidopsis Thaliana.” Current Opinion in Plant Biology 4 (5): 447–56.
doi:10.1016/S1369-5266(00)00199-0.
Sun, Wenxian, Yangrong Cao, Kristin Jansen Labby, Pascal Bittel, Thomas Boller, and Andrew F
Bent. 2012. “Probing the Arabidopsis Flagellin Receptor: FLS2-FLS2 Association and the
Contributions of Specific Domains to Signaling Function.” The Plant Cell Online 24 (3):
1096–1113. doi:10.1105/tpc.112.095919.
Szabo, L J, and W R Bushnell. 2001. “Hidden Robbers: The Role of Fungal Haustoria in
Parasitism of Plants.” Proceedings of the National Academy of Sciences of the United
States of America 98 (14): 7654–55. doi:10.1073/pnas.151262398.
Takahashi, Akira, Catarina Casais, Kazuya Ichimura, and Ken Shirasu. 2003. “HSP90 Interacts
with RAR1 and SGT1 and Is Essential for RPS2-Mediated Disease Resistance in
Arabidopsis.” Proceedings of the National Academy of Sciences 100 (20): 11777–82.
doi:10.1073/pnas.2033934100.
Takken, F L W, and W I L Tameling. 2009. “To Nibble at Plant Resistance Proteins.” Science
(New York, N.Y.) 324 (5928): 744–46. doi:10.1126/science.1171666.
Takken, Frank L W, and Aska Goverse. 2012. “How to Build a Pathogen Detector: Structural
Basis of NB-LRR Function.” Current Opinion in Plant Biology 15 (4): 375–84.
doi:10.1016/j.pbi.2012.05.001.
Takken, Frank LW, Mario Albrecht, and Wladimir IL Tameling. 2006. “Resistance Proteins:
Molecular Switches of Plant Defence.” Current Opinion in Plant Biology 9 (4): 383–90.
doi:10.1016/j.pbi.2006.05.009.
Tameling, Wladimir I. L, Sandra D. J Elzinga, Patricia S Darmin, Jack H Vossen, Frank L. W
Takken, Michel A Haring, and Ben J. C Cornelissen. 2002. “The Tomato R Gene Products
I-2 and Mi-1 Are Functional ATP Binding Proteins with ATPase Activity.” The Plant Cell
Online 14 (11): 2929–39. doi:10.1105/tpc.005793.
Tameling, Wladimir I. L., and David C. Baulcombe. 2007. “Physical Association of the NB-LRR
Resistance Protein Rx with a Ran GTPase–Activating Protein Is Required for Extreme

115
Resistance to Potato Virus X.” The Plant Cell Online 19 (5): 1682–94.
doi:10.1105/tpc.107.050880.
Tameling, Wladimir I. L., Claudia Nooijen, Nora Ludwig, Marta Boter, Erik Slootweg, Aska
Goverse, Ken Shirasu, and Matthieu H. A. J. Joosten. 2010. “RanGAP2 Mediates
Nucleocytoplasmic Partitioning of the NB-LRR Immune Receptor Rx in the Solanaceae,
Thereby Dictating Rx Function.” The Plant Cell Online 22 (12): 4176–94.
doi:10.1105/tpc.110.077461.
Tapping, Richard I. 2009. “Innate Immune Sensing and Activation of Cell Surface Toll-like
Receptors.” Seminars in Immunology 21 (4): 175–84. doi:10.1016/j.smim.2009.05.003.
Thaler, Jennifer S., Parris T. Humphrey, and Noah K. Whiteman. 2012. “Evolution of Jasmonate
and Salicylate Signal Crosstalk.” Trends in Plant Science 17 (5): 260–70.
doi:10.1016/j.tplants.2012.02.010.
Thomma, Bart P. H. J., Kristel Eggermont, Iris A. M. A. Penninckx, Brigitte Mauch-Mani, Ralph
Vogelsang, Bruno P. A. Cammue, and Willem F. Broekaert. 1998. “Separate Jasmonate-
Dependent and Salicylate-Dependent Defense-Response Pathways in Arabidopsis Are
Essential for Resistance to Distinct Microbial Pathogens.” Proceedings of the National
Academy of Sciences 95 (25): 15107–11.
Thomma, Bart P. H. J., Kristel Eggermont, Koenraad F. M.-J. Tierens, and Willem F. Broekaert.
1999a. “Requirement of Functional Ethylene-Insensitive 2Gene for Efficient Resistance of
Arabidopsis to Infection by Botrytis Cinerea.” Plant Physiology 121 (4): 1093–1101.
doi:10.1104/pp.121.4.1093.
———. 1999b. “Requirement of Functional Ethylene-Insensitive 2Gene for Efficient Resistance
of Arabidopsis to Infection by Botrytis Cinerea.” Plant Physiology 121 (4): 1093–1101.
doi:10.1104/pp.121.4.1093.
Thordal-Christensen, Hans, Ziguo Zhang, Yangdou Wei, and David B. Collinge. 1997.
“Subcellular Localization of H2O2 in Plants. H2O2 Accumulation in Papillae and
Hypersensitive Response during the Barley—powdery Mildew Interaction.” The Plant
Journal 11 (6): 1187–94. doi:10.1046/j.1365-313X.1997.11061187.x.
Toutenhoofd, S L, and E E Strehler. 2000. “The Calmodulin Multigene Family as a Unique Case
of Genetic Redundancy: Multiple Levels of Regulation to Provide Spatial and Temporal
Control of Calmodulin Pools?” Cell Calcium 28 (2): 83–96. doi:10.1054/ceca.2000.0136.
Traw, M. Brian, and Joy Bergelson. 2003. “Interactive Effects of Jasmonic Acid, Salicylic Acid,
and Gibberellin on Induction of Trichomes in Arabidopsis.” Plant Physiology 133 (3):
1367–75. doi:10.1104/pp.103.027086.
Tsuda, Kenichi, Akira Mine, Gerit Bethke, Daisuke Igarashi, Christopher J Botanga, Yayoi Tsuda,
Jane Glazebrook, Masanao Sato, and Fumiaki Katagiri. 2013. “Dual Regulation of Gene
Expression Mediated by Extended MAPK Activation and Salicylic Acid Contributes to
Robust Innate Immunity in Arabidopsis Thaliana.” PLoS Genetics 9 (12): e1004015.
doi:10.1371/journal.pgen.1004015.
Tyler, Brett M, Shiv D Kale, Qunqing Wang, Kai Tao, Helen R Clark, Kelly Drews, Vincenzo
Antignani, et al. 2013. “Microbe-Independent Entry of Oomycete RxLR Effectors and
Fungal RxLR-like Effectors into Plant and Animal Cells Is Specific and Reproducible.”
Molecular Plant-Microbe Interactions: MPMI 26 (6): 611–16. doi:10.1094/MPMI-02-13-
0051-IA.
Ueda, Hirokazu, Yube Yamaguchi, and Hiroshi Sano. 2006. “Direct Interaction between the
Tobacco Mosaic Virus Helicase Domain and the ATP-Bound Resistance Protein, N Factor

116
during the Hypersensitive Response in Tobacco Plants.” Plant Molecular Biology 61 (1-2):
31–45. doi:10.1007/s11103-005-5817-8.
Underwood, William, Maeli Melotto, and Sheng Yang He. 2007. “Role of Plant Stomata in
Bacterial Invasion.” Cellular Microbiology 9 (7): 1621–29. doi:10.1111/j.1462-
5822.2007.00938.x.
Underwood, William, Shuqun Zhang, and Sheng Y He. 2007. “The Pseudomonas Syringae Type
III Effector Tyrosine Phosphatase HopAO1 Suppresses Innate Immunity in Arabidopsis
Thaliana.” The Plant Journal: For Cell and Molecular Biology 52 (4): 658–72.
doi:10.1111/j.1365-313X.2007.03262.x.
Van der Biezen, E A, and J D Jones. 1998. “The NB-ARC Domain: A Novel Signalling Motif
Shared by Plant Resistance Gene Products and Regulators of Cell Death in Animals.”
Current Biology: CB 8 (7): R226–227.
Van der Biezen, E, and J Jones. 1998. “Plant Disease-Resistance Proteins and the Gene-for-Gene
Concept.” Trends in Biochemical Sciences 23 (12): 454–56.
Van Der Hoorn, Renier A. L, and Sophien Kamoun. 2008. “From Guard to Decoy: A New Model
for Perception of Plant Pathogen Effectors.” The Plant Cell Online 20 (8): 2009–17.
doi:10.1105/tpc.108.060194.
Vazquez, Franck, Hervé Vaucheret, Ramya Rajagopalan, Christelle Lepers, Virginie Gasciolli,
Allison C. Mallory, Jean-Louis Hilbert, David P. Bartel, and Patrice Crété. 2004.
“Endogenous Trans-Acting siRNAs Regulate the Accumulation of Arabidopsis mRNAs.”
Molecular Cell 16 (1): 69–79. doi:10.1016/j.molcel.2004.09.028.
Veitia, Reiner A. 2007. “Exploring the Molecular Etiology of Dominant-Negative Mutations.” The
Plant Cell Online 19 (12): 3843–51. doi:10.1105/tpc.107.055053.
Vlot, A Corina, D’Maris Amick Dempsey, and Daniel F Klessig. 2009. “Salicylic Acid, a
Multifaceted Hormone to Combat Disease.” Annual Review of Phytopathology 47: 177–
206. doi:10.1146/annurev.phyto.050908.135202.
Voegele, Ralf T., and Kurt Mendgen. 2003. “Rust Haustoria: Nutrient Uptake and beyond.” New
Phytologist 159 (1): 93–100. doi:10.1046/j.1469-8137.2003.00761.x.
Voinnet, Olivier. 2009. “Origin, Biogenesis, and Activity of Plant MicroRNAs.” Cell 136 (4):
669–87. doi:10.1016/j.cell.2009.01.046.
Wagner, Stephan, Johannes Stuttmann, Steffen Rietz, Raphael Guerois, Elena Brunstein, Jaqueline
Bautor, Karsten Niefind, and Jane E. Parker. 2013. “Structural Basis for Signaling by
Exclusive EDS1 Heteromeric Complexes with SAG101 or PAD4 in Plant Innate
Immunity.” Cell Host & Microbe 14 (6): 619–30. doi:10.1016/j.chom.2013.11.006.
Walker, J E, M Saraste, M J Runswick, and N J Gay. 1982. “Distantly Related Sequences in the
Alpha- and Beta-Subunits of ATP Synthase, Myosin, Kinases and Other ATP-Requiring
Enzymes and a Common Nucleotide Binding Fold.” The EMBO Journal 1 (8): 945–51.
Wan, Jinrong, Xue-Cheng Zhang, David Neece, Katrina M Ramonell, Steve Clough, Sung-Yong
Kim, Minviluz G Stacey, and Gary Stacey. 2008. “A LysM Receptor-Like Kinase Plays a
Critical Role in Chitin Signaling and Fungal Resistance in Arabidopsis.” The Plant Cell
Online 20 (2): 471–81. doi:10.1105/tpc.107.056754.
Wang, Zheng, Pei Meng, Xiaoyan Zhang, Dongtao Ren, and Shuhua Yang. 2011. “BON1 Interacts
with the Protein Kinases BIR1 and BAK1 in Modulation of Temperature-Dependent Plant
Growth and Cell Death in Arabidopsis.” The Plant Journal 67 (6): 1081–93.
doi:10.1111/j.1365-313X.2011.04659.x.
Ward, E. R., S. J. Uknes, S. C. Williams, S. S. Dincher, D. L. Wiederhold, D. C. Alexander, P.
Ahl-Goy, J. P. Metraux, and J. A. Ryals. 1991. “Coordinate Gene Activity in Response to

117
Agents That Induce Systemic Acquired Resistance.” The Plant Cell Online 3 (10): 1085–
94. doi:10.1105/tpc.3.10.1085.
Wawra, Stephan, Mark Agacan, Justin A. Boddey, Ian Davidson, Claire M. M. Gachon, Matteo
Zanda, Severine Grouffaud, et al. 2012. “Avirulence Protein 3a (AVR3a) from the Potato
Pathogen Phytophthora Infestans Forms Homodimers through Its Predicted Translocation
Region and Does Not Specifically Bind Phospholipids.” The Journal of Biological
Chemistry 287 (45): 38101–9. doi:10.1074/jbc.M112.395129.
Wei, Hai-Lei, Suma Chakravarthy, Jay N Worley, and Alan Collmer. 2012. “Consequences of
Flagellin Export through the Type III Secretion System of Pseudomonas Syringae Reveal a
Major Difference in the Innate Immune Systems of Mammals and the Model Plant
Nicotiana Benthamiana.” Cellular Microbiology, October. doi:10.1111/cmi.12059.
Wienkoop, Stefanie, Sacha Baginsky, and Wolfram Weckwerth. 2010. “Arabidopsis Thaliana as a
Model Organism for Plant Proteome Research.” Journal of Proteomics 73 (11): 2239–48.
doi:10.1016/j.jprot.2010.07.012.
Wiermer, Marcel, Bart J Feys, and Jane E Parker. 2005. “Plant Immunity: The EDS1 Regulatory
Node.” Current Opinion in Plant Biology 8 (4): 383–89. doi:10.1016/j.pbi.2005.05.010.
Wigge, Philip A., and Detlef Weigel. 2001. “Arabidopsis Genome: Life without Notch.” Current
Biology 11 (3): R112–R114. doi:10.1016/S0960-9822(01)00043-4.
Wildermuth, M C, J Dewdney, G Wu, and F M Ausubel. 2001. “Isochorismate Synthase Is
Required to Synthesize Salicylic Acid for Plant Defence.” Nature 414 (6863): 562–65.
doi:10.1038/35107108.
Williams, Simon J, Pradeep Sornaraj, Emma deCourcy-Ireland, R Ian Menz, Bostjan Kobe, Jeffrey
G Ellis, Peter N Dodds, and Peter A Anderson. 2011. “An Autoactive Mutant of the M Flax
Rust Resistance Protein Has a Preference for Binding ATP, Whereas Wild-Type M Protein
Binds ADP.” Molecular Plant-Microbe Interactions: MPMI 24 (8): 897–906.
doi:10.1094/MPMI-03-11-0052.
Wilton, Mike, Rajagopal Subramaniam, James Elmore, Corinna Felsensteiner, Gitta Coaker, and
Darrell Desveaux. 2010. “The Type III Effector HopF2 Pto Targets Arabidopsis RIN4
Protein to Promote Pseudomonas Syringae Virulence.” Proceedings of the National
Academy of Sciences 107 (5): 2349–54. doi:10.1073/pnas.0904739107.
Wirthmueller, Lennart, Yan Zhang, Jonathan D.G. Jones, and Jane E. Parker. 2007. “Nuclear
Accumulation of the Arabidopsis Immune Receptor RPS4 Is Necessary for Triggering
EDS1-Dependent Defense.” Current Biology 17 (23): 2023–29.
doi:10.1016/j.cub.2007.10.042.
Wu, T. H., and M. G. Marinus. 1994. “Dominant Negative Mutator Mutations in the mutS Gene of
Escherichia Coli.” Journal of Bacteriology 176 (17): 5393–5400.
Wu, Yue, Di Zhang, Jee Yan Chu, Patrick Boyle, Yong Wang, Ian D Brindle, Vincenzo De Luca,
and Charles Després. 2012. “The Arabidopsis NPR1 Protein Is a Receptor for the Plant
Defense Hormone Salicylic Acid.” Cell Reports 1 (6): 639–47.
doi:10.1016/j.celrep.2012.05.008.
Xia, Shitou, Zhaohai Zhu, Lin Hao, Jin-Gui Chen, Langtao Xiao, Yuelin Zhang, and Xin Li. 2009.
“Negative Regulation of Systemic Acquired Resistance by Replication Factor C Subunit3
in Arabidopsis.” Plant Physiology 150 (4): 2009–17. doi:10.1104/pp.109.138321.
Xiang, Tingting, Na Zong, Yan Zou, Yong Wu, Jie Zhang, Weiman Xing, Yan Li, et al. 2008.
“Pseudomonas Syringae Effector AvrPto Blocks Innate Immunity by Targeting Receptor
Kinases.” Current Biology: CB 18 (1): 74–80. doi:10.1016/j.cub.2007.12.020.

118
Xie, Dao-Xin, Bart F. Feys, Sarah James, Manuela Nieto-Rostro, and John G. Turner. 1998.
“COI1: An Arabidopsis Gene Required for Jasmonate-Regulated Defense and Fertility.”
Science 280 (5366): 1091–94. doi:10.1126/science.280.5366.1091.
Xing, Denghui, and Zhixiang Chen. 2006. “Effects of Mutations and Constitutive Overexpression
of EDS1 and PAD4 on Plant Resistance to Different Types of Microbial Pathogens.” Plant
Science 171 (2): 251–62. doi:10.1016/j.plantsci.2006.03.022.
Xu, Qixian, and Lingrui Zhang. 2009. “Plant Caspase-like Proteases in Plant Programmed Cell
Death.” Plant Signaling & Behavior 4 (9): 902–4.
Xu, Xinping, Chunhong Chen, Baofang Fan, and Zhixiang Chen. 2006. “Physical and Functional
Interactions between Pathogen-Induced Arabidopsis WRKY18, WRKY40, and WRKY60
Transcription Factors.” The Plant Cell 18 (5): 1310–26. doi:10.1105/tpc.105.037523.
Yan, Nieng, Jijie Chai, Eui Seung Lee, Lichuan Gu, Qun Liu, Jiaqing He, Jia-Wei Wu, et al. 2005.
“Structure of the CED-4–CED-9 Complex Provides Insights into Programmed Cell Death
in Caenorhabditis Elegans.” Nature 437 (7060): 831–37. doi:10.1038/nature04002.
Yang, Shuhua, Huijun Yang, Paula Grisafi, Siraprapha Sanchatjate, Gerald R Fink, Qi Sun, and
Jian Hua. 2006. “The BON/CPN Gene Family Represses Cell Death and Promotes Cell
Growth in Arabidopsis.” The Plant Journal: For Cell and Molecular Biology 45 (2): 166–
79. doi:10.1111/j.1365-313X.2005.02585.x.
Yang, Tianbao, and B Poovaiah. 2002. “A Calmodulin-binding/CGCG Box DNA-Binding Protein
Family Involved in Multiple Signaling Pathways in Plants.” The Journal of Biological
Chemistry 277 (47): 45049–58. doi:10.1074/jbc.M207941200.
Yi, Hankuil, and Eric J. Richards. 2007. “A Cluster of Disease Resistance Genes in Arabidopsis Is
Coordinately Regulated by Transcriptional Activation and RNA Silencing.” The Plant Cell
19 (9): 2929–39. doi:10.1105/tpc.107.051821.
———. 2009. “Gene Duplication and Hypermutation of the Pathogen Resistance Gene SNC1 in
the Arabidopsis Bal Variant.” Genetics 183 (4): 1227–34.
doi:10.1534/genetics.109.105569.
Yoda, Hiroshi, Yube Yamaguchi, and Hiroshi Sano. 2003. “Induction of Hypersensitive Cell Death
by Hydrogen Peroxide Produced through Polyamine Degradation in Tobacco Plants.” Plant
Physiology 132 (4): 1973–81. doi:10.1104/pp.103.024737.
Yoo, So Yeon, Kirsten Bomblies, Seung Kwan Yoo, Jung Won Yang, Mi Suk Choi, Jong Seob
Lee, Detlef Weigel, and Ji Hoon Ahn. 2005. “The 35S Promoter Used in a Selectable
Marker Gene of a Plant Transformation Vector Affects the Expression of the Transgene.”
Planta 221 (4): 523–30. doi:10.1007/s00425-004-1466-4.
Yucel, I, Y X Xiao, and S W Hutcheson. 1989. “Influence of Pseudomonas Syringae Culture
Conditions on Initiation of the Hypersensitive Response of Culture Tobacco Cells.”
Applied and Environmental Microbiology 55 (7): 1724–29.
Zhai, Jixian, Dong-Hoon Jeong, Emanuele De Paoli, Sunhee Park, Benjamin D Rosen, Yupeng Li,
Alvaro J González, et al. 2011. “MicroRNAs as Master Regulators of the Plant NB-LRR
Defense Gene Family via the Production of Phased, Trans-Acting siRNAs.” Genes &
Development 25 (23): 2540–53. doi:10.1101/gad.177527.111.
Zhang, Lei, Liqun Du, Chenjia Shen, Yanjun Yang, and B.w. Poovaiah. 2014. “Regulation of Plant
Immunity through Ubiquitin-Mediated Modulation of Ca2+-Calmodulin-AtSR1/CAMTA3
Signaling.” The Plant Journal, n/a–n/a. doi:10.1111/tpj.12473.
Zhang, Yan, Stephan Dorey, Michal Swiderski, and Jonathan D G Jones. 2004. “Expression of
RPS4 in Tobacco Induces an AvrRps4-Independent HR That Requires EDS1, SGT1 and

119
HSP90.” The Plant Journal: For Cell and Molecular Biology 40 (2): 213–24.
doi:10.1111/j.1365-313X.2004.02201.x.
Zhang, Yuelin, Sandra Goritschnig, Xinnian Dong, and Xin Li. 2003. “A Gain-of-Function
Mutation in a Plant Disease Resistance Gene Leads to Constitutive Activation of
Downstream Signal Transduction Pathways in Suppressor of npr1-1, Constitutive 1.” The
Plant Cell Online 15 (11): 2636–46. doi:10.1105/tpc.015842.
Zhang, Zhibin, Yaling Wu, Minghui Gao, Jie Zhang, Qing Kong, Yanan Liu, Hongping Ba,
Jianmin Zhou, and Yuelin Zhang. 2012. “Disruption of PAMP-Induced MAP Kinase
Cascade by a Pseudomonas Syringae Effector Activates Plant Immunity Mediated by the
NB-LRR Protein SUMM2.” Cell Host & Microbe 11 (3): 253–63.
doi:10.1016/j.chom.2012.01.015.
Zhao, Youfu, Roger Thilmony, Carol L. Bender, Andreas Schaller, Sheng Yang He, and Gregg A.
Howe. 2003. “Virulence Systems of Pseudomonas Syringae Pv. Tomato Promote Bacterial
Speck Disease in Tomato by Targeting the Jasmonate Signaling Pathway.” The Plant
Journal 36 (4): 485–99. doi:10.1046/j.1365-313X.2003.01895.x.
Zhao, Yue, Jieling Yang, Jianjin Shi, Yi-Nan Gong, Qiuhe Lu, Hao Xu, Liping Liu, and Feng
Shao. 2011. “The NLRC4 Inflammasome Receptors for Bacterial Flagellin and Type III
Secretion Apparatus.” Nature 477 (7366): 596–600. doi:10.1038/nature10510.
Zheng, Zuyu, Synan Abu Qamar, Zhixiang Chen, and Tesfaye Mengiste. 2006. “Arabidopsis
WRKY33 Transcription Factor Is Required for Resistance to Necrotrophic Fungal
Pathogens.” The Plant Journal: For Cell and Molecular Biology 48 (4): 592–605.
doi:10.1111/j.1365-313X.2006.02901.x.
Zhou, Bo, Shaohong Qu, Guifu Liu, Maureen Dolan, Hajime Sakai, Guodong Lu, Maria Bellizzi,
and Guo-Liang Wang. 2006. “The Eight Amino-Acid Differences Within Three Leucine-
Rich Repeats Between Pi2 and Piz-T Resistance Proteins Determine the Resistance
Specificity to Magnaporthe Grisea.” Molecular Plant-Microbe Interactions 19 (11): 1216–
28. doi:10.1094/MPMI-19-1216.
Zhou, Fasong, Stephen Mosher, Miaoying Tian, Giovanna Sassi, Jane Parker, and Daniel F
Klessig. 2008. “The Arabidopsis Gain-of-Function Mutant ssi4 Requires RAR1 and SGT1b
Differentially for Defense Activation and Morphological Alterations.” Molecular Plant-
Microbe Interactions: MPMI 21 (1): 40–49. doi:10.1094/MPMI-21-1-0040.
Zhu, Shifeng, Rae-Dong Jeong, Srivathsa C. Venugopal, Ludmila Lapchyk, DuRoy Navarre,
Aardra Kachroo, and Pradeep Kachroo. 2011. “SAG101 Forms a Ternary Complex with
EDS1 and PAD4 and Is Required for Resistance Signaling against Turnip Crinkle Virus.”
PLoS Pathog 7 (11): e1002318. doi:10.1371/journal.ppat.1002318.
Zhu, Ying, Baijuan Du, Jun Qian, Baohong Zou, and Jian Hua. 2013. “Disease Resistance Gene-
Induced Growth Inhibition Is Enhanced by rcd1 Independent of Defense Activation in
Arabidopsis.” Plant Physiology 161 (4): 2005–13. doi:10.1104/pp.112.213363.
Zhu, Zhaohai, Fang Xu, Yaxi Zhang, Yu Ti Cheng, Marcel Wiermer, Xin Li, and Yuelin Zhang.
2010. “Arabidopsis Resistance Protein SNC1 Activates Immune Responses through
Association with a Transcriptional Corepressor.” Proceedings of the National Academy of
Sciences 107 (31): 13960–65. doi:10.1073/pnas.1002828107.
Zipfel, Cyril, Gernot Kunze, Delphine Chinchilla, Anne Caniard, Jonathan D.G. Jones, Thomas
Boller, and Georg Felix. 2006. “Perception of the Bacterial PAMP EF-Tu by the Receptor
EFR Restricts Agrobacterium-Mediated Transformation.” Cell 125 (4): 749–60.
doi:10.1016/j.cell.2006.03.037.

120
Zurbriggen, Matias D, Nestor Carrillo, and Mohammad-Reza Hajirezaei. 2010. “ROS Signaling in
the Hypersensitive Response.” Plant Signaling & Behavior 5 (4): 393–96.

121
Manuscript
A Dominant Negative NB-LRR screen uncover suppressors of
autoimmunity in camta3.
Christiaan Greeff, Signe Lolle, Klaus Petersen, Michael Krogh Jensen, John Mundy and
Morten Petersen.
Section for Functional Genomics, Department of Biology, University of Copenhagen, Copenhagen,
Denmark.
Abstract
A small set of Resistance proteins (R-proteins), guards plants against a large set of pathogen effector proteins
that can suppress or subvert plant defense responses. The guard model attempts to solve this discrepancy
by proposing that a major function of R proteins is to monitor host effector targets. In response to effector
mediated changes on targets, R proteins trigger the hypersensitive response.
We have proposed that a corollary to this 'guard model' is that forms of plant autoimmunity are due to
inappropriate R protein activation. For example, we showed that a knockout of Accelerated Cell Death
11 (acd11) leads to inappropriate activation of hypersensitive cell death. We have previously performed a
large-scale survival screen for suppressors of acd11 and found that cell death in acd11 is suppressed by
mutations in a gene encoding an R protein. We have thus proposed that loss of ACD11 results in HR cell
death because LAZ5 directly or indirectly guards it. The LAZ5 alleles we first found were dominant negative
(laz5-DN). The laz-DN allele mutation was found in a conserved functionally important ATP binding region,
the P-loop. Site-directed DN mutant alleles can be made for other R genes, as we have recently found that
transgenics with similarly mutated rpm1-DN alleles lose resistance to Pseudomonas syringae expressing
the AvrRpm1 effector. Accordingly, we have constructed a collection of 100 R-DN alleles and transformed
them into other autoimmune mutants including camta3. CAMTA3 was previously shown to be a negative
regulator of plant defense by inhibiting transcription of EDS1 and NDR1, important downstream signaling
components of R-protein signaling. We found that dominant negative alleles of DSC-D can suppress all
tested camta3-1 phenotypes. We hypothesize that like acd11 and other autoimmune mutants, the increased
levels of defense genes like EDS1 in camta3-1 might be a consequence of R protein activation and not merely
as a result of negative regulation of plant defense responses as was previously proposed. DSC and CAMTA3
are part of a nuclear localized complex supporting the possibility that DSC is directly guarding CAMTA3.

122
Introduction
Both plants and animals employ pattern recognition receptors (PRRs) to detect pathogens
associated molecular patterns (PAMPS) such as bacterial flagellin and fungal chitin. PAMP
perception triggers a defence response collectively known as PAMP triggered immunity
(PTI)(Jones and Dangl 2006). Successful pathogens can deploy effectors into the host cell
that modify host targets to suppress PTI (Gao et al. 2009). Adapted plants can detect such
effectors via cytosolic immune receptors termed resistance (R) proteins which monitor and
guards host effector targets. In response to effector mediated changes in host targets, R
proteins elicit strong defence and cell death programs collectively called the hypersensitive
response (HR) or effector triggered immunity (ETI) (Bray Speth, Lee, and He 2007). The
most prevalent type of plant R proteins belongs to the nucleotide binding site (NB)-leucine-
rich repeat (LRR) class that can be divided into Toll/Interleukin-1 receptor (TIR)- or coiled-
coil (CC)-NB-LRR R proteins (Dangl and Jones, 2006).
The HR is defined as a rapid localised programmed cell death following
pathogen infection (Heath 1998). Active metabolism and protein synthesis is required for
establishment of HR distinguishing it from necrotic cell death (Mur et al. 2008).
Pseudomonas syringae (Pst) inject some 40 effectors via a type III secretion system to
establish infection in Arabidopsis (Buell et al. 2003). These effectors include avrRpm1, that
targets and phosphorylates the host protein RIN4 and avrRps2, that cleave RIN4. These
changes of RIN4 are detected by two R proteins, RPM1 and RPS2, that elicit ETI upon
phosphorylation or in the absence of RIN4, respectively (Belkhadir et al. 2004). Importantly,
loss of RIN4 results in autoimmunity that is dependent on RPS2. Several genetic
requirements for HR have also been described and include ENHANCED DISEASE
SUSEPTABILITY1 (EDS1) and PHYTOALEXIN DEFICIENT4 (PAD4) required for HR

123
mediated by TIR-NBS-LRR and NON-RACE SPECIFIC DISEASE RESISTANCE1 (NDR1)
important for CC-NBS-LRR triggered HR (Aarts et al. 1998).
In screens for regulators of HR several mutants that exhibit spontaneous HR and
autoimmunity in the absence of pathogen infection have been isolated (Lorrain et al.
2003)(Takahashi et al. 1999)(Greenberg and Ausubel 1993)(Dietrich et al.
1994)(Greenberg et al. 1994). In line with the autoimmune phenotype, the genes mutated
in these mutants have in many cases been suggested to be negative regulators of HR cell
death and defence responses (Dietrich et al. 1997)(Petersen et al. 2000)(Du et al. 2009).
Interestingly, the genetic requirement for the autoimmunity in many of these mutants are
largely the same as that found for the HR. For example, most autoimmune mutants exhibit
EDS1/PAD4 or NDR1 dependent phenotypes including stunted growth, accumulation of
reactive oxygen species (ROS) and defence gene expression (Lorrain et al. 2003)
(Brodersen et al. 2002). Like pathogen triggered HR, the spontaneous HR in autoimmune
mutants can also often be suppressed by high temperature(Zhang et al. 2012) Cheng et al.
2013). In addition, autoimmune mutants pheno-copy both over-expression of R genes (Li
et al. 2010:1)(Stokes, Kunkel, and Richards 2002) and constitutive gain of function mutants
caused by specific mutations in R genes (Gao et al. 2011)(Williams et al. 2011).
Recent reports implicate R proteins in the development of autoimmunity. For example,
ACD11 is a sphingolipid transfer protein and acd11 mutants that shows spontaneous cell
death at two week stage. We found that autoimmunity in acd11 is caused by inappropriate
activation of the TIR-NBS-LRR protein we named LAZ5. Acd11 is rescued by laz5 and laz5-
D2 a dominant negative allele (Palma et al. 2010). This suggests that ACD11 or complexes

124
including ACD11 represents an effector target(s) guarded by LAZ5. Similarly, the double
KO of two MAP kinase kinases MKK1 and MKK2 also leads to autoimmunity that can be
suppressed by mutations in the R protein SUMM2. The mkk1/mkk2 pathway is known to
be a target of the HopAI1 effector and this effector activity triggers SUMM2. mkk1/2 double
mutants mimics HopAI1 effector action triggering SUMM2 mediated defence (Zhang et al.
2012). Additionally autoimmune mutant snc-1 has been shown to be a gain of function
mutation in a TIR-NBS-LRR R gene. The snc-1 mutant shows PAD4-dependent
autoimmunity (Zhu et al. 2010). Thus, it is possible that other autoimmune mutants also
represent mutations in favoured effector targets.
CAMTA3, is a calmodulin binding transcription activator and there are 6 related
members of the CAMTA family in Arabidopsis, CAMTA1-6. CAMTA1, CAMTA2 and
CAMTA3 appears to work together to regulate gene expression (Kim et al. 2013) but, loss
of CAMTA3 is sufficient to cause autoimmunity (Du et al. 2009). CAMTA3 has been
implicated in negatively regulating SA mediated defense responses including the negative
regulation of EDS1 expression (Galon et al. 2008). In line with this observation, CAMTA3
was shown capable of binding a CGCG box (vCGCGb) in the EDS1 promoter region and
camta3 exhibit PAD4-dependent autoimmunity (Du et al. 2009). That autoimmunity in
camta3, might be R gene dependent was proposed based on rescue by high temperature
and mutations in PAD4 (Du et al. 2009) .
We aimed to screen for R gene dependant autoimmunity by transforming autoimmune
mutants with dominant negative (DN) versions of some 100 R genes. The NBS domain,
also called NB-ARC domain is part of the signal transduction ATPases with numerous
domains (STAND) family which includes NOD, APAF-1 and CED4. These domains are well

125
conserved and generally function as molecular switches. The NBS domain contains an P-
loop or Walker A motif, GXXXXGKT(T/S)(Leipe et al. 2002) that is important in coordinating
nucleotide binding. Mutations in this motif often cause loss of function. RPS2, Rx, N, Mla10,
M, L6 and I2 function is known to be abolished by P-loop mutations (Tao et al. 2000;
Bendahmane et al. 2002; Mestre and Baulcombe 2006; Williams et al. 2011; Bai et al. 2012;
Takken, Albrecht, and Tameling 2006; Howles et al. 2005). Additionally, N (Dinesh-Kumar,
Tham, and Baker 2000), ADR1(Roberts et al. 2013). and LAZ5 (Palma et al. 2010) P-loop
mutants are dominant negative.
Thus, by transforming autoimmune mutants with our collection of DN R genes, we can link
R genes to specific autoimmunity through a suppressor screen. Using this approach would
also be less labor intensive that crossing autoimmune mutants to R gene knock out lines
and avoid off target problems encountered with RNAi screens (Senthil-Kumar and Mysore
2011). We first show that transgenic plants expressing RPM1 DN are indistinguishable
from rpm1-3 knockout mutants in terms of HR and gene-for-gene resistance and
importantly, we established that the DN mutation does not interfere with common CC-NBS-
LRR or TIR-NBS-LRR signaling pathways.
After introducing our collection of DN genes into autoimmune mutants, we found a DN
mutant of At4g12010 we call dominant negative suppressor of camta3 (DSC-D) that is able
to suppress camta3 autoimmunity. Activation of this R protein can explain much of the
camta3 autoimmunity. This suggests CAMTA3 could represent a ‘gaurdee’ that is being
monitored by this R protein. BiFC and FRET assays support this idea, showing that
CAMTA3 and DSC may be part of a nuclear localised complex.

126
Results
P-loop mutant specificity
Because we and others have observed that specific mutations in the P-loop of R proteins
can display DN effects (Palma et al. 2010; Dinesh-Kumar, Tham, and Baker 2000:-;
Roberts et al. 2013) we decided to introduce P-loop mutations in a large collection of R
genes found in the Arabidopsis genome. RPM1 represent a well-studied R protein and we
first examined the effect of P-loop mutations on the function of RPM1. To this end, P-loop
mutant of RPM1 (GK,221,222,AA) which we named RPM1-D was cloned into a 35S plant
expression vector and introduced into Col-0 background by Agrobacterium-mediated
transformation. Infection with Pst DC3000 (AvrRpm1) triggers cell death in plants
expressing RPM1 and we first decided to compare cell death responses in 4-week-old Col-
0, rpm1-3 and two independent RPM1-D transformants, RPM1-D1 and RPM1-D2. HR cell
death was quantified using an electrolyte leakage assay (Mackey et al., 2003), as cell death
causes release of electrolytes measured as changes in the conductance of a bath solution.
These assays showed that Pst (DC3000) (AvrRpm1) induced cell death caused an increase
in conductance in Col-0 that was apparent already after 3 to 4 hours (Figure 1A). This
increase was significantly suppressed in RPM1-D1 and RPM1-D2 transgenic plants that
did not have statistically different ion leakage profiles from the rpm1 plants (Figure 1A). To
extend our analysis, 4-week-old short-day-grown rosette leaves were syringe-infiltrated
with Pst DC3000 (AvrRpm1) and bacterial growth measured on day 0 and day 3 (Figure
1B) post-infiltration. Time-course assays revealed that after 3 days bacterial growth was
almost 100 times higher in RPM1-D1, RPM1-D2 and rpm1-3 plants compared to Col-0
(Figure 1A). Thus, expression of dominant negative versions of RPM1 in wild type plants
compromise the function of RPM1 and breaks resistance.

127
Next, we decided to test the specificity of the dominant suppression in transgenic plants
expressing the RPM1-D construct. To accomplish this, we examined if another R protein,
the TIR-NBS-LRR RPS4 that recognize AvrRps4, was unaffected by the presence of
RPM1-D. This analysis revealed that transgenic RPM1-D1 and RPM1-D2 plants had similar
bacterial growth of Pst DC3000 (AvrRps4) as Col-0 and rpm1-3 (Figure 1C) and eds1-2,
included as susceptible control, had a significantly higher growth (Figure 1C). Likewise, the
CC-NBS-LRR R protein, RPS2, showed normal responses to Pst DC3000 (AvrRpt2) thus
the presence of dominant negative versions of RPM1 did not inhibit recognition of AvrRpt2
(Figure 1D). The ndr1 included as a susceptible control in this experiment, was significantly
higher than all other samples (Figure 1D). Thus, dominant negative versions of RPM1 do
not influence the function of other R proteins and function indistinguishable from rpm1-null
mutants.
DN library construction and LMM Screens
Since acd11 and mpk4 autoimmune mutants can be suppressed by mutations in specific R
genes (Palma et al., 2010; Zhang et al., 2012), it is possible that this can be attributed to
others as well. We decided to take advantage of our discovery that dominant versions of R
genes function like null alleles and mutated 73 TIR-NBS-LRR R-genes. More specifically,
the conserved NBS P-loop motif GXXXXGKT(T/S) was mutated to GXXXXAAT(T/S) by
employing mismatched primers and a USER based multi fragment cloning (Geu-Flores et
al. 2007) .
We transformed the sequenced clones into Agrobacterium PGV3101 and transformed a
collection of autoimmune mutants. Since our collection of TIR-NBS-LRR DN mutants do
not represent all TIR-NBS-LRR genes found in Col-0 we transformed multiple autoimmune

128
mutants to improve our chances of identifying suppressors. Seeds from 10 independent T1
(T2 seeds) were collected after BASTA selection on soil and screened for suppression of
growth and chlorosis or early flowering phenotypes associated with autoimmunity. Out of
73 DN constructs one, At4g12010 DN, was able to suppress all camta3-1 growth and
chlorosis phenotypes (Figure 2A). No other DN constructs were able to suppress camta3-
1 phenotypes. We named At4g12010 DSC (Dominant suppressor of camta3). At4g12010
(Sup figure 2A) encode a typical TIR-NBS-LRR R protein proposed to be part of an R
protein pair (Narusaka et al. 2009). However crossing camta3 to two DSC T-DNA insertion
lines, SALK_099668 and Sail_49_C05 did however not give us similar suppression (S
Figure 1).
Phenotypic analysis
To examine the suppression in more detail, T3 lines homozygous for DSC-D were selected.
6 week old camta3-1 mutants grown in SD conditions shows stunted growth and necrosis
in older leaves (Figure2A) and (Du et al. 2009). In contrast camta3-1/DSC-D1 grown under
the same conditions showed WT growth. DSC-D3 had similar fresh growth weight as Col-
0 grown under the same conditions (Figure2A). In addition, microscopic analysis of camta3-
1 mutant leaves stained with trypan blue to detect dead cells showed massive cell death of
individual mesophyll cells. This contrasted camta3-1/DSC-D1/D2 in which there was a
complete absence of cell death (Figure 2B). Finally, DAB staining also indicated that the
increased ROS produced in camta3-1 is absent in camta3-1 /DSC-D1/D2 lines (Figure 2C).
We next examined if suppression of growth retardation and cell death could be translated
to suppression of the enhanced defence phenotype of camta3-1 (Galon et al. 2008:3). To
this end, we first spray inoculated 4 week old plants with Pst DC3000 and Pst

129
DC3000(avrRPS4) and measured bacterial growth after 3 days. Increased resistance to
both strains could be seen in camta3-1 (Figure 3A, B). However in camta3-1/DSC-D1 and
camta3-1/DSC-D2 bacterial growth was restored to wild type levels for both virulent Pst
DC3000 (Figure 3A) and avirulent Pst DC3000(AvrRps4) (Figure 3B). Importantly, DN
versions of At4g12010 transformed into wild type Col-0,DSC-D3 and DSC-D4, also
supported the same Pst DC3000 bacterial growth level as Col-0 and thus, DSC-D by itself
did not alter Pst infectivity. The eds1-2, was included as susceptible control and had a
significantly higher growth of Pst DC3000 (avrRPS4) (Figure 3B) (Heidrich et al. 2011).
Defence gene induction
Camta3-1 mutants also constitutively express defence genes including the well-established
Pathogenesis-Related1 (PR1) gene (Du et al. 2009). We therefore analysed mRNA levels
of PR1 by QPCR. Figure 4A shows PR1 defence marker gene expression in Col-0, camta3-
1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4. PR1 gene expression was
completely restored to WT levels in camta3-1/DSC-D1 and camta3-1/DSC-D2. No
statistical significant differences were seen between camta3-1/DSC-D1, camta3-1/DSC-
D2, DSC-D3, DSC-D4 and Col-0 (Figure 4A).
Since its been proposed that CAMTA3 is a negative regulator of EDS1 expression, we also
analysed levels of EDS1 (Du et al. 2009). In agreement with this finding, a 6 fold change
in EDS1 mRNA levels between camta3-1 and Col-0 was seen at 6 week growth stage when
cell death phenotype is well established (Figure 4B). However, camta3-1/DSC-D1 and
camta3-1/DSC-D2 showed WT EDS1 mRNA levels. Moreover, no statistical significant
differences were seen for induction of EDS1 expression in 4 week old plants after Pst

130
DC3000 (avrRPS4) inoculation, in all lines tested. Thus the presence of DSC-D does not
appear to cause a general suppression of EDS1 mRNA expression. Interestingly, we could
not detect differences in the abundance of EDS1 transcript levels in camta3-1 and Col-0 in
4 week old plants (Figure 4C) and no statistical significant differences were seen in the
induction of EDS1 levels by avrRPS4 in of Col-0, camta3-1, camta3-1/DSC-D1, camta3-
1/DSC-D2, DSC-D3 and DSC-D4 after 4 hrs (Figure 4C or D?)
CAMTA3 and DSC1 exist in nuclear complexes
The suppression of camta3-1 by DSC-D could indicate that CAMTA3 is under surveillance
and autoimmunity is triggered by DSC in the absence of CAMTA3. A possibility that could
imply DSC and CAMTA3 could be found in complexes. We therefore first examined the
cellular localization of CAMTA3 fused to CFP and DSC or DSC-D1 fused to YFP.
Interestingly, all showed nuclear localisation when transiently expressed in N.
benthamiana. (Figure 5). Thus, the P-loop mutation did not change localisation pattern of
DSC (Figure 5 B and C) and co-expression of CAMTA3 and DSC did not influence
localisation (Figure 5A). Since DSC and CAMTA3 are nuclear localised it is possible that
they interact similar to RPS2 and RIN4 (Belkhadir et al. 2004) and we therefore tested their
physical interaction in vivo using BiFC (Figure 6). Nuclear localised BiFC signal was
consistently seen with co-infiltrated N-terminal-nYFP-CAMTA3 and N-terminal-cYFP-DSC
constructs in N. benthamiana (Figure 6A). Additionally BiFC signal was also seen with N-
terminal nYFP-CAMTA3 and N-terminal-cYFP-DSC-D construct (Figure 6B). Therefore the
p-loop mutation does not appear to interfere with this complex. Transient expression of
CAMTA3 with another nuclear localised NB-LRR SUMM2 control constructs showed no

131
detectable BiFC signal (S Figure 4A). In another control we co-infiltrated cYFP-DSC with
nuclear localised nYFP-MPK4 but again, no detectable BiFC signal was detected (S figure
4B) (Andreasson et al. 2005). We extended our analysis and conducted Fosters resonance
energy transfer (FRET) analysis in N. benthamiana leaves to confirm the results obtained
from the BiFC expreriment. By using CFP-CAMTA as donor and YFP-DSC as acceptor,
clear increased florescence of donor could be seen after photo bleaching of YFP (Figure 7
A). This change in intensity is shown as a heat map (Figure 7A). The same FRET efficacy
was not seen when using YFP-SUMM2 as acceptor (Figure 7B). FRET signal was obtained
for both CFP-CAMTA3/YFP-DSC and CAMTA3-CFP/YFP-DSC. Largest change in
florescent intensity appears located close to the nuclear membrane. Figure 7B shows the
average FRET efficiency from 5 separate FRET analyses. In summary, these results
indicate that DSC and CAMTA3 exist in nuclear complexes in plants and could represent
guard and gaurdee.
Discussion Autoimmune mutants typically display phenotypes and genetic requirements similar to R
gene activation (Lorrain et al. 2003) and a proportion of these autoimmune mutants could
represent mutations in favored effector targets guarded by R proteins. We previously
discovered that a DN P-loop mutation in LAZ5 was sufficient to suppress acd11
autoimmunity (Palma 2010). In line with this P-loop mutations in other R genes also seems
to suppress their function (Ref). We confirmed that we could compromise the function of
the well studied RPM1 resistant protein without disrupting the function of other R genes
(Figure 1 C and D). Abolishment of RPM1 function by p-loop mutation has been shown

132
previously but not the dominant negative function (Tornero et al. 2002; Chung et al. 2011).
In addition, the structural and functional conservation in the STAND family (Leipe, Koonin,
and Aravind 2004) in plants and animals supports the specific DN behaviour of the p-loop
mutants are well conserved in the relatively closely related NB-LRR family (Maekawa et al.
2012). We introduced P-loop mutations in a large collection of TIR-NB-LRRs and in a
forward genetic screen we transformed these DN genes into camta3 to examine the
possibility that autoimmunity in this mutant might be caused by R gene activation. We found
that one DN R gene, DCS-D, could suppress all the reported camta3-1 defence associated
phenotypes (Galon et al. 2008)(Du et al. 2009). Thus, DSC-D was capable of suppressing
the stunted growth, cell death, increased ROS production and elevated levels of PR1 and
EDS1 mRNA transcripts.
Importantly the increased resistance towards virulent and avirulent strains of Pst DC3000
seen in camta3-1 was also restored in camta3-1/DSC-D (Figure 3A and B). Given a role as
a negative regulator of the expression of EDS1 and subsequent immunity this finding is
somehow surprising. There is abundant evidence that CAMTA3 is a negative regulator of
EDS1. Importantly CAMTA3 has been shown to recognise promoter elements of EDS1 and
that these same promoter elements are responsible for the suppression of LUC reporter
genes expression (Du et al. 2009). Additionally, chromatin-immunoprecipitations in
protoplasts with transiently expressed GFP tagged CAMTA3 shows enrichment of EDS1
promoter elements. Thus CAMTA3 can bind EDS1 promoter elements (Nie et al. 2012). In
line with a negative regulatory function over-expression of CAMTA3 result is increases
susceptibility to pathogens and a dominant mutation in CAMTA3 had a similar effect (Jing
et al. 2011). However the absence of elevated EDS1 mRNA in camta3-1/DSC-D (Figure
4B) and restoration of EDS1 regulation upon infection could indicate that in camta3 single

133
mutants DSC triggers EDS1-dependent immunity. But how can decreased resistance
caused by over-expression and dominant mutations in CAMTA3 fit into such a model? And,
if CAMTA3 is under DSC surveillance because it is an effector target with specific functions
in immunity why are camta3/DSC-D mutants not immunosuppressed? To answer the last
question first, CAMTA3 belongs to a small family of some 6 transcriptions factors in
Arabidopsis and its homologues in may function redundantly during PTI. In support of this
it is known that the stunted growth phenotype increases in severity in double
camta1/camta3 and camta2/camta3 mutants and even more so in triple
camta1/camta2/camta3 mutants. An alternative explanation is that we simply have not yet
tested a pathogen whose infection strategy could reveal a role of CAMTA3 in immunity. To
address the first question raised above, the other CAMTA members may partially also
explain this. CAMTA3 has been shown to collaborate with CAMTA1 and 2 in regulating of
gene expression (Kim et al. 2013) and massive over-expression of CAMTA3 might interfere
with this balance and somehow functions a plug and hinder whatever positive role these
transcription factors could posses in immunity. This explanation goes well in hand with the
observation that specific mutated versions of CAMTA3 increase susceptibility.
Here, we also note that other autoimmune mutants, like mpk4 and acd11, also display
elevated levels of EDS1 (Brodersen et al. 2006)(Brodersen et al. 2002) overexpression of
EDS1 alone does not cause HR in Arabidopsis (Xing and Chen 2006).
A further possible explanation for lower EDS1 levels in camta3-1/DSC-D1 and camta3-
1/DSC-D2 could be that DSC-D supressed EDS1 mRNA expression. However, none of the
DSC-D lines tested in this study was statistically significantly affected in EDS1 regulation
(Figure 4C).

134
Since DSC is implicated through suppression by DSC-D as the NB-LRR triggered by loss
of CAMTA3, two independent T-DNA knockouts of DSC were crossed to camta3-1. We
were not able to show similar phenotypic suppression (S. Figure 1) as seen in camta3-
1/DSC-D. It is possible that another R protein contributes to the camta3 phenotype since
dominant p-loop mutants like DSC-D can overcome redundancy. This is seen in
acd11/laz5-D2 that can fully supresses acd11 phenotype although acd11 laz5-1 still
displays significant cell death and activated defence (Palma et al. 2010). Dominant
mutations are known to typically be more effective than single gene knock out mutants or
point mutations in genetic screens (Sheppard 1994). ACD11 is not the only protein
suggested to be guarded by multiple NB-LRRs. RIN4 is guarded by 3 NB-LRRs. Two of
these NB-LRR guard against a single effector, avrB. AvrB is recognised in Col-0 by RPM1
the loss of RPM1 still triggers HR in response to this effector. Double RPM1 and TAO1 R
proteins knock-out results in loss of a HR to avrB (Eitas, Nimchuk, and Dangl 2008). We
will attempt to uncover other NB-LRRs activated in camta3-1.
Elevated expression of R proteins can lead to autoimmunity (Tang et al. 1999) but we did
not detect elevated DSC mRNA in camta3-1. (Sup figure 3B) and DSC does not contain a
CGCG box and are therefore unlikely to be directly transcriptionally regulated by CAMTA3
(TAIR). Aberrant regulation in DSC therefore does not explain camta3-1 autoimmunity.
In support of a model were CAMTA3 or a complex containing CAMTA3 is under DSC
surveillance both proteins was shown to be nuclear localised (Figure 5A) and interacts as
indicated by BiFC (Figure 6) and FRET analysis (Figure 7). Many examples exist of R
proteins interacting with possible gaurdees (Tameling and Baulcombe 2007) (Mucyn et al.
2006)(Caplan et al. 2008). A primary example is RPS2 that interacts with RIN4. Loss of
RIN leads to lethality caused by activated RPS2 (Belkhadir et al. 2004:2). CAMTA3 and

135
DSC could be part of a nuclear complex which is disrupted by loss of CAMTA3. This could
result in activation of DSC leading to observed phenotypes that are consistent with
constitutively activated ETI (Hammond-Kosack and Jones 1996)(Tameling et al.
2006)(Bendahmane et al. 2002). These results supports the model where DSC is gaurding
a CAMTA3 containing complex.
From our interaction data of CAMTA3 and DSC, we conclude that CAMTA3 could possibly
be a gaurdee of DSC. This would be beneficial to plant defence because hypothetical
effectors targeting and activating a negative regulator of defence like CAMTA3 would be to
the advantage of pathogens. If this is the case plants could have evolved R proteins to
guard against such effectors.
Many lesion mimic mutants display elevated levels of defence genes including EDS1 and
possibly many pleiotropic effects that could be mistakenly attributed to a lost gene product.
R gene activation could be the underlying cause of many other autoimmune mutants that
display requirements for EDS1 or NDR1 and has a temperature sensitive phenotype. Since
there are around 150 NB-LRR in Arabidopsis that potentially guard integrity of defence
responses it naturally follows that disrupting many immunity related proteins may lead
conditions that resemble pathogen effector action and as a result activate NB-LRRs. This
could make functional genomic studies related to defence misleading. If we can eliminate
underlying pleotropic defence effects caused by inappropriate activation of R genes we can
study real effects of genetic changes unhindered.

136
Materials and methods
Statistical analysis
Multiple ANOVA analysis was performed, aided by commercial Graphpad prism software.
Significance of results is indicated by * for p value>0,05 , ** for p between 0,05 and 0,01
and *** for p<0,01, with reference to the sample marked with a black dot.
Plants growth conditions.
Salk lines obtained from the Nottingham Arabidopsis Seed Centre and primers from isect
tool (http://signal.salk.edu/isectprimers.html) used to genotype lines for homozygousity.
Camta3-1/SALK_001152. SALK_009668, Sail_49_C05, Col-0 ecotype, ndr1-1(Century,
Holub, and Staskawicz 1995:1), eds1-2(Aarts et al. 1998), rpm1-3(),
Maintenance of Pathogen Isolates.
Pseudomonas syringae pv. tomato DC3000 containing the avirulence genes
avrRps4 (Hinsch and Staskawicz 1996), avrRpm1 (Grant et al. 1995), avrRpt2
(Bent et al. 1994), in the broad host range vector pVSP61 or DC3000
containing empty pVSP61 were cultured at 28 °C on NYG plates.
Ion leakage assay
Conductivity assays were conducted as previously described using 0.2OD600 Pst DC3000
(avrRpm1) (Aviv et al. 2002) in 4 week old soil grown plants.

137
Cloning and generation of transgenic plants
P-loop TIR-NBS-LRRs were created from genomic DNA by USER mutagenesis as
described in (Nour-Eldin et al. 2006) and cloned into modified pCAMBIA-3300 as described,
using a uracil-excision based cloning technique (USER, New England Biolabs). Cloning
primers were tagged with 5′-ggcttaaU-X3′ for the forward primer and 5′-ggtttaaUX-3′ for the
reverse primer. Mutant primers were made containing the mutation GKTT to AATT of the
p-loop motif and appropriate uracil incorporated to give seamless overlap of two fragments
(Geu-Flores et al. 2007).
WT DSC, DSC-D mutant and CAMTA3 was amplified from genomic DNA (Col-0) or plasmid
template, cloned into pENTR/D-TOPO (Invitrogen) and transferred to Gateway-compatible
constitutive expression vectors by LR recombination reaction (Invitrogen). Destination
Vectors used were pGWB644(35S pro C-Terminal CFP), pGWB645 (35S pro N-Terminal
CFP)(Nakamura et al. 2010), pGWB541(35S pro C-Terminal YFP), pGWB542 (35S pro N-
Terminal YFP)(Nakagawa et al. 2007), Split YFP vectors used were nEYFP/pUGW2 (35S
pro, C-nEYFP), cEYFP/pUGW2(35S pro, C-cEYFP), nEYFP/pUGW0(35S pro, N-nEYFP)
and cEYFP/pUGW0(35S pro, N-cEYFP) (Hino et al. 2011). GFP Antibody (B-2): sc-9996
(santa cruz) was used to detect expression of both YFP fragments.
The final constructs were verified by sequencing, electroporated into Agrobacterium
tumefaciens strain GV3101 and used to transform camta3-1 or wild type plants by floral dip
method (Clough and Bent 1998). Transgenic plants were selected on soil with BASTA (120
mg/ml).

138
Trypan blue staining
Staining for cell death was done as described in (Koch and Slusarenko 1990) on 6 Week
old soil grown plants.
Bacterial growth assays
Performed as described in (Katagiri, Thilmony, and He 2002) by syringe infiltration of 0.001
OD600 or spray inoculation of 0.2 OD600 plus 0.02% Silvet-L77 (Lehle seeds) of 4 week
old soil grown plants. Day 0 was analyzed in infiltrated leaves to be sure no statistical
difference is present at day 0 and that day3 shows positive growth.
Microscopy
Agrobacterium strains containing plant expression vector was grown in LB media overnight
at 28°C. Relevant cultures were spun down and suspended in (10mM MgCl/10mM MES
pH 5.6. ) After two days leaves were infiltrated with water and leaf disks sampled. Confocal
microscopy and FRET-AB was performed on a SP5-X confocal microscope equipped with
a HCX lambda blue PL APO 320 objective (0.7 numerical aperture). Excitation of
Fluorophores was done with White light or argon laser at the respective absorption maxima.
QPCR
PR1, EDS1, DSC+DSC2 and CAMTA mRNA levels were evaluated by QPCR in 6 week
old plants. Induction of EDS1 by 0.005 OD Pst DC3000 (avrRPS4) mRNA was done by
infiltrating 3 large proximal leaves on 4 week old plants. Distal leaves of Pst DC3000
(avrRPS4) inoculated and MgCl2 infiltrated leaves were harvested and frozen in Liquid
nitrogen. Total RNA was extracted with Trizol reagent (Sigma) or RNA extraction kit
(Machenery nagel) and integrity confirmed by agarose gel electrophoreses. Turbo DNase
treatment (ambion) was performed. cDNA synthesis was done using polydT primers and

139
ReverseAid First Strand cDNA Synthesis Kit (Thermo Scientific). QPCR was performed
with DyNAmo ColorFlash SYBR Green qPCR Kit (Thermo Scientific). QPCR Primers for
EDS1, PR1, DSC, DSC2, CAMTA3 and UBQ10 quantification is shown in S. Table 2.
Refrences
Aarts, N, M Metz, E Holub, et al.
1998 Different Requirements for EDS1 and NDR1 by Disease Resistance Genes Define at
Least Two R Gene-Mediated Signaling Pathways in Arabidopsis. Proceedings of the
National Academy of Sciences of the United States of America 95(17): 10306–10311.
Andreasson, Erik, Thomas Jenkins, Peter Brodersen, et al.
2005 The MAP Kinase Substrate MKS1 Is a Regulator of Plant Defense Responses. The
EMBO Journal 24(14): 2579–2589.
Aviv, Daniel H., Christine Rustérucci, Ben F. Holt Iii, et al.
2002 Runaway Cell Death, but Not Basal Disease Resistance, in lsd1 Is SA- and
NIM1/NPR1-Dependent. The Plant Journal 29(3): 381–391.
Bai, Shiwei, Jie Liu, Cheng Chang, et al.
2012 Structure-Function Analysis of Barley NLR Immune Receptor MLA10 Reveals Its
Cell Compartment Specific Activity in Cell Death and Disease Resistance. PLoS Pathogens
8(6). http://dx.doi.org/10.1371/journal.ppat.1002752.
Belkhadir, Youssef, Zachary Nimchuk, David A. Hubert, David Mackey, and Jeffery L.
Dangl
2004 Arabidopsis RIN4 Negatively Regulates Disease Resistance Mediated by RPS2 and
RPM1 Downstream or Independent of the NDR1 Signal Modulator and Is Not Required for
the Virulence Functions of Bacterial Type III Effectors AvrRpt2 or AvrRpm1. The Plant
Cell Online 16(10): 2822–2835.
Bendahmane, Abdelhafid, Garry Farnham, Peter Moffett, and David C Baulcombe
2002 Constitutive Gain-of-Function Mutants in a Nucleotide Binding Site-Leucine Rich
Repeat Protein Encoded at the Rx Locus of Potato. The Plant Journal: For Cell and
Molecular Biology 32(2): 195–204.
Bent, A F, B N Kunkel, D Dahlbeck, et al.
1994 RPS2 of Arabidopsis Thaliana: A Leucine-Rich Repeat Class of Plant Disease
Resistance Genes. Science (New York, N.Y.) 265(5180): 1856–1860.
Bray Speth, Elena, Young Nam Lee, and Sheng Yang He
2007 Pathogen Virulence Factors as Molecular Probes of Basic Plant Cellular Functions.
Current Opinion in Plant Biology 10(6): 580–586.

140
Brodersen, Peter, Morten Petersen, Henrik Bjørn Nielsen, et al.
2006 Arabidopsis MAP Kinase 4 Regulates Salicylic Acid- and Jasmonic Acid/ethylene-
Dependent Responses via EDS1 and PAD4. The Plant Journal 47(4): 532–546.
Brodersen, Peter, Morten Petersen, Helen M. Pike, et al.
2002 Knockout of Arabidopsis ACCELERATED-CELL-DEATH11 Encoding a
Sphingosine Transfer Protein Causes Activation of Programmed Cell Death and Defense.
Genes & Development 16(4): 490–502.
Buell, C. Robin, Vinita Joardar, Magdalen Lindeberg, et al.
2003 The Complete Genome Sequence of the Arabidopsis and Tomato Pathogen
Pseudomonas Syringae Pv. Tomato DC3000. Proceedings of the National Academy of
Sciences 100(18): 10181–10186.
Caplan, J.L., P. Mamillapalli, T.M. Burch-Smith, K. Czymmek, and S.P. Dinesh-Kumar
2008 Chloroplastic Protein NRIP1 Mediates Innate Immune Receptor Recognition of a
Viral Effector. Cell 132(3): 449–462.
Century, K. S., E. B. Holub, and B. J. Staskawicz
1995 NDR1, a Locus of Arabidopsis Thaliana That Is Required for Disease Resistance to
Both a Bacterial and a Fungal Pathogen. Proceedings of the National Academy of Sciences
92(14): 6597–6601.
Cheng, Cheng, Xiquan Gao, Baomin Feng, et al.
2013 Plant Immune Response to Pathogens Differs with Changing Temperatures. Nature
Communications 4.
http://www.nature.com/ncomms/2013/130926/ncomms3530/full/ncomms3530.html,
accessed October 2, 2013.
Chung, Eui-Hwan, Luis da Cunha, Ai-Jiuan Wu, et al.
2011 Specific Threonine Phosphorylation of a Host Target by Two Unrelated Type III
Effectors Activates a Host Innate Immune Receptor in Plants. Cell Host & Microbe 9(2):
125–136.
Clough, Steven J., and Andrew F. Bent
1998 Floral Dip: A Simplified Method forAgrobacterium-Mediated Transformation
ofArabidopsis Thaliana. The Plant Journal 16(6): 735–743.
Dietrich, R A, M H Richberg, R Schmidt, C Dean, and J L Dangl
1997 A Novel Zinc Finger Protein Is Encoded by the Arabidopsis LSD1 Gene and
Functions as a Negative Regulator of Plant Cell Death. Cell 88(5): 685–694.
Dietrich, Robert A., Terrence P. Delaney, Scott J. Uknes, et al.
1994 Arabidopsis Mutants Simulating Disease Resistance Response. Cell 77(4): 565–577.
Dinesh-Kumar, S P, W H Tham, and B J Baker

141
2000 Structure-Function Analysis of the Tobacco Mosaic Virus Resistance Gene N.
Proceedings of the National Academy of Sciences of the United States of America 97(26):
14789–14794.
Du, Liqun, Gul S. Ali, Kayla A. Simons, et al.
2009 Ca2+/calmodulin Regulates Salicylic-Acid-Mediated Plant Immunity. Nature
457(7233): 1154–1158.
Eitas, Timothy K, Zachary L Nimchuk, and Jeffery L Dangl
2008 Arabidopsis TAO1 Is a TIR-NB-LRR Protein That Contributes to Disease Resistance
Induced by the Pseudomonas Syringae Effector AvrB. Proceedings of the National
Academy of Sciences of the United States of America 105(17): 6475–6480.
Feys, Bart J., Lisa J. Moisan, Mari-Anne Newman, and Jane E. Parker
2001 Direct Interaction between the Arabidopsis Disease Resistance Signaling Proteins,
EDS1 and PAD4. The EMBO Journal 20(19): 5400–5411.
Galon, Yael, Roy Nave, Joy Boyce, et al.
2008 Calmodulin-Binding Transcription Activator (CAMTA) 3 Mediates Biotic Defense
Responses in Arabidopsis. FEBS Letters 582(6): 943–948.
Gao, Minghui, Xia Wang, Dongmei Wang, et al.
2009 Regulation of Cell Death and Innate Immunity by Two Receptor-like Kinases in
Arabidopsis. Cell Host & Microbe 6(1): 34–44.
Gao, Zhiyong, Zhiyoug Gao, Eui-Hwan Chung, Timothy K Eitas, and Jeffery L Dangl
2011 Plant Intracellular Innate Immune Receptor Resistance to Pseudomonas Syringae Pv.
Maculicola 1 (RPM1) Is Activated At, and Functions On, the Plasma Membrane.
Proceedings of the National Academy of Sciences of the United States of America 108(18):
7619–7624.
Geu-Flores, Fernando, Hussam H. Nour-Eldin, Morten T. Nielsen, and Barbara A. Halkier
2007 USER Fusion: A Rapid and Efficient Method for Simultaneous Fusion and Cloning
of Multiple PCR Products. Nucleic Acids Research 35(7): e55.
Grant, M R, L Godiard, E Straube, et al.
1995 Structure of the Arabidopsis RPM1 Gene Enabling Dual Specificity Disease
Resistance. Science (New York, N.Y.) 269(5225): 843–846.
Greenberg, J T, A Guo, D F Klessig, and F M Ausubel
1994 Programmed Cell Death in Plants: A Pathogen-Triggered Response Activated
Coordinately with Multiple Defense Functions. Cell 77(4): 551–563.
Greenberg, Jean T., and Frederick M. Ausubel
1993 Arabidopsis Mutants Compromised for the Control of Cellular Damage during
Pathogenesis and Aging. The Plant Journal 4(2): 327–341.

142
Hammond-Kosack, K. E., and J. D. Jones
1996 Resistance Gene-Dependent Plant Defense Responses. The Plant Cell Online 8(10):
1773–1791.
Heath, Michèle C.
1998 Apoptosis, Programmed Cell Death and the Hypersensitive Response. European
Journal of Plant Pathology 104(2): 117–124.
Heidrich, Katharina, Lennart Wirthmueller, Céline Tasset, et al.
2011 Arabidopsis EDS1 Connects Pathogen Effector Recognition to Cell Compartment-
Specific Immune Responses. Science (New York, N.Y.) 334(6061): 1401–1404.
Hino, Takeshi, Yuji Tanaka, Makoto Kawamukai, et al.
2011 Two Sec13p Homologs, AtSec13A and AtSec13B, Redundantly Contribute to the
Formation of COPII Transport Vesicles in <I>Arabidopsis thaliana</I>. Bioscience,
Biotechnology, and Biochemistry 75(9): 1848–1852.
Hinsch, M, and B Staskawicz
1996 Identification of a New Arabidopsis Disease Resistance Locus, RPs4, and Cloning of
the Corresponding Avirulence Gene, avrRps4, from Pseudomonas Syringae Pv. Pisi.
Molecular Plant-Microbe Interactions: MPMI 9(1): 55–61.
Howles, Paul, Greg Lawrence, Jean Finnegan, et al.
2005 Autoactive Alleles of the Flax L6 Rust Resistance Gene Induce Non-Race-Specific
Rust Resistance Associated with the Hypersensitive Response. Molecular Plant-Microbe
Interactions 18(6): 570–582.
Jing, Beibei, Shaohua Xu, Mo Xu, et al.
2011 Brush and Spray: A High-Throughput Systemic Acquired Resistance Assay Suitable
for Large-Scale Genetic Screening. Plant Physiology 157(3): 973–980.
Jones, Jonathan D. G., and Jeffery L. Dangl
2006 The Plant Immune System. Nature 444(7117): 323–329.
Katagiri, Fumiaki, Roger Thilmony, and Sheng Yang He
2002 The Arabidopsis Thaliana-Pseudomonas Syringae Interaction. The Arabidopsis Book
/ American Society of Plant Biologists 1.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3243347/, accessed November 21, 2013.
Kim, Yongsig, Sunchung Park, Sarah Gilmour, and Michael Thomashow
2013 Roles of CAMTA Transcription Factors and Salicylic Acid in Configuring the Low
Temperature Transcriptome and Freezing Tolerance of Arabidopsis. The Plant Journal : For
Cell and Molecular Biology. http://dx.doi.org/10.1111/tpj.12205.
Koch, E., and A. Slusarenko

143
1990 Arabidopsis Is Susceptible to Infection by a Downy Mildew Fungus. The Plant Cell
Online 2(5): 437–445.
Leipe, Detlef D, Eugene V Koonin, and L Aravind
2004 STAND, a Class of P-Loop NTPases Including Animal and Plant Regulators of
Programmed Cell Death: Multiple, Complex Domain Architectures, Unusual Phyletic
Patterns, and Evolution by Horizontal Gene Transfer. Journal of Molecular Biology 343(1):
1–28.
Leipe, Detlef D, Yuri I Wolf, Eugene V Koonin, and L Aravind
2002 Classification and Evolution of P-Loop GTPases and Related ATPases. Journal of
Molecular Biology 317(1): 41–72.
Li, Yingzhong, Shuxin Li, Dongling Bi, et al.
2010 SRFR1 Negatively Regulates Plant NB-LRR Resistance Protein Accumulation to
Prevent Autoimmunity. PLoS Pathog 6(9): e1001111.
Lorrain, Séverine, Fabienne Vailleau, Claudine Balagué, and Dominique Roby
2003 Lesion Mimic Mutants: Keys for Deciphering Cell Death and Defense Pathways in
Plants? Trends in Plant Science 8(6): 263–271.
Maekawa, Takaki, Barbara Kracher, Saskia Vernaldi, Emiel Ver Loren van Themaat, and
Paul Schulze-Lefert
2012 Conservation of NLR-Triggered Immunity across Plant Lineages. Proceedings of the
National Academy of Sciences of the United States of America 109(49): 20119–20123.
Mestre, Pere, and David Baulcombe
2006 Elicitor-Mediated Oligomerization of the Tobacco N Disease Resistance Protein. The
Plant Cell 18(2): 491–501.
Mucyn, Tatiana S, Alfonso Clemente, Vasilios M. E Andriotis, et al.
2006 The Tomato NBARC-LRR Protein Prf Interacts with Pto Kinase in Vivo to Regulate
Specific Plant Immunity. The Plant Cell Online 18(10): 2792–2806.
Mur, Luis A J, Paul Kenton, Amanda J Lloyd, Helen Ougham, and Elena Prats
2008 The Hypersensitive Response; the Centenary Is upon Us but How Much Do We
Know? Journal of Experimental Botany 59(3): 501–520.
Nakagawa, Tsuyoshi, Takamasa Suzuki, Satoko Murata, et al.
2007 Improved Gateway Binary Vectors: High-Performance Vectors for Creation of
Fusion Constructs in Transgenic Analysis of Plants. Bioscience, Biotechnology, and
Biochemistry 71(8): 2095–2100.
Nakamura, Shinya, Shoji Mano, Yuji Tanaka, et al.
2010 Gateway Binary Vectors with the Bialaphos Resistance Gene, <I>bar</I>, as a
Selection Marker for Plant Transformation. Bioscience, Biotechnology, and Biochemistry
74(6): 1315–1319.

144
Narusaka, Mari, Ken Shirasu, Yoshiteru Noutoshi, et al.
2009 RRS1 and RPS4 Provide a Dual Resistance-Gene System against Fungal and
Bacterial Pathogens. The Plant Journal 60(2): 218–226.
Nie, Haozhen, Chunzhao Zhao, Guangheng Wu, et al.
2012 SR1, a Calmodulin-Binding Transcription Factor, Modulates Plant Defense and
Ethylene-Induced Senescence by Directly Regulating NDR1 and EIN3. Plant Physiology
158(4): 1847–1859.
Nour-Eldin, Hussam H., Bjarne G. Hansen, Morten H. H. Nørholm, Jacob K. Jensen, and
Barbara A. Halkier
2006 Advancing Uracil-Excision Based Cloning towards an Ideal Technique for Cloning
PCR Fragments. Nucleic Acids Research 34(18): e122–e122.
Palma, Kristoffer, Stephan Thorgrimsen, Frederikke Gro Malinovsky, et al.
2010 Autoimmunity in Arabidopsis acd11 Is Mediated by Epigenetic Regulation of an
Immune Receptor. PLoS Pathog 6(10): e1001137.
Petersen, M, P Brodersen, H Naested, et al.
2000 Arabidopsis Map Kinase 4 Negatively Regulates Systemic Acquired Resistance. Cell
103(7): 1111–1120.
Roberts, Melinda, Saijun Tang, Anna Stallmann, Jeffery L Dangl, and Vera Bonardi
2013 Genetic Requirements for Signaling from an Autoactive Plant NB-LRR Intracellular
Innate Immune Receptor. PLoS Genetics 9(4): e1003465.
Senthil-Kumar, Muthappa, and Kirankumar S Mysore
2011 Caveat of RNAi in Plants: The off-Target Effect. Methods in Molecular Biology
(Clifton, N.J.) 744: 13–25.
Sheppard, D
1994 Dominant Negative Mutants: Tools for the Study of Protein Function in Vitro and in
Vivo. American Journal of Respiratory Cell and Molecular Biology 11(1): 1–6.
Stokes, Trevor L., Barbara N. Kunkel, and Eric J. Richards
2002 Epigenetic Variation in Arabidopsis Disease Resistance. Genes & Development
16(2): 171–182.
Takahashi, A, T Kawasaki, K Henmi, et al.
1999 Lesion Mimic Mutants of Rice with Alterations in Early Signaling Events of
Defense. The Plant Journal: For Cell and Molecular Biology 17(5): 535–545.
Takken, Frank LW, Mario Albrecht, and Wladimir IL Tameling
2006 Resistance Proteins: Molecular Switches of Plant Defence. Current Opinion in Plant
Biology 9(4): 383–390.

145
Tameling, Wladimir I. L, Jack H Vossen, Mario Albrecht, et al.
2006 Mutations in the NB-ARC Domain of I-2 That Impair ATP Hydrolysis Cause
Autoactivation. Plant Physiology 140(4): 1233–1245.
Tameling, Wladimir I. L., and David C. Baulcombe
2007 Physical Association of the NB-LRR Resistance Protein Rx with a Ran GTPase–
Activating Protein Is Required for Extreme Resistance to Potato Virus X. The Plant Cell
Online 19(5): 1682–1694.
Tang, Xiaoyan, Mingtang Xie, Young Jin Kim, et al.
1999 Overexpression of Pto Activates Defense Responses and Confers Broad Resistance.
The Plant Cell Online 11(1): 15–29.
Tao, Y, F Yuan, R T Leister, F M Ausubel, and F Katagiri
2000 Mutational Analysis of the Arabidopsis Nucleotide Binding Site-Leucine-Rich
Repeat Resistance Gene RPS2. The Plant Cell 12(12): 2541–2554.
Tornero, Pablo, Ryon A. Chao, William N. Luthin, Stephen A. Goff, and Jeffery L. Dangl
2002 Large-Scale Structure -Function Analysis of the Arabidopsis RPM1 Disease
Resistance Protein. The Plant Cell 14(2): 435–450.
Williams, Simon J, Pradeep Sornaraj, Emma deCourcy-Ireland, et al.
2011 An Autoactive Mutant of the M Flax Rust Resistance Protein Has a Preference for
Binding ATP, Whereas Wild-Type M Protein Binds ADP. Molecular Plant-Microbe
Interactions: MPMI 24(8): 897–906.
Xing, Denghui, and Zhixiang Chen
2006 Effects of Mutations and Constitutive Overexpression of EDS1 and PAD4 on Plant
Resistance to Different Types of Microbial Pathogens. Plant Science 171(2): 251–262.
Zhang, Zhibin, Yaling Wu, Minghui Gao, et al.
2012 Disruption of PAMP-Induced MAP Kinase Cascade by a Pseudomonas Syringae
Effector Activates Plant Immunity Mediated by the NB-LRR Protein SUMM2. Cell Host &
Microbe 11(3): 253–263.
Zhu, Ying, Baijuan Du, Jun Qian, Baohong Zou, and Jian Hua
2013 Disease Resistance Gene-Induced Growth Inhibition Is Enhanced by rcd1
Independent of Defense Activation in Arabidopsis. Plant Physiology 161(4): 2005–2013.
Zhu, Zhaohai, Fang Xu, Yaxi Zhang, et al.
2010 Arabidopsis Resistance Protein SNC1 Activates Immune Responses through
Association with a Transcriptional Corepressor. Proceedings of the National Academy of
Sciences 107(31): 13960–13965.

146
Figure legends
Figure 1. P-loop suppression and specificity.
All assays were done on 4 week old soil grown plants. RPM1-D lines have the same Pst
DC3000(avrRpm1) bacterial growth and ion leakage as rpm1-3 KO lines and the effect is
R-gene specific. A, Ion leakage profiles Col-0, rpm1-3, RPMI-D1 and RPM1-D2 in response
to Pst DC3000 (avrRpm1) 0.2OD 600 up to 7 hours post infiltration. Mean and S.D. from 6
leaf disk in 4 replicates within an experiment. B, Day 0 and day 3 log C.F.U./cm² bacterial
growth of Pst DC3000 (avrRpm1) 0.001OD 600 in Col-0, rpm1-3, RPMI-D1 and RPM1.
Mean of 4 replicates. C, Day 0 and day 3 log C.F.U./cm² bacterial growth of Pst DC3000
(avrRps4) 0.001OD 600 in Col-0, rpm1-3, RPMI-D1 and RPM1. Mean of 4 replicates. D,
Day 0 and day 3 log C.F.U./cm² bacterial growth of Pst DC3000 (avrRpt2) 0.001OD 600 in
Col-0, rpm1-3, RPMI-D1 and RPM1. Mean of 4 replicates. A,B,C, Significance of results is
indicated by * for p value>0,05 , ** for p between 0,05 and 0,01 and *** for p<0,01, with
reference to the sample marked with a black dot.
Figure 2. Phenotype suppression of camta3-1 and DSC-D suppressor in short day
grown 6 week old plants at 22 °C.
A, Phenotypes of 6 week old short day grown Col-0, camta3-1,camta3-1/DSC-D1 and DSC-
D3. B, Trypan blue stained 6 week old leaves of Col-0, camta3-1,camta3-1/DSC-D1 and
DSC-D3. C, DAB stained 6 week old leaves of Col-0, camta3-1,camta3-1/DSC-D1 and
DSC-D3.
Figure 3: Enhanced disease resistance of camta3-1 is abolished by DSC-D to both
(a) DC3000 and (b) DC3000 (avrRPS4).

147
A, log C.F.U./cm² bacterial growth 3 days after spray inoculation with DC3000 0.2OD600
in 0.02% silwet. B, log C.F.U./cm² bacterial growth 3 days after spray inoculation with
DC3000 (avrRps4) 0.2OD600 in 0.02% silwet. Significance of results is indicated by * for p
value>0,05 , ** for p between 0,05 and 0,01 and *** for p<0,01, with reference to the sample
marked with a black dot.
Figure 4: Analysis of defence gene expression in Col-0, camta3-1, camta3-1/DSC-D1,
camta3-1/DSC-D2, DSC-D3 and DSC-D4.
A, QPCR of basal PR1 transcript levels, relative to UBQ10, in Col-0, camta3-1, camta3-
1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 in 6 week old S.D. grown plants. B,
QPCR of basal EDS1 transcript levels, relative to UBQ10, in Col-0, camta3-1, camta3-
1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 in 6 week old S.D. grown plants.. C,
QPCR of basal and Pst DC3000 (avrRps4) 0.005 OD 600 induced EDS1 transcript levels,
relative to UBQ10, in Col-0, camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and
DSC-D4 in 4 week old S.D. grown plants. A,B,C, Significance of results is indicated by * for
p value>0,05 , ** for p between 0,05 and 0,01 and *** for p<0,01, with reference to the
sample marked with a black dot.
Figure 5 Subcellular localisation of CAMTA3, DSC and DSC-D.
A, Confocal images of N. benthamiana leaves infiltrated with N terminal YFP-DSC and N
terminal CFP-CAMTA3. Images shows expression in nucleus and Merged image of
Chlorophyll, florescent and DIC channels on far right. B, Confocal images of N.
benthamiana leaves infiltrated with N terminal YFP-DSC. Images shows expression in
nucleus and merged image of Chlorophyll, florescent and DIC channels on far right. C,

148
Confocal images of N. benthamiana leaves infiltrated with N terminal YFP-DSC-D and N
terminal CFP-CAMTA3. Images shows expression in nucleus and merged image of
chlorophyll, florescent and DIC channels on far right.
Figure 6 BiFC analysis of CAMTA3/DSC and CAMTA3/DSC-D interaction.
A, BiFC signal form N. benthamiana expressing N-terminal tagged, C-terminal-YFP-DSC
and N-Terminal tagged, NTerminal-YFP-CAMTA3. Chlorophyll, DIC channels and merged
image follows in order. B, BiFC signal form N. benthamiana expressing N-terminal tagged,
C-terminal-YFP-DSC-D and N-Terminal tagged NTerminal-YFP-CAMTA3. Chlorophyll,
DIC channels and merged image follows in order.
Figure 7 FRET analysis of CAMTA3/DSC and CAMTA3/DSC-D interaction.
A, Confocal images of N. Benthamiana leaves expressing N terminal YFP-DSC and N
terminal CFP-CAMTA3. The first panel shown YFP-DSC localisation before bleach, second
panel shows CFP-CAMTA3 pre YFP bleach. Third panel is CFP-CAMTA3 intensity post
YFP bleach. Last panel is heat map of change in CFP florescence indicating FRET.
B, Table summarising average for multiple fret analysis and average FRET for controls.
S Figure 1 T-DNA Phenotype of DSC knockout lines
Phenotypes of DSC homozygous T-DNA knockout lines SALK-009668 and Sail_49_C05
T-DNA KO show same phenotype as camta3-1.
S Figure 2 Gene structure of CAMTA3

149
A, Camta3/At2g22300 gene structure showing different domains. B, DSC-D insert
genotyping, using two different 35S primers and two different DSC reverse primers. All
DSC-lines show presence of insert where Col-0 line does not.
S Figure 3 Ethylene and flg22 responses
A and B, Triple response in Col-0, camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-
D3 and DSC-D4 seedlings germinated in dark. A, Shows MS only plates and B, shows
0.5mM ACC MS plates. camta3-1/DSC-D1, camta3-1/DSC-D2 triple response does not
differ significantly from DSC-D3 and DSC-D4. C, Average of weight of 12 seedlings grown
in MS supplemented with flg22. Average seedling growth inhibition of Col-0, camta3-1,
camta3-1/DSC-D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4 from 0, 2,5nM ,25 nM and
100nM flg22 is shown. Bar graph shows % growth relative to untreated. Growth inhibition
is comparable to Col-0 in all cases.
S Figure 4 QPCR of CAMTA3, DSC, and DSC2
A, QPCR levels of CAMTA3 transcripts in Col-0, camta3-1, camta3-1/DSC-D1, camta3-
1/DSC-D2, DSC-D3 and DSC-D4. Camta3-1, camta3-1/DSC-D1, camta3-1/DSC-D2
shows loss of CAMTA3 mRNA and confirms that these lines are genotypic camta3-1 lines.
B, QPCR levels of DSC including DSC-D transcripts in Col-0, camta3-1, camta3-1/DSC-
D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4. DSC is not elevated in camta3-1 and no
significant increase in DSC is seen from 35S construct. No DSC gene silencing in seen.
C, QPCR levels of DSC2 including DSC2-D transcripts in Col-0, camta3-1, camta3-1/DSC-
D1, camta3-1/DSC-D2, DSC-D3 and DSC-D4. DSC2 is not elevated in camta3-1 and no
significant increase in DSC is seen from 35S construct. No DSC gene silencing in seen.

150
S Figure 5 BiFC controls.
A, Negative BiFC form N. benthamiana expressing N-terminal tagged, C-terminal-YFP-
SUMM2 and N-Terminal tagged, NTerminal-YFP-CAMTA3. Chlorophyll, DIC channels and
merged image follows in order. B, Negative BiFC form N. benthamiana expressing N-
terminal tagged, C-terminal-YFP-DSC and N-Terminal tagged N-Terminal-YFP-MPK4.
Chlorophyll, DIC channels and merged image follows in order. C, Positive control BiFC
signal form N. benthamiana expressing N-terminal tagged, C-terminal-YFP-MIP and N-
Terminal tagged NTerminal-YFP-MPK4. Chlorophyll, DIC channels and merged image
follows in order.
S Table 1
Table of all cloned p-loop mutants TNLs
S table 2
List of primers used in this study.
Accession numbers
At3g48090 (EDS1) NM_114678. At3g20600 (NDR1): NP_188696. At3g07040 (RPM1):
NP_187360. At4g26090 (RPS2): NP_194339. At5g45250 (RPS4):
Conflicting interests
Publications are needed for career advancement and used as metrics to award research
funding.

151
Acknowledgements
Imaging data was collected at the Center for Advanced Bioimaging (CAB) Denmark,
University of Copenhagen. Thanks to Suksadwa

152

153

154
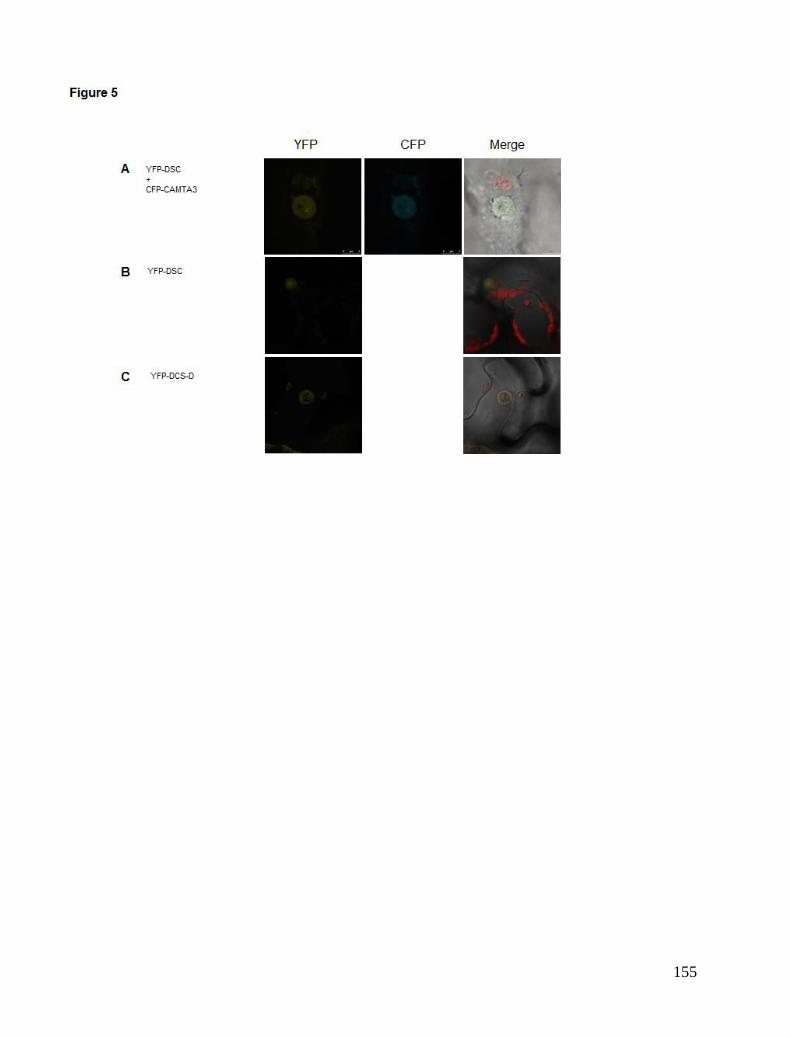
155
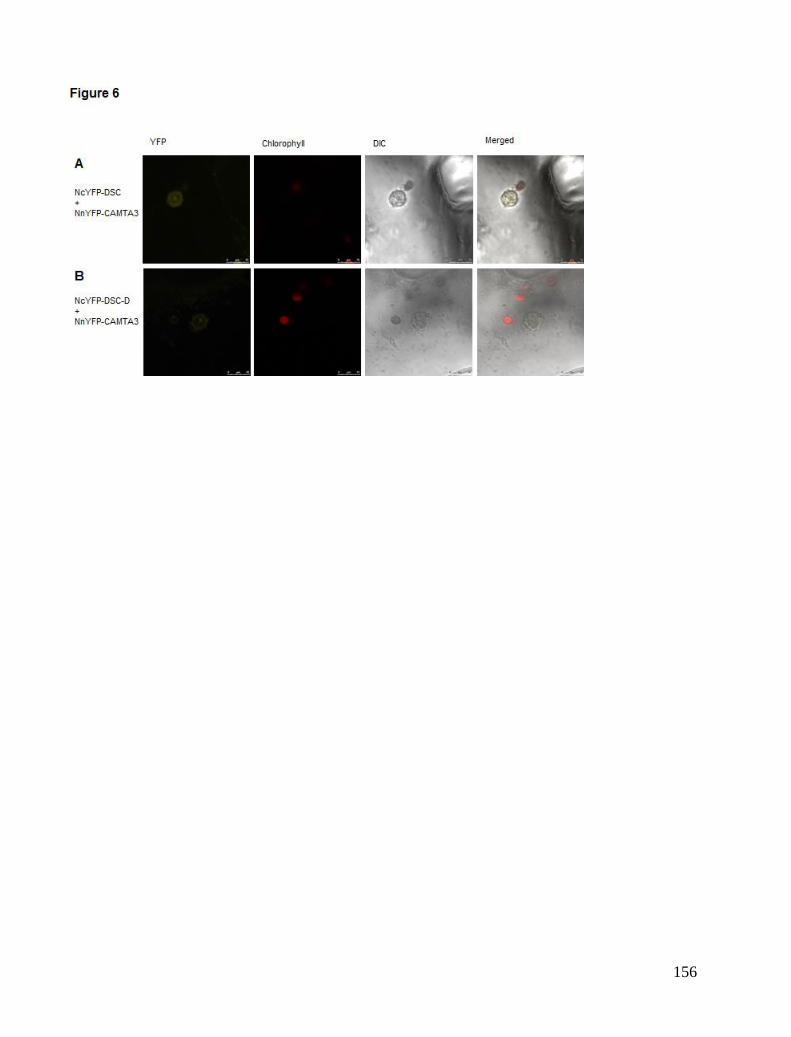
156
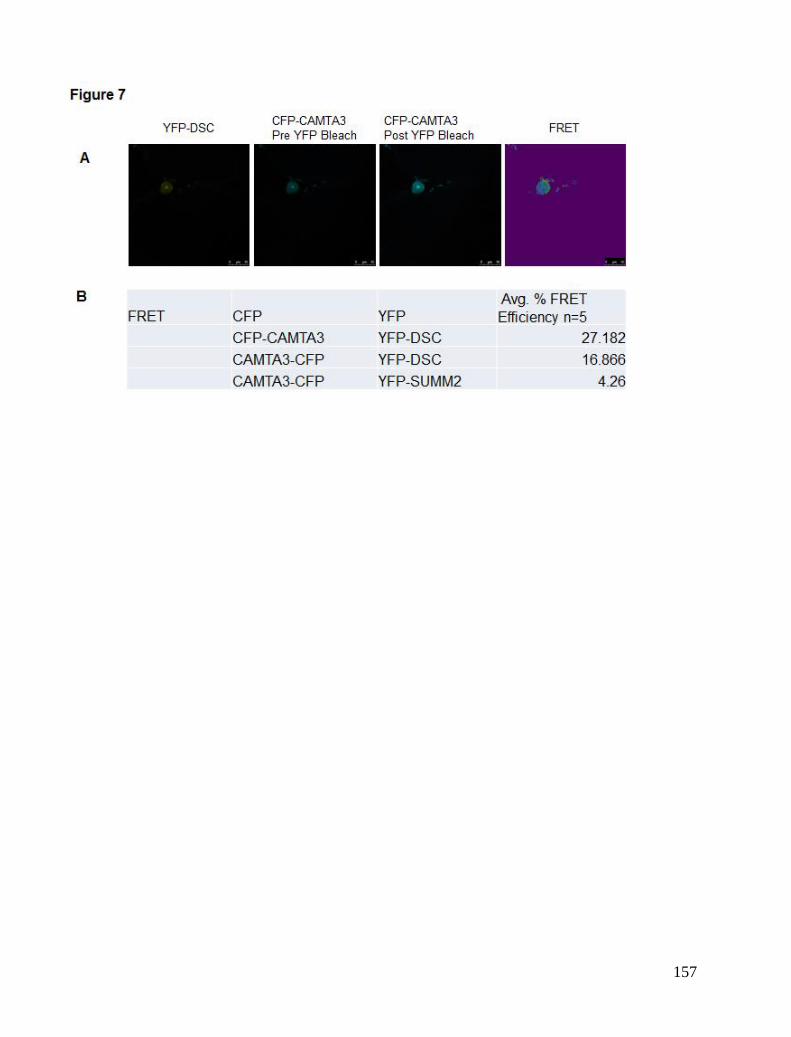
157

158

159

160
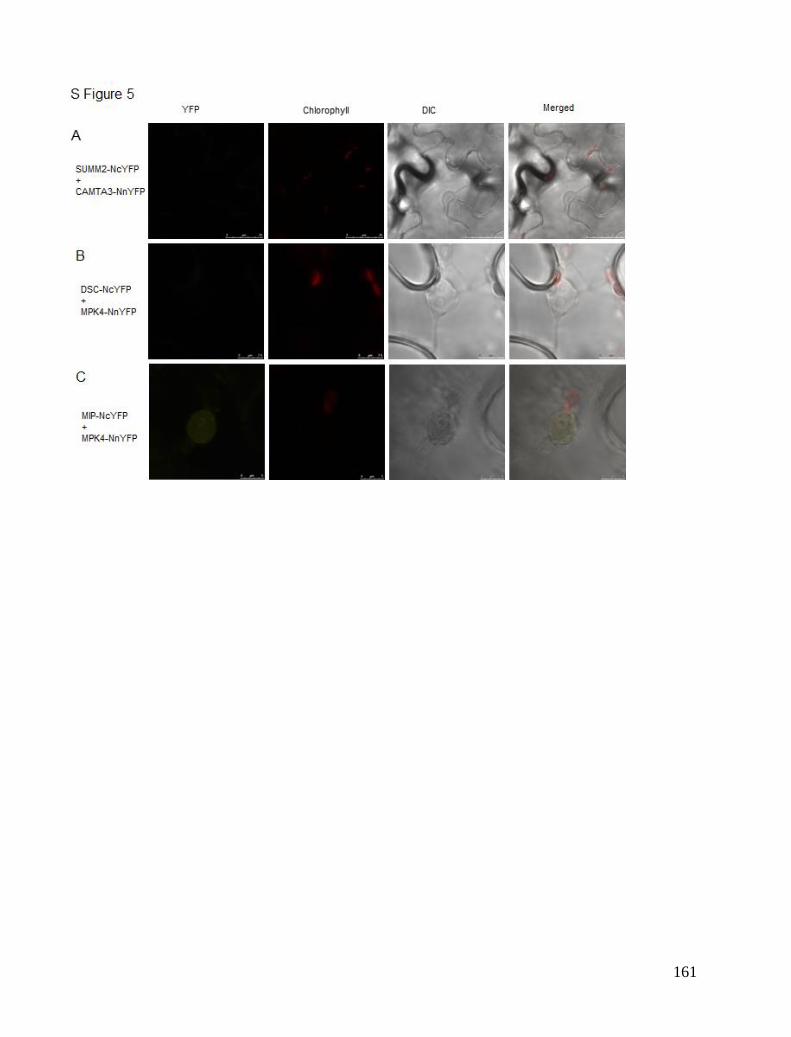
161

162
Published Review
Receptor-like kinase complexes in plant innate immunity
Christiaan Greeff, Milena Roux, John Mundy and Morten Petersen* Department of Biology, Copenhagen University, Copenhagen, Denmark
Edited by: Jacqueline Monaghan, The Sainsbury Laboratory, UK Reviewed by: Pascal Braun, Technical University of Munich, Germany Lennart Wirthmueller, John Innes Centre, UK *Correspondence: Morten Petersen, Department of Biology, Copenhagen University, Biocenter 3.1.45, Ole Maaløes Vej 5, Copenhagen, Denmark. e-mail: [email protected]
Receptor-like kinases (RLKs) are surface localized, transmembrane receptors comprising a large family of well-studied kinases. RLKs signal through their transmembrane and juxtamembrane domains with the aid of various interacting partners and downstream components. The N-terminal extracellular domain defines ligand specificity, and RLK families are sub-classed according to this domain. The most studied of these subfamilies include those with (1) leucine-rich repeat (LRR) domains, (2) LysM domains (LYM), and (3) the Catharanthus roseus RLK1-like (CrRLK1L) domain. These proteins recognize distinct ligands of microbial origin or ligands derived from intracellular protein/carbohydrate signals. For example, the pattern-recognition receptor (PRR) AtFLS2 recognizes flg22 from flagellin, and the PRR AtEFR recognizes elf18 from elongation factor (EF-Tu). Upon binding of their cognate ligands, the aforementioned RLKs activate generic immune responses termed pattern-triggered immunity (PTI). RLKs can form complexes with other family members and engage a variety of intracellular signaling components and regulatory pathways upon stimulation. This review focuses on interesting new data about how these receptors form protein complexes to exert their function.
Keywords: receptor-like kinases, complexes, plant immunity, signaling, defense
INTRODUCTION
Autotrophs, like plants, are the source of nutrients for
heterotrophs. Plants are members of complex communities and
have co-evolved commensal and pathological relationships with
microbes. A fine balancing act is required to effectively combat
invasion by pathogenic heterotrophs while effectively guarding
resources for vegetative and reproductive growth (King and
Roughgarden, 1982). This entails appropriately timed activation
of defense responses to conserve energy for producing numerous
healthy progeny, thus increasing evolutionary fitness through this
adaptive plasticity (Sultan,2000). Detecting harmful heterotrophs
and converting this recognition to intracellular signals aimed at
combating the intruder and alerting surrounding tissue, is a major
challenge, especially since pathogens co-evolve with their hosts
to elude discovery (Frank, 1992; Lehti-Shiu etal., 2009). Plant genomes encode a large number of surface receptorlike
kinases (RLKs) to perceive different signals from both distal cells
responding to stresses such as herbivore feeding or to the presence
of pathogens through detection of non-self molecules (Shiu and
Bleecker, 2001). RLKs generally have an extracellular ligand-
binding domain, a membrane-spanning region, a juxtamembrane
(JM) domain, and a serine/threonine kinase domain. Equivalent
mammalian receptors from the Pelle/IRAK family differ in
usually employing a cytosolic tyrosine kinase domain (Gish and
Clark, 2011). A conserved aspartate in the catalytic loop of most
kinases is required for catalytic activity. Ser/Thr kinases mostly
have an arginine preceding this catalytic aspartate. Kinases with
such residues are termed RD kinases, although most RLKs
implicated in microbial detection are non-RD kinases, lacking an
arginine preceding the catalytic aspartate. They in general require
additional proteins to modulate their function (Johnson etal.,
1996; Dardick and Ronald, 2006). An important example is
BAK1, which interacts with many Arabidopsis RLKs, and is
required for their activity (discussed below). The plant RLK family has more than 600 members in
Arabidopsis (Shiu etal., 2004). RLKs are divided into 44 sub-
families depending on their N-terminal domains. While RLKs
have been implicated in many biologically important processes
(Gish and Clark, 2011), this review focuses on RLKs involved in
pathogen detection. Plant RLKs involved in immunity are so-called
patternrecognition receptors (PRRs) that detect pathogen-
associated molecular patterns (PAMPs) and, upon binding of their
cognate elicitors, initiate a well-characterized set of defense
responses termed PAMP-triggered immunity (PTI). Features of
PTI include reactive oxygen species (ROS) production, callose
deposition, generation of secondary messengers, and defense gene
expression (Jones and Dangl, 2006). RLK elicitation also leads to
activation of several mitogen-activated protein (MAP) kinases
(Suarez-Rodriguez etal., 2007; Mithoe etal., 2011). PAMPs, and
more broadly, microbial-associated molecular patterns (MAMPs)
and damage-associated molecular patterns (DAMPs), can activate
RLKs(Lerougeetal.,1990; Gómez-GómezandBoller,2000; Zipfel
etal., 2006; Krol etal., 2010). Binding of PAMPS and DAMPS to
their specific receptors leads to a broad range of downstream
signaling events and effects. Figures1A–C gives an overview of
some of the complexes of Xa21,FlS2,and EF-Tu receptor (EFR)
that will be discussed in this review. Figure 1D shows biological
effects of FLS2, Xa21 and EFR.
THE LRR FAMILY
The best-studied members of the leucine-rich repeat (LRR)-RLK
family are the non-RD kinases AtFLS2, AtEFR, and OsXa21
(Park etal., 2010a) and the RD kinase BAK1 (Chinchilla etal.,

163
2007; Heese etal., 2007). These receptors present the core of our
current knowledge regarding RLKs involved in defense.
Xa21
The Xa21 extracellular domain is composed of 23 LRRs and was
one of the first eukaryotic RLKs found to be involved in resistance
(Song etal.,1995; Wang etal.,2006). Xa21 binds the Xanthomonas
oryzae pv. oryzae (Xoo)secretedtyrosine(Tyr)O-
sulfonationpeptide AxYS22 (Lee etal., 2009). Much has been
learned about the function of Xa21. For example, the amino acids
Ser686, Thr688, and Ser689 in the cytosolic JM domain are
important for stability and for endoplasmic reticulum (ER)
processing (Xu etal., 2006; Park etal., 2010a). Phosphorylation of
residues in the JM domain is also critical for the activation of
Xa21 and binding of at least four Xa21-binding proteins named
XB3, XB15, XB24, and XB10 (OsWRKY62; Park etal., 2010b)
associated with Xa21 via the JM domain. These interactions are
all dependent on Thr705 since mutation of this JM domain residue
abolishes XB-Xa21 binding (Chen etal., 2010a). XB3 is an E3 ligase important for Xa21 accumulation and is a
substrateforXa21kinaseactivity,althoughthebiologicalrelevance
of this relationship is still unclear. After Xa21 binds AxYS22,
XB3 is activated by transphosphorylation and likely leads to
cleavage of a negative regulator of defense or even of itself,
allowing other interactions to take place (Wang etal., 2006). Xa21 is regulated by two proteins through phosphorylation;
XB15, a protein phosphatase 2C (PP2C) and XB24, a protein with
intrinsic ATPase activity (Park etal., 2008). XB15
dephosphorylates Xa21 and XB15 over-expression reduces Xoo
resistance while xb15 null-mutants exhibit increased cell death
and resistance to Xoo. This would point to a negative regulatory
role of XB15. On the other hand, XB24 promotes
autophosphorylation of Xa21 and may be required to prevent
proteolytic cleavage of Xa21. The complex between XB24 and
Xa21 dissociates upon Xoo infection or AxYS22 binding (Chen
etal., 2010b). Phosphorylation, especially in the JM domain, plays
a critical role in Xa21 stability. It is clear that autophosphorylation
of certain residues in Xa21 promotes an inactive state but the exact
changes in phosphorylation status upon pathogen infection remain
largely unknown. Xa21 binds to the WRKY transcription factor XB10 and this
binding requires an active Xa21 kinase domain. Binding of the
AxYS22 peptide to Xa21 leads to translocation of a Xa21 kinase
domain-GFPfragmenttothenucleuswhereitinteractswithXB10.
The nuclear translocation is important for Xoo resistance and the
Xa21 kinase domain/XB10 complex probably affects defense

164
gene expression (Park and Ronald, 2012). Whether this or a
similar mechanism also applies to other RLKs is currently
unknown, but future studies will likely address this issue.
Recently, a largescale yeast two-hybrid study revealed yet another
set of Xa21 interacting partners (Seo etal., 2011). Although the
biological significance of these discoveries in signaling remains
to be seen, they may provide interesting clues to the functions of
Xa21 and other RLKs. To help proteins fold properly, the ER contains a number of
chaperones including BiPs (binding immunoglobulin protein) that
bind N-glycosylated proteins and direct them to the ER (Molinari
and Helenius, 2000). Xa21 is also N-glycosylated and interacts
with BiP3, an HSP70-like ATPase located in the ER, and this is
important for correct folding and functioning of the protein (Park etal., 2010a). While a pool of Xa21 locates to the PM where
AxYS22 ligand is perceived, the majority of the receptor is found
in the ER.
AtFLS2
The FLS2 (flagellin sensing 2) receptor recognizes the
wellconserved protein flagellin from a broad class of bacterial
plant pathogens including Pseudomonas syringae pv. tomato
(Pto) DC3000 (Gómez-Gómez and Boller, 2000). Direct binding
of the flagellin-derived peptide flg22 has been shown using 125I-
labeled peptides (Chinchilla etal., 2006), but a recent report also
implicates FLS2 in unsulfonated Xoo Ax21 peptide perception.
These two peptides are not sequence related, which makes the
finding quite astonishing (Danna etal., 2011). FLS2 was recently shown to form homo-dimers independently
of flg22 binding, but whether this dimerization is important for
receptor function is not known (Sun etal., 2012). However, it is
well-established that FLS2 forms heterodimers with
BRI1associated kinase 1 (BAK1) (Chinchilla etal., 2007; Schulze
etal., 2010) in the presence of bound flg22. BAK1 is a common
component in many RLK signaling complexes, and was first
identified for its requirement in brassinosteroid signaling via the
receptor BRI1 (Li etal., 2002). The essential role of BAK1 in
flg22 sensing was revealed by the marked reduction of flg22-
induced responses in bak1 plants (Chinchilla etal., 2007; Heese
etal., 2007). Importantly, the BAK1–FLS2 interaction most likely
does not compete with other known BAK1 interactors such as
BRI1, and the BAK1– FLS2 interaction is therefore not
responsible for BR-mediated PAMP defense suppression
(Albrecht etal.,2012). BAK1 is a member of the somatic
embryogenesis receptor kinase (SERK) family comprising 5
members, SERK1, SERK2, BAK1/SERK3, BAK1like
(BKK1)/SERK4, and SERK5. FLS2 interactions with SERK1,
SERK2, SERK5, and BKK have been detected, but its
predominant association is with BAK1. BAK1 and BKK1 are
thought to act cooperatively in PAMP signaling and resistance to
biotrophic pathogens (Roux etal., 2011). BAK1 and FLS2 also interact with Botrytis-induced kinase 1
(BIK1), which is a receptor-like cytoplasmic kinase (RLCK)
implicated in resistance to necrotrophic pathogens (Veronese
etal., 2006). BAK1 and FLS2 phosphorylate BIK1 (Lu etal., 2010)
and BIK1 in turn phosphorylates both FLS2 and BAK1. This is
thought to be an important signal amplification mechanism.
However, since FLS2 has been shown to have very low catalytic
activity in vitro (Schwessinger etal., 2011), BAK1 probably
possesses the predominant kinase activity influencing BIK1
phosphorylation. The BIK1–FLS2/BAK1 association is
decreased after flg22 sensing, suggesting that BIK1 is released to
activate downstream signaling components (Lu etal., 2010).
BIK1’s role in PTI is dependent on complex interactions with
major immune response regulators and may thus provide RLK
signaling complexes with the ability to discriminate between
biotrophic and necrotrophic pathogens (Laluk etal., 2011).
Importantly, bik1 mutants display enhanced susceptibility to Pto
DC3000, reduced flg22 responsiveness, as well as compromised
flg22-induced resistance to virulent Pto DC3000. The BIK1-
related kinases, PBS-like kinase 1 (PBL1) and PBL2 also interact
with FLS2 and BAK1. pbl1 mutants show less reduction in PTI
responses but the effect seems to be additive to BIK1 function
(Zhang etal., 2010). BAK1, BKK1, SERK1, and SERK2 have also been shown to
interact with BIR1 (BAK1-interacting receptor-like kinase 1), an
activeproteinkinase. Thebir1mutantexhibitsincreasedresistance to
biotrophic Pto DC3000 and Hyaloperonospora arabidopsidis
Noco2, due to apparent R protein activation (Wang etal., 2011).
The bir1 phenotype is partially rescued in bir1 pad4 double
mutants, and is completely rescued in the bir1 pad4 sobir
(suppressor of bir1-1) triple mutant. Phytoalexin deficient 4
(PAD4) is one of the critical components required for
Toll/interleukin-1 receptor (TIR) R protein signaling. Many
constitutively active defense phenotypes that result from activated
TIR R proteins are suppressed by PAD4 loss of function (Wiermer
etal., 2005; Palma etal., 2010; Zhang etal., 2012). The
aforementioned results thus indicate that the bir1 phenotype is
partly dependent upon R protein activation, although the majority
of defense induction in bir1 occurs through SOBIR1. SOBIR1 is
also a RLK, and over-expression of SOBIR1 leads to activation
of cell death. SOBIR1 does not function in flg22 sensing and does
not interact with BIR1. Exactly how loss of BIR1 activates
SOBIR1 is a mystery (Gao etal., 2009), and it is still uncertain
whether BIR1 has a role in the PAMP signaling pathway. Kinase-associated protein phosphatase (KAPP) interacts with
the FLS2 kinase domain (Gómez-Gómez and Boller, 2000), and
this interaction may be important for receptor endocytosis upon
activation as was found for AtSERK1 (Shah etal., 2002). KAPP
has also been found in complexes with other RLKs (Williams
etal., 1997; Stone etal., 1998) but whether it functions as a general
regulator of a broader spectrum of RLKs needs to be explored. FLS2 also interacts with E3 ligases that polyubiquitinate the
receptor after flg22 signaling. FLS2 is subsequently degraded by
the proteasome, which might constitute a mechanism for
attenuation as has been described for the mammalian Toll-like
receptor 4 (TLR4) and TLR9 (Chuang and Ulevitch, 2004). Plant
U-Box 12 (PUB12) and PUB13, both E3 ubiquitin ligases, have
been shown to be BAK1 phosphorylation targets, and this
modification is required for their association with FLS2. This
phosphorylation is reminiscent of the previously
mentionedXa21phosphorylationof XB3.
PUB12andPUB13control flg22-dependent, proteasome-mediated
degradation of FLS2 (Lu etal., 2011), making this system
important for FLS2 signaling attenuation, together with receptor
endocytosis (Salomon and Robatzek, 2006).

165
Despite being a transmembrane protein, FLS2 does not depend
critically on N-glycosylation for its function as has been found for
EFR (Nekrasov etal., 2009; Saijo etal., 2009; Häweker etal.,
2010). However, FLS2 has recently been shown to interact with
the reticulon-like proteins RTNLB1 and RTNLB2. RTNLB1/2
are together involved in regulating FLS2 transport from the ER to
the plasma membrane (Lee etal., 2011). In addition, stomatal
cytokinesis defective 1 (SCD1) was identified by mass
spectrometry as an FLS2 interaction partner. Scd1 mutants display
SA-dependent enhanced resistance to infection with Pto DC3000,
as well as enhanced accumulation of PR1 transcripts and
hydrogen peroxide. However, the same mutants are less sensitive
to PAMPs, with reduced seedling growth inhibition and ROS
production in response to flg22 or elf18 (Korasick etal., 2010).
EF-Tu RECEPTOR
EF-Tu receptor is a LRR-RLK that recognizes the peptide elf18
from bacterial elongation factor (EF)-Tu. EFR and BAK1 have
also been shown to interact in a ligand-dependent manner (Roux
etal., 2011). Indeed, many of the signaling components
downstream of EFR and FLS2 are shared. While both EFR and
FLS2 are capable of associating with all members of the SERK
family, BKK1, SERK1,
SERK2haveastrongerassociationwithEFRthanwithFLS2(Roux
etal., 2011). This might allow EFR to avoid pathogen effector
action on the single SERKs. Studies of SERK function have been
difficultduetotheirapparentredundancyandthelethalityof some
double mutants such as serk1 serk2 and bak1-4 bkk1-1
(Colcombet etal.,2005; Heetal.,2007). However,the discovery of
a novelallele of bak1, bak1-5, enabled study of non-lethal bak1-5
bkk1 double mutants. This revealed that BAK1 and BKK1 act
cooperatively in PAMP signaling (Roux etal., 2011;
Schwessinger etal., 2011). BIK1 is phosphorylated upon elf18 and flg22 treatment (Lu
etal., 2010). Given the many parallels between FLS2 and EFR, it
is possible that transphosphorylation of the EFR/BAK1 complex
also occurs, although direct proof is still lacking. In contrast to
FLS2, but similarly to Xa21, N-glycosylation is critical for EFR
function and EFR is subject to ER quality control that requires
several chaperones involved in ER-QC for full activity (Häweker
etal., 2010).
PEPR1
In contrast to the three receptors described above, Pep1 receptor
1 (PEPR1) binds AtPep1 (Yamaguchi etal., 2006) a DAMP
derived from the precursor gene PROPEP1. PEPR1 and PEPR2
act redundantly to perceive AtPep1. BAK1 was shown to interact
with PEPR1 like FLS2 and EFR (Postel etal., 2010). PEPR1
possesses a putative guanylyl cyclase (GC) domain and cGMP
production by the purified RLK was shown in vitro (Qi etal.,
2010). Interestingly, a GC domain is also present in BRI1 and was
shown to have a catalytic function in vitro (Kwezi etal., 2007).
This cGMP generated after elicitation may trigger a cyclic
nucleotide-activated Ca2+ channel as part of its signaling activity
(Ali etal., 2007).
LysM FAMILY
Chitin elicitor receptor kinase 1 (CERK1) is the best studied
Arabidopsis LysM-RLK (Kaku etal.,2006; Miya etal.,2007; Wan
etal., 2008), and direct binding of chitin to CERK1 has been
detected (Iizasa etal., 2010; Petutschnig etal., 2010). Unlike FLS2
and EFR, CERK1’s perception of fungal chitin is BAK1-
independent. In rice, Chitin elicitor-binding protein (CeBIP), a
LysM domaincontaining protein, associates with OsCerk1 and
these proteins function together in a hetero-oligomer receptor
complex to elicit chitin signaling in a ligand-dependent manner
(Shimizu etal., 2010).
TwoLysMdomainproteins,LYM1andLYM3,haverecently been
shown to be important for peptidoglycan (PGN), but not chitin
recognition. LYM1 and LYM3 are not functionally redundant,
and it has been proposed that LYM1, LYM3 and CERK1 may
form a complex or complexes. cerk1 is hypersusceptible to Pto
DC3000 and shows reduced sensitivity to PGN, phenocopying
lym1/lym3, however CERK1 does not bind to PGN. Further, given
the fact that neither LYM1 nor LYM3 contain a cytoplasmic
domain, a LYM1/LYM3/CERK1 complex seems likely
(Willmann etal., 2011). RLKs often hetero-oligomerize for
optimal functioning as seen in the co-operativity of FLS2/BAK1,
EFR/BAK1 and PEPR1/PEPR2.
CrRLK1L FAMILY
Another RLK, FERONIA (FER) was first shown to control pollen
tube reception (Escobar-Restrepo etal., 2007). However, the
expressionof FERthroughouttheplantsuggestsageneralfunction
not strictly associated with root development or pollen tube
reception. Indeed, FER has more recently been shown to aid
powdery mildew (PM) penetration into host cells (Kessler etal.,
2010) and
toberesponsibleforsusceptibilitytotheoomyceteH.arabidopsidis
(Nibau and Cheung, 2011). It is suspected that FER might play a
role in controlling localization of MLO family proteins, known to
be important for PM infection (Consonni etal., 2006), as it does
for NTA during pollen tube reception. This however still needs to
be shown, as well as whether ROS signaling has an effect on MLO
localization. Given the many roles of FER it is not surprising to
find that it is important for disease resistance as well. FER appears to exert is signaling functions by controlling ROS
production. FER was shown to interact with guanine nucleotide
exchange factors (GEFs) that regulate RHO GTPases
(RAC/ROPs). RAC/ROP is known play important roles in
stressinduced responses. In rice, the binding of a RAC/ROP called
Rac GTPase to NADPH oxidases has been characterized, and Rac
GTPase was show to be required for PAMP-mediated ROS
production (Wong etal., 2007). In Arabidopsis, Rop2 was shown
to co-immunoprecipitate with FER. In addition, transgenic
plantsexpressingconstitutivelyactive,GTP-boundRop2displayed
increased ROS production (Cheung and Wu,2011). This indicates
that a FER-GEF-RAC/ROP complex is likely able to effect ROS
production. While ROS play a role in root development, there are
hintsthatFERisinvolvedinROSproductionduringPAMPsignaling
in leaves. For example, FER is enriched in detergent-resistant
membranes (DRMs) after flg22 treatment, and FER shows

166
flg22induced phosphorylation (Benschop etal., 2007). Fer
mutants also exhibit enhanced ROS production, and aberrant
stomatal responsesuponflg22treatment(Keinathetal.,2010).
Theincrease in ROS production in the fer mutant is puzzling given
the reduced Rho GTPase activity in this mutant (Duan etal.,
2010). The relationship between FLS2 and FER in the control of
ROS production is very interesting and should attract attention in
the near future.
CONCLUDING REMARKS
There have been enormous advancements in our knowledge about
RLK signaling in the last decade, but many questions still remain
unanswered. For example, the link between the PRR receptors
and production of ROS and activation of MAP kinases is still
missing. Nevertheless, a quite comprehensive picture of the route
from receptor activation to enhanced defense gene expression has
emerged for Xa21 and similar data for FLS2 and EFR are sure to
come to light.
ACKNOWLEDGMENTS
This work was supported by grants to Morten Petersen from the
Danish Research Council for Technology and Production (11-
106302) and the Strategic Research Council (09-067148).

REFERENCES
Albrecht, C., Boutrot, F., Segonzac, C., Schwessinger, B., GimenezIbanez, S., Chinchilla, D., Rathjen, J. P., De Vries, S. C., and Zipfel, C. (2012). Brassinosteroids inhibit pathogen-associated molecular pattern-triggered immune signaling independent of the receptor kinase BAK1. Proc.
Natl. Acad. Sci. U.S.A. 109, 303–308. Ali, R., Ma, W., Lemtiri-Chlieh, F., Tsaltas, D., Leng, Q., von Bodman, S., and Berkowitz, G. A. (2007). Death don’t have no mercy and neither does calcium:
Arabidopsis CYCLIC NUCLEOTIDEGATEDCHANNEL2 and innate immunity. Plant Cell 19, 1081–1095. Benschop, J. J., Mohammed, S., O’Flaherty, M., Heck, A. J. R., Slijper, M., and Menke, F. L. H. (2007). Quantitative phosphoproteomics of early elicitor signaling
in Arabidopsis. Mol. Cell Proteomics 6, 1198–1214. Chen, X., Chern, M., Canlas, P. E., Jiang, C., Ruan, D., Cao, P., and Ronald, P. C. (2010a). A conserved threonine residue in the juxtamembrane domain of the
XA21 pattern recognition receptor is critical for kinase autophosphorylation and XA21-mediated immunity. J. Biol. Chem. 285, 10454–10463. Chen, X., Chern, M., Canlas, P. E., Ruan, D., Jiang, C., and Ronald, P. C. (2010b). An ATPase promotes autophosphorylation of the pattern recognition receptor
XA21 and inhibits XA21-mediated immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 8029–8034. Cheung, A. Y., and Wu, H.-M. (2011). THESEUS 1, FERONIA and relatives: a family of cell wall-sensing receptor kinases? Curr. Opin. Plant Biol. 14, 632–
641. Chinchilla, D., Bauer, Z., Regenass, M., Boller, T., and Felix, G. (2006). The Arabidopsis receptor kinase FLS2 binds flg22 and determines the specificity
of flagellin perception. Plant Cell 18, 465–476. Chinchilla, D., Zipfel, C., Robatzek, S., Kemmerling, B., Nürnberger, T., Jones, J. D. G., Felix, G., and Boller,
T. (2007). A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448, 497–500. Chuang, T.-H., and Ulevitch, R. J. (2004). Triad3A, an E3 ubiquitinprotein ligase regulating Toll-like receptors. Nat. Immunol. 5, 495–502. Colcombet, J.,
Boisson-Dernier, A., Ros-Palau, R., Vera, C. E., and Schroeder, J. I. (2005). Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASES1 and 2 are
essential for tapetum development and microspore maturation. Plant Cell 17, 3350–3361. Consonni, C., Humphry, M. E., Hartmann, H. A., Livaja, M., Durner, J., Westphal, L., Vogel, J., Lipka,
V., Kemmerling, B., Schulze-Lefert, P., Somerville, S. C., Panstruga, R. (2006). Conserved requirement for a plant host cell protein in powdery mildew
pathogenesis. Nat. Genet. 38, 716–720. Danna, C. H., Millet, Y. A., Koller, T., Han, S.-W., Bent, A. F., Ronald, P. C., and Ausubel, F. M. (2011). The Arabidopsis flagellin receptor FLS2 mediates the
perception of Xanthomonas Ax21 secreted peptides. Proc. Natl. Acad. Sci. U.S.A. 108, 9286–9291. Dardick, C., and Ronald, P. (2006). Plant and animal pathogen recognition receptors signal through nonRD kinases. PLoS Pathog. 2, e2. doi:
10.1371/journal.ppat.0020002 Duan, Q., Kita, D., Li, C., Cheung,
A. Y., and Wu, H.-M. (2010). FERONIA receptor-like kinase regulates RHO GTPase signaling of root hair development. Proc. Natl. Acad. Sci. U.S.A. 107,
17821–17826. Escobar-Restrepo, J.-M., Huck, N., Kessler, S., Gagliardini, V., Gheyselinck, J., Yang, W.-C., and Grossniklaus, U. (2007). The FERONIA receptor-like kinase
mediates male-female interactions during pollen tube reception. Science 317, 656–660. Frank, S. A. (1992). Models of plantpathogen coevolution. Trends Genet. 8, 213–219. Gao, M., Wang, X., Wang, D., Xu, F., Ding, X., Zhang, Z., Bi, D., Cheng,
Y. T., Chen, S., Li, X., and Zhang, Y. (2009). Regulation of cell death and innate immunity by two receptorlike kinases in Arabidopsis. Cell Host Microbe 6,
34–44. Gish, L. A., and Clark, S. E. (2011). The RLK/Pelle family of kinases. Plant J. 66, 117–127. Gómez-Gómez, L., and Boller, T. (2000). FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol.
Cell 5, 1003–1011. Häweker, H., Rips, S., Koiwa, H.,
Salomon, S., Saijo, Y., Chinchilla, D., Robatzek, S., and von Schaewen, A. (2010). Pattern recognition receptors require N-glycosylation to mediate plant
immunity. J. Biol. Chem. 285, 4629–4636. He,K.,Gou,X.,Yuan,T.,Lin,H.,Asami, T., Yoshida, S., Russell, S. D., and Li, J. (2007). BAK1 and BKK1 regulate brassinosteroid-dependent growth and
brassinosteroid-independent cell-death pathways. Curr. Biol. 17, 1109–1115. Heese, A., Hann, D. R., GimenezIbanez, S., Jones, A. M. E., He,
K., Li, J., Schroeder, J. I., Peck, S. C., and Rathjen, J. P. (2007). The receptor-like kinase SERK3/BAK1 is a central regulator of innate immunity in plants.
Proc. Natl. Acad. Sci. U.S.A. 104, 12217–12222. Iizasa, E., Mitsutomi, M., and Nagano,
Y. (2010). Direct binding of a plant LysM receptor-like kinase, LysM RLK1/CERK1, to chitin in vitro. J. Biol. Chem. 285, 2996–3004. Johnson, L. N., Noble, M. E. and Owen, D. J. (1996). Active and inactive protein kinases: structural basis for regulation. Cell 85, 149–158. Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. Kaku, H., Nishizawa, Y., IshiiMinami, N., Akimoto-Tomiyama, C., Dohmae, N., Takio, K., Minami, E., and Shibuya, N. (2006). Plant cells recognize chitin
fragments for defense signaling through a plasma membrane receptor. Proc. Natl. Acad. Sci. U.S.A. 103, 11086–
11091. Keinath, N. F., Kierszniowska, S., Lorek, J., Bourdais, G., Kessler, S.
A., Shimosato-Asano, H., Grossniklaus, U., Schulze, W. X., Robatzek, S., and Panstruga, R. (2010). PAMP (pathogen-associated molecular pattern)-induced
changes in plasma membrane compartmentalization reveal novel components of plant immunity. J. Biol. Chem. 285, 39140–39149. Kessler, S. A., Shimosato-Asano, H., Keinath, N. F., Wuest, S. E., Ingram, G., Panstruga, R., and Grossniklaus, U. (2010). Conserved molecular components for
pollen tube reception and fungal invasion. Science 330, 968–971. King, D., and Roughgarden, J. (1982). Multiple switches between vegetative and reproductive growth in annual plants. Theor. Popul. Biol. 21, 194–204. Korasick, D. A., McMichael, C., Walker, K. A.,Anderson, J. C., Bednarek, S.Y., andHeese,A.(2010). Novelfunctions of stomatal cytokinesis-defective 1 (SCD1)
in innate immune responses against bacteria. J. Biol. Chem. 285, 23342–23350. Krol, E., Mentzel, T., Chinchilla, D., Boller, T., Felix, G., Kemmerling, B., Postel, S., Arents, M., Jeworutzki, E., Al-Rasheid, K. A. S., Becker, D., and Hedrich,
R. (2010). Perception of the Arabidopsis danger signal peptide 1 involves the pattern recognition receptor AtPEPR1 and its close homologue AtPEPR2. J. Biol.
Chem. 285, 13471–13479.

8
Kwezi, L., Meier, S., Mungur, L., Ruzvidzo, O., Irving, H., and Gehring, C. (2007). The Arabidopsis thaliana brassinosteroid receptor (AtBRI1) contains a domain
that functions as a guanylyl cyclase in vitro. PLoS ONE 2, e449. doi: 10.1371/journal.pone.0000449
Laluk, K., Luo, H., Chai, M., Dhawan, R., Lai, Z., and Mengiste, T. (2011). Biochemical and genetic requirements for function of the immune response regulator
BOTRYTISINDUCED KINASE1 in plant growth, ethylene signaling, and PAMP-triggered immunity in Arabidopsis. Plant Cell 23, 2831–
2849. Lee, H. Y., Bowen, C. H., Popescu, G. V., Kang, H.-G., Kato, N., Ma, S., Dinesh-Kumar, S., Snyder, M., and Popescu, S. C. (2011). Arabidopsis RTNLB1 and
RTNLB2 reticulonlike proteins regulate intracellular trafficking and activity of the FLS2 immune receptor. Plant Cell 23, 3374–3391. Lee, S.-W., Han, S.-W., Sririyanum, M., Park, C.-J., Seo, Y.-S., and Ronald, P. C. (2009). A Type Isecreted, sulfated peptide triggers XA21-mediated innate
immunity. Science 326, 850–853. Lehti-Shiu, M. D., Zou, C., Hanada, K., and Shiu, S.-H. (2009). Evolutionary history and stress regulation of plant receptor-likekinase/Pellegenes. Plant Physiol.
150, 12–26. Lerouge, P., Roche, P., Faucher, C., Maillet, F., Truchet, G., Promé, J. C., and Dénarié, J. (1990). Symbiotic host-specificity of Rhizobium meliloti is
determined by a sulphated and acylatedglucosamineoligosaccharide signal. Nature 344, 781–784. Li, J., Wen, J., Lease, K. A., Doke, J. T., Tax, F. E., and Walker,
J. C. (2002). BAK1, an Arabidopsis LRR receptorlike protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 110, 213–222. Lu, D.,
Lin, W., Gao, X., Wu, S., Cheng, C., Avila, J., Heese, A., Devarenne, T. P., He, P., and Shan, L. (2011). Direct ubiquitination of pattern recognition receptor FLS2 attenuates plant innate
immunity. Science 332, 1439–1442. Lu, D., Wu, S., Gao, X., Zhang, Y., Shan, L., and He, P. (2010). A receptorlike cytoplasmic kinase, BIK1, associates with a flagellin receptor complex to initiate
plant innate immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 496–501. Mithoe, S. C., Boersema, P. J., Berke, L., Snel, B., Heck, A. J. R., and Menke, F. L. H. (2011). Targeted quantitative phosphoproteomics approach for the detection
of phospho-tyrosine signaling in plants. J. Proteome Res. 11, 438–448. Miya, A., Albert, P., Shinya, T.,
Desaki, Y., Ichimura, K., Shirasu, K., Narusaka, Y., Kawakami, N., Kaku, H., and Shibuya, N. (2007). CERK1, aLysMreceptorkinase,isessentialfor chitin
elicitor signaling in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 104, 19613–19618. Molinari, M., and Helenius, A.
(2000). Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science 288, 331–333. Nekrasov, V., Li, J., Batoux, M., Roux,
M.,Chu,Z.-H.,Lacombe,S.,Rougon, A., Bittel, P., Kiss-Papp, M., Chinchilla, D., van Esse, H. P., Jorda, L., Schwessinger, B., Nicaise, V., Thomma, B. P., Molina, A., Jones, J. D., and Zipfel, C.
(2009). Control of the pattern-recognition receptor EFR by an ER protein complex in plant immunity. EMBO J. 28, 3428– 3438. Nibau, C., and Cheung, A. Y. (2011). New insights into the functional roles of CrRLKs in the control of plant cell growth and development. Plant Signal. Behav.
6, 655–659. Palma, K., Thorgrimsen, S., Malinovsky, F. G., Fiil, B. K., Nielsen, H. B., Brodersen, P., Hofius, D., Petersen, M., and Mundy, J. (2010).
Autoimmunity in Arabidopsis acd11 is mediated by epigenetic regulation of an immune receptor. PLoS Pathog. 6, e1001137. doi: 10.1371/journal.
ppat.1001137 Park, C.-J., Bart, R., Chern, M., Canlas, P. E., Bai, W., and Ronald, P. C. (2010a). Overexpression of the endoplasmic reticulum chaperone BiP3 regulates XA21-
mediated innate immunity in rice. PLoS ONE 5, e9262. doi: 10.1371/journal. pone.0009262 Park, C.-J., Han, S.-W., Chen, X., and Ronald, P. C. (2010b). Elucidation of XA21-mediated innate immunity. Cell. Microbiol. 12, 1017–1025. Park, C.-J., Peng, Y., Chen, X., Dardick, C., Ruan, D., Bart, R., Canlas, P. E., and Ronald, P. C. (2008). Rice XB15,a protein phosphatase 2C,negatively regulates
cell death and XA21mediated innate immunity. PLoS Biol. 6, e231. doi: 10.1371/journal. pbio.0060231
Park, C.-J., and Ronald, P. C. (2012). Ligand induced cleavage and nuclear localization of the rice XA21 immune receptor. Nature Precedings. Available at:
http://hdl.handle.net/10101/ npre.2012.6776.1. Petutschnig, E. K., Jones, A. M. E., Serazetdinova, L., Lipka, U., and Lipka, V. (2010). The lysin motif receptorlike kinase (LysM-RLK) CERK1 is a major chitin-
binding protein in Arabidopsis thaliana and subject to chitin-induced phosphorylation. J. Biol. Chem. 285, 28902–28911. Postel, S., Küfner, I., Beuter, C., Mazzotta, S., Schwedt, A., Borlotti, A., Halter, T., Kemmerling, B., and Nürnberger, T. (2010). The multifunctional leucine-rich
repeat receptor kinase BAK1 is implicated in Arabidopsis development and immunity. Eur. J. Cell Biol. 89, 169–174. Qi, Z., Verma, R., Gehring, C., Yamaguchi, Y., Zhao, Y., Ryan, C. A., and Berkowitz, G. A. (2010). Ca2+ signaling by plant Arabidopsis thaliana Pep peptides
depends on AtPepR1, a receptor with guanylyl cyclase activity, and cGMP-activated Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 107, 21193–21198. Roux, M., Schwessinger, B., Albrecht, C., Chinchilla, D., Jones, A., Holton,
N., Malinovsky, F. G., Tör, M., de Vries, S., and Zipfel, C. (2011). The Arabidopsis leucine-rich repeat receptor–like kinases BAK1/SERK3 and BKK1/SERK4
are required for innate immunity to hemibiotrophic and biotrophic pathogens. Plant Cell 23, 2440–2455. Saijo, Y., Tintor, N., Lu, X., Rauf, P., Pajerowska-Mukhtar, K., Häweker, H., Dong, X., Robatzek, S., and Schulze-Lefert, P. (2009). Receptor quality control in
the endoplasmic reticulum for plant innate immunity. EMBO J. 28, 3439–3449. Salomon, S., and Robatzek, S. (2006). Induced endocytosis of the receptor kinase FLS2. Plant Signal. Behav. 1, 293–295. Schulze, B., Mentzel, T., Jehle, A. K., Mueller, K., Beeler, S., Boller, T., Felix, G., and Chinchilla, D. (2010). Rapid heteromerization and phosphorylation of
ligand-activated plant transmembrane receptors and their associated kinase BAK1. J. Biol. Chem. 285, 9444–9451. Schwessinger, B., Roux, M., Kadota,
Y., Ntoukakis, V., Sklenar, J., Jones, A., and Zipfel, C. (2011). Phosphorylation-dependent differential regulation of plant growth, cell death, and innate
immunity by the regulatory receptor-like kinase BAK1. PLoS Genet. 7, e1002046. doi: 10.1371/journal.pgen.1002046 Seo, Y.-S., Chern, M., Bartley, L.
E., Han, M., Jung, K.-H., Lee, I., Walia, H., Richter, T., Xu, X., Cao, P., Bai, W., Ramanan, R., Amonpant, F., Arul, L., Canlas, P. E., Ruan, R., Park, C. J., Chen, X., Hwang, S., Jeon, J. S., and
Ronald, P. C. (2011). Towards establishment of a rice stress response interactome. PLoS Genet. 7, e1002020. doi: 10.1371/journal.pgen.1002020 Shah, K., Russinova, E., Gadella, T. W. J. Jr, Willemse, J., and De
Vries, S. C. (2002). The Arabidopsis kinase-associated protein phosphatase controls internalization of the somatic embryogenesis receptor kinase 1. Genes Dev.
16, 1707–

9
1720. Shimizu, T., Nakano, T., Takamizawa, D., Desaki, Y., Ishii-Minami, N., Nishizawa, Y., Minami, E., Okada, K., Yamane, H., Kaku, H., and
Shibuya, N. (2010). Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J. 64, 204–214. Shiu, S.-H., and Bleecker, A. B. (2001). Plant receptor-like kinase gene family: diversity, function, and signaling. Sci. STKE 2001, re22. Shiu, S.-H., Karlowski, W. M., Pan, R.,
Tzeng, Y.-H., Mayer, K. F. X., and Li, W.-H. (2004). Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 16, 1220–1234.
Song, W.-Y., Wang, G.-L., Chen, L.- L., Kim, H.-S., Pi, L.-Y., Holsten, T., Gardner, J., Wang, B., Zhai, W.X., Zhu, L.-H., Fauquet, C., and Ronald, P. (1995). A receptor kinaselike protein encoded by the rice disease resistance
gene, Xa21. Science 270, 1804–1806. Stone, J. M., Trotochaud, A. E., Walker, J. C., and Clark, S. E. (1998). Controlof meristemdevelopment by CLAVATA1 receptor kinase and kinase-associated protein phosphatase interactions. Plant Physiol. 117, 1217–
1225. Suarez-Rodriguez, M. C., AdamsPhillips, L., Liu, Y., Wang, H., Su,
S.-H., Jester, P. J., Zhang, S., Bent, A. F., and Krysan, P. J. (2007). MEKK1 is required for flg22-induced MPK4 activation inArabidopsis plants. Plant Physiol.
143, 661–669. Sultan, S. E. (2000). Phenotypic plasticity for plant development, function and life history. Trends Plant Sci. 5, 537–542. Sun, W., Cao, Y., Jansen, K. L., Bittel, P., Boller, T., and Bent, A. F. (2012). Probing the Arabidopsis flagellinreceptor: FLS2-FLS2association and the
contributions of specific domains to signaling function. Plant Cell 24, 1096–1113. Veronese, P., Nakagami, H., Bluhm, B.,
AbuQamar, S., Chen, X., Salmeron, J., Dietrich, R. A., Hirt, H., and Mengiste, T. (2006). The membraneanchored BOTRYTIS-INDUCED KINASE1 plays distinct roles in Arabidopsis resistance to necrotrophic and biotrophic pathogens. Plant Cell 18, 257–273.
Wan, J., Zhang, X.-C., Neece, D., Ramonell, K. M., Clough, S., Kim, S.-Y., Stacey, M. G., and Stacey, G. (2008). A LysM receptor-like kinase plays a critical
role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 20, 471–481. Wang, Y.-S., Pi, L.-Y., Chen, X.,
Chakrabarty, P. K., Jiang, J., De Leon, A. L., Liu, G.-Z., Li, L., Benny, U., Oard, J., Ronald, P. C., and Song, W. Y. (2006). Rice XA21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell 18, 3635–3646.
Wang, Z., Meng, P., Zhang, X., Ren, D., and Yang, S. (2011). BON1 interacts with the protein kinases BIR1 and BAK1 in modulation of temperature-
dependentplantgrowth and cell death in Arabidopsis. Plant J. 67, 1081–1093. Wiermer, M., Feys, B. J., and Parker, J. E. (2005). Plant immunity: the EDS1 regulatory node. Curr. Opin. Plant Biol. 8, 383–389. Williams, R. W., Wilson, J. M., and Meyerowitz, E. M. (1997). A possible role for kinase-associated protein phosphatase in the Arabidopsis CLAVATA1 signaling
pathway. Proc. Natl. Acad. Sci. U.S.A. 94, 10467– 10472. Willmann, R., Lajunen, H. M., Erbs, G., Newman, M.-A., Kolb, D., Tsuda, K., Katagiri, F., Fliegmann, J., Bono, J.-J., Cullimore, J. V., Jehle, A. K., Götz,
F., Kulik, A., Molinaro, A., Lipka, V., Gust, A. A., and Nürnberger, T. (2011). Arabidopsis lysin-motif proteins LYM1 LYM3 CERK1 mediate bacterial
peptidoglycan sensing and immunity to bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 108, 19824– 19829. Wong, H. L., Pinontoan, R., Hayashi, K., Tabata, R., Yaeno, T., Hasegawa, K., Kojima, C., Yoshioka, H., Iba, K., Kawasaki, T., and Shimamoto, K. (2007).
Regulation of rice NADPH oxidase by binding of Rac GTPase to its N-terminal extension. Plant Cell 19, 4022–4034. Xu, W., Wang, Y., Liu, G., Chen, X.,
Tinjuangjun, P., Pi, L., and Song, W. (2006). The autophosphorylated Ser686, Thr688, and Ser689 residues in the intracellular juxtamembrane domain of XA21 are implicated in stability control
of rice receptor-like kinase. Plant J. 45, 740–751. Yamaguchi, Y., Pearce, G., and Ryan, C. A. (2006). The cell surface leucine-rich repeat receptor for AtPep1, an endogenous peptide elicitor in Arabidopsis, is
functional intransgenictobaccocells. Proc. Natl. Acad. Sci. U.S.A. 103, 10104–10109. Zhang, J., Li, W., Xiang, T., Liu, Z., Laluk, K., Ding, X., Zou, Y., Gao, M., Zhang, X., Chen,
S.,Mengiste, T., Zhang, Y., and Zhou, J. M. (2010). Receptor-like cytoplasmic kinases integrate signaling from multiple plant immune receptors and are targeted
by a Pseudomonas syringae effector. Cell Host Microbe 7, 290–301. Zhang, Z., Wu, Y., Gao, M., Zhang, J., Kong, Q., Liu, Y., Ba, H., Zhou, J., and Zhang, Y. (2012). Disruption of PAMP-induced MAP kinase cascade by a
Pseudomonas syringae effector activatesplantimmunitymediatedby the NB-LRR protein SUMM2. Cell Host Microbe 11, 253–263. Zipfel, C., Kunze, G., Chinchilla, D., Caniard, A., Jones, J. D. G., Boller, T., and Felix, G. (2006). Perception of the bacterial PAMP EF-Tu by the
receptorEFRrestrictsAgrobacteriummediated transformation. Cell 125, 749–760. Conflict of Interest Statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be
construed as a potential conflict of interest.
Received: 03 May 2012; paper pending published: 01 June 2012; accepted: 16 August 2012; published online: 24 August 2012. Citation: Greeff C, Roux M, Mundy J and Petersen M (2012) Receptor-like kinase complexes in plant innate immunity. Front. PlantSci. 3:209. doi: 10.3389/
fpls.2012.00209 This article was submitted to Frontiers in Plant Proteomics, a specialty of Frontiers in Plant Science. Copyright © 2012 Greeff, Roux, Mundy and Petersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License,
which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices
concerning any third-party graphics etc.

10
Appendix I
at1g10920nUF GGCTTAAUATGAAGAGTTTAGGAATACAAGAGATTATC
at1g12210nUF GGCTTAAUATGGGCGGTTGTGTCTCGGTCTCGTTGTCA
at1g12220nUF GGCTTAAUATGGGAGGTTGTTTCTCTGTTTCATTGCCA
at1g12280nUF GGCTTAAUATGGGAGCTTGTTTAACACTCTCGTTCTCT
at1g12290nUF GGCTTAAUATGGGTGGTTGTGTCTCTGTCCAAGTATCA
at1g15890nUF GGCTTAAUATGGGAAACTGTGTAGCACTAGAAATATCT
at1g33560nUF GGCTTAAUATGGCTTCGTTCATAGATCTTTTCGCCGGC
at1g50180nUF GGCTTAAUATGGCAGAGGCTATTGTTTCTGTTACAGTG
at1g53350nUF GGCTTAAUATGGCTGAGGCAGTTGTATCATTTGGAGTT
at1g58390nUF GGCTTAAUATGGCTGGAGAACTTGTGTCGTTTGGAATA
at1g58400nUF GGCTTAAUATGGTCGAGGCAATTGTTTCATTTGGAGTA
at1g58410nUF GGCTTAAUATGGAACTTGTGTCGTTTGGAGTAGAAAAG
at1g59124nUF GGCTTAAUATGGCTGGGGAACTTATTTCGTTTGGTATA
at1g59218nUF GGCTTAAUATGGCTGGGGAACTTATTTCGTTTGGTATA
at1g59620nUF GGCTTAAUATGGCTGAGACACTTTTGTCATTTGGAGTC
at1g59780nUF GGCTTAAUATGCAGGACTTATATATGGTTGATTCAATT
at1g61180nUF GGCTTAAUATGGGGAGTTGTTTTTCTCTTCAAGTTAGT
at1g61190nUF GGCTTAAUATGGGAAATTTTGTGTGTATAGAAATTTCT
at1g61300nUF GGCTTAAUATGGGGTGTTGTTTTTCTGTCCAGTTTTCT
at1g61310nUF GGCTTAAUATGGGGAGTTGTTTTTCTTTTCAAATTGCT
at1g62630nUF GGCTTAAUATGGGTATTTCTTTCTCGATACCCTTTGAT
at1g63350nUF GGCTTAAUATGGGTATCTCTTTCTCGATACCCTTTGAT
at1g63360nUF GGCTTAAUATGGGTATTTCTTTCTCGATACCCTTTGAT
at3g07040nUF GGCTTAAUATGGCTTCGGCTACTGTTGATTTTGGGATC
at3g14460nUF GGCTTAAUATGGCGAACTCCTATTTATCAAGTTGTGCA
at3g14470nUF GGCTTAAUATGACCGGCATAGGAGAGATGTTCCTTGCA
at3g15700nUF GGCTTAAUATGGGGAAAGATTTCAAGAGTATGGTGACA
at3g50950nUF GGCTTAAUATGGTGGACGCTGTTGTAACAGTGTTTTTA
at4g10780nUF GGCTTAAUATGGGTAGTTGTATCTCTCTCCAAATATCA
at4g14610nUF GGCTTAAUATGGGAGGTTGTATCTCGGTCTCGGTGTCA
at4g26090nUF GGCTTAAUATGGATTTCATCTCATCTCTTATCGTTGGC
at4g27190nUF GGCTTAAUATGGAGTGTTGCGCCCCTGTCATTGGTGAG
at4g27220nUF GGCTTAAUATGTTCAGATCGAACGCAAGAGCGTTGAAT
at4g33300nUF GGCTTAAUATGGCCATCACCGATTTTTTCGCCGGTGAA
at5g04720nUF GGCTTAAUATGGCAGATATAATCGGCGGCGAAGTTGTG
at5g05400nUF GGCTTAAUATGGGAGCCTGCTTCTCTGTGGCAATATCC
at5g35450nUF GGCTTAAUATGGCTGAAGGAGTTGTGTCGTTTGGAGTT
at5g43730nUF GGCTTAAUATGGTGGACTGGCTTTCACTATTACCCTGG
at5g43740nUF GGCTTAAUATGCTGGGCTGGCTTGTAATACCCTGGAAT
at5g45510nUF GGCTTAAUATGTCTGATCCGTTGAAGATGGCGGCCGAG
at5g47250nUF GGCTTAAUATGAATTGCTGTTGGCAGGTAGTTGAGCCA
at5g47260nUF GGCTTAAUATGGGAAATAATTTCTCAGTTGAATCTCCA
at5g48620nUF GGCTTAAUATGGCTGAAGGATTTGTGTCGTTTGGGCTT

11
at5g63020nUF GGCTTAAUATGGGAGGTTGTGTCTCTGTATCAATATCA
at5g66630nUF GGCTTAAUATGCCGATCTCGGATGTCGCTTCTTTGGTT
at5g66900nUF GGCTTAAUATGAACGATTGGGCTAGTTTGGGAATAGGT
at5g66910nUF GGCTTAAUATGGTCGTGGTCGATTGGCTTGGTTTGGGA
at1g52660nUF GGCTTAAUATGGGAAAAGATTTCAAGAGTTTGGTTACT
at4g19060nUF GGCTTAAUATGGATATTGCTAAGAAGTTCATATCTGAA
At1g58807nUF GGCTTAAUATGGCTGGGGAACTTATTTCGTTTGGTATA
AT1G51480nUF GGCTTAAUATGGTGCAGCACTGGGATGGTGACCCAGCT
at5g47280nUF GGCTTAAUATGCTTTTTAATTTGAACGATGAGGCAAGA
AT1G58848nUF GGCTTAAUATGGCTGGGGAACTTATTTCGTTTGGTATA
at3g46530nUF GGCTTAAUATGGTAGATGCGATCACGGAGTTCGTTGTG
at5g43470nUF GGCTTAAUATGGCTGAAGCATTTGTGTCGTTTGGACTT
at1g10920-nURGGTTTAAUTCAAAGCACGAAGAAGTGGCATCGCCCTTC
at1g12210-nURGGTTTAAUTCAACGCAGCCTACAAGTAGGTAAGAAACG
at1g12220-nURGGTTTAAUTTATGTTTCTCTCCACCGCCACCTGGATGA
at1g12280-nURGGTTTAAUCTACCGCACATAACTAACTTGCCATTCAGG
at1g12290-nURGGTTTAAUTTATCGATTACACAAAACCAACTTGCATGA
at1g15890-nURGGTTTAAUTTATTCATTGGCATGTCGCGGAAACTCAGT
at1g33560-nURGGTTTAAUCTAATCGTCAAGCCAATCCACGGTGAAGCA
at1g50180-nURGGTTTAAUTTAAAGTTCACAGTTCTCAAAAACAACACA
at1g53350-nURGGTTTAAUTCAATAAGAAGATATACTAGGATTATGTCT
at1g58390-nURGGTTTAAUTCATTTGAGGTAGCCTCCTATGAATTCAAC
at1g58400-nURGGTTTAAUTTATTTGTAGTCCTTTTCGAATTTAACAGA
at1g58410-nURGGTTTAAUTTAAGAACTGAATTTAATAAAAGGAATGTT
at1g59124-nURGGTTTAAUCTAGAGTCGGCAATGGATCTTTCTCCAAAC
at1g59218-nURGGTTTAAUCTAGTAGAATTCAACAGAAGGAATGTGTTG
at1g59620-nURGGTTTAAUTTAAAGAAATCGAACAAGAGGAATGTGTTG
at1g59780-nURGGTTTAAUTCAGATGATTGGACTAGGGAAAGAATATAT
at1g61180-nURGGTTTAAUTCATGCTAACCTTGAAAGAAACATTAGCAA
at1g61190-nURGGTTTAAUCTAAACGCATAAACTTTTGATTGTGTAAAA
at1g61300-nURGGTTTAAUCTACTAAAGAAGAAGAAAATGAAACAATGT
at1g61310-nURGGTTTAAUTCAATTATACAATCCAAATGCTCTACAAGA
at1g62630-nURGGTTTAAUCTAGTCTTCAAAGATTGTTTCACACCTTTC
at1g63350-nURGGTTTAAUTTAAAGATGGAAGCAGTTCATATCACGAGA
at1g63360-nURGGTTTAAUTCATTTAAGAGAAATCAATTGGCATGAAGG
at3g07040-nURGGTTTAAUCTAAGATGAGAGGCTCACATAGAAAGAGCC
at3g14460-nURGGTTTAAUTCAAGAGAAGATCTCGCCATCGATTTCCAC
at3g14470-nURGGTTTAAUTTAGTCATATCTTGAAGATGAACGTGAATC
at3g15700-nURGGTTTAAUTTATAGAATTGGACCTGAGAAATTAGGAGG
at3g50950-nURGGTTTAAUTCAGGTTCTGTGCAATGGTGTTTTCATCCA
at4g10780-nURGGTTTAAUCTAGGAGAGGATATACACCAGTTTGGTTGA at4g14610Stopadd-nURGGTTTAAUAdded stop TCATGAATGTTTTAGTTTTAGAGTAATCAA
at4g26090-nURGGTTTAAUTCAATTTGGAACAAAGCGCGGTAAATAACA
at4g27190-nURGGTTTAAUTTATAGCATTTGAGTTGTTGCATCCGCTAT

12
at4g27220-nURGGTTTAAUTTACCCTAGAGTCCTAGACATGTATTCCCA
at4g33300-nURGGTTTAAUTTATTCGTCAAGCCAGTCTAGGCTGAAGCA
at5g04720-nURGGTTTAAUCTAATCGTCGAGCCAATCCCTGCTGAAAGA
at5g05400-nURGGTTTAAUTTAGCTTGGGAAAAAACGCTCCTTAGTCGC
at5g35450-nURGGTTTAAUTTATTCTCTTTGTTCGTCGTCACAGTTGAA
at5g43730-nURGGTTTAAUTCACCCTTTCAGCTTGGGAAAATTAGCTAT
at5g43740-nURGGTTTAAUTTAAAAGAAAAAAAAACGTGAAGGACTCTT
at5g45510-nURGGTTTAAUTCAGCATTCATGTGGTGGAGGTACATCACT
at5g47250-nURGGTTTAAUTTATAATGGACGTTGATGAAGATTCGGGCA
at5g47260-nURGGTTTAAUTTAGGTATAACATAAAAGGAAATTCGTACC
at5g48620-nURGGTTTAAUCTACAGGTCACAGTTGATAAATTGAACATC
at5g63020-nURGGTTTAAUTCAGAAGGAAATCTCATAGCCGTCAGCGTC
at5g66630-nURGGTTTAAUTTACCTCCGGCGAAGAATCTCCTTGAGGGT
at5g66900-nURGGTTTAAUTTAAAACATTTGAAGCAAGTTCAGGTTATG
at5g66910-nURGGTTTAAUTTAAAACGTTAGAAGCAACTTCAGGTTATG
at1g52660-nURGGTTTAAUCTAGTTCCTGTCCAGTAACTTTAGAACTGT
at4g19060-nURGGTTTAAUTTAGAAGCTCTGCTTCCTATCTATGTTTCC
At1g58807-nURGGTTTAAUTCATCTCATCAAGAGTCTCCCAGTTATATC
AT1G51480-nURGGTTTAAUTTATTCCTCTGCATGCATGGGAAAATTAGC
at5g47280-nURGGTTTAAUCTACTCGTCGGGCCAAGTCATGTTGAACCA
AT1G58848-nURGGTTTAAUCTAGTAGAATTCAACAGAAGGAATGTGTTG
at3g46530-nURGGTTTAAUTCAGCAGTAGATTTGACCAAACGCTCGCAA
at5g43470-nURGGTTTAAUCTACTGGTCACAGTTGATAAATTGAACATC
at1g12280UmF ACCACCCZTCTCACACGAATCA
at1g12290UmF ACAACCCZTCTCACTCAGATCA
at1g15890UmF ACCACGCZCTTAGCTTCTATTA
at1g50180UmF ACAACTTZAGCCAAACAGATTT
at1g53350UmF ACGACACZCGCGAGACAAGTTT
at1g58390UmF ACCACACZTGCTAGACAGGTTT
at1g58400UmF ACCACCCZTGCAAGACAAGTTT
at1g58410UmF ACCACCCZTGCTAGACAAGTTT
at1g59218UmF ACCACACZTGCTAAACAGGTTT
at1g59780UmF ACCACCCZTGCACGGCAAATTT
mcg_UmF-0950 ACTACAAZCGCTCAAGAAGTGTTCAATG
mcg_UmF-2210 ACCACCCZTCTCACGCAGATCAACAATAA
mcg_UmF-3300 AAACCACZCTTGCCAAAGAACTTCAACGGG
mcg_UmF-6530 ACTGCACZTGCTAGGAAGCTCTACAACTC
mcg_UmF1g10920 AAACCACZCTCGCAAGACAAGTCTTTCATC
mcg_UmF1g12220 ACGACACTACTCACGAAGATCAACAATAAG
mcg_UmF1g33560 AAACCACZCTTGCAATAGAGCTTTCAAAGG
mcg_UmF1g59124 ACCACACZTGCTAAACAGGTTTTTAACCAT
mcg_UmF1g59620 ACCACCCZTGCAAGACAAGTTTTCAATCAT
mcg_UmF1g61180 ACAACCCZTTTCAAGAAAATCCACAACAAG
mcg_UmF3g07040 ACTACACZCTCAGCGAATATCTTCAAGTCT
mcg_UmF3g14470 ACCACACZTTCACAACTCCTTTACAATGAT

13
mcg_UmF5g04720 ACCACTCZTGCCAAAGAGCTTGCCCGGGAC
mcg_UmF5g47280 ACCATTCZTGCCAAGGAGCTTGCGCGGGAC
mcg_UmF5g43470 ACCACTCZGGCTAGACAAGTCTTTCATCAT
61190UmF ACCCTTTZCAAGAAAATCCACAATAAGTTCGCTG
1300UmF ACCCTTTZCAAGAAAATCCACAATAAGTTCGCTAA
1310UmF ACCCTTTZCAAGAAAATCCACAACAAGTTCGCT
2630UmF ACACTTCZCACTCAGCTCTTCAATATGTTCA
3360UmF ACACTTCZCACTCAGCTCTACAATATGTTCAATAA
4460UmF ACTACCTZGACAGAGATAGTTTTTAATGACTAC
5700UmF ACGACGGZGTTGACTCAGGTAAACAACAGGTTGCTTC
O780UmF ACCACCCZTCTCACGCAGATTCACAATACGTTACATG
7190UmF ACGACATZGGTCAGGACGCTGAACAATAAGCTTCGG
7220UmF ACAACACZTGTCAGGACGCTTAACAATGATCTCTTGA
5400UmF ACCACCCZCCTCTCTCAAATCAACAACAAGTTCCGCA
5450UmF ACCACTTZGGCCAGACAAATCTTTCATCATGATTTAG
at5g43730UmF ACCACCCZCTTAGAAAGTCTCAACAACAAATTTGTTG
at5g43740UmF ACCACCCZCTTAGAAAGTCTCAACAACAAATTTGTTG
at5g45510UmF ACAAGGAZGGCACAAATGGTTGACAAAGAAGCCTCTA
at5g47250UmF ACTACCCZCCTCACTCTAATTAACAACAAGTTCGTTG
at5g47260UmF ACCACCCZTCTTACTAAACTCAGAAACAAGTTACTTG
at5g48620UmF ACCACTCZGGCTAGACAAGTCTTTCATCATGATTTAG
at5g63020UmF ACTACACZTCTCAGTCATATCAACAATAGATTCTCTA
at5g66900UmF ACCACGCZGGTTAGTCGGCTTTGTGACGATCCAGATA
at5g66910UmF ACCACGCZGGTTACTAAGCTTTGTGACGACCCAGAGA
at1g52660UmF AGACGACZGTTTTGACTCAGGTTAATAACAGGTTGCT
at4g19060UmF ACGACATZGTGTCAGGCTGTGTTCAACGACGAAGATG
AT1G51480UmF ACCACACZCTTAGCTTGCATCAACAACAAATTCGTTG
AT1G58848UmF ACCACACZTGCTAAACAGGTTTTTAACCATGAGGAT
at1g12280UmR AGGGTGGZTttGccattTCCACCCATGCC
at1g12290UmR AGGGTTGZTttTccattTCCACCCATGCC
at1g15890UmR AGCGTGGZTttAccattTCCCCCCATACC
at1g50180UmR AAAGTTGZTttCccattACCACCCATTCC
at1g53350UmR AGTGTCGZTttAccattACCCCCCATACC
at1g58390UmR AGTGTGGZTttAccattACCACCCATCCC
at1g58400UmR AGGGTGGZTttAccattACCTCCCATCCC
at1g58410UmR AGGGTGGZTttAccattACCACCCATCCC
at1g59218UmR AGTGTGGZTttAccattACCACCCATCCC
at1g59780UmR AGGGTGGZTttGccattACCACCCAAACC
mcg_UmR-0950 ATTGTAGZTttAccattCCCTCCCATCCC
mcg_UmR-2210 AGGGTGGZTttGccattTCCACCCATACC
mcg_UmR-3300 AGTGGTTZTAccattACCGCCCATTCC
mcg_UmR-6530 AGTGCAGZCtttccattGCCTCCCATACC
mcg_UmR1g10920 AGTGGTTZtAccattACCACCCATCCC
mcg_UmR1g12220 AGTGTCGZTttGccattTCCCCCCATA
mcg_UmR1g33560 AGTGGTTZtCccattACCGCTCATTCC

14
mcg_UmR1g59124 AGTGTGGZTttAccattACCACCCATA
mcg_UmR1g59620 AGGGTGGZTttAccattACCGCCCATC
mcg_UmR1g61180 AGGGTTGZTttGccattACCACCCATA
mcg_UmR3g07040 AGTGTAGZTttCccattACCGCCCATC
mcg_UmR3g14470 AGTGTGGZTttTccattCCCTCCAATT
mcg_UmR5g04720 AGAGTGGZTttCccattACCACTCATC
mcg_UmR5g47280 agaatggZtttcccattaccgatcatc
mcg_UmR5g43470 AGAGTGGZTttAccattACCGCCCATC
61190UmR AAAAGGGZTGTTttGccattACCACCCATACC
1300UmR AAAAGGGZGGTTttTccattACCACCCATACC
1310UmR AAAAGGGZTGTTttGccattACCACCCATACC
2630UmR AGAAGTGZTGTCttCccattACCGCCCATACC
3360UmR AGAAGTGZTGTCttCccattACCGCCCATACC
4460UmR AAGGTAGZCttTccattCCCTGGCATACC
5700UmR ACCGTCGZCttCccattACCTTCCACGCC
O780UmR AGGGTGGZTttGccattTCCCCCCATACC
7190UmR AATGTCGZTttAccattTCCTCCCATGCC
7220UmR AGTGTTGZCttCccattACCGCCCATACC
5400UmR AGGGTGGZCttGccattTCCCCCCATACC
5450UmR AAAGTGGZTttAccattACCGCCCATCCC
at5g43730UmR AGGGTGGzTttTccattTCCCCCCATACC
at5g43740UmR AGGGTGGzTttTccattTCCCCCCATACC
at5g45510UmR atccttgzcttcccgttcccagcttcgccaaccaG
at5g47250UmR AGGGTAGzTttGccattGCCTCCCATACC
at5g47260UmR AGGGTGGzTttGccattTCCACCCCTACC
at5g48620UmR AGAGTGGzTttAccattACCGCCCATCCC
at5g63020UmR AGTGTAGzTttAccattTCCACCCATACC
at5g66900UmR AGCGTGGzTttcccgttACCAGGAGGAGC
at5g66910UmR AGCGTGGzCttCccattACCAGGAGGACC
at1g52660UmR AGTCGTCztTccattACCTTCCACACC
at4g19060UmR AATGTCGzTttCccattTCCGTATTTCCC
AT1G51480UmR AGTGTGGzTttTccattTCCCCCCATACC
AT1G58848UmR AGTGTGGzTttAccattACCACCCATCCC

15





























