Embed Size (px)
Citation preview

FEATURE REVIEW
Pharmacogenetics in psychiatry: translating researchinto clinical practiceAK Malhotra1,2, J-P Zhang1,2 and T Lencz1,2
1Division of Psychiatry Research, The Zucker Hillside Hospital, Glen Oaks, NY, USA and 2The Feinstein Institute for MedicalResearch, Manhasset, NY, USA
Pharmacogenetic/pharmacogenomic (PGx) approaches to psychopharmacology aim to identifyclinically meaningful predictors of drug efficacy and/or side-effect burden. To date, however, PGxstudies in psychiatry have not yielded compelling results, and clinical utilization of PGx testing inpsychiatry is extremely limited. In this review, the authors provide a brief overview on the statusof PGx studies in psychiatry, review the commercialization process for PGx tests and thendiscuss methodological considerations that may enhance the potential for clinically applicablePGx tests in psychiatry. The authors focus on design considerations that include increasedascertainment of subjects in the earliest phases of illness, discuss the advantages of drug-induced adverse events as phenotypes for examination and emphasize the importance ofmaximizing adherence to treatment in pharmacogenetic studies. Finally, the authors discussunique aspects of pharmacogenetic studies that may distinguish them from studies of othercomplex traits. Taken together, these data provide insights into the design and methodologicalconsiderations that may enhance the potential for clinical utility of PGx studies.Molecular Psychiatry (2012) 17, 760–769; doi:10.1038/mp.2011.146; published online 15 November 2011
Keywords: pharmacogenetics; antipsychotics; antidepressants; gene; treatment
Pharmacogenetic/pharmacogenomic (PGx) approachesto psychopharmacology aim to identify clinicallymeaningful predictors of drug efficacy and/orside-effect burden. Given the clinical heterogeneityand prognostic uncertainty associated with mostpsychiatric disorders, the prospect of pharmaco-genetically informed individualized treatment holdsconsiderable appeal. Equally appealing are thepragmatic advantages of PGx approaches, includingthe ease of obtaining appropriate blood or salivasamples for DNA extraction, the immutability ofgenotype information and the increasingly low costof genotyping assays. More broadly, PGx data mayhelp illuminate the still-obscure molecular substratesof psychotropic drug action.
To date, however, PGx studies in psychiatry havenot yielded compelling results, and clinical utiliza-tion of PGx testing in psychiatry is extremely limited.In this review, we will provide a brief overview on thestatus of PGx studies in psychiatry, review thecommercialization process for PGx tests and thendiscuss methodological considerations that mayenhance the potential for clinically applicable PGxtests in psychiatry.
Pharmacogenetic studies of psychotropicdrug response
PGx studies have been carried out with multipleclasses of psychotropic drugs, with a primary focuson the interindividual variation in drug efficacy. Thevast majority of studies, which have been reviewedextensively elsewhere,1–7 have utilized a candidategene approach, usually based upon the receptorpharmacology of the psychotropic agent. The proofof principle for this approach is provided by PGxstudies of the DRD2 gene, which codes for thedopamine D2 receptor, the common site of action ofall commercially available drugs with antipsychoticpotency.8 A recent meta-analysis indicates that afunctional polymorphism in the DRD2 promoterregion, which modulates levels of gene expression,significantly influences antipsychotic drug efficacy.9
Similarly, a promoter region polymorphism in thegene coding for the serotonin transporter, the com-mon site of action for the selective serotonin reuptakeinhibitor class of antidepressants, has been impli-cated by meta-analyses of selective serotonin reuptakeinhibitor efficacy.10 In both instances, carriers of acommon yet deficient genetic variant demonstratebetween half and two-thirds of the response rate ofnoncarriers. Although these effect sizes are statisti-cally robust, they do not yet yield adequate sensitivityand specificity to reliably guide clinical practice.Moreover, alternative treatment strategies for poor-prognosis genetic carriers have not been empirically
Received 19 December 2010; revised 10 August 2011; accepted3 October 2011; published online 15 November 2011
Correspondence: Dr AK Malhotra, The Zucker Hillside Hospital,75-59 263rd Street, Glen Oaks, NY 11004, USA.E-mail: [email protected]
Molecular Psychiatry (2012) 17, 760–769& 2012 Macmillan Publishers Limited All rights reserved 1359-4184/12
www.nature.com/mp

tested, a task made more difficult by the fact that mostwidely used psychotropic medications in each majorclass have similar primary targets.
Pharmacokinetic genetic studies of psychotropicdrug response are supported by the identification ofmultiple functional variants with well-defined effectson drug metabolism. For example, the gene for thecytochrome P450 2D6 enzyme, which is responsiblefor the metabolism of several psychotropic agents,contains > 100 genetic variants (as cataloged by thewebsite: http://www.cypalleles.ki.se), many of whichyield nonfunctional or reduced-function enzymes.There are four phenotypes of CYP2D6 produced bycombinations of various alleles with different degreesof enzymatic activities: poor metabolizer, intermedi-ate metabolizer, extensive metabolizer and ultrarapidmetabolizer. Compared with extensive metabolizerswith normal CYP2D6 enzyme activity, poor metabo-lizers and intermediate metabolizers have minimal orreduced activity, respectively. Ultrarapid metaboli-zers have duplicate or multiple copies of the gene thatresult in increased enzyme activity. Approximately7–10% of Caucasians and 1–2% of Asians are poormetabolizers,11 who tend to accumulate higher druglevels in blood and, theoretically, require lower dosesto achieve therapeutic effects. Ultrarapid metaboli-zers, in contrast, consist of only 1% of the populationand may require higher doses because of fasterelimination of the drug.
Despite the functional significance of these var-iants, relatively fewer studies have focused on theCYP450 system in psychiatric PGx,5 as comparedwith pharmacodynamic studies. In part, this has beenbecause of the lack of compelling empirical supportfor a relationship between plasma drug levels andpsychotropic drug efficacy. Multiple early reports onthe tricyclic antidepressant nortryptiline suggested a‘therapeutic window’ for efficacy with plasma druglevels between 50 and 150 ng ml–1;12 for the prototypicatypical antipsychotic clozapine, plasma levels of atleast 200 ng ml–1 were considered a potential thresh-old level required for clinical response.13 However,consistent data in this regard and for other psycho-tropic drugs are limited. The lack of a clear relation-ship between drug levels and efficacy suggests thatpharmacokinetic genetic variation may not signifi-cantly predict differential drug efficacy, although apotential role in side-effect prediction (discussed ingreater detail below) has led some authors to suggestclinical guidelines for dosing based upon CYP450genotype.14
A final approach that has been as yet uncommon inpsychiatric PGx research is the utilization of genome-wide association studies (GWASs). A major challengeto GWASs in pharmacogenetics are the large samplesizes presumed to be necessary to overcome thestatistical penalty incurred by the genotyping ofhundreds of thousands of single-nucleotide poly-morphisms.15 Although difficult to achieve fordisease susceptibility studies, sample sizes in thethousands (or even tens of thousands) are essentially
out of reach for prospective PGx trials. PGx data setshave traditionally been ‘piggybacked’ off of ongoingclinical trials, which are often not optimally designedfrom a pharmacogenetic perspective because of theinclusion of multiple treatment arms, ethnicallyand clinically heterogeneous samples and high dis-continuation rates. An exception is the GENDEP(Genome-Based Therapeutic Drugs for Depression)project—which was specifically designed to achievepharmacogenetic aims.16 All subjects were drawnfrom a single ethnic group, there were only twotreatment arms (escitalopram and nortryptyline), drugselection was based upon distinct biological hypoth-eses on the mechanism of drug action (serotonergicversus noradrenergic) and the sample size (n = 811)was much larger than most academic clinical trials.Unfortunately, GENDEP, as with other GWAS dataon antidepressant response from the STAR*D(Sequenced Treatment Alternatives to Relieve Depres-sion) study17 (n = 1491) and the Munich Antidepres-sant Response Study18 (n = 339), did not detect anygenome-wide significant or clinically useful predic-tors of antidepressant response. Similarly, the GWASof lithium treatment response in the STEP-BD(Systematic Treatment Enhancement Program forBipolar Disorder)19 (n = 1177) and several GWASsof antipsychotic drug responses from the CATIE(Clinical Antipsychotic Trials of Intervention Effec-tiveness)20–23 (n = 738) did not find any geneticmarkers that can be readily used in clinical settings.The limitations of these studies include use ofchronic patients, lack of medication adherence mon-itoring and ambiguity of phenotype definition.
Clinical pharmacogenetic testing in psychiatry
The lack of compelling data from pharmacogeneticstudies has hampered the development of clinicalPGx tests. To be approved by the US Food and DrugAdministration (FDA), clinical tests must achievehigher levels of sensitivity and specificity thanobserved with PGx results to date. For this reason,most commercially available PGx tests in psychiatryhave not undergone FDA approval, but have soughtalternative paths into the commercial marketplace.
Currently, clinical genetic testing services in theUnited States are regulated by the Clinical LaboratoryImprovements Amendments of 1988 (CLIA) (Code ofFederal Regulations, Title 42, Part 493, 1995). CLIAcertification is primarily focused on quality controland laboratory procedures, and does not provide data-based review of the clinical utility of a particular test.Most PGx tests offered by various companies andlaboratories achieved CLIA certification, but did notundergo FDA approval. Therefore, laboratories maydevelop tests (lab-developed tests) that are CLIAcertified as biologically accurate assays, but may haverelatively limited predictive power for any clinicalphenotype. These tests may be marketed to physi-cians or, in some cases, directly to potential con-sumers, but cannot be sold for use by other
Pharmacogenetics in psychiatryAK Malhotra et al
761
Molecular Psychiatry

laboratories.24 The lab-developed tests are not routi-nely subject to post-marketing surveillance by federalauthorities, and therefore limited data are availableon the long-term impact of the introduction of thesetests into clinical practice.
Alternatively, testing companies can pursue FDAapproval for commercial testing products. Moderate-risk tests undergo 510(k) clearance, which justrequires review of the data that support the clinicalclaim about the test. Higher-risk tests, in which apotentially serious intervention could be consideredbased upon test results, may be required to provideclinical trial evidence in support of the utility of thetest. It should be noted, however, that the regulatoryenvironment for genetic testing is undergoing scru-tiny and new legislation may be proffered thatsignificantly alters the current regulatory frameworkfor PGx tests.24
To date, there is only one FDA-approved pharma-cogenetic test for use in psychiatry. Roche Diagnosticsreceived FDA 510(k) clearance in 2004 for a micro-array-based product, the AmpliChip R CYP450 Test,which assesses 27 alleles in CYP2D6 and 3 alleles inCYP2C19. This product received approval based upondata indicating that CYP450 genotype can influencedrug efficacy and safety via effects on drug metabo-lism.25 The limited marketing upon the release of thetest emphasized the relationship between thesegenotypes and clinical response to psychotropicdrugs (such as the antipsychotic drug risperidone)that are primarily metabolized by CYP2D6 orCYP2C19, but the test was not associated with anyspecific drug or class of drugs and no specific clinicalclaims were granted. The clinical uptake of thisproduct has been modest, perhaps because of clin-ician concerns about the interpretation of test results,the paucity of prospective data suggesting that testutilization influences clinical outcome and the lack ofreimbursement for an expensive test. Moreover, withrapid progress in genotyping technology, severalcompanies have produced genotyping platformsspecifically designed for PGx, focusing on geneticmarkers associated with drug absorption, distribu-tion, metabolism and enzymes (ADME). Illumina’sVeraCode ADME core panel assays markers in34 genes, one-third of which code for cytochromeP450 enzymes (www.illumina.com/products/veracode_adme_core_panel.ilmn). The DMET Plus Premier Packfrom Affymetrix provides 1936 drug metabolismmarkers in B230 genes (www.affymetrix.com).Other companies have also developed lab-developedtests that assess CYP450 genotypes, and thesemay compete for market utilization. However,adoption of these platforms for clinical practicehas been sparse. High cost and long turnaround time(at least 5–7 days) hinder their practical application.Several other non-FDA approved PGx tests thatwere specifically designed for psychotropic drugresponses have been made commercially available,including tests for prediction of response to anti-depressant and antipsychotic drugs; however, these
tests have also had limited utility and clinicalapplications.26
Design considerations to enhance pharmacogeneticstudies
Clearly, stronger data will be needed to achieveregulatory approval of clinically useful PGx testingin psychiatry. To accomplish this goal, it may beworthwhile to consider specific study design featuresthat may enhance the power and precision ofpharmacogenetic studies.
Focus on early phases of illnessThe majority of PGx data sets in psychiatry arederived from ongoing clinical trials. Unfortunately,the patients who enter into these trials are oftenchronically ill with lengthy prior treatment histories;highly responsive patients may not present for thesestudies, as they do not seek changes to treatment ifadequate response has already been attained. Studysamples drawn from trials in chronic subjects maytherefore be systematically biased toward inclusion ofpatients who are not fully responsive to treatment orwho are nonadherent with treatment (or both), andnot represent the full spectrum of treatment out-comes.27 Consistent with this, overall response ratesin most clinical trials of antipsychotic drugs areroutinely lower than in trials that focus on patients inthe earliest phases of illness, in which remission rateshave been reported as high as 80% of enrolledsubjects.28,29 Chronically ill cohorts are also markedby increased duration of psychotic symptoms, sub-stance abuse and functional/social disabilities; eachfactor may influence drug response rates and intro-duce increased variance into data analyses. For thesereasons, studies that concentrate on early-phase orfirst-episode patients may provide enhanced powerfor pharmacogenetic studies of drug efficacy; forexample, our recent antipsychotic PGx meta-analysisdemonstrated a 50% greater effect size for the DRD2promoter polymorphism in studies containing first-episode patients compared with studies of chronicpatients.9
Utilization of additional phenotypes; adverse eventsA drawback of pharmacogenetic studies that focus onclinical efficacy as the primary phenotype is theinherent method variance introduced by the use ofclinical symptom ratings. Standard ratings scales aredependent upon subjective patient report and ratersensitivity, and tend to be decreasingly reliable assample sizes and study site number increase. Theeffects of diminished rating reliability on study powerwere quantified in a report by Perkins et al.,30 withdramatic results. As shown in Figure 1 (data inter-polated from Perkins et al.30), study power is reduced,and sample size requirements concomitantly in-creased, as the reliability of the dependent measure(phenotype) falls. These effects are equally valid forPGx studies that compare two groups defined by
Pharmacogenetics in psychiatryAK Malhotra et al
762
Molecular Psychiatry
genotype as for clinical trials that compare twotreatment conditions. Thus, contrary to current trendsin genetics studies of other complex traits, it is possiblethat large PGx studies can be deceptively underpow-ered, whereas smaller trials with rigorously collectedphenotypic information may be more robust.
Another approach to enhancing signal to noise inPGx studies is to focus on treatment-related adverseeffects as the phenotype of interest. In other branchesof medicine, there have been notable successes inusing PGx to identify powerful predictors of drug-induced adverse events. A specific human leukocyteantigen (HLA) allele markedly increases the risk forliver injury as a result of fluvoxacillin treatment (oddsratio (OR) = 80.6).31 Furthermore, the same HLA allele(HLA-B*5701) provides 100% specificity for devel-opment of an immunologically confirmed hypersen-sitivity reaction to abacavir, a widely used treatmentfor AIDS;32 the hypersensitivity reaction developedin every one of 23 carriers of the *5701 allele. Incontrast, the overall occurrence rate is < 3% innoncarriers.32 More recently, HLA genetic markerswere also found to be associated with antiepileptic(for example, lamotrogine) drug-induced Stevens–Johnson syndrome.33,34
Rare, yet critical, adverse effects may also occurwith psychotropic drugs. Most notably, clozapine isthe only antipsychotic with demonstrated superiorityfor treatment-resistant schizophrenia,35,36 yet it re-mains clinically underutilized in part because of itsassociation with agranulocytosis, a potentially fatalblood dyscrasia observed in < 1% of patients.37 Theconcomitant burden of routine blood monitoringcould potentially be lifted by the identification of aPGx biomarker enabling clinicians to ascertain risk foragranulocytosis a priori. A recent candidate genestudy demonstrated some promise in this regard,detecting a replicated association of an allele at theHLA-DQB1 locus with the risk of agranulocytosis intwo small clozapine-treated cohorts.38 The ORs wereextremely high (OR = 16.86), especially compared
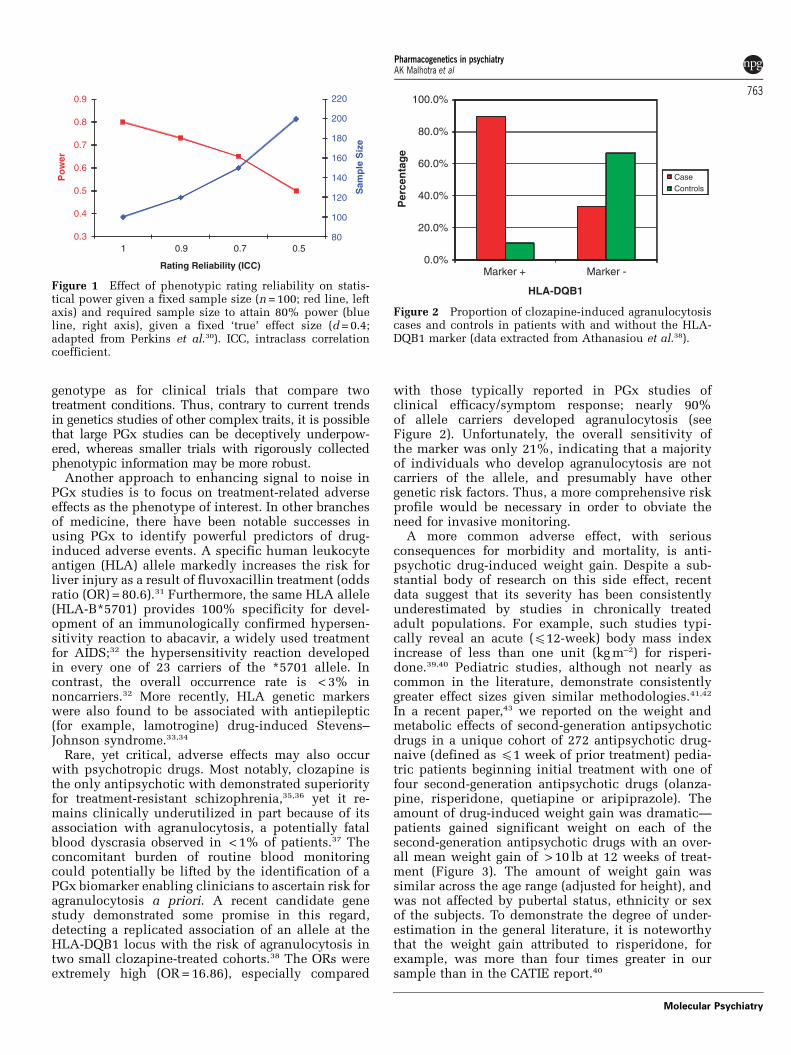
with those typically reported in PGx studies ofclinical efficacy/symptom response; nearly 90%of allele carriers developed agranulocytosis (seeFigure 2). Unfortunately, the overall sensitivity ofthe marker was only 21%, indicating that a majorityof individuals who develop agranulocytosis are notcarriers of the allele, and presumably have othergenetic risk factors. Thus, a more comprehensive riskprofile would be necessary in order to obviate theneed for invasive monitoring.
A more common adverse effect, with seriousconsequences for morbidity and mortality, is anti-psychotic drug-induced weight gain. Despite a sub-stantial body of research on this side effect, recentdata suggest that its severity has been consistentlyunderestimated by studies in chronically treatedadult populations. For example, such studies typi-cally reveal an acute (p12-week) body mass indexincrease of less than one unit (kg m–2) for risperi-done.39,40 Pediatric studies, although not nearly ascommon in the literature, demonstrate consistentlygreater effect sizes given similar methodologies.41,42
In a recent paper,43 we reported on the weight andmetabolic effects of second-generation antipsychoticdrugs in a unique cohort of 272 antipsychotic drug-naive (defined as p1 week of prior treatment) pedia-tric patients beginning initial treatment with one offour second-generation antipsychotic drugs (olanza-pine, risperidone, quetiapine or aripiprazole). Theamount of drug-induced weight gain was dramatic—patients gained significant weight on each of thesecond-generation antipsychotic drugs with an over-all mean weight gain of > 10 lb at 12 weeks of treat-ment (Figure 3). The amount of weight gain wassimilar across the age range (adjusted for height), andwas not affected by pubertal status, ethnicity or sexof the subjects. To demonstrate the degree of under-estimation in the general literature, it is noteworthythat the weight gain attributed to risperidone, forexample, was more than four times greater in oursample than in the CATIE report.40
0.0%
20.0%
40.0%
60.0%
80.0%
100.0%
Marker + Marker -
Perce
ntage
HLA-DQB1
CaseControls
Figure 2 Proportion of clozapine-induced agranulocytosiscases and controls in patients with and without the HLA-DQB1 marker (data extracted from Athanasiou et al.38).
80
100
120
140
160
180
200
220
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1 0.9 0.7 0.5
Sam
ple
Siz
e
Po
wer
Rating Reliability (ICC)
Figure 1 Effect of phenotypic rating reliability on statis-tical power given a fixed sample size (n = 100; red line, leftaxis) and required sample size to attain 80% power (blueline, right axis), given a fixed ‘true’ effect size (d = 0.4;adapted from Perkins et al.30). ICC, intraclass correlationcoefficient.
Pharmacogenetics in psychiatryAK Malhotra et al
763
Molecular Psychiatry

These data suggest that pharmacogenetic studies ofweight gain that include previously treated patientsare not optimally designed. Variable histories of priordrug exposure between subjects may confoundattempts to identify subtle genetic effects on acomplex phenotype such as weight gain. Consistentwith this hypothesis, Reynolds et al.44 studied 123antipsychotic drug-naive schizophrenic Chinesepatients and found that a promoter region polymorph-ism—759 C/T in the 5-HT2C receptor gene—signifi-cantly influenced weight gain followingantipsychotic treatment. Study participants with theT allele at this locus gained significantly less weightthan subjects with the C allele at 6 and 10 weeks oftreatment. This effect was observed in patientsreceiving risperidone or chlorpromazine, regardlessof gender, and remained significant after exclusion ofsubjects who were either underweight or obese atbaseline. Moreover, none of the 27 subjects carryingthe T allele met criteria for severe weight gain ( > 7%increase from baseline body weight) after 6 weeks oftreatment, compared with 28% of the 96 subjectswithout the T allele. Templeman et al.45 reportedsimilar results in a small first-episode cohort treatedwith a mix of antipsychotic medications includingolanzapine. As expected, subsequent studies inpreviously treated patients have been less robust,2
although meta-analysis confirms that the effect is notlikely to be false positive.46
Similar results were recently reported for thefunctional promoter region variant in DRD2 in astudy of antipsychotic-induced weight gain in first-episode patients with minimal prior antipsychoticexposure.47 Carriers of a single-nucleotide deletion inthe DRD2 promoter (–141C Ins/Del) demonstratedsubstantially more weight gain than noncarriers after6 weeks of treatment, regardless of medication(risperidone or olanzapine). Again, the effect sizeswere relatively large; mean weight gain in deletioncarriers at 6 weeks was B6 lb higher than innoncarriers.
Adherence in pharmacogenetic studiesIt is well recognized by clinical researchers thattreatment adherence is poor among psychiatricpatients. A recent review demonstrated that the ratesof nonadherence are 28 to 52% in patients with majordepressive disorder, 20 to 50% in patients withbipolar disorder and 20 to 72% in those withschizophrenia.48 Typical methods of assessing adher-ence include patients’ self-report, family report, pillcount, blood levels and pharmacy refill records, all ofwhich may still underestimate the scope of theproblem. In the CATIE trial, 74% of patients stoppedtheir initially assigned medications within 18months, and almost 30% stopped medication becauseof ‘patient’s decision’.40
Not only can medication nonadherence result insymptom relapse clinically, but it can also lead toweakened signals in research studies of adverseevents. However, this important issue has often beenoverlooked in PGx research, and the effect ofnonadherence on statistical power has not beenquantified. Intuitively, if a substantial proportion ofsubjects are nonadherent with treatment, it wouldbe very difficult to detect a significant genotype–phenotype (that is, response to an administereddrug) relationship regardless of the strength of theeffect of the genetic marker. This could be particularlyimportant for studies of side effects such asweight gain, as almost all subjects with documentedadherence to second-generation antipsychotics gainsome degree of weight;43 a PGx study that failedto formally assess medication adherence wouldmisclassify nonadherent subjects as impervious toweight gain.
To quantify the effects of medication nonadherenceon statistical power in PGx studies, we conducted aMonte Carlo simulation study using the R statisticalprogramming language.49 In this simulation, modeledafter a recently published PGx weight gain study byour group,47 we assumed that a specific geneticmarker increased one’s susceptibility to weight gainduring 16 weeks of antipsychotic drug treatment.The primary analysis compares two genotypic groups(for example, marker carriers vs noncarriers). Medica-tion nonadherent subjects are assumed to gain noweight in average,43 and are randomly distributedbetween both genotype groups. After settingappropriate statistical parameters (that is, mean ands.d. of weight gain in each group, sample size,frequency of rare genotype, percentage of nonadher-ence in the sample and so on), the R programrandomly generated 200 000 samples and performeda t-test on the difference between means of the twogenotype groups in each sample. Power was calcu-lated as the percentage of samples with a significantt-test (P < 0.05).
Figure 4 shows the power curves for differentsample sizes as a function of nonadherence rate inthe sample, with a 20% frequency of risk genotypeand a moderate true effect size (that is, Cohen’sd = 0.50), which could represent a typical scenario in
0
1
2
3
4
5
6
7
8
9
10
Aripiprazole Olanzapine Quetiapine Risperidone Untreated
Wei
gh
t C
han
ge
(kg
)
Antipsychotic Drug
Figure 3 Weight gain observed after 12 weeks of exposureto second-generation antipsychotics in drug-naive youth(adapted from Correll et al.43).
Pharmacogenetics in psychiatryAK Malhotra et al
764
Molecular Psychiatry
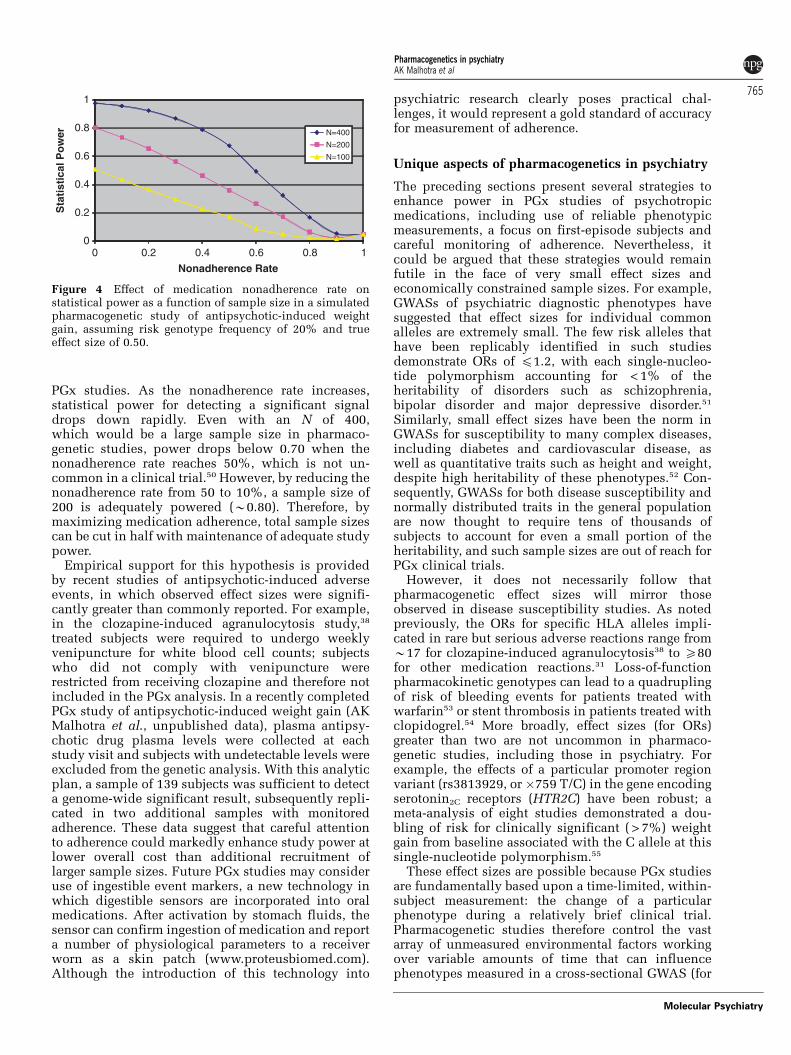
PGx studies. As the nonadherence rate increases,statistical power for detecting a significant signaldrops down rapidly. Even with an N of 400,which would be a large sample size in pharmaco-genetic studies, power drops below 0.70 when thenonadherence rate reaches 50%, which is not un-common in a clinical trial.50 However, by reducing thenonadherence rate from 50 to 10%, a sample size of200 is adequately powered (B0.80). Therefore, bymaximizing medication adherence, total sample sizescan be cut in half with maintenance of adequate studypower.
Empirical support for this hypothesis is providedby recent studies of antipsychotic-induced adverseevents, in which observed effect sizes were signifi-cantly greater than commonly reported. For example,in the clozapine-induced agranulocytosis study,38
treated subjects were required to undergo weeklyvenipuncture for white blood cell counts; subjectswho did not comply with venipuncture wererestricted from receiving clozapine and therefore notincluded in the PGx analysis. In a recently completedPGx study of antipsychotic-induced weight gain (AKMalhotra et al., unpublished data), plasma antipsy-chotic drug plasma levels were collected at eachstudy visit and subjects with undetectable levels wereexcluded from the genetic analysis. With this analyticplan, a sample of 139 subjects was sufficient to detecta genome-wide significant result, subsequently repli-cated in two additional samples with monitoredadherence. These data suggest that careful attentionto adherence could markedly enhance study power atlower overall cost than additional recruitment oflarger sample sizes. Future PGx studies may consideruse of ingestible event markers, a new technology inwhich digestible sensors are incorporated into oralmedications. After activation by stomach fluids, thesensor can confirm ingestion of medication and reporta number of physiological parameters to a receiverworn as a skin patch (www.proteusbiomed.com).Although the introduction of this technology into
psychiatric research clearly poses practical chal-lenges, it would represent a gold standard of accuracyfor measurement of adherence.
Unique aspects of pharmacogenetics in psychiatry
The preceding sections present several strategies toenhance power in PGx studies of psychotropicmedications, including use of reliable phenotypicmeasurements, a focus on first-episode subjects andcareful monitoring of adherence. Nevertheless, itcould be argued that these strategies would remainfutile in the face of very small effect sizes andeconomically constrained sample sizes. For example,GWASs of psychiatric diagnostic phenotypes havesuggested that effect sizes for individual commonalleles are extremely small. The few risk alleles thathave been replicably identified in such studiesdemonstrate ORs of p1.2, with each single-nucleo-tide polymorphism accounting for < 1% of theheritability of disorders such as schizophrenia,bipolar disorder and major depressive disorder.51
Similarly, small effect sizes have been the norm inGWASs for susceptibility to many complex diseases,including diabetes and cardiovascular disease, aswell as quantitative traits such as height and weight,despite high heritability of these phenotypes.52 Con-sequently, GWASs for both disease susceptibility andnormally distributed traits in the general populationare now thought to require tens of thousands ofsubjects to account for even a small portion of theheritability, and such sample sizes are out of reach forPGx clinical trials.
However, it does not necessarily follow thatpharmacogenetic effect sizes will mirror thoseobserved in disease susceptibility studies. As notedpreviously, the ORs for specific HLA alleles impli-cated in rare but serious adverse reactions range fromB17 for clozapine-induced agranulocytosis38 to X80for other medication reactions.31 Loss-of-functionpharmacokinetic genotypes can lead to a quadruplingof risk of bleeding events for patients treated withwarfarin53 or stent thrombosis in patients treated withclopidogrel.54 More broadly, effect sizes (for ORs)greater than two are not uncommon in pharmaco-genetic studies, including those in psychiatry. Forexample, the effects of a particular promoter regionvariant (rs3813929, or �759 T/C) in the gene encodingserotonin2C receptors (HTR2C) have been robust; ameta-analysis of eight studies demonstrated a dou-bling of risk for clinically significant ( > 7%) weightgain from baseline associated with the C allele at thissingle-nucleotide polymorphism.55
These effect sizes are possible because PGx studiesare fundamentally based upon a time-limited, within-subject measurement: the change of a particularphenotype during a relatively brief clinical trial.Pharmacogenetic studies therefore control the vastarray of unmeasured environmental factors workingover variable amounts of time that can influencephenotypes measured in a cross-sectional GWAS (for
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8 1
Sta
tist
ical
Po
wer
Nonadherence Rate
N=400
N=200
N=100
Figure 4 Effect of medication nonadherence rate onstatistical power as a function of sample size in a simulatedpharmacogenetic study of antipsychotic-induced weightgain, assuming risk genotype frequency of 20% and trueeffect size of 0.50.
Pharmacogenetics in psychiatryAK Malhotra et al
765
Molecular Psychiatry

example, of body mass index) conducted in thegeneral population. Similarly, although there may bea multiplicity of subtle genetic and environmentalpathways for development of psychiatric illness, it isreasonable that a few key genes in relevant pharma-cokinetic and pharmacodynamic pathways can exertrelatively large effect sizes in a clinical trials context.Thus, pharmacogenetic GWASs may be sufficientlypowered with sample sizes measured in thehundreds, rather than in the tens of thousands.
Moreover, pharmacologic effects may also beindependent of diagnosis or other historical andclinical characteristics of patients, permitting thepooling of studies of specific pharmacologic agentsacross multiple diagnostic groups. For example, thephenotype of antipsychotic-induced weight gain doesnot appear to differ in patients with schizophrenia,bipolar disorder or nonpsychotic behavioral distur-bances,41,43 and initial pharmacogenetic data suggestno difference across diagnostic groups as well (AKMalhotra et al., unpublished data). This considerationmay mitigate, to some extent, the difficulties inrecruiting and following well-characterized clinicaltrial samples.
Finally, for PGx to be clinically useful, large PGxclinical trials of head-to-head drug comparisons areneeded to validate the strategy of selecting and dosingdrugs based on genetic testing. Genetic markers needto be shown as true moderators of differentialtreatment and clinical outcome to be capable ofguiding drug selection.56 CYP2D6 is a good example.If a schizophrenic patient is a poor metabolizer, theclinician may choose quetiapine or ziprasidone,instead of risperidone or aripiprazole, which aremetabolized primarily by CYP2D6. However, therehave been no prospective data to test alternativetreatment based on genetic markers. Furthermore, it iseven more challenging in the case of selectingantipsychotic drugs based on genetic variants ofdopamine receptors. The Del allele of �141C Ins/Delin DRD2 is associated with poor response to anti-psychotic drugs,9 and also increases the liability ofweight gain induced by antipsychotics drugs.47
However, all available antipsychotics to date exerttheir effect by D2 blockade. Even if a patient has theDel allele, there is no fundamentally alternative drugtreatment. Future research should focus on develop-ing new effective drugs without D2 antagonism, andthus provide more options in clinical managementwhen a patient is a poor responder because of variantsin the DRD2 gene.
Optimizing PGx studies
Unlike other branches of medicine, prospective PGxstudies are lacking in psychiatry. A rare example is arecent study of treating severe alcohol abuse withondansetron, in which patients were randomized bygenotypes of the serotonin transporter gene (SLC6A4)and the treatment effect was significantly greater inthe L/L genotype group as compared with the L/S and
S/S groups.57 This study highlighted an essentialcharacteristic of a prospective pharmacogenetic clin-ical trial,58 that is, randomization by genotype.However, to best position PGx testing for clinicalpractice, studies that match individuals with aparticular genotype with a specific effective treat-ment, that is, testing the genotype� treatment inter-action,56 would be ideal. The drawback of thisapproach is that multiple medication arms will berequired, increasing sample size requirements andstudy cost and therefore diminishing feasibility.
To optimize a PGx clinical trial, several key issuesshould be considered. Table 1 summarizes the prosand cons and alternative options associated with eachkey issue, many of which have already been dis-cussed in previous sections. In the ideal scenario, weattempt to design a PGx study to examine the efficacyof alternative treatments associated with a hypothe-tical genetic marker. It is hypothesized that medica-tion treatment A is efficacious for patients with aparticular genotype of the marker, but not for patientswithout the genotype. In contrast, medication treat-ment B is efficacious for patients without theparticular genotype, but not for patients with thegenotype. Patients would preferably be in their firstepisode of illness with minimal prior medicationexposure, and could be genotyped at baseline.Minimal turnaround time for genotyping will beneeded, as many patients will require rapid initiationof treatment and randomization cannot be delayed formore than 24–48 h in many cases. Patients are thenrandomized into either A or B treatment, stratified ongenotypes. Hence, this is a 2� 2 randomized factorialdesign. The primary outcome is efficacy, that is,symptom reduction, which could be rated by acentralized rating system to increase assessmentreliability. Medication adherence should be carefullymonitored by plasma drug levels, or perhaps by noveltechniques such as digestive event markers.
It should be recognized, of course, that manypractical and logistical problems prevent a singlestudy to achieve the perfect design. Nevertheless, theissues listed in Table 1 should be carefully consideredin designing a PGx clinical trial. Short of a perfectPGx study, there are still many opportunities inpsychiatric PGx that could be conducted to provideinformative data for clinical application. For example,given that the Del allele of �141C Ins/Del in DRD2 isassociated with poor response to antipsychotic drugsin schizophrenia,9 a clinical trial may be conducted toexamine whether patients carrying the Del allele canbenefit from early clozapine treatment, instead ofgoing through multiple unsuccessful trials of non-clozapine antipsychotics. Taking advantage of thefinding that the �759 C/T polymorphism in HTR2C isassociated with antipsychotic-induced weight gain,46
a study could be designed such that risk allele carriersare randomly assigned to regular antipsychotic drugtreatment plus a weight loss drug such as metformin59
versus antipsychotic drug plus placebo. Finally, aprospective, randomized clinical trial is needed to
Pharmacogenetics in psychiatryAK Malhotra et al
766
Molecular Psychiatry

demonstrate the clinical utility of CYP2D6 genotyp-ing. For CYP2D6 poor metabolizers, dosing risper-idone at lower levels and/or slower titrationsschedules, or choosing alternative drugs that are notmetabolized by the CYP2D6 pathway, may be able toincrease efficacy, minimize side effects and poten-tially shorten inpatient stays, resulting in decreasedcosts that could offset any costs associated with PGxtesting.
Conclusions
It is important to consider what goals are reasonablyexpected of PGx research. Individually tailoredtherapies remain a somewhat distant goal, if for noother reason that our armamentarium is mechanisti-
cally limited. As noted above, as all antipsychoticshave effects at the D2 receptor, it may not be possibleto recommend a different form of treatment forindividuals with DRD2 genotypes indicative of poorresponse and increased weight gain liability.9,47
However, such individuals might be given priorityfor additional clinical attention, given increasingconstraints on physician time and hospital length ofstay. Similarly, individuals at risk for increasedweight gain or other adverse events can be givenlower doses, adjunctive therapies (psychosocial orpharmacologic) and/or increased monitoring. Ananalogy can again be made to the pharmacogeneticsof warfarin,60 in which it was recently demonstratedthat the use of genetic markers to guide dosing cut therate of adverse events in half. Although pharmaco-genetic markers are unlikely to attain perfect
Table 1 Major decision points in designing a pharmacogenetic/pharmacogenomic (PGx) clinical trial
Issues Options Pros Cons
Sampling First-episode/drug-naive patients
Eliminate confounding byprior drug exposurem Effect size and power
Difficult to recruitPotential loss of generalizability
Chronic patients Easy to recruit Confound by prior drug history, substanceabuse, psychosocial issuesStructural/functional brain changesassociated with long-term treatment andillnessPatients may be resistant to treatment
Choosing aphenotype
Drug efficacy/effectiveness
Most clinically relevant Symptom rating scales are subjective tobias and reliability issues
Adverse drug response Relatively easy to assess withgood reliability
Not major focus of clinical intervention
Genotyping Commercial lab Professional management andavailability
Slower turnaround time (with someexceptions)Difficult to get customized genotyping
In-house genotyping Able to customize genotypingFast turnaround time
More expensive for rapid testing just forthe study
Designingintervention
One arm, one drug Maximized sample sizem Homogeneity
Unable to test moderator effect of a geneticmarker
Multiple arms,multiple drugs
Able to test moderator effectof a genetic markerMost relevant in answeringclinical questions
Smaller sample size for each drugm Heterogeneity
Adherencemonitoring
Blood drug level Easy to obtain Post hoc ascertainment of adherenceleading to smaller sample size
Digestible event marker Real-time ascertainment ofadherence enabling interventionto improve adherence andm sample size
More expensivePatients may have difficulty acceptingthe technology
Minimizingplaceboresponse
Centralized rating system m ReliabilityAble to access more sites
Technology requiredPotential loss of confidentiality
Placebo lead-in phase Minimize true placebo response Loss of treatment timem Trial length
Pharmacogenetics in psychiatryAK Malhotra et al
767
Molecular Psychiatry

sensitivity and specificity in the foreseeable future,they can still usefully inform clinical decisionmaking, clarify prognosis and ultimately help guidethe development of novel medication strategies.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the NIH GrantsP50MH080173 (to AK Malhotra), P30MH090590(JM Kane) and R01MH79800-01 (to AK Malhotra).
References
1 Arranz MJ, de Leon J. Pharmacogenetics and pharmacogenomics ofschizophrenia: a review of last decade of research. Mol Psychiatry2007; 12: 707–747.
2 Zhang JP, Malhotra AK. Pharmacogenetics and antipsychotics:therapeutic efficacy and side effects prediction. Expert Opin DrugMetab Toxicol 2011; 7: 9–37.
3 Kato M, Serretti A. Review and meta-analysis of antidepressantpharmacogenetic findings in major depressive disorder. MolPsychiatry 2010; 15: 473–500.
4 Sturgess JE, George TP, Kennedy JL, Heinz A, Muller DJ.Pharmacogenetics of alcohol, nicotine and drug addiction treat-ments. Addict Biol 2011; 16: 357–376.
5 Fleeman N, Dundar Y, Dickson R, Jorgensen A, Pushpakom S,McLeod C et al. Cytochrome P450 testing for prescribingantipsychotics in adults with schizophrenia: systematic reviewand meta-analyses. Pharmacogenomics J 2011; 11: 1–14.
6 Taylor MJ, Sen S, Bhagwagar Z. Antidepressant response and theserotonin transporter gene-linked polymorphic region. Biol Psy-chiatry 2010; 68: 536–543.
7 McCarthy MJ, Leckband SG, Kelsoe JR. Pharmacogenetics oflithium response in bipolar disorder. Pharmacogenomics 2010;11: 1439–1465.
8 Kapur S, Mamo D. Half a century of antipsychotics and still acentral role for dopamine D2 receptors. Prog Neuropsychophar-macol Biol Psychiatry 2003; 27: 1081–1090.
9 Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation andclinical response to antipsychotic drug treatment: a meta-analysis.Am J Psychiatry 2010; 167: 763–772.
10 Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis ofserotonin transporter gene promoter polymorphism (5-HTTLPR)association with selective serotonin reuptake inhibitor efficacy indepressed patients. Mol Psychiatry 2007; 12: 247–257.
11 Mizutani T. PM frequencies of major CYPs in Asians andCaucasians. Drug Metab Rev 2003; 35: 99–106.
12 Ulrich S, Lauter J. Comprehensive survey of the relationshipbetween serum concentration and therapeutic effect of amitripty-line in depression. Clin Pharmacokinet 2002; 41: 853–876.
13 Conley RR. Optimizing treatment with clozapine. J Clin Psychiatry1998; 59(Suppl 3): 44–48.
14 de Leon J, Armstrong SC, Cozza KL. Clinical guidelines forpsychiatrists for the use of pharmacogenetic testing for CYP4502D6 and CYP450 2C19. Psychosomatics 2006; 47: 75–85.
15 Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, Kelsoe Jet al. Genomewide association studies: history, rationale, andprospects for psychiatric disorders. Am J Psychiatry 2009; 166:540–556.
16 Uher R, Perroud N, Ng MY, Hauser J, Henigsberg N, Maier W et al.Genome-wide pharmacogenetics of antidepressant response in theGENDEP project. Am J Psychiatry 2010; 167: 555–564.
17 Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GDet al. A genomewide association study of citalopram response inmajor depressive disorder. Biol Psychiatry 2011; 67: 133–138.
18 Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S et al.A genomewide association study points to multiple loci thatpredict antidepressant drug treatment outcome in depression.Arch Gen Psychiatry 2009; 66: 966–975.
19 Perlis RH, Smoller JW, Ferreira MA, McQuillin A, Bass N,Lawrence J et al. A genomewide association study of response tolithium for prevention of recurrence in bipolar disorder. Am JPsychiatry 2009; 166: 718–725.
20 Aberg K, Adkins DE, Bukszar J, Webb BT, Caroff SN, Miller DDet al. Genomewide association study of movement-related adverseantipsychotic effects. Biol Psychiatry 2010; 67: 279–282.
21 Adkins DE, Aberg K, McClay JL, Bukszar J, Zhao Z, Jia P et al.Genomewide pharmacogenomic study of metabolic side effects toantipsychotic drugs. Mol Psychiatry 2011; 16: 321–332.
22 Alkelai A, Greenbaum L, Rigbi A, Kanyas K, Lerer B. Genome-wide association study of antipsychotic-induced parkinsonismseverity among schizophrenia patients. Psychopharmacology(Berl) 2009; 206: 491–499.
23 McClay JL, Adkins DE, Aberg K, Stroup S, Perkins DO,Vladimirov VI et al. Genome-wide pharmacogenomic analysis ofresponse to treatment with antipsychotics. Mol Psychiatry 2011;16: 76–85.
24 McGuire AL, Evans BJ, Caulfield T, Burke W. Science andregulation. Regulating direct-to-consumer personal genome test-ing. Science (New York, NY) 2010; 330: 181–182.
25 Kirchheiner J, Brosen K, Dahl ML, Gram LF, Kasper S, Roots I et al.CYP2D6 and CYP2C19 genotype-based dose recommendations forantidepressants: a first step towards subpopulation-specificdosages. Acta Psychiatr Scand 2001; 104: 173–192.
26 de Leon J. Pharmacogenomics: the promise of personalizedmedicine for CNS disorders. Neuropsychopharmacology 2009;34: 159–172.
27 Malhotra AK, Lencz T, Correll CU, Kane JM. Genomics and thefuture of pharmacotherapy in psychiatry. Int Rev Psychiatry(Abingdon, England) 2007; 19: 523–530.
28 Lieberman JA, Phillips M, Gu H, Stroup S, Zhang P, Kong L et al.Atypical and conventional antipsychotic drugs in treatment-naivefirst-episode schizophrenia: a 52-week randomized trial ofclozapine vs chlorpromazine. Neuropsychopharmacology 2003;28: 995–1003.
29 Robinson DG, Woerner MG, Alvir JM, Geisler S, Koreen A,Sheitman B et al. Predictors of treatment response from a firstepisode of schizophrenia or schizoaffective disorder. Am JPsychiatry 1999; 156: 544–549.
30 Perkins DO, Wyatt RJ, Bartko JJ. Penny-wise and pound-foolish:the impact of measurement error on sample size requirements inclinical trials. Biol Psychiatry 2000; 47: 762–766.
31 Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe’er I, Floratos Aet al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009; 41:816–819.
32 Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic Jet al. HLA-B*5701 screening for hypersensitivity to abacavir.N Engl J Med 2008; 358: 568–579.
33 Hung SI, Chung WH, Liu ZS, Chen CH, Hsih MS, Hui RC et al.Common risk allele in aromatic antiepileptic-drug inducedStevens-Johnson syndrome and toxic epidermal necrolysis inHan Chinese. Pharmacogenomics 2010; 11: 349–356.
34 Kazeem GR, Cox C, Aponte J, Messenheimer J, Brazell C, NelsenAC et al. High-resolution HLA genotyping and severe cutaneousadverse reactions in lamotrigine-treated patients. PharmacogenetGenomics 2009; 19: 661–665.
35 Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for thetreatment-resistant schizophrenic. A double-blind comparisonwith chlorpromazine. Arch Gen Psychiatry 1988; 45: 789–796.
36 McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY,Rosenheck RA et al. Effectiveness of clozapine versus olanzapine,quetiapine, and risperidone in patients with chronic schizophre-nia who did not respond to prior atypical antipsychotic treatment.Am J Psychiatry 2006; 163: 600–610.
37 Kelly DL, Dixon LB, Kreyenbuhl JA, Medoff D, Lehman AF,Love RC et al. Clozapine utilization and outcomes by race in apublic mental health system: 1994-2000. J Clin Psychiatry 2006;67: 1404–1411.
Pharmacogenetics in psychiatryAK Malhotra et al
768
Molecular Psychiatry

38 Athanasiou MC, Dettling M, Cascorbi I, Mosyagin I, Salisbury BA,Pierz KA et al. Candidate gene analysis identifies a polymorphismin HLA-DQB1 associated with clozapine-induced agranulocytosis.J Clin Psychiatry 2011; 72: 458–463.
39 Parsons B, Allison DB, Loebel A, Williams K, Giller E, Romano Set al. Weight effects associated with antipsychotics: a comprehen-sive database analysis. Schizophr Res 2009; 110: 103–110.
40 Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA,Perkins DO et al. Effectiveness of antipsychotic drugs in patientswith chronic schizophrenia. N Engl J Med 2005; 353: 1209–1223.
41 Pandina GJ, Aman MG, Findling RL. Risperidone in the manage-ment of disruptive behavior disorders. J Child Adolesc Psycho-pharmacol 2006; 16: 379–392.
42 Sikich L, Frazier JA, McClellan J, Findling RL, Vitiello B, Ritz Let al. Double-blind comparison of first- and second-generationantipsychotics in early-onset schizophrenia and schizo-affectivedisorder: findings from the treatment of early-onset schizophreniaspectrum disorders (TEOSS) study. Am J Psychiatry 2008; 165:1420–1431.
43 Correll CU, Manu P, Olshanskiy V, Napolitano B, Kane JM,Malhotra AK. Cardiometabolic risk of second-generation antipsy-chotic medications during first-time use in children and adoles-cents. JAMA 2009; 302: 1765–1773.
44 Reynolds GP, Zhang ZJ, Zhang XB. Association of antipsychoticdrug-induced weight gain with a 5-HT2C receptor gene poly-morphism. Lancet 2002; 359: 2086–2087.
45 Templeman LA, Reynolds GP, Arranz B, San L. Polymorphisms ofthe 5-HT2C receptor and leptin genes are associated withantipsychotic drug-induced weight gain in Caucasian subjectswith a first-episode psychosis. Pharmacogenet Genomics 2005; 15:195–200.
46 Sicard MN, Zai CC, Tiwari AK, Souza RP, Meltzer HY, LiebermanJA et al. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacoge-nomics 2010; 11: 1561–1571.
47 Lencz T, Robinson DG, Napolitano B, Sevy S, Kane JM, Goldman Det al. DRD2 promoter region variation predicts antipsychotic-induced weight gain in first episode schizophrenia. Pharmaco-genet Genomics 2010; 20: 569–572.
48 Julius RJ, Novitsky Jr MA, Dubin WR. Medication adherence: areview of the literature and implications for clinical practice.J Psychiatr Pract 2009; 15: 34–44.
49 Zhang JP, Lencz T, Malhotra AK. Enhancing power in pharmaco-genetic studies: maximizing adherence and meta-analytic strate-gies. Symposium Presented at the Society of Biological PsychiatryAnnual Meeting. San Francisco, CA, 13th May 2011.
50 Velligan DI, Weiden PJ, Sajatovic M, Scott J, Carpenter D, Ross Ret al. The expert consensus guideline series: adherence problemsin patients with serious and persistent mental illness. J ClinPsychiatry 2009; 70(Suppl 4): 1–46; quiz 47-48.
51 Sullivan PF. The psychiatric GWAS consortium: big science comesto psychiatry. Neuron 2010; 68: 182–186.
52 Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA,Hunter DJ et al. Finding the missing heritability of complexdiseases. Nature 2009; 461: 747–753.
53 Higashi MK, Veenstra DL, Kondo LM, Wittkowsky AK, Srinouan-prachanh SL, Farin FM et al. Association between CYP2C9 geneticvariants and anticoagulation-related outcomes during warfarintherapy. JAMA 2002; 287: 1690–1698.
54 Mega JL, Simon T, Collet JP, Anderson JL, Antman EM, Bliden Ket al. Reduced-function CYP2C19 genotype and risk of adverseclinical outcomes among patients treated with clopidogrel pre-dominantly for PCI: a meta-analysis. JAMA 2010; 304: 1821–1830.
55 De Luca V, Mueller DJ, de Bartolomeis A, Kennedy JL. Associationof the HTR2C gene and antipsychotic induced weight gain: a meta-analysis. Int J Neuropsychopharmacol 2007; 10: 697–704.
56 Simon GE, Perlis RH. Personalized medicine for depression: canwe match patients with treatments? Am J Psychiatry 2010; 167:1445–1455.
57 Johnson BA, Ait-Daoud N, Seneviratne C, Roache JD, Javors MA,Wang XQ et al. Pharmacogenetic approach at the serotonintransporter gene as a method of reducing the severity of alcoholdrinking. Am J Psychiatry 2011; 168: 265–275.
58 Kirchheiner J, Fuhr U, Brockmoller J. Pharmacogenetics-basedtherapeutic recommendations–ready for clinical practice? Nat RevDrug Discov 2005; 4: 639–647.
59 Wu RR, Zhao JP, Jin H, Shao P, Fang MS, Guo XF et al. Lifestyleintervention and metformin for treatment of antipsychotic-in-duced weight gain: a randomized controlled trial. JAMA 2008; 299:185–193.
60 Lenzini PA, Grice GR, Milligan PE, Dowd MB, Subherwal S, DeychE et al. Laboratory and clinical outcomes of pharmacogenetic vs.clinical protocols for warfarin initiation in orthopedic patients.J Thromb Haemost 2008; 6: 1655–1662.
Pharmacogenetics in psychiatryAK Malhotra et al
769
Molecular Psychiatry





























