Embed Size (px)
Citation preview

PCR on slide: advantages in environmental and clinical microbiology
J.-C. Avarre1, E. Masseret
2, M. Alunni-Fabbroni
3, P. Hartmann
3, W. Mann
3, N. Häfner
4, M. Dürst
4, C.
Woudstra5, P. Fach
5, G. Cerovic
6, M. Rattier
6, A. Le bras
6, R. Melizzi
6, J.-L. Grabias
6 and C. Weisbuch
6
1 IRD, UR175 CAVIAR, 361 Rue Jean-François Breton, BP5095, 34196 Montpellier cedex 05, France 2 Université Montpellier II, CNRS, Ifremer, IRD, UMR 5119 Ecosystèmes lagunaires, cc093, place E. Bataillon, 34095
Montpellier, France 3 Beckman Coulter Biomedical GmbH, Sauerbruchstrasse 50, 81377München, Germany 4 Klinik für Frauenheilkunde, Universitätsklinikum Jena, Friedrich-Schiller-Universität Jena, Bachstr. 18, 07740 Jena,
Germany 5 AFSSA LERQAP, IdentyPath platform, 23 Av du Général De Gaulle, 94706 Maisons Alfort Cedex, France 6 Genewave SAS, 172 Rue de Charonne, Immeuble le Dorian, 75011 Paris, France
Polymerase Chain Reaction (PCR) has become an indispensable tool for the analysis of DNA. Though it is not the only
method for nucleic acid amplification, its success is due to its simplicity. PCR is conventionally performed in tubes or in
microplate wells, in volumes ranging from 10 to 100 µl. With the advent of microfluidic and lab-on-a-chip technologies,
varying formats of PCR systems have been developed, with the aim of reducing both reaction volumes and reaction times,
while keeping at least the same sensitivity. Among these formats, an original on-slide system has been developed, which
allows nucleic acid extraction and PCR amplification in a single low-volume “reaction site”, on 48 samples in parallel.
DNA can be detected either off-line by regular post-amplification gel electrophoresis, or on-line by measuring
fluorescence of up to 5 dyes simultaneously. After a brief description of the features and characteristics of this analysis
format, its advantages over conventional PCR systems will be illustrated through three different applications highly
relevant for clinical and environmental microbiology, i.e.: analysis of the progression of human papillomavirus (HPV)-
induced lesions at the molecular level, identification of phytoplankton cells from water samples and detection of
Clostridium botulinum types C and D from naturally contaminated samples.
Keywords: on-slide PCR, AmpliGrid, real-time, microorganism, detection, quantification, identification, single-cell
analysis
1. Introduction
The polymerase chain reaction (PCR) is a simple technique that amplifies a DNA template to produce specific DNA
fragments in vitro. It was originally developed to amplify short segments of a longer DNA molecule [1]. The simplicity,
low cost and versatility of PCR made this technique extremely valuable for a wide range of applications, not only in
basic research, but also in commercial uses, including genetic identity testing, forensics, industrial quality control and in
vitro diagnostics. As a result, PCR has become commonplace in most molecular biology labs. However, PCR has
evolved far beyond simple amplification and detection, and many extensions of the original PCR format have been
described.
PCR is conventionally performed in tubes or in microplate wells, with "large" volumes generally ranging from 10 to
100 µl. Over the last years, considerable efforts have been made to miniaturize PCR systems [2]. One of the motivations
for the development of integrated miniaturised PCR is to process samples of small volume. Based on this idea,
microfluidic devices, or lab-on-a-chip, have witnessed impressive progress and on-chip PCR reaction volumes have
dropped down to nl [3, 4]. The miniaturization of PCR protocols offers many advantages including short assay time,
high precision and sensitivity, high-throughput, low reagent consumption and portability [5]. Yet, though on-chip PCR
is likely to become the 'next-generation PCR', translating microfluidics to biology labs is still limited by its cost and the
expertise it requires.
In this context, an intermediary on-slide system has been developed, which allows sensitive and rapid nucleic acid
extraction and PCR amplification in single 1-µl “reaction sites”. The system, called AmpliGrid (Beckman Coulter,
Advalytix Products, Munich, Germany), enables to process 48 samples in parallel. Amplifications are achieved with a
dedicated thermal cycler, AmpliSpeed (Beckman Coulter, Advalytix Products). Results can be analyzed off-line or in
real-time by fluorescence measurement with another dedicated instrument, AmpliHyb (Genewave, Paris, France).
This chapter describes this new analysis format and its advantages for microbiological applications, as tested by
different research groups in 3 different applications focusing on the detection and/or identification of microorganisms.
_______________________________________________________________________________________
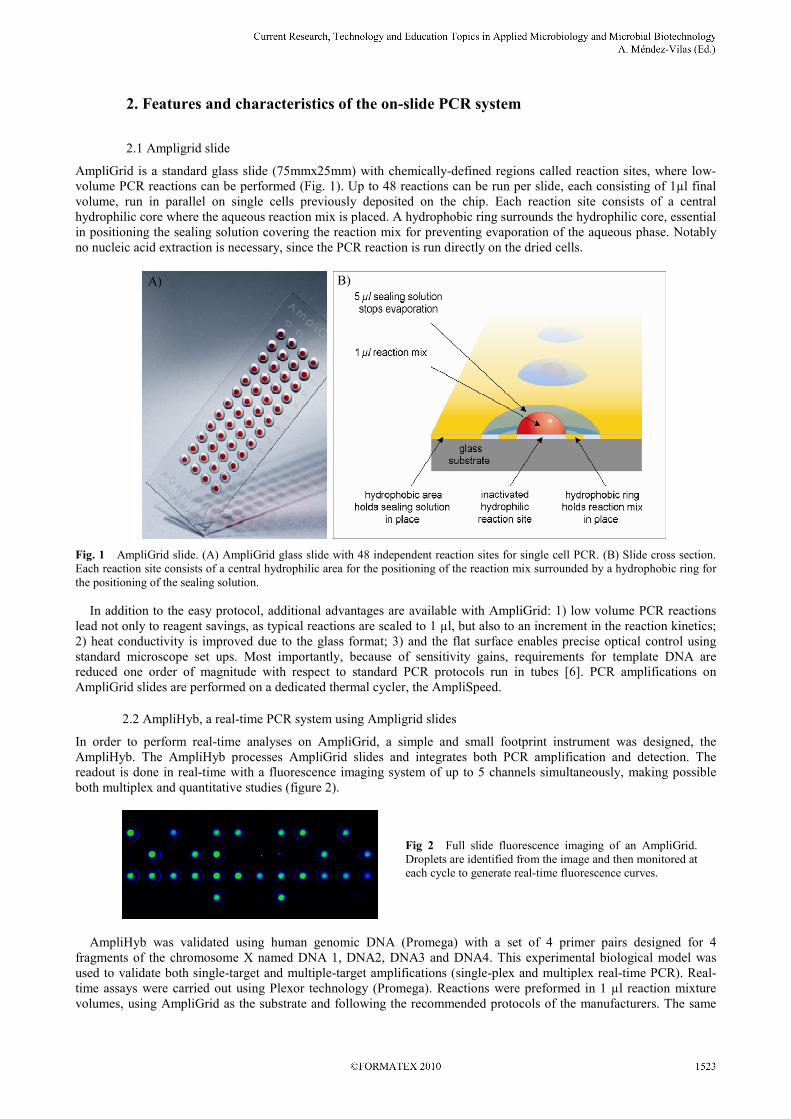
2. Features and characteristics of the on-slide PCR system
2.1 Ampligrid slide
AmpliGrid is a standard glass slide (75mmx25mm) with chemically-defined regions called reaction sites, where low-
volume PCR reactions can be performed (Fig. 1). Up to 48 reactions can be run per slide, each consisting of 1µl final
volume, run in parallel on single cells previously deposited on the chip. Each reaction site consists of a central
hydrophilic core where the aqueous reaction mix is placed. A hydrophobic ring surrounds the hydrophilic core, essential
in positioning the sealing solution covering the reaction mix for preventing evaporation of the aqueous phase. Notably
no nucleic acid extraction is necessary, since the PCR reaction is run directly on the dried cells.
Fig. 1 AmpliGrid slide. (A) AmpliGrid glass slide with 48 independent reaction sites for single cell PCR. (B) Slide cross section.
Each reaction site consists of a central hydrophilic area for the positioning of the reaction mix surrounded by a hydrophobic ring for
the positioning of the sealing solution.
In addition to the easy protocol, additional advantages are available with AmpliGrid: 1) low volume PCR reactions
lead not only to reagent savings, as typical reactions are scaled to 1 µl, but also to an increment in the reaction kinetics;
2) heat conductivity is improved due to the glass format; 3) and the flat surface enables precise optical control using
standard microscope set ups. Most importantly, because of sensitivity gains, requirements for template DNA are
reduced one order of magnitude with respect to standard PCR protocols run in tubes [6]. PCR amplifications on
AmpliGrid slides are performed on a dedicated thermal cycler, the AmpliSpeed.
2.2 AmpliHyb, a real-time PCR system using Ampligrid slides
In order to perform real-time analyses on AmpliGrid, a simple and small footprint instrument was designed, the
AmpliHyb. The AmpliHyb processes AmpliGrid slides and integrates both PCR amplification and detection. The
readout is done in real-time with a fluorescence imaging system of up to 5 channels simultaneously, making possible
both multiplex and quantitative studies (figure 2).
AmpliHyb was validated using human genomic DNA (Promega) with a set of 4 primer pairs designed for 4
fragments of the chromosome X named DNA 1, DNA2, DNA3 and DNA4. This experimental biological model was
used to validate both single-target and multiple-target amplifications (single-plex and multiplex real-time PCR). Real-
time assays were carried out using Plexor technology (Promega). Reactions were preformed in 1 µl reaction mixture
volumes, using AmpliGrid as the substrate and following the recommended protocols of the manufacturers. The same
Fig 2 Full slide fluorescence imaging of an AmpliGrid.
Droplets are identified from the image and then monitored at
each cycle to generate real-time fluorescence curves.
B) A)
_______________________________________________________________________________________

PCR conditions were used for simplex and multiplex PCR in all presented results: 10 min at 95°C followed by 45
cycles of 30 sec at 95°C, 60 sec at 64°C, 60 sec at 72°C and a final elongation of 10 min at 72°C.
For simplex validation, serial 10-fold dilutions of human genomic DNA (corresponding to 2-20,000 copies) were
subjected to amplification with primers designed to DNA4. Amplification curves showed a linear correlation with the
template DNA concentration in the range tested (R2 = 0.9956 over 3 replicates), indicating that the equivalent of 2
copies were reliably detected (Fig. 3). The same experiment performed with LC green instead of Plexor reached very
similar results (data not shown).
Cycle
PCR computedData
a) b)
Fig 3 Amplification of human genomic DNA with primers designed to DNA4. 10-fold dilutions (2-20,000 copies) were amplified
by the Plexor technology (a), and individual quantification cycle (Cq) values were plotted against the log of DNA concentration
(n=3) (b).
Validation of the multiplex assay was achieved by comparing the performance of simplex and multiplex assays using
PCR primers labeled with the following fluorophores: FAM, TAMRA, Cy5 and TexasRed. PCR conditions were as
described previously. Generally, a successful multiplex real-time PCR reaction is expected when multiplex and single-
plex assays performed simultaneously on the same AmpliGrid slide produce similar quantification cycle (Cq) values [7]
for the amplification of a particular gene, and when the exponential phase of the multiplex reaction can be
superimposed over that of the simplex reactions, i. e. amplification efficiencies are nearly identical.
In order to investigate whether single-plex Cq values correspond to multiplex ones, 200 pg of human genomic DNA
was used to test the 4 different targets (DNA 1, DNA2, DNA3 and DNA4). In each experiment, single-plex and
different combinations of multiplexing were performed in parallel on the same AmpliGrid slide: 2-plex, 3-plex and 4-
plex reactions. Obtained results showed that exponential phase of both single-plex and multiplex reactions could be
superimposed. In addition, multiplex and single-plex Cq values were in accordance with each other (as illustrated in
Table 1 for 4-plex assays). Similar results were obtained for 2- and 3-plex assays (data not shown).
In order to detect possible unspecific amplification, or primer-dimer formation, which may both result in false
positive signals, a melting procedure was realized after each amplification (both Plexor and LC Green chemistries are
well adapted for this type of analysis). Plotting the second derivative of each melting profile revealed the presence of
one single peak with average Tm of 85.7, 87.0, 86.9 and 84.8 °C for the 4 amplicons DNA1, DNA2, DNA3 and DNA4,
respectively.
Table 1 Comparison of Cq values (mean± SD) between single-plex (n=2) and 4-plex (n=11) PCR.
Target DNA Single-Plex Cq Four-plex Cq
DNA1 29.25 ±0.38 30.44 ±0.54
DNA2 30.74 ±0.59 30.82 ±0.36
DNA3 31.19 ±0.78 31.10 ±1.28
DNA4 33.59 ±1.22 32.19 ±0.54
These results demonstrate that AmpliHyb combined with the AmpliGrid platform allows reliable and sensitive real-
time PCR, in both simplex and multiplex formats. They also show that PCR in small reaction volumes is well adapted
for melting analysis.
3. Applications to clinical and environmental microbiology
3.1 Single cell PCR opens new opportunities to analyze the progression of HPV-induced lesions at the
molecular level
_______________________________________________________________________________________

3.1.1 Introduction
Single cell analysis is a suitable platform to address a number of basic questions related to virus induced cancer.
Although the etiological role of high-risk human papillomavirus (HR-HPV) in genital cancer, in particular cervical
cancer, is established, the process of HPV-induced carcinogenesis at the molecular level is far from being fully
understood [8, 9]. In a generally accepted model for natural HPV infection, the virus infects cells of the basal or
parabasal epithelial layer of the cervix uteri via micro fissures. The virus may then persist without causing clinically
evident lesions or it may induce a virus-productive lesion termed CIN1 (cervical intraepithelial neoplasia) which may
progress to a precancerous lesion (CIN2, CIN3) and invasive cervical cancer. To date in situ hybridization analyses
have provided insight into the molecular events that take place at different stages of disease in a histological context. It
became evident that HPV DNA replication is tightly linked to epithelial cell differentiation. In contrast, precancerous
lesions and invasive carcinoma, which are characterized by a lack of differentiation, are not permissive for virus
replication. Moreover, viral gene expression is suppressed in the basal and parabasal cells of CIN1 but not in advanced
lesions [10]. Thus in situ hybridization is an extremely valuable tool to analyze viral DNA and RNA in order to relate
these observations to the pathological state of the tissue but it suffers from limited sensitivity. Copy numbers as low as
1-2 molecules, be it viral DNA or RNA, cannot be reproducibly detected in tissue sections (unpublished data). These
limitations can be overcome by the use of single cell real-time PCR technology in combining laser capture
microdissection or other methods for cell separation. Questions pertinent to the understanding of HPV-induced
carcinogenesis can now be addressed. For example: are epithelial stem cells the targets of HPV infection? Can HPV
DNA be detected in basal cells of all CIN1? And if so, is viral gene expression down-regulated or even completely
suppressed in these cells? Could the absence of HPV DNA in CIN1 basal cells be an explanation for the self-limitation
of a lesion? Is there a correlation between HPV copy number and the level of viral gene expression? Is there a
correlation between the state of cellular differentiation and different viral transcript species?
3.1.2 Results
To address these questions, the AmpliGrid platform was used for the identification of a single viral molecule in a single
HPV infected SiHa cell spotted on the reaction site, alone or in the presence of a HPV-negative C33 cell background.
SiHa is an established human cervical carcinoma cell line positive for 1-2 copies of integrated HPV16 DNA per cell.
Identification of these cells was achieved by PCR analysis of two viral genes, E6 and E2, yielding products of 124bp
and 110bp, respectively. SiHa cells were micromanipulated on the slide reaction sites in the presence or the absence of
background C33 cells and let dry completely. The PCR reaction mixture including primer pairs for the detection of E6
and E2 was deposited on top of the reaction site and the PCR program consisted in an initial denaturation of 10 min at
95°C followed by 40 cycles of denaturation (95°C, 30 sec), annealing (58°C, 60 sec) and extension (72°C, 60 sec) and a
final extension step of 10 min at 72°C. Once PCR was finished the products were directly loaded and separated on an
8% polyacrylamide gel. As shown in Fig. 4, single gene copies could be efficiently amplified by PCR on AmpliGrid
starting from single cells. Both products are showing the expected size while the C33 negative control cells did not
generate any kind of PCR product.
500400
300
100
200
200
500400
300
100
E2 E6
C33
bp bp
SiHa
A B
Fig. 4 E2 and E6 amplification in SiHa and C33 cells. (A) An increasing number of HPV negative C33 cells (from 0 to 1000) was
deposited on different AmpliGrid reaction sites and analysed by PCR. E2 and E6 products were never detectable. (B) Single HPV
positive SiHa cells were deposited on different AmpliGrid reaction sites. E6 and E2 products were visible with the expected size
(124bp and 110bp respectively).
_______________________________________________________________________________________

When SiHa cells were deposited on AmpliGrid reaction site in the presence of 10, 100 or 1000 C33 cells, the HPV
negative cell line used as negative control, no variation in the efficiency was observed (Fig. 5).
bp
500400
300
200
100
In conclusion, these findings clearly indicate that the sensitivity of the AmpliGrid platform easily allows the
detection of single viral gene copies within a single cell, even in the background of hundreds of different cells. The
questions outlined above can thus be addressed by using the basic platform, i.e. detection of amplified DNA by gel
electrophoresis. A higher throughput, yet, could be achieved by the simultaneous real-time detection of PCR products
using different fluorescent dyes.
3.2. Identification of phytoplankton cells from water samples
3.2.1 Context and aims of the study
Dinoflagellates belonging to the genus Alexandrium are often involved in harmful algal blooms, which are frequently
associated with the paralytic shellfish-poisoning syndrome. These blooms are considered as one of the main ecological
problems at a world scale in coastal regions [11]. In Thau lagoon (French Mediterranean coast), two ribotypes are
frequently observed during such blooms, which cannot be formally differentiated by microscopic observation:
Alexandrium catenella and A. tamarense [12]. For this reason, a semi-multiplex PCR test targeting the 18S-28S rDNA-
ITS region was developed to differentiate the two species according to their amplicon size, i.e 126 bp for A. tamarense
vs. 240 bp for A. catenella [12]. Based on regular PCR, this test involved the establishment of monoclonal cultures from
environmental samples. This procedure was not only time-consuming but sometimes failed to yield sufficient material
for DNA extraction and amplification. The AmpliGrid PCR system was therefore tested to evaluate the possibility of
identifying these dinoflagellate species directly from single cells, as this would greatly improve the speed and accuracy
of Alexandrium detection from natural samples, by avoiding the laborious establishment of monoclonal cultures.
3.2.2 Results
The protocol used in this study combined cell lysis/DNA extraction with PCR amplification in a single 1.5-µl “reaction
site”. One-µl drops of biological sample were distributed in the wells of 48-well AmpliGrid chips. Samples were dried
at room temperature and layered with 5 µl of sealing solution, which allowed shaping “reaction sites” on the slide (Fig.
6).
Fig. 5 Single-plex amplification of E2 (lines 1-4) and E6
(lines 5, 7-9) or duplex amplification (E2+E6, lane 10) in SiHa
cells in the presence of HPV negative C33 cells. Single SiHa
cells were deposited on different AmpliGrid reaction sites in
the presence of different amount of HPV negative C33 cells
(from 0 to 1000). E2 and E6 were amplified with comparable
efficiency despite the presence of unspecific cellular
background.
_______________________________________________________________________________________

a) b)
c)
Fig.6 Microscope observation of an Alexandrium catenella single cell deposited in a reaction site of a 48-well AmpliGrid prior to
cell lysis/DNA extraction. Visualization of the cell in a well (a, X25) and further enlargements of the cell prior (b, X200) and after
drying at room temperature (c, X250). The arrow points to the cell.
For cell lysis/DNA extraction, 0.75 µl of cell extraction/working solution was added to each reaction site. The cell
extraction procedure, consisting of 5 min at 75°C followed by 2 min at 95°C, was performed on an AmpliSpeed slide
cycler (Advalytix). Then, 0.75 µl of a 2X PCR mix containing all necessary reagents was added to the reaction sites.
After amplification with the AmpliSpeed slide cycler, PCR products were then visualized by regular gel electrophoresis
(for more details, see [13]).
The ability of this method to identify A. catenella and A. tamarense from single cells was assessed on fresh
monoclonal cultures that were established from water samples collected in Thau lagoon (French Mediterranean coast)
during May 2007. Cells in cultures were numerated and serial dilutions were realized in order to reach a final
concentration of ~1 cell µl-1
. Quantity and quality of the template material deposited in the AmpliGrid wells was
inspected under an inverted microscope prior to cell lysis. Amplifications were performed on 1 µl of such preparations.
The actual number of intact cells varied from 0 to 14 per well. Many wells were devoid on intact cells and contained
only damaged cells, cellular debris and/or empty theca. Positive controls consisted in ~10 ng of DNA from pure
cultures of A. catenella or A. tamarense, and negative controls consisted in H2O. All of the wells loaded with samples
were positive for either A. catanella or A. tamarense (Table 1).
Table 2 PCR amplification results and microscopic cell numeration control on AmpliGrid slides (reproduced from [13])
# cells / well # positive amplifications / # wells
A. catenella A. tamarense
0 intact cell 37/37 21/21
1 cell 25/25 10/10
2 cells 17/17 8/8
3 cells 12/12 3/3
4 cells 9/9 3/3
5 cells 2/2 -
6 cells 2/2 -
7 cells 2/2 -
8 cells 1/1 -
9 cells 1/1 -
_______________________________________________________________________________________

10 cells 1/1 -
14 cells 1/1 -
Total 110/110 45/45
Amplification also occurred on samples devoid of intact cells. When cultures were mixed, amplifications yielded two
bands corresponding to the 2 dinoflagellate species, even though there were no intact cells present in the reactions (Fig.
7). These results tend to indicate that this protocol is highly sensitive and is able to amplify trace amounts of DNA
contained in cellular debris.
M 654321
Fig. 7 PCR amplification results of samples consisting of a 1:1 mix of the two cultures and devoid of intact cells. M: 100-bp
molecular weight marker. Lanes 1-2: positive controls for A.catenella and A. tamarense strains, respectively; lane 3: negative control;
lanes 4-6: wells loaded with a 1:1 mix of the two cultures. A.catenella and A. tamarense products were visible with the expected size
(240 and 129 bp, respectively).
Another series of experiments aimed at testing the method directly on environmental samples. These latter consisted
in 3 water samples collected in Thau lagoon during two recent blooms (autumn 2008 and spring 2009). Their original
Alexandrium sp. concentration was estimated by microscope to ~260, ~770 and ~46,900 cells.l-1
. Water was filtered-
concentrated such that cells were concentrated ~10,000 times at the end. The resulting cell suspensions were diluted
1000 times because of the high abundance of detritus that may interfere with DNA amplification, and one µl was used
as PCR template, without checking for the potential presence of Alexandrium sp. cells in the wells. Results showed that
DNA was detected in the 3 natural samples, albeit at different rates. If all the wells tested (8) for the most concentrated
sample were positive, only 4/8 and 3/9 wells were positive for the two least concentrated samples. Furthermore, both
dinoflagellate species were detected in 2 of the 3 samples. Including the different steps in sample preparation, the final
Alexandrium sp. cell concentration was ~2.6x10-3
, ~7.7x10-3
and ~0.46 cells µl-1
in the three samples, respectively.
Considering that the number of rDNA copies per Alexandrium sp. cell is approximately 200-2000 [14, 15], this suggests
that almost one single copy of rDNA gene could be detected in a reaction site [13]. Such results confirm that low
volumes are appropriate to enhance the sensitivity and efficiency of DNA amplification, as already stated [16, 17].
In conclusion, this on-slide PCR method proved very sensitive and could successfully identify Alexandrium catenella
and A. tamarense at the infra-single cell level. Compared to other described methods used for the genetic
characterization of single phytoplankton cells [18, 19], this procedure presents several major advantages and/or
improvements: i) it is very easy and quick, as identification can be obtained within less than 3 hrs; ii) DNA loss is
impossible since extraction and amplification occur in the same reaction site; iii) the on-slide format allows for an easy
visual control of both quality and quantity of the templates using standard microscopy, ensuring a quality control when
necessary, e.g. for taxonomic studies; iv) its exceptional sensitivity makes possible to skip the uneasy microscopic
selection of phytoplankton cells for monitoring applications.
3.3 Detection of Clostridium botulinum types C and D from naturally contaminated samples
3.3.1 Introduction
Botulism is a severe flaccid paralytic disease caused by seven different neurotoxin subtypes (BoNT A-G) produced by
bacterial species such as Clostridium botulinum [20]. All BoNT subtypes act at the neuromuscular junction blocking the
release of acetylcholine thus leading to flaccid paralysis [21]. BoNT types A, B, E and more rarely F cause human
_______________________________________________________________________________________

botulism. Toxin types C and D are mainly responsible for animal botulism [22]. The economic, medical and alimentary
consequences of animal botulism can be catastrophic in the event of an epizooty, underlying the necessity of rapid
identification. Thus research has focused on the development of specific and reliable techniques to identify BoNT-
producing clostridia [23]. There are several reports on the detection of type C (bont/C) and type D (bont/D) genes by
PCR [24], but only one by real-time PCR [25]. Real-time PCR presents the advantages of being more specific and
sensitive than conventional PCR. We developed a real-time PCR (Rt PCR) assay for detection of type C and type D C.
botulinum bont genes for veterinary and epizootic study purposes. The test was developed on the AmpliHyb system, a
new device of great technical interest, capable of monitoring micro-volume Rt PCR assays on microscope slides.
3.3.2 Primers and probes for nucleic acid amplification
The sensitivity, specificity and reproducibility of an Rt PCR-based assay to detect microorganisms in naturally
contaminated samples are largely dependent on the design of appropriate primers and probes. Unfortunately C.
botulinum has a low GC content and its bont genes have a high degree of similarity; these features make it challenging
to find acceptable primers and probes. C2 and D3 oligonucleotides were designed in homologous regions of subtypes
bont/C and subtypes bont/D respectively and in non-homologous regions of C. botulinum subtypes A, B, E, and F for
toxin-gene specific identification. All probes were 5’-labelled with 6-carboxy-x-rhodamine (ROX) and 3’-labelled with
Black Hole Quencher (BHQ-2). All primers and probes were purchased from Sigma Aldrich (St. Quentin Fallavier,
France). Specificity was first evaluated in silico against sequences from the GenBank database using the BLAST
algorithm (http://www.ncbi.nih.gov/BLAST), which revealed no cross reaction. It was then tested with DNA from pure
microbial strains.
3.3.3 Real-time PCR assays
Real-time PCR amplifications were carried out with the AmpliHyb. Ampligrid reaction sites were preloaded with 1µl of
extracted DNA from reference strains or naturally contaminated samples and air-dried at room temperature. One µl of
master mix was placed on each reaction site for a final concentration of 600 nM primers and 400 nM probes.
Evaporation was prevented by covering the aqueous phase with 5µl of a special sealing solution. Rt PCR was then
performed on the AmpliHyb with 40 cycles in about 2h45 min by acquiring the images that correspond exactly to the
elementary arrays. Positive samples were identified by a fluorescence signal greater than or equal to 0.05 units at the
beginning of the exponential phase on a scale from 0 to 1 arbitrary units and a quantification cycle below 38 Cq.
3.3.4 Specificity study
Specificity was evaluated on eleven C. botulinum type C (strain 850131 as bont/C reference) and six type D (strain
1873 as bont/D reference). Twenty three Clostridium strains (BoNT/A, B, AB, E, F) were tested as BoNT-producing
negative controls. Twenty one Clostridium strains were used as non-BoNT-producing negative controls. Twenty three
strains of other bacterial species were analyzed as non-Clostridium negative controls. C2 and D3 oligonucleotides gave
positive signals for respectively all type C and type D C. botulinum strains and no signals for the negative controls,
BoNT-producing/non-BoNT-producing Clostridium negative controls, and the other non-Clostridium bacteria.
3.3.5 Sensitivity study
The limit of detection (LOD) was determined with serial 10-fold dilutions of genomic DNA over a range of six orders
of magnitude. The LOD is the lowest amount of C. botulinum in a test sample that has been reproducibly detected in at
least three experiment sets. LOD was converted to a genomic copy number based on the total genomic DNA content of
each strain. DNA from C. botulinum type C (strain NCTC 850131) and type D (strain NCTC 1873) was extracted and
tested as a reference. Extracted DNA was quantified prior to serial dilution with the NanoDrop ND-1000
spectrophotometer (Thermo Scientific, Wilmington, USA). PCR efficiency was evaluated for each primer/probe pair
with serial dilution in accordance with the correlation coefficient (R²). bont/C2 oligonucleotides gave sensitivity results
of 44 genome copies and bont/D3 oligonucleotides gave sensitivity results of 20 genome copies with PCR efficiency of
respectively 92% and 91%. All real-time PCR assays showed a strong linear correlation (R²>0.99) between Cq values
and the template concentration (Fig.8).
_______________________________________________________________________________________

a) b)
c) d)
Fig. 8 Sensitivity study of bont/C2 and bont/D3 oligonucleotides. bont/C2 permits detection until 140 fg of type C C. botulinumstrain 850131 corresponding to 44 equivalent genomes, with a correlation coefficient of 0.9963 (a, b). bont/D3 permits detection until50 fg of type D C. botulinum strain 1873 corresponding to 20 equivalent genomes, with a correlation coefficient of 0.9998 (c, d).
3.3.6 Investigation of suspected naturally contaminated samples
One hundred and twenty five suspected botulism cases were collected by the Analysis and Development Laboratory 22(LDA22, Brittany, France) during animal botulism epizootic events in Brittany, France, in 2009. Samples wereincubated in anaerobic conditions in pre-reduced tryptone-glucose-yeast extract (TGY). DNA was extracted with theDNeasy blood and tissue kit (Qiagen, Hilden, Germany). Samples were typed for presence of the type C and type Dgene by standard Rt PCR considered as the reference in this study, and by the AmpliHyb real-time PCR array. Fiftyseven out of the 125 samples were detected positive for either the bont/C or bont/D gene by conventional Rt PCR.Among them, 38 were type C, 16 were type D, and 3 were detected both as type C and D. Regarding the AmpliHybresults, 33 out of the 125 samples were found positive by real-time PCR array. Twenty were type C, 11 were type D andtwo were detected both as type C and D. The results of the AmpliHyb and conventional Rt PCR matched to 80.80%. Todecrease the level of discrepancy observed between the AmpliHyb and conventional Rt PCR, a pre-amplification stepprior to the AmpliHyb assay was investigated. Applied BioSystems markets the “Taqman Preamp Master Mix Kit”which allows an increase in specific DNA targets. Starting material is increased prior to PCR and the resulting pre-amplification product is then used for Rt PCR. With the addition of this pre-amplification step, the concordance resultsbetween AmpliHyb and conventional PCR matched to 96.80% (Table 3).
Table 3 Concordance results between conventional Rt PCR and AmpliHyb assay
N = 125 AmpliHyb PCR N = 125 AmpliHyb PCRwithout pre-amplification Positive Negative with pre-amplification Positive Negative
ConventionalRt PCR
Positive33 24 57
ConventionalRt PCR
Positive 53 4 5726.40% 19.20% 45.60% 42.40% 3.20% 45.60%
Negative0 68 68
Negative 0 68 680.00% 54.40% 54.40% 0.00% 54.40% 54.40%
33 92 50 7226.40% 73.60% 42.40% 57.60%
Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology A. Méndez-Vilas (Ed.)
1530 ©FORMATEX 2010
_______________________________________________________________________________________

3.3.7 Conclusions
Animal botulism is a worldwide problem that causes significant economic losses as it concerns cattle [26] and several
varieties of birds [27]. A real-time PCR assay for the detection of the type C and type D C. botulinum toxin genes was
developed on the AmpliHyb system. Oligonucleotides C2 and D3 were found to be specific and sensitive for the
detection of bont/C and D genes of BoNT-producing clostridia, as no cross detection was observed with C. botulinum
BoNT/A, B, E and F producing strains, other Clostridia or Enterobacteriaceae species which are frequently isolated
from environmental samples. These results indicate that bont/C2 and bont/D3 oligonucleotides are suitable for the rapid
and specific characterization of toxigenic bont-positive type C and D Clostridium strains. The applicability of the
bont/C2 and bont/D3 assay to botulism investigation in complex samples was analyzed in 125 naturally contaminated
samples. DNA was extracted and tested blindly on the AmpliHyb against conventional real-time PCR considered as the
reference method. Data obtained correlated quite well, with 80.80% of concordance, and after a pre-amplification step it
was possible to increase the concordance to 96.8%. The AmpliHyb system showed comparable specificity and
sensitivity to conventional real-time PCR on pure DNA microbial strains, but necessitated a pre-amplification step on
complex DNA samples. The benefits of using this device are the low level of genetic material needed, as the limit of
detection reached 50 fg for C. botulinum type D, and the smaller volumes of reaction components which substantially
reduces running costs. As the experiments demonstrated good concordance results with conventional methods, the
AmpliHyb system promises to be an efficient alternative to conventional microwell plate-based systems for real-time
PCR.
4. General Conclusion
The accuracy, sensitivity and robustness of an on-slide PCR system has been tested and compared with conventional
PCR. Results showed high reproducibility and sensitivity. As described above, this system enables to dramatically
reduce the amount of starting genetic material and to detect DNA at the infra-single cell level. The AmpliGrid format
allows an easy visual control of both quality and quantity of the templates using standard microscopy, ensuring a quality
control when necessary, e.g. for taxonomic studies. The real-time version based on the AmpliHyb offers the possibility
to perform quantitative PCR amplifications in a very short time. Combined with the reduced amount of reagents needed,
this on-slide PCR format therefore constitutes a powerful cost- and time-effective alternative to conventional end-point
and real-time PCR systems. It should prove extremely valuable in both environmental and clinical microbiology.
Acknowledgments We would like to thank Dr. Buffereau’s team (LDA-22, Brittany, France) for providing naturally C. botulinum
contaminated samples. The research on phytoplankton cells was funded by a grant from the French National Program 'Ecosphère
Continentale et Côtière' (EC2CO-PNEC) and by the ALCAT program of Ifremer.
References
[1] Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of betaglobin genomic
sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350.
[2] Auroux PA, Koc Y, deMello A, Manz A, Day PJR. Miniaturised nucleic acid analysis. Lab Chip. 2004; 4(6):534-546.
[3] Zhang CS, Xing D. Miniaturized PCR chips for nucleic acid amplification and analysis: latest advances and future trends.
Nucleic Acids Research. 2007; 35(13):4223-4237.
[4] Zhang YH, Ozdemir P. Microfluidic DNA amplification-A review. Analytica Chimica Acta. 2009; 638(2):115-125.
[5] Baker M. Clever PCR: more genotyping, smaller volumes. Nature Methods. 2010; 7(5):351-355.
[6] Lutz-Bonengel S, Sanger T, Heinrich M, Schon U, Schmidt U. Low volume amplification and sequencing of mitochondrial DNA
on a chemically structured chip. International Journal of Legal Medicine. 2007; 121(1):68-73.
[7] Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M et al. The MIQE Guidelines: Minimum information for
publication of quantitative real-time PCR experiments. Clinical Chemistry. 2009; 55(4):611-622.
[8] Hafner N, Driesch C, Gajda M, Jansen L, Kirchmayr R, Runnebaum IB, et al. Integration of the HPV16 genome does not
invariably result in high levels of viral oncogene transcripts. Oncogene. 2008; 27(11):1610-1617.
[9] zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. 2009; 384(2):260-265.
[10] Durst M, Glitz D, Schneider A, zur Hausen H. Human Papillomavirus type-16 (HPV-16) gene expression and DNA-replication
in cervical neoplasia - analysis by in situ hybridization. Virology. 1992; 189(1):132-140.
[11] Maso M, Garces E. Harmful microalgae blooms (HAB); problematic and conditions that induce them. Marine Pollution
Bulletin. 2006; 53(10-12):620-630.
[12] Genovesi B, Masseret E, Shin-Grzebyk MS, Grzebyk D, Berrebi P, Gagnaire PA, et al. Co-occurrence of two Alexandrium
species in Thau Lagoon. Harmful Algal News. 2008; 37.
[13] Masseret E, Enquebecq M, Laabir M, Genovesi B, Vaquer A, Avarre JC. A simple and innovative method for species
identification of phytoplankton cells on minute quantities of DNA. Environmental Microbiology Reports. 2010; In press: DOI:
10.1111/j.1758-2229.2010.00164.x
_______________________________________________________________________________________

[14] Galluzzi L, Penna A, Bertozzini E, Vila M, Garces E, Magnani M. Development of a real-time PCR assay for rapid detection
and quantification of Alexandrium minutum (a dinoflagellate). Applied and Environmental Microbiology. 2004; 70(2):1199-
1206.
[15] Galluzzi L, Bertozzini E, Penna A, Perini F, Garces E, Magnani M. Analysis of rRNA gene content in the Mediterranean
dinoflagellate Alexandrium catenella and Alexandrium taylori: implications for the quantitative real-time PCR-based
monitoring methods. Journal of Applied Phycology. 2010; 22(1):1-9.
[16] Gaines ML, Wojtkiewicz PW, Valentine JA, Brown CL. Reduced volume PCR amplification reactions using the AmpF rho
STR (R) profiler plus (TM) kit. Journal of Forensic Sciences. 2002; 47(6):1224-1237.
[17] Kricka LJ, Wilding P. Microchip PCR. Analytical and Bioanalytical Chemistry. 2003; 377(5):820-825.
[18] Frommlet JC, Iglesias-Rodriguez MD. Microsatellite genotyping of single cells of the dinoflagellate species Lingulodinium
polyedrum (Dinophyceae): A novel approach for marine microbial population genetic studies. Journal of Phycology. 2008;
44(5):1116-1125.
[19] Lynn DH, Pinheiro M. A Survey of Polymerase Chain Reaction (PCR) amplification studies of unicellular protists using single-
cell PCR. Journal of Eukaryotic Microbiology. 2009; 56(5):406-412.
[20] Sobel J. Botulism. Clinical Infectious Diseases. 2005; 41(8):1167-1173.
[21] Poulain B, Lonchamp E, Jover E, Popoff MR, Molgo J. Mechanisms of action of botulinum toxins and neurotoxins. Annales De
Dermatologie Et De Venereologie. 2009; 136:S73-S76.
[22] Herreros J, Lalli G, Montecucco C, Schiavo G. Pathophysiological properties of clostridial neurotoxins. In: Alouf JE, Freer JH,
eds. The comprehensive sourcebook of bacterial protein toxins, 2nd edition. London, Academic Press; 1999.
[23] Lindstrom M, Korkeala H. Laboratory diagnostics of botulism. Clinical Microbiology Reviews. 2006; 19(2):298-314
[24] Fach P, Gibert M, Griffais R, Popoff MR. Investigation of animal botulism outbreaks by PCR and standard methods. FEMS
Immunology and Medical Microbiology. 1996; 13(4):279-285.
[25] Kouguchi H, Suzuki T, Hasegawa K, Mutoh S, Watanabe T, Niwa K, et al. Quantitative detection of gene expression and toxin
complex produced by Clostridium botulinum serotype D strain 4947. Journal of Microbiological Methods. 2006; 67(3):416-
423.
[26] Whitlock R. Botulism toxicosis of cattle. Proceedings of the Annual Conference of the American Association of Bovine
Practitioners. 1999; 32:45–53.
[27] Neimanis A, Gavier-Widen D, Leighton F, Bollinger T, Rocke T, Morner T. An outbreak of type c botulism in Herring Gulls
(Larus argentatus) in southeastern Sweden. Journal of Wildlife Diseases. 2007; 43(3):327-336.
_______________________________________________________________________________________





























