Embed Size (px)
DESCRIPTION
Pattern Detection and Co-methylation Analysis of Epigenetic Features in Human Embryonic Stem Cells. Ben Niu , Qiang Yang, Jinyan Li, Hong Xue, Simon Chi-keung Shiu, Weichuan Yu, Huiqing Liu, Sankar Kumar Pal HKPolyU. Computational Epigenetics. - PowerPoint PPT Presentation
Citation preview

Pattern Detection and Co-methylation Analysis of Epigenetic Features in Human
Embryonic Stem Cells
Ben Niu, Qiang Yang, Jinyan Li, Hong Xue, Simon Chi-keung Shiu, Weichuan Yu, Huiqing Liu, Sankar Kumar Pal
HKPolyU

Computational Epigenetics
An emerging and most exciting area incorporating the state of the artMachine learning Molecular biology
Aims to understand the epigenetic process in gene transcriptional regulation
Advance our knowledge to the medical arsenal in treating human diseases.

The Research
Human Epigenome project (HEP): the next wave to the Human Genome Project (HGP)
Started in 2003 after completion of the Human Genome Project. HEP aims to identify the epigenetic markers associated with human diseases ‘Journal of Epigenetics’ has been released: first journal dedicated to the
communications in Epigenetics, started in 2006.
Series of publications in highly cited journals in 2005-07: Nature
Focus issue on epigenetics, Nature Review Genetics, April, 2007. Cell
Special issue on epigenetics, Cell, Feburary, 2007. J. Bioinformatics
We are jointly invited to write a review paper on computational epigenetics to the Journal of bioinformatics.

The Industry Epigenetics open a rapidly growing market of
epigenetic medical services (diagnostic, drugs) According to 2007 report of MarketResearch, as shown in the figure,
the global market of epigenetic applications (i.e., drug+ diagnostic services) will be 4 billion US$, by 2012, the annual Growth rate at present time is 60.4%.
0500
10001500200025003000350040004500
2005 2006 2007 2008 2009 2010 2011 2012
global Market(Million U.S.$)
Promising direction!

What we know
Basically: Genes can be turned on/ off through Cytosine methylation or
Histone modifications, a reversible process The epigenetic events is heritable, can change the cell’s phen
otypes without altering its sequence Functionally:
Dominate the growth of cancer and embryonic stem cells These two type of cells are of great medical interests
Cancer is the leading cause of human death hESCs are the answer to the regenerative treatments
For the two points see: Nature Insight: Epigenetics Vol. 447, 2007.

What we don’t know
The logic behind DNA methylation underlying cells’ behaviors remains unclear
How DNA methylation concerts the product of molecular machineries for cell functions
In the context of epigenetics, we need to address two issues: What are the rules of DNA methylation differing the
cancer, the normal, the human ES cells from each other.
Uncover the interactive patterns of the genes in these cells. The role of methylation in coordinating the activities of genes.

State of the art in Methylation Analysis
SVMs, ANNs have been successfully applied to predict the epigenetic events, for example,
Methylation status of CpG sites Computational prediction of methylation status in human genomic sequence, PNAS, Vol.
103(28), 2006.
CpG islands/ promoter regions in DNA sequence CpG island mapping by Epigenome prediction’, Plos Computational Biology, Volume 3(6), 2007. Promoter prediction analysis on the whole human genome’, Nature Biotechnology, Vol. 22, 2004.
Cancers Tumour class prediction and discovery by microarray-based DNA methylation analysis, NAR, Vol.
30, 2002. Co-regulation analysis through clustering
Clustering of methylation arrays Marjoram P, Chang J, Laird PW, Siegmund KD: Cluster analysis for DNA methylation profiles
having a detection threshold. BMC Bioinformatics Vol. 7, 2006.

2 Problems
1. Traditional methods, SVMs, ANNs are ‘black box’ models Knowledge extracted are characterized by the
connection weights, and Support Vectors. hard to understand for biologists
2. Investigate the co-methylation patterns Cancer cells human Embryonic stem cells (hESCs) Co-methylation analysis can help to uncover the h
idden pathways leading to new drug design

Methodogy
Two computational methods proposed1. Adaptive Cascade Sharing Trees (ACS4) fo
r problem 1 To learn the human understandable DNA methy
lation rules
2. Adaptive clustering for problem 2 To highlight the orchestration of genes for functi
on through the methylation mechanism

ACS4 method (1)
Promoters are regulatory elements upstream the 5’ end of TSS.
Methylation of promoter CpGs remodels the chromatin structure for gene expression
Methylated CpG methyl-binding proteins (MeCP)
methyltransferaseHistone deacetylases
(HDAC)

ACS4 method (2)
Methylation levels of promoters can be measured using Microarrays
Each spot on the array corresponds to a promoter CpG sites.
The methylation intensity is a numerical value between 0 and 1.

ACS4 method (3)
Objective: learn human understandable rules that define the epigenetic process in cancer and embryonic stem cells
Idea: Adaptively partition the numeric attributes into a
set of the linguistic domains, e.g., ‘high’, ‘very high’, ‘Medium’, ‘Low’, ‘Very Low’ .
Train a committee of trees to select the most salient features and predict through voting.

ACS4 method (4)

ACS4 method (5)

ACS4 method (6)

ACS4 method (7)
We have learned k rules Given a testing sample,
compute pi
Rules are weighted according to their Coverage, i.e., the number of matched samples
Overall prediction is made by voting across the rules.

ACS4 method (8) Dataset:
37 hESC, 33 non-hESC, 24 cancer cell lines, 9 normal cell lines. 1,536 attributes
Result Just 2 attributes are enough to separate the 3 cell types No need of 40 attributes by using fisher’s score in [1]. Wet lab cost can be reduced by testing on 2 attributes only, instead of 40. Accuracy is better, except when compared with SVM, but SVM cannot tell us ‘why’. Rules can be easily understood to biologist to conceive new biological experiments
seeking in wet lab proof.
[1] ‘Human embryonic stem cells have a unique epigenetic signature‘, Genome Research, Vol. 16, 2006

ACS4:Biological interpretation(1)
Example: IF PI3-504 is ‘High’ THEN hESC IF PI3-504 is ‘Low’ AND NPY-1009 is ‘Low’ T
HEN Normal IF PI3-504 is ‘Low’ AND NPY-1009 is ‘High’ T
HEN Cancer

ACS4:Biological interpretation(2)
The two marker genes
PI3(PI 3-kinases )-activate the cell growth, proliferation, differentation, motility, intracellular trafficking
Down-regulated in hESCs maintain stable state Keep from growth, proliferation, diff
erentiation…
Neuropeptide Y (NPY)- signal protein produced by nerves
[Immunology:Stress and Immunity, Science, Vol. 311, 2006.]
Experiment shows deficiency of NPY cause immune defects
Consistent to our computational result

ACS4: Biological interpretation(3)
Example: IF PI3-504 is ‘High’ THEN hESC
PI3 gene is silenced to maintain a stable cell context in hESCs IF PI3-504 is ‘Low’ AND NPY-1009 is ‘Low’ THEN Nor
mal Normal cells can grow, and grow safely with immune defenses
IF PI3-504 is ‘Low’ AND NPY-1009 is ‘High’ THEN Cancer
Cancer cells grow, and grow out of control, due to the immune deficiency

Adaptive clustering (1)
Co-methylation of genes are importantBecause we want to know how genes are c
o-working in the epigenetic frameworkClustering should reflect the true distribution
of the gene space.assuming data are normally distributed, which is
usually the case in real world applicationsFisher’s criterion is computed to validate the res
ult of clustering, and choose the best one.

Adaptive clustering (2)
For embryonic and cancer cells we optimally cluster the 1536 genes for each round of clustering with k-Means, we start from differ
ent # of initial centers. Candidate clustering result with the largest Fisher’s discrimi
nant score qualifies for further analysis. Each cluster of genes can be functionally related, and partici
pate in the same pathway of DNA methylation. By further analysis of the sequences, we can find out the feat
ure binding sites for each cluster of genes, and discover the epigenetic binding factors unknown before.
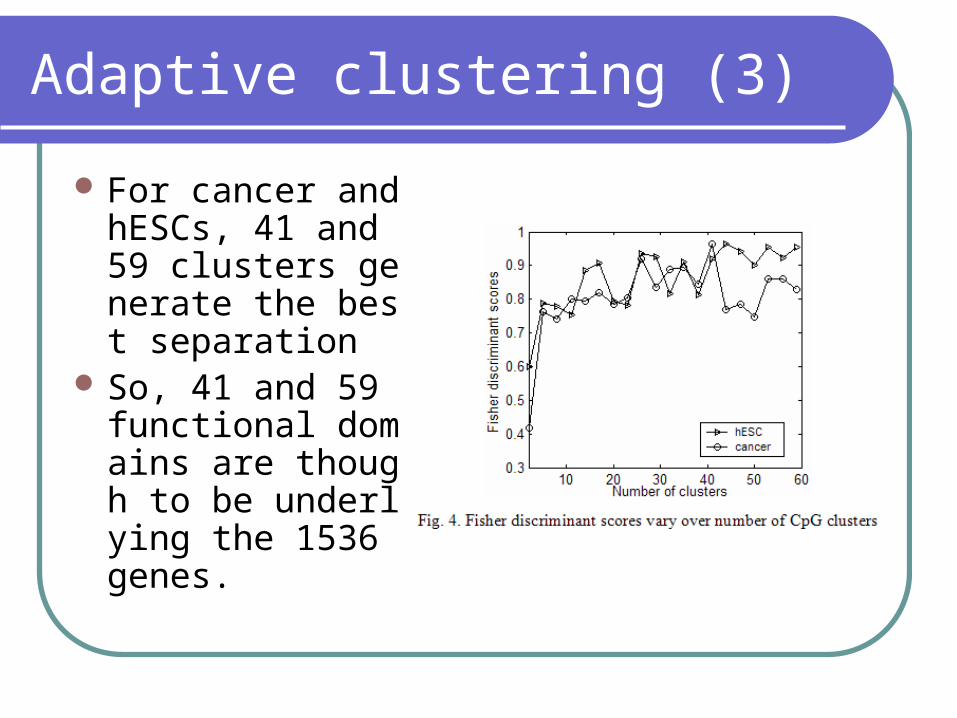
Adaptive clustering (3)
For cancer and hESCs, 41 and 59 clusters generate the best separation
So, 41 and 59 functional domains are though to be underlying the 1536 genes.

Adaptive clustering (4)
In experiments: The distance measure d is based on Pearson’s correlatio
n score. N = 60.

Adaptive clustering (5)
For hESC the formed clusters of the co-methylated genes, e.g., MAGEA1, STK23, EFNB1, MKN3, TMEFF2, AR, FMR1, are most related to differentiation, self-renewal, and migration of hESC activities.

Adaptive clustering (6)
For cancer cells, the formed clusters of the co-methylated genes, e.g., RASGRF1, MYC, and CFTR, are highly involved in cell apoptosis, DNA repair, tumour suppressing, and ion transportation, which are typically the immunological activities of cells against DNA damages.

Adaptive clustering (7)
Particularly, we discover: gene CFTR (7q31), long in focus in medical research, is co-
methylated with MT1A (16q13) and KCNK4 (11q13). CFTR defects contribute to the disease of Cystic Fibrosis (CF). One in twenty-two people of European descent carry one gene for
CF, making it the most common and lethal genetic disease of still no cure at the present time among such people.
The CFTR and KCNK4 proteins form the ion channels across cell membranes, while MT1A proteins bind with the ions as the transporters. They are all related to the transportation of ions across cell membrane, functionally related.
The can participate in the same pathway, the breakdown of which can explain the process of turmogenesis

Adaptive clustering (8)
Two summarize: Co-methylation occurs widely across the whole
genome It dominates the growth and development of
various types of cells Different cells exhibit different patterns of co-
methylation Our adaptive clustering algorithm can naturally
capture the group-wise activities in these cells.

Conclusion
Genome wide Epigenetic analysis: promising direction to research and industry
The logic of DNA methylation can be learned and interpreted by using our proposed ACS4 algorithm Just 2 attributes are good enough to separate the 3 cell types No need of 40 attributes by using fisher’s score in G.R. paper. Wet lab cost can be reduced by testing on just 2 attributes, instead of
40, lab cost is significantly reduced, more cost - effective. More accurate by adaptively partition the attribute domain Knowledge learned are human understandable, to assist biologist d
esign in wet lab test for further investigations Adaptive clustering
Epigenetic events are highly active in cancer and hESCs. Functionally related genes are co-methylated patterns of co-methylation are much different in cancer and hESCs,
highlighting the versatile roles of Epigenetic events in cell function.

Thanks!



















![Maternal Factors that Induce Epigenetic Changes Contribute ...Epigenetic pathways have transformed our understanding of molecular genetics [28]. The DNA CpG methylation blueprints](https://img.dokumen.tips/doc/110x75/5e8559e636e51d5ee66b7f83/maternal-factors-that-induce-epigenetic-changes-contribute-epigenetic-pathways.jpg)









